

Abstract
Multiple system atrophy (MSA) is a demyelinating neurodegenerative disorder characterized by accumulation of aggregated α-synuclein (aSyn) inside oligodendrocyte precursors, mature oligodendroglia, and neurons. MSA dysfunction is associated with loss of trophic factor production by glial and neuronal cells. Here, we report that recombinant wild type human aSyn uptake by OLN-93, an oligodendroglia cell-line, reduced brain-derived neurotrophic factor (BDNF) expression. Furthermore, OLN-93 cells stably transfected with human wild type or an MSA-associated mutant aSyn, A53E that produces neuronal and glial inclusions, reduced BDNF mRNA to nearly unmeasurable qPCR levels. Curiously, another MSA-associated aSyn mutant, G51D that also produces neuronal and glial inclusions, caused only a trend toward BDNF mRNA reduction in transfected OLN-93 cells. This suggests that oligodendrocyte-associated BDNF loss occurs in response to specific aSyn types. Treating OLN-93 cells with 160 nM FTY720 (Fingolimod, Gilenya®), a Food and Drug Administration (FDA) approved therapeutic for multiple sclerosis, counteracted BDNF downregulation in all aSyn OLN-93 cells. FTY720 also restored BDNF mRNA in OLN-93 cells treated with recombinant aSyn, as measured by qPCR or semiquantitatively on agarose gels. Immunoblots confirmed that FTY720 increased histone 3 acetylation in OLN-93, and chromatin immunoprecipitation assays showed increased acetylated histone 3 at BDNF promoter 1 after FTY720. Moreover, OLN-93 cells treated with valproic acid, a classic histone deacetylase inhibitor, confirmed that increasing acetylated histone 3 levels increases BDNF expression. Cumulatively, the data suggest that FTY720-associated histone deacetylase inhibition stimulates BDNF expression in oligodendroglial cells, raising the possibility that MSA patients may also benefit by treatment with FTY720.
Keywords: Anti-inflammatory, Glioprotective, Multiple system atrophy, Neuroprotective, Restorative effects
1. Introduction
Synucleinopathies are aging-related neurodegenerative disorders characterized by the accumulation of aSyn aggregates inside neuronal and glial cells. These pathologies include Parkinson’s disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) (Goedert and Spillantini, 1998; Spillantini, 1999; Spillantini and Goedert, 2000). In PD and DLB, aSyn accumulates inside neuronal cells, the very cells that make it. However, in MSA, aSyn accumulates in the myelin-producing oligodendroglia (OLG) cells, which normally do not express aSyn (Reyes et al., 2014). Among the synucleinopathies, MSA stands out because it is a demyelinating neurodegenerative disorder that can progress from diagnosis to death within 5–10 years (Jellinger, 2014; Stefanova and Wenning, 2016). The hallmark lesions of MSA are the so-called Papp-Lantos bodies, also called glial cytoplasmic inclusions (GCI) that are enriched in aggregated aSyn in the cytosol of OLG cells (Jellinger, 2014; Miller et al., 2004; Wakabayashi et al., 1998). Similar lesions are also present in the oligodendrocyte precursor cells (OPC) that normally give rise to mature OLGs throughout life (May et al., 2014). GCIs in MSA are believed to cause OLG cell death, which leads to neuronal demyelination and subsequent neurodegeneration (Jellinger, 2014; Stefanova and Wenning, 2016). As is the case for all demyelinating disorders, the loss of axonal myelination in MSA is accompanied by a loss of other supportive factors that OLGs normally provide to neurons, such as neurotrophic factors (Ettle et al., 2016; Ubhi et al., 2012).
In the past ten years, accumulating evidence suggests that aSyn is secreted by neurons and can spread cell-to-cell in a prion-like manner (Emmanouilidou et al., 2010; Goedert et al., 2010), though there are conflicting data regarding whether aSyn is normally expressed by OLGs (Djelloul et al., 2015; Miller et al., 2005). Abundant data suggest that MSA pathology occurs after aSyn uptake by OLG cells (Ettle et al., 2014; Kisos et al., 2012; May et al., 2014; Pukass and Richter-Landsberg, 2014), and that the source of aSyn inside OLGs is likely neuronally-derived (Reyes et al., 2014). Also, multiple lines of evidence show that aSyn accumulation in neurons and OLGs can induce abnormal gene expression, independently of aSyn-aggregation-related cell death (Goers et al., 2003; Kim et al., 2014; Ma et al., 2014; May et al., 2014; Siddiqui et al., 2012; Yuan et al., 2010; Zhou et al., 2013). The pattern of expression of BDNF in MSA brain suggests that atypical BDNF expression contributes to MSA pathology (Kawamoto et al., 1999). Furthermore, BDNF levels are reduced in MSA mice that express human aSyn under OLG- or OPC-specific promoters (Ubhi et al., 2010), and aSyn overexpressing OPC cells also downregulate BDNF expression (May et al., 2014). Thus, aSyn accumulation in OLGs is thought to cause a loss of trophic support to neurons.
Because many of the MSA models described above relied on aSyn overexpression in OPC or OLG cells, which may or may not be a natural mechanism for aSyn accumulation (Djelloul et al., 2015; Miller et al., 2005; Reyes et al., 2014), we sought to measure the impact of aSyn uptake versus aSyn overexpression on the expression of BDNF by OLG cells. Furthermore, we sought to establish whether deleterious effects of aSyn accumulation in OPC/OLG cells was specific, or might also be triggered by the synuclein family member, beta-synuclein (bSyn) (Jakes et al., 1994). In addition, we evaluated the pre-clinical efficacy of FTY720 (2-Amino-2-[2-(4-octyl-phenyl)-ethyl]-propane-1,3-diol hydrochloride) in OLG cells. This drug stands out as a first-line pharmacological intervention for MSA by its BDNF stimulatory effects in vitro and in vivo (Deogracias et al., 2012; Di Pardo et al., 2014; Efstathopoulos et al., 2015; Hait et al., 2015; Heinen et al., 2015; Miguez et al., 2015; Noda et al., 2013; Vargas-Medrano et al., 2014) and also because as an FDA approved drug, it could be fast-tracked as a neuroprotective MSA therapy as recently done for pediatric Rett syndrome patients (ClinicalTrials.gov identifier NCT02061137). With this in mind, we assessed the impact of FTY720 on BDNF expression in OLN-93 cells as it relates to MSA, while modulating aSyn accumulation by either uptake or overexpression. Finally, we evaluated the mechanism of action by which FTY720 influences BDNF expression in OLGs.
2. Materials and methods
2.1. Cell culture
OLN-93 cells were generously provided by Dr. Jeffrey D. Macklis (Harvard Stem Cell Institute, Boston, MA) and Christiane Richter-Landsberg (Carl von Ossietzky Universitat; Oldenburg, Germany). OLN-93 cells were grown in Dulbecco’s Modified Eagle’s Medium with high glucose (Sigma-Aldrich, St. Louis, MO, Cat.D5648-10X1L), supplemented with 10% heat-inactivated fetal bovine serum (FBS) (GE Healthcare Life Sciences HyClone Laboratories, Logan, Utah, Cat. SH300071.03). Non-transfected OLN-93 cells were grown in medium containing 50 U/mL penicillin and 50 μg/mL streptomycin. Transfected OLN-93 cell lines were grown in medium with added G418 sulfate (0.8 mg/mL) (Corning Life Sciences Inc., Corning, NY, Cat. 61-234-RG). Incubation was maintained at 37 °C with 10% CO2.
2.2. Site-directed mutagenesis and transfection to establish stable aSyn OLN-93 cell lines
In order to generate plasmids to express G51D-aSyn and A53E-aSyn mutant forms, we used a pcDNA3.1 plasmid containing human wild type aSyn (WT-aSyn) that we have used before (Perez et al., 2002). Single amino acid substitutions were achieved using a QuickChange XL Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA, Cat. 200516) following the manufacturer’s protocol. The G51D-aSyn and A53E-aSyn mutations were then verified by automated DNA sequencing (University of Texas at El Paso Genomic Analysis Core Facility, Department of Biology).
OLN-93 cells were transfected in 6 wells plates using 1 μg of plasmid vector (pcDNA3.1) as a control or human aSyn plasmids (WT-aSyn, A53E-aSyn, or G51D-aSyn) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, Cat. 11668-019) and Opti-MEM (Thermo Fisher Scientific, Waltham, MA, Cat. 22600-134). A separate well was mock-transfected using no plasmid. Selection of stably transfected clonal cell lines was accomplished using G418 sulfate (1 mg/mL) with clones chosen after all mock transfected cells had died.
Monoclonal cell lines were obtained by the limiting dilution selection method. At 3–5 days after the initial seeding into 96 well plates, wells containing a single colony were identified and selected for transfer and expansion. Stably transfected OLN-93 clonal aSyn lines were then selected for comparison based on aSyn levels measured by immunoblotting and immunocytochemistry.
2.3. Drugs and aSyn or bSyn treatment
For all assays that included drugs or recombinant proteins treatments, cells were seeded and grown in antibiotic-free medium during the entire assay. FTY720 (AbMole BioScience, Kowloon, Hong Kong, Cat. M1712) was prepared in ethanol and cells were treated at 160 nM as in our earlier studies using MN9D dopaminergic cells (Vargas-Medrano et al., 2014). Similarly, valproic acid (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, Cat. sc-202378) was prepared in double distilled sterile water and cells were treated with 150 μM or 75 μM concentrations. Full length, non-tagged, recombinant human wild type aSyn and recombinant human wild type bSyn expressed in E. coli were purchased from rPeptide LLC (Bogart, GA, Cat. S-1001-2 and S-1003-2, respectively) and prepared as instructed by the manufacturer. Briefly, lyophilized recombinant aSyn and bSyn were reconstituted to a 1 mg/mL (~69 μM) solution using double distilled sterile water. Cells were treated with recombinant human aSyn or bSyn at 1 μM final concentrations.
2.4. Immunoblots
Protein concentrations in cell lysates were determined by the bicinchoninic acid assay (Smith et al., 1985). Total proteins (25–50 μg per lane) were separated by SDS-PAGE, transferred to nitrocellulose membranes, blocked with 5% non-fat dry milk, and then incubated with primary antibodies overnight at 4 °C. Primary antibodies for immunoblotting included anti-aSyn (Santa Cruz Biotechnology Inc., Cat. sc-7011-R) (1:200 dilution), anti-bSyn (Novus Biologicals, Littleton, CO, Cat. NB100-79903) (1:1000 dilution), anti-AcH3 (Lys9/Lys14) (Cell Signaling Technology, Inc., Danvers, MA, Cat. 9677) (1:500 dilution), anti-total histone H3 (Cell Signaling Technology Inc., Cat. 96C10) (1:500 dilution), anti-phosphorylated ERK1/2 (Tyr204) (Santa Cruz Biotechnology Inc., Cat. sc-7383) (1:200 dilution), anti-total ERK1/2 (Santa Cruz Biotechnology Inc., Cat. sc-93) (1:200 dilution) and anti-β-actin (Cell Signaling Technology Inc., Cat. 3700 or 4970) (1:1000 dilution). All blots were imaged using the Odyssey system (LiCor Biosciences, Lincoln, NE, model # 9210) and quantified with Image Studio software (LiCor Biosciences).
2.5. Immunocytochemistry
OLN-93 cells were seeded on 8-well chamber slides (Nalge Nunc International, Rochester, NY, Cat. 154534) previously coated with poly-L-lysine (Sigma-Aldrich, Cat. P1274-100 MG) and grown overnight. For aSyn uptake assays, cells were then treated with 1 μM recombinant human wild type aSyn or vehicle (PBS) for 12 h. After respective treatments, cells were washed gently with PBS, fixed with 4% paraformaldehyde for 25 min at room temperature (RT) and subsequently incubated for 30 min at RT in a permeabilization/blocking solution containing 1% bovine serum albumin and 0.1% triton-X 100 in PBS. Cells were then incubated with the primary anti-aSyn antibody Syn-1 (BD Biosciences, San Jose, CA, Cat. 610787) (1:100 dilution) overnight at 4 °C. Cells were then incubated with Cy5-conjugated anti-mouse secondary antibody (Thermo Fisher Scientific, Cat. A10523) (1:1000 dilution) for 1 h at RT and subsequently washed and incubated with ActinGreen 488 following manufacturer’s instructions (Thermo Fisher Scientific, Cat. R37110). Samples were coverslipped using Vectashield mounting medium plus DAPI (Vector Laboratories, Burlingame, CA, Cat. H-1500). Images were obtained using the Olympus FluoView-1000 confocal microscope (Olympus, Center Valley, PA).
2.6. Gene expression assessment
Total mRNAs were extracted from OLN-93 cells using the RNeasy Plus Mini Kit (Qiagen Inc., Valencia, CA, Cat. 74134) and retro-transcribed with a High Capacity RNA-to-cDNA Kit (Thermo Fisher Scientific, Cat. 4387406), as per manufacturer’s instructions. RNA concentration and purity was assessed using NanoDrop 2000 spectrophotometry (Thermo Fisher Scientific). RNA integrity and genomic DNA contamination were assessed using 28 s/18 s rRNAs band ratios in an RNA “bleach” gel as described by Aranda et al. (2012). The mRNAs were measured using real time quantitative PCR (qPCR) in a RealPlex Mastercycler 2 (Eppendorf, Hauppauge, NY). Relative expression of mRNAs was evaluated using Taqman probe assays (Thermo Fisher Scientific) for rat BDNF (Cat. Rn02531967_s1), rat NGF (Cat. Rn01533872_m1) and rat GAPDH (Cat. Rn01775763_g1) and eukaryotic 18 S ribosomal RNA (Cat. 4332641) as internal controls. Reactions were carried out in triplicate using GoTaq qPCR Master Mix (Promega, Madison, WI, Cat. A6001). For qualitative PCR assessment of BDNF and GAPDH expression, the amplicons from the qPCR assays were loaded into a 1% agarose gel containing ethidium bromide for UV imaging. According to the manufacturer, the 142 bp and 174 bp bands are the correct respective sizes of the amplicons produced by rat BDNF and Gapdh in Taqman assays.
2.7. Chromatin immunoprecipitation with qPCR (ChIP-qPCR)
Chromatin immunoprecipitation (ChIP) was performed using the Pierce Magnetic ChIP Kit (Thermo Fisher Scientific, Cat. 26157) following manufacturer’s instructions with one modification: all incubations with buffers requiring supplementation with protease/phosphatase inhibitors cocktail were also supplemented with 100 μM valproic acid in order to inhibit residual activity of histone deacetylases (HDAC) and to preserve the levels of acetylated histones throughout remaining incubations. Immunoprecipitation was performed with overnight incubation at 4 °C with the ChIP-validated anti-AcH3 antibody (same used for immunoblots, section 2.4) in a 1:50 dilution. Immunoprecipitation with a normal rabbit IgG antibody was also performed as negative ChIP control. Previous to immunoprecipitation, 10% of every sample was separated and later used as input. Relative abundance of sequences of Gapdh and BDNF promoters in immunoprecipitated DNA was quantified by ChIP-qPCR using ChIP-validated primers for rat Gapdh promoter (fwd-TGAGAGAGGCCCAGCTACTC and rev-AGGGCTGCAGTCCGTATTTA), rat BDNF promoter 1 (fwd-TTCGATT-CACGCAGTTGTTC and rev-GCAGCCTCTCTGAGCCAGT) and rat BDNF promoter 4 (fwd-GCGCGGAATTCTGATTCTGGTAAT and rev-GAGAGGGCTCCACGCTGCCTTGACG) (Koo et al., 2015). ChIP-qPCR reactions were carried out in triplicate in a RealPlex Mastercycler 2 using USB HotStart-IT Sybr Green qPCR Master Mix (Thermo Fisher Scientific, Cat. 75760). Results of the ChIP-qPCR values for immunoprecipitated samples were normalized to the percentages of their respective inputs from 3 independent experiments.
2.8. Statistics
Histograms represent mean ± standard deviation (SD) from 3 or more independent experiments. Unpaired Student’s t-tests or one-way ANOVA were performed using Prism 6 (GraphPad Software Inc., La Jolla, CA), with significance set to p < 0.05. Relative mRNA expression was calculated and statistically analyzed using the comparative Ct method (2−ΔΔCt) and the Relative Expression Software Tool (REST-2009) (Qiagen Inc.) (Pfaffl et al., 2002). Whisker box plots generated by REST-2009 for relative mRNA expression demonstrate the median (midline inside the box), interquartile ranges 1 and 3 (upper and lower edges of the box), as well as maximum and minimum expression values (top and bottom whiskers) (Pfaffl et al., 2002).
3. Results
3.1. Uptake of recombinant human wild type aSyn and bSyn by OLN-93 cells
Modeling OLG aSyn uptake after Reyes et al. (2014), we exposed OLN-93 cells to 1 μM recombinant human wild type aSyn or vehicle for 12 h. We also treated cells with 1 μM recombinant human wild type bSyn for 12 h, to control for the effects of another member of the synuclein family. Treated cells were then prepared for evaluation of aSyn and bSyn uptake. Immunoblots of cell lysates confirmed the presence of aSyn and bSyn only in cells treated with recombinant human wild type aSyn or bSyn respectively (Fig. 1A and B). Immunocytochemistry of OLN-93 cells treated with recombinant human wild type aSyn confirmed the presence of aSyn signal (Fig. 1C, red signal in upper right frame); while cells treated with vehicle only had no aSyn immunoreactivity (Fig. 1C). Taking into account previous reports showing interaction between aSyn and β-actin (Bellani et al., 2010; Esposito et al., 2007; Zhou et al., 2004), we confirmed aSyn internalization in OLG cells by confocal microscopy, which showed co-localization of aSyn (red) with β-actin (green) producing yellow merged signal only in aSyn treated cells (Fig. 1C, at arrowheads). We next assessed whether aSyn or bSyn internalization produced similar effects on BDNF expression in OLN-93 cells.
Fig. 1. Uptake of recombinant wild type human aSyn and bSyn by OLN-93 cells.
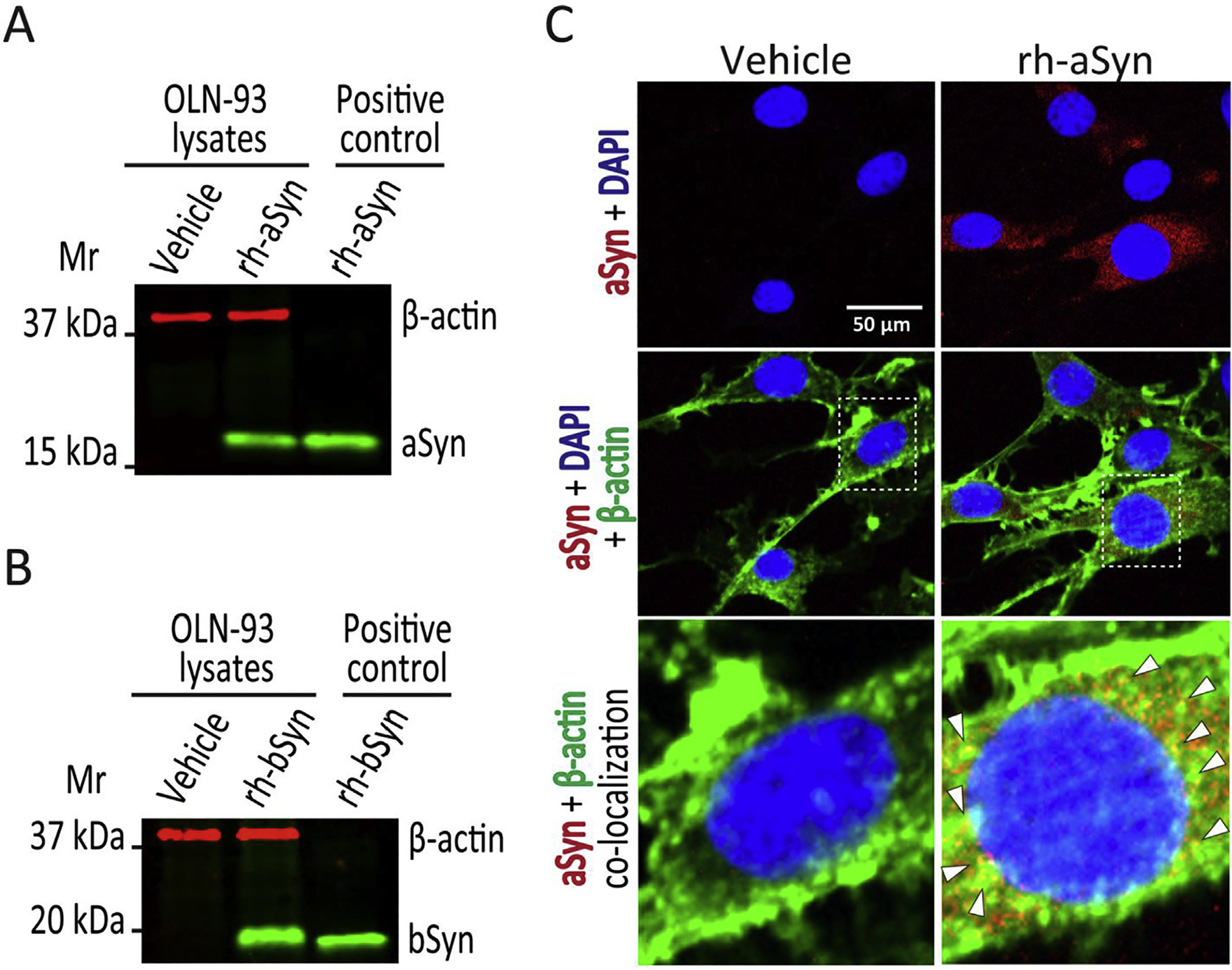
(A). Immunoblot of OLN-93 cell lysates after 12 h treatment with vehicle (lane 1) or 1 μM recombinant human wild type aSyn (rh-aSyn, lane 2), and recombinant human aSyn (25 ng) as positive control (lane 3). A 19 kDa aSyn band is apparent only in OLN-93 cells exposed to recombinant human aSyn and the positive control, with none noted for vehicle treated cells. (B). Immunoblot of OLN-93 cell lysates after 12 h treatment with vehicle (lane 1) or 1 μM recombinant human wild type bSyn (rh-bSyn, lane 2), and recombinant human bSyn (25 ng) as positive control (lane 3). A 20 kDa bSyn band is apparent only in OLN-93 cells exposed to recombinant human bSyn and the positive control, with none noted for vehicle treated cells. (C). Confocal microscopic images of OLN-93 cells treated with vehicle or 1 μM recombinant human aSyn for 12 h. Cells were stained for aSyn (Syn-1 antibody 1:100 dilution, red signal) and nuclei (DAPI, blue signal) (upper frames) show that aSyn is present only in OLN-93 cells treated with recombinant human aSyn (rh-aSyn, right frames). Merged images showing aSyn, DAPI plus β-actin (ActinGreen 488, green signal) (middle frames) show that aSyn signal is mainly confined to the cytosolic compartment. Enlarged images of nuclear and perinuclear area (lower frames) show co-localization of aSyn and β-actin (arrowheads pointing to yellow spots) mainly in the perinuclear area. Scale bar = 50 μm.
3.2. aSyn, but not bSyn internalization, downregulates BDNF mRNA which is counteracted by FTY720 in OLN-93 cells
Using qPCR, we saw that OLN-93 cells treated with 1 μM recombinant human aSyn for 12 h had significant reduction in BDNF mRNA as compared to vehicle-treated cells (Fig. 2, white whisker box). However, cells treated for 12 h with 1 μM recombinant human bSyn produced no significant change in BDNF mRNA as compared to vehicle-treated cells (Fig. 2, light gray whisker box). Also, to determine if aSyn uptake may have affected other BDNF-related neurotrophic factors, we assessed the relative expression of another key member of the neurotrophin family, nerve growth factor (NGF) (Park and Poo, 2013). Interestingly, NGF mRNA in OLN-93 cells was not significantly affected by recombinant human aSyn internalization (1.43 mean fold change, 95% confidence interval 0.77–2.5, p = 0.2), suggesting a relatively specific effect of aSyn uptake on BDNF in OLGs.
Fig. 2. aSyn but not bSyn uptake affects BDNF mRNA in OLN-93 cells treated ± FTY720.
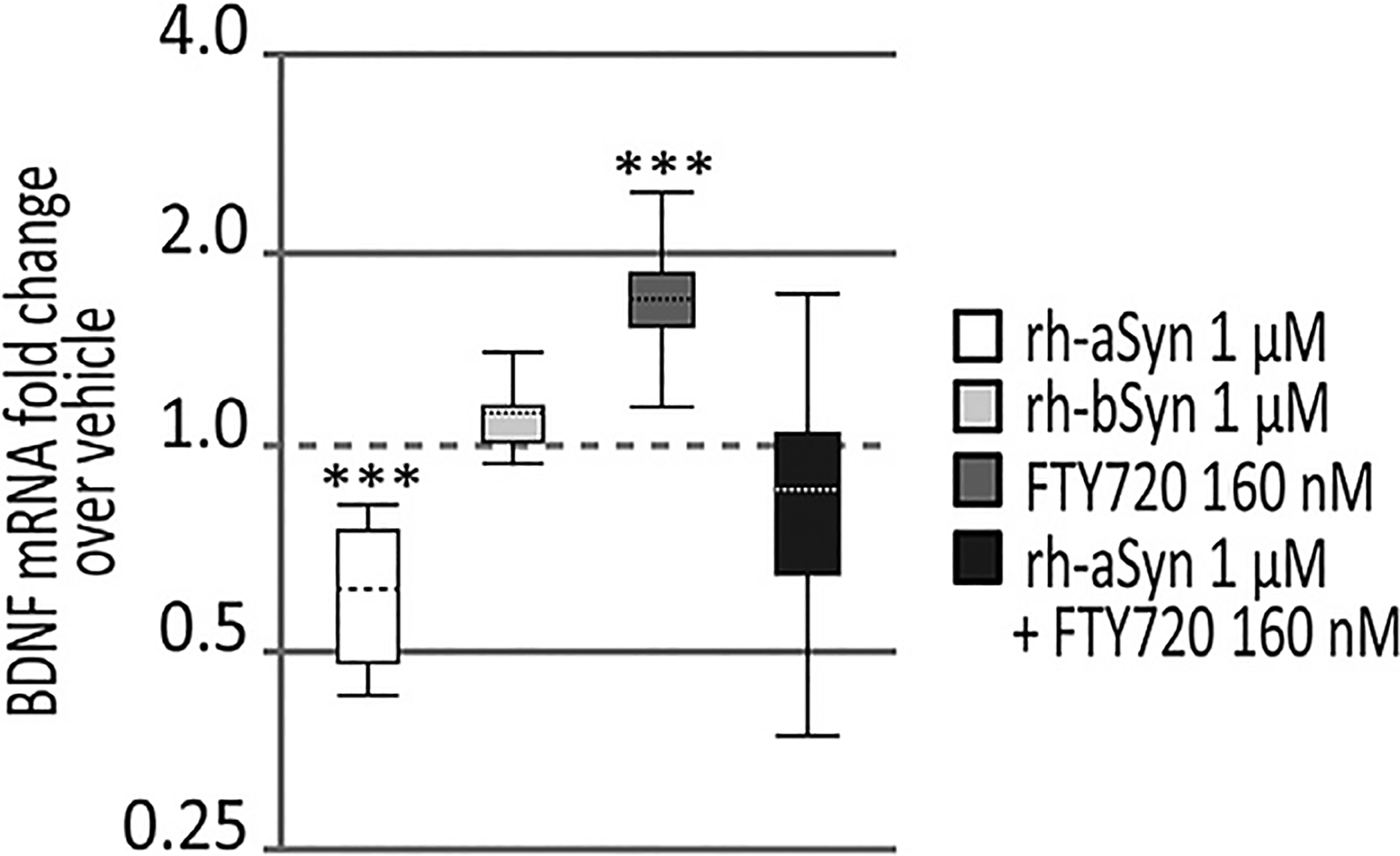
Analysis of qPCR (n = 3 assays/condition) show that OLN-93 cells exposed to 1 μM recombinant human wild type aSyn (rh-aSyn) for 12 h had significantly less BDNF expression (white whisker box). OLN-93 cells exposed to 1 μM recombinant human wild type bSyn (rh-bSyn) for 12 h had no change in BDNF expression (light gray whisker box). OLN-93 cells treated with 160 nM FTY720 for 12 h had significantly increased BDNF expression (dark gray whisker box) and OLN-93 cells double treated with 1 μM recombinant human aSyn + 160 nM FTY720 for 12 h (black whisker box) returned to normal levels as noted in vehicle-treated cells. Dashed line represents vehicle-treated baseline values, at 1.0. Statistical analysis and whisker box plots were generated using REST-2009 suite (as described in Section 2.8). ***p < 0.001.
Similar to our earlier assessment in neuronal cells (Vargas-Medrano et al., 2014), we found that using 160 nM FTY720 treatment of OLN-93 cells for 12 h significantly increased BDNF expression as measured using qPCR (Fig. 2, dark gray whisker box). To assess if FTY720 might also counteract the BDNF downregulation in response to recombinant human aSyn uptake, we treated OLN-93 cells with 160 nM FTY720 in the presence of 1 μM recombinant human aSyn for 12 h. Cells exposed to this double treatment returned to baseline BDNF levels seen in vehicle-treated cells (Fig. 2, black whisker box). This suggests that FTY720 has the ability to counteract the negative effects of aSyn accumulation in OLGs with regard to BDNF expression.
3.3. aSyn can be stably expressed in OLN-93 cells
In stably transfected OLN-93 aSyn-cells, we confirmed the absence of aSyn in our empty vector transfected cells and equivalent aSyn levels in cell lysates prepared from the various OLN-93 cell lines as assessed by immunoblot (Fig. 3A). We also confirmed intracellular aSyn (red signal) in OLN-93 cells stably transfected with wildtype (WT-aSyn), G51D-aSyn, or A53E-aSyn as evaluated by immunocytochemistry. As expected, the empty vector control cells were negative for intracellular aSyn immunoreactivity (Fig. 3B).
Fig. 3. Expression of aSyn in the OLN-93 stably transfected cell lines.
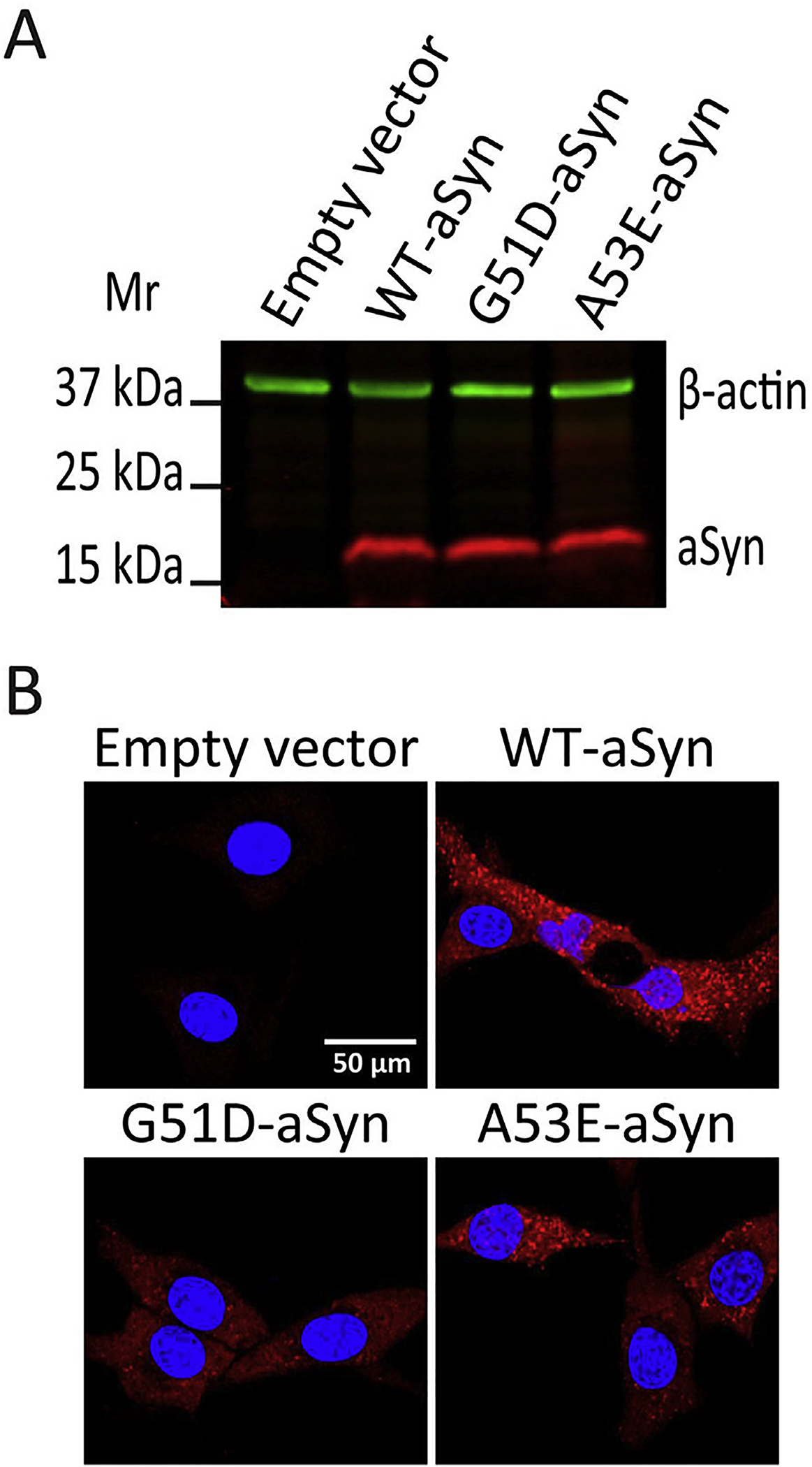
(A). Immunoblot of lysates of OLN-93 cell lines expressing empty vector, WT-aSyn, G51D-aSyn, or A53E-aSyn show aSyn only in cells transfected with aSyn-plasmids. (B). Confocal microscopic images of OLN-93 cell lines expressing empty vector, WT-aSyn, G51D-aSyn, or A53E-aSyn show aSyn immunoreactivity (Syn-1 antibody 1:100 dilution, red signal) only in aSyn-transfected cell lines. Stained nuclei are also shown in blue (DAPI). Scale bar = 50 μm.
3.4. WT-aSyn and A53E-aSyn expression in OLN-93 cells blocks BDNF expression
Using qPCR to measure BDNF expression in stably transfected OLN-93 aSyn cells, we were surprised to find that empty vector cells and G51D-aSyn clonal cells had similar BDNF expression (1.05 mean fold change, 95% confidence interval 0.78–1.57, p = 0.9), while two OLN-93 clonal lines that expressed WT-aSyn or A53E-aSyn had no BDNF amplification even after the full 40 cycles of qPCR. This lack of amplification prevented quantitative assessment of BDNF expression in those cell lines using qPCR. However, we demonstrate the differences in BDNF expression in OLN-93 aSyn clonal cells using a semi-quantitative approach in which we visualized the amplification products generated by qPCR on agarose gels (Fig. 4A). This allowed us to demonstrate the 142 base pair (bp) BDNF cDNA amplicon in both empty vector and G51D-aSyn transfected cells (Fig. 4A, lanes 1 and 4), which was essentially absent in the qPCR products from WT-aSyn or A53E-aSyn cell lines at baseline (Fig. 4A, lanes 2 and 6). When data were quantified from multiple experiments, the near complete absence of BDNF expression by WT-aSyn and A53E stable cell lines was evident (Fig. 4B). As an internal control, we saw that 174 bp Gapdh amplicons were equivalent in all stably transfected OLN-93 cell lines (Fig. 4A). Moreover, similar to our findings after aSyn uptake (section 3.2), NGF expression was not significantly altered in any of our aSyn stably transfected OLN-93 cell lines as compared to empty vector control cells as assessed using qPCR (Fig. 5).
Fig. 4. Impact of aSyn and FTY720 on BNDF expression in stably transfected OLN-93 cell lines.
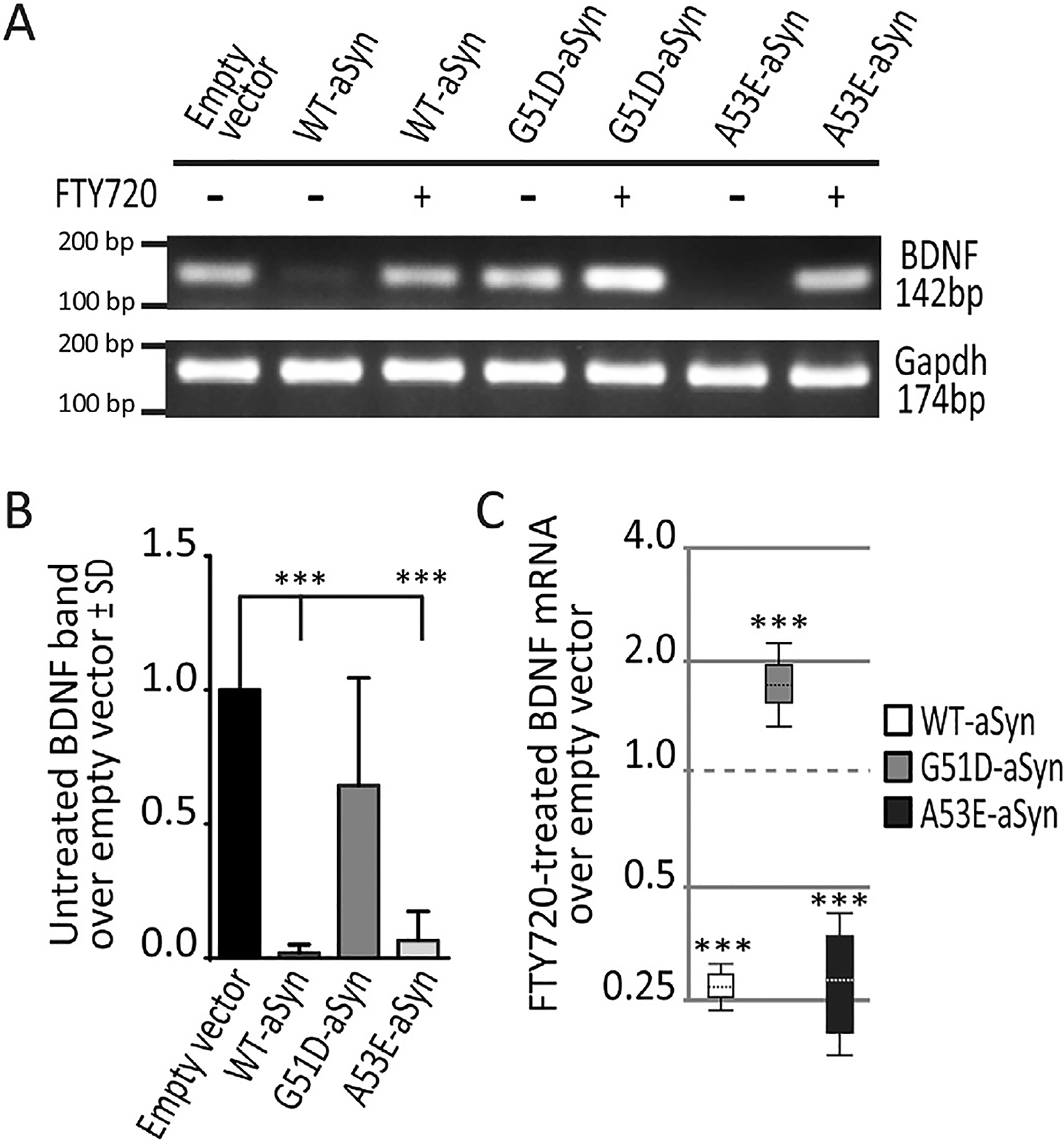
(A). Representative agarose gel of qPCR data showing BDNF cDNA amplicon (142 bp) in untreated (−) and 12 h FTY720-treated (+) cells. Data are shown as follows: OLN-93 cells expressing empty vector (lane 1), WT-aSyn (lanes 2 and 3), the G51D-aSyn mutant (lanes 4 and 5) and A53E-aSyn mutant (lanes 6 and 7). BDNF amplicons are absent in WT-aSyn that were untreated (lane 2) but present in FTY720-treated WT-aSyn cells (lane 3). Likewise, BDNF amplicons are absent in untreated A53E-aSyn cells (lane 6) but present in FTY720-treated A53E-aSyn cells (lane 7). BDNF amplicons are present in G51D-aSyn cells at baseline (lane 4) and increased after FTY720 treatment (lane 5). Gapdh amplicons (174 bp) are equivalent for all conditions. (B). Untreated cell BDNF amplicon band intensities from multiple agarose gels (n = 3) confirm a significant loss of signal in WT-aSyn and A53E-aSyn OLN-93 cell lines. One-way ANOVA; ***p < 0.001. (C). The qPCR for BDNF in FTY720-treated cells shows that BDNF expression remained significantly lower in WT-aSyn cells (white whisker box) and A53E-aSyn cells (black whisker box) compared to untreated empty vector cells (dashed line at 1.0). BDNF expression in FTY720-treated G51D-aSyn cells (gray whisker box) is increased above untreated empty vector cells (dashed line at 1.0). Statistical analysis and whisker box plots were generated using REST-2009 suite (as described in Section 2.8). ***p < 0.001.
Fig. 5. Impact of aSyn on NGF expression in stably transfected OLN-93 cell lines.
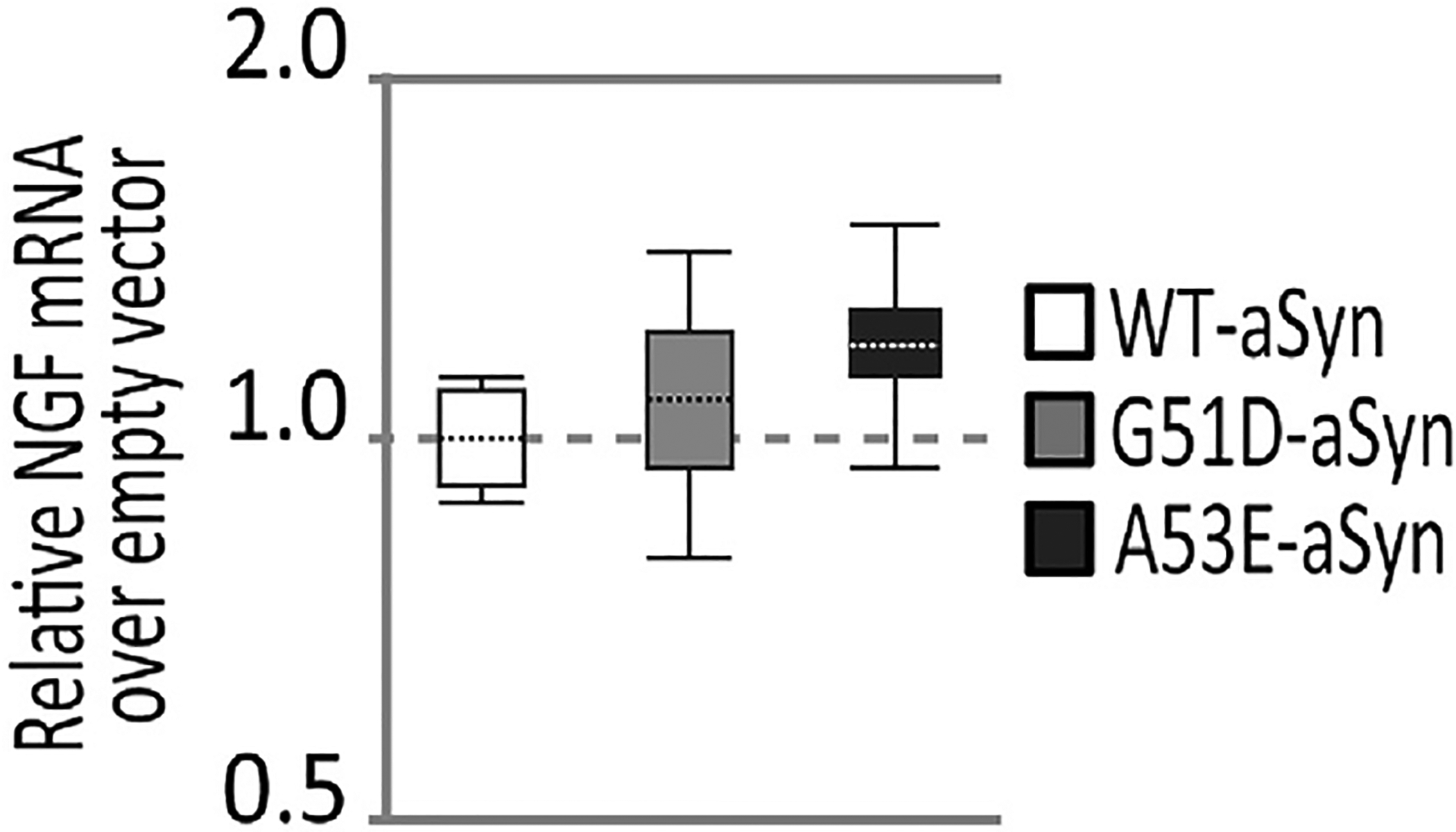
Analysis of qPCR (n = 3/cell line) for NGF expression in OLN-93 cell lines shows that all the aSyn expressing cell lines (whisker boxes) have similar levels of NGF expression compared to empty vector transfected cells (dashed line at 1.0). Statistical analysis and whisker box plots were generated using REST-2009 suite (as described in Section 2.8).
3.5. FTY720 significantly increases BDNF expression in OLN-93 clonal cell lines
Empty vector transfected cells expressed BDNF (Fig. 4A, lane 1) and 12 h treatments with 160 nM FTY720 significantly increased BDNF expression in all stably transfected aSyn OLN-93 cell lines (Fig. 4A, lanes 3, 5 and 7), visualized on agarose gels showing the BDNF amplicons generated by qPCR. Only after FTY720 treatment, was BDNF amplification observable in stably transfected WT-aSyn and A53E-aSyn cell lines (Fig. 4A, lanes 5 and 7 as compared to lanes 4 and 6, respectively). Importantly, the fact that FTY720 increased BDNF mRNA in all OLG aSyn lines allowed us to analyze BDNF expression in WT-aSyn and A53E-aSyn cell lines by qPCR as demonstrated in Fig. 4C, with the value of empty vector control cells shown as the dashed line set at 1.0. Even after FTY720 treatment, BDNF mRNA levels remained significantly lower in WT-aSyn (Fig. 4C, white whisker box) and A53E-aSyn (Fig. 4C, black whisker box) cells as compared to empty vector control cells. Similar to what we observed in non-transfected OLN-93 cells (Fig. 2), stably transfected G51D-aSyn cells had an significant increase in BDNF expression in response to FTY720 (Fig. 4A, lane 5 as compared to lane 4; Fig. 4C, gray whisker box).
3.6. FTY720 and valproic acid increase global levels of AcH3 in OLN-93 cells
On immunoblots we noted that 12 h of 160 nM FTY720 treatment of OLN-93 cells produced a significant increase in global acetylation of histone 3 (AcH3) (Fig. 6A, lane 2 and 6B). We saw no significant increase in phosphorylated/active ERK1/2 (pERK1/2) levels at 12 h after FTY720 treatment (1.42 ± 0.34 SD, p = 0.1, Student’s t-test). Others have reported that short term (5–45 min) treatments with 1 μM FTY720 induce a transient increase of pERK1/2 levels in OLG cells, while 100 nM FTY720 treatments do not elicit such response (Coelho et al., 2007). In order to test a potential transient elevation of pERK1/2 levels with 160 nM FTY720, we treated the OLN-93 cells for 5, 15 and 45 min. These short term treatments with 160 nM FTY720 for 5, 15 and 45 min, did not change pERK1/2 levels in OLN-93 cells (5 min 1.31 ± 0.31 SD, p = 0.43; 15 min 0.89 ± 0.17 SD, p = 0.57; 45 min 0.96 ± 0.03 SD, p = 0.23; Student’s t-test). This suggests that FTY720-associated effects in OLN-93 cells may have occurred by increasing histone acetylation, as FTY720 is a confirmed inhibitor of HDACs (Hait et al., 2014, 2015). This encouraged us to perform the following control experiments in which we measured the impact of valproic acid on AcH3. To parallel our FTY720 experiments, we treated OLN-93 cells for 12 h with 75 μM or 150 μM valproic acid. Immunoblots show that both doses of valproic acid increased AcH3 levels in OLN-93 cells (Fig. 6A, lanes 3 and 4), which when quantified from multiple experiments confirmed increased AcH3 in response to both FTY720 and valproic acid (Fig. 6B).
Fig. 6. Impact of FTY720, valproic acid and aSyn uptake on AcH3 levels in OLN-93 cells.
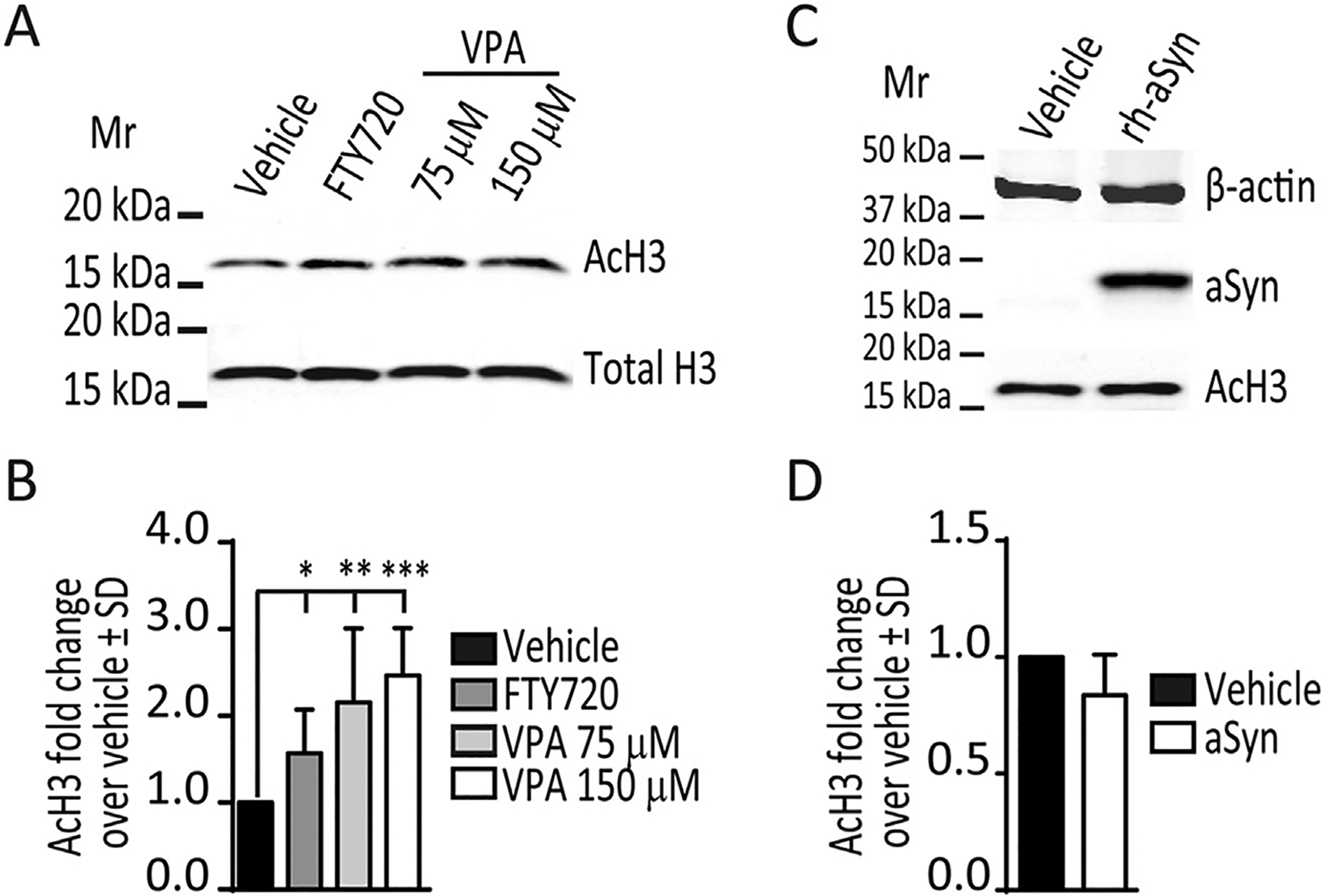
(A). Representative immunoblot showing levels of AcH3 and total histone H3 in lysates from OLN-93 cells treated with vehicle, FTY720 (160 nM), or valproic acid (VPA, 75 μM or 150 μM) 12 h treatments. (B). Histogram of AcH3 levels (n ≥ 3/condition) in OLN-93 cells treated with vehicle, FTY720 160 nM, VPA 75 μM or VPA 150 μM for 12 h. One-way ANOVA; *p < 0.05, **p < 0.01 and ***p < 0.001 (C). Representative immunoblot showing levels of AcH3 and aSyn in lysates from OLN-93 cells treated with vehicle or 1 μM recombinant human aSyn (rh-aSyn) for 12 h (D). Histogram of AcH3 levels (n = 3/condition) in OLN-93 cells treated with vehicle or 1 μM recombinant human aSyn for 12 h show no difference between treatments. Student’s t-test.
3.7. HDAC inhibition increases BDNF expression in OLN-93 cells
Valproic acid is a prototypical HDAC inhibitor that has previously been shown to induce BDNF expression in neuronal and glial cells (Fukuchi et al., 2009; Wu et al., 2008; Yasuda et al., 2009). In our experiments in which OLN-93 cells were treated with 75 μM and 150 μM valproic acid, we noted that only the higher dose (150 μM) significantly increased BDNF expression (1.6 mean fold change, 95% confidence interval 1.31–1.86, p < 0.001) at 12 h in OLN-93 cells, by qPCR. This provides support that increased acetylation of histone 3 in OLN-93 cells contributes to increasing BDNF expression.
3.8. FTY720 specifically increases histone acetylation at the BDNF promoter in OLN-93 cells
Chromatin immunoprecipitation (ChIP) assays are the gold-standard to determine if a pharmacological or a molecular change in histone acetylation functionally influences BDNF gene expression (Fukuchi et al., 2009; Tian et al., 2010; Wu et al., 2008; Yasuda et al., 2009; Zeng et al., 2011). To assess this in our cells, we used ChIP assays to evaluate the relationship between parallel increases in BDNF expression and AcH3 in response to FTY720 at BDNF promoter 1 (BDNF-P1) and promoter 4 (BDNF-P4), as both of these promoters respond to changes in histone acetylation by HDAC inhibition (Fukuchi et al., 2009; Yasuda et al., 2009). We measured AcH3 levels in OLN-93 cells at BDNF-P1 and BDNF-P4 after 12 h treatments with 160 nM FTY720 or vehicle. ChIP-qPCR assays for both of these promoters revealed that the relative AcH3 level at BDNF-P1 was significantly increased by FTY720; however, AcH3 levels at BDNF-P4 showed only a trend toward an increase (Fig. 7). In order to control for potential effects on other promoters or nonspecific binding of the anti-AcH3 antibody, we also measured AcH3 levels at the Gapdh promoter, as this promoter is known to have high AcH3 levels but is not typically affected by treatment with HDAC inhibitors (Khobta et al., 2010; Makarona et al., 2014; To et al., 2008). This allowed us to show that AcH3 levels in the Gapdh promoter were not changed in response to FTY720 (Fig. 7). ChIP-qPCR assays of the same BDNF and Gapdh promoters, using normal rabbit IgG as a negative control, showed extremely low (<0.15% of input) in fact nonspecific IgG binding to these promoters (Fig. 7). Thus, we conclude that the changes in histone acetylation induced by 12 h FTY720 treatment in OLN-93 cells effectively and specifically remodeled chromatin of at least one of the promoters that regulate BDNF expression.
Fig. 7. FTY720 specifically increases BDNF promoter 1 histone 3 acetylation (AcH3) in OLN-93 cells relative to controls.
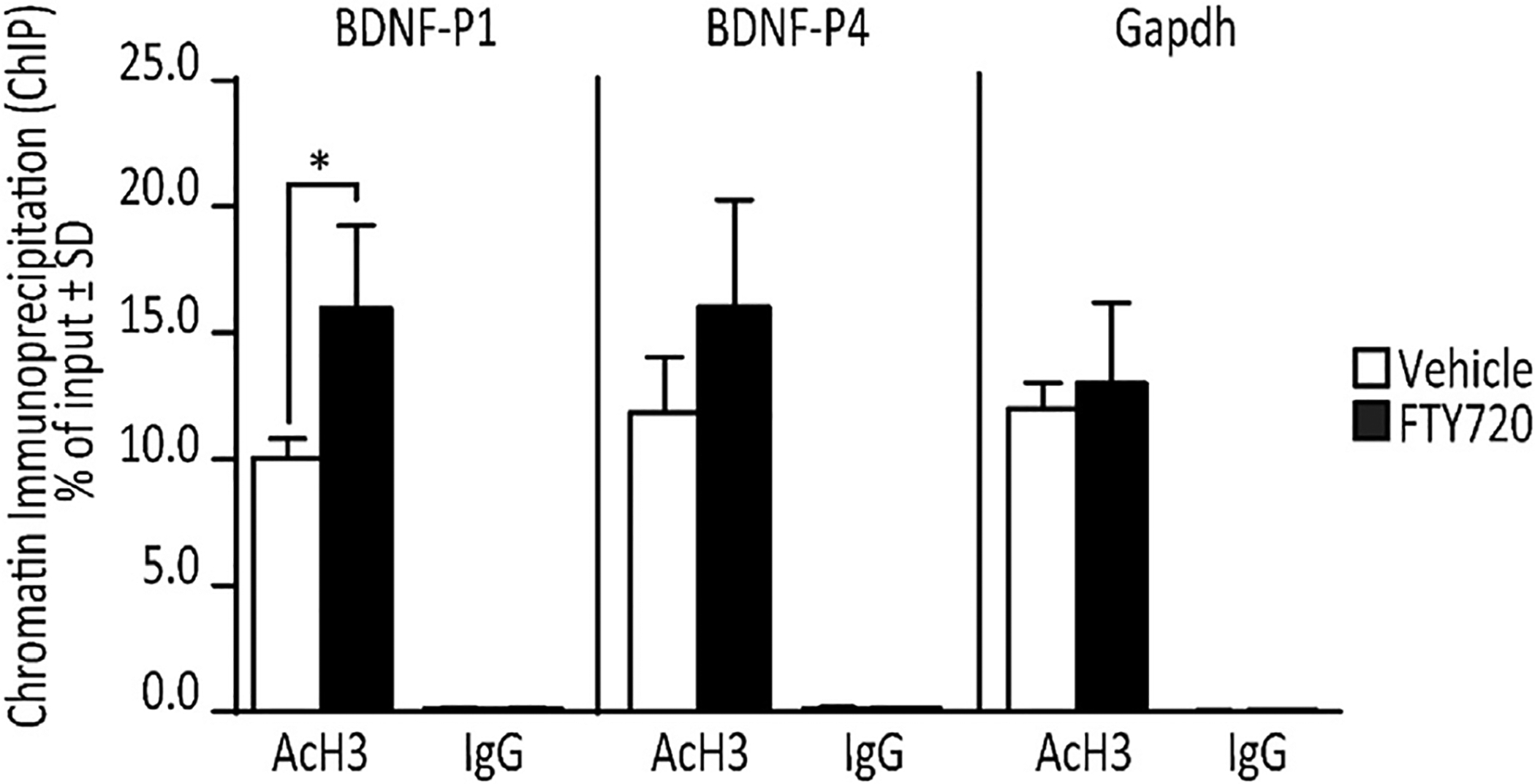
ChIP-qPCR analysis (n = 3/condition) normalized as percentage of input, shows that 12 h 160 nM FTY720 significantly increases AcH3 levels at BDNF promoter 1 (BDNF-P1), but not BDNF promoter 4 (BDNF-P4), which shows a trend toward an increase as compared to vehicle treated cells. FTY720 does not change AcH3 levels at the Gapdh promoter. Control ChIP-qPCR assays using normal rabbit IgG (IgG) antibody shows only extremely low nonspecific binding for all conditions. Student’s t-test; *p < 0.05.
3.9. Uptake of aSyn has no effect on AcH3 levels in OLN-93 cells
Since aSyn can affect gene expression by reducing histone acetylation in neuronal models (Goers et al., 2003; Kontopoulos et al., 2006; Siddiqui et al., 2012), we explored this mechanism as a potential cause for the BDNF downregulation observed in our cells. However, on immunoblots we noted that 12 h after 1 μM recombinant human aSyn exposure, OLN-93 cells showed no significant differences in global AcH3 levels (Fig. 6C and D).
4. Discussion
aSyn pathology occurs in several neurodegenerative disorders, in which aSyn forms Lewy-like protein aggregates in neurons of PD and DLB brains. In contrast, in MSA, aSyn accumulates in OLG cells. As previously mentioned, multiple lines of evidence support the hypothesis that the aSyn in GCIs in MSA brain derives from aSyn secreted by neurons. In this study, we used OLN-93 cells to assess if: (1) there are differences in response to aSyn uptake or aSyn overexpression in OLG cells regarding BDNF expression; (2) if MSA mutant aSyn similarly affects BDNF expression in OLN-93 cells; (3) FTY720, the FDA-approved multiple sclerosis drug, can counteract the negative effects of aSyn in OLGs; and (4) the contribution of FTY720-induced histone acetylation to stimulate BDNF expression.
Abnormal BDNF expression occurs in MSA patients and MSA models (Kawamoto et al., 1999; May et al., 2014; Nishimura et al., 2005; Ubhi et al., 2010). We found that uptake of WT recombinant human aSyn produced a marked downregulation of BDNF expression in OLN-93 cells. Previous studies of aSyn uptake models in OPC/OLG cells used aSyn concentrations ranging from 0.7 to 5 μM recombinant aSyn protein (Ettle et al., 2014; Kisos et al., 2012; May et al., 2014; Pukass and Richter-Landsberg, 2014; Reyes et al., 2014). For example, after 1 μM recombinant aSyn uptake, Ettle et al. (2014) showed that aSyn accumulation in differentiating OPCs impairs their expression of myelin basic protein. Control experiments of bSyn uptake, in which we did not detect changes in BDNF expression, also suggest that the observed BDNF downregulation is a specific response to aSyn accumulation. In fact, both aSyn uptake and aSyn overexpression downregulated BDNF expression in OLN-93 cells, with more profound effects seen in WT-aSyn and A53E-aSyn stably transfected lines. The recently discovered A53E aSyn mutation was found in a family with early onset PD and MSA (Ghosh et al., 2014; Pasanen et al., 2014). Another recently identified MSA aSyn mutant, G51D (Fares et al., 2014; Kiely et al., 2013), also decreased BDNF expression in OLN-93 stable cell lines, though less profoundly than WT or A53E aSyn. Others report a loss of BDNF in OLG cell lines overexpressing human wild type aSyn, that may cause failure of progenitors, the OPC cells, to differentiate into mature OLG cells (May et al., 2014). The same report noted that OPC differentiation impairment occurs in human MSA brain (May et al., 2014). The degree of BDNF downregulation seen in our studies (moderate vs undetectable) suggest that aSyn overexpression may intensify the BDNF loss compared to aSyn uptake, however, cumulatively the data suggest that aSyn effects on BDNF can be appropriately modeled either by aSyn overexpression or aSyn uptake. Whether particular aSyn species typically have greater effects on BDNF expression will be important to further evaluate when recombinant A53E or G51D aSyn proteins are available for use in aSyn OLG uptake models.
As a control we also assessed NGF expression in OLN-93 cells in response to aSyn uptake and aSyn overexpression. It is important to note that aSyn did not significantly alter the expression of NGF (Fig. 5), another major neurotrophin in OLG cells. This strongly suggests that, at least with regard to neurotrophins, there is not a global effect induced by aSyn accumulation in OPC/OLG cells.
While our aSyn uptake and stable expression models used an OPC/OLG cell line and thus include potential differences with regard to primary cells, the OLN-93 cell line provides a model with intrinsic characteristics resembling late pre-oligodendrocytes (immature cells) even when undifferentiated (De Vries and Boullerne, 2010). Further, the differentiation necessary for primary OPC cells often requires use of pro-differentiating trophic factors, including BDNF (Coelho et al., 2007; Xiao et al., 2012), which could have introduced confounding variables regarding the effects of aSyn accumulation and FTY720 treatment. Furthermore, the use of an immortalized OPC/OLG cell line also allowed us to generate and study stably-transfected cells that represent a more homogenous model than transient transfections would achieve in primary cell cultures.
Regarding the ability of FTY720 to increase BDNF expression, our findings add another type of neural cell (OPC/OLG) to the list in which this response is observed (Deogracias et al., 2012; Efstathopoulos et al., 2015; Heinen et al., 2015; Noda et al., 2013; Vargas-Medrano et al., 2014). Similarly, FTY720 was even able to stimulate BDNF expression above baseline noted for control OLGs in the G51D-aSyn mutant OLN-93 cell line, which was the only aSyn stable cell line that did not undergo massive BDNF downregulation. Importantly, FTY720-associated increases in BDNF completely counteracted the response to recombinant human WT aSyn uptake and partially reversed the near complete downregulation of BDNF in WT-aSyn and A53E-aSyn stably transfected OLN-93 cell lines. These findings provide vital support for FTY720 repurposing for patients with MSA.
To evaluate the pharmacological mechanisms by which FTY720 increases BDNF expression in our cells we assessed independent pharmacological targets of FTY720 that are proposed to have BDNF-inducing properties, (1) activation of S1PRs (Deogracias et al., 2012) and (2) inhibition of HDACs (Hait et al., 2014). This allowed us to show that induction of BDNF expression in OLN-93 cells in response to FTY720 was not accompanied by sustained increase in pERK1/2, which is a downstream effector of S1PRs. Furthermore, the early transient changes in pERK1/2 associated with S1PR-mediated signaling in OLG cells (Coelho et al., 2007) were not detected in our cells after 5, 15 or 45 min treatment with 160 nM FTY720. These experiments suggest that the observed effect of FTY720 in OLN-93 cells does not primarily involve S1PR-mediated signaling at the 160 nM concentration evaluated in our study. On the other hand, 160 nM FTY720 for 12 h significantly increased AcH3 levels, in a manner commonly associated with HDAC inhibition. Furthermore, the elevated AcH3 levels were detected specifically at promoter BDNF-P1, suggesting that changes in histone acetylation are responsible for the observed induction of BDNF expression by FTY720 in our studies. Moreover, treatment with valproic acid, as a positive control, confirmed that HDAC inhibition alone, without S1PR activation, was sufficient to increase BDNF expression in OLN-93 cells. Taken together, our molecular and pharmacological assessments suggest that FTY720 directly contributes to BDNF induction in OLN-93 cells by inhibiting HDAC activity.
In summary, our data also suggest that FTY720 has the potential to treat MSA by its ability to increase BDNF expression as well as by counteracting BDNF downregulation in response to aSyn accumulation in OPC/OLG cells. Furthermore, research on FTY720 in multiple sclerosis has identified other key benefits of FTY720, including a reduction in neuroinflammation and the promotion of remyelination (Cui et al., 2014; Jung et al., 2007; Miron et al., 2008, 2010; Noda et al., 2013; Zhang et al., 2015), both of which would also be beneficial for those with MSA.
Acknowledgements
The authors thank Drs. Macklis and Richter-Landsberg for providing OLN-93 cells, and for guidance for their maintenance and transfection. We are also grateful to the Fogarty International Center - U.S./Costa Rica Neuropsychiatric Genetics Research Training Program (NCOD-5D43TW008333) and the Texas Tech El Paso Graduate School of Biomedical Sciences for tuition and stipend support (to ISU). This work was also supported by funding from the MSA Coalition (243335), Lizanell and Colbert Coldwell Foundation (243036), Hoy Family Research, and gifts from Ms. Anna Mae Doyle (to RGP). This work is dedicated in loving memory of Mr. Steven McLellan Hoy and to all who are affected by Multiple System Atrophy.
Abbreviations:
- AcH3
acetylated histone 3
- aSyn
α-synuclein
- BDNF
brain-derived neurotrophic factor
- bp
base pair
- DLB
Dementia with Lewy Bodies
- FDA
Food and Drug Administration
- GCI
glial cytoplasmic inclusions
- HDAC
histone deacetylase
- MSA
Multiple System Atrophy
- PD
Parkinson’s disease
- rh-aSyn
recombinant human wild type α-synuclein
- WT-aSyn
wild type human α-synuclein
- OLG
oligodendroglia
- OPC
oligodendrocyte precursor cells
- RT
room temperature
Footnotes
Conflict of interest statement
The authors declare that they have no actual or potential conflicts regarding any aspect of the work submitted for publication.
References
- Aranda PS, LaJoie DM, Jorcyk CL, 2012. Bleach gel: a simple agarose gel for analyzing RNA quality. Electrophoresis 33, 366–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellani S, Sousa VL, Ronzitti G, Valtorta F, Meldolesi J, Chieregatti E, 2010. The regulation of synaptic function by alpha-synuclein. Commun. Integr. Biol 3, 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho RP, Payne SG, Bittman R, Spiegel S, Sato-Bigbee C, 2007. The immunomodulator FTY720 has a direct cytoprotective effect in oligodendrocyte progenitors. J. Pharmacol. Exp. Ther 323, 626–635. [DOI] [PubMed] [Google Scholar]
- Cui QL, Fang J, Kennedy TE, Almazan G, Antel JP, 2014. Role of p38MAPK in S1P receptor-mediated differentiation of human oligodendrocyte progenitors. Glia 62, 1361–1375. [DOI] [PubMed] [Google Scholar]
- Deogracias R, Yazdani M, Dekkers MP, Guy J, Ionescu MC, Vogt KE, Barde YA, 2012. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U. S. A 109, 14230–14235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pardo A, Amico E, Favellato M, Castrataro R, Fucile S, Squitieri F, Maglione V, 2014. FTY720 (fingolimod) is a neuroprotective and disease-modifying agent in cellular and mouse models of Huntington disease. Hum. Mol. Genet 23, 2251–2265. [DOI] [PubMed] [Google Scholar]
- Djelloul M, Holmqvist S, Boza-Serrano A, Azevedo C, Yeung Maggie S, Goldwurm S, Frisèn J, Deierborg T, Roybon L, 2015. Alpha-synuclein expression in the oligodendrocyte lineage: an in vitro and in vivo study using rodent and human models. Stem Cell Rep 5, 174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efstathopoulos P, Kourgiantaki A, Karali K, Sidiropoulou K, Margioris AN, Gravanis A, Charalampopoulos I, 2015. Fingolimod induces neurogenesis in adult mouse hippocampus and improves contextual fear memory. Transl. Psychiatry 5, e685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K, 2010. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci 30, 6838–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito A, Dohm CP, Kermer P, Bahr M, Wouters FS, 2007. alpha-synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiol. Dis 26, 521–531. [DOI] [PubMed] [Google Scholar]
- Ettle B, Reiprich S, Deusser J, Schlachetzki JC, Xiang W, Prots I, Masliah E, Winner B, Wegner M, Winkler J, 2014. Intracellular alpha-synuclein affects early maturation of primary oligodendrocyte progenitor cells. Mol. Cell Neurosci 62, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettle B, Schlachetzki JC, Winkler J, 2016. Oligodendroglia and myelin in neurodegenerative diseases: more than just bystanders? Mol. Neurobiol 53, 3046–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares MB, Bouziad NA, Dikiy I, Mbefo MK, Jovicic A, Kiely A, Holton JL, Lee SJ, Gitler AD, Eliezer D, Lashuel HA, 2014. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of alpha-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet 23, 4491–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi M, Nii T, Ishimaru N, Minamino A, Hara D, Takasaki I, Tabuchi A, Tsuda M, 2009. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neurosci. Res 65, 35–43. [DOI] [PubMed] [Google Scholar]
- Ghosh D, Sahay S, Ranjan P, Salot S, Mohite GM, Singh PK, Dwivedi S, Carvalho E, Banerjee R, Kumar A, Maji SK, 2014. The newly discovered Parkinson’s disease associated finnish mutation (A53E) attenuates alpha-synuclein aggregation and membrane binding. Biochemistry 53, 6419–6421. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, 1998. Lewy body diseases and multiple system atrophy as alpha-synucleinopathies. Mol. Psychiatry 3, 462–465. [DOI] [PubMed] [Google Scholar]
- Goedert M, Clavaguera F, Tolnay M, 2010. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci 33, 317–325. [DOI] [PubMed] [Google Scholar]
- Goers J, Manning-Bog AB, McCormack AL, Millett IS, Doniach S, Di Monte DA, Uversky VN, Fink AL, 2003. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 42, 8465–8471. [DOI] [PubMed] [Google Scholar]
- Hait NC, Wise LE, Allegood JC, O’Brien M, Avni D, Reeves TM, Knapp PE, Lu J, Luo C, Miles MF, Milstien S, Lichtman AH, Spiegel S, 2014. Active, phosphorylated fingolimod inhibits histone deacetylases and facilitates fear extinction memory. Nat. Neurosci 17, 971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait NC, Avni D, Yamada A, Nagahashi M, Aoyagi T, Aoki H, Dumur CI, Zelenko Z, Gallagher EJ, Leroith D, Milstien S, Takabe K, Spiegel S, 2015. The phosphorylated prodrug FTY720 is a histone deacetylase inhibitor that reactivates ERalpha expression and enhances hormonal therapy for breast cancer. Oncogenesis 4, e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen A, Beyer F, Tzekova N, Hartung HP, Kury P, 2015. Fingolimod induces the transition to a nerve regeneration promoting Schwann cell phenotype. Exp. Neurol 271, 25–35. [DOI] [PubMed] [Google Scholar]
- Jakes R, Spillantini MG, Goedert M, 1994. Identification of two distinct synucleins from human brain. FEBS Lett 345, 27–32. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, 2014. Neuropathology of multiple system atrophy: new thoughts about pathogenesis. Mov. Disord 29, 1720–1741. [DOI] [PubMed] [Google Scholar]
- Jung CG, Kim HJ, Miron VE, Cook S, Kennedy TE, Foster CA, Antel JP, Soliven B, 2007. Functional consequences of S1P receptor modulation in rat oligodendroglial lineage cells. Glia 55, 1656–1667. [DOI] [PubMed] [Google Scholar]
- Kawamoto Y, Nakamura S, Akiguchi I, Kimura J, 1999. Increased brain-derived neurotrophic factor-containing axons in the basal ganglia of patients with multiple system atrophy. J. Neuropathol. Exp. Neurol 58, 765–772. [DOI] [PubMed] [Google Scholar]
- Khobta A, Anderhub S, Kitsera N, Epe B, 2010. Gene silencing induced by oxidative DNA base damage: association with local decrease of histone H4 acetylation in the promoter region. Nucleic Acids Res 38, 4285–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, Proukakis C, Quinn N, Lees AJ, Hardy J, Revesz T, Houlden H, Holton JL, 2013. alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol 125, 753–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Park JM, Moon J, Choi HJ, 2014. Alpha-synuclein interferes with cAMP/PKA-dependent upregulation of dopamine beta-hydroxylase and is associated with abnormal adaptive responses to immobilization stress. Exp. Neurol 252, 63–74. [DOI] [PubMed] [Google Scholar]
- Kisos H, Pukass K, Ben-Hur T, Richter-Landsberg C, Sharon R, 2012. Increased neuronal alpha-synuclein pathology associates with its accumulation in oligodendrocytes in mice modeling alpha-synucleinopathies. PLoS ONE 7, e46817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontopoulos E, Parvin JD, Feany MB, 2006. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet 15, 3012–3023. [DOI] [PubMed] [Google Scholar]
- Koo JW, Mazei-Robison MS, LaPlant Q, Egervari G, Braunscheidel KM, Adank DN, Ferguson D, Feng J, Sun H, Scobie KN, Damez-Werno DM, Ribeiro E, Pena CJ, Walker D, Bagot RC, Cahill ME, Anderson SA, Labonte B, Hodes GE, Browne H, Chadwick B, Robison AJ, Vialou VF, Dias C, Lorsch Z, Mouzon E, Lobo MK, Dietz DM, Russo SJ, Neve RL, Hurd YL, Nestler EJ, 2015. Epigenetic basis of opiate suppression of Bdnf gene expression in the ventral tegmental area. Nat. Neurosci 18, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma KL, Song LK, Yuan YH, Zhang Y, Han N, Gao K, Chen NH, 2014. The nuclear accumulation of alpha-synuclein is mediated by importin alpha and promotes neurotoxicity by accelerating the cell cycle. Neuropharmacology 82, 132–142. [DOI] [PubMed] [Google Scholar]
- Makarona K, Caputo VS, Costa JR, Liu B, O’Connor D, Iskander D, Roper D, Robertson L, Bhatnagar N, Terpos E, Georgiou E, Papaioannou M, Layton DM, Luzzatto L, Roberts I, Karadimitris A, 2014. Transcriptional and epigenetic basis for restoration of G6PD enzymatic activity in human G6PD-deficient cells. Blood 124, 134–141. [DOI] [PubMed] [Google Scholar]
- May VE, Ettle B, Poehler AM, Nuber S, Ubhi K, Rockenstein E, Winner B, Wegner M, Masliah E, Winkler J, 2014. alpha-Synuclein impairs oligodendrocyte progenitor maturation in multiple system atrophy. Neurobiol. Aging 35, 2357–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguez A, Garcia-Diaz Barriga G, Brito V, Straccia M, Giralt A, Gines S, Canals JM, Alberch J, 2015. Fingolimod (FTY720) enhances hippocampal synaptic plasticity and memory in Huntington’s disease by preventing p75NTR up-regulation and astrocyte-mediated inflammation. Hum. Mol. Genet 24, 4958–4970. [DOI] [PubMed] [Google Scholar]
- Miller DW, Cookson MR, Dickson DW, 2004. Glial cell inclusions and the pathogenesis of neurodegenerative diseases. Neuron Glia Biol 1, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DW, Johnson JM, Solano SM, Hollingsworth ZR, Standaert DG, Young AB, 2005. Absence of alpha-synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J. Neural Transm 112, 1613–1624. [DOI] [PubMed] [Google Scholar]
- Miron VE, Jung CG, Kim HJ, Kennedy TE, Soliven B, Antel JP, 2008. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann. Neurol 63, 61–71. [DOI] [PubMed] [Google Scholar]
- Miron VE, Ludwin SK, Darlington PJ, Jarjour AA, Soliven B, Kennedy TE, Antel JP, 2010. Fingolimod (FTY720) enhances remyelination following demyelination of organotypic cerebellar slices. Am. J. Pathol 176, 2682–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura M, Kuno S, Kaji R, Kawakami H, 2005. Brain-derived neurotrophic factor gene polymorphisms in Japanese patients with sporadic Alzheimer’s disease, Parkinson’s disease, and multiple system atrophy. Mov. Disord 20, 1031–1033. [DOI] [PubMed] [Google Scholar]
- Noda H, Takeuchi H, Mizuno T, Suzumura A, 2013. Fingolimod phosphate promotes the neuroprotective effects of microglia. J. Neuroimmunol 256, 13–18. [DOI] [PubMed] [Google Scholar]
- Park H, Poo MM, 2013. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci 14, 7–23. [DOI] [PubMed] [Google Scholar]
- Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Poyhonen M, Paetau A, 2014. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 35 (2180), e2181–2185. [DOI] [PubMed] [Google Scholar]
- Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ, 2002. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci 22, 3090–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW, Horgan GW, Dempfle L, 2002. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukass K, Richter-Landsberg C, 2014. Oxidative stress promotes uptake, accumulation, and oligomerization of extracellular alpha-synuclein in oligodendrocytes. J. Mol. Neurosci 52, 339–352. [DOI] [PubMed] [Google Scholar]
- Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E, 2014. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 62, 387–398. [DOI] [PubMed] [Google Scholar]
- Siddiqui A, Chinta SJ, Mallajosyula JK, Rajagopolan S, Hanson I, Rane A, Melov S, Andersen JK, 2012. Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: implications for Parkinson’s disease. Free Radic. Biol. Med 53, 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC, 1985. Measurement of protein using bicinchoninic acid. Anal. Biochem 150, 76–85. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, 1999. Parkinson’s disease, dementia with Lewy bodies and multiple system atrophy are alpha-synucleinopathies. Park. Relat. Disord 5, 157–162. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M, 2000. The alpha-synucleinopathies: parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann. N. Y. Acad. Sci 920, 16–27. [DOI] [PubMed] [Google Scholar]
- Stefanova N, Wenning GK, 2016. Review: multiple system atrophy: emerging targets for interventional therapies. Neuropathol. Appl. Neurobiol 42, 20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F, Marini AM, Lipsky RH, 2010. Effects of histone deacetylase inhibitor Trichostatin A on epigenetic changes and transcriptional activation of Bdnf promoter 1 by rat hippocampal neurons. Ann. N. Y. Acad. Sci 1199, 186–193. [DOI] [PubMed] [Google Scholar]
- To KK, Polgar O, Huff LM, Morisaki K, Bates SE, 2008. Histone modifications at the ABCG2 promoter following treatment with histone deacetylase inhibitor mirror those in multidrug-resistant cells. Mol. Cancer Res 6, 151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubhi K, Rockenstein E, Mante M, Inglis C, Adame A, Patrick C, Whitney K, Masliah E, 2010. Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J. Neurosci 30, 6236–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubhi K, Inglis C, Mante M, Patrick C, Adame A, Spencer B, Rockenstein E, May V, Winkler J, Masliah E, 2012. Fluoxetine ameliorates behavioral and neuropathological deficits in a transgenic model mouse of alpha-synucleinopathy. Exp. Neurol 234, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Medrano J, Krishnamachari S, Villanueva E, Godfrey WH, Lou H, Chinnasamy R, Arterburn JB, Perez RG, 2014. Novel FTY720-based compounds stimulate neurotrophin expression and phosphatase activity in dopaminergic cells. ACS Med. Chem. Lett 5, 782–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries GH, Boullerne AI, 2010. Glial cell lines: an overview. Neurochem. Res 35, 1978–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H, 1998. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett 249, 180–182. [DOI] [PubMed] [Google Scholar]
- Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, Kinyamu H, Lu N, Gao X, Leng Y, Chuang DM, Zhang W, Lu RB, Hong JS, 2008. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int. J. Neuropsychopharmacol 11, 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Ferner AH, Wong AW, Denham M, Kilpatrick TJ, Murray SS, 2012. Extracellular signal-regulated kinase 1/2 signaling promotes oligodendrocyte myelination in vitro. J. Neurochem 122, 1167–1180. [DOI] [PubMed] [Google Scholar]
- Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM, 2009. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol. Psychiatry 14, 51–59. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Sun J, Zhao M, Hu J, Wang X, Du G, Chen NH, 2010. Overexpression of alpha-synuclein down-regulates BDNF expression. Cell Mol. Neurobiol 30, 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Tan M, Kohyama J, Sneddon M, Watson JB, Sun YE, Xie CW, 2011. Epigenetic enhancement of BDNF signaling rescues synaptic plasticity in aging. J. Neurosci 31, 17800–17810. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang J, Zhang ZG, Li Y, Ding X, Shang X, Lu M, Elias SB, Chopp M, 2015. Fingolimod treatment promotes proliferation and differentiation of oligodendrocyte progenitor cells in mice with experimental autoimmune encephalomyelitis. Neurobiol. Dis 76C, 57–66. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Gu G, Goodlett DR, Zhang T, Pan C, Montine TJ, Montine KS, Aebersold RH, Zhang J, 2004. Analysis of alpha-synuclein-associated proteins by quantitative proteomics. J. Biol. Chem 279, 39155–39164. [DOI] [PubMed] [Google Scholar]
- Zhou M, Xu S, Mi J, Ueda K, Chan P, 2013. Nuclear translocation of alpha-synuclein increases susceptibility of MES23.5 cells to oxidative stress. Brain Res 1500, 19–27. [DOI] [PubMed] [Google Scholar]