

Abstract
The epithelial-mesenchymal transition (EMT) is the process by which epithelial cells lose their tightly packed polarized characteristics and acquire a migratory mesenchymal phenotype. EMT plays a pivotal role in embryonic development, wound healing, tissue regeneration, organ fibrosis and cancer progression. The basic helix-loop-helix (bHLH) transcription factors TWIST1/2 are key EMT-inducing transcription factors that govern transcription of EMT-associated genes. Although regulation of TWIST1 activity and stability has been well studied, little is known about how TWIST2 is post-translationally regulated. Here we have identified ZNF451, a SUMO2/3 specific E3 ligase, as a novel regulator of TWIST2 in promoting its stability. ZNF451 directly binds to and SUMOylates TWIST2 at K129 residue, and consequently blocks ubiquitination and proteasome-dependent degradation of TWIST2. Ectopic expression of ZNF451 increases the protein level of TWIST2 in mammary epithelial cells, leading to increased expression of mesenchymal markers, whereas depletion of ZNF451 suppresses mesenchymal phenotypes. Collectively, our findings demonstrate that ZNF451 plays a vital role in EMT through SUMOylation-dependent stabilization of TWIST2.
Keywords: Zinc finger protein 451, TWIST2, SUMOylation, EMT
Introduction
EMT is a physiological and multi-step process, by which the epithelial cells gradually lose cell polarity and cell-cell contacts, and acquire motile and invasive characteristics of mesenchymal cells [1,2]. EMT is induced by several extracellular signals including components of growth factors such as TGF-β, NF-κB, FGF and EGF, hypoxia, and acidity in microenvironment [1,3-8]. EMT plays a vital role in embryogenesis and development, including formation of mesoderm in gastrulation, postnatal engagement in mammary gland development, and wound healing. EMT is also viewed as a pathologic phenomenon that occurs in fibrosis, chronic inflammation, and cancer [6,9,10]. EMT has been observed in various tumor tissues, such as breast cancer, lung cancer, prostate cancer and gastric cancer. In particular, EMT is associated with cancer metastasis, during which the carcinoma cells disseminate from the site of primary tumors and establish secondary tumors in distant organs [2,11,12]. EMT also contributes to resistance against conventional therapies due to its critical role in maintaining cancer stem cell state [13-15].
E-Cadherin was considered as a suppressor of tumor progression. Decreased expression of E-Cadherin is associated with loss of cell differentiation and poor prognosis in patients [16]. During the process of EMT, E-Cadherin is crucially repressed at the transcriptional level, which is mainly triggered by elevated expression of key transcriptional factors of bHLH family (TWIST1/2), Snail family (Snail/Slug), or ZEB factors (ZEB1/2) [1,6,17-19]. The regulatory roles of TWIST1 and TWIST2 in EMT initiation have been thoroughly studied. TWIST1/2 bind to the conserved E-box sequences of E-Cadherin promoter and recruit the co-repression factors such as HDAC2 to repress the transcription of E-Cadherin [20-23]. Consistent with this, overexpression of TWIST1/2 induces the expression of mesenchymal markers such as Fibronectin and N-Cadherin. Although TWIST1 and TWIST2 share sequence homology and have demonstrated functional similarity, they have distinct functions. For example, mutations of TWIST1 and TWIST2 are found in different hereditary diseases. TWIST2 recessive mutations cause Setleis syndrome, while dominant mutations of TWIST1 cause Saethre-Chotzen syndrome [24,25], indicating that these two genes exhibit non-redundant functions in skin and bone development. Ectopic expression of TWIST2, but not TWIST1, in mammary epithelial cells and breast cancer cells increases the percentage of CD44high/CD24low sub-populations, promotes the expression of stem cell markers, and enhances the self-renewal of stem-like cells. Moreover, TWIST2 results in the constitutive activation of STAT3, which exists in more than 50% of primary breast tumors and tumor-derived cell lines [26].
Post-translational modifications of transcription factors that are involved in the process of EMT such as Snail/Slug, ZEB1/2 and TWIST1 have been reported to be one of the critical mechanisms for regulating their capacity to induce EMT [10,27,28]. Several ubiquitin E3 ligases have been identified to promote proteasomal degradation of TWIST1. For example, p53-Pirh2 complex promotes TWIST1 degradation and attenuates EMT [29]. Imipramine blue halts EMT by promoting FBXL14-mediated TWIST1 degradation [30]. Tumor suppressor PAQR3 suppresses EMT by promoting the complex formation between TWIST1 and its E3 ubiquitin ligase BTRC, which in turn increases proteasome-mediated TWIST1 degradation [31]. IKKβ-mediated phosphorylation of TWIST1 also enhances its degradation by increasing the binding affinity of BTRC to TWIST1 [32]. Furthermore, MAPK-mediated phosphorylation of TWIST1 on S68 residue enhances its stabilization and drives invasion and metastasis of cancer cells [33], while SCP1-mediated dephosphorylation of pS68 accelerates TWIST1 degradation [34]. However, till now, the functions and mechanisms of post-translational modification of the highly related TWIST2 were not defined.
SUMO is a small ubiquitin-like protein. Like ubiquitination, SUMOylation also targets lysine residues for conjugation with SUMO molecules as a post-translational modification of target protein. One way that SUMO modification regulates protein function is to block ubiquitin conjugation to the same lysine residue, resulting in blockade of proteasome-mediated substrate degradation [35,36]. ZNF451 (also called ZATT), which is associated with promyelocytic leukemia bodies, is a SUMO2/3-specific E3 ligase [37]. The known function of ZNF451 is to bind to and SUMOylate TOP2 cleavage complex (TOP2cc), which recruits tyrosyl-DNA phosphodiesterase 2 (TDP2) on stalled TOP2cc, thus helping to resolve the genotoxic TOP2 DNA-protein cross-links [38]. ZNF451 harbors two SUMO-Interacting Motifs (SIMs) and interacts with SUMO E2 conjugating enzyme Ubc9 or SUMOs [39,40]. Thus, ZNF451 exerts its functions via SUMO modification machinery.
In this study, we identified ZNF451 as a positive regulator of EMT through its SUMO E3 ligase activity. Functionally, overexpression of ZNF451 promotes EMT, while its knockdown profoundly inhibits EMT. Moreover, TWIST2 depletion attenuates ZNF451-mediated EMT process, suggesting that ZNF451 enhances EMT through TWIST2. Mechanistically, ZNF451 interacts with TWIST2 and SUMOylates TWIST2 at K129 residue. SUMOylation deficiency mutant of TWIST2 exhibits decreased ubiquitination and increased stability, indicating that the same K129 serves as the target residue for both SUMOylation and ubiquitination. Furthermore, ZNF451-induced SUMOylation of TWIST2 depends on its SIM motifs and its SUMO E3 ligase activity. Hence, findings from this study identified a novel function of ZNF451 in inducing the cell EMT response. Our studies also revealed a novel post-translational modification of TWIST2 by SUMO through the E3 ligase activity of ZNF451, and discovered a novel mechanism by which ZNF451 regulates the activity of TWIST2 during EMT.
Material and methods
Plasmids
Expression plasmids for full-length ZNF451, its domain mutants (SIM1, SIM2) and deletion mutants (1-256 and 56-end) were obtained by PCR and cloned into different mammalian expression vectors to fuse with various tags. Their sequence integrity was confirmed by sequencing. Plasmids expressing wild-type TWIST2 and its mutants (K32R, K52R, K53R, K91R, K108R, and K129R) were generated by PCR and PCR-based mutagenesis. For lentiviral expression vector for ZNF451, ORF of ZNF451 or its mutant was subcloned into pWPI vector. For RNAi experiment, oligonucleotides targeting ZNF451 shRNA were annealed and inserted into pLKO.1 vector.
Antibodies and reagents
Primary antibodies used in this study are as following: N-Cadherin (BD Biosciences), ubiquitin (Santa Cruz), ZNF451 and FLAG tag (Sigma), TWIST1 and TWIST2 (Abcam), E-Cadherin, Fibronectin, Vimentin and HA tag (Cell Signaling Technology). Puromycin and Cycloheximide (CHX) were purchased from Sigma and Merck, respectively.
Cell culture, cell transfection and lentiviral transduction
MCF10A cells were maintained in Dulbecco’s modified Eagle medium/nutrient mixture F-12 (5% horse serum, 20 ng/ml EGF, 0.5 mg/ml hydrocortisone, 100 ng/ml cholera toxin and 10 μg/ml insulin). MCF10A were transfected with plasmids mixed with X-tremeGENE™ HP DNA Transfection Reagent (Roche) at the ratio of 1:2.5. HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium plus 10% fetal bovine serum. HEK293T cells were transfected with plasmids mixed with Polyethylenimine (Polyscience) at the ratio of 1:3. HaCaT cells were maintained in minimum Eagle’s medium supplemented with 10% fetal bovine serum.
For stable expression of ZNF451, HEK293T cells were transfected with lentiviral expression plasmids together with lentiviral packaging plasmid psPAX2 and envelope plasmid pMD2.G. 48 h after transfection, lentiviruses were collected from the medium, purified by centrifugation and then used to infect host cells. Stable cells were selected in the presence of 2 ng/ml of puromycin.
Immunoprecipitation and western blotting
Immunoprecipitation (IP) was performed as previously described. Briefly, plasmids of HA-ZNF451 and Myc-TWIST1/2 were transiently co-transfected into HEK239T cells. 36 h after transfection, cell lysates were harvested by lysis buffer (150 mM NaCl, 20 mM Tris-HCl pH 8.0, 1% NP-40, 2 mM EDTA). Protein A Sepharose CL-4B (GE Healthcare) and anti-HA antibodies were added into the cell lysates and incubate at 4°C for 6 h. After 3x washes, immunoprecipitated proteins were separated by SDS-PAGE, transferred to polyvinylidene difluoride membrane, immunostained by indicated primary antibodies, and finally detected by horseradish peroxidase-conjugated secondary antibodies and visualized by chemiluminescence (Pierce).
In vitro GST pull-down assay
GST pull-down experiments were carried out as previously described [41]. GST fusion protein of TWIST1/2 was prepared from E. coli strain DE3. In vitro translation of FLAG-ZNF451 was carried out using Quick Coupled Transcription/Translation System (Promega).
SUMOylation assay in vivo
SUMOylation of TWIST2 was carried out as previously described [42]. Briefly, HEK293T cells were transiently transfected with TWIST2 (HA- or His-tag) and SUMO2/3. HA-TWIST2 was IP’ed using anti-HA antibody or His-TWIST2 was pulled down using Ni-NTA beads (Pierce) under a denature condition. Precipitates were analyzed using anti-SUMO (or epitope tag on SUMO) antibodies by western blotting assays.
RNA interference
Small interference RNAs (siRNAs) targeting human TWIST2 were synthesized by RiboBio Co (target sequence: nt 305-323 of coding region, GCAAGATCCAGACGCTCAA). Cells were transfected with siControl or siTWIST2 using Lipofectamine RNAi MAX (Invitrogen). Small hairpin RNA (shRNA) targeting human ZNF451 were designed as the following: shZNF451 target sequence, nt 810-828 of coding region, GCATATGTCTGGAAAGAAT.
Quantitative reverse transcription-PCR (qRT-PCR)
Total RNA (1 μg) isolated from cells using TRIzol Reagent (Invitrogen) was reverse-transcribed to complementary DNA using Transcriptor Reverse Transcriptase (Takara). Complementary DNA was then diluted and used for quantification by real-time PCR using Power SYBR® Green PCR Master Mix (Applied Biosystems) and 7500 real-time PCR system (Applied Biosystems). qRT-PCR primers were listed in the following: E-Cadherin, 5’-GACAACAAGCCCGAATT-3’ (forward) and 5’-GGAAACTCTCTCGGTCCA-3’ (reverse); N-Cadherin, 5’-CGGGTAATCCTCCCAAATCA-3’ (forward) and 5’-CTTTATCCCGGCGTTTCATC-3’ (reverse); Vimentin, 5’-GAGAACTTTGCCGTTGAAGC-3’ (forward) and 5’-GCTTCCTGTAGGTGGCAATC-3’ (reverse); Fibronectin, 5’-CAGTGGGAGACCTCGAGAAG-3’ (forward) and 5’-TCCCTCGGAACATCAGAAAC-3’ (reverse); GAPDH, 5’-AGCCACATCGCTCAGACAC-3’ (forward) and 5’-GCCCAATACGACCAAATCC-3’ (reverse).
Results
ZNF451 promotes EMT
We initially identified ZNF451 as an interacting protein of Smad4, the central mediator in TGF- signal transduction. We have previously reported that ZNF451 binds to Smad4 and inhibits Smad4-mediated, TGF-β-induced growth inhibitory function [41]. Since TGF-β activity is a strong inducer of EMT, we expected that ZNF451 would inhibit EMT. To test this, we generated cell clones that stably expressed FLAG-ZNF451 in MCF10A and HaCaT cell line, the in vitro model cell systems to study the process of EMT. MCF10A and HaCaT cell are immortalized human mammary epithelial and keratinocyte cell lines, respectively. To our surprise, we found that ectopic expression of ZNF451 induced EMT, converting the epithelial cell morphology to mesenchymal cell morphology. As shown in Figure 1A, FLAG-ZNF451-expressing cells began to lose their polarized epithelial cell morphology and became scattered and spindle-like, resembling mesenchymal cell morphology in both cell lines (Figure 1A). Western blot analysis of typical EMT markers revealed that, in comparison to control vector-transfected cells, ectopic expression of ZNF451 decreased the level of epithelial marker E-Cadherin and increased the level of mesenchymal markers such as N-Cadherin, Vimentin and Fibronectin in both MCF10A and HaCaT cells (Figure 1B). Immunofluorescence staining of control and ZNF451-expressing cells confirmed the down regulation of E-Cadherin and the up regulation of Vimentin and Fibronectin in ZNF451-expressing cells (Figure 1C, and data not shown). Further analysis with qRT-PCR revealed that the ZNF451-mediated regulation of EMT marker expression was at the transcriptional level (Figure 1D).
Figure 1.
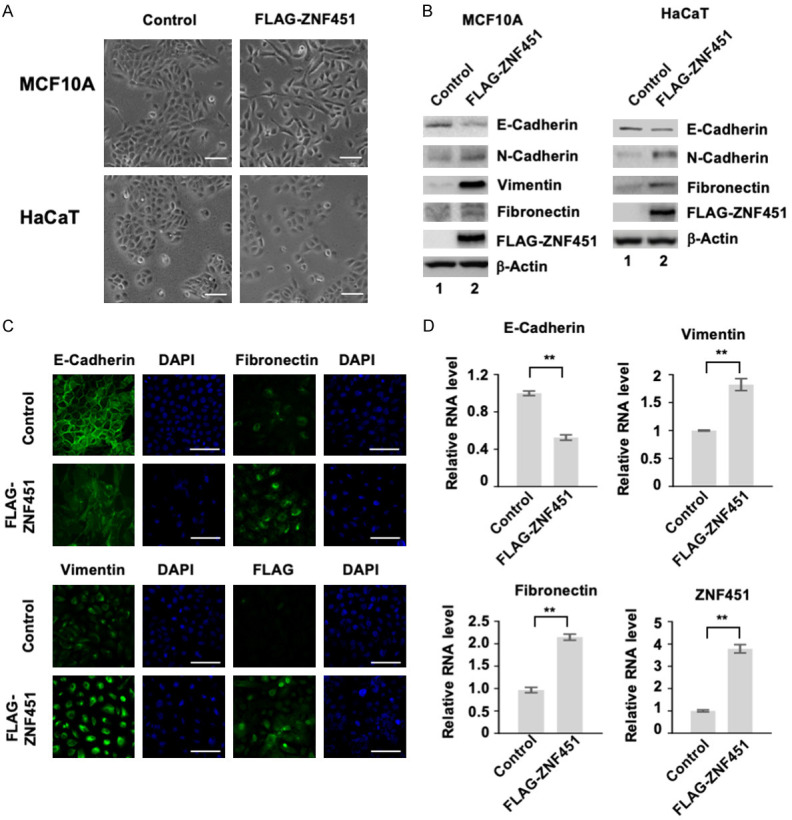
Ectopic expression of ZNF451 enhances EMT in MCF10A and HaCaT cells. A. Overexpression of ZNF451 promotes mesenchymal phenotype in MCF10A and HaCaT cells. Scale bars, 100 μm. B. ZNF451 increases the expression of mesenchymal markers, but decreases that of E-Cadherin, an epithelial marker. Protein levels of N-Cadherin, E-Cadherin, Fibronectin, Vimentin and transfected ZNF451 were analyzed in indicated cells by western blotting. β-Actin is an internal control. C. ZNF451 promotes EMT phenotype. Immunofluorescence staining assays were performed in control and ZNF451 expressing MCF10A cells. E-Cadherin, Fibronectin, Vimentin and transfected ZNF451 proteins were immunostained (green) and DAPI marks the nucleus (blue). D. ZNF451 promotes transcriptional changes of EMT markers. Total mRNAs from MCF10A cells stably expressing ZNF451 or control were extracted for qRT-PCR analysis. Target genes include E-Cadherin, Fibronectin, Vimentin and ZNF451. Data are shown in triplicates as mean + s.e.m. *P < 0.05, **P < 0.01.
To confirm the above observation obtained from ZNF451 ectopic expression experiments and determine the physiological functions of ZNF451 in EMT regulation, we used both CRISPR/Cas9-mediated gene knockout and lentivirus-mediated specific shRNA knockdown approaches to deplete the expression of ZNF451 in MCF10A and HaCaT and to study the effect of ZNF451 depletion on cell morphology and EMT. The representative morphology of control and ZNF451-depleted cells was shown in Figure 2A. We found that MCF10A and HaCaT cells with depletion of ZNF451 exhibited more columnar-like and packed morphology (Figure 2A). Western blot analysis and qRT-PCR analysis of EMT markers confirmed the morphological changes that knockdown of ZNF451 increased the expression of epithelial marker (E-Cadherin) and reduced the expression of mesenchymal markers (N-Cadherin, Vimentin and Fibronectin) at both protein and mRNA levels (Figure 2B, 2C, and data not shown). Immunofluorescence staining further confirmed the expression of E-Cadherin was enhanced whereas that of Fibronectin and Vimentin was decreased in ZNF451-depleted MCF10A and HaCaT cells (Figure 2D and data not shown). Knockout of ZNF451 gene by CRISPR/Cas9 in HaCaT also led to the same results (Figure 2E, 2F). Taken together, our results from ZNF451 ectopic expression and depletion of ZNF451 expression approaches suggest that ZNF451 promotes the process of EMT.
Figure 2.
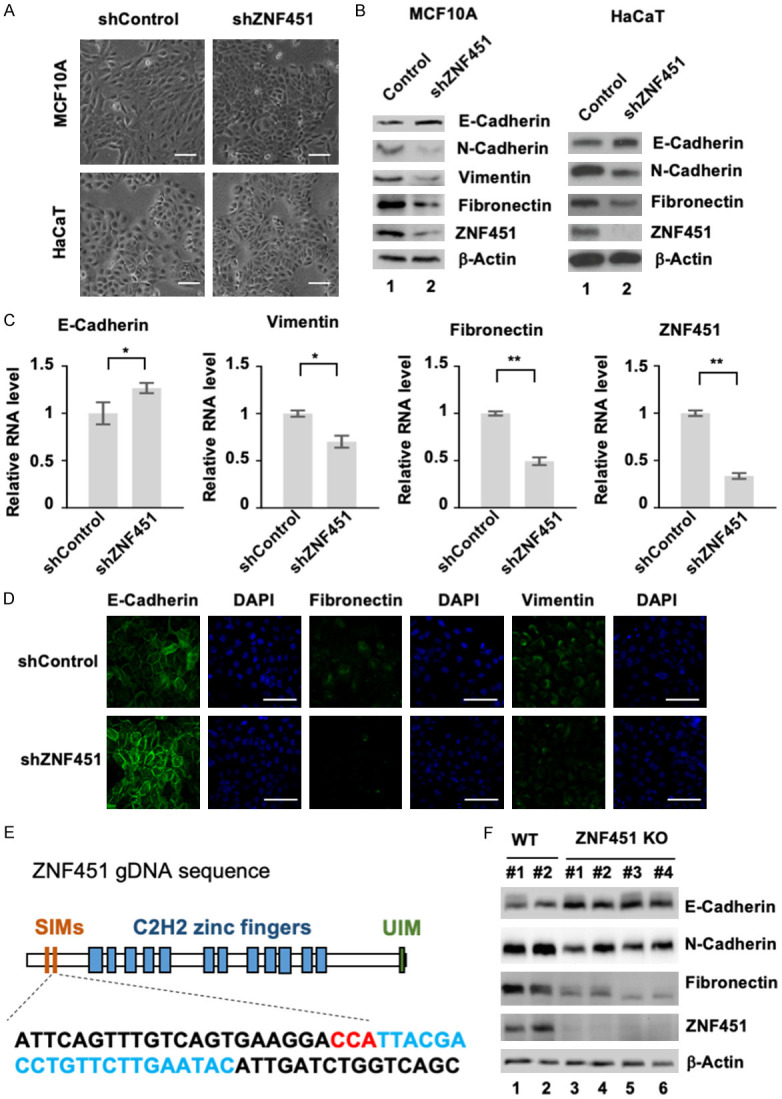
Depletion of ZNF451 expression attenuates EMT morphology. A. Knockdown of ZNF451 enhances epithelial morphology in MCF10A and HaCaT cells. Scale bars, 100 μm. B. Knockdown of ZNF451 enhances the expression of epithelial marker, but inhibits that of mesenchymal markers. Whole cell lysates from shControl and shZNF451 were harvested for western blotting analysis with N-Cadherin, E-Cadherin, Vimentin, Fibronectin and ZNF451. β-Actin is an internal reference. C. Knockdown of ZNF451 reverses transcriptional changes for EMT. Total mRNAs from MCF10A cells with shZNF451 or control were extracted for qRT-PCR analysis of target genes such as E-Cadherin, Fibronectin, Vimentin and endogenous ZNF451. Data are shown in triplicates as mean + s.e.m. *P < 0.05, **P < 0.01. D. Expression of ZNF451 promotes epithelial features. Immunofluorescence staining assay was performed in MCF10A cells stably carrying shZNF451 and shControl. E-Cadherin, Fibronectin, and Vimentin proteins were immunostained (green). DAPI marks the nucleus (blue). E. CRISPR/Cas9 gDNA sequence targeting second SIM domain of ZNF45. The gDNA target is shown with targeted sequence in cyan and PAM in red. F. Knockout of ZNF451 in HaCaT cells increases the expression of E-Cadherin, and decreases that of Fibronectin and N-Cadherin. Four clones of ZNF451 KO and two clones of control were used. Whole cell lysates were harvested for western blotting analysis with antibodies against N-Cadherin, E-Cadherin, Fibronectin and ZNF451. β-Actin is an internal reference.
ZNF451-induced EMT does not require Smad4
It has been reported that EMT can be induced or regulated by various growth factors, among which TGF-β has been well documented as a major inducer of EMT during embryogenesis, cancer progression and fibrosis [4,5,43]. Although we previously showed that ZNF451 is a negative regulator of TGF-β activity through interacting with Smad4 to attenuate TGF-β-induced growth inhibition [41], the ability of ZNF451 to induce EMT is obviously opposite to its role as a negative regulator of TGF-β signaling. Thus, we sought to understand this paradoxical role of ZNF451 in TGF-β-induced EMT in MCF10A cells. Upon TGF-β treatment (1 ng/ml, 24 h), MCF10A control cells began to undergo EMT as expected and became scattered, while FLAG-ZNF451-expressing MCF10A cells had more dispersive distribution and spindle-like morphology, much closer to that of mesenchymal cells after TGF-β treatment, suggesting a synergistic effect of ZNF451 with TGF-β in inducing EMT (Figure 3A). Western blot assays of standard EMT markers confirmed that ZNF451 overexpression modulated the expression of EMT markers to the similar extent as achieved by TGF-β treatment (Figure 3B, Lane 2 and 3). Moreover, the effect of ZNF451 in suppressing the expression of E-Cadherin and inducing the expression of N-Cadherin and Fibronection was further enhanced by TGF-β treatment (Figure 3B, Lane 4). As EMT represents a process in which epithelial cells gain the features of invasive mesenchymal cells, in addition to examining the protein levels of EMT markers, we also assessed the effect of ZNF451 in cell migration by using wound-healing assays in MCF10A cells. As shown in Figure 3C, TGF-β treatment induced the migration of vector-transfected control cells, and ectopic expression of ZNF451 induced cell migration similarly. However, importantly, expression of ZNF451 rendered cells to be more migratory in response to TGF-β. To validate the data obtained from ZNF451 overexpression experiments, we examined the effects of ZNF451 on TGF-β-induced expression of EMT markers in ZNF451 knockdown cells (Figure 3D). As expected, TGF-β treatment inhibited the expression of E-Cadherin while simultaneously inducing the expression of N-Cadherin and Fibronectin in shControl cells, which was attenuated in shZNF451 cells (Figure 3D). To determine if the shZNF451 cells were responsive to TGF-β, expression of p21, which is normally induced to enable TGF-β growth inhibitory response, was measured in shZNF451 cells. TGF-β-induced expression of p21 was enhanced in shZNF451 cells, which is consistent with our previous finding that ZNF451 actually suppressed TGF-β-induced p21 expression [41].
Figure 3.

ZNF451 enhances TGF-β-induced EMT and promotes migration in epithelial cells. A. ZNF451 promotes TGF-β-induced morphological changes. MCF10A cells stably expressing ZNF451 or vector control were treated with TGF-β (2 ng/ml) for 48 h. Scale bars, 100 μm. B. Stable expression of ZNF451 enhances TGF-β-induced downregulation of E-Cadherin expression and upregulation of mesenchymal gene expression. Stable expression of ZNF451 or control was treated with 2 ng/ml TGF-β or without TGF-β treatment for 36 h. Levels of E-Cadherin, N-Cadherin, Fibronectin and FLAG-ZNF451 were analyzed by western blotting. β-Actin is an internal reference. C. ZNF451 accelerates TGF-β-induced cell migration. Confluent monolayers of MCF10A cells stably expressing ZNF451 or control were scratched with a pipette tip to create a cell-free space. The wounded cell monolayers were pre-incubated with or without 2 ng/ml of TGF-β for 12 h. The wound healing was recorded by microphotography of the same region after 12 h. Quantitative analysis of wound healing was performed. Data are shown in triplicates as mean + s.e.m. *P < 0.05, **P < 0.01. D. Knockdown of ZNF451 in MCF10A cells attenuates TGF-β-induced EMT marker expression. Cells with stable knockdown of ZNF451 or control were treated with 2 ng/ml TGF-β for different time (12, 24, 36 or 48 h) or without TGF-β treatment. Levels of N-Cadherin, E-Cadherin, Fibronectin, p21 and endogenous ZNF451 were analyzed by western blotting. β-Actin is an internal reference. SB431542 is a selective and potent inhibitor of the TGF-β pathway that inhibits ALK5.
Smad4, the central mediator for TGF-β signal transduction, plays an essential role in TGF-β-induced growth inhibitory function and EMT. Since we identified ZNF451 initially as a Smad4-binding protein and that ZNF451 inhibited Smad4/TGF-β-induced growth inhibition, we questioned the role of Smad4 in ZNF451-induced EMT. For this purpose, we constructed FLAG-ZNF451 and shZNF451 in Smad4 KO HaCaT cells. As shown in Figure 4A, Smad4 protein expression was diminished in Smad4 KO cells. However, ectopic expression of ZNF451 in Smad4 KO cells still retained the ability to induce EMT markers such as a decreased expression of E-Cadherin and an increased expression of N-Cadherin and Fibronectin (Figure 4B, Lane 3 and 4). Conversely, knockdown of ZNF451 in Smad4 KO cell could still increase the expression of E-Cadherin and suppress the expression of N-Cadherin and Fibronectin (Figure 4B, Lane 1 and 2, Figure 4C). The effect of ZNF451 overexpression and ZNF451 knockdown on the expression of EMT markers obtained from western blot analysis was further confirmed by immunofluorescence staining of E-Cadherin and Fibronectin in Smad4 KO cells (Figure 4D). qRT-PCR assays for the transcript levels of EMT markers in ZNF451 overexpression cells and in ZNF451 knockdown cells also showed the same results that the expression level of ZNF451 exhibited a positive correlation with those of mesenchymal markers and a negative correlation with epithelial markers (Figure 4E), suggesting that Smad4 was not required for ZNF451-induced EMT.
Figure 4.

ZNF451 enhances EMT independent of Smad4. A. Western blot assays indicates the knockout efficiency of Smad4 in Smad4 KO HaCaT cells. β-Actin is an internal reference. B. ZNF451 retains the ability to promote EMT in Smad4 KO HaCaT cells. B. The expression of EMT markers was examined in Smad4 KO HaCaT cells stably expressing ZNF451 or shZNF451 and their control cells. Levels of E-Cadherin, N-Cadherin, Fibronectin and ZNF451 were analyzed by western blotting. β-Actin is an internal reference. C. Depletion of ZNF451 inhibits EMT in Smad4 KO HaCaT cells. Levels of E-Cadherin, N-Cadherin and endogenous ZNF451 were analyzed by western blotting. β-Actin is an internal reference. D. ZNF451 is still able to decrease the expression of E-Cadherin and increase the expression of Fibronectin in Smad4 KO HaCaT cells. Immunofluorescence staining assay was performed in ZNF451-expressing or control and shZNF451-expressing or control Smad4 KO HaCaT cells. E-Cadherin and Fibronectin were immunostained (green). DAPI marks the nucleus (blue). E. ZNF451 increases, while shZNF451 decreases, the mRNA levels of mesenchymal markers in Smad4 KO HaCaT cells. Total mRNAs from Smad4 KO HaCaT cells with stable expression or knockdown of ZNF451 or control were extracted for qRT-PCR analysis. Data are shown as mean + s.e.m. (triplicate assays). *P < 0.05, **P < 0.01.
ZNF451 promotes EMT through its SUMO E3 ligase activity
What is the molecular mechanism underlying the function of ZNF451 in inducing cellular EMT process? Since ZNF451 belongs to the new class of SUMO2/3-specific ligases that execute catalysis via a tandem SUMO-interaction motif (SIM) region [44], we first wondered if the effect of ZNF451 in inducing EMT requires its SUMO E3 ligase activity. To test this, we generated several ZNF451 mutation constructs, including the ZNF45-SIM2 mutant with a point mutation in the SIM2 region which resulted in the loss of SUMO E3 ligase activity, and two deletion mutants: ZNF451 1-256 (N-terminal 1-256 amid acids only) and ZNF451 56-end (N-terminal deletion), both of which lost SUMOylation activity (Figure 5A). When we ectopically expressed these ZNF451 mutants in HaCaT cells, we found that only ZNF451 wildtype (Figure 5B, Lane 4) retained while all of the SUMO E3 ligase mutants lost the ability to down regulate the expression of E-Cadherin and up regulate that of mesenchymal markers (e.g. N-Cadherin, Vimentin and Fibronectin), indicating that these mutants fail to induce EMT responses and the SUMO E3 ligase activity was required for ZNF451 function (Figure 5B). We further performed phalloidine staining to evaluate the accumulation and distribution of F-Actin during EMT in the presence or absence of TGF-β treatment. Expression of ZNF451 apparently augmented TGF-β-induced actin cytoskeleton rearrangement associated with EMT, whereas ZNF451 SIM2 mutant that lost SUMO E3 ligase activity abolished the assembly of F-Actin fiber (Figure 5C). These results suggest that ZNF451 promotes EMT dependent on its SUMO E3 ligase activity.
Figure 5.
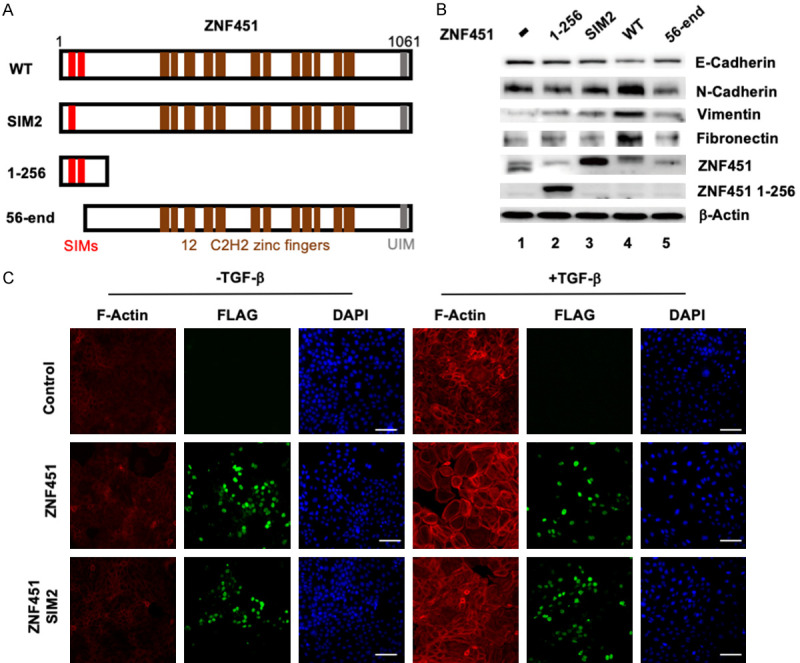
SUMO E3 ligase activity of ZNF451 is essential in promoting EMT morphology. A. The schematic diagram of ZNF451 wildtype and mutants. Structural domains are indicated. ZNF451 SIM2 mutant carries a mutation in the second SIM domain leading to the loss of E3 ligase activity. ZNF451 1-256 mutant consists of first 256 aa including the SIM1/2 domains. ZNF451 56-end mutant represents a truncation lacking the N-terminal 56 amino acids that delete the two SIM domains. B. ZNF451-induced EMT requires the integrity of its SUMOylation activity. Only wildtype ZNF451 (Lane 4) can induce the suppression of epithelial marker E-Cadherin while increase the expression of mesenchymal markers N-Cadherin, Vimentin and Fibronectin. Cell lysates from control, ZNF451 WT and mutants were harvested for western blotting analysis of different EMT makers and ZNF wildtype and mutants. β-Actin is an internal reference. C. ZNF451 increases TGF-β-induced F-Actin formation. HaCaT cells stably expressing wild-type ZNF451 or its E3 ligase inactive mutant ZNF451-SIM2 or control were stained by phalloidine. Red represents F-Actin, green represents FLAG tag fusion protein and blue represents cell nucleus.
ZNF451 binds to TWIST1/2 and stabilizes TWIST2
ZNF451 is a nuclear protein localized in the nuclear PML bodies where it forms scaffolds with both transcriptional co-activators and repressors. Components of PML bodies are assembled to enrich or trap transcription factors and SUMO modifications can act as the determinants in this assembly [35]. As EMT initiation and progression are tightly controlled by transcription factors such as Snail/Slug and TWIST1/2 [21], we were wondering if ZNF451 might induce EMT through modulating these transcription factors. To identify which transcription factor(s) was directly regulated by ZNF451, we performed co-immunoprecipitation screen to identify ZNF451-associated transcription factors (data not shown). We found that ZNF451 interacted with TWIST1 and TWIST2 in HEK293T cells (Figure 6A, Lane 4 and 5). When we immunoprecipitated ZNF451 from cell lysates with HA antibody, we could detect the presence of TWIST1 and TWIST2 in the immunoprecipitated complex as determined by western blot assay with anti-Myc antibody. RanBP3 was also co-transfected in this experiment to serve as a negative binding control (Figure 6A, Lane 6). To determine whether ZNF451 associated with TWIST1 and TWIST2 directly or indirectly through other binding partners, we performed an in vitro GST pulldown assay. We observed a direct interaction of ZNF451 with TWIST1 and TWIST2. As shown in Figure 6B, both purified GST-TWIST1 and GST-TWIST2 could retrieve the in vitro translated FLAG-ZNF451 in the binding assay.
Figure 6.
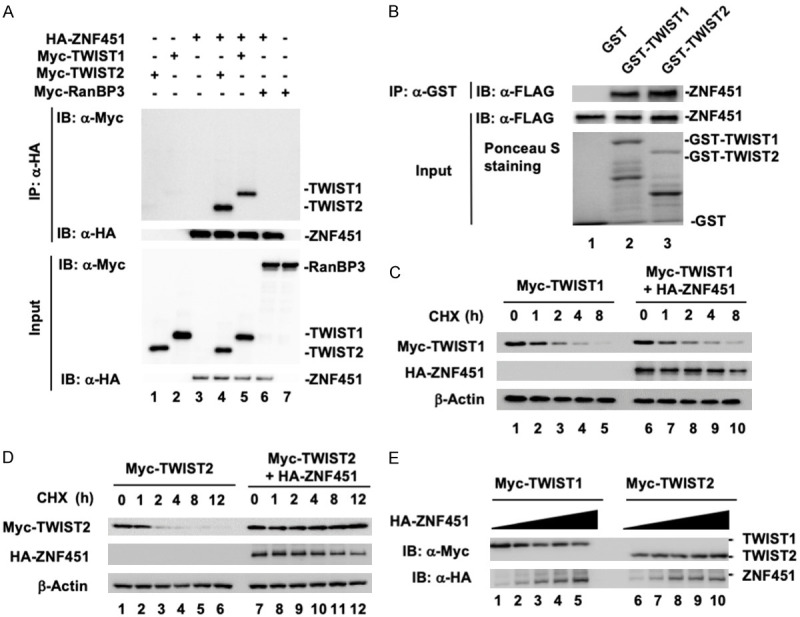
ZNF451 binds to TWIST1 and TWIST2, but specifically stabilizes TWIST2. (A) ZNF451 interacts with TWIST1/2 in vivo. HEK293T cells were transiently transfected with the indicated plasmids. ZNF451 was immunoprecipitated (IP) with anti-HA antibody and then subjected to SDS-PAGE and western blotting with anti-Myc antibody to detect ZNF451-bound TWIST1/2. RanBP3 protein was used as negative binding control. (B) Direct interaction between ZNF451 and TWIST1/2 in vitro. GST-TWIST1 or GST-TWIST2 fusion protein was expressed in E. coli and purified by glutathione beads. In vitro translated FLAG-ZNF451 was incubated with purified glutathione bead-bound GST protein or GST-TWIST1/2. The retrieved complex was subjected to SDS-PAGE and the presence of ZNF451 was detected by western bloting with anti-FLAG antibody. The input of GST, GST-TWIST1, or GST-TWIST2 was shown by Ponceau S staining. (C) ZNF451 has minimal effect on the half-life of TWIST1. TWIST1 was co-transfected with HA-ZNF451 in HEK293T cells. After 36 h, cells were treated with CHX (10 μg/ml) for the indicated time. Whole cell lysates were harvested for western blotting analysis. (D) ZNF451 prolongs the half-life of TWIST2. Transfection, CHX treatment and western blotting analysis were done as described in (C). (E) ZNF451 stabilizes TWIST2 in a dose-dependent manner. The same amounts of TWIST1 or TWIST2 were transfected with an increasing amount of HA-ZNF451 in HEK293T cells. After 36 h, cell lysates were harvested for western blotting analysis.
As TWIST1/2 are highly unstable proteins with a very short half-life around 45 min [21], we also tested the effect of ZNF451 on the protein stability of TWIST1/2 in HEK293T cells. In the cycloheximide (CHX) chase assay to determine the half-life of TWIST1/2, we found that ectopic expression of FLAG-ZNF451 had no effect on the half-life of TWIST1 (Figure 6C). In contrast, ectopic expression of FLAG-ZNF451 dramatically increased the protein stability of TWIST2 (Figure 6D). In addition, FLAG-ZNF451 increased the protein level of TWIST2 in a dose-dependent manner (Figure 6E). These results suggest that ZNF451 specifically controls the stability of TWIST2, but not TWIST1.
ZNF451 SUMOylates TWIST2 at K129 site and decreases its ubiquitination
Since the SUMO E3 ligase activity of ZNF451 drives EMT and stabilizes TWIST2, we wondered if ZNF451 could SUMOylate TWIST2. We used TWIST2 and ZNF451 co-transfection experiments in HEK293T cells. As shown in Figure 7A, we found that TWIST2 was SUMO modified by the co-expression of ZNF451 (Figure 7A, Lane 4). However, when SUMO E3 ligase inactive form of ZNF451 SIM1 or SIM2 mutant was co-expressed, the SUMO modification of TWIST2 was not detected (Figure 7B, Lane 5 and 6), comparing to wildtype ZNF451 transfected sample (Figure 7B, Lane 4). These data suggest that ZNF451 caused SUMOylation of TWIST2, which required its E3 ligase activity.
Figure 7.
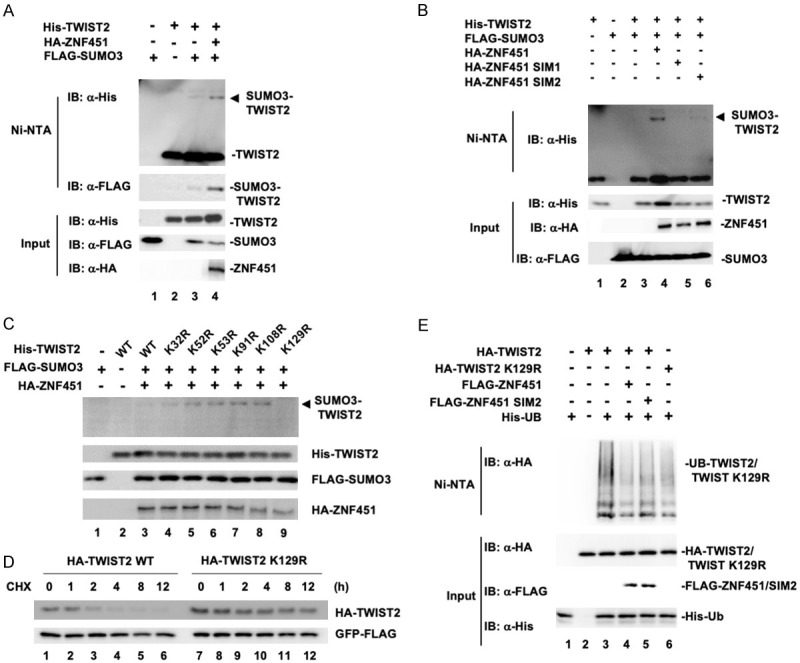
ZNF451 SUMOylates TWIST2 at K129 residue and decreases the ubiquitination level of TWIST2. (A) ZNF451 drives SUMO3 modification of TWIST2. HEK293T cells were transiently transfected with the indicated plasmids and the SUMOylation of TWIST2 was examined with Ni-NTA precipitation, followed by western blotting. (B) ZNF451 requires its SUMO E3 activity to drive SUMO3 modification of TWIST2. HEK293T cell transfection with wildtype ZNF451 or E3 ligase inactive mutants of ZNF451 SIM1 or SIM2, Ni-NTA precipitation and western blotting were done as in (C). (C) Lys-129 is the SUMOylation acceptor of TWIST2. HEK293T cells were transfected with indicated plasmids. Whole cell lysates were tested for SUMOylation of TWIST2 WT or its mutants (K32R, K52R, K53R, K91R, K108R, K129R). (D) TWIST2 K129R mutant has a prolonged half-life. TWIST2 WT or TWIST2 K129 mutant was transfected in HEK293T cells. After 36 h, cells were treated with CHX (10 μg/ml) for indicated time above. Whole cell lysates were harvested for western blotting analysis. GFP was co-transfected and examined by Western blot in all samples as an internal control. (E) ZNF451 decreases TWIST2 ubiquitination. HEK293T cells were transfected with indicated plasmids. His-Ub was retrieved by using Ni-NTA precipitation and then subjected to anti-HA western blotting analysis for TWIST2 ubiquitination detection.
We next sought to identify the specific SUMOylation sites on TWIST2. TWIST2 contains six putative lysine residues that were predicted for SUMO modification. We then generated mutation constructs in which each of these lysine (K) residues were exchanged to arginine (R). By co-transfecting individually each of the mutants with ZNF451 and SUMO3, we found that the SUMOylation band of TWIST2 completely vanished when K129 was mutated to R129 (Figure 7C, Lane 9), indicating that K129 residue of TWIST2 is the main SUMOylation site. By using CHX pulse-chase experiment to compare the stability of wildtype and K129R mutant of TWIST2, we found that the TWIST2 K129R mutant was much more stable than wildtype TWIST2 (Figure 7D), suggesting that the K129 site was important for TWIST2 degradation. In this experiment, GFP was transfected in all the samples and GFP expression level was examined by western blot assay to serve as an internal negative control. GFP level was uniform in all the samples (Figure 7D). Since TWIST2 degradation is partially mediated by the ubiquitin-proteasome pathway, we then examined the ubiquitination of TWIST2 proteins. We found that wildtype ZNF451 strongly decreased the ubiquitination of TWIST2 (Figure 7E, comparing Lane 3 and 4), and this inhibition was partially attenuated in SIM2 mutant of ZNF451-expressing cells (Figure 7E, Lane 5). Furthermore, the ubiquitination of TWIST2 K129R mutant was greatly reduced even in the absence of ZNF451 (Figure 7E, Lane 6). Hence, these results suggested that K129 site in TWIST2 was the major site for SUMO modification and ubiquitination, and that ZNF451-mediated SUMOylation at the K129 residue might antagonize the ubiquitination of TWIST2 on the same lysine acceptor.
ZNF451 promotes EMT through TWIST2
To directly demonstrate the functional importance of ZNF451-regulated stability of TWIST2 in EMT, we examined the effect of TWIST2 knockdown in ZNF451-induced EMT in MCF10A cells. As shown in Figure 8A, ectopic expression of ZNF451 dramatically increased the endogenous level of TWIST2, but not TWIST1 (Figure 8A, Lane 2), and induced the expression of EMT marker: e.g. downregulation of E-Cadherin expression and upregulation of N-Cadherin and Fibronectin. However, when TWIST2 was knocked down by siTWIST2, ZNF451-induced EMT phenotype was attenuated: e.g. the protein levels of Fibronectin and N-Cadherin were markedly decreased, while the protein level of E-Cadherin was partially recovered (Figure 8A, Lane 3). Consistently, wound healing assays that measured the cell migratory ability showed the same results; the ZNF451-expressing cells exhibited more motility, which could be blocked by siTWIST2 (Figure 8B). To further demonstrate the important role of TWIST2 SUMOylation in ZNF451-mediated EMT, wildtype or SIM2 mutant of ZNF451 was co-transfected with TWIST2 in an E-Cadherin-luc reporter assay to measure the repressing activity of TWIST2 on the E-Cadherin promoter. We found that wildtype ZNF451 facilitated the repressive effect of TWIST2 on E-Cadherin promoter (Figure 8C), whereas ZNF451 SIM2 mutant partially lost this repressive activity on E-Cadherin promoter. Consistently, analysis of the Cancer Genome Atlas (TCGA) data found that the ZNF451 mRNA level was higher in hepatocellular carcinoma (HCC) than that in normal tissues (Figure 8D). Furthermore, TCGA data also showed a positive correlation between ZNF451 and N-Cadherin expression (Figure 8E).
Figure 8.
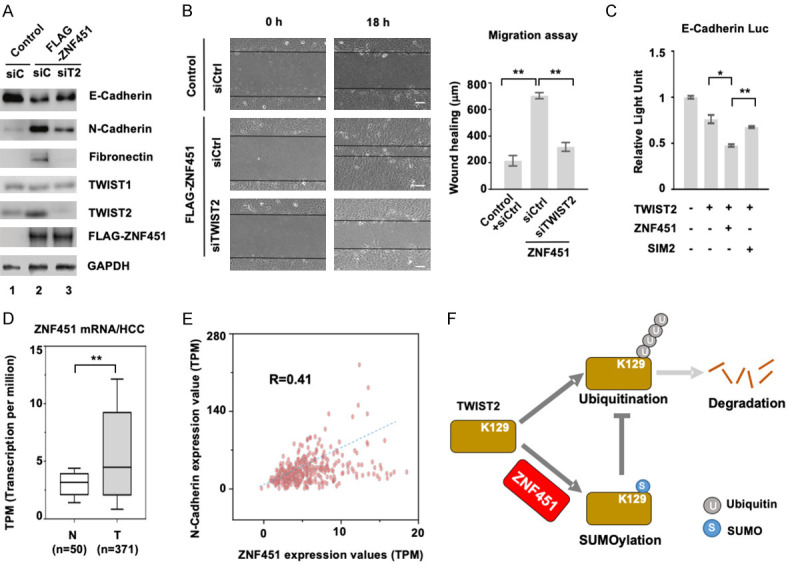
ZNF451 mediates EMT through TWIST2. A. Knockdown of TWIST2 attenuates ZNF451-induced EMT in MCF10A. siTWIST2 or siControl was transfected into MCF10A cell lines harboring FLAG-ZNF451 or control. After 48 h, whole cell lysates were harvested for western blotting analysis to determine the expression of EMT markers. B. Knockdown of TWIST2 compromises ZNF451-induced wound healing response in MCF10A. siTWIST2 or siControl were transfected into MCF10A cell lines harboring FLAG-ZNF451 or control. Confluent monolayers of cells were scratched with a pipette tip to create a cell-free space. The wound healing was recorded by microphotography of the same region during 18 h period. Quantitative analysis of wound healing was performed. Data are shown as mean + s.e.m. (triplicate assays). *P < 0.05, **P < 0.01. C. Wildtype ZNF451 but not E3 ligase inactive mutant of ZNF451 SIM2 facilitates TWIST2-induced suppression of E-Cadherin promoter. Indicated plasmids were co-transfected with E-Cadherin luciferase plasmid, and the luciferase activity was measured to represent the transcription of E-Cadherin. Data are shown as mean + s.e.m. (triplicate assays). *P < 0.05, **P < 0.01. D. ZNF451 mRNA level is elevated in HCC tumors in comparison to normal tissues. HCC data in TCGA was analyzed. **P < 0.01. E. Expression levels of ZNF451 and N-Cadherin mRNAs are positively correlated in HCC. HCC data in TCGA was analyzed. F. A working model of ZNF451-mediated EMT in epithelial cells.
Taken together, we proposed that ZNF451 regulates EMT response, which requires its SUMO E3 ligase activity and its effect on TWIST2 SUMOylation and ubiquitination (Figure 8F).
Discussion
We previously identified ZNF451 as a transcriptional co-repressor of TGF-β signaling. ZNF451 protein is localized in PML bodies. Although the function of PML bodies is largely unknown, ZNF451 interacts with CREB-binding protein, death domain-associated protein (Daxx), HDACs and SUMOs, suggesting its potential role in transcriptional regulation [45,46]. ZNF451 also interacts with Smad4 proteins and blocks the recruitment of transcription co-activator p300 to the Smad complex, consequently inhibiting TGF-β-induced growth inhibitory responses [41]. In addition to inducing growth inhibitory genes, TGF-β-induced transcription potently represses epithelial genes (e.g. E-Cadherin, ZO-1) and activates mesenchymal genes (e.g. N-Cadherin, Vimentin) during EMT [47]. During the course of our study on the role of ZNF451 in TGF-β signaling, we noticed that while ZNF451 functionally blocked Smad-dependent transcription of genes involved in cell cycle arrest, it surprisingly did not block TGF-β-induced EMT and instead appeared to induce EMT by itself. Overexpression of ZNF451 in epithelial cells such as MCF10A and HaCaT cells accelerates the morphological changes towards mesenchymal cells, accompanied with upregulation of mesenchymal markers (e.g. N-Cadherin, Vimentin and Fibronectin) and downregulation of epithelial markers (e.g. E-Cadherin). Conversely, knockdown of ZNF451 results in an opposite effect, which demonstrates the essential role of ZNF451 in modulating EMT. This interesting observation of the paradoxical role of ZNF451 prompted us to further investigate the functions and mechanism of ZNF451 in inducing EMT.
In our study, ZNF451 behaves as an EMT promoter through stabilization of TWIST2, a key transcription factor in inducing EMT. In this case, ZNF451 does not act a transcriptional co-repressor of Smad proteins, but instead functions as a SUMO E3 ligase to SUMOylate TWIST2 during EMT. SUMOylation has been demonstrated to regulate gene transcription, nuclear transport, and activation of signal transduction pathways. Unlike K48-polyubiquitination that tags substrates for proteasomal or lysosomal degradation, protein SUMOylation has distinct outcomes, including transcription repression, subcellular localization, and protein stability [48-51]. The ability of ZNF451 to promote TWIST2 stability is through SUMO2/3 modification at K129 residue of TWIST2, which directly blocks ubiquitination of TWIST2. This differs from a previous report that ZNF451 promoted the degradation of PML through SUMO2/3 modification [52]. Although highly related TWIST1 and TWIST2 have similar sequences around K129 (in TWIST2), ZNF451 selectively enhances the stability of TWIST2, but not that of TWIST1. Indeed, ZNF451 induces EMT in a manner dependent of TWIST2. This suggests that ZNF451 induces EMT through SUMOylation-dependent stabilization of TWIST2.
EMT represents morphological changes upon transcriptional reprogramming, which is mostly achieved by epigenetic regulation of cell type-specific gene transcription. Key EMT transcription factors like Snail/Slug, TWIST1/2 and ZEB1/2 play important roles in this epigenetic reprogramming. It has also been well established that post-translational modifications of these transcription factors play important roles in regulating their activity. Post-translational modifications occur on the specific amino acid side chains and are often catalyzed by enzymes including kinases and ubiquitin ligases. These enzymes regulate the function of different proteins and eventually maintain the dynamic balance of intracellular environment. Phosphorylation and ubiquitination of Snail/Slug, TWIST1/2, and ZEB1/2 have been identified to consequently impact EMT.
SUMO modification at lysine residues is typically observed within a minimal consensus motif ΨKxE/D (where Ψ is a hydrophobic residue, x is any residue). Interestingly, the sequence around K129 of TWIST2 apparently does not fit this motif, but rather represents an inverted motif E/DxKΨ. Although TWIST1 contains the same consensus inverted motif located around K175, it appears to not be SUMOylated. It is previously reported that K175 is a critical ubiquitination site for TWIST1 degradation. TWIST1 K175R mutant exhibits decreased ubiquitination level and increased stability in vivo. Consistently, our results show the SUMOylation of TWIST2 competitively inhibits its ubiquitination and proteasome-mediated degradation.
In conclusion, our studies have identified ZNF451 as a positive regulator of EMT, which depends on its SUMO ligase activity. ZNF451 SUMOylates TWIST2, increases its stabilization, and promotes TWIST2-mediated progression of EMT.
Acknowledgements
We thank the laboratory members for helpful discussion and technical assistance. We appreciate Emily Feng for her editing of this manuscript. This research was partly supported by grants from NSFC (31730057, 91540205, 31571447), MOST 973 Program (2015CB553803), and the Fundamental Research Funds for the Central Universities.
Disclosure of conflict of interest
None.
References
- 1.Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20:69–84. doi: 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- 2.Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7:re8. doi: 10.1126/scisignal.2005189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Derynck R, Muthusamy BP, Saeteurn KY. Signaling pathway cooperation in TGF-beta-induced epithelial-mesenchymal transition. Curr Opin Cell Biol. 2014;31:56–66. doi: 10.1016/j.ceb.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kahata K, Dadras MS, Moustakas A. TGF-beta family signaling in epithelial differentiation and epithelial-mesenchymal transition. Cold Spring Harb Perspect Biol. 2018;10:a022194. doi: 10.1101/cshperspect.a022194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Derynck R, Muthusamy BP, Saeteurn KY. Signaling pathway cooperation in TGF-β-induced epithelial-mesenchymal transition. Curr Opin Cell Biol. 2014;31:56–66. doi: 10.1016/j.ceb.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kahata K, Dadras MS, Moustakas A. TGF-β family signaling in epithelial differentiation and epithelial-mesenchymal transition. Cold Spring Harb Perspect Biol. 2018;10:a022194. doi: 10.1101/cshperspect.a022194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aiello NM, Kang Y. Context-dependent EMT programs in cancer metastasis. J Exp Med. 2019;216:1016–1026. doi: 10.1084/jem.20181827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu W, Kang Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev Cell. 2019;49:361–374. doi: 10.1016/j.devcel.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019;29:212–226. doi: 10.1016/j.tcb.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Campbell K. Contribution of epithelial-mesenchymal transitions to organogenesis and cancer metastasis. Curr Opin Cell Biol. 2018;55:30–35. doi: 10.1016/j.ceb.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–629. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Staalduinen J, Baker D, Ten Dijke P, van Dam H. Epithelial-mesenchymal-transition-inducing transcription factors: new targets for tackling chemoresistance in cancer? Oncogene. 2018;37:6195–6211. doi: 10.1038/s41388-018-0378-x. [DOI] [PubMed] [Google Scholar]
- 15.Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014;13:497–512. doi: 10.1038/nrd4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 17.Stemmler MP, Eccles RL, Brabletz S, Brabletz T. Non-redundant functions of EMT transcription factors. Nat Cell Biol. 2019;21:102–112. doi: 10.1038/s41556-018-0196-y. [DOI] [PubMed] [Google Scholar]
- 18.Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25:675–686. doi: 10.1016/j.tcb.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diepenbruck M, Christofori G. Epithelial-mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr Opin Cell Biol. 2016;43:7–13. doi: 10.1016/j.ceb.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Franco HL, Casasnovas J, Rodríguez-Medina JR, Cadilla CL. Redundant or separate entities?--roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res. 2011;39:1177–1186. doi: 10.1093/nar/gkq890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu J, Qin L, He T, Qin J, Hong J, Wong J, Liao L, Xu J. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011;21:275–289. doi: 10.1038/cr.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vesuna F, Lisok A, Kimble B, Domek J, Kato Y, van der Groep P, Artemov D, Kowalski J, Carraway H, van Diest P, Raman V. Twist contributes to hormone resistance in breast cancer by downregulating estrogen receptor-alpha. Oncogene. 2012;31:3223–3234. doi: 10.1038/onc.2011.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah P, Gau Y, Sabnis G. Histone deacetylase inhibitor entinostat reverses epithelial to mesenchymal transition of breast cancer cells by reversing the repression of E-cadherin. Breast Cancer Res Treat. 2014;143:99–111. doi: 10.1007/s10549-013-2784-7. [DOI] [PubMed] [Google Scholar]
- 24.Howard TD, Paznekas WA, Green ED, Chiang LC, Ma N, Ortiz de Luna RI, Garcia Delgado C, Gonzalez-Ramos M, Kline AD, Jabs EW. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15:36–41. doi: 10.1038/ng0197-36. [DOI] [PubMed] [Google Scholar]
- 25.Tukel T, Šošić D, Al-Gazali LI, Erazo M, Casasnovas J, Franco HL, Richardson JA, Olson EN, Cadilla CL, Desnick RJ. Homozygous nonsense mutations in TWIST2 cause Setleis syndrome. Am J Hum Genet. 2010;87:289–296. doi: 10.1016/j.ajhg.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang X, Cai Y, Liu J, Wang Z, Wu Q, Zhang Z, Yang CJ, Yuan L, Ouyang G. Twist2 contributes to breast cancer progression by promoting an epithelial-mesenchymal transition and cancer stem-like cell self-renewal. Oncogene. 2011;30:4707–4720. doi: 10.1038/onc.2011.181. [DOI] [PubMed] [Google Scholar]
- 27.Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16:488–494. doi: 10.1038/ncb2976. [DOI] [PubMed] [Google Scholar]
- 28.Sánchez-Tilló E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A, Postigo A. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012;69:3429–3456. doi: 10.1007/s00018-012-1122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang-Hartwich Y, Tedja R, Roberts CM, Goodner-Bingham J, Cardenas C, Gurea M, Sumi NJ, Alvero AB, Glackin CA, Mor G. p53-Pirh2 complex promotes twist1 degradation and inhibits EMT. Mol Cancer Res. 2019;17:153–164. doi: 10.1158/1541-7786.MCR-18-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang WH, Su YH, Hsu WH, Wang CC, Arbiser JL, Yang MH. Imipramine blue halts head and neck cancer invasion through promoting F-box and leucine-rich repeat protein 14-mediated Twist1 degradation. Oncogene. 2016;35:2287–2298. doi: 10.1038/onc.2015.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo W, You X, Xu D, Zhang Y, Wang Z, Man K, Wang Z, Chen Y. PAQR3 enhances Twist1 degradation to suppress epithelial-mesenchymal transition and metastasis of gastric cancer cells. Carcinogenesis. 2016;37:397–407. doi: 10.1093/carcin/bgw013. [DOI] [PubMed] [Google Scholar]
- 32.Zhong J, Ogura K, Wang Z, Inuzuka H. Degradation of the transcription factor Twist, an oncoprotein that promotes cancer metastasis. Discov Med. 2013;15:7–15. [PMC free article] [PubMed] [Google Scholar]
- 33.Li NY, Weber CE, Wai PY, Cuevas BD, Zhang J, Kuo PC, Mi Z. An MAPK-dependent pathway induces epithelial-mesenchymal transition via Twist activation in human breast cancer cell lines. Surgery. 2013;154:404–410. doi: 10.1016/j.surg.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 34.Sun T, Fu J, Shen T, Lin X, Liao L, Feng XH, Xu J. The small c-terminal domain phosphatase 1 inhibits cancer cell migration and invasion by dephosphorylating Ser(P)68-twist1 to accelerate twist1 protein degradation. J Biol Chem. 2016;291:11518–11528. doi: 10.1074/jbc.M116.721795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin X, Liang M, Liang YY, Brunicardi FC, Feng XH. SUMO-1/Ubc9 promotes nuclear accumulation and metabolic stability of tumor suppressor Smad4. J Biol Chem. 2003;278:31043–31048. doi: 10.1074/jbc.C300112200. [DOI] [PubMed] [Google Scholar]
- 36.Bies J, Markus J, Wolff L. Covalent attachment of the SUMO-1 protein to the negative regulatory domain of the c-Myb transcription factor modifies its stability and transactivation capacity. J Biol Chem. 2002;277:8999–9009. doi: 10.1074/jbc.M110453200. [DOI] [PubMed] [Google Scholar]
- 37.Koidl S, Eisenhardt N, Fatouros C, Droescher M, Chaugule VK, Pichler A. The SUMO2/3 specific E3 ligase ZNF451-1 regulates PML stability. Int J Biochem Cell Biol. 2016;79:478–487. doi: 10.1016/j.biocel.2016.06.011. [DOI] [PubMed] [Google Scholar]
- 38.Schellenberg MJ, Lieberman JA, Herrero-Ruiz A, Butler LR, Williams JG, Muñoz-Cabello AM, Mueller GA, London RE, Cortés-Ledesma F, Williams RS. ZATT (ZNF451)-mediated resolution of topoisomerase 2 DNA-protein cross-links. Science. 2017;357:1412–1416. doi: 10.1126/science.aam6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karvonen U, Jääskeläinen T, Rytinki M, Kaikkonen S, Palvimo JJ. ZNF451 is a novel PML body- and SUMO-associated transcriptional coregulator. J Mol Biol. 2008;382:585–600. doi: 10.1016/j.jmb.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 40.Eisenhardt N, Chaugule VK, Koidl S, Droescher M, Dogan E, Rettich J, Sutinen P, Imanishi SY, Hofmann K, Palvimo JJ, Pichler A. A new vertebrate SUMO enzyme family reveals insights into SUMO-chain assembly. Nat Struct Mol Biol. 2015;22:959–967. doi: 10.1038/nsmb.3114. [DOI] [PubMed] [Google Scholar]
- 41.Feng Y, Wu H, Xu Y, Zhang Z, Liu T, Lin X, Feng XH. Zinc finger protein 451 is a novel Smad corepressor in transforming growth factor-β signaling. J Biol Chem. 2014;289:2072–2083. doi: 10.1074/jbc.M113.526905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu S, Long J, Yuan B, Zheng M, Xiao M, Xu J, Lin X, Feng XH. SUMO modification reverses inhibitory effects of smad nuclear interacting protein-1 in TGF-β responses. J Biol Chem. 2016;291:24418–24430. doi: 10.1074/jbc.M116.755850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 44.Cappadocia L, Pichler A, Lima CD. Structural basis for catalytic activation by the human ZNF451 SUMO E3 ligase. Nat Struct Mol Biol. 2015;22:968–975. doi: 10.1038/nsmb.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Leo C, Zhu J, Wu X, O’Neil J, Park EJ, Chen JD. Sequestration and inhibition of Daxx-mediated transcriptional repression by PML. Mol Cell Biol. 2000;20:1784–1796. doi: 10.1128/mcb.20.5.1784-1796.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu WS, Vallian S, Seto E, Yang WM, Edmondson D, Roth S, Chang KS. The growth suppressor PML represses transcription by functionally and physically interacting with histone deacetylases. Mol Cell Biol. 2001;21:2259–2268. doi: 10.1128/MCB.21.7.2259-2268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 48.Lyst MJ, Stancheva I. A role for SUMO modification in transcriptional repression and activation. Biochem Soc Trans. 2007;35:1389–1392. doi: 10.1042/BST0351389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8:947–956. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- 50.Verger A, Perdomo J, Crossley M. Modification with SUMO. A role in transcriptional regulation. EMBO Rep. 2003;4:137–142. doi: 10.1038/sj.embor.embor738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gill G. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 2004;18:2046–2059. doi: 10.1101/gad.1214604. [DOI] [PubMed] [Google Scholar]
- 52.Zhong S, Salomoni P, Pandolfi PP. The transcriptional role of PML and the nuclear body. Nat Cell Biol. 2000;2:E85–90. doi: 10.1038/35010583. [DOI] [PubMed] [Google Scholar]