

Abstract
It is becoming increasingly clear that mechanical stress in adhesive junctions plays a significant role in dictating the fate of cell–cell attachment under physiological conditions. Targeted disruption of cell–cell junctions leads to multiple pathological conditions, among them the life-threatening autoimmune blistering disease pemphigus vulgaris (PV). The dissociation of cell–cell junctions by autoantibodies is the hallmark of PV, however, the detailed mechanisms that result in tissue destruction remain unclear. Thus far, research and therapy in PV have focused primarily on immune mechanisms upstream of autoantibody binding, while the biophysical aspects of the cell–cell dissociation process leading to acantholysis are less well studied. In work aimed at illuminating the cellular consequences of autoantibody attachment, it is reported that externally applied mechanical stress mitigates antibody-induced monolayer fragmentation and inhibits p38 MAPK phosphorylation activated by anti-Dsg3 antibody. Further, it is demonstrated that mechanical stress applied externally to cell monolayers enhances cell contractility via RhoA activation and promotes the strengthening of cortical actin, which ultimately mitigates antibody-induced cell–cell dissociation. The study elevates understanding of the mechanism of acantholysis in PV and shifts the paradigm of PV disease development from a focus solely on immune pathways to highlight the key role of physical transformations at the target cell.
Keywords: autoantibodies, cell–cell adhesion, desmosome, mechanical stress, pemphigus
1. Introduction
Pemphigus vulgaris (PV) is a potentially life-threatening autoimmune blistering disease that affects the skin and mucous membranes and serves as an excellent model for the study of human organ-specific autoimmunity.[1] PV is characterized by the presence of autoantibodies (autoAbs) directed against keratinocyte surface antigens,[2] primarily the desmosomal cadherins desmoglein 3 (Dsg3) and Dsg1.[3,4] Binding of these autoAbs leads to desmosome internalization, keratin retraction, cell dissociation, and ultimately blister formation.[5] Several signaling pathways induced by autoAb binding in PV have been identified, including protein kinase C (PKC),[6,7] p38 mitogen-activated protein kinase (MAPK),[8,9] and Rho GTPases family proteins such as RhoA.[10,11] Nevertheless, there remain large gaps in knowledge concerning the precise mechanisms through which autoAb-binding induces blister formation.[12] The bulk of research in PV to date has focused on immune mechanisms upstream of autoAb binding (i.e., antibody specificity, B-cell and T-cell functions). Consequently, therapeutic interventions in PV have mostly relied on nonspecific immunosuppression, which is often accompanied by detrimental long-term side effects.[13] However, the biophysical aspects of the cell behavior after autoAb binding are only recently being explored thanks to the advancement of innovative technologies applied to fundamental biological questions at the cellular level.[14–16] There is already compelling evidence that mechanical stress in adhesive junctions plays a significant role in dictating the fate of cell–cell attachment.[17,18] We posit that mechanical stress may thus govern the cell dissociation process in PV, and the detailed investigation of these processes will broaden our understanding of disease pathomechanisms at the target cell–cell adhesion sites.
Cell–cell adhesion is mechanically maintained by a concerted effort from both desmosomes and adherens junctions (AJs), which anchor intermediate filaments (IF) and actin filaments, respectively, at cell–cell contacts.[19] Downstream of autoAb binding, cellular responses and signaling pathways, mediated by p38 MAPK, PKC, RhoA and others, are known to have major effects on the AJ complex, cell–extracellular matrix (ECM) adhesion, and cytoskeleton remodeling,[20–23] thus impacting mechanical stress within cell–cell junctions. It has been suggested mechanical stress may, in turn, regulate PV pathogenesis either by direct physical intervention through pulling or pushing cell-cell attachments and/or indirectly by modulating signaling pathways.[24,25] Specifically, disruption of desmosomes by anti-Dsg3 antibodies (Abs) leads to an upregulation of myosin II, a potent cell contractility promoter and actin stress enhancer, and the elevated stress within the actin filaments increases tissue fragility.[24] We and others have shown that removing the desmosome/IF linkage reduces cell–cell junction force and relaxes cell contractility, while strengthening this linkage increases junction forces,[26] and can ameliorate PV autoAb-mediated loss of cell–cell adhesion.[25]
Therefore, it is evident that mechanical stress plays a regulatory role in orchestrating the effector molecules downstream of PV-associated Ab binding and may thus determine keratinocyte fate in PV. However, to date, the role of mechanical stress in PV pathomechanisms has not been directly explored. Leveraging nanoscale technologies to investigate biological processes, we previously correlated ultrastructural changes at cell–cell adhesion structures with mechanical responses following desmosome disruption.[15,16] Furthermore, we have shown that desmosome disruption modulates cell adhesion strength and cell contractility, and elevates mechanical stress levels within AJs.[26] Based on these findings, we postulate that cell mechanical stress is altered after PV-associated Ab-induced structural changes at the desmosome, invoking mechanotransduction at the AJs in an attempt to stabilize the cell–cell adhesion. However, under disease conditions, the AJ-driven compensatory mechanical stress response is insufficient to overcome cell–cell contact destabilization and complete cell dissociation ensues. We thus hypothesized that the supply of external mechanical stress to keratinocytes at critical time points after anti-Dsg3 autoAb attachment can interfere with cell signaling pathways involved in promoting the disruption of cell–cell contacts to stabilize the compromised cell–cell adhesion. We report here a protective mechanism against anti-Dsg3 Ab-induced acantholysis through the enhancement of the cellular contractility at the actin filament networks. Specifically, we show that externally applied cyclic stretch can suppress anti-Dsg3 Ab-activated signaling molecule expression, enhance the cortical actin network, stabilize the E-cadherin (E-cad) based AJs and ultimately mitigate the anti-Dsg3 Ab-induced cell–cell dissociation, potentially through the activation of the RhoA pathway. We believe that this response mechanism to mechanical stress may act in a similar fashion as in normal homeostasis to promote cell proliferation and wound healing. Our data reinforce the key regulatory role of sequential mechanical stress variations in the maintenance and disruption of cell–cell adhesion that continues to shift the paradigm of PV disease development from a focus solely on immune pathways to a deeper understanding of the key role in cytomechanics in pathology at the target cell level.
2. Results
2.1. Mechanical Stretch Protects Cell Monolayers from Anti-Dsg3 Ab-Induced Cell Dissociation
The conventional dispase-based keratinocyte dissociation assay, in which cell monolayers are exposed to shear stress by pipetting to reveal weaknesses in or loss of intracellular adhesion, was performed to examine the effect of external mechanical stress on induced cell dissociation by AK23, a mouse anti-human Dsg3 antibody that is sufficient to disrupt intercellular adhesion in human keratinocytes and has been widely used in studies on the downstream effect of antibody binding in Pemphigus (Figure 1a).[27,28] We selected a cyclic stretch regimen with 10% strain and 1 Hz frequency on the Flexcell 5000 system based on its effects in promoting cell proliferation and wound healing in keratinocytes.[29,30] Monolayers of the HaCaT cell line grown under control conditions (medium only) maintained relative integrity, breaking only to around 20 particles. However, with 2 μg mL−1 and 10 μg mL−1 anti-Dsg 3 pathogenic monoclonal antibody (AK23 mAb) treatment for 4 h, the monolayer was fragmented into significantly more particles (in the range of 80 to 100), around 4.5 fold more than controls. Importantly, cyclic stretch loading at 10% strain and 1 Hz frequency applied to the HaCaT cell monolayer in the presence of AK23 mAb inhibits cell dissociation induced this Ab. Specifically, compared with AK23 mAb treated groups, a significant reduction (over 60%) in the number of cell sheet fragments was found in cyclic stretch groups for both concentrations (AK23(10 μg mL−1)+CS and AK23(2 μg mL−1)+CS) (Figure 1b). This result provides direct evidence that external mechanical stress may counter the detrimental effects in cell monolayers induced by anti-Dsg3 Abs. We next sought to examine if this protective effect is facilitated by alterations in key PV pathways.
Figure 1.
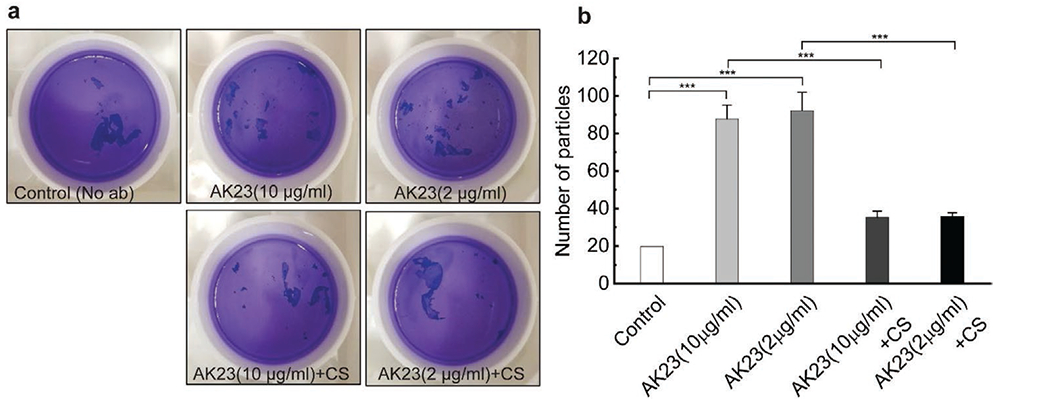
Dispase-based dissociation assay shows reduced cell fragmentation after cyclic mechanical stretch with the presence of AK23 mAb. HaCaT cells were exposed to two concentrations of AK23 mAb with and without 10% 1 Hz cyclic stretch for 4 h. Released cell sheets were subsequently pipetted to induce rupture, and the number of particles was determined to evaluate cell–cell adhesive strength. a) Dissociation assay images of HaCaT monolayers after culture in regular medium (Control), or treatment with 10 μg mL−1 AK23 mAb (AK 23(10 μg mL−1)), 2 μg mL−1 AK23 mAb (AK23(2 μg mL−1)), 10 μg mL−1 AK23 mAb and 10% 1 Hz cyclic stretch (AK23(10 μg mL−1) + CS), and 2 μg mL−1 AK23 mAb and 10% 1 Hz cyclic stretch (AK23(2 μg mL−1) + CS); b) Quantitative result of dissociation assay images. All values are mean ± SD (n = 4). ***p < 0.005.
2.2. Mechanical Stretch Suppresses Anti-Dsg3 Ab-Activated p38 Pathways
Based on the inhibition of anti-Dsg3 Ab-induced cell dissociation observed after application of external mechanical stretch, the modulation effect of external mechanical stress on PV-associated pathways was further examined. The expression level of p38 MAPK, a key molecule in PV-induced pathology, was quantified by Western blot in monolayers of HaCaT cells subjected to AK23 mAb treatment with and without mechanical stretch. A significant induction of phosphorylation of p38 (p-p38) MAPK was observed after a 4 h AK23 mAb treatment compared to untreated controls, while the total p38 MAPK remains unchanged (Figure 2). The ratio of p-p38 MAPK to p38 MAPK from AK23 mAb-treated groups increased 0.5 fold (10 μg mL−1) and 1.5 fold (2 μg mL−1) compared to the control, consistent with previous research.[31] Importantly, the AK23 mAb-induced activation of p-p38 MAPK was suppressed by cyclic stretch (AK23+CS in Figure 2a,b). The reduction of p-p38/p38 ratio for 10 and 2 μg mL−1 AK23 mAb with cyclic stretch groups was nearly 478% and 53.3%, respectively, suggesting that external mechanical stress has a modulating effect on p38 MAPK signaling, at least at the 4 h time point. The results demonstrate that activation of p38 MAPK by anti-Dsg3 Abs can be suppressed by the application of mechanical stress and suggest that this may alter downstream pathways and lead to the reversed course in cell–cell adhesion loss.
Figure 2.
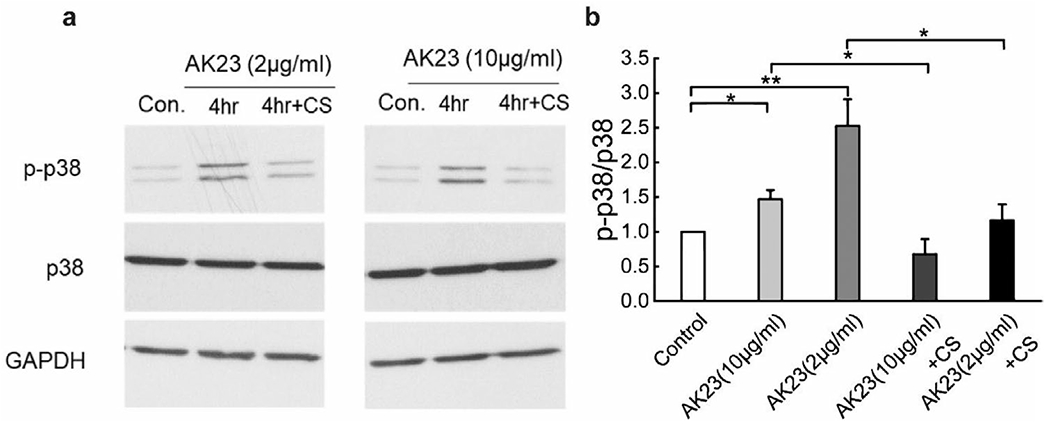
Cyclic mechanical stretch suppresses anti-Dsg3 Ab-induced p38 phosphorylation. Expression of p38 and p-p38 were quantified with Western blot in cells exposed to AK23 mAb with and without cyclic stretch (10% strain, 1 Hz). a) Western blot results of p38 and p-p38 with different treatments: control, 10 μg mL−1 AK23 mAb (AK23(10 μg mL−1)), 2 μg mL−1 AK23 mAb (AK23(2 μg mL−1)), 10 μg mL−1 AK23 mAb with cyclic stretch (AK23(10 μg mL−1) + CS) and 2 μg mL−1 AK23 mAb with cyclic stretch (AK23(2 μg mL−1) + CS). GAPDH is shown as a loading control. b) The ratio of band intensity, p-p38/p38, as compared with the control set at 1. All values are mean ± SEM (n = 5). *p < 0.05, **p < 0.01.
2.3. Mechanical Stretch Modulates Desmosomes and AJs
It is apparent that the application of mechanical stretch can reverse some internal biochemical changes initiated by exposure to anti-Dsg3 Abs. To further understand the interplay between these PV-associated Abs and mechanical stretch, we investigated several cell structural and adhesion proteins with relevance to both the desmosome as well as the AJ complex. We utilized immunofluorescence imaging to evaluate changes in protein distribution and structure in addition to quantifying overall levels with Western blotting. The Flexcell 5000 system was used for the cell dissociation assays and to quantify protein expressions by Western blot. However, it is not suitable for microscopic imaging. Therefore, a new stretching system with molded PDMS stretching chambers was built and used for fluorescent imaging examination (see Experimental Section). It is worth mentioning that each system applies uniaxial stretch with the same strain and strain rate to the cell monolayer. In addition to cyclic stretch (CS, 10% strain, 1 Hz, 4 h), a static stretch condition (SS, 10% strain, 4 h) was added to the study to compare differences between time-varying and constant stretch to determine which conditions have the most significant impact. As we had observed no significant difference between treatments with 2 or 10 μg mL−1 AK23 mAb in the keratinocyte dissociation assay, a concentration of 2 μg mL−1 was used for subsequent studies.
The staining intensity of Dsg3 under cyclic stretch increased significantly compared with unstretched controls, which indicates that mechanical stress may stimulate cells to strengthen desmosomal junctions (Figure 3). In addition, longer strand fibrous networks were observed in CS groups than in controls (Figure 3b IF). Interestingly, in static stretch groups the fluorescence intensity of Dsg3 was reduced (Figure 3c Dsg3). We also observed a breakdown of long strand IFs with static stretch (Figure 3c IF), suggesting a possible detrimental effect of long-term exposure to sustained strain. The introduction of the anti-Dsg3 Ab altered the distribution of Dsg3 protein (Figure 3d Dsg3, 3h) and rearranged the IF structures (Figure 3d IF). An intensity increase in Dsg3 was found in AK23 groups (Figure 3g), but Dsg3 molecules were less concentrated at sites of cell–cell contact, suggesting an internalization of Dsg3 from the cell membrane to the cytoplasm. It is worth mentioning that the distribution of Dsg3 molecules were quantified by recording the intensity value along a straight line drawn across the cell–cell border (Figure S1, Supporting Information). The peak intensity value on the border was then normalized against the baseline value in the cytoplasm. This normalized value was plotted as relative intensity and is the same for E-cad and RhoA in the following sections. As for IFs, distinct changes occurred in their structure. Shorter strands of IFs were observed with AK23 mAb treatment as compared with IF fibers continuously stretching across entire cells in controls (Figure 3d IF), suggesting IF fibers depolymerized and retracted from cell–cell contacts in response to anti-Dsg3 Ab exposure.
Figure 3.
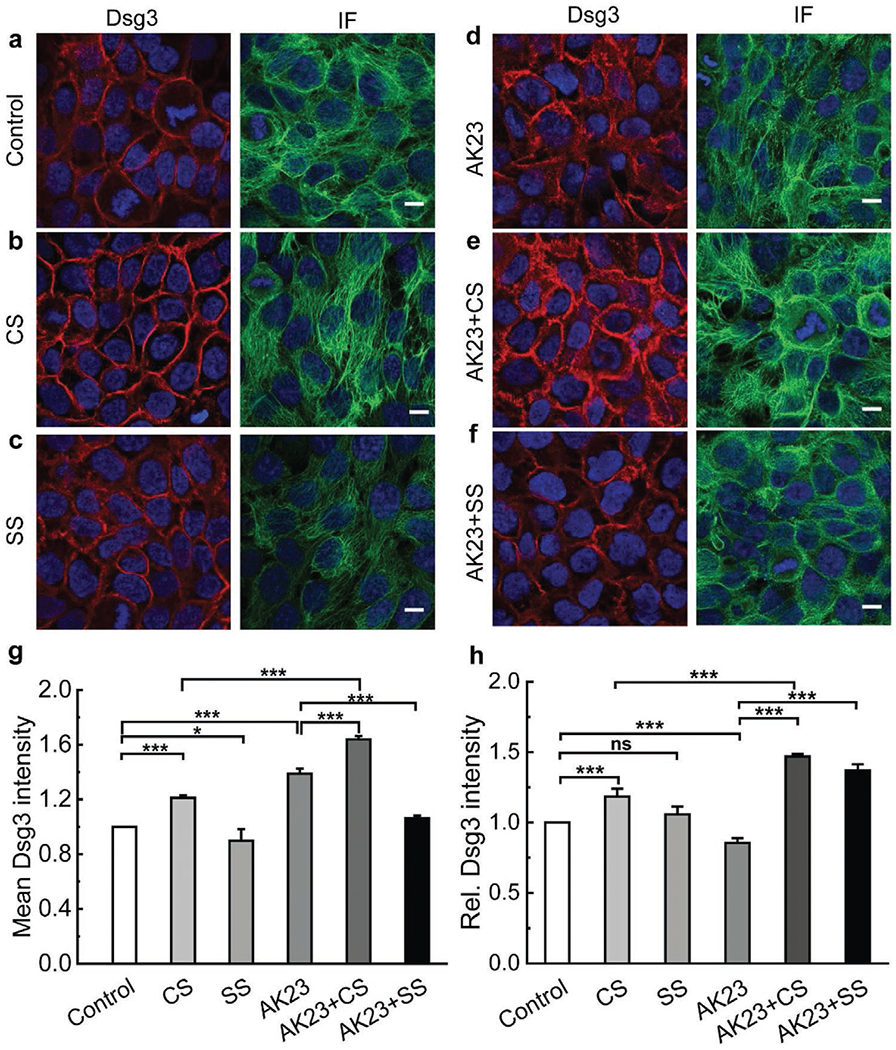
Mechanical stretch protects desmosomes from anti-Dsg3 Ab-induced instability. Confluent keratinocytes were subjected to combinations of AK23 mAb treatment with and without cyclic and static stretch. Cells were then stained and imaged to investigate how expression and distribution of selected proteins related to desmosomes (Dsg3, IF) changed with different treatments. The studied conditions were a) untreated control; b) cyclic stretch (10% strain, 1 Hz); c) static stretch (10% strain); d) AK23 mAb (2 μg mL−1); e) AK23 mAb (2 μg mL−1) with cyclic stretch; and f) AK23 mAb (2 μg mL−1) with static stretch; g) Average intensity of Dsg3 based on five images for each test condition over five separate experiments; all values are mean ± SEM (n = 5); h) A total of 10 lines were drawn across the cell–cell contact on each fluorescence images of Dsg3 under control, CS, SS, AK23+CS, and AK23+SS, and the peak intensity of each line normalized against the baseline intensity at the cytoplasm from each group were plotted as relative intensity (Rel. Dsg3 Intensity). Images are representative of more than five experiments for each condition; all values are normalized against control (control set to 1). all values are mean ± SEM (n = 10). * p < 0.05, **p < 0.01, ***p < 0.005, ns p > 0.05. Scale bar: 10 μm.
To examine the inhibitory effect of mechanical stress on changes induced by anti-Dsg3 Abs, cells were stretched either cyclically or statically in the presence of AK23 mAb. Results from AK23+CS groups show that cyclic stretch significantly increased the intensity of Dsg3 at the cell–cell contact (Figure 3e Dsg3, 3g,h), indicating that Dsg3 was redistributed to the junction to stabilize cell–cell adhesion. In addition, IF fibers were long and continuous, similar to controls (Figure 3e, IF). These results show that cyclic stretch mitigates the desmosome instability induced by anti-Dsg3 Ab. For the AK23+SS group, Dsg3 was found to cluster more at the cell periphery when compared to AK23 mAb treatment alone, but to a lesser degree than the AK23+CS group (Figure 3f Dsg3, 3h). In addition, there was some extent of restoration of the fibrous IF in the AK23+SS group compared with AK23-treated cells (Figure 3f IF), but to a significantly lesser degree compared with AK23+CS, suggesting that static stretch does not stabilize the cell junction at levels seen with cyclic stretch.
Similar to desmosomes, AJs are also strengthened under cyclic stretch, evident from a significant increase in E-cad intensity at the cell–cell border when compared to unstretched controls (Figure 4a,b E-cad, 4g,h, and Figure S2 in the Supporting Information). E-cad increased and clustered at the cell boundary under static stretch, but to a lesser degree than under cyclic stretch (Figure 4c E-cad, 4g,h). The same conclusion was also drawn from the comparison between AK23+CS and AK23+SS groups. Compared with controls, E-cad intensity increased under AK23 mAb treatment in a potential attempt to compensate desmosome destabilization (Figure 4d E-cad, 4g). However, compared to AK23 groups, the introduction of cyclic stretch elevated the E-cad intensity and promoted its clustering at the cell–cell contact (Figure 4e E-cad, 4g,h). Again, these findings indicate the inhibitory effect of cyclic stretch on anti-Dsg3 Ab-induced cell–cell adhesion destabilization. On the other hand, the benefit from static stretch to counter this destabilization is limited with no enhancement of E-cad at the cell–cell contact (Figure 4f E-cad, 4g,h).
Figure 4.
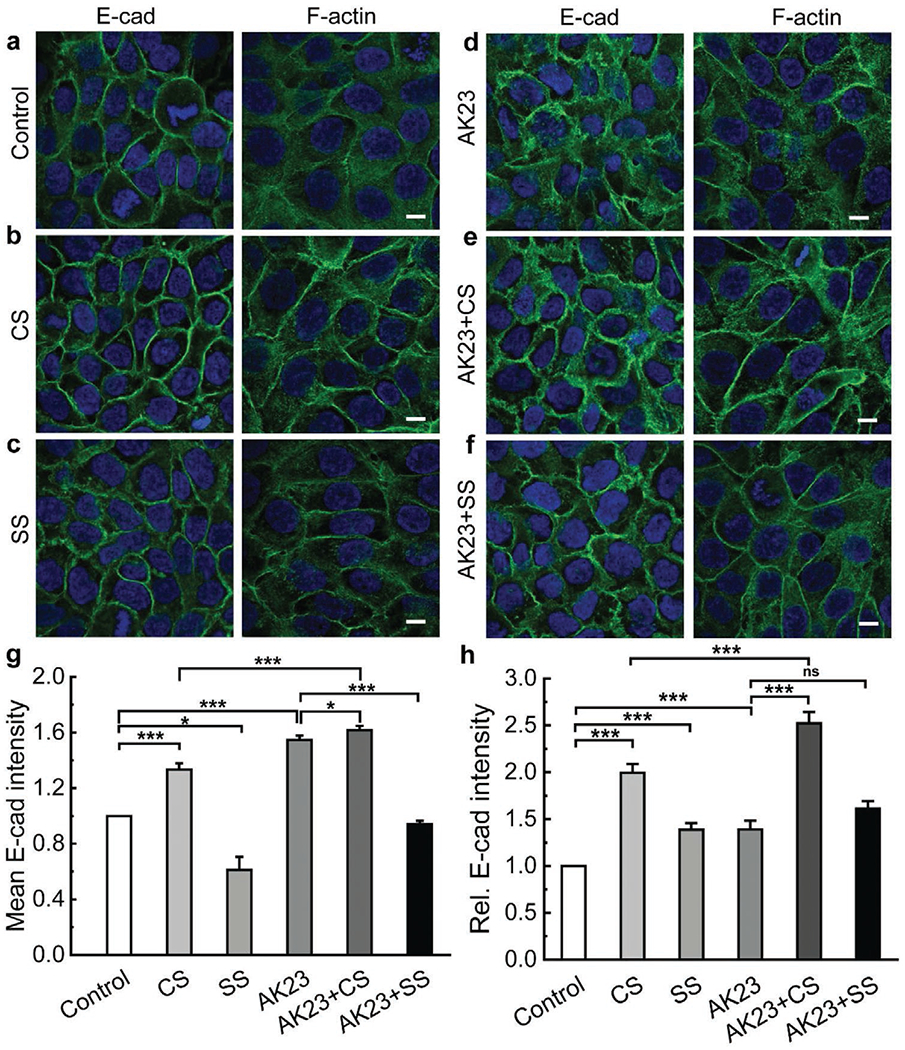
Mechanical stretch protects AJs from anti-Dsg3 Ab-induced instability. Confluent keratinocytes were subjected to combinations of AK23 mAb treatment with and without cyclic and static stretch. Cells were then stained and imaged to investigate how expression and distribution of selected proteins related to AJs (E-cad, F-actin) changed with different treatments. The studied conditions were a) untreated control; b) cyclic stretch (10% strain, 1 Hz); c) static stretch (10% strain); d) AK23 mAb (2 μg mL−1); e) AK23 mAb (2 μg mL−1) with cyclic stretch; and f) AK23 mAb (2 μg mL−1) with static stretch; g) Average intensity of E-cad based on five images for each test condition over five separate experiments; all values are mean ± SEM (n = 5); h) A total of 10 lines were drawn across the cell-cell contact on each fluorescence images of E-cad under control, CS, SS, AK23+CS, and AK23+SS, and the peak intensity of each line normalized against the baseline intensity at the cytoplasm from each group were plotted as relative intensity (Rel. E-cad Intensity); all values are mean ± SEM (n = 10). Images are representative of over five experiments for each condition; all values are normalized against control (control set to 1). *p < 0.05, **p < 0.01, ***p < 0.005, ns p > 0.05. Scale bar: 10 μm.
Cyclic stretch, not static stretch, enhanced F-actin with and without the presence of AK23 mAb (Figure 4a–f, F-actin, quantitative data in Figure S5 in the Supporting Information) compared with control groups. Importantly, the changes in F-actin is more pronounced in terms of its structures and distributions. When treated with AK23 mAb, F-actin was also observed to remodel. Specifically, cell–cell boundaries became less well-defined and blurry, while the distribution of F-actin became dispersed throughout the cytoplasm with a clear tendency towards uniformity (Figure 4d, F-actin). However, this uniform distribution was altered by mechanical stretch, and a return to well-defined boundaries and a clustering of F-actin at the cell–cell contact were promoted by both cyclic and static stretch (Figure 4e,f, F-actin). These results show that mechanical stretch induces F-actin reorganization and maintains cell–cell adhesion stability, even in the presence of instability invoked by anti-Dsg3 Ab.
2.4. Mechanical Stress Stimulates AJs via RhoA Activation
The RhoA pathway has been shown to be triggered from the onset of strain in AJs.[32] Thus, in order to examine the effect of external stress on the RhoA pathway, we subjected cell monolayers to cyclic and static stretch and analyzed RhoA expression by Western blot and immunofluorescence imaging. Both stretch regimens resulted in an elevated level of RhoA as compared to controls (Figure 5a). Quantitative data show that the relative expression of RhoA in cell monolayers increased by about 40% for both mechanical stretch conditions (Figure 5c), suggesting a potential activation of the RhoA pathway under mechanical stress.
Figure 5.
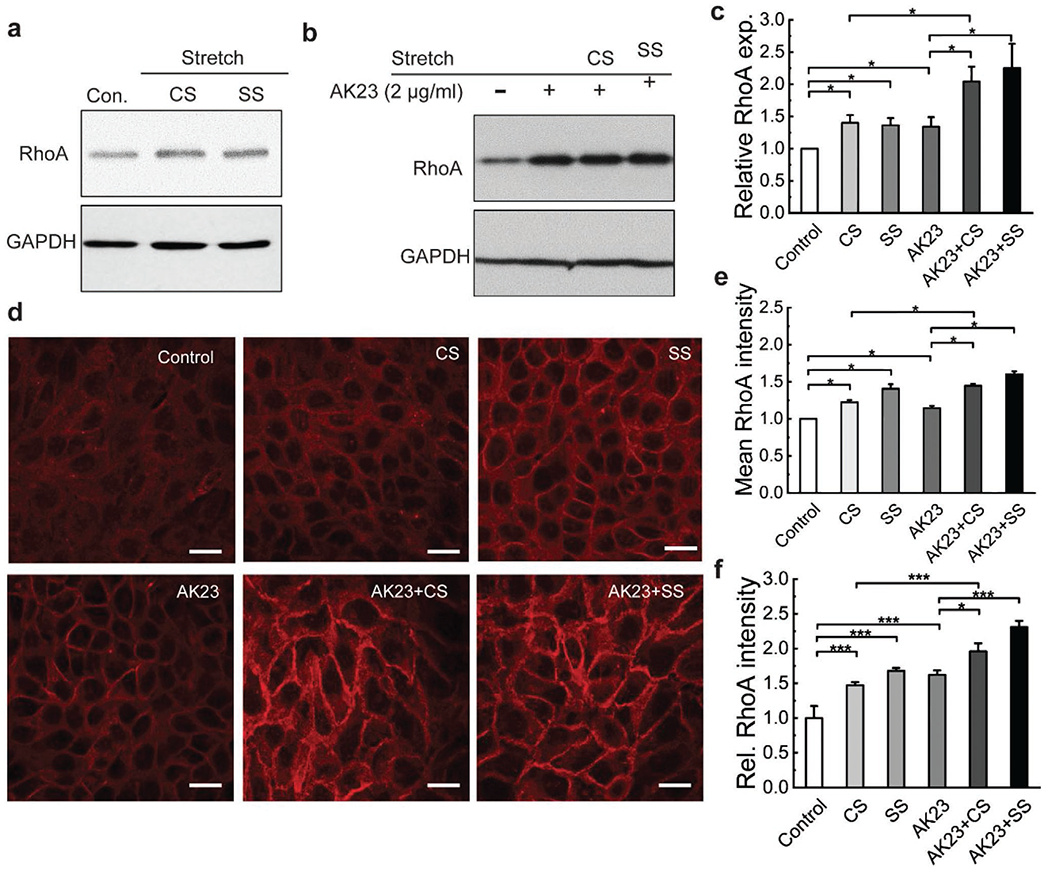
Mechanical stretch increases RhoA expression and localization at cell–cell adhesion without and with AK23 mAb treatment. Expression of RhoA was investigated with western blot and immunofluorescence imaging after exposure to both cyclic stretch (10% strain, 1 Hz) and static stretch (10% strain) to determine if mechanical stretch influences the RhoA pathway. a) Western blot analysis of RhoA, comparing expression in control to cells treated with cyclic and static stretch; GAPDH is shown as a loading control. Images are representative of over three experiments for each condition; b) Western blot results comparing RhoA expression of control to treatment with AK23 mAb and AK23 mAb with CS and SS. GAPDH is shown as a loading control. c) Quantitative Western blot data from (a) and (b), with all values are normalized against control (control set to 1). All values are mean ± SEM (n = 3). d) Fluorescent images of RhoA expression under corresponding test conditions: no anti-Dsg3 Ab and mechanical stretch (Control), cyclic stretch (CS), static stretch (SS), AK23 mAb (AK23), AK23 mAb and cyclic stretch (AK23+CS), AK23 mAb and static stretch (AK23+SS); Images are representative of over five experiments for each condition; e) Average cell intensity of based on five images for each test condition over five separate experiments; all values are mean ± SEM (n = 5); f) A total of 30 lines were drawn across the cell-cell contact on each fluorescence images under each condition, and the peak intensity of each line normalized against the baseline intensity at the cytoplasm from each group were plotted as relative intensity (Rel. RhoA Intensity); all values are mean ± SEM (n = 10). *p < 0.05, **p < 0.01, ***p < 0.005. Scale bar: 20 μm.
To further study the activation of the RhoA pathway under external stress, monolayers subjected to each stretch regimen were stained for RhoA and imaged by confocal microscopy (Figure 5d). The fluorescent intensities of RhoA in CS and SS groups were qualitatively observed to be higher than that of the unstretched control, which was confirmed through a quantitative analysis of average RhoA immunofluorescence intensity (Figure 5e). In addition, the average intensity of the SS group was slightly higher than the CS group, indicating that static stretch triggers a stronger response from the RhoA pathway. The distribution of RhoA was also altered by mechanical stretch. Specifically, the fluorescent signal in control groups was uniform, while the fluorescence intensity peaks at the cell-cell border in CS groups, and even more evidently in SS groups (Figure S3, Supporting Information). A statistical analysis also shows that both stretch regimens trigger concentration of RhoA at the cell–cell contact with static stretch to a larger extent than cyclic stretch (Figure 5f).
The RhoA pathway was also examined in the presence of AK23 mAb when cells were subjected to mechanical stretch (Figure 5). Compared with the control, RhoA expression in AK23 treated groups increased, but to a significantly lesser extent than when cells were also stretched (AK23+CS, AK23+SS) (Figure 5b,c). The confocal images show that RhoA fluorescence intensity increased with the treatment of anti-Dsg3 Ab (Figure 5d), which agrees well with the Western blot results. Significant increase in RhoA levels can be found in AK23+CS and AK23+SS groups (Figure 5e), indicating that mechanical stress promotes cytosolic RhoA expression in cell monolayers. Importantly, RhoA redistribution can also be found in AK23+CS and AK23+SS groups in a similar fashion as in mechanical stretch alone (Figure 5f). This indicates that RhoA redistributes after mechanical stretch and there is increased RhoA clustering at cell–cell contacts. It is worth mentioning that since we control cell density in our experiments, the appearance of difference in cell culture is likely attributable to the rearrangement of the cells along the axial direction of the mechanical stretching.
2.5. Anti-Dsg3-Induced Keratinocyte Dissociation Mitigated by RhoA Activation
To examine the extent to which activation of the RhoA pathway is critical in the inhibitory effects observed from mechanical stretch, Rho Activator I (CN01) was added as a treatment condition to directly stimulate the RhoA pathway. The keratinocyte dissociation assay showed that AK23-induced increase in HaCaT monolayer fragmentation can be significantly mitigated by CN01 (Figure S6, Supporting Information), suggesting that the RhoA activation can partly protect keratinocytes from the effect of AK23. Importantly, the expression and distribution of Dsg3, E-cad, and RhoA were compared between control samples, AK23 mAb treated samples, and AK23+CN01-treated samples. In control samples, Dsg3 can be seen to localize tightly at cell-cell borders (Figure 6a Control). Dsg3 was less tight at the cell–cell border with AK23 mAb treatment, indicating Dsg internalization (arrow in Figure 6a AK23, 6g, and Figure S4 in the Supporting Information), as reported in other studies.[33] With the addition of CN01, however, some of these negative effects were reversed, with Dsg3 relocating more tightly to the cell–cell border (arrow in Figure 6a AK23+CN01, Figure 6g). These observations match closely to those from the application of mechanical stretch, especially cyclic stretch. Treatment with CN01 also impacts the expression of E-cad. Compared with control samples, monolayers exposed to AK23 mAb experienced an increase in the expression of E-cad (arrow in Figure 6b AK23, Figure 6e), possibly in an attempt to compensate for loss in desmosomal cell–cell adhesion. The addition of CN01 further increased the expression of E-cad at cell borders, possibly as a response from pathways triggered by CN01 (Figure 6h). As expected, the overall expression of total RhoA also increased when AK23 mAb was added to cells, and further increased with the addition of CN01 (Figure 6c,f,i). Interestingly, Ak23 treatment (2 μg mL−1 for 4 h) seemed to lead to a redistribution of active RhoA (GTP-bound Rho) away from the cell border, and was restored by the addition of CN01, which elevates the active RhoA localization at the cell–cell contact (Figure S7, Supporting Information). These data suggest that AK23 action can also be mitigated by interfering with active RhoA distribution.
Figure 6.
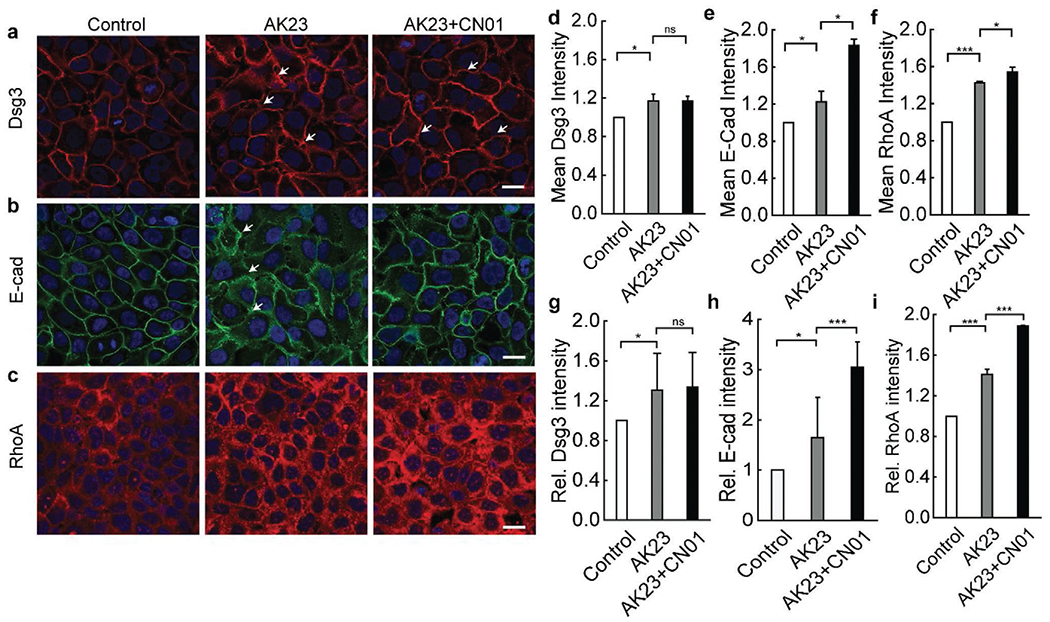
Activation of RhoA pathway with CN01 triggers changes in cell adhesion and structure proteins similar to mechanical stretch. Cells were studied under the influence of AK23 mAb with and without RhoA pathway activation via CN01 using immunofluorescence imaging. a) Dsg3 localization to cell-cell junctions triggered by AK23 mAb is increased with exposure to CN01; b) E-cad expression and localization to cell-cell junctions is increased with addition of CN01; c) RhoA expression is increased with exposure to AK23 mAb, and further increased with addition of CN01, while localization largely remains unchanged; Images are representative of over five experiments for each condition; d–f) Average intensity of Dsg3, E-cad, and RhoA, respectively, based on five images for each test condition over five separate experiments; all values are mean ± SEM (n = 5); g–i) A total of 30 lines were drawn across the cell-cell contact on each fluorescence images of Dsg3, E-cad, and RhoA, respectively, under control, AK23, and AK23+CN01 conditions, and the peak intensity of each line normalized against the baseline intensity at the cytoplasm from each group were plotted as relative intensity (Rel. Dsg3, E-cad, and RhoA Intensity); all values are mean ± SEM (n = 10). *p < 0.05, ***p < 0.005, ns: p > 0.05. Scale bar: 20 μm.
3. Discussion
Mechanical stretch has been shown to induce a spectrum of physiological transformations in many cell types, and activate intracellular biochemical signals and alter gene expression.[34] The most widely reported effects are stress fiber reorganization in endothelial cells[35] and in muscles cells[36] through Rho-dependent stress fiber assembly from myosin light chain phosphorylation.[37,38] The cytoskeleton reorganization represents an important mechanism for cells to adapt to an increased mechanical stress and for preparation for cell proliferation.[39] In human skin, it has been shown that elevated mechanical stress may lead to epidermal development[40] and hyperproliferation.[41] In epidermal keratinocytes, mechanical stretch is reported to promote cell proliferation via EGFR and ERK1/2[29] phosphorylation.[41] Elevated mechanical stress may also alter cytokine production in keratinocytes in the process to stimulate cell proliferation.[42] Our study shows that mechanical stretch-induced cellular physiological changes can provide a protective mechanism for cell–cell adhesion to maintain stability against autoAb-induced desmosome disassembly. We believe that this mechanism is facilitated by the same set of pathways that prepare keratinocytes for proliferation[41,42] and for wound healing.[30] Placed in this context, the seemingly counterintuitive finding of the protective effect of externally applied mechanical stress in a disease state may represent the consequences of enhancing normal homeostatic mechanisms that are operative under physiological conditions.
Specifically, we showed that HaCaT cell monolayers are significantly less fragmented after being subjected to cyclic stretch when treated with anti-Dsg3 Abs in conventional dispase based dissociation assays, a clear indication that periodic mechanical stress exposure leads to cell–cell adhesion stability. This protective effect is further underscored by the fact that cyclic mechanical stretch suppresses anti-Dsg3 Ab-induced p38 phosphorylation. As one of the key molecules in cell–cell dissociation in the context of PV, p38 activation also leads to phosphorylation, destabilization, and subsequent retraction of keratin IFs from the cell–cell border[43,44] and regulates the reorganization of actin filaments, which contributes to the cell–cell dissociation process in PV.[45] Thus, abrogation of the phosphorylation of p38 MAPK by external mechanical stress lends support to the notion that external mechanical stress fundamentally alters PV-associated pathways and thus changes the pathological outcome.[8] It is worth mentioning that the quantification of p38 was conducted at the 4 h time point, and there are reports showing that p38 expression may exhibit a two-peak fluctuation at 4 and 24 h time points.[31] Thus, the sequence and timing of mechanical stress alterations under disease and experimental conditions is critical in determining cellular outcomes.
Central to the mechanotransduction response of cells to mechanical stretch and PV pathology is the RhoA pathway, which is triggered from increased tension within AJs.[32] RhoA has been suggested to play a crucial role in the AJ-desmosome crosstalk[21] and is among the pathways activated by p38 MAPK. Our results show that the expression of RhoA was upregulated by both cyclic and static stretch (and by CN01 activation), with increased expression at cell–cell borders. Similarly, exposure to an anti-Dsg3 Ab results in increased RhoA expression and localization to cell–cell borders, but to a lesser extent. This increase contrasts with the RhoA reduction observed 24 h post PV IgG incubation in other studies,[46] and may be due to the transient compensating effect from AJs with the compromised desmosome.[26] When mechanical stretch is applied to cells treated with the anti-Dsg3 Ab, the expression of RhoA is further increased, suggesting that the RhoA pathway can be enhanced through mechanical stretching without rupturing cell–cell adhesions, similar to stretch-induced cellular responses in other cells.[35,36] RhoA activation is closely related to the remodeling of actin filaments, a process crucial to cell–cell detachment. For instance, activation of RhoA blocks the retraction of keratin IFs as well as cell adhesion loss in PV,[11] while inhibition of RhoA leads to blister formation.[10] It is suspected that the protective effect of RhoA in PV pathogenesis may stem from its role in regulation of the anchorage of desmosomal proteins to the IF cytoskeleton and the remodeling of actin filaments.[45]
The protective effect of mechanical stretch is also exhibited through cytoskeleton reorganization. Keratinocytes subjected to cyclic stretch show upregulations in Dsg3, IF, E-Cad, and F-actin expression. The depolymerization of IF fibrous networks and cortical F-actin are typical cytoskeletal modifications in PV.[45,47] Whereas the reappearance of continuous long strand fibrous networks and upregulation of F-actin, accompanied with the increase and redistribution to the cell–cell contact of Dsg3 and E-cad, in cells subjected to cyclic mechanical stretch with the presence of anti-Dsg3 Ab demonstrate an evident protective effect of the applied external stress. Compared with cyclic stretch (periodic strain), we find that static stretch (sustained strain) is less effective in maintaining cell–cell adhesion stability and cytoskeleton reorganization in cells under conditions of anti-Dsg3 Ab treatment. Dsg3 and E-cad show negligible changes under static stretch. Interestingly though, static stretch can elicit elevated RhoA signals and promotes F-actin expression as well as distribution to the cell–cell contact, in agreement with previous reports.[48] The difference in the protective effect of cyclic stretch and static stretch may stem from the overall magnitude of the applied strain. Cyclic strain allows more time for cells to adapt via signaling molecule-facilitated cytoskeleton reorganization, while a sustained high strain level of static stretch may simply cause permanent damage to the cell–cell adhesion and fail to elicit a proper cellular response. In addition, it has been shown recently that acute mechanical stress (equiaxial strain, 10% for 10 min) activates RhoA at the AJ and promotes E-cadherin enhancement at the cell–cell junction.[49] Whether the mitigation effects we observed here with cyclic mechanical stretch, particularly the E-cadherin increase at the cell–cell contact, is RhoA dependent will need to be explored further. This requires a systematic study with varied strain levels as well as frequencies (for cyclic stretch) in the context of RhoA inhibition.[35]
In summary, our work underscores the key role of mechanical stress at the tissue level in autoAb-mediated diseases, potentially shifting conventional paradigms regarding pathomechanisms in PV. Specifically, we undertook a series of investigations to describe the novel finding that modulation of mechanical stress can mitigate anti-Dsg3 Ab-induced cell–cell dissociation. Our work reveals mechanotransduction pathways relevant to cell–cell adhesion as potential therapeutic targets, particularly those coordinated through RhoA-activated actomyosin contractility. We propose that the mechanism of the mitigating effect of mechanical stretch on PV pathology is as follows: The introduction of anti-Dsg3 Ab causes initial desmosomal instability in part through phosphorylation of p38 MAPK and its activation of downstream effectors. As desmosomes are structurally altered, this leads to reduced desmosomal resistance and decreased tension in AJs.[26] This is followed by a reactive temporary RhoA upregulation and transient increased tension in AJs as a compensatory response in an attempt to maintain cell–cell contact. However, RhoA is not upregulated to the degree needed to entirely combat the initial autoAb assault on desmosomes, and the increased tension in AJs ultimately triggers a broader stress response in keratinocytes with the upregulation of Rac1, EGFR, and Src.[12] This leads to further phosphorylation of p38 MAPK, and the cycle repeats itself until complete acantholysis. When cyclic mechanical stress is applied, the RhoA pathway is activated to a level sufficient enough to further elevate tension in AJs and to facilitate the formation of new AJs, which inhibits PV pathology by offsetting desmosome instability and interrupting the vicious cycle. This mechanism is orchestrated by sequential changes in mechanical stress at the cell–cell junction and within the cytoskeleton network, as illustrated in Figure 7. It is worth mentioning that the uniaxial stretch we applied may also produce other effects in the cell monolayer, such as reorientation of the cytoskeleton.[50,51] Nevertheless, we speculate that the strain/stress condition changes at the cell–cell junction resulted from the uniaxial stretch should be sufficient to elicit the mitigation effects we observed, and that we may see similar effects with other stretching modes, such as biaxial stretch. It remains to be determined if the beneficial effect of mechanical loading also receives contributions from mechanobiological pathways other than RhoA that have not yet been extensively investigated in Pemphigus. In terms of disease development, it will be interesting to evaluate if PV patients are particularly compromised, perhaps based on genetic susceptibility, in their ability to mount a compensatory RhoA activation response by AJs following autoAb attack, rendering them at increased risk for blister development, providing at least one explanation for why certain individuals that harbor PV-associated Abs in their blood do not progress to disease.[3] Finally, we acknowledge that stretch in itself may alter the binding affinity of anti-Dsg3 antibodies. However, our data comparing the effect of cyclic stretch both before and during treatment with AK23 and the observation that both conditions protect keratinocytes from AK23 induced p38 phosphorylation suggest that the protective effects we demonstrate are unlikely to be related to AK23 binding affinity.
Figure 7.
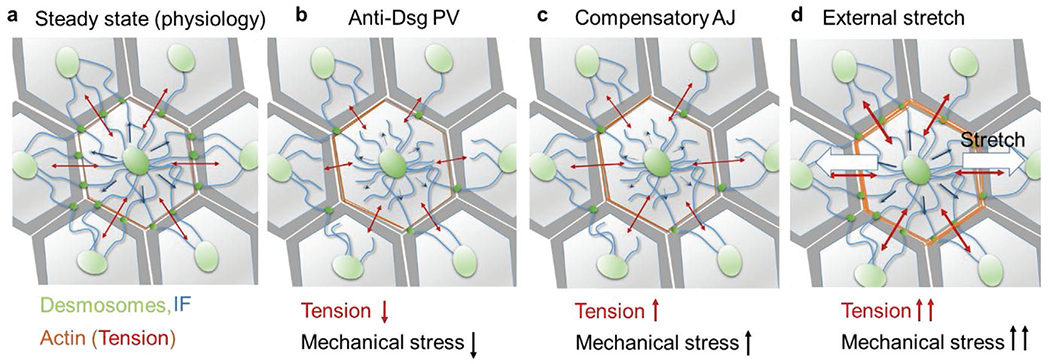
Protective mechanism of applied external stretch against anti-Dsg3-induced cell disassociation. a) Under normal physiology, IFs in keratinocytes provide resistance (blue arrows) to tension in the actin network (red arrows).[26] b) This equilibrium is interrupted by anti-Dsg3 Ab-induced desmosome disassembly, which reduces the resistance capacity of the IF network and lowers the tension in the AJs and actin network and mechanical stress at the cell–cell adhesion. c) A transient tensional compensation is invoked in AJs via RhoA activation and localization to the cell–cell border, resulting in a temporary increase in tension and mechanical stress. However, a continued assault from anti-Dsg3 Ab ultimately results in the complete cell dissociation. d) An externally applied uniaxial cyclic stretch (white arrows) can activate mechanosensitive pathways that upregulate RhoA, which elevates tension in AJs, thickens cortical actin, and stabilizes the cell–cell adhesion.
4. Experimental Section
Cell Culture:
Cultured human keratinocyte (HaCaT) cells were seeded into stretch chambers and 24-well glass bottom plates (Cellvis) containing Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher) with 10% fetal bovine serum (FBS) (Thermo Fisher) and 1% penicillin–streptomycin (P/S) (Thermo Fisher) in an incubator (Thermo Fisher) at 37 °C and 5% CO2, and cultured to near 80% confluency. Stretch chambers were treated with 10 μg mL−1 fibronectin (Cytoskeleton, Inc) overnight before cell seeding to promote cell adhesion.
Antibodies and Immunostaining:
HaCaT cells after AK23 mAb (2 μg mL−1, D219-3, MBL) treatment or stretching were fixed with 10% trichloroacetic acid (TCA) (Sigma-Aldrich) for RhoA staining for 15 min. For desmoglein3 (Dsg3), E-cadherin (E-cad) and F-actin protein fixation, 4% paraformaldehyde was added for 10 min. The intermediate filament protein (IF) was fixed by cold methanol-acetone (1:1) for 5 min. After fixation, the fixing reagents were removed and cells were washed with phosphate buffer saline (PBS) (Thermo Fisher) three times for 4 min each, followed by permeabilization with 0.1% Triton X-100 for 5 min. Cells were again washed three times with PBS for 4 min each. After permeabilization, RhoA antibody 26C4 (Santa Cruz) (1:100), active RhoA GTP (NewEast Biosciences) (1:200), Pan-Keratin (C11) Mouse mAb (1:200, Cell Signaling Technology), Dsg3 antibody (produced in our lab [52]), E-cad antibody (4A2, Cell Signaling Technologies), Alexa Fluor 488 Goat Anti-Mouse IgG Antibodies (1:50, Thermo Fisher), and Alexa Fluor 594 Goat Anti-Mouse IgG Antibodies (1:250, Thermo Fisher) were used in corresponding protein staining. Cells were exposed to these antibodies for 1 h at room temperature. For F-actin protein immunofluorescent staining, Alexa Fluor 488 Phalloidin (1:100, Thermo Fisher) was used for 30 min. After immunofluorescent antibody treatment, the staining reagents were removed and cells were washed with PBS three times for 4 min each, followed by nucleus staining with DAPI (1:1000, Thermo Fisher) for 10 min. The immunofluorescent labelling process was finished after washing with PBS three times for 4 min.
Dispase Based Cell–Cell Dissociation Assay:
HaCaT cells were seeded on six-well plates under growth media (DMEM with 10% FBS and 1% PS). After reaching confluency, cells were incubated with AK23 mAb (2 or 10 μg mL−1), AK23 mAb (2 μg mL−1) and RhoA activator (CN01, 1 unit mL−1, from Cytoskeleton) or subjected to cyclic stretch (10% strain, 1Hz) with AK23 mAb for 4 h. The control cells were incubated under growth media without AK23 mAb or cyclic stretch treatment. After washing with PBS three times, cells were incubated with dispase (Gibco Dispase II, 2.4 U mL−1) for 20 min to release the cells as a cell sheet. After that, dispase was neutralized with PBS and the cell sheets were carefully washed with PBS three times. The released cell sheets were then subjected to mechanical stress by 1 mL pipetting with a pipette tip coated with FBS. Cell fragments were fixed with 10% formaldehyde for 20–30 min and stained with 20 μL of 0.02% crystal violet at room temperature. Images were taken with a camera and the number of particles was analyzed with ImageJ software.
Antibody Treatment and RhoA Activation Assay:
After HaCaT cells reached confluency in DMEM, fresh DMEM was added in each well. For AK23 groups, anti-Dsg3 (Mouse) mAb (AK23 mAb) (2 μg mL−1, MBL) was added to DMEM. For AK23+CN01 groups, AK23 mAb and RhoA activator (CN01) (1 unit per mL, Cytoskeleton, Inc) were added in DMEM. After antibody treatment, cells were cultured at 37 °C for 4 h before staining and imaging process.
Stretching with Flexcell 5000 and Western Blotting:
For immunoblotting, HaCaT cells were seeded for 24 h on Bioflex cell stretch plates (Type-I collagen coated, Flexcell International Corp.) and subjected to cyclic (10% strain, 1 Hz frequency, 4 h) or static stretch (10% strain, 4 h). For unstretched control, cells were seeded on the same stretch plate but not exposed to stretch loading. During the stretching experiment, cells were incubated in AK23 mAb. After stretching, cells were lysed with RIPA buffer (50 × 10−3 m Tris-HCl, pH 7.4, 150 × 10−3 m NaCl, 5 × 10−3 m EDTA, 1 × 10−3 m PMSF and 1% Triton X-100) containing a protease-inhibitor cocktail (S8830; Sigma) and phosphatase inhibitor cocktail (P2850; Sigma). Proteins were separated by SDS-PAGE using 12% gels and blotted onto PVDF (polyvinylidene fluoride) membranes. The blots were incubated overnight at 4 °C with anti-p-p38 (SC-166182; Santa Cruz Biotechnology), anti-p38 (#9212; Cell Signaling), anti-RhoA (#2117; Cell signaling) or anti-GAPDH (SC-32233; Santa Cruz Biotechnology). Blots were then washed and incubated with HRP-conjugated anti-mouse or anti-rabbit IgG (Jackson ImmunoResearch), followed by washing and detection of immunoreactivity with enhanced chemiluminescence (Santa Cruz Biotechnology).
The Flexcell 5000 system was used for the dispase assay and quantifying protein expression by Western blot under the influence of stretch and the anti-Dsg3 antibody. The system has a large chamber and is designed for Western blot analysis after stretching. However, it is not compatible for imaging in a microscope, so a new stretch chamber was designed and fabricated that could be imaged along with a new system for stretching cells in the chamber. This new device was used for all experiments that include fluorescent images.
PDMS Stretch and Imaging Chamber:
Cells were cultured in PDMS stretch chambers until they were 80% confluent. To create the stretch chambers, a mold was designed with SolidWorks and was fabricated using an Objet500 Connex3 PolyJet 3D Printer with VeroClear (Stratasys). The molds were baked in an oven for 2 h at 80 °C to allow them to outgas, which allows PDMS to fully crosslink. PDMS (Dow) was made by mixing the base and crosslinker at a 1:10 ratio and desiccating to remove bubbles. The PDMS was then cast into the mold and placed into an oven for 2 h at 80 °C to facilitate crosslinking, after which the stretch chamber was removed from the mold. A 1 × 1 in piece of 0.005 in silicone (Specialty Manufacturing) was then bonded to the stretch chamber using an oxygen plasma treatment. The chamber and silicone sheet were placed in a plasma cleaner (Harrick Plasma) and ran using ambient air for 45 s, after which the two pieces were joined and baked in an oven at 80 °C for 1 h to promote crosslinking between the two parts.
Cell Stretcher Design for Confocal Microscopy Imaging:
To stretch the stretch chambers, a cell stretching system was designed. The system uses a NEMA 23 Power Plus Lead Screw Assembly (PBC Linear) controlled with an Arduino Uno and an STR4 stepper motor driver. A stepper motor was selected to allow for precise, consistent control of strain applied to the stretch chamber. The Arduino Uno interfaces with a user interface programmed in Matlab that allows the user to specify stretching parameters, such as static versus cyclic strain and the strain level and frequency (for cyclic stretching).
Microscopy Imaging:
Samples were imaged with a ZEISS LSM800 Confocal laser scanning microscope with an incubation system using a 40× NA 1.3 oil immersion objective and a 63× NA 1.4 oil immersion objective. To image cells in stretch chambers, an insert was designed and placed in the chamber to apply a slight stretch to the bottom of the chamber to flatten it, and was then placed on the slide platform to image. Imaging parameters, including laser power, master gain, and objective pinhole diameter, were optimized for each protein and kept consistent between groups for the same protein.
Immunofluorescence Image Quantification:
Mean Intensity Calculation:
In immunofluorescence images, relative protein expression was calculated based on pixel intensity within the cell membrane and cytoplasm of each cell. To identify the cell membrane and cytoplasm of cells in an image, a custom CellProfiler (Broad Institute) pipeline was created to analyze images. First, the nucleus of each cell was identified as primary objects using the DAPI image. Then, using these primary object locations and the corresponding protein immunofluorescence image to be analyzed, the cell membrane of each cell was identified as secondary objects. The intensity of a single cell was calculated as the average intensity of pixels that were within the cell boarder but outside of the nucleus. Data on the average intensity of each individual cell in each image was then exported to an Excel file, and a Matlab script was used to compile this data to find the average cell intensity within an individual image as well as across a set of images in an experiment.
Immunofluorescence Image Quantification:
Intensity Distribution:
The relative Dsg3, E-cad, F-actin, and RhoA expression were calculated based on pixel intensity on the cell–cell border. A Matlab script was used to collect the intensity of each pixel along a line drawn on the immunofluorescence image and export it to an Excel file. For the curve of relative Dsg3, E-cad, and RhoA intensity, data was obtained from one line drawn on the image. For relative distribution, the peak intensity from ten lines was divided by the mean intensity from 20 pixels at each end of each line to control for background noise. Statistical analysis based on these data in the Excel file was performed.
Statistical Analysis:
Statistical analyses were performed using student t-test, and one-way ANOVA was used for more than two samples. Statistical significance was determined at p < 0.05.
Supplementary Material
Acknowledgements
X.J., J.R., and E.K. contributed equally to this work. The authors acknowledge the funding support from the NSF (Awards #1826135 and #1936065), the NIH National Institutes of General Medical Sciences P20GM113126 (Nebraska Center for Integrated Biomolecular Communication), P20GM104320 (Nebraska Center for the Prevention of Obesity Diseases), 1U54GM115458-01 (Great Plains IDeA-CTR), R15AR072959, and P30GM127200 (Nebraska Center for Nanomedicine). The authors also acknowledge funding support from the Nebraska Collaborative Initiative and Nebraska EPSCoR FIRST award.
Footnotes
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/adbi.202000159.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Xiaowei Jin, Department of Mechanical and Materials Engineering, University of Nebraska-Lincoln, Lincoln, NE 68588, USA.
Jordan Rosenbohm, Department of Mechanical and Materials Engineering, University of Nebraska-Lincoln, Lincoln, NE 68588, USA.
Eunju Kim, Department of Mechanical and Materials Engineering, University of Nebraska-Lincoln, Lincoln, NE 68588, USA.
Amir Monemian Esfahani, Department of Mechanical and Materials Engineering, University of Nebraska-Lincoln, Lincoln, NE 68588, USA.
Kristina Seiffert-Sinha, Department of Dermatology, University at Buffalo, Buffalo, NY 14203, USA.
James K. Wahl, III, Department of Oral Biology University of Nebraska Medical Center Lincoln, NE 68583, USA.
Jung Yul Lim, Mary and Dick Holland Regenerative Medicine Program University of Nebraska Medical Center Omaha, NE 68198, USA.
Animesh A. Sinha, Department of Dermatology, University at Buffalo, Buffalo, NY 14203, USA
Ruiguo Yang, Nebraska Center for Integrated Biomolecular Communication University of Nebraska-Lincoln Lincoln, NE 68588, USA.
References
- [1].Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, Amagai M, Nat. Rev. Dis. Primers 2017, 3, 17026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Beutner EH, Lever WF, Witebsky E, Jordon R, Chertock B, J. Am. Med. Assoc 1965, 192, 682. [DOI] [PubMed] [Google Scholar]
- [3].Sajda T, Hazelton J, Patel M, Seiffert-Sinha K, Steinman L, Robinson W, Haab BB, Sinha AA, Proc. Natl. Acad. Sci. USA 2016, 113, 1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sinha AA, Sajda T, Front. Med 2018, 5, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Amagai M, Klaus-Kovtun V, Stanley JR, Cell 1991, 67, 869. [DOI] [PubMed] [Google Scholar]
- [6].Osada K, Seishima M, Kitajima Y J. Invest. Dermatol 1997, 108, 482. [DOI] [PubMed] [Google Scholar]
- [7].Spindler V, Endlich A, Hartlieb E, Vielmuth F, Schmidt E, Waschke J, Am. J. Pathol 2011, 179, 1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS, Proc. Natl. Acad. Sci. USA 2006, 103, 12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bektas M, Jolly PS, Berkowitz P, Amagai M, Rubenstein DS, J. Biol. Chem 2013, 288, 9447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Waschke J, Spindler V, Bruggeman P, Zillikens D, Schmidt G, Drenckhahn D, J. Cell Biol 2006, 175, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Spindler V, Waschke J, Ann. Anat.-Anat. Anz 2011, 193, 177. [DOI] [PubMed] [Google Scholar]
- [12].Sajda T, Sinha AA, Front. Immunol 2018, 9, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kridin K, Ther. Clin. Risk Manage 2018, 14, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Iskratsch T, Wolfenson H, Sheetz MP, Nat. Rev. Mol. Cell Biol 2014, 15, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Seiffert-Sinha K, Yang R, Fung CK, Lai KW, Patterson KC, Payne AS, Xi N, Sinha AA, PLoS One 2014, 9, e106895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fung CKM, Seiffert-Sinha K, Lai KWC, Yang R, Panyard D, Zhang J, Xi N, Sinha AA, Nanomed. Nanotechnol. Biol. Med 2010, 6, 191. [DOI] [PubMed] [Google Scholar]
- [17].Charras G, Yap AS, Curr. Biol 2018, 28, R445. [DOI] [PubMed] [Google Scholar]
- [18].Broussard JA, Yang RG, Huang CJ, Nathamgari SSP, Beese AM, Godsel LM, Hegazy MH, Lee S, Zhou F, Sniadecki NJ, Green KJ, Espinosa HD, Mol. Biol. Cell 2017, 28, 3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Green KJ, Simpson CL, J. Invest. Dermatol 2007, 127, 2499. [DOI] [PubMed] [Google Scholar]
- [20].Selby JC, in Mechanobiology of Cell-Cell and Cell-Matrix Interactions, Springer, New York: 2011, p. 169. [Google Scholar]
- [21].Godsel LM, Dubash AD, Bass-Zubek AE, Amargo EV, Klessner JL, Hobbs RP, Chen X, Green KJ, Mol. Biol. Cell 2010, 21, 2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fogh BS, Multhaupt HA, Couchman JR, J. Histochem. Cytochem. 2014, 62, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Saika S, Okada Y, Miyamoto T, Yamanaka O, Ohnishi Y, Ooshima A, Liu C-Y, Weng D, Kao WW-Y, Invest. Ophthalmol. Visual Sci 2004, 45, 100. [DOI] [PubMed] [Google Scholar]
- [24].Sumigray K, Zhou K, Lechler T, PLoS One 2014, 9, e101824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dehner C, Rötzer V, Waschke J, Spindler V, Am. J. Pathol 2014, 184, 2528. [DOI] [PubMed] [Google Scholar]
- [26].Broussard JA, Yang R, Huang C, Nathamgari SSP, Beese AM, Godsel LM, Hegazy MH, Lee S, Zhou F, Sniadecki NJ, Green KJ, Espinosa HD, Mol. Biol. Cell 2017, 28, 3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, Koyasu S, Nishikawa T, Amagai M, J. Immunol 2003, 170, 2170. [DOI] [PubMed] [Google Scholar]
- [28].Walter E, Vielmuth F, Rotkopf L, Sárdy M, Horváth ON, Goebeler M, Schmidt E, Eming R, Hertl M, Spindler V, Sci. Rep 2017, 7, 3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schmidt JB, Chen K, Tranquillo RT, Cell. Mol. Bioeng 2016, 9, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Takei T, Han O, Ikeda M, Male P, Mills I, Sumpio BE, J. Cell. Biochem 1997, 67, 327. [DOI] [PubMed] [Google Scholar]
- [31].Mao XM, Sano Y, Park JM, Payne AS, J. Biol. Chem 2011, 286, 1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Acharya BR, Nestor-Bergmann A, Liang X, Gupta S, Duszyc K, Gauquelin E, Gomez GA, Budnar S, Marcq P, Jensen OE, Dev. Cell 2018, 47, 439. [DOI] [PubMed] [Google Scholar]
- [33].Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, Rubenstein DS, J. Biol. Chem 2005, 280, 23778. [DOI] [PubMed] [Google Scholar]
- [34].Monemian Esfahani A, Rosenbohm J, Reddy K, Jin X, Bouzid T, Riehl B, Kim E, Lim JY, Yang R, Tissue Eng., Part C 2019, 25, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kaunas R, Nguyen P, Usami S, Chien S, Proc. Natl. Acad. Sci. USA 2005, 102, 15895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Smith P, Am. J. Respir. Cell Mol. Biol 2003, 28, 436. [DOI] [PubMed] [Google Scholar]
- [37].ChrzanowskaWodnicka M, Burridge K, J. Cell Biol 1996, 133, 1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ridley AJ, Hall A, Cell 1992, 70, 389. [DOI] [PubMed] [Google Scholar]
- [39].Gudipaty SA, Lindblom J, Loftus PD, Redd MJ, Edes K, Davey C, Krishnegowda V, Rosenblatt J, Nature 2017, 543, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tokuyama E, Nagai Y, Takahashi K, Kimata Y, Naruse K, PLoS One 2015, 10, e0141989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yano S, Komine M, Fujimoto M, Okochi H, Tamaki K, J. Invest. Dermatol 2004, 122, 783. [DOI] [PubMed] [Google Scholar]
- [42].Qiao P, Guo W, Ke Y, Fang H, Zhuang Y, Jiang M, Zhang J, Shen S, Qiao H, Dang E, J. Invest. Dermatol. 2019, 139, 1470. [DOI] [PubMed] [Google Scholar]
- [43].Woll S, Windoffer R, Leube RE, J. Cell Biol 2007, 177, 795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ku NO, Azhar S, Omary MB, J. Biol. Chem 2002, 277, 10775. [DOI] [PubMed] [Google Scholar]
- [45].Gliem M, Heupel WM, Spindler V, Harms GS, Waschke J, Am. J. Physiol. Cell Physiol 2010, 299, C606. [DOI] [PubMed] [Google Scholar]
- [46].Waschke J, Spindler V, Bruggeman P, Zillikens D, Schmidt G, Drenckhahn D, J. Cell Biol 2006, 175, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Spindler V, Waschke J, Ann. Anat 2011, 193, 177. [DOI] [PubMed] [Google Scholar]
- [48].Rens EG, Merks RM, Biophys. J 2017, 112, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Acharya BR, Nestor-Bergmann A, Liang X, Gupta S, Duszyc K, Gauquelin E, Gomez GA, Budnar S, Marcq P, Jensen OE, Bryant Z, Yap AS, Dev. Cell 2018, 47, 439. [DOI] [PubMed] [Google Scholar]
- [50].Rabbani M, Janmaleki M, Tafazzoli-Shadpour M, Teymoori M, Rezvaninejad S, Tissue Eng. Regener. Med 2016, 13, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chen Y, Pasapera AM, Koretsky AP, Waterman CM, Proc. Natl. Acad. Sci. USA 2013, 110, E2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wahl JK 3rd, Hybridoma Hybridomics 2002, 21, 37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.