

Abstract
Breast cancer is the leading cause of cancer-related deaths in women worldwide. Several studies have indicated that abnormal chondroitin sulfate (CS) chains accumulate in breast cancer tissues; however, the functions and dysregulation of CS synthases are largely unknown. Here, we demonstrate that chondroitin polymerising factor (CHPF) is frequently upregulated in breast cancer tissues and that its high expression is positively associated with tumor metastasis, high stages, and short survival time. CHPF modulates CS formation in breast cancer cells. Additionally, we found that CHPF promotes tumor growth and metastasis accompanied by an increase in G-CSF levels and the number of myeloid-derived suppressor cells in tumor tissue. We revealed that tumor cell-derived G-CSF is co-localised with CS on the cell surface. Interestingly, our study is the first to identify that syndecan-4 (SDC4) is modified by CHPF and that it is involved in CHPF-mediated phenotypes. Moreover, breast cancer patients with high expression of both SDC4 and CHPF had worse overall survival compared to other subsets, which implied the synergistic effects of these two genes. In summary, our results indicated that the upregulation of CHPF in breast cancer contributes to the malignant behaviour of cancer cells, thereby providing novel insights on the significance of CHPF-modified SDC4 in breast cancer pathogenesis.
Keywords: Breast cancer, chondroitin sulfate, CHPF, tumor microenvironment, syndecan-4
Introduction
Although the recent advancements in breast cancer therapies using novel mechanisms, involving actionable cancer mutations and the immune system, breast cancer remains the leading cause of cancer death in women worldwide [1,2]. Accumulating evidence indicates that patients with advanced tumor stages, characterised by large tumor size, lymph node invasion, skin invasion, or distal metastasis, are tightly regulated by the tumor microenvironment (TME) [3,4]. Moreover, the cellular and extracellular components of TME directly govern the outcomes of chemotherapy and immunotherapy in breast cancer [5-8]. Thus, a thorough understanding of the change in the TME is crucial for improving the survival of breast cancer patients.
Glycosaminoglycans (GAGs) are unbranched polysaccharide chains which can exist as free chains, such as hyaluronan, or covalently link to core proteins such as proteoglycans (PGs). GAGs and PGs are major components of extracellular matrix in both normal and tumor tissue, and they are also abundant on the surface of cancer cells. Chondroitin sulfate (CS) is a member of the GAG family, which is covalently linked to the core protein, forming chondroitin sulfate proteoglycans (CSPGs). Increasing evidence on the relationship between dysregulation of CSPGs and breast cancer cell proliferation, invasion, and metastasis has been reported. For instance, versican is deposited in the tumor stroma, which is associated with aggressive phenotypes and relapse in node-negative breast cancer [9,10]. In contrast, low levels of decorin in invasive breast carcinomas are associated with poor outcomes, and decorin has been shown to possess anti-angiogenic activities [11,12]. Importantly, CSPG4 (NG2) is considered as a target for the antibody-based immunotherapy of triple-negative breast cancer and several other types of cancer [13,14]. The CS side chains of CSPGs are known to exert bioactivity; however, the alternations of CS chains on the PGs or the dysregulation of CS-modifying enzymes require further elucidation.
In humans, the biosynthesis of CS chains is initiated by the linkage of N-acetylgalactosamine (GalNAc) to a tetrasaccharide structure, glucuronic acid (GlcA)-GalNAc-GalNAc-xylose, on a core protein. Next, the elongation (polymerization) of CS chain is catalyzed by a group of bifunctional CS synthases (CHSY1, CHPF, CHPF2, and CHSY3), which have dual β1-3 glucuronosyltransferase and β1-4 N-acetylgalactosaminyltransferase activities. One single CS chain can be composited by up to 50 repeats of GlcA-GalNAc disaccharide units, and each CS unit could be further classified according to their modifications [15-17]. For instance, CS is often O-sulfated at C-4 (4-O-sulfated CS) or at C-6 (6-O-sulfated CS) on the GalNAc residue by sulfotransferases (CHST family) catalysis. Depending on the spectrotemporal expression of the CS synthases and CS sulfotransferases, a single CS chain usually consists of a series of variably sulfated units.
The biological functions of CS chains on CSPG are endorsed by their affinity to various adhesion molecules, trans-membrane proteins, growth factors, and cytokines [16,18]. We have previously found that CHSY1 promotes the formation of aggressive phenotypes of HCC cells by activating the Hedgehog signalling pathway. Inhibition of this pathway with vismodegib decreased the CHSY1-induced migration, invasion, and lung metastasis of HCC cells [19]. Our study also indicated that dermatan sulfate epimerase 1 modulated the tumor-infiltrated immune cell population and regulated CCL5 signalling in HCC cells [20]. In glioma cells, we used a human phosphor-receptor tyrosine kinase array to identify the selective regulation of PDGFRA signalling by CHSY1 [21]. These studies suggest that the differential expression of CS synthases and CS-modifying enzymes directly changes the content and structure of CS in the TME, which may modulate the immune response and regulate diverse cellular signalling pathways in tumor tissues.
In this study, we focused on the CS polymerisation enzymes, which build the elemental structure of CS in breast cancer. We found that CHPF is frequently overexpressed in breast cancer and its high expression is associated with poor survival. Therefore, we hypothesised that CHPF can regulate the malignant growth of breast cancer cells by modulating the functions of CSPG in the TME.
Materials and methods
Cell culture and transfection
MCF7, SK-BR-3, MDA-MB-468, MDA-MB-435, MDA-MB-231, HS578T, 4T1, and JC were obtained from the American Type Culture Collection (Manassas, VA) in 2014, and cultured in DMEM (Life Technologies) containing 0.1 mM sodium pyruvate, 10% FBS (5% FBS for 4T1 cells), 2 mM L-glutamine, 100 IU/mL penicillin, and 100 μg/mL streptomycin. Empty pCMV6 and Chpf-pCMV6 plasmids were transfected to 4T1 cells using TOOLstrong Transfection Reagent (BIOTOOLs, TW). The transfected cells were selected with 600 μg/mL of G418. For gene silence experiments, ON-TARGETplus SMARTpool siRNA against CHPF or Sdc4, and non-targeting control siRNA were purchased from Dharmacon. Cells were transfected with 20 nmol of siRNA using Lipofectamine RNAiMAX (Invitrogen). For shRNA transfection, pLKO/CHPF-shRNA plasmids (5’-GCTGTGGCCTCCACGTATTTA-3’), and nontargeting pLKO plasmids were purchased from National RNAi Core Facility (Academia Sinica, Taipei, Taiwan). The short hairpin RNA (shRNA) plasmids were transfected and selected with 1.0 μg/ml of puromycin for 10 days.
Reagents and antibodies
Full length Chpf cDNA clone were purchased from OriGene. Chondroitinase ABC, β-D-xylopyranoside, and CCK8 reagent was purchased from Sigma-Aldrich. Mouse monoclonal antibody against CHPF (sc-376183) and Syndecan-4 (sc-12766) was purchased from Santa Cruz. Antibodies against ZO-1, E-cadherin, β-catenin, N-cadherin, Vimentin were purchased from Cell Signaling Technology. Antibodies against chondroitin sulfate (CS56) and ACTB were purchased from GeneTex, Inc. The antibodies against mouse F4/80-FITC, Ly6G-Alex488, CD45-PerCP, Gr1-Violet 510, CD11b-PE, and I-A/I-E-FITC (MHCII) were all purchased Biolegend. G-CSF antibody (bs-1023R) was purchased from Bioss Inc.
Tissue array and immunohistochemistry
Paraffin-embedded human breast cancer tissue microarrays were purchased from Shanghai Outdo Biotech. Arrays were incubated with CHPF antibody (1:200) in 5% bovine serum albumin/PBS and 0.1% Triton X-100 (Sigma) for 16 h at 4°C. UltraVision Quanto Detection System (Thermo Fisher Scientific Inc.) was used to amplify primary antibody signal. The specific immunostaining was visualized with 3,3-diaminobenzidine (DAB) and counterstained with hematoxylin (Sigma). The staining intensity were graded by microscopy by two scorers blinded to the clinical parameters. Images were obtained by Tissue FAX Plus Cytometer.
Immunoprecipitation and CSPG identification
For immunoprecipitation assay, cell lysates (0.8 mg) were incubated with 4 µg of antibody at 4°C for 16 h. Protein L sepharose beads (BioVision Inc) was then added to lysates for 3 h for pulldown CS56 antibody (mouse IgM). Protein A/G sepharose beads (Thermo Fisher Scientific Inc) was used to capture SDC4 antibody (mouse IgG). The pull-downed proteins were then separate by 7.5% or 12% SDS-PAGE, and visualized by Stain-Free Imaging Technology (Bio-Rad) or applied to Western blotting. For CSPG identification, protein bands were in-gel digestion by trypsin chromatography based (LC-based) and applied to tandem mass spectrometry (LC MS/MS, Dionex Ultimate 3000 RSLCnano system Hybrid mass spectrometer). The data files obtained following LC-MS/MS analysis were processed in the SwissProt, Mascot version 2.5, and Percolator1,2.
Animal experiments
The 4T1 orthotopic mouse model of spontaneous breast cancer metastasis was performed referring to a previous report [22]. In brief, 1×105 cells will injected into BALB/c mouse mammary fat pad, and the primary tumors were removed after 3 weeks. Lung metastasis was measured one month after the surgery. All animal experiments in this study were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Chung Shan Medical University Experimental Animal Center.
Flow cytometry and myeloid-derived suppressor cells (MDSC) isolation
Breast cancer cells were detached with 10 mM EDTA and stained with CS56 antibody at 1:100 dilutions on ice for 30 min. Cells were incubated with FITC-conjugated anti-mouse IgM antibody on ice for 30 min. For analyzing tumor infiltrated myeloid cells, 100 mg of primary 4T1 tumor tissues were excised at day 21 (n = 7 for each group). Disaggregation cell suspension was filtered through cell strainer to remove cell clumps and briefly treated with ACK buffer to remove red blood cells. Cells were spin down and blocking by anti-CD16/32 for 15 minutes before surface antigen staining. The MDSC population (CD45+MHCII-CD11b+Gr1hi) was isolated by cell sorter (BD FACSAria).
Cell viability assay
Cells (2 × 103) were seeded into 96-well plates with culture medium. Cell viability was analyzed by CCK8 assay at 0, 24, 48, and 72 hours following manufacture’s protocol (Sigma-Aldrich). Four wells per group of each time point were measured by OD 450 nm, and two wells of only media were used to measure the background absorbance. The experiments were repeated for three times, and relative fold changes were shown.
Cell invasion assay
Transwell inserts for 24-well plate (Corning) with Matrigel (BD Biosciences) coated porous filters (pore size 8 μm) were used to evaluate cell cell invasion. 2 × 104 cells in 0.2 ml serum-free DMEM were seeded into inserts, and 0.6 ml DMEM containing 10% FBS was added in lower part of the well. For measuring MDSC-induced cell invasion, 4T1 cells (2 × 104) in 5% FBS-DMEM were seeded into inserts, freshly isolated MDSC (5 × 104) in 5% FBS-DMEM was added in lower part of the well. Cells were stained by crystal violet and counted after 24 hour incubation. Independent experiments were repeated for three times, and average number of cells per microscopic field over three fields of each filter.
Confocal microscopy
The z-stacked confocal images of immunofluorescence staining were captured with a confocal microscope, Leica TCS SP8. Each confocal image illustrated is the stack of five series of scans in cell or tumor tissue section (1.5 μm in total thickness).
Statistical analysis
Data analysis was performed using GraphPad Prism 6. Two-sided Fisher exact test was used for comparisons between CHPF expression and clinicopathologic features of breast cancer tissue array. Kaplan-Meier analysis and the log-rank test were used to estimate survival rate. P < 0.05 was considered statistically significant.
Results
Upregulation of CHPF in breast cancer tissues is associated with decreased patient progression-free survival, overall survival, and metastasis
Abnormal expression of CS synthases leads to dysregulation of CS formation in cancer cells and the TME. Several independent breast cancer datasets in the ONCOMINE database indicated that gene expression of CHPF was significantly upregulated in cancer tissues (Figure S1A). Analysis of gene expression of three major bifunctional CS synthases (CHSY1, CHPF, and CHPF2) was performed using Betastasis (http://www.betastasis.com/). High expression of CHPF was significantly associated with poor overall survival (P = 0.036, n = 225, Figure S1B). Thus, we further evaluated the expression of CHPF in different subtypes and tumor stages using The Cancer Genome Atlas (TCGA) dataset, which contains 104 normal tissues and 947 tumor tissues. The results indicated that all breast cancer subtypes (luminal A, normal-like, luminal B, HER2-enriched, and basal-like) express higher CHPF levels than the normal tissues (Figure 1A and Table S1). Additionally, the expression of CHPF in each stage (stages I, II, III, and IV) was significantly higher than that in the normal tissues. The mean expression levels of CHPF gradually increased with no statistical significance in stages III and IV (Figure 1B and Table S2). Moreover, we found that the expression of CHPF was not associated with lymph node metastasis but was significantly increased in patients with distant metastasis (Figure 1C and 1D). Further analysis of overall survival (OS) and progression-free survival (PFS) revealed that high expression of CHPF was significantly associated with decreased overall survival and PFS. Interestingly, significant differences appeared in the subsets of patients in stages III and IV (n = 227) but not those in stages I and II (n = 720, Figure 1E and 1F). We further analyzed OS and PFS in each breast cancer subtype. High expression of CHPF is associated with decrease of OS in basal-like patients and decrease of PFS in luminal B patients (Figure S2). To validate TCGA data and examine protein expression of CHPF in cancer tissue, we performed immunohistochemistry (IHC) with CHPF antibody on a tissue array that contained 133 primary breast cancer tissue samples from another independent cohort. Under our staining conditions, we found that CHPF was expressed mainly in the paranuclear cytoplasm of cancer cells. The intensity of the staining was scored according to the percentage of CHPF-positive cells in each sample (0: negative; +1: < 20%; +2: 20%-50%; +3: > 50%; Figure 1G). Our results indicated that 37.6% (50/133) of the tested samples revealed strong CHPF staining (+2 and +3) and that CHPF intensity is positively associated with high tumor stage (Table 1). Consistently, high expression of CHPF was associated with poor patient OS (Figure 1H). Overall, these results suggested that CHPF is frequently upregulated in breast cancer patients and its expression correlates with poor prognosis.
Figure 1.
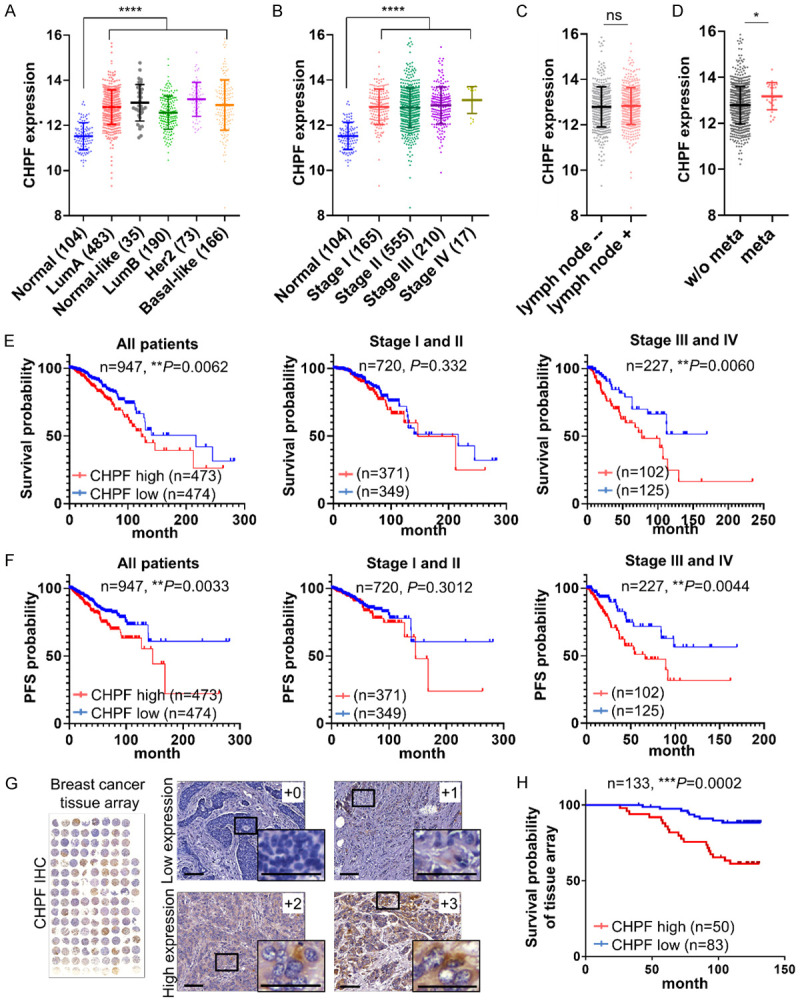
CHPF is frequently upregulated in human breast cancer and associated with poor prognosis. (A) Comparison of CHPF gene expression in breast cancer subtypes and normal breast tissue in TCGA dataset. ****P < 0.0001. (B) Comparison of gene expression of CHPF in patients with different stages of breast cancer in TCGA dataset. ****P < 0.0001. (C) Gene expression of CHPF in patients with or without lymph node invasion. (ns: non-significant). (D) Gene expression of CHPF in patients with or without distance metastasis. *P < 0.05. Survival analysis (E) and progression-free survival (F) of CHPF expression in breast cancer patients in TCGA dataset (n = 947), low stages (I and II) and high stages (III and IV) subsets. The median FPKM value of CHPF was used. (G) Immunohistochemistry of CHPF on a breast cancer tissue array. The staining was visualised in brown colour using a 3,3-diaminobenzidine liquid substrate system. All sections were counterstained with haematoxylin. Representative images of different staining intensities are shown. Amplified images are shown at the bottom right of each image. Scale bars: 50 μm. (H) Kaplan-Meier analysis of the overall survival for breast cancer tissue array cases (n = 133, P = 0.0002).
Table 1.
Correlation of CHPF expression with clinicopathological features of Breast cancer tissue array
| Factor | CHPF expression | P value (Two-sided Fisher’s exact test) | ||
|---|---|---|---|---|
|
| ||||
| Low (83) | High (50) | |||
| (0 and +1) | (+2 and +3) | |||
| Age | < 50 years | 28 | 14 | 0.566 |
| ≥ 50 years | 55 | 36 | ||
| AJCC Stages | I + II | 63 | 26 | 0.007** |
| III | 20 | 24 | ||
| Grade | Grade II | 63 | 31 | 0.116 |
| Grade III | 20 | 19 | ||
| ER (IHC) | Positive | 61 | 29 | 0.085 |
| Negative | 22 | 21 | ||
| PR (IHC) | Positive | 47 | 23 | 0.283 |
| Negative | 36 | 27 | ||
| HER2 (IHC) | Positive | 33 | 22 | 0.717 |
| Negative | 50 | 28 | ||
P < 0.01.
CHPF modulates CS formation and enhances the malignant phenotypes of breast cancer cells
To investigate the function of CHPF in breast cancer, we first analysed its expression in human and mouse breast cancer cell lines. The expression level of CHPF was relatively high in MDA-MB-231 cells and low in 4T1 and JC cells (Figure 2A). Then, we stably overexpressed Chpf in 4T1 and JC cells and silenced CHPF in MDA-MB-231 cells (Figure 2B). Flow cytometry with CS56 antibody revealed that the overexpression of CHPF increased the CS-positive cell population, while CHPF silencing moderately decreased the CS56-positive cell population (Figure 2C). These results indicated that CHPF modulated CS formation in breast cancer cells.
Figure 2.

CHPF regulates CS formation and malignant phenotypes in breast cancer cells. A. Western blots of CHPF in breast cancer cell lines. Total protein staining was considered as a loading control. B. Stable overexpression of CHPF in 4T1 cells and shRNA knockdown of CHPF in MDA-MB-231 cells. The expression levels of CHPF were analysed using western blotting. ACTB and total protein staining were used as the loading control. C. Flow cytometry using CS56 antibody. Percentage of CS56-positive population is shown. D. CCK8 assay reveals the relative cell viability of Chpf-overexpressed clones and CHPF-silenced clones. Data are represented as the mean ± SD from three independent experiments. *P < 0.05, **P < 0.01. E. Transwell invasion assay reveals relative cell mobility of Chpf-overexpressed clones and CHPF-silenced clones. Representative images are shown on the right. Data are expressed as the mean ± SD from three independent experiments. **P < 0.01, ***P < 0.001. F. Western blotting of ZO-1, E-cadherin (E-cad), β-catenin, N-cadherin (N-cad), and vimentin in Chpf-overexpressed 4T1 and JC cells. Representative images are shown. ACTB was used as the loading control. The relative signal intensities are shown on the right. **P < 0.01 and ***P < 0.001 compared to control (mock).
Phenotypic analysis showed that overexpression of Chpf significantly enhanced cell viability in 4T1 and JC cells, while the silencing of CHPF suppressed cell viability in MDA-MB-231 and MDA-MB-468 cells. Additionally, overexpression of Chpf drastically enhanced transwell cell invasion, while the silencing of CHPF inhibited cell invasion (Figure 2E). Since breast cancer invasion is strongly associated with the reactivation of epithelial-mesenchymal transition (EMT) [23,24], we examined the expression of epithelial markers ZO-1, E-cadherin, and β-catenin, and mesenchymal markers, N-cadherin and vimentin, using western blotting. Our results revealed that overexpression of Chpf promoted EMT in both 4T1 and JC cells (Figure 2F).
CHPF promotes tumor growth, metastasis, and MDSC accumulation in tumor tissues
We used orthotopic transplantation breast cancer animal models to evaluate the effects of CHPF on tumor growth, metastasis, and cytokine expression (36). Primary tumors were surgically removed at the third week and spontaneous lung metastasis was measured four weeks after tumor removal. The results indicated that overexpression of Chpf increased tumor weight (Figure 3A) and spontaneous lung metastasis was significantly enhanced (Figure 3B). Several studies have shown that CS chains can modulate the binding of cytokines and chemokines in the TME [16,25]. Thus, we profiled the quantities of important cytokines and chemokines in dissected primary tumor tissue using multiplex immunoassay. The data showed that several cytokines (IL-1a, IL-6, IL-12, and G-CSF) and chemokines (CCL2, CCL3, CCL4, and CCL5) are increased in Chpf tumors (Figure 3C). Among these secretory factors, G-CSF was dramatically surged in Chpf-overexpressing tumors. G-CSF is a key inflammatory component that facilitates granulocytic MDSC accumulation in various human tumors [26-28]. Consistent with the aforementioned sentence, an increased number of MDSCs (MHCII-CD11b+Gr1+) in Chpf-overexpressing tumor tissues were observed using flow cytometry (Figures 3D and S3). The tissue sections were further stained with macrophage (F4/80) and granulocyte markers (Ly6G) and G-CSF to identify G-CSF producers. We found a low number of macrophages and granulocytes that were G-CSF positive, and also found that 4T1 cancer cells could be the major contributors to G-CSF accumulation in the tumor tissue (Figure 3E and 3F).
Figure 3.

CHPF promotes tumor growth, metastasis, and MDSC accumulation in tumor tissue. Overexpression of Chpf increases tumor weight (A) and lung metastasis (B) of 4T1 tumor model (n = 6; *P < 0.05). Images of dissected tumors and lungs are shown on the left. Arrows indicate lung tumor nodules. Scale bars: 1.0 cm. (C) Cytokine and chemokine levels in 4T1 tumor tissues. Tumor tissue lysate (100 mg) is analysed using multiplex immunoassay. *P < 0.05, **P < 0.01. (D) Chpf increases infiltrated MDSC numbers in tumor tissue. Representative images of gating Gr1 high (Gr1h) and Gr1 intermediate (Gr1int) MDSCs are shown on the right. The mean ± SD are shown on the left (n = 7). *P < 0.05, **P < 0.01. (E) Immunostaining of F4/80 and G-CSF in 4T1 tumor sections. (F) Immunostaining of Ly6G and G-CSF in 4T1 tumor sections. Amplified images are shown at the bottom right of each image. Scale bars: 20 μm.
CHPF enhances G-CSF binding to the cell surface CS and activates MDSC-induced cell invasion
Previous studies indicate that invasive breast cancer cells, such as 4T1 and MDA-MB-231, constitutively express high levels of G-CSF [28,29]. Thus, we used these cell lines to analyse the effects of CHPF on G-CSF production. Quantitative reverse transcription PCR (qRT-PCR) showed that the gene expression of G-CSF (CSF3) was not regulated by CHPF levels (Figure 4A). However, overexpression of CHPF increased G-CSF protein levels in 4T1 cells. In contrast, silencing of CHPF decreased G-CSF levels in MDA-MB-231 cells (Figure 4B). We suggest that CHPF-modified CS may facilitate G-CSF accumulation surrounding the cancer cells. The distribution of cell surface CS and G-CSF was analysed using confocal microscopy. The results indicated that overexpression of Chpf significantly increased the co-localisation of CS and G-CSF on the cell surface. In addition, cell surface co-localisation of CS and G-CSF decreased when β-D-xylopyranoside was used to suppress CS formation (Figure 4C).
Figure 4.

CHPF regulates G-CSF protein levels in breast cancer cells. A. mRNA levels of CHPF and G-CSF in Chpf-overexpressed and CHPF-silenced breast cancer cells. ***P < 0.001 compared to control groups. B. Protein levels of G-CSF in Chpf-overexpressed and CHPF-silenced breast cancer cells. C. Co-localisation of cell surface CS and G-CSF on 4T1 cells. Mock, CHPF clone, and β-D-xylopyranoside (β-XP)-treated cells were stained with CS56 antibody (green) and G-CSF antibody (red). Amplified images are shown on the right. Dashed circles indicate the area of the nucleus. Arrows point the co-localised (yellow) spots on the cells. Scale bars: 20 μm. D. Co-culture of MDSCs and 4T1 cells induced 4T1 cell invasion. Mock or Chpf-overexpressed cells in the transwell were co-cultured with or without freshly isolated MDSCs and incubated in 5% FBS-DMEM for 24 h. Invasive cells were counted and the representative images are shown on the right. Data are expressed as the mean ± SD from three independent experiments. ***P < 0.001.
Previous studies indicate that tumor-derived G-CSF not only promotes the generation of MDSCs but also facilitates the formation of a premetastatic niche [28,30]. To evaluate the effects of MDSCs on the invasion of cancer cells, we isolated MDSCs (MHCII-CD11b+Gr1hi) from 4T1 tumor tissues and co-cultured them with 4T1-mock or Chpf-overexpressing cells. The results revealed that overexpression of CHPF significantly promoted MDSC-induced cell mobility (Figure 4D).
Syndecan-4 is modified by CHPF and contributes to CHPF-mediated malignancy in breast cancer
It can be seen in Figure 2C that CHPF regulates cell surface CS formation. To identify the PGs that were modified by CHPF, protein lysates from mock and Chpf-overexpressed cells were immunoprecipitated using CS56 antibody. The protein bands from SDS-PAGE were purified and analysed using mass spectrometry (Figure 5A). Using our experimental conditions, we identified that Syndecan-4 (SDC4) was the only PG with increased levels in Chpf-overexpressed 4T1 cells, and SDC4 was detected in MDA-MB-231 cell lysates after CS56 immunoprecipitation (IP). SDC4 is generally considered as a heparan sulfate proteoglycan (HSPG); however, it can carry CS chains depending on the cellular enzyme expression profile [31]. We digested all types of CS in the protein lysate using chondroitinase ABC and found that the molecular weight of SDC4 shifted to 60 kDa in the Chpf-overexpressing lysate, suggesting that CHPF added CS chains on SDC4 (Figure 5B). We also confirmed that the overexpression of Chpf did not modulate the gene expression of SDC4 (Figure 5C). To examine whether SDC4 participated in CHPF-induced cell invasion, the expression of SDC4 was silenced by siRNA in mock and Chpf-overexpressed clones. The results indicated that the silencing of SDC4 suppressed CHPF-induced cell invasion. Additionally, the silencing of SDC4 decreased the CHPF-enhanced cellular G-CSF in protein levels but not in mRNA levels (Figure 5E and 5F). Next, SDC4 IP assay was used to evaluate G-CSF binding to SDC4. The results demonstrated that G-CSF was captured by immobilized SDC4 in Chpf-overexpressing lysate (Figure 5G). These data suggest that CS modification on SDC4 is critical for CHPF-mediated cell invasion and G-CSF binding to SDC4.
Figure 5.

Syndecan-4 is involved in CHPF-mediated breast cancer malignancy. A. Immunoprecipitation (IP) of 4T1 cell lysate using CS56 antibody. The immunoprecipitated protein is separated using 8% SDS-PAGE and visualised using stain-free technology (Bio-Rad). The red arrowheads indicate the CHPF-enhanced proteins after IP. The black arrowhead indicates the heavy chains of the antibody. B. CHPF modifies Syndecan-4 (SDC4). Western blots of SDC4 in mock or Chpf-overexpressed cell lysates. Chondroitinase ABC (ChABC) was used to digest CS in the cell lysate. C. Relative mRNA expression of SDC4 in mock or Chpf-overexpressed 4T1 cells. D. Silencing of SDC4 by siRNA suppresses CHPF-induced cell invasion. The mock or Chpf-overexpressed cells were transfected with non-targeting control siRNAs (Ctr si) or SDC4 specific siRNAs (SDC4 si) and subjected to transwell invasion assay. Data are expressed as the mean ± SD from three independent experiments. ***P < 0.001, *P < 0.05. The representative images are shown on the right. E. Silencing of SDC4 decreases CHPF-enhanced G-CSF accumulation. Protein levels of G-CSF and SDC4 were examined using western blotting. ACTB was used as loading the control. F. Relative mRNA expression of CHPF, SDC4, and G-CSF in the transfected cells. ***P < 0.001 compared to their control. G. CHPF enhanced G-CSF binding to SDC4. Immunoprecipitation (IP) of mock and Chpf-overexpressed 4T1 cell lysates with anti-SDC4 antibody or non-specific IgG control for 18 h at 4°C. Western blots of G-CSF and SDC4 after IP, and 40 μg of input proteins were shown at right. H. High expression of SDC4 with CHPF is associated with poor overall survival in breast cancer patients. Kaplan-Meier analysis of overall survival was performed according to gene expression. The median survival is shown on the right.
Next, we studied the relationship between CHPF and SDC4 in human breast cancer samples. Interestingly, although there was no significant difference in the overall survival between patients with SDC4 high and SDC4 low expressions in TCGA breast cancer patients, the subset with high expression of both SDC4 and CHPF was significantly associated with poor overall survival and short median survival time compared to that of the subset with low expression of SDC4 and CHPF (Figure 5G).
Discussion
Alterations in the biosynthesis of glycans usually occur when glycosyltransferases are dysregulated, which results in abnormal glycans in the TME, directing crucial physiological functions in cancer progression in all types of malignant tumors [32-34]. In this study, we found that the upregulation of CHPF exacerbated poor prognostic breast cancer population, especially in high tumor stages and triple-negative (basal-like) subsets. We proposed novel functions of CHPF in promoting cancer cell malignancies as well as increasing G-CSF in the TME. Importantly, our study is the first to demonstrate that CHPF modifies CS on SDC4, which is involved in CHPF-mediated phenotypes. Breast cancer patients with high expression of both SDC4 and CHPF had the worst overall survival compared to other subsets, which implied the synergistic effects of these two genes.
A few years ago, a distinct type of CS structure, which was detected exclusively in the human placenta using a recombinant malaria parasite protein (rVAR2CSA), has been found to appear in 90% of breast tumors [35,36]. The crucial CS-modifying enzymes that contribute to rVAR2CSA binding have been identified, including CSGALNACTs, CHSY1, CHPF, and CHST11 [37]. However, only CHST11, the CS 4-O-sulphotransferase, has been reported to be associated with poor overall survival of breast cancer patients and is involved in the formation of surface P-selectin ligands in aggressive breast cancer cells [35,38]. Here, we propose that CHPF is frequently upregulated in breast cancer tissues, which could be a CS synthase that facilitates abnormal CS formation. We searched for CHPF co-expressed genes using cBioPortal (1904 samples of breast cancer) and found that CHST3 (CS 6-O-sulphotransferase) is significantly associated with CHPF and not with CHST11 [39]. These findings imply that upregulation of CHPF may favour 6-O-sulfated CS formation in breast cancer cells, since CS56 immunoreactivity requires CHST3-mediated 6-O-sulfated CS [37]. Our results from CS56 immunostaining and flow cytometry should partially reflect the CHPF-modified CS in breast cancer cells. In addition, a previous study proposed that strong CS56 staining of cancer cells, not in the tumor stroma, correlates with a decrease in recurrence-free survival and overall survival of breast cancer patients [40]. It might be worthy to further investigate the relationship between CHPF and CHST3 in breast cancer tissues.
G-CSF produced by tumor cells is known to stimulate tumor progression by facilitating tumor angiogenesis, promoting metastasis, and increasing MDSC-mediated immunosuppression in TME [41,42]. Wnt1-FGFR-induced mTOR activation has been proposed to enhance G-CSF expression in breast cancer models [43]. Although CS is suggested to regulate various growth factor signalling [16,44], we did not detect the gene expression of G-CSF that was regulated by the overexpression or silencing of CHPF, thereby suggesting that CHPF-regulated G-CSF levels could be post-translational.
The negatively charged CS and HS chains display various affinities for growth factors and cytokines. Differential density and sulfation patterns of PGs generate concentration gradients for these secretory factors [16,25]. Our data demonstrated that CHPF increased the levels of several inflammatory cytokines in 4T1 orthotopically transplanted tumor tissues, and the increase in G-CSF was the most significant factor among the tested factors. Further, using immunostaining and western blotting, we showed that the increase in G-CSF was cancer cell derived. Additionally, elevated G-CSF levels accompany increasing MDSCs in CHPF-overexpressing tumor tissues. These results are consistent with those of previous studies in breast cancer models [28,29]. Furthermore, confocal microscopy revealed an increase in G-CSF/CS56 co-localisation on the cell surface and western blotting showed that G-CSF protein levels were regulated by CHPF. These data suggested a novel interaction between CHPF-modulated CS and G-CSF in the breast cancer TME.
Interestingly, we identified SDC4 as one of the CHPF-modified PGs in breast cancer cells using CS56 IP and protein identification approaches. Syndecan family (SDC1, SDC2, SDC3, and SDC4) are type I transmembrane PGs that are found on the surface in a development-, cell-type-, and tissue-specific manner [45]. A previous study indicated that the SDC4 bearing CS and HS in mammary gland cells and the highly sulfated CS on SDC4 display a strong affinity for growth factors [31]. Our western blot data indicated that expression of CHPF in breast cancer cells enhanced CS chains on SDC4, and a partial decrease in G-CSF protein levels in SDC4-silenced cells (Figure 5E). In addition, IP of SDC4 indicated CHPF enhances G-CSF binding to SDC4. Although we cannot exclude that other CSPGs also participated in the accumulation of G-CSF on cell surface and tumor tissue, our results suggested that CHPF-modified SDC4 is at least one of the CSPGs trapping G-CSF on the cell surface.
Several studies have indicated that SDC4 is overexpressed in a subset of breast cancer [46,47] and can regulate breast cancer cell mobility [48,49]. We found that silence of SDC4 did not influence cell invasion in the low Chpf-expressing 4T1 cells (mock transfectants), but inhibited CHPF-induced cell invasion to a great extent. These in vitro studies suggested that there are G-CSF/MDSC independent mechanisms participates in CHPF-mediated phenotypical changes, and the CS bearing SDC4 may have crucial functions in breast cancer cell malignancy. Importantly, the significant decrease in overall survival in breast cancer patients with high expression of both CHPF and SDC4 is in agreement with our hypothesis.
In conclusion, the results obtained in this study suggest that CHPF may enhance tumor malignancy in multiple ways and SDC4 could be a crucial mediator involved in CHPF-induced malignant behaviours of breast cancer cells. This study not only shows a pathophysiological role of CHPF in breast cancer cells but also contributes to the understanding of their synergistic effects with SDC4 in breast cancer progression. These findings suggest a possible prognostic factor and therapeutic target for breast cancer.
Acknowledgements
This study was supported by Ministry of Science and Technology, Taiwan, MOST-109-2320-B-040-007-MY3 and MOST-106-2320-B-040-009-MY3 (CH Liu), MOST-109-2320-B-040-009 (WC Liao). The bioinformatics analysis was performed by the Bioinformatics Core Laboratory, Molecular Medicine Research Center, Chang Gung University, Taiwan (grant CLRPD1J0012). Confocal microscopy was performed through the use of the Medical Research Core Facilities Center, Office of Research & Development at China medical University, Taichung, Taiwan. TissueFAX Plus Cytometer, flow cytometry, ZEISS Axio Imager A2 microscope, and quantitative real-time PCR were performed in the Instrument Center of Chung Shan Medical University, which is supported by Ministry of Science and Technology, Ministry of Education, and Chung Shan Medical University.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Role for immune therapy in advanced breast cancer. Cancer Discov. 2018;8:132–133. doi: 10.1158/2159-8290.CD-NB2017-176. [DOI] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Schrijver WAME, Suijkerbuijk KPM, van Gils CH, van der Wall E, Moelans CB, van Diest PJ. Receptor conversion in distant breast cancer metastases: a systematic review and meta-analysis. J Natl Cancer Inst. 2018;110:568–580. doi: 10.1093/jnci/djx273. [DOI] [PubMed] [Google Scholar]
- 4.Massagué J, Obenauf AC. Metastatic colonization by circulating tumor cells. Nature. 2016;529:298–306. doi: 10.1038/nature17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy SM, Reuben A, Barua S, Jiang H, Zhang S, Wang L, Gopalakrishnan V, Hudgens CW, Tetzlaff MT, Reuben JM, Tsujikawa T, Coussens LM, Wani K, He Y, Villareal L, Wood A, Rao A, Woodward WA, Ueno NT, Krishnamurthy S, Wargo JA, Mittendorf EA. Mittendorf, poor response to neoadjuvant chemotherapy correlates with mast cell infiltration in inflammatory breast cancer. Cancer Immunol Res. 2019;7:1025–1035. doi: 10.1158/2326-6066.CIR-18-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gebremeskel S, Lobert L, Tanner K, Walker B, Oliphant T, Clarke LE, Dellaire G, Johnston B. Natural killer T-cell immunotherapy in combination with chemotherapy-induced immunogenic cell death targets metastatic breast cancer. Cancer Immunol Res. 2017;5:1086–1097. doi: 10.1158/2326-6066.CIR-17-0229. [DOI] [PubMed] [Google Scholar]
- 7.Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–68. doi: 10.1016/j.canlet.2016.01.043. [DOI] [PubMed] [Google Scholar]
- 8.Clift R, Souratha J, Garrovillo SA, Zimmerman S, Blouw B. Remodeling the tumor microenvironment sensitizes breast tumors to anti-programmed death-ligand 1 immunotherapy. Cancer Res. 2019;79:4149–4159. doi: 10.1158/0008-5472.CAN-18-3060. [DOI] [PubMed] [Google Scholar]
- 9.Ricciardelli C, Brooks JH, Suwiwat S, Sakko AJ, Mayne K, Raymond WA, Seshadri R, LeBaron RG, Horsfall DJ. Regulation of stromal versican expression by breast cancer cells and importance to relapse-free survival in patients with node-negative primary breast cancer. Clin Cancer Res. 2002;8:1054–60. [PubMed] [Google Scholar]
- 10.Suwiwat S, Ricciardelli C, Tammi R, Tammi M, Auvinen P, Kosma VM, LeBaron RG, Raymond WA, Tilley WD, Horsfall DJ. Expression of extracellular matrix components versican, chondroitin sulfate, tenascin, and hyaluronan, and their association with disease outcome in node-negative breast cancer. Clin Cancer Res. 2004;10:2491–8. doi: 10.1158/1078-0432.ccr-03-0146. [DOI] [PubMed] [Google Scholar]
- 11.Neill T, Painter H, Buraschi S, Owens RT, Lisanti MP, Schaefer L, Iozzo RV. Decorin antagonizes the angiogenic network: concurrent inhibition of Met, hypoxia inducible factor 1alpha, vascular endothelial growth factor A, and induction of thrombospondin-1 and TIMP3. J Biol Chem. 2012;287:5492–506. doi: 10.1074/jbc.M111.283499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Troup S, Njue C, Kliewer EV, Parisien M, Roskelley C, Chakravarti S, Roughley PJ, Murphy LC, Watson PH. Reduced expression of the small leucine-rich proteoglycans, lumican, and decorin is associated with poor outcome in node-negative invasive breast cancer. Clin Cancer Res. 2003;9:207–14. [PubMed] [Google Scholar]
- 13.Wang X, Osada T, Wang Y, Yu L, Sakakura K, Katayama A, McCarthy JB, Brufsky A, Chivukula M, Khoury T, Hsu DS, Barry WT, Lyerly HK, Clay TM, Ferrone S. Ferrone, CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J Natl Cancer Inst. 2010;102:1496–512. doi: 10.1093/jnci/djq343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ilieva KM, Cheung A, Mele S, Chiaruttini G, Crescioli S, Griffin M, Nakamura M, Spicer JF, Tsoka S, Lacy KE, Tutt ANJ, Karagiannis SN. Chondroitin sulfate proteoglycan 4 and its potential as an antibody immunotherapy target across different tumor types. Front Immunol. 2018;8:1911. doi: 10.3389/fimmu.2017.01911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugahara K, Mikami T. Chondroitin/dermatan sulfate in the central nervous system. Curr Opin Struct Biol. 2007;17:536–45. doi: 10.1016/j.sbi.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 16.Djerbal L, Lortat-Jacob H, Kwok J. Chondroitin sulfates and their binding molecules in the central nervous system. Glycoconj J. 2017;34:363–376. doi: 10.1007/s10719-017-9761-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwok JC, Warren P, Fawcett JW. Chondroitin sulfate: a key molecule in the brain matrix. Int J Biochem Cell Biol. 2012;44:582–6. doi: 10.1016/j.biocel.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Mikami T, Kitagawa H. Biosynthesis and function of chondroitin sulfate. Biochim Biophys Acta. 2013;1830:4719–33. doi: 10.1016/j.bbagen.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Liu CH, Lan CT, Chou JF, Tseng TJ, Liao WC. CHSY1 promotes aggressive phenotypes of hepatocellular carcinoma cells via activation of the hedgehog signaling pathway. Cancer Lett. 2017;403:280–28. doi: 10.1016/j.canlet.2017.06.023. [DOI] [PubMed] [Google Scholar]
- 20.Liao WC, Yen HR, Liao CK, Tseng TJ, Lan CT, Liu CH. DSE regulates the malignant characters of hepatocellular carcinoma cells by modulating CCL5/CCR1 axis. Am J Cancer Res. 2019;9:347–362. [PMC free article] [PubMed] [Google Scholar]
- 21.Liao WC, Liao CK, Tseng TJ, Ho YJ, Chen YR, Lin KH, Lai TJ, Lan CT, Wei KC, Liu CH. Chondroitin sulfate synthase 1 enhances proliferation of glioblastoma by modulating PDGFRA stability. Oncogenesis. 2020;9:9. doi: 10.1038/s41389-020-0197-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paschall AV, Liu K. An orthotopic mouse model of spontaneous breast cancer metastasis. J Vis Exp. 2016:54040. doi: 10.3791/54040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben-Baruch A. Host microenvironment in breast cancer development: inflammatory cells, cytokines and chemokines in breast cancer progression: reciprocal tumor-microenvironment interactions. Breast Cancer Res. 2003;5:31–6. doi: 10.1186/bcr554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 25.Theocharis AD, Karamanos NK. Proteoglycans remodeling in cancer: underlying molecular mechanisms. Matrix Biol. 2019;75-76:220–259. doi: 10.1016/j.matbio.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 26.Li W, Zhang X, Chen Y, Xie Y, Liu J, Feng Q, Wang Y, Yuan W, Ma J. G-CSF is a key modulator of MDSC and could be a potential therapeutic target in colitis-associated colorectal cancers. Protein Cell. 2016;7:130–40. doi: 10.1007/s13238-015-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawano M, Mabuchi S, Matsumoto Y, Sasano T, Takahashi R, Kuroda H, Kozasa K, Hashimoto K, Isobe A, Sawada K, Hamasaki T, Morii E, Kimura T. The significance of G-CSF expression and myeloid-derived suppressor cells in the chemoresistance of uterine cervical cancer. Sci Rep. 2015;5:18217. doi: 10.1038/srep18217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waight JD, Hu Q, Miller A, Liu S, Abrams SI. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS One. 2011;6:e27690. doi: 10.1371/journal.pone.0027690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hollmén M, Karaman S, Schwager S, Lisibach A, Christiansen AJ, Maksimow M, Varga Z, Jalkanen S, Detmar M. G-CSF regulates macrophage phenotype and associates with poor overall survival in human triple-negative breast cancer. Oncoimmunology. 2015;5:e1115177. doi: 10.1080/2162402X.2015.1115177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pickup MW, Owens P, Gorska AE, Chytil A, Ye F, Shi C, Weaver VM, Kalluri R, Moses HL, Novitskiy SV. Novitskiy, development of aggressive pancreatic ductal adenocarcinomas depends on granulocyte colony stimulating factor secretion in carcinoma cells. Cancer Immunol Res. 2017;5:718–729. doi: 10.1158/2326-6066.CIR-16-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deepa SS, Yamada S, Zako M, Goldberger O, Sugahara K. Chondroitin sulfate chains on syndecan-1 and syndecan-4 from normal murine mammary gland epithelial cells are structurally and functionally distinct and cooperate with heparan sulfate chains to bind growth factors. A novel function to control binding of midkine, pleiotrophin, and basic fibroblast growth factor. J Biol Chem. 2004;279:37368–76. doi: 10.1074/jbc.M403031200. [DOI] [PubMed] [Google Scholar]
- 32.Mereiter S, Balmaña M, Campos D, Gomes J, Reis CA. Glycosylation in the era of cancer-targeted therapy: where are we heading? Cancer Cell. 2019;36:6–16. doi: 10.1016/j.ccell.2019.06.006. [DOI] [PubMed] [Google Scholar]
- 33.Chandler KB, Costello CE, Rahimi N. Glycosylation in the tumor microenvironment: implications for tumor angiogenesis and metastasis. Cells. 2019;8:544. doi: 10.3390/cells8060544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peixoto A, Relvas-Santos M, Azevedo R, Santos LL, Ferreira JA. Ferreira, protein glycosylation and tumor microenvironment alterations driving cancer hallmarks. Front Oncol. 2019;9:380. doi: 10.3389/fonc.2019.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salanti A, Clausen TM, Agerbæk MØ, Al Nakouzi N, Dahlbäck M, Oo HZ, Lee S, Gustavsson T, Rich JR, Hedberg BJ, Mao Y, Barington L, Pereira MA, LoBello J, Endo M, Fazli L, Soden J, Wang CK, Sander AF, Dagil R, Thrane S, Holst PJ, Meng L, Favero F, Weiss GJ, Nielsen MA, Freeth J, Nielsen TO, Zaia J, Tran NL, Trent J, Babcook JS, Theander TG, Sorensen PH, Daugaard M. Daugaard, targeting human cancer by a glycosaminoglycan binding malaria protein. Cancer Cell. 2015;28:500–514. doi: 10.1016/j.ccell.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agerbæk MØ, Bang-Christensen SR, Yang MH, Clausen TM, Pereira MA, Sharma S, Ditlev SB, Nielsen MA, Choudhary S, Gustavsson T, Sorensen PH, Meyer T, Propper D, Shamash J, Theander TG, Aicher A, Daugaard M, Heeschen C, Salanti A. The VAR2CSA malaria protein efficiently retrieves circulating tumor cells in an EpCAM-independent manner. Nat Commun. 2018;9:3279. doi: 10.1038/s41467-018-05793-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen YH, Narimatsu Y, Clausen TM, Gomes C, Karlsson R, Steentoft C, Spliid CB, Gustavsson T, Salanti A, Persson A, Malmström A, Willén D, Ellervik U, Bennett EP, Mao Y, Clausen H, Yang Z. The GAGOme: a cell-based library of displayed glycosaminoglycans. Nat Methods. 2018;15:881–888. doi: 10.1038/s41592-018-0086-z. [DOI] [PubMed] [Google Scholar]
- 38.Cooney CA, Jousheghany F, Yao-Borengasser A, Phanavanh B, Gomes T, Kieber-Emmons AM, Siegel ER, Suva LJ, Ferrone S, Kieber-Emmons T, Monzavi-Karbassi B. Chondroitin sulfates play a major role in breast cancer metastasis: a role for CSPG4 and CHST11 gene expression in forming surface P-selectin ligands in aggressive breast cancer cells. Breast Cancer Res. 2011;13:R58. doi: 10.1186/bcr2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Svensson KJ, Christianson HC, Kucharzewska P, Fagerström V, Lundstedt L, Borgquist S, Jirström K, Belting M. Chondroitin sulfate expression predicts poor outcome in breast cancer. Int J Oncol. 2011;39:1421–8. doi: 10.3892/ijo.2011.1164. [DOI] [PubMed] [Google Scholar]
- 41.Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, Meng YG, Ferrara N. Ferrara, G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106:6742–7. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, Yu L, Ross J, Korsisaari N, Cao T, Bou-Reslan H, Kallop D, Weimer R, Ludlam MJ, Kaminker JS, Modrusan Z, van Bruggen N, Peale FV, Carano R, Meng YG, Ferrara N. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A. 2010;107:21248–55. doi: 10.1073/pnas.1015855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, Holdman XB, Herschkowitz JI, Pond A, Xie G, Kurley S, Nguyen T, Liao L, Dobrolecki LE, Pang L, Mo Q, Edwards DP, Huang S, Xin L, Xu J, Li Y, Lewis MT, Wang T, Westbrook TF, Rosen JM, Zhang XH. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumor initiation. Nat Cell Biol. 2016;18:632–44. doi: 10.1038/ncb3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shipp EL, Hsieh-Wilson LC. Profiling the sulfation specificities of glycosaminoglycan interactions with growth factors and chemotactic proteins using microarrays. Chem Biol. 2007;14:195–208. doi: 10.1016/j.chembiol.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 45.Cheng B, Montmasson M, Terradot L, Rousselle P. Syndecans as cell surface receptors in cancer biology. A focus on their interaction with PDZ domain proteins. Front Pharmacol. 2016;7:10. doi: 10.3389/fphar.2016.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baba F, Swartz K, van Buren R, Eickhoff J, Zhang Y, Wolberg W, Friedl A. Syndecan-1 and syndecan-4 are overexpressed in an estrogen receptor-negative, highly proliferative breast carcinoma subtype. Breast Cancer Res Treat. 2006;98:91–8. doi: 10.1007/s10549-005-9135-2. [DOI] [PubMed] [Google Scholar]
- 47.Lendorf ME, Manon-Jensen T, Kronqvist P, Multhaupt HA, Couchman JR. Syndecan-1 and syndecan-4 are independent indicators in breast carcinoma. J Histochem Cytochem. 2011;59:615–29. doi: 10.1369/0022155411405057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim HC, Multhaupt HA, Couchman JR. Cell surface heparan sulfate proteoglycans control adhesion and invasion of breast carcinoma cells. Mol Cancer. 2015;14:15. doi: 10.1186/s12943-014-0279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang H, Jin H, Rapraeger AC. Syndecan-1 and syndecan-4 capture epidermal growth factor receptor family members and the alpha3beta1 integrin via binding sites in their ectodomains: novel synstatins prevent kinase capture and inhibit alpha6beta4-integrin-dependent epithelial cell motility. J Biol Chem. 2015;290:26103–13. doi: 10.1074/jbc.M115.679084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.