

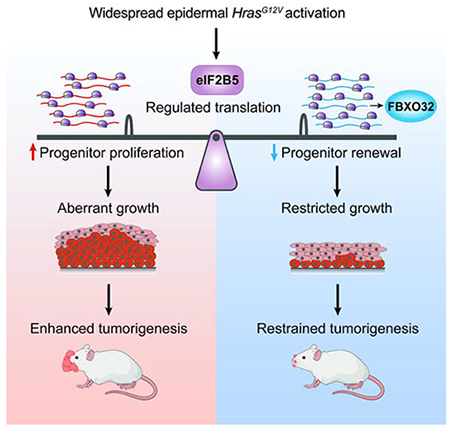
Summary
Human skin tolerates a surprisingly high burden of oncogenic lesions. While adult epidermis can suppress the expansion of individual mutant clones, the mechanisms behind tolerance to oncogene activation across broader regions of tissue are unclear. Here, we uncover a dynamic translational mechanism that coordinates oncogenic HRAS-induced hyperproliferation with loss of progenitor self-renewal to restrain aberrant growth and tumorigenesis. We identify translation initiator eIF2B5 as a central co-regulator of HRAS proliferation and cell fate choice. By coupling in vivo ribosome profiling with genetic screening, we provide direct evidence that oncogene-induced loss of progenitor self-renewal is driven by eIF2B5-mediated translation of ubiquitination genes. Ubiquitin ligase FBXO32 specifically inhibits epidermal renewal without affecting overall proliferation, thus restraining HRAS-driven tumorigenesis while maintaining normal tissue growth. Thus, oncogene-driven translation is not necessarily inherently tumor promoting but instead can manage widespread oncogenic stress by steering progenitor fate to prolong normal tissue growth.
Graphical Abstract
eTOC
Developing epidermis has remarkable ability to suppress aberrant growth despite widespread oncogenic insult. Cai et al. uncover translation initiation factor eIF2B5 as a central coordinator of HRAS progenitor behavior. Functional dissection of the oncogenic translatome reveals a dynamic translational mechanism that inhibits renewal during oncogenic hyperproliferation to restrain tumorigenesis.
Introduction
The skin possesses remarkable ability to tolerate genetic and structural abnormalities. Surprisingly, this extends to mutations in known cancer-driving genes, which are frequently found in physiologically normal human skin (Martincorena et al., 2015), suggesting that the tissue has adaptive mechanisms to restrain the expansion of mutant cell populations and protect against progression to cancer. We recently observed through direct intravital imaging that the adult epidermis can entirely resolve abnormal growth of mutant cell clones following activation of Hras1 or β-catenin (Brown et al., 2017). We further uncovered that oncogenic Pik3ca epidermal clones can be completely blocked from expansion and eventually expelled from the adult tissue through loss of growth-sustaining progenitor cells (Ying et al., 2018). However, these studies only examined the epidermis’ growth-restrictive potential in the context of individual clones arising from a single cell. The mechanisms behind tissue tolerance to widespread oncogene activation, as seen in oncogene-driven congenital overgrowth disorders (Keppler-Noreuil et al., 2016; Rauen, 2013) and in field cancerization where broad areas of genetically altered tissue are asymptomatic (Curtius et al., 2017), remain unexplored.
Tissues can employ various cell-autonomous strategies to block the proliferation of single clones with somatic mutations, including apoptosis and senescence (Braig et al., 2005; Fearnhead et al., 1998). Neighboring wildtype (WT) cells can also facilitate oncogene tolerance through non-cell-autonomous events that restrict expansion or displace mutant clones from the tissue (Brown et al., 2017; Ying et al., 2018). An illustrative case is the recent finding that WT cells surrounding mutant Pik3ca clones maintain pro-renewal JNK signaling, allowing WT cells to outcompete and expel highly differentiating mutant clones (Ying et al., 2018). However, this mechanism for oncogene tolerance is not feasible when a large proportion of the tissue carries the same lesion, abolishing the growth-suppressive potential of WT neighbors. Furthermore, since the epidermis requires frequent cell turnover for its development and function (Fuchs and Raghavan, 2002; van der Flier and Clevers, 2009), extensive elimination of mutant cells or a complete proliferation block would significantly disrupt tissue architecture and integrity. How oncogenic epidermis preserves the rapid physiological growth needed for tissue development while restraining pathological overgrowth remains a fundamental question.
The embryonic murine interfollicular epidermis (IFE) is an ideal system to explore oncogene-induced stem cell behaviors in the context of rapid tissue growth (Beronja et al., 2013; 2010; Williams et al., 2011). Its well-defined structure features frequently proliferating basal progenitor cells, which have the unique ability to self-renew, and their suprabasal differentiated progeny, which are post-mitotic and provide the form and function of skin (Fuchs, 2007). Therefore, the IFE basal progenitors, which are thought to undergo stochastic fate decisions to self-renew or differentiate, are the key regulators of epidermal growth and maintenance. Here, we report that broad activation of oncogenic HrasG12V during epidermal development induces a translational landscape that inhibits progenitor cell renewal to contain tissue overgrowth. We uncover translation initiation factor eIF2B5 as a central regulator of HrasG12V growth and coordinator of progenitor cell response to oncogene-induced hyperproliferation. This coordination is achieved through eIF2B5-mediated translation of distinct proliferation and renewal regulators. Notably, while previous studies have indirectly linked translation rate with cell fate choice (Blanco et al., 2016; Liakath-Ali et al., 2018; Signer et al., 2014), our analysis provides direct functional evidence that oncogene-driven renewal inhibition operates through eIF2B5-mediated translation of ubiquitination genes, resulting in striking restriction of HRAS tumorigenesis. Our study therefore uncovers a dynamic translational mechanism by which the epidermis manages widespread oncogenic stress to prolong normal tissue growth.
Results
Broad oncogenic HRAS activation induces translationally regulated progenitor cell behaviors
To probe mechanisms that restrain aberrant growth during widespread oncogene activation, we employed a conditional knock-in mouse that activates oncogenic HrasG12V at physiological levels upon transduction with lentivirus containing Cre recombinase (LV-Cre; Figures S1A and S1B) (Chen et al., 2009). We used an in utero lentiviral injection method (Beronja et al., 2010) to target the ectoderm of E9.5 embryos with LV-Cre, which resulted in broad epidermal transduction reported by the expression of mGFP [Rosa26mT/mG Cre reporter (Muzumdar et al., 2007)] or YFP [Rosa26YFP/YFP Cre reporter (Srinivas et al., 2001)] (Figure S1C). We first assessed progenitor cell proliferation and found that epidermal HrasG12V activation induced a ~2-fold increase in IFE basal cell proliferation rate (Figure 1A). During normal development, progenitor proliferation is accompanied by a high probability of self-renewal (renewal probability=0.7), which sustains physiological epidermal expansion (Beronja et al., 2013; Ying et al., 2018). We therefore expected that the elevated proliferation rate in HrasG12V basal progenitors would have a profound effect on epidermal growth. We modelled theoretical tissue expansion from a single ectodermal cell at E9.5 to E18.5, using observed proliferation rates in HrasG12V and WT basal cells and a renewal probability=0.7 following epidermal stratification at E15.5 (Sotiropoulou and Blanpain, 2012) (Figure 1B). Our model successfully predicted the experimentally observed tissue expansion in WT epidermis (Beronja et al., 2013), and further suggested that HrasG12V epidermis would undergo 25-fold the growth seen in WT epidermis over the same timeframe. However, this prediction was not compatible with the limited tissue expansion observed in E18.5 HrasG12V epidermis (Figures S1D and S1E). Quantification of clonal expansion from E9.5 to E18.5 revealed that HrasG12V expression induces only ~4-fold the growth seen in WT epidermis (Figures 1B and S1F). Therefore, increased proliferation of HrasG12V epidermis appears to be counteracted by a potent growth-suppressive mechanism.
Figure 1. Oncogenic HRAS activation induces translationally regulated progenitor cell behaviors.
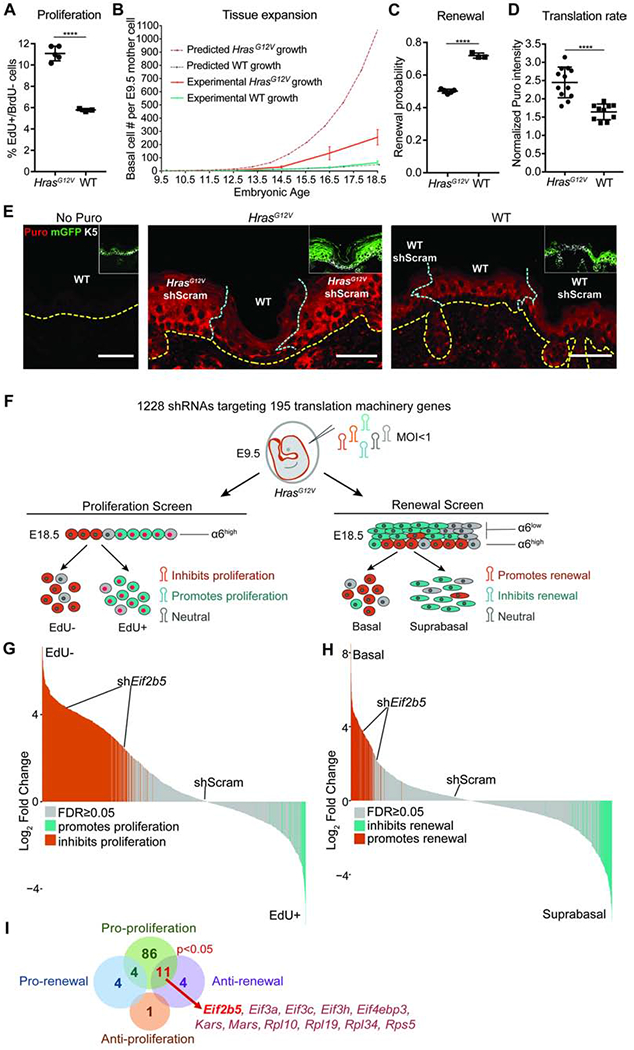
A) Proliferation rate in IFE basal progenitor cells transduced with control shRNA. n=4 HrasG12V and 3 WT animals. Approx. 100 basal cells were scored per animal.
B) Predicted (dashed lines) and experimentally observed (solid lines) tissue growth derived from a single progenitor cell at E9.5. Predicted tissue growth is based on observed progenitor cell proliferation rates (Figure 1A) and renewal probability=0.7 (Ying et al., 2018). n=3 animals per timepoint per condition.
C) Progenitor cell renewal probability in IFE basal cells transduced with control shRNA. n=4 HrasG12V and 3 WT animals. Approx. 100 cell divisions were scored per animal.
D) Translation rate as measured by in vivo puromycin incorporation. HrasG12V and WT epidermis were transduced with control shRNA. Puromycin intensity in transduced IFE basal cells was normalized to underlying dermal puromycin intensity. n=12 HrasG12V and 11 WT imaging fields containing epidermis and dermis. 3 animals per condition were assessed.
E) Representative immunofluorescence staining of puromycin incorporation (red) in transduced (mGFP+, green, inset) IFE basal cells (K5+, white, inset) at E18.5. HrasG12V and WT epidermis were transduced with control shRNA. No-puro control used skin from vehicle-only injected WT mice. Yellow dashed line marks basement membrane; blue dashed line marks border of transduced epidermis. Scale bars, 50μm.
F) In vivo genetic screens to identify epidermal progenitor proliferation and renewal regulators. Assays are based on enrichment (orange) or depletion (blue) of shRNAs in non-dividing EdU− relative to dividing EdU+ epidermal progenitors (proliferation screen) or α6-Integrinhigh basal progenitors relative to α6-Integrinlow differentiated suprabasal cells (renewal screen).
G-H) Needle plots show fold change for individual shRNAs in (G) proliferation and (H) renewal screens. n=3 biological replicates (DESeq2 FDR<0.05).
I) Overlap of renewal and proliferation screens reveals regulators of HrasG12V basal progenitor behaviors. Hypergeometric test used to analyze significance of gene overlap.
Mean and SD are shown. Unpaired two-tailed t test used to determine statistical differences. ****:P-value<0.0001. See also Figure S1 and Table S1.
While apoptosis and senescence occur at very low rates and are not significant factors in limiting epidermal growth (Beronja et al., 2013), we recently demonstrated that strong inhibition of progenitor cell renewal can reduce the fitness of mutant clones relative to WT neighbors, resulting in their complete ablation from the epidermis (Ying et al., 2018). Though this clonal expulsion mechanism cannot be employed during widespread oncogene activation, we hypothesized that moderate suppression of basal progenitor renewal may offset hyperproliferation to limit expansion of HrasG12V epidermis. To test the rate of progenitor renewal following HrasG12V activation, we used our EdU/BrdU pulse-chase incorporation assay (Ying et al., 2018) to distinctly mark replicating progenitors and score the fate of their progeny after cell division. We found a significant reduction in the probability of IFE progenitor cell renewal in broadly activated HrasG12V epidermis (Figure 1C). Importantly, although reduced, HrasG12V renewal probability remained at 0.5, which allows for tissue maintenance while limiting excess growth. We hypothesized that a switch from high renewal and low proliferation in WT tissue to low renewal and high proliferation in HrasG12V epidermis is indicative of a molecular mechanism that can coordinate stem cell response to widespread oncogene activation and contribute to the fundamental plasticity of epidermal development.
Intriguingly, protein synthesis has recently been implicated in regulating both stem cell proliferation and fate choice (Buszczak et al., 2014) and may be a critical coordinator of the epidermal response to oncogene activation. To probe translation’s potential role in oncogene tolerance, we first examined the effect of HrasG12V activation on progenitor cell translation rate. We employed the SUnSET method, where in vivo protein synthesis is quantified by detecting puromycin incorporation into nascent peptides (Schmidt et al., 2009), which revealed a ~2-fold increase in protein synthesis rate in HrasG12V basal cells (Figure 1D). This translation rate elevation is illustrated at the border regions containing activated HrasG12V epidermis and neighboring untransduced WT epidermis (Figure 1E). Elevated translation rate was also seen in HrasG12V primary keratinocytes cultured from FACS isolated E18.5 basal cells (Figure S1G), suggesting that HrasG12V-induced translation upregulation can occur independently of dermal signaling.
We hypothesized that elevated translation rate in HrasG12V basal progenitors reflected an interactive relationship with the translation apparatus. We performed two in vivo functional screens to identify the translation machinery components that regulate HrasG12V progenitor proliferation and renewal (Figure 1F). We transduced E9.5 HrasG12V epidermis at clonal density (MOI<1) with a lentivirus pool of 1228 shRNAs targeting 195 translation initiation factors, elongation factors, ribosomal proteins, tRNA synthetases, and poly(A) binding proteins (Bhat et al., 2015; Moffat et al., 2006) (Figure 1F). Our screens utilized the validated principle (Ying et al., 2018) that shRNAs targeting proliferation promoters are enriched in the non-dividing relative to dividing cell population, while shRNAs targeting renewal promoters are enriched in the suprabasal relative to basal cell population (Figure 1F). We classified genes as proliferation or renewal regulators if at least two of their shRNAs were significantly changed with consistent directionality (Beronja et al., 2013).
We uncovered 101 translation genes as proliferation promoters (Figures 1G and 1I, Table S1), consistent with the essential role of translation in cell cycle progression (A. S. Y. Lee et al., 2015; Stumpf et al., 2013). In contrast, our screens identified only 15 translation genes as renewal inhibitors and 8 as renewal promoters (Figures 1H and 1I, Table S1). Notably, a significant subset of 11 genes simultaneously promoted HrasG12V progenitor proliferation and inhibited self-renewal (p<0.05; Figure 1I). Amongst these dual regulators, translation initiation factor Eif2b5 was substantially altered across epithelial cancers and implicated in a congenital disorder marked by tissue specific defects (W. Li et al., 2004; Scali et al., 2006; Gao et al., 2013). We focused our downstream studies on Eif2b5 to dissect the interplay between HrasG12V activation and protein synthesis control.
RAS progenitor cell behaviors and tissue growth are dependent on eIF2B5
To elucidate how eIF2B5 regulates HrasG12V stem cell behaviors, we first assessed its role in HrasG12V-driven translation. We examined basal cell translation rate in transduced (mGFP+) epidermis (Rosa26mT/mG Cre reporter (Muzumdar et al., 2007)) and found that shRNA depletion of Eif2b5 (Figures S2A and S2B) rescued HrasG12V translation rate to WT levels (Figures 2A and 2B). Interestingly, comparable Eif2b5 depletion in WT epidermis did not significantly affect translation rate, suggesting that HrasG12V epidermis has increased dependency on eIF2B5 (Figures 2A and 2B). Of note, the elevated translation rate observed in cultured HrasG12V primary keratinocytes was also dependent on eIF2B5 (Figure S2C) and was not confounded by differences in proteasome activity between genetic backgrounds (Figure S2D). Observed translational differences were not due to induction of secondary cell stress, as indicated by eIF2α phosphorylation on serine 51 (Figure S2C) (Koromilas, 2015). Instead, Ser51 p-eIF2α was decreased upon Eif2b5 depletion (Figure S2C), which may act as a compensatory mechanism to rescue translation.
Figure 2. Oncogenic HRAS-induced progenitor cell behaviors and tissue growth are dependent on eIF2B5.
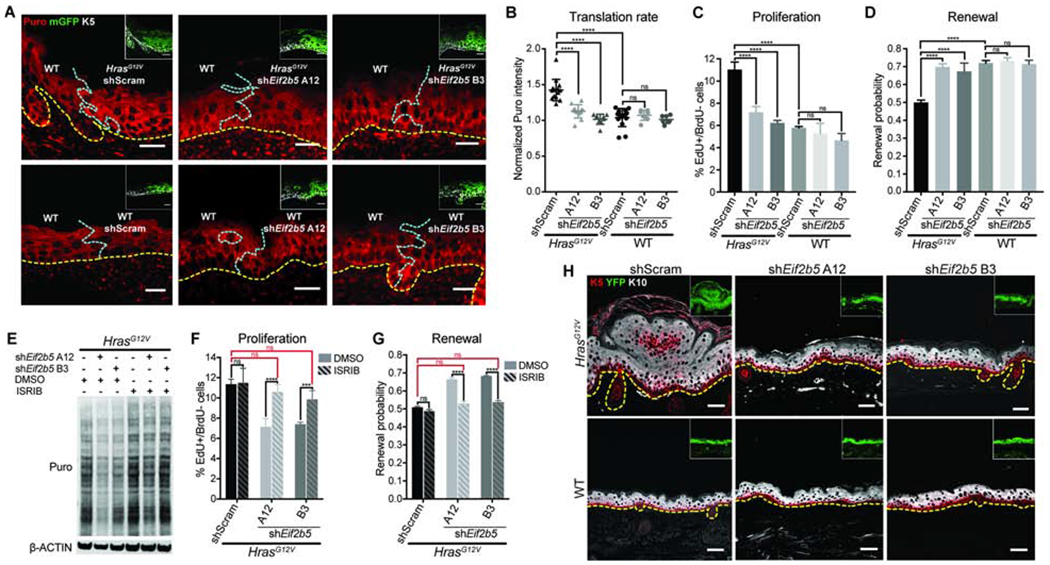
A-B) Representative (A) immunofluorescence staining and (B) quantification of puromycin incorporation (red) in transduced (mGFP+, green, inset) IFE basal cells (K5+, white, inset), normalized to neighboring untransduced basal cells (K5+mGFP-). Yellow dashed line marks basement membrane; blue dashed line marks border of transduced epidermis. n=12 HrasG12V+shScram, 12 HrasG12V+shEif2b5 A12, 9 HrasG12V+shEif2b5 B3, 13 WT+shScram, 9 WT+shEif2b5 A12, and 9 WT+shEif2b5 B3 imaging fields containing transduced and untransduced epidermis. 3 animals per condition were assessed. Scale bars, 25 μm.
C-D) (C) Proliferation rate and (D) renewal probability in transduced IFE basal progenitor cells. n=3 animals per condition. Approx. 100 basal cells (for proliferation assay) or 100 cell divisions (for renewal assay) were scored per animal.
E) Western blot of puromycin incorporation (Puro) in transduced primary keratinocytes treated with DMSO or ISRIB.
F-G) (F) Proliferation rate and (G) renewal probability in transduced IFE basal progenitor cells treated with DMSO or ISRIB. n=3 animals per condition. Approx. 100 basal cells (for proliferation assay) or 100 cell divisions (for renewal assay) were scored per animal. Key comparisons shown in red.
H) Representative immunofluorescence staining of keratin 5 (K5, red) and keratin 10 (K10, white) in transduced E18.5 epidermis. Cre reporter YFP (green) marks the transduced epidermis (inset). Dashed line marks the basement membrane. Scale bars, 50μm.
Mean and SD are shown. ANOVA with Tukey’s range test for multiple comparisons used to determine statistical differences. *:P-value<0.05; ***:P-value<0.001; ****:P-value<0.0001; ns=not significant. See also Figure S2.
Next, we explored whether HrasG12V-induced progenitor cell behaviors are also dependent on eIF2B5. Upon Eif2b5 depletion, HrasG12V progenitor cell proliferation rate and renewal probability were both rescued to WT levels (Figures 2C and 2D). In contrast, comparable Eif2b5 depletion in WT tissue did not significantly affect progenitor proliferation or renewal, suggesting that eIF2B5 has an oncogene-specific regulatory effect on stem cell behavior (Figures 2C and 2D). eIF2B5 is the catalytic subunit of the eIF2B complex, a guanine exchange factor that returns eIF2 to its active state, allowing eIF2 to form the ternary complex for translation initiation (Figure S2E) (Pavitt, 2005). We next tested if HrasG12V progenitor cell behaviors were dependent on eIF2B complex activity. We treated Eif2b5-depleted HrasG12V keratinocytes with ISRIB, a small molecule that promotes eIF2B dimerization and activity (Sidrauski et al., 2013; 2015), and found that ISRIB largely rescued HrasG12V translation rate (Figure 2E). We reasoned that if eIF2B5 is regulating HrasG12V progenitor cell behavior through its canonical eIF2B complex role, ISRIB treatment should suppress Eif2b5 knockdown phenotypes by increasing eIF2B activity. Indeed, Eif2b5 depletion in HrasG12V embryos followed by ISRIB treatment of maternal mice completely restored the rapid proliferation rate and low renewal probability in HrasG12V epidermis (Figures 2F and 2G).
We hypothesized that eIF2B5-dependent proliferation rate and renewal probability were able to account for the HrasG12V growth phenotype, as no difference in apoptosis was detected across any conditions (Figure S2F). HrasG12V mice displayed grossly thickened and raised skin (Figure S2G), indicative of epidermal overgrowth. Tissue cross-sections of fully transduced (YFP+) epidermis (expressing Rosa26YFP/YFP Cre reporter (Srinivas et al., 2001)) revealed hyperplastic expansion in both basal progenitor (K5+) and suprabasal differentiated (K10+) layers (Figure 2H). Remarkably, Eif2b5 depletion in HrasG12V epidermis restored the smooth appearance and stratified architecture found in WT skin (Figures 2H and S2G). HrasG12V primary keratinocytes also exhibited eIF2B5-dependent growth rate elevation, further reproducing the behavior of basal cells in vivo (Figure S2H).
Together, these studies establish eIF2B5, through its role in eIF2B, as a central regulator of HRAS-induced progenitor cell behaviors and tissue growth.
eIF2B5 controls the translation of a specific subset of HrasG12V-dependent mRNAs
To dissect molecular mechanisms by which eIF2B5 drives HrasG12V progenitor cell behaviors, we used in vivo ribosome profiling to identify eIF2B5-dependent translational changes in HrasG12V progenitors (Figure 3A) (Ingolia et al., 2009; Sendoel et al., 2017). We transduced E9.5 HrasG12V and WT epidermis with control shRNA or shEif2b5 B3, our most efficient shRNA targeting Eif2b5. The virus was titrated to ensure comparable transduction rate across conditions, which suppressed HrasG12V epidermal overgrowth without tissue damage (Figure S3A). E18.5 skin was enzymatically dissociated to generate a basal cell-enriched population (Figure S3B) with preserved polysomes and depleted Eif2b5 (Figures S3C and S3D). Our ribosome footprint libraries displayed expected enrichment of 28nt reads, triplet periodicity, and alignment to coding regions (Figures S3E–G). Across biological replicates, our sequencing libraries were highly reproducible (Pearson r2 > 0.9) (Figures S3H and S3I).
Figure 3. eIF2B5 regulates translation of a specific subset of HrasG12V-dependent mRNAs.
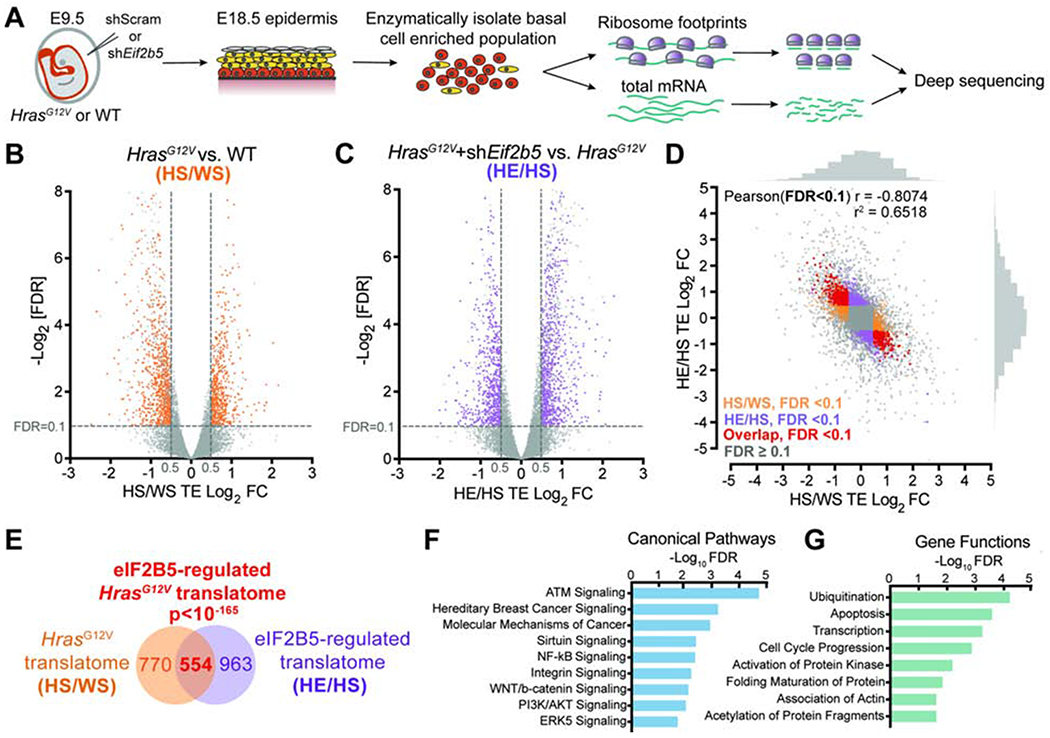
A) In vivo basal cell ribosome profiling strategy. Transduced epidermis was enzymatically digested at E18.5 to yield a basal cell enriched population for profiling.
B-C) Volcano plots of translation efficiency (TE) changes in E18.5 basal cells comparing (B) HrasG12V+shScram (HS) with WT+shScram (WS) and (C) HrasG12V+shEif2b5 (HE) with HrasG12V+shScram (HS). Significant genes (colored) have Log2TE fold change>0.5, FDR<0.1 (dashed lines) by Xtail analysis and insignificant transcriptional changes by EdgeR analysis (FDR>0.1 or Log2FC<0.5). n=3 biological replicates.
D-E) Comparison of genes with significant translation efficiency changes in (B) (HS/WS; in orange) and (C) (HE/HS; in purple) reveals (D) negative correlation and (E) significant overlap (in red). The overlapping gene set comprises the eIF2B5-regulated HrasG12V translatome. Pearson’s X2 test used to analyze correlation of genes with significant translation efficiency, and hypergeometric test used to analyze overlap of gene sets.
F-G) Ingenuity pathway analysis of overlapping genes for enriched (F) canonical pathways and (G) gene functions.
To assess differences in active translation independent of transcriptional changes, we identified all genes with significantly altered translation efficiencies (Log2FC>0.5, FDR<0.1) (Xiao et al., 2016) and excluded genes whose transcripts were also significantly changed (Log2FC>0.5, FDR<0.1) (Robinson et al., 2010). To identify the HrasG12V translatome, we compared HrasG12V+shScram to WT+shScram (HS/WS) profiling and identified 1324 genes that were translationally altered upon HrasG12V activation (Figure 3B, Table S2). We then compared HrasG12V+shEif2b5 to HrasG12V+shScram (HE/HS) profiling and identified 1517 genes whose translation was regulated by eIF2B5 (Figure 3C, Table S3). Strikingly, a large portion of the HrasG12V translatome was significantly rescued by Eif2b5 depletion, resulting in strong negative correlation between significantly altered genes in HS/WS and HE/HS analyses (Pearson r = −0.8074, r2 = 0.6518) (Figures 3D and S3J, Table S4). Overall, we found 554 shared genes between the two analyses, representing the subset of the HrasG12V translatome that is regulated by eIF2B5 (Figure 3E). This gene set is enriched for cancer-related pathways and functions involving transcription, cell cycle progression, and post-translational gene regulation (Figures 3F and 3G) (Krämer et al., 2013).
To independently assess the validity of our profiling approach, we analyzed the polysome distribution of a set of candidate mRNAs (Figure S3K). We observed that Mgarp, Sdhaf1, Hmgn1, Nfu1, Lmna, Zc3h18, and Cbx5 mRNAs were enriched in heavier polysome fractions in HrasG12V compared to WT basal progenitors. Each gene exhibited mRNA re-distribution into lighter polysome fractions after eIF2B5 depletion, which was not seen in a control mRNA (Figure S3K), validating our in vivo polysome profiling results. To investigate how translation specificity may be regulated, we analyzed the 5’ untranslated region (5’ UTR) composition of HrasG12V (HS/WS) and eIF2B5 (HE/HS) translationally dependent mRNAs. We observed higher GC content and lower thermodynamic stability of these 5’ UTRs compared to 19,009 control genes (Figures S3L and S3M) with no significant difference in their length (Figures S3N). This suggested that HrasG12V- and eIF2B5-dependent mRNAs may possess a cis-regulatory element encoded within their 5’ UTRs. Motif analysis revealed a guanine-enriched sequence with high incidence of guanine at position 20, similar to a translation initiation sensitive motif called the Guanine Rich Translational Element (GRTE) previously described in prostate cancer (Liu et al., 2019) (Figure S3O). The motif was present in 62.48% of HrasG12V translationally upregulated mRNAs versus 26.86% of genomic 5’ UTR sequences (P<2.2e-16). Importantly, this enrichment was also observed in eIF2B5-dependent mRNAs (70.97% versus 26.86%, P<2.2e-16) (Figure S3O).
Collectively, these studies unveil a distinct regulon of genes within the HrasG12V translatome that are selectively translated by eIF2B5.
Oncogenic HRAS-induced loss of renewal is driven by eIF2B5-regulated ubiquitination
A molecular mechanism that irretrievably couples proliferation with loss of renewal is problematic for the epidermis, which requires rapid baseline proliferation for its development and function. We therefore hypothesized that eIF2B5 co-regulates HrasG12V stem cell behaviors through separate translational networks that independently drive proliferation or inhibit renewal. Such functional segregation would allow the epidermis to dynamically adapt its cell fate choices without compromising its physiological need for continual proliferation. To separate the eIF2B5-regulated oncogenic translatome into independent proliferation promoters or renewal inhibitors, we utilized our in vivo functional screening strategy (Figure 1F). We screened a lentivirus pool of 3607 shRNAs targeting the 554 eIF2B5-regulated genes identified by ribosome profiling (Figures 4A and 4B), which revealed 72 genes that specifically promoted proliferation and 40 genes that inhibited renewal in HrasG12V progenitor cells (Figure 4C, Table S5). Supporting the validity of our screens, negative regulators of p53, Mdm2 and Dnajb9, were identified as top proliferation promoters (Figure 4A) (H. J. Lee et al., 2014; Wade et al., 2013). In our renewal screen, Sox4 was the top renewal promoter (Bergsland et al., 2006; Lourenço and Coffer, 2017), while peroxisome biogenesis factor Pex16 was a renewal inhibitor, supporting the recent finding that peroxisome function is required for epidermal differentiation (Figure 4B) (Asare et al., 2017). In addition to expected mitotic cell division genes (Duronio and Xiong, 2013), proliferation-specific regulators were enriched for transcription factors (Figure S4A). This finding is consistent with the transcriptional changes found in our ribosome profiling, where a large proportion of transcriptionally altered genes was upregulated in HrasG12V progenitor cells and rescued by Eif2b5 depletion (Figure S4B).
Figure 4. eIF2B5 regulates oncogenic translation of genes that specifically promote proliferation or inhibit renewal.
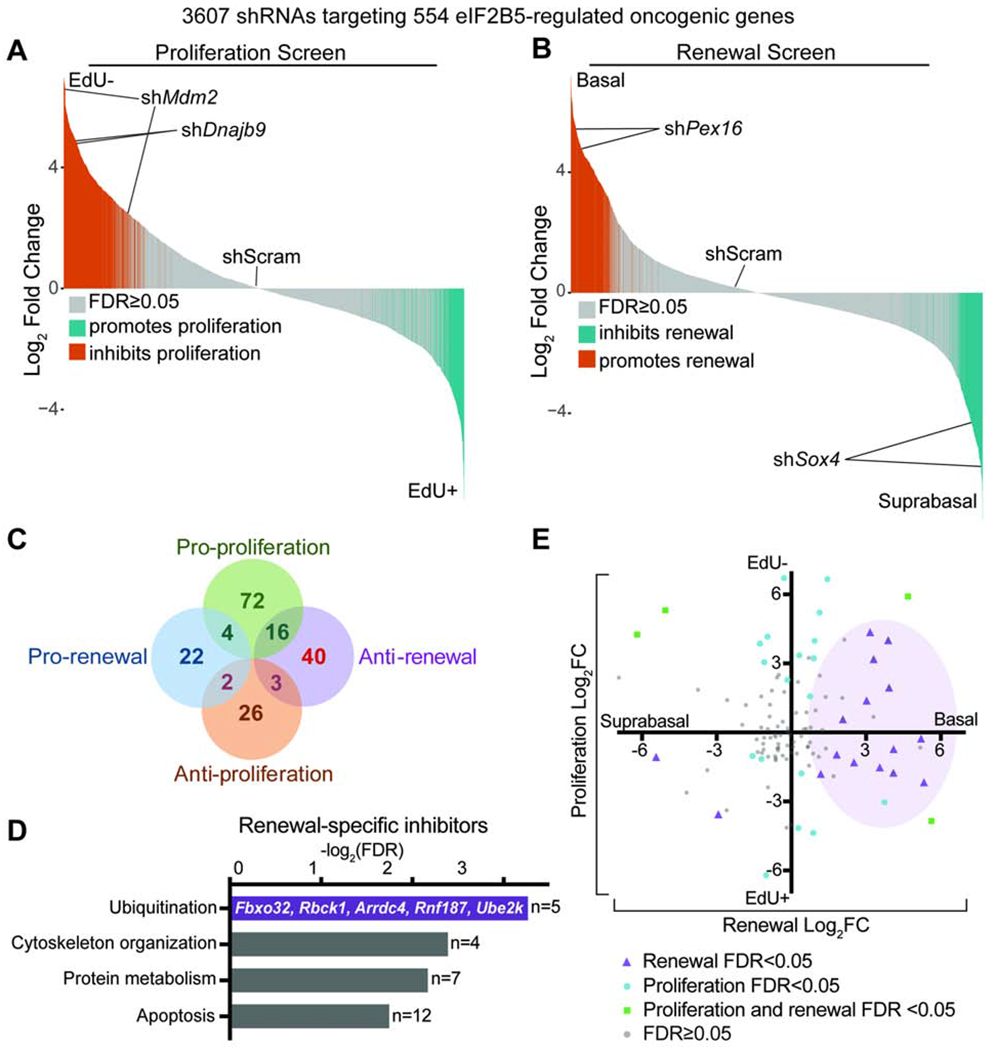
A-C) In vivo (A) proliferation and (B) renewal screens using an shRNA lentivirus pool targeting eIF2B5-regulated HrasG12V-dependent genes reveal (C) a distinct of genes that specifically inhibit renewal without affecting proliferation (red). n=3 biological replicates (DESeq2 FDR<0.05).
D) Ingenuity pathway analysis of renewal inhibitors identifies enrichment for ubiquitination genes.
E) Screen results for individual shRNAs targeting all ubiquitination genes in the screening pool. Purple shading demonstrates clustering of significantly altered shRNAs towards the pro-renewal axis.
In contrast to transcriptional control of cell proliferation, specific inhibitors of renewal were enriched for genes involved in ubiquitination and protein metabolism (Figure 4D). Specifically, shRNAs targeting the ubiquitination genes in our screening pool showed strong bias towards promoting progenitor cell renewal (Figure 4E). This suggests the provocative idea that the epidermis, which relies on stochastic cell fate choice and frequent turnover, employs highly dynamic post-translational gene regulation to adapt cell fate choice to oncogenic stress (Mesa et al., 2017; Park et al., 2017; Rompolas et al., 2016). Moreover, translationally controlled ubiquitination genes that specifically inhibit renewal may represent a growth-suppressive signaling core within the oncogenic translational landscape.
eIF2B5-regulated FBXO32 drives HRAS-induced loss of renewal
Among the eIF2B5-regulated ubiquitination genes that specifically inhibit HrasG12V basal cell renewal, we chose to further investigate Fbxo32 (also known as Mafbx/Atrogin1). FBXO32 is the substrate-recognition subunit of the Skp1-Cullin1-Fbox (SCF) E3 ubiquitin ligase complex (Figure S5A) (Zheng et al., 2016) and has been extensively studied in skeletal and cardiac muscle development, where it is positively associated with muscle atrophy (Al-Yacoub et al., 2016; Bodine and Baehr, 2014; Gomes et al., 2001). In cancer, FBXO32 has been implicated in both tumor suppression and oncogenesis (Tanaka et al., 2016; Mei et al., 2015; Sahu et al., 2017; Zhou et al., 2017). However, its epidermal function and potential role as a stem cell regulator have not been explored.
We began by testing if FBXO32 was indeed regulated by eIF2B5. FBXO32 protein was specifically elevated in HrasG12V compared to WT basal cells, and Eif2b5 depletion in HrasG12V progenitors reduces FBXO32 protein without affecting transcript levels (Figures 5A and 5B). Moreover, Fbxo32 mRNA resides in heavier polysome fractions in HrasG12V primary keratinocytes and shifts to lighter fractions after Eif2b5 depletion (Figure 5C). Together, this suggests that FBXO32 translation in HrasG12V progenitors is mediated by eIF2B5. Next, we sought to determine what makes FBXO32 uniquely sensitive to alterations in HRAS activity and eIF2B5. The 5’ UTR of Fbxo32 mRNA contains the GRTE motif that is enriched in HrasG12V and eIF2B5 translationally-dependent mRNAs (Figure S5B). To test if the GRTE can modulate translation rates, we cloned the Fbxo32 5’ UTR into a luciferase reporter construct (Fbxo32WT) and also generated a GRTE-deleted mutant (Fbxo32GRTEdel) (Figure S5C). In HrasG12V primary keratinocytes, the Fbxo32WT reporter exhibited significantly more translation than the Fbxo32GRTEdel mutant (Figure S5C). Remarkably, this difference was completely abolished in WT cells (Figure S5C). These findings establish the GRTE as a functional 5’ UTR cis-regulatory element critical for efficient translation of distinct mRNAs when translation initiation is aberrantly active during oncogene expression. This further supports our ribosomal profiling results and establishes FBXO32 as an HRASG12V-driven and eIF2B5 translation-dependent molecule.
Figure 5. FBXO32 is translationally regulated by eIF2B5 and drives loss of renewal in HrasG12V epidermal progenitors.
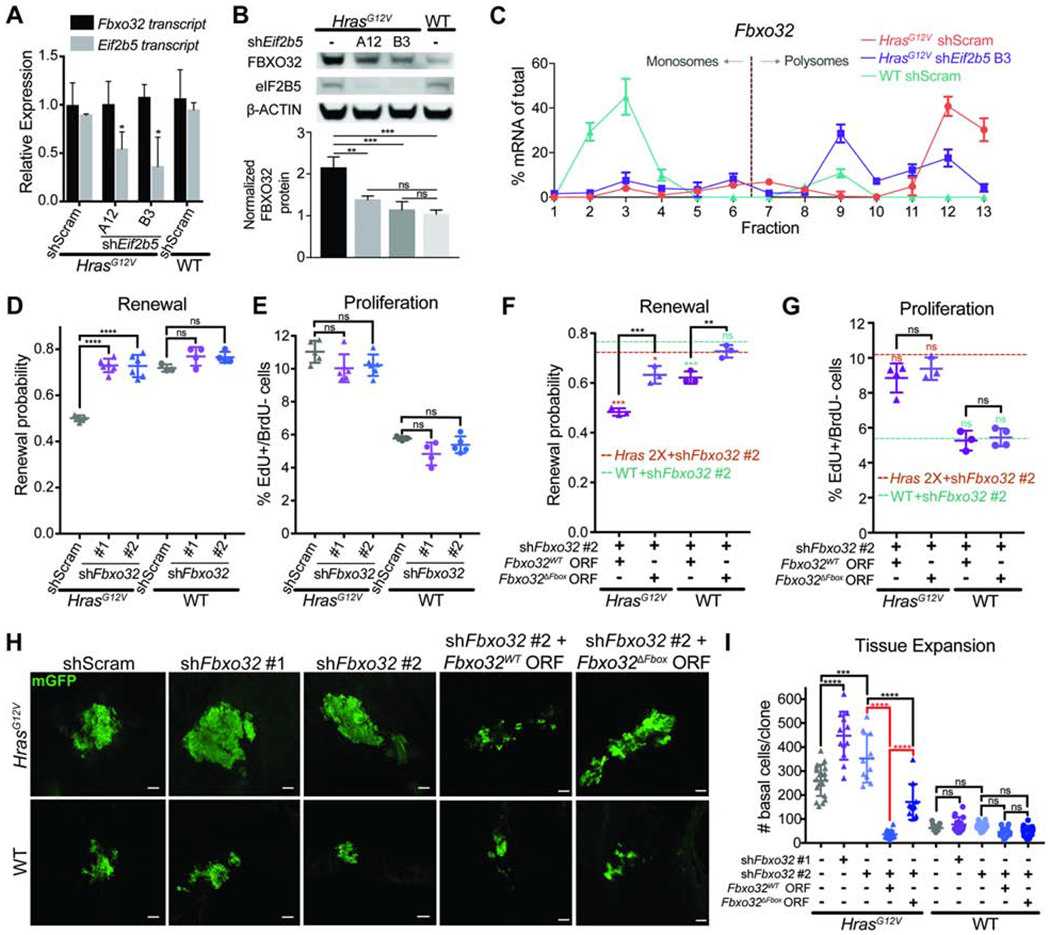
A) mRNA expression analysis by qPCR in E18.5 basal cells. n=3 animals per condition.
B) Representative western blot in transduced basal cells. FBXO32 protein level was normalized to ß-ACTIN for quantification. n=3 biological replicates.
C) qPCR of polysome fractions for Fbxo32 in primary HrasG12V keratinocytes transduced with control shRNA or shE/72b5.
D-E) (D) Renewal probability and (E) proliferation rate in transduced IFE basal cells. n=5 HrasG12V+shScram, 5 HrasG12V+shFbxo32 #1, 6 HrasG12V+shFbxo32 #2, 3 WT+shScram, 4 WT+shFbxo32 #1, and 5 WT+shFbxo32 #2 animals. Approx. 100 cell divisions (for renewal assay) or 100 basal cells (for proliferation assay) were scored per animal.
F-G) (F) Renewal probability and (G) proliferation rate in IFE basal cells transduced with shFbxo32 #2 co-expressing Fbxo32WT or Fbxo32ΔFbox ORF. Dashed lines represent average renewal probability or proliferation rate when HrasG12V (orange) or WT (green) epidermis is transduced with shFbxo32 #2 alone. Colored asterisks represent significant differences when compared to dataset indicated by colored dashed lines. n=3 animals per condition. Approx. 100 cell divisions (for renewal assay) or 100 basal cells (for proliferation assay) were scored per animal.
H-I) Representative (H) immunofluorescence staining and (I) quantification of basal cell numbers per clone in transduced E18.5 epidermis. mGFP (green) marks transduced cells. n=18 HrasG12V+shScram, 12 HrasG12V+shFbxo32 #1, 11 HrasG12V+shFbxo32 #2, 18 HrasG12V+shFbxo32 #2+Fbxo32WT, 11 HrasG12V+shFbxo32 #2+Fbxo32ΔFbox, 15 WT+shScram, 16 WT+shFbxo32 #1, and 12 WT+shFbxo32 #2, 15 WT+shFbxo32 #2+Fbxo32WT, and 16 WT+shFbxo32 #2+Fbxo32ΔFbox clones. 3 animals were assessed per condition. Key comparisons shown in red. Scale bars, 50μm.
Mean and SD are shown. ANOVA with Tukey’s range test for multiple comparisons used to determine statistical differences. *:P-value<0.05; **:P-value<0.01; ***:P-value<0.001; ****:P-value<0.0001; ns=not significant. See also Figure S5.
To test if FBXO32 can regulate epidermal progenitor cell behaviors, we depleted Fbxo32 in vivo using two efficient shRNAs (Figures S5D and S5E). Fbxo32 depletion completely rescued HrasG12V basal cell renewal probability to WT levels without altering HrasG12V hyperproliferation (Figures 5D and 5E). This demonstrates that the two progenitor cell behaviors downstream of HRAS and eIF2B5 can be separately regulated. Next, we asked if the ubiquitin ligase activity of FBXO32 was necessary for its renewal inhibitor function. To abrogate its ability to bind to SKP1 and form the SCF complex, we deleted the F-box domain in FBXO32 (FBXO32ΔFbox) (Figure S5F) (Jin et al., 2004). Lentivirus co-expression of shFbxo32 #2, targeting the 3’UTR of Fbxo32, with Fbxo32WT or Fbxo32ΔFbox ORF allowed us to deplete endogenous FBXO32 and rescue with either wild type FBXO32WT or mutant FBXO32ΔFbox (Figures S5G and S5H). In vivo, HrasG12V and WT progenitor cell renewal probabilities were completely rescued by expression of Fbxo32WT, but not Fbxo32ΔFbox, while proliferation rates remained unaffected across test conditions (Figures 5F and 5G). This suggests that the ubiquitin ligase function of FBXO32 drives its specific anti-renewal role in stem cell regulation. Together, we have established that eIF2B5-regulated FBXO32 ubiquitin ligase function is necessary for HRAS-induced loss of renewal.
Since FBXO32 specifically promotes HrasG12V-mediated suppression of progenitor renewal, we reasoned that FBXO32 would restrict clonal expansion and tissue growth. Fbxo32 depletion in individual HrasG12V basal cells at E9.5 resulted in a significant increase in the number of basal cells per clone by E18.5 (Figures 5H and 5I), indicating accelerated clonal expansion. Importantly, expansion could be completely suppressed by overexpression of the wild type FBXO32WT but not mutant FBXO32ΔFbox (Figures 5H and 5I), demonstrating that the ubiquitin ligase function of FBXO32 is necessary to limit HrasG12V clonal expansion. Fbxo32 depletion in WT progenitors only had a small effect on renewal rate and did not significantly affect clonal expansion (Figures 5H and 5I), suggesting that eIF2B5-regulated renewal inhibition via FBXO32 has oncogene-specificity.
FBXO32-mediated loss of renewal restrains HRAS tumor initiation and growth
The ability of eIF2B5-mediated translation to co-regulate stem cell fate choice and proliferation suggests that it acts as an adaptive coordinator of tissue response to oncogenic insult. Post-development, translational control may remain a critical mechanism for adjusting cell fate choice in response to oncogenic hyperproliferation. While the majority of skin in adult HrasG12V animals is tolerant to oncogene activation and does not form tumors, some permissive regions such as the muzzle can develop squamous papillomas (Beronja et al., 2013; Chen et al., 2009). We hypothesized that (i) tumorigenesis reflects a failure of the renewal block in these permissive regions; (ii) additional loss of anti-renewal signaling could further exacerbate tumor formation; and (iii) reactivation of anti-renewal signaling could make permissive regions more tumor resistant. Therefore, we explored whether modulating the cell fate arm of the eIF2B5-mediated translation program through FBXO32 could impact stem cell equilibrium and tumorigenesis.
To suppress renewal inhibition without affecting proliferation, we depleted Fbxo32 in HrasG12V animals and tracked them long-term for skin morphology changes. Fbxo32-depleted animals displayed significantly faster papilloma initiation, accelerated tumor growth, and increased tumor burden (Figures 6A–C). Furthermore, tumors from Fbxo32-depleted HrasG12V animals exhibited elevated progenitor cell renewal with no significant change in proliferation rate (Figures 6D and 6E). Therefore, specific ablation of the cell fate axis of translational co-regulation hinders the skin’s ability to manage oncogenic lesions, resulting in accelerated tumor formation and growth in permissive areas. To test for the specificity of this effect, we performed a rescue experiment by simultaneously depleting endogenous Fbxo32 and overexpressing Fbxo32WT ORF. This abolished the rapid tumorigenesis seen upon Fbxo32 depletion and actually made tumor prone regions in HrasG12V epidermis less tumor permissive (Figures 6A–C). Furthermore, Fbxo32WT ORF fully rescued renewal inhibition in Fbxo32-depleted HrasG12V progenitors, suggesting that its cell fate regulation is sufficient to restrict tumorigenesis (Figures 6D and 6E).
Figure 6. FBXO32 is necessary to restrain HrasG12V tumor initiation and growth.
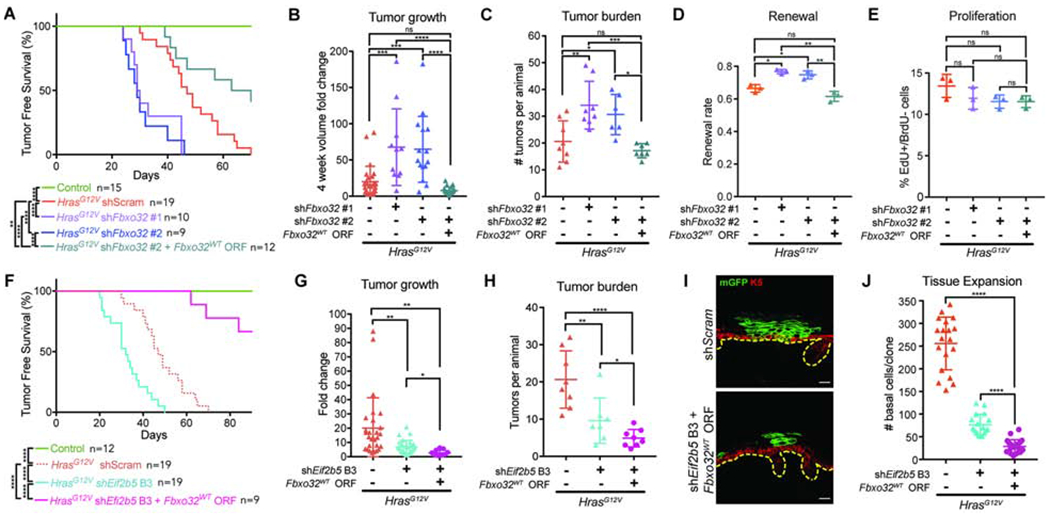
A) Tumor-free survival of WT (control) and transduced HrasG12V animals. Log-rank test used for significance analyses.
B) Tumor volume fold change 4 weeks after tumor initiation. n=27 HrasG12V+shScram, 11 HrasG12V+shFbxo32 #1, 15 HrasG12V+shFbxo32 #2, and 21 HrasG12V+shFbxo32 #2+Fbxo32WT tumors. 4 animals were assessed per condition.
C) Number of tumors per animal within 4 weeks of first tumor initiation. n=8 HrasG12V+shScram, 8 HrasG12V+shFbxo32 #1, 6 HrasG12V+shFbxo32 #2, and 7 HrasG12V+shFbxo32 #2+Fbxo32WT animals.
D-E) (D) Renewal probability and (E) proliferation rate in tumors of transduced HrasG12V animals. n=3 animals per condition. Approx. 100 cell divisions (for renewal assay) or 100 basal cells (for proliferation assay) were scored per animal.
F) Tumor-free survival of WT (control) or transduced HrasG12V animals. Log-rank test used for significance analyses.
G) Tumor volume fold change 4 weeks after tumor initiation. n=27 HrasG12V+shScram, 27 HrasG12V+shEif2b5 B3, and 15 HrasG12V+shEif2b5 B3+Fbxo32WT tumors. 4 animals were assessed per condition.
H) Number of tumors per animal within 4 weeks of first tumor initiation. n=8 HrasG12V+shScram, 7 HrasG12V+shEif2b5 B3, and 8 HrasG12V+shEif2b5 B3+Fbxo32WT animals.
I) Representative cross-section of transduced HrasG12V clone. mGFP (green) marks transduced epidermal cells, K5 (red) marks basal progenitors, and dashed line marks basement membrane. Scale bars, 25μm.
J) Basal cell numbers per clone in transduced E18.5 epidermis. n=18 HrasG12V+shScram, 17 HrasG12V+shEif2b5 B3, and 21 HrasG12V+shEif2b5 B3+Fbxo32WT clones. 3 animals were assessed per condition.
Mean and SD are shown. ANOVA with Tukey’s range test for multiple comparisons used to determine statistical differences. *:P-value<0.05; **:P-value<0.01; ***:P-value<0.001; ****:P-value<0.0001; ns=not significant. See also Figure S5.
To explore whether FBXO32 is a central regulator of tumor suppression in HrasG12V epidermis, we first examined the effect of Eif2b5 depletion on tumorigenesis. Eif2b5 knockdown, despite reducing HrasG12V progenitor proliferation rate, resulted in accelerated tumor initiation on par with what was observed with Fbxo32 depletion (Figure 6F). This was accompanied by reduced tumor growth and burden per animal (Figures 6G and 6H), suggesting that tumor initiation is primarily a function of progenitor cell renewal probability, whereas post-initiation, tumor growth is highly dependent on proliferation rate. We further hypothesized that simultaneous Eif2b5 depletion and Fbxo32 overexpression should exploit the anti-proliferative axis of eIF2B5 loss while preserving its pro-differentiation function through FBXO32. We co-expressed shEif2b5 B3 with Fbxo32WT ORF, which restored FBXO32 protein level in Eif2b5 depleted keratinocytes (Figures S5I and S5J). In vivo, we observed that most transduced clones were differentiated out of the basal epidermis and into suprabasal layers by E18.5 (Figure 6I). While this prevented us from directly assaying progenitor cell proliferation and renewal rates, we found that Fbxo32WT overexpression in Eif2b5-depleted HrasG12V epidermis resulted in substantial loss of clonal expansion (Figure 6J). Importantly, compared to Eif2b5 depletion alone, concomitant Fbxo32WT overexpression resulted in significant inhibition of HrasG12V tumor initiation (Figure 6F), slower tumor growth (Figure 6G), and lower tumor burden per animal (Figure 6H), demonstrating the additive anti-growth effect of targeting both proliferation and renewal axes. Together, these studies establish that translationally coordinated renewal inhibition in response to oncogenic hyperproliferation, through regulation of FBXO32, is a potent mechanism of tumor suppression in skin epithelium.
Discussion
How tissues tolerate oncogenic lesions to restrain aberrant growth and tumorigenesis is of fundamental interest to cancer biology (Greaves and Maley, 2012; Lowe et al., 2004). While suppression of mutant clones has been described in homeostatic adult epidermis (Brown et al., 2017; Martincorena et al., 2015; Ying et al., 2018), our studies uncover translationally-regulated inhibition of progenitor renewal as a central mechanism of tolerance to broad oncogene activation. eIF2B5 coordinates loss of renewal in response to oncogenic hyperproliferation, which limits the tissue’s pathological growth while preserving the proliferation necessary for epidermal development. Despite a transition in progenitor cell dynamics from high renewal probability and low proliferation rate in WT tissue to low renewal probability and high proliferation rate in HrasG12V tissue, epidermal development remained robust, demonstrating the significant plasticity within development as long as balanced stem cell coordination is maintained. It is tempting to speculate that such plasticity operates in RASopathies, which arise from germline mutations in RAS/MAPK signaling pathway genes, including HRAS (Rauen, 2013). Although associated with numerous morphological defects and a predisposition to cancer (Hafner and Groesser, 2014), patients with RASopathies have intact epidermis at birth, and additional skin phenotypes do not present until adolescence or adulthood (Digilio et al., 2011; Pierpont et al., 2014). This suggests that epidermal development in RASopathy is non-pathological. While the corrective potential of eIF2B5-dependent translation in human development under oncogenic stress is yet to be explored, our work proposes a mechanistic framework for tissue homeostasis through balanced translation of both pro-proliferative and pro-differentiation factors.
Elevated translation rates have recently been correlated with reduced renewal (Blanco et al., 2016; Liakath-Ali et al., 2018; Signer et al., 2014), but little is known about how this fundamental process drives discreet stem cell behaviors. We coupled in vivo ribosome profiling with unbiased and systematic screening to functionally dissect the translational networks regulated by eIF2B5 following oncogene activation. Our analyses revealed that oncogene-induced progenitor cell phenotypes were controlled by differential translation of distinct networks of mRNAs. Moreover, we observe that translation specificity is mediated in part through a cis- regulatory element encoded within the 5’ UTRs of target transcripts. As such, mRNA specific translation provides a gene specific mechanism for the control of cell proliferation and renewal downstream of eIF2B5.
While stemness has commonly been considered a transcriptionally regulated process (Bardin et al., 2010; Dai and Segre, 2004; Kashyap et al., 2009), this view is not fully compatible with the dynamic nature of epidermal progenitor cells. In vivo lineage tracing has revealed that cell fate is not determined at the time of cell division but instead occurs afterwards through stochastic delamination (Rompolas et al., 2016). This process is rapidly adaptable to tissue stresses like oncogene activation or wounding (Brown et al., 2017; Mesa et al., 2017; Park et al., 2017), suggesting that the mechanisms governing cell fate choice are also highly responsive. Intriguingly, our discovery of an unexpected enrichment of ubiquitination and protein metabolism genes amongst self-renewal inhibitors suggests that post-translational processes, including proteasome-dependent degradation, may provide a dynamic mechanism to modulate cell fate choice in rapidly growing tissue. We identified E3 ubiquitin ligase FBXO32 as a robust inhibitor of epidermal progenitor cell renewal. Ongoing research in the context of its well-characterized role in muscle atrophy has identified several substrates of FBXO32, including myosin heavy chain, vimentin, and desmin, and MyoD (Lokireddy et al., 2011; Tapscott, 2005; Tintignac et al., 2005). FBXO32 target substrates and their function in other tissues remain largely unknown and warrant future investigation.
We establish translationally-regulated renewal inhibition as a bona fide tumor-suppressive mechanism. This suggests that reactivating the anti-renewal signaling behind tissue-wide oncogene tolerance may be a therapeutic strategy to counteract field cancerization, which is often seen in skin and digestive epithelia (Curtius et al., 2017). Notably, terminal differentiation that removes excess cells from the progenitor pool is not a common feature of all epithelia, but instead seems specific for high turnover tissues like the skin, esophageal, and intestinal epithelia, which require continual proliferation and terminal differentiation for their self-maintenance (Fuchs, 2007; Srinivas et al., 2001; van der Flier and Clevers, 2009). Like skin, physiologically normal human esophagus also harbors unexpectedly high prevalence of oncogenic driver mutations (Martincorena et al., 2018), suggesting that other epithelia could also employ renewal inhibition to control field cancerization and tumorigenesis.
The discovery of oncogene-induced translation deregulation has instigated broad efforts to develop therapies that target the translation machinery (Bhat et al., 2015; Hsieh and Ruggero, 2010; Xu et al., 2019). This strategy is particularly appealing because the translation apparatus integrates most oncogenic pathways and its components are often upregulated in cancer (Ruggero, 2013; Truitt and Ruggero, 2016), suggesting that rapidly proliferating oncogenic cells are addicted to elevated protein synthesis and can be selectively targeted. However, our finding that translation coordinates cell fate alongside proliferation to balance growth provides a cautionary tale. Broad translation inhibition may initially limit cancer cell proliferation and tumor growth but could facilitate disease maintenance through its pro-renewal effect. Moreover, tissue physiology may change throughout development to preferentially bias one regulatory axis over the other, causing unbalanced shifts upon broad translation inhibition. Instead, functional understanding of the specific genes that are differentially translated during oncogene activation will allow us to more precisely target the cancer translatome through its downstream effectors.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Slobodan Beronja (beronja@fredhutch.org).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Ribosome profiling sequencing data can be accessed at NCBI Gene Expression Omnibus (GSE 126660). All other data are available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
HrasG12V/G12V (Chen et al., 2009), Rosa26YFP/YFP, and Rosa26mT/mG Cre-reporter mice (Jackson Laboratories) were on C56BL/6 or C56BL/6-Tyrc-2J/J backgrounds. All animals were immunocompetent and not previously used in any experimental procedures or treatments. We used equal numbers of male and female animals throughout the study. In utero injections were performed at E9.5 with maternal mice aged 2-8 months. In utero injections were performed under isoflurane anesthesia. Postoperatively, maternal mice were monitored daily for 5 days. Tumorigenesis studies were performed with embryonically transduced mice and were monitored every 2-3 days until 12 weeks of age. Mice were housed and cared for in an AAALAC-accredited facility at the Fred Hutchinson Cancer Research Center, and all animal experiments were conducted in accordance with ethical regulations of the Fred Hutchinson Cancer Research Center IACUC-approved protocol, project license number 50814.
Primary cell cultures
Basal cells were isolated as previously described (Nowak and Fuchs, 2009). Briefly, E18.5 skin was digested in 2mg/ml dispase (Corning) at 37°C for 1 hr to isolate the epidermis and 0.25% trypsin-EDTA for 10 min release single cells. Basal cells were stained with CD49f/α6-Integrin-PerCP (GoH3, 1:50; BioLegend) and FACS sorted using BD FACSAria II (BD Biosciences). Isolated basal cells were co-cultured on mitomycin C-treated NIH/3T3 feeder cells in 0.05 mM Ca2+ E media (Nowak and Fuchs, 2009) supplemented with 15% chelexed (BioRad) fetal bovine serum (Gibco) for 5 passages to generate primary keratinocyte cell culture lines. Primary keratinocyte lines were maintained in 0.05 mM Ca2+ E media with 15% chelexed FBS.
METHOD DETAILS
Lentivirus production and transduction
Large-scale production and concentration of lentivirus were performed as previously described (Beronja et al., 2010). Briefly, 293TN cells (System Biosciences, LV900A-1) cultured in DMEM+10% FBS (Gibco) were transfected using calcium phosphate with lentiviral backbone and helper plasmids pMD2.G and psPAX2 (Addgene plasmids 12259 and 12260) for 16 hr, then incubated in UltraCULTURE media (Lonza) for 48 hr. Supernatant was collected and concentrated using Centricon Plus-70 centrifugal filter unit (100 kDa, Millipore), then further pelleted by ultracentrifugation at 45,000 rpm for 1 hr using a MLS 50 rotor (Beckman Coulter). Viral pellet was resuspended in Viral Resuspension Buffer (VRB; 20 mM Tris pH 8.0, 250 mM NaCl, 10 mM MgCl2, 5% sorbitol) to generate lentiviral stock for intra-amniotic injections. Lentiviral transduction of primary keratinocytes in culture and of embryonic ectoderm in utero were performed as previously described with modifications (Beronja et al., 2013; 2010; Beronja and Fuchs, 2012). Briefly, primary keratinocytes were plated on 6-well plates and incubated in E media with 20% chelexed FBS, lentivirus, and 40 μg/ml polybrene at 37°C for 30 min, then centrifuged at 37°C and 1100xg for 30min. Centrifuged plates were rinsed with PBS and cultured in E media with 15% FBS for 2 days before processing for analyses. For in utero lentiviral transduction of embryonic ectoderm, pregnant mice were imaged one day before surgery using a Vevo 1100 animal ultrasound imager with a MS550D transducer (Visualsonics) to precisely stage the embryos. This ensured that lentiviral transduction was performed on E9.25–9.75 day embryos. Under ultrasound guidance, 1uL of lentivirus was injected into the amniotic cavity on the ventral side of each embryo with a Nanoject II microinjector (Drummond).
Lentiviral constructs
RNA interference-mediated gene depletion was achieved using pLKO.1 shRNA vectors from the mouse TRC1.0 shRNA library (Sigma-Aldrich). Additional shRNA sequences targeting genes of interest were obtained from The RNAi Consortium Library and cloned into the pLKO.1 vector (Addgene plasmid 10878; Table S6). To construct the lentiviral pools and ensure equal lentivirus representation, plasmids were mixed together in equal molar ratios. Cre-shRNA expression was achieved using pLKO.1-Cre vectors (Addgene plasmid 25997) as previously described (Beronja et al., 2013; 2010). Expression of Cre-recombinase with EF1α promoter-driven ORF overexpression was achieved using a pLX-Cre-EF1α vector modified from pLX302 (Addgene plasmid 25896) as previously described (Ying et al., 2018), in which the following were modified: 1) Cre was inserted downstream of the PGK promoter using Kpn1/Xba1 sites, and 2) CMV promoter between Xho1/Nde1 sites was replaced with an EF1α promoter from pEF-BOS (Addgene plasmid 21924). ORFs encoding genetic lesions were subcloned into entry vectors and recombined into pLX-Cre-EF1α using LR clonase (Invitrogen). Co-expression of shRNA in tandem with ORF overexpression in the pLX-Cre-EF1α vector was achieved by inserting hU6 promoter-driven shRNA downstream of the Rev-responsive element (RRE) using AleI/AscI sites.
Immunofluorescence
Mouse skin and tumor tissues were paraformaldehyde-fixed, OCT embedded, and cut into 8μm-thick sections. Sections were incubated in 0.1% PBST containing 2% goat serum, 2% donkey serum, 1% gelatin, and 1% BSA for 1hr at room temperature. If necessary, sections were incubated with M.O.M. mouse blocking reagent (Vector) for 1 hr at room temperature. Section were then incubated with primary antibodies overnight at 4°C, followed by incubation with Alexa-Fluor conjugated secondary antibodies (Invitrogen) for 1 hr at room temperature. Slides were mounted using ProLong Gold Antifade Mountant (Thermo Fisher). For EdU-BrdU proliferation and pulse-chase renewal assays, tissue sections were first processed for EdU Click-iT according to manufacturer’s instructions (Thermo Fisher). Next, tissues were fully processed for K10 and GFP immunofluorescence detection. Lastly, tissue sections were incubated in 2N HCl at 37°C for 30 min to denature DNA, quenched with 0.1M sodium borate pH 8.5 twice, and processed for BrdU immunofluorescence detection. Confocal images were taken on a Zeiss LSM700 system, using Plan-Apochromat 20x/0.8 dry objective. Images were processed using Zeiss Zen and ImageJ software (Abramoff et al., 2004). The following primary antibodies were used: chicken anti-GFP (ab13970, 1:1000; Abcam); mouse anti-BrdU (MoBU-1, 1:100; Invitrogen); rabbit anti-Keratin 10 (Poly19054, 1:1000; BioLegend); mouse anti-Puromycin (12D10, 1:500; Millipore); guinea pig anti-Keratin 5 (BP5006, 1:1000; Origene).
EdU-BrdU proliferation assay
Basal cell proliferation rate was quantified based on nucleoside incorporation as previously described (Ying et al., 2018). 50mg/kg EdU (Invitrogen) was administered intraperitoneally, followed by 100mg/kg BrdU (Invitrogen) injection 2 hours later, before assessing nucleoside incorporation by immunofluorescence 6 hours post-BrdU injection. Cells that complete S-phase during the first 2 hours incorporate EdU only, while cells subsequently going through S-phase incorporate both EdU and BrdU. Therefore, cells that divide within the initial 2 hours have EdU+ BrdU− labelled progeny. One cell division gives rise to two nucleoside-labeled daughter cells, so the number of dividing cells in the initial 2 hours is (total EdU+BrdU− cells)/2. In the epidermis, only basal cells have the potential to divide. Therefore, the proliferation rate of basal cells that divided in the initial 2 hours is ((total EdU+BrdU− cells)/2)/(total basal cell number).
EdU-BrdU pulse-chase renewal assay
Basal cell renewal probability was quantified based on nucleoside incorporation and differentiation marker expression as previously described (Ying et al., 2018). Since we can uniquely label a population of epidermal progenitor cells (EdU+ BrdU−) that have undergone division within the initial 2 hours as described above, we can also assess the differentiation state of their daughter cells based on expression of differentiation marker keratin 10 (K10). 50mg/kg EdU (Invitrogen) was administered intraperitoneally, followed by 100mg/kg BrdU (Invitrogen) injection 2 hours later, before assessing nucleoside incorporation by immunofluorescence 6 hours post-BrdU injection. We calculated the probability of progenitor cell renewal during the first 2 hours using the following equation: Renewal probability= (number of EdU+ BrdU− K10− cells) / (total number of EdU+ BrdU− cells).
Translation rate assay
Translation rate assay was adapted from previous protocol (Goodman and Hornberger, 2013). Animals were transduced at E9.5 by in utero injection of lentivirus. At E18.5, maternal mice were injected intraperitoneally with 50mg/kg puromycin (Thermo Fisher) and euthanized after 30 min to collect E18.5 embryos. Headskin was fixed in 4% paraformaldehyde and paraffin-embedded. Sections were de-waxed and stained using the immunofluorescence procedure described above with the following modifications: antigen retrieval was performed using citrate-based antigen unmasking solution (Vector) at 95°C for 30 min. Sections were subjected to incubation with puromycin antibody (12D10, Millipore) followed by incubation with secondary antibody conjugated to Alexa-Fluor 594 (Invitrogen). Puromycin intensity in transduced epidermal basal cells was quantified by (Alexa-Fluor 594 total fluorescence intensity)/(surface area) in K5+GFP+ regions of tissue cross-sections using Imaris software (Bitplane). To normalize for potential differences in puromycin administration or incorporation between animals, we employed the following internal controls: 1) In partially transduced tissues, puromycin intensity in K5+GFP+ transduced basal cells was normalized to puromycin intensity in neighboring K5+ GFP− untransduced WT basal cells in each tissue section; and 2) In fully transduced tissues, puromycin intensity in K5+ epidermal basal cells was normalized to puromycin intensity in the immediate underlying dermis in each tissue section.
Tissue expansion assay
E9.5 animals expressing Rosa26mT/mG Cre-reporter (Jackson Laboratories) were transduced by in utero lentiviral injection at MOI<<1. At E12.5, 14.5, 16.5, or 18.5, whole-mount headskin was fixed in 4% paraformaldehyde and stained using the immunofluorescence procedure described above with chicken anti-GFP (ab13970, 1:500; Abcam) overnight at 4°C, followed by incubation with a secondary antibody conjugated to Alexa-Fluor 488 (Invitrogen) overnight at 4°C. Tissues were optically cleared using the SeeDB method as previously described (Ke et al., 2013) for confocal imaging. Briefly, tissues were sequentially incubated in 20% (w/v), 40%, 60%, 80%, 100%, and SeeDB fructose solutions at room temperature until clear.
Drug treatments
Animals were transduced by in utero lentiviral injection at E9.5. Maternal mice were injected daily, starting at E14.5, with 1mg/kg ISRIB (Sigma-Aldrich) or equivalent volume DMSO. Animals were processed at E18.5 for renewal and proliferation assays as described above. For primary keratinocyte cell culture, cells were transduced with lentivirus 48 hr prior to drug treatment. Cells were treated with 500nM ISRIB or equivalent volume DMSO for 24 hr prior to processing for western blot analysis.
In vivo genetic screens and data analyses
To quantify construct abundance in the proliferation and renewal screens, tissue was processed as previously described (Ying et al., 2018). Briefly, headskin of mice at E18.5 was digested in 2mg/ml dispase (Corning) at 37 C° for 1 hour to separate epidermis from dermis. Epidermal tissue was further digested with 0.25% trypsin for 20 min into single cells. Headskins from 8 animals were pooled together to make one biological replicate, thus achieving a ~40 fold pool coverage. For the proliferation screen, single epidermal cells were subjected to Click-iT EdU detection (Invitrogen) followed by CD49f/α6-Integrin-PerCP (GoH3, 1:50; Biolegend) staining. Cell populations of interest were isolated using BD FACSAria II machine (BD Biosciences). For the renewal screen, single epidermal progenitor cells were stained with CD326/EpCAM-APC (G8.8, 1:50; BD Biosciences) and CD49f/a6-Integrin-PerCP (GoH3, 1:50; Biolegend). gDNA from all samples was extracted using the DNeasy Blood & Tissue Kit (Qiagen). shRNA pre-amplification, sequencing (HiSeq 2500, Illumina), and data analysis were performed as previously described (Beronja et al., 2013). Briefly, raw Illumina reads were trimmed to 21nt hairpin sequences using the FASTX-Toolkit and aligned to a custom reference genome of all screened shRNA sequences with BWA (H. Li and Durbin, 2009). Mapped sequences were indexed with Samtools (H. Li et al., 2009) to generate read counts per shRNA. Differential enrichment or depletion of shRNAs in each condition was identified using DESeq2 (Love et al., 2014). For each screened shRNA, a statistical cutoff of FDR<0.05 was used (minimum read count of 5). Genes identified as putative proliferation or renewal regulators must have at least 2 shRNAs significantly enriched/depleted with the same directionality across all significantly altered shRNAs.
Western blot analyses
Cells were lysed for 30 min on ice in RIPA Lysis Buffer supplemented with phosphatase and protease inhibitor cocktails (Santa Cruz Biotechnology). Lysates were cleared by centrifugation at 17000g for 10 min at 4°C. Supernatants were removed and assayed for protein concentration using the Pierce BCA Protein Assay Kit (Thermo Fisher). Western blotting was performed using Novex system (Invitrogen). Equal amounts of proteins were subjected to SDS-PAGE and transferred to PVDF membranes (Thermo Fisher). Membranes were incubated with primary antibodies at 4°C overnight. Membranes were washed in TBS-Tween and then incubated with HRP conjugated anti-mouse or anti-rabbit secondary antibodies (Jackson Immunoresearch) for 1 hr at room temp and developed using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Fisher). Signal detection was captured using Odyssey Fc system (LI-COR; Figure S6). The following primary antibodies were used: mouse anti-eIF2B5 (B-7, 1:50; Santa Cruz Biotechnology); mouse anti-β-Actin (2D4H5, 1:3000 for WB; Proteintech); mouse anti-Puromycin (12D10, 1:1000; Millipore); mouse anti-EIF2α (L57A5, 1:500; Cell Signaling); rabbit anti-phospho-EIF2α Ser51 (119A11, 1:500; Cell Signaling); rabbit anti-FBXO32 (EPR9148(2), 1:500; Abcam); mouse anti-V5 (V5-10, 1:3000 for WB; Sigma-Aldrich).
Quantitative PCR
Total RNA was isolated using the RNeasy Plus Mini Kit (Qiagen) and reverse-transcribed with iScript Reverse Transcription Supermix (Bio-Rad). cDNA was diluted 1:5 with water. Quantitative PCR was performed with 1 μl cDNA using SYBR Green PCR Master Mix (Thermo Fisher) with primer sets specific for Eif2b5, Fbxo32, and Ppib as a control (Table S6). Normalized mRNA expression levels were calculated using comparative Ct.
Proteasome activity assay
Primary keratinocytes were transduced with lentivirus and plated in triplicate per condition at 5000 cells/well/90uL in a 96 well plate. Cells were treated for 4 hr with DMSO control, 1mg/mL puromycin, or 100nM bortezomib (Sigma-Aldrich) before assaying for proteasome activity using the 20S Proteasome Activity Assay Kit (APT280, Millipore) according to the manufacturer’s protocol. Cells were incubated with the Proteasome Assay Loading Solution (LLVY-R110 peptide substrate) at 37 °C for 2 hr. Fluorescence was measured with a microtiter plate reader (Biotek Synergy HT).
In vitro proliferation assay
To measure in vitro cell proliferation, the IncuCyte ZOOM™ proliferation assay was used (Essen Bioscience). Primary keratinocytes were transduced with lentivirus expressing H2B-GFP to mark nuclei and seeded at 5000 cells/well in 500 μl of media each in a 24-well plate. Plates were then placed in the IncuCyte ZOOM™ and live cell time-lapse imaging without labels was performed. Cell proliferation was monitored by counting cell nuclei in well images over time.
Apoptosis assay
Animals transduced with lentivirus at E9.5 were processed at E18.5 to isolate epidermal cells as described above (Nowak and Fuchs, 2009) and stained for active caspase 3 expression according to manufacturer’s instructions (BD Pharmingen). Flow cytometric analysis of caspase 3 staining in YFP+ transduced cells was performed on BD LSR II.
In vivo ribosome profiling and data analyses
The in vivo basal cell ribosome profiling sample preparation is adapted from previously described methodology (Sendoel et al., 2017). Headskins of E18.5 mice were collected immediately after euthanization and digested in 5mg/mL dispase (Corning) supplemented with 8 mg/ml cycloheximide (Sigma-Aldrich) for 20 min at 37°C. Epidermis was separated from dermis and placed immediately in 0.25% trypsin supplemented with 4 mg/ml cyclohexamide and incubated for 8 min at 37°C. The resulting cell suspension, enriched for basal epidermal keratinocytes, was filtered through a 40-μm cell strainer, spun down, and lysed in mammalian lysis buffer supplemented with 0.1 mg/ml cyclohexamide according to the TruSeq Ribo Profile (Mammalian) protocol (Illumina). Cells were lysed on ice for 10 min, centrifuged at 16,000g for 10 min at 4°C, and supernatant was flash-frozen in liquid nitrogen. 20 headskins were pooled per biological replicate. The supernatant was used to isolate both total RNA and ribosome bound fractions as previously described (Ingolia et al., 2009) using the TruSeq Ribo Profile Mammalian kit (Illumina). Briefly, lysates were treated with TruSeq RP nuclease for 45 min at room temperature to generate ribosome-protected fragments, which were isolated using sephacryl S400 columns (GE Healthcare). rRNAs were removed from ribosome-protected fragments and total RNA using Ribo-Zero Gold (Illumina, MRZH11124). Quality of the RNA was determined using Agilent 2100 Bioanalyzer, with all samples passing the quality threshold of RNA integrity numbers (RIN) > 9. Barcodes were used to generate pooled libraries. Ribosome-protected fragments and total mRNA libraries were amplified in 7–9 PCR cycles. Libraries were analyzed on the Agilent 2100 Bioanalyzer before sequencing. The pools were sequenced on a HiSeq 2500 platform using the SR50 protocol. Profiling analysis was adapted from a previously described analysis pipeline (Ingolia et al., 2009). The raw sequences were trimmed using the FASTX-Toolkit to remove the 3′ adaptor sequence (AGATCGGAAGAGCACACGTCT). Trimmed sequence reads were aligned to mouse rRNA reference using Bowtie and mapped rRNA sequences were discarded (Langmead et al., 2009). TopHat2 was used to align non-rRNA reads to the mouse genome mm10 and counted for gene associations against the UCSC genes database with HTSeq (Anders et al., 2015; Kim et al., 2013). R/Bioconductor package Xtail was used to find differentially expressed genes at the translational level using both ribosome-bound and mRNA samples (Xiao et al., 2016). Differential expression analysis of total mRNA reads was performed using R/Bioconductor package edgeR (Robinson et al., 2010). R/Bioconductor package riboseqR was used to calculate triplicate periodicity in all samples (Chung et al., 2015). For each analysis, a statistical cutoff of FDR<0.1 and log2Fold Change>0.5 was used (minimum read count of 5). Genes with significant differential translation efficiency were uploaded into Ingenuity Pathway Analysis (Qiagen), and a core analysis was run to identify canonical pathways and functional enrichment.
Polysome analyses
HrasG12V and WT primary keratinocytes were transduced with lentivirus 48 hrs prior to processing. Cells were incubated in 100μg/mL cycloheximide for 5 minutes before rinsing and lysing in polysome lysis buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 10 mM MgCl2, 1 mM DTT, 100 μg/ml cycloheximide, 200 μg/ml heparin, 1% Triton X-100, 40 U/ml SUPERase In RNase inhibitor (Thermo Fisher), and 10U/mL DNAse I). Clarified lysates were loaded on 10-50% sucrose gradients prepared in 1 mM DTT, 100 μg/ml cycloheximide, and 200 μg/ml heparin. Gradients were fractionated using a Piston Gradient Fractionator (Biocomp). Firefly luciferase mRNA (Fluc; Promega) spike-in was added to each fraction before RNA extraction. RNA was extracted using RNeasy Plus Mini Kit (Qiagen) and cDNA was synthesized using iScript Reverse Transcription Supermix (Bio-Rad). qPCR was performed as described above for Mgarp, Sdhaf1, Hmgn1, Nfu1, Lmna, Zc3h18, Cbx5, Fbxo32, Ppib, and Fluc (Table S6). To account for differences in RNA extraction efficiency, relative mRNA abundances in each fraction were normalized to Fluc mRNA. Normalized mRNA abundances in each fraction were expressed as a proportion of the total transcript level for each gene.
5’UTR analysis and motif discovery
5’ UTRs of the 1324 genes that are translationally upregulated in HrasG12V and the subset of 554 genes that are dependent on eIF2B5 were obtained using the known gene ID from the UCSC Genome Browser (mm10). Target versus non-target mRNAs were compared for 5’ UTR length, %G+C content, and folding energy by the Wilcoxon two-sided test. The MEME suite was used for motif discovery and analysis (Bailey et al., 2009). 5’UTRs were inputted into the MEME pipeline (default settings, output of 15 motifs) to discover the guanine-rich translational element (GRTE). The GRTE position weighted map was entered into FIMO to determine the prevalence of the GRTE in translationally upregulated mRNAs (Grant et al., 2011). A list of all 5’ UTRs from the mouse genome (n=19009) was obtained from UCSC and used as a control dataset. Enrichment was calculated using Pearson’s χ2 test, counting genes with at least one occurrence of the GRTE as successes.
GRTE luciferase reporter assay
The Fbxo32WT 5’UTR construct was cloned by inserting the full-length (232 bp) Fbxo32 5’UTR into a CMV-Luc2CP backbone (Addgene plasmid 62857 with ARE removed) immediately upstream of the firefly luciferase CDS and downstream of the CMV TSS. From this construct, the Fbxo32GRTEdel 5’UTR construct was cloned by using a Q5 mutagenesis kit (NEB) to remove the 41bp GRTE (CAGGAGGCGACCTTCCCCAACGCCTGCGCCCCTGTGAGTGC). WT and HrasG12V keratinocytes were seeded in 12-well plates for luciferase assays and qPCR. 24h post-seeding, they were transfected with 1 μg Fbxo32WT or Fbxo32GRTEdel construct using Lipofectamine 3000 (Thermo Fisher). 48h post-transfection, cells were collected for luciferase assays and qPCR. Luciferase assays were performed using Promega’s Dual-Luciferase® Reporter Assay System to the manufacturer’s specifications and read using a BioTek Synergy 2 microplate reader. qPCR was performed as described above with primers targeting Luc2CP. Luciferase RLU values were normalized to relative luciferase mRNA expression to quantify translation levels.
Tumor analyses
Animals were transduced at E9.5 with low-titer lentivirus containing Cre and shRNA against scrambled control or test genes. Transductions were confirmed by fluorescence of Cre-reporter YFP in ear clippings, and the remaining animals were monitored for 12 weeks post-birth. Animals were examined every 2–3 days and scored positive when tumors were larger than 2mm in diameter. Tumor-bearing animals were tracked weekly to measure individual tumors along their length, width, and depth for up to 5 weeks post-tumor initiation. Tumor volume was calculated using formula V=(π/6)x(LxWxD) (Tomayko and Reynolds, 1989). Tumor burden is the total number of tumors that arise per animal during the 4 weeks after first tumor initiation. Log-rank test was used to analyze significant differences in tumor-free survival.
QUANTIFICATION AND STATISTICAL ANALYSIS
All quantitative data were collected from experiments performed at least three independent times. All sample numbers (n) represent biological replicates and all data are expressed as mean ± s.d. Statistical details are as indicated in figure legends. Differences between two groups were analyzed with two-tailed Student’s t-test, and differences between more than two groups were analyzed with analysis of variance (ANOVA) with Tukey’s range test for multiple comparisons. Overlap between gene sets was analyzed using hypergeometric test. Significant differences were considered when p<0.05, as indicated by asterisks. Animals were randomly assigned to experimental groups. No blinding methods were used. No datapoints were excluded from analyses. Statistical and graphical data analyses were performed in R and Prism 7 (Graphpad).
Supplementary Material
Supplemental Table S1. Regulators of HrasG12V progenitor proliferation and renewal within the translation machinery, Related to Figure 1
Candidates for proliferation and renewal regulation as identified by DESeq2 analysis of shRNA enrichment in non-dividing (EdU-) relative to dividing (EdU+) basal progenitors (for proliferation screen), or in basal progenitor relative to the differentiated suprabasal cells (for renewal screen). Positive fold-change value indicates shRNA enrichment in the EdU− population (for proliferation screen) or in basal progenitors (for renewal screen).
Supplemental Table S2: Translationally altered genes in HrasG12V epidermal progenitors -- HrasG12V+shScram vs. WT+shScram (HS/WS), Related to Figure 3
Genes with translational efficiency changes were identified by Xtail analysis and excluded genes with significant transcriptional changes (as determined by EdgeR). Positive fold-change value indicates translational upregulation in HrasG12V+shScram basal progenitors.
Supplemental Table S3: Translationally altered genes regulated by eIF2B5 in HrasG12V epidermal progenitors -- HrasG12V+shEif2b5 to HrasG12V+shScram (HE/HS), Related to Figure 3
Genes with translational efficiency changes were identified by Xtail analysis and excluded genes with significant transcriptional changes (as determined by EdgeR). Positive fold-change value indicates translational upregulation in HrasG12V+shEif2b5 basal progenitors.
Supplemental Table S4: Translationally altered genes in HrasG12V epidermal progenitors that are rescued by shEif2b5, Related to Figure 3
Genes with translational efficiency changes were identified by Xtail analysis and excluded genes with significant transcriptional changes (as determined by EdgeR). Positive fold-change value indicates translational upregulation in HrasG12V progenitors.
Supplemental Table S5: Regulators of HrasG12V progenitor proliferation and renewal within the eIF2B5-regulated oncogenic translatome, Related to Figure 4
Candidates for proliferation and renewal regulation as identified by DESeq2 analysis of shRNA enrichment in non-dividing (EdU−) relative to dividing (EdU+) basal progenitors (for proliferation screen), or in basal progenitor relative to the differentiated suprabasal cells (for renewal screen). Positive fold-change value indicates shRNA enrichment in the EdU− population (for proliferation screen) or in basal progenitors (for renewal screen).
Supplemental Table S6: shRNA and primer sequences used in this study, Related to STAR Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| chicken anti-GFP | Abcam | ab13970; RRID:AB_300798 |
| mouse anti-BrdU (MoBU-1) | Invitrogen | B35128; RRID:AB_2536432 |
| rabbit anti-Keratin 10 (Poly19054) | BioLegend | 905403; RRID:AB_2749902 |
| mouse anti-Puromycin (12D10) | Millipore | MABE343; RRID:AB_2566826 |
| guinea pig anti-Keratin 5 | Origene | BP5006 |
| CD49f/α6-Integrin-PerCP (GoH3) | BioLegend | 313617 RRID:AB_1575054 |
| CD326/EpCAM-APC (G8.8) | BD Biosciences | 563478; RRID:AB_2738234 |
| mouse anti-EIF2B5 (B-7) | Santa Cruz Biotechnology | Sc-55558; RRID:AB_831253 |
| mouse anti-β-Actin (2D4H5) | Proteintech | 66009; RRID:AB_2687938 |
| mouse anti-EIF2α (L57A5) | Cell Signaling | 2103S; RRID:AB_836874 |
| rabbit anti-phospho-EIF2α Ser51 (119A11) | Cell Signaling | 3597S; RRID:AB_390740 |
| rabbit anti-FBXO32 (EPR9148(2)) | Abcam | ab168372 |
| mouse anti-V5 (V5-10) | Sigma-Aldrich | V8012; RRID:AB_261888 |
| Bacterial and Virus Strains | ||
| TRC1.0 Lentiviral Mouse Genome shRNA Library | Sigma-Aldrich | Discontinued, now MISSION shRNA Library |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | Gibco | 11995-065 |
| UltraCULTURE media | Lonza | 12-725F |
| E media | Nowak and Fuchs, 2009 | N/A |
| Fetal bovine serum | Gibco | 16000-044 |
| Polybrene | Sigma | 107689-10G |
| Trypsin-EDTA | GIBCO | 25200-056 |
| Chelex 100 | Bio-Rad | 1422822 |
| M.O.M. Blocking Reagent | Vector | MKB-22113 |
| ProLong Gold Antifade Mountant | ThermoFisher | P36930 |
| Antigen Unmasking Solution, Citric Acid Based | Vector | H-3300-250 |
| RIPA Lysis Buffer System | Santa Cruz Biotech | Sc-24948 |
| ISRIB | Sigma-Aldrich | SML0843 |
| SuperSignal Wester Pico PLUS Chemiluminescent Substrate | Thermo Fisher | 34577 |
| iScript Reverse Transcription Supermix | Bio-Rad | 1708840 |
| SYBR Green PCR Master Mix | Thermo Fisher | 4309155 |
| Lipofectamine 3000 | Thermo Fisher | L3000001 |
| BrdU | Invitrogen | B23151 |
| EdU | Invitrogen | A10044 |
| Puromycin | Thermo Fisher | A11138-03 |
| Cyclohexamide | Sigma-Aldrich | C7698 |
| Bortezomib | Sigma-Aldrich | 504314 |
| Dispase | Corning | 354235 |
| Mitomycin C | Sigma-Aldrich | M4287 |
| Critical Commercial Assays | ||
| Click-iT EdU Alexa Fluor 555 Imaging Kit | Invitrogen | C10338 |
| 20S Proteasome Activity Assay | Millipore | APT280 |
| PE Active Caspase-3 Apoptosis Kit | BD Biosciences | 550914 |
| DNeasy Blood & Tissue Kit | Qiagen | 69504 |
| RNeasy Plus Mini Kit | Qiagen | 74134 |
| Q5 Site-Directed Mutagenesis Kit | NEB | E0554S |
| Pierce BCA Protein Assay Kit | Thermo Fisher | 23225 |
| Dual-Luciferase Reporter Assay System | Promega | E1910 |
| TruSeq Ribo Profile (Mammalian) Library Prep Kit | Illumina | RPHMR12126 |
| Ribo-Zero rRNA Removal Kit (Human/Mouse/Rat) | Illumina | MRZH11124 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO GSE 126660 |
| Mouse reference genome NCBI build 38 (mm10) | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/ |
| Experimental Models: Cell Lines | ||
| Mouse: WT primary keratinocytes | This paper | N/A |
| Mouse: HrasG12V primary keratinocytes | This paper | N/A |
| Mouse: NIH/3T3 cells | ATCC | CRL-1658; RRID:CVCL_0594 |
| Human: 293TN cells | System Biosciences | LV900A-1 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Rosa26YFPFYP | The Jackson Laboratory | 006148; RRID:IMSR_JAX:006148 |
| Mouse: Rosa26mT/mG | The Jackson Laboratory | 007676; RRID:IMSR_JAX:007676 |
| Mouse: FR-HrasG12V | Donated by James A. Fagin | N/A |
| Oligonucleotides | ||
| See Table S6 for shRNA and PCR primer sequences | ||
| Recombinant DNA | ||
| Plasmid: pLKO.1 | Addgene | 10878; RRID:Addgene_10878 |
| Plasmid: pLKO.1-Cre | Addgene | 25997; RRID:Addgene_25997 |
| Plasmid: pLX-Cre-EF1α ORF vector | Ying et al., 2018 | N/A |
| Plasmid: pLX-shRNA-Cre-EF1α ORF vector | This paper | N/A |
| Plasmid: pMD2.G | Addgene | 12259; RRID:Addgene_12259 |
| Plasmid psPAX2 | Addgene | 12260; RRID:Addgene_12260 |
| Plasmid: CMV-Luc2CP/ARE | Addgene | 62857; RRID:Addgene_62857 |
| Software and Algorithms | ||
| FASTX-Toolkit | Hannon Lab | http://hannonlab.cshl.edu/fastx_toolkit/ |
| BWA | Li et al., 2009 | http://bio-bwa.sourceforge.net/ |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Bowtie | Langmead et al., 2009 | https://bioconductor.org/packages/release/bioc/html/Rbowtie.html |
| TopHat2 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| HTSeq | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.10.0/ |
| Xtail | Xiao et al., 2016 | https://github.com/xryanglab/xtail |
| edgeR | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| riboseqR | Chung et al., 2015 | http://bioconductor.org/packages/release/bioc/html/riboSeqR.html |
| Ingenuity Pathway Analysis | Qiagen | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ |
| Imaris | Bitplane | https://imaris.oxinst.com/ |
| ImageJ | Abramoff et al., 2004 | https://imagej.nih.gov/ij/ |
| Prism 7 | GraphPad | https://www.graphpad.com/ |
| MEME | Bailey et al., 2009 | http://meme-suite.org/tools/meme |
| FIMO | Grant et al., 2011 | http://meme-suite.org/tools/fimo |
| ZEN | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html |
Highlights.
Epidermis manages widespread oncogenic stress by inhibiting progenitor renewal
HRAS induces aberrant translation and alters progenitor behavior through eIF2B5
eIF2B5 coordinates cell fate with proliferation through distinct translation networks
eIF2B5-regulated ubiquitin ligase FBXO32 blocks renewal to restrain aberrant growth
Acknowledgements
We thank J. Fagin for sharing the HrasG12V fl/fl knock-in mouse. We thank Y. Lim for assistance with translation assays and ribosome profiling. We thank T. Gujral for assistance with Incucyte cell proliferation assay. We thank FHCRC Comparative Medicine (AAALAC accredited; G. Roble, director) for care of mice in accordance with National Institutes of Health (NIH) guidelines, Genomics (J. Delrow, director) for sequencing, Experimental Histopathology (S. Chanthaphavong, director) for tissue processing, and Flow Cytometry (A. Berger, director) for flow cytometry and FACS. This research was funded by NIH awards R01-AR070780 (S.B.), F30-CA224951 (E.Y.C) and R01-GM135362 (S.B. and A.C.H.), and in part by the NIH/NCI Cancer Center Support Grant P30 CA015704.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing financial interests.
References
- Abramoff MD, Magalhaes PJ, Ram SJ 2004. Image processing with ImageJ. Biophotonics International 11(7), 36–42. [Google Scholar]
- Al-Yacoub N, Shaheen R, Awad SM, Kunhi M, Dzimiri N, Nguyen HC, Xiong Y, Al-Buraiki J, Al-Habeeb W, Alkuraya FS, Poizat C, 2016. FBXO32, encoding a member of the SCF complex, is mutated in dilated cardiomyopathy. Genome Biol. 17, 2347. doi: 10.1186/s13059-015-0861-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W, 2015. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asare A, Levorse J, Fuchs E, 2017. Coupling organelle inheritance with mitosis to balance growth and differentiation. Science 355, eaah4701. doi: 10.1126/science.aah4701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS, 2009. MEME Suite: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–208. doi: 10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardin AJ, Perdigoto CN, Southall TD, Brand AH, Schweisguth F, 2010. Transcriptional control of stem cell maintenance in the Drosophila intestine. Development 137, 705–714. doi: 10.1242/dev.039404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsland M, Werme M, Malewicz M, Perlmann T, Muhr J, 2006. The establishment of neuronal properties is controlled by Sox4 and Sox11. Genes Dev. 20, 3475–3486. doi: 10.1101/gad.403406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beronja S, Fuchs E, 2012. RNAi-Mediated Gene Function Analysis in Skin, in: Stem Cells in Regenerative Medicine, Methods in Molecular Biology. Humana Press, Totowa, NJ, pp. 351–361. doi: 10.1007/978-1-62703-227-8_23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beronja S, Janki P, Heller E, Lien WH, Keyes BE, Oshimori N, Fuchs E, 2013. RNAi screens in mice identify physiological regulators of oncogenic growth. Nature 501, 185–190. doi: 10.1038/nature12464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beronja S, Livshits G, Williams S, Fuchs E, 2010. Rapid functional dissection of genetic networks via tissue-specific transduction and RNAi in mouse embryos. Nat. Med 16, 821–827. doi: 10.1038/nm.2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I, 2015. Targeting the translation machinery in cancer. Nat Rev Drug Discov 14, 261–278. doi: 10.1038/nrd4505 [DOI] [PubMed] [Google Scholar]
- Blanco S, Bandiera R, Popis M, Hussain S, Lombard P, Aleksic J, Sajini A, Tanna H, Cortes-Garrido R, Gkatza N, Dietmann S, Frye M, 2016. Stem cell function and stress response are controlled by protein synthesis. Nature 534, 335–340. doi: 10.1038/nature18282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, Baehr LM, 2014. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. American Journal of Physiology-Endocrinology and Metabolism 307, E469–E484. doi: 10.1152/ajpendo.00204.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AHFM, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA, 2005. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436, 660–665. doi: 10.1038/nature03841 [DOI] [PubMed] [Google Scholar]
- Brown S, Pineda CM, Xin T, Boucher J, Suozzi KC, Park S, Matte-Martone C, Gonzalez DG, Rytlewski J, Beronja S, Greco V, 2017. Correction of aberrant growth preserves tissue homeostasis. Nature 548, 334–337. doi: 10.1038/nature23304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buszczak M, Signer RAJ, Morrison SJ, 2014. Cellular differences in protein synthesis regulate tissue homeostasis. Cell 159, 242–251. doi: 10.1016/j.cell.2014.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Mitsutake N, LaPerle K, Akeno N, Zanzonico P, Longo VA, Mitsutake S, Kimura ET, Geiger H, Santos E, Wendel HG, Franco A, Knauf JA, Fagin JA, 2009. Endogenous expression of Hras(G12V) induces developmental defects and neoplasms with copy number imbalances of the oncogene. Proc. Natl. Acad. Sci. U.S.A 106, 7979–7984. doi: 10.1073/pnas.0900343106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung BY, Hardcastle TJ, Jones JD, Irigoyen N, Firth AE, Baulcombe DC, Brierley I, 2015. The use of duplex-specific nuclease in ribosome profiling and a user-friendly software package for Ribo-seq data analysis. RNA 21, 1731–1745. doi: 10.1261/rna.052548.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtius K, Wright NA, Graham TA, 2017. An evolutionary perspective on field cancerization. Nat. Rev. Cancer 18, 19–32. doi: 10.1038/nrc.2017.102 [DOI] [PubMed] [Google Scholar]
- Dai X, Segre JA, 2004. Transcriptional control of epidermal specification and differentiation. Curr. Opin. Genet. Dev 14, 485–491. doi: 10.1016/j.gde.2004.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digilio MC, Lepri F, Baban A, Dentici ML, Versacci P, Capolino R, Ferese R, De Luca A, Tartaglia M, Marino B, Dallapiccola B, 2011. RASopathies: Clinical Diagnosis in the First Year of Life. Mol Syndromol 1, 282–289. doi: 10.1159/000331266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duronio RJ, Xiong Y, 2013. Signaling Pathways that Control Cell Proliferation. Cold Spring Harb Perspect Biol 5, a008904–a008904. doi: 10.1101/cshperspect.a008904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnhead HO, Rodriguez J, Govek EE, Guo W, Kobayashi R, Hannon G, Lazebnik YA, 1998. Oncogene-dependent apoptosis is mediated by caspase-9. Proceedings of the National Academy of Sciences 95, 13664–13669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E, 2007. Scratching the surface of skin development. Nature 445, 834–842. doi: 10.1038/nature05659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E, Raghavan S, 2002. Getting under the skin of epidermal morphogenesis. Nat. Rev. Genet 3, 199–209. doi: 10.1038/nrg758 [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N, 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6(269), pI1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL, 2001. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proceedings of the National Academy of Sciences 98, 14440–14445. doi: 10.1073/pnas.251541198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CA, Hornberger TA, 2013. Measuring protein synthesis with SUnSET: a valid alternative to traditional techniques? Exerc Sport Sci Rev 41, 107–115. doi: 10.1097/JES.0b013e3182798a95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS, 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. doi: 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves M, Maley CC, 2012. Clonal evolution in cancer. Nature 481, 306–313. doi: 10.1038/nature10762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner C, Groesser L, 2014. Mosaic RASopathies. Cell Cycle 12, 43–50. doi: 10.4161/cc.23108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Ruggero D, 2010. Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin. Cancer Res 16, 4914–4920. doi: 10.1158/1078-0432.CCR-10-0433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS, 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. doi: 10.1126/science.1168978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW, 2004. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 18, 2573–2580. doi: 10.1101/gad.1255304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap V, Rezende NC, Scotland KB, Shaffer SM, Persson JL, Gudas LJ, Mongan NP, 2009. Regulation of Stem Cell Pluripotency and Differentiation Involves a Mutual Regulatory Circuit of the Nanog, OCT4, and SOX2 Pluripotency Transcription Factors with Polycomb Repressive Complexes and Stem Cell microRNAs. Stem Cells and Development 18, 1093–1108. doi: 10.1089/scd.2009.0113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke MT, Fujimoto S, Imai T, 2013. SeeDB: a simple and morphology-preserving optical clearing agent for neuronal circuit reconstruction. Nature Neuroscience 16, 1154–1161. doi: 10.1038/nn.3447 [DOI] [PubMed] [Google Scholar]
- Keppler-Noreuil KM, Parker VER, Darling TN, Martinez-Agosto JA, 2016. Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 172, 402–421. doi: 10.1002/ajmg.c.31531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL, 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. doi: 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koromilas AE, 2015. Roles of the translation initiation factor eIF2α serine 51 phosphorylation in cancer formation and treatment. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 1849, 871–880. doi: 10.1016/j.bbagrm.2014.12.007 [DOI] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J Jr, Tugendreich S, 2013. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. doi: 10.1093/bioinformatics/btt703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL, 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ASY, Kranzusch PJ, Cate JHD, 2015. eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 522, 111–114. doi: 10.1038/nature14267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Kim JM, Kim KH, Heo JI, Kwak SJ, Han JA, 2014. Genotoxic stress/p53-induced DNAJB9 inhibits the pro-apoptotic function of p53. Cell Death & Differentiation 22, 86–95. doi: 10.1038/cdd.2014.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R, 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup, 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wang X, van der Knaap MS, Proud CG, 2004. Mutations Linked to Leukoencephalopathy with Vanishing White Matter Impair the Function of the Eukaryotic Initiation Factor 2b Complex in Diverse Ways. Mol. Cell. Biol 24, 3295–3306. doi: 10.1128/MCB.24.8.3295-3306.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liakath-Ali K, Mills EW, Sequeira I, Lichtenberger BM, Pisco AO, Sipila KH, Mishra A, Yoshikawa H, Wu CC-C, Ly T, Lamond AI, Adham IM, Green R, Watt FM, 2018. An evolutionarily conserved ribosome-rescue pathway maintains epidermal homeostasis. Nature 556, 376–380. doi: 10.1038/s41586-018-0032-3 [DOI] [PubMed] [Google Scholar]
- Liu Y, Horn JL, Banda K, Goodman AZ, Lim Y, Jana S, Arora S, Germanos AA, Wen L, Hardin WR, Yang YC, Coleman IM, Tharakan RG, Cai EY, Uo T, Pillai SPS, Corey E, Morrissey C, Chen Y, Carver BS, Plymate SR, Beronja S, Nelson PS, Hsieh AC, 2019. The androgen receptor regulates a druggable translational regulon in advanced prostate cancer. Science Translational Medicine 11, eaaw4993. doi: 10.1126/scitranslmed.aaw4993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokireddy S, McFarlane C, Ge X, Zhang H, Sze SK, Sharma M, Kambadur R, 2011. Myostatin induces degradation of sarcomeric proteins through a Smad3 signaling mechanism during skeletal muscle wasting. Mol. Endocrinol 25, 1936–1949. doi: 10.1210/me.2011-1124 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lourenço AR, Coffer PJ, 2017. SOX4: Joining the Master Regulators of Epithelial-to-Mesenchymal Transition? Trends in Cancer 3, 571–582. doi: 10.1016/j.trecan.2017.06.002 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S, 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Cepero E, Evan G, 2004. Intrinsic tumour suppression. Nature 432, 307–315. doi: 10.1038/nature03098 [DOI] [PubMed] [Google Scholar]
- Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, Fitzgerald RC, Handford PA, Campbell PJ, Saeb-Parsy K, Jones PH, 2018. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917. doi: 10.1126/science.aau3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, Stebbings L, Menzies A, Widaa S, Stratton MR, Jones PH, Campbell PJ, 2015. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886. doi: 10.1126/science.aaa6806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Z, Zhang D, Hu B, Wang J, Shen X, Xiao W, 2015. FBXO32 Targets c-Myc for Proteasomal Degradation and Inhibits c-Myc Activity. J. Biol. Chem 290, 16202–16214. doi: 10.1074/jbc.M115.645978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesa KR, Kawaguchi K, Gonzalez DG, Cockburn K, Boucher J, Xin T, Klein AM, Greco V, 2017. Epidermal stem cells self-renew upon neighboring differentiation. doi: 10.1101/155408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, Carpenter AE, Foo SY, Stewart SA, Stockwell BR, Hacohen N, Hahn WC, Lander ES, Sabatini DM, Root DE, 2006. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. doi: 10.1016/j.cell.2006.01.040 [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L, 2007. A global double-fluorescent Cre reporter mouse. genesis 45, 593–605. doi: 10.1002/dvg.20335 [DOI] [PubMed] [Google Scholar]
- Nowak JA, Fuchs E, 2009. Isolation and Culture of Epithelial Stem Cells, in: Stem Cells in Regenerative Medicine, Methods in Molecular Biology. Humana Press, Totowa, NJ, pp. 215–232. doi: 10.1007/978-1-59745-060-7_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Gonzalez DG, Guirao B, Boucher JD, Cockburn K, Marsh ED, Mesa KR, Brown S, Rompolas P, Haberman AM, Bella’iche Y, Greco V, 2017. Tissue-scale coordination of cellular behaviour promotes epidermal wound repair in live mice. Nat. Cell Biol 19, 155–163. doi: 10.1038/ncb3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavitt GD, 2005. eIF2B, a mediator of general and gene-specific translational control. Biochemical Society Transactions 33, 1487–1492. doi: 10.1042/BST20051487 [DOI] [PubMed] [Google Scholar]
- Pierpont MEM, Magoulas PL, Adi S, Kavamura MI, Neri G, Noonan J, Pierpont EI, Reinker K, Roberts AE, Shankar S, Sullivan J, Wolford M, Conger B, Santa Cruz M, Rauen KA, 2014. Cardio-facio-cutaneous syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 134, e1149–62. doi: 10.1542/peds.2013-3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauen KA, 2013. The RASopathies. Annual Review of Genomics and Human Genetics 14, 355–369. doi: 10.1146/annurev-genom-091212-153523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK, 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompolas P, Mesa KR, Kawaguchi K, Park S, Gonzalez D, Brown S, Boucher J, Klein AM, Greco V, 2016. Spatiotemporal coordination of stem cell commitment during epidermal homeostasis. Science 352, 1471–1474. doi: 10.1126/science.aaf7012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D, 2013. Translational control in cancer etiology. Cold Spring Harb Perspect Biol 5, a012336–a012336. doi: 10.1101/cshperspect.a012336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu SK, Tiwari N, Pataskar A, Zhuang Y, Borisova M, Diken M, Strand S, Beli P, Tiwari VK, 2017. FBXO32 promotes microenvironment underlying epithelial-mesenchymal transition via CtBPI during tumour metastasis and brain development. Nat Commun 8, 1438. doi : 10.1038/s41467-017-01366-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scali O, Di Perri C, Federico A, 2006. The spectrum of mutations for the diagnosis of vanishing white matter disease. Neurological Sciences 27, 271–277. doi: 10.1007/s10072-006-0683-y [DOI] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, Pierre P, 2009. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 6, 275–277. doi: 10.1038/nmeth.1314 [DOI] [PubMed] [Google Scholar]
- Sendoel A, Dunn JG, Rodriguez EH, Naik S, Gomez NC, Hurwitz B, Levorse J, Dill BD, Schramek D, Molina H, Weissman JS, Fuchs E, 2017. Translation from unconventional 5’ start sites drives tumour initiation. Nature 541, 494–499. doi: 10.1038/nature21036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK-H, Wilson C, Okreglak V, Ashkenazi A, Hann B, Nader K, Arkin MR, Renslo AR, Sonenberg N, Walter P, 2013. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2, 7193. doi: 10.7554/eLife.00498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Tsai JC, Kampmann M, Hearn BR, Vedantham P, Jaishankar P, Sokabe M, Mendez AS, Newton BW, Tang EL, Verschueren E, Johnson JR, Krogan NJ, Fraser CS, Weissman JS, Renslo AR, Walter P, 2015. Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. Elife 4, e07314. doi: 10.7554/eLife.07314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signer RAJ, Magee JA, Salic A, Morrison SJ, 2014. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54. doi: 10.1038/nature13035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulou PA, Blanpain C, 2012. Development and homeostasis of the skin epidermis. Cold Spring Harb Perspect Biol 4, a008383–a008383. doi: 10.1101/cshperspect.a008383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin C-S, William CM, Tanabe Y, Jessell TM, Costantini F, 2001. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Developmental Biology 2001 1:1 1, 4. doi: 10.1186/1471-213X-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf CR, Moreno MV, Olshen AB, Taylor BS, Ruggero D, 2013. The Translational Landscape of the Mammalian Cell Cycle. Mol. Cell 52, 574–582. doi: 10.1016/j.molcel.2013.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Kosaka T, Miyazaki Y, Mikami S, Niwa N, Otsuka Y, Minamishima YA, Mizuno R, Kikushi E, Miyajima A, Sabe H, Okada Y, Ohlen P, Suematsu M, Oya M, 2016. Acquired platinum resistance involves epithelial to mesenchymal transition through ubiquitin ligase FBXO32 dysregulation. jCi Insight 1, 4147. doi: 10.1172/jci.insight.83654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott SJ, 2005. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development 132, 2685–2695. doi: 10.1242/dev.01874 [DOI] [PubMed] [Google Scholar]
- Tintignac LA, Lagirand J, Batonnet S, Sirri V, Leibovitch MP, Leibovitch SA, 2005. Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J. Biol. Chem 280, 2847–2856. doi : 10.1074/jbc.M411346200 [DOI] [PubMed] [Google Scholar]
- Tomayko MM, Reynolds CP, 1989. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemotherapy and Pharmacology 24, 148–154. doi: 10.1007/BF00300234 [DOI] [PubMed] [Google Scholar]
- Truitt ML, Ruggero D, 2016. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 16, 288–304. doi: 10.1038/nrc.2016.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier LG, Clevers H, 2009. Stem Cells, Self-Renewal, and Differentiation in the Intestinal Epithelium. Annual Review of Physiology 71,241–260. doi: 10.1146/annurev.physiol.010908.163145 [DOI] [PubMed] [Google Scholar]
- Wade M, Li Y-C, Wahl GM, 2013. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 13, 83–96. doi: 10.1038/nrc3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SE, Beronja S, Pasolli HA, Fuchs E, 2011. Asymmetric cell divisions promote Notch-dependent epidermal differentiation. Nature 470, 353–358. doi: 10.1038/nature09793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Zou Q, Liu Y, Yang X, 2016. Genome-wide assessment of differential translations with ribosome profiling data. Nat Commun 7, 11194. doi: 10.1038/ncomms11194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y, Stumpf CR, Xue L, Devericks E, So L, Nguyen HG, Griselin A, Gordan JD, Umetsu SE, Reich SH, Worland ST, Asthana S, Barna M, Webster KR, Cunningham JT, Ruggero D, 2019. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat. Med 39, 1. doi: 10.1038/s41591-018-0321-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, Sandoval M, Beronja S, 2018. Oncogenic activation of PI3K induces progenitor cell differentiation to suppress epidermal growth. Nat. Cell Biol 20, 1256–1266. doi: 10.1038/s41556-018-0218-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Zhou Q, Wang Z, Wei W, 2016. Recent advances in SCF ubiquitin ligase complex: Clinical implications. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1866, 12–22. doi: 10.1016/j.bbcan.2016.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Liu Y, Zhu R, Ding F, Wan Y, Li Y, Liu Z, 2017. FBXO32 suppresses breast cancer tumorigenesis through targeting KLF4 to proteasomal degradation. Oncogene 36, 3312–3321. doi: 10.1038/onc.2016.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1. Regulators of HrasG12V progenitor proliferation and renewal within the translation machinery, Related to Figure 1
Candidates for proliferation and renewal regulation as identified by DESeq2 analysis of shRNA enrichment in non-dividing (EdU-) relative to dividing (EdU+) basal progenitors (for proliferation screen), or in basal progenitor relative to the differentiated suprabasal cells (for renewal screen). Positive fold-change value indicates shRNA enrichment in the EdU− population (for proliferation screen) or in basal progenitors (for renewal screen).
Supplemental Table S2: Translationally altered genes in HrasG12V epidermal progenitors -- HrasG12V+shScram vs. WT+shScram (HS/WS), Related to Figure 3
Genes with translational efficiency changes were identified by Xtail analysis and excluded genes with significant transcriptional changes (as determined by EdgeR). Positive fold-change value indicates translational upregulation in HrasG12V+shScram basal progenitors.
Supplemental Table S3: Translationally altered genes regulated by eIF2B5 in HrasG12V epidermal progenitors -- HrasG12V+shEif2b5 to HrasG12V+shScram (HE/HS), Related to Figure 3
Genes with translational efficiency changes were identified by Xtail analysis and excluded genes with significant transcriptional changes (as determined by EdgeR). Positive fold-change value indicates translational upregulation in HrasG12V+shEif2b5 basal progenitors.
Supplemental Table S4: Translationally altered genes in HrasG12V epidermal progenitors that are rescued by shEif2b5, Related to Figure 3
Genes with translational efficiency changes were identified by Xtail analysis and excluded genes with significant transcriptional changes (as determined by EdgeR). Positive fold-change value indicates translational upregulation in HrasG12V progenitors.
Supplemental Table S5: Regulators of HrasG12V progenitor proliferation and renewal within the eIF2B5-regulated oncogenic translatome, Related to Figure 4
Candidates for proliferation and renewal regulation as identified by DESeq2 analysis of shRNA enrichment in non-dividing (EdU−) relative to dividing (EdU+) basal progenitors (for proliferation screen), or in basal progenitor relative to the differentiated suprabasal cells (for renewal screen). Positive fold-change value indicates shRNA enrichment in the EdU− population (for proliferation screen) or in basal progenitors (for renewal screen).
Supplemental Table S6: shRNA and primer sequences used in this study, Related to STAR Methods
Data Availability Statement
Ribosome profiling sequencing data can be accessed at NCBI Gene Expression Omnibus (GSE 126660). All other data are available from the Lead Contact upon request.