

Abstract
Most of screening-detected prostate cancer (PCa) are indolent and not lethal. Biomarkers that can predict aggressive diseases independently of clinical features are needed to improve risk stratification of localized PCa patients and reduce overtreatment. We aimed to identify leukocyte DNA methylation differences between clinically defined aggressive and non-aggressive PCa. We performed whole genome DNA methylation profiling in leukocyte DNA from 287 PCa patients with Gleason Score (GS) 6 and ≥8 using Illumina 450k methylation arrays. We observed a global hypomethylation in GS≥8 patients compared to GS=6 PCa patients; in contrast, the methylation level in core promoter and exon 1 region was significantly higher in GS≥8 patients than GS=6 PCa. We then performed 5-fold cross validated random forest model training on 1,459 differentially methylated CpG Probes (DMPs) with false discovery rate (FDR) <0.01 between GS=6 and GS≥8 groups. The power of the predictive model was further reinforced by ranking the DMPs with Decreased Gini and re-train the model with the top 97 DMPs (Testing AUC=0.920, predict accuracy =0.847). In conclusion, we identified a CpG methylation signature in leukocyte DNA that is associated with aggressive clinical features of PCa at diagnosis.
Keywords: Prostate cancer, aggressive disease, Gleason score, whole genome DNA methylation, peripheral blood leukocytes
Introduction
Prostate cancer (PCa) is the most common cancer and second leading cause of cancer death in American men [1]. There will be an estimated 191,930 new cases and 33,330 deaths from PCa in the United States in 2020 [1]. PCa presents no symptoms until it becomes advanced or metastatic. The wide use of prostate-specific antigen (PSA) testing for the screening and early detection has contributed to the greatly improved survival of PCa [2]. However, many of PSA screening-detected PCa are indolent and pose little threat to the survival of patients. Commonly used clinical variables, including PSA level, Gleason score (GS), and tumor stage, are not sufficient to predict which patients will have aggressive diseases and which will have indolent diseases. Thus, the majority of men with localized PCa receive upfront aggressive treatments (radical prostatectomy and radiotherapy), which are often associated with significant side effects, causing overdiagnosis and overtreatment. Biomarkers that can predict aggressive diseases are needed to improve risk stratification of PCa patients for better-informed clinical management.
Compared with other cancer types, genetic mutations are less common in PCa tumors [3]. Epigenetic changes including DNA methylation play a prominent role in prostate carcinogenesis and progression [4]. Global hypomethylation and site-specific hypermethylation in promoter regions of tumor suppressor genes have been frequently observed in most cancers including PCa [5,6]. Recently, there has been growing interest in using DNA methylation in peripheral blood leukocytes as predictors of cancer risks and clinical outcomes [7-19]. Specific CpG site methylation in leukocyte DNA has been shown to be associated with the risk of PCa [15-17] but no study has systemically investigated the role of leukocyte DNA methylation in predicting aggressive PCa.
In this study, we performed a genome-wide CpG methylation profiling in leukocyte DNA from a large number of PCa patients and identified specific leukocyte CpG methylation pattern among GS=6 and GS≥8 patients.
Materials and method
Study population
This study included 287 non-Hispanic white men with histologically confirmed adenocarcinoma of prostate from the University of Texas MD Anderson Cancer Center. Blood specimens were collected from the patients at diagnosis before any treatments. Clinical and follow-up data were abstracted from patient medical records by clinical coding specialists; these data included date of diagnosis, performance status, clinical stage, histological grade and pathological stage, treatment (active surveillance, prostatectomy, radiotherapy, and hormone therapy), and progression (biochemical recurrence and metastasis). The MD Anderson Tumor Registry conducts annual vital status follow-ups for all cancer patients. All patients signed an informed consent form. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Institutional Review Board of MD Anderson Cancer Center. We also included publicly available global DNA methylation data of healthy people (GSE85210) as control group.
DNA extraction, bisulfite treatment and Illumina 450k beadchip
DNA was extracted with Qiagen mini kit (Qiagen, Germany) according to the manufacturer’s protocol. One microgram of genomic DNA was treated with sodium bisulfite using the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA) according to the manufacturer’s protocol. In order to minimize the batch effects, similar numbers of samples with GS=6 and GS≥8 were put on the same chip for hybridization. Briefly, whole genome DNA methylation profiling was performed on 500 ng of bisulfite-treated DNA using the Illumina infimum Human Methylation 450k Beadchip (Illumina, Inc., San Diego, CA, USA) following standardized protocols and manufacturer’s instructions. The 450k beadchip contains 485,577 cytosine positions in human genome, among which 365,934 CpG sites are located within known gene regions such as promoter, gene body or untranslated regions (UTRs), 119,830 are in intergenic regions [20]. Beadchips were scanned on an Illunima HiScan SQ that has two-color laser fluorescent scanner with a 0.375 um spatial resolution. The intensities of the images were extracted using Genome Studio Methylation Module.
Data analysis
Data analyses were performed with R version 3.4.3, Bioconductor packages, Chip Analysis Methylation Pipeline (ChAMP) [21] and bash scripts. Raw intensity data (idat files) were organized as the initial loading files. The methylation status of each specific CpG site was shown as β-values, calculated as the ratio of the fluorescence intensity signals of the methylated (M) and unmethylated (U) alleles [22]. β values range between 0 (non-methylated) and 1 (completely methylated). The probe detection p-value threshold was set as 0.01 and any samples showing high fraction of failed probes (>0.05) was removed. Any probes with less than 3 detected beads in at least 5% of samples were also removed. Non-CpG probes also were removed. Y chromosomes were not ruled out since our dataset contains only male patients. Only one sample from GS=6 group was removed due to high percentage of failed probes. We also carried out normalization of our dataset in order to remove the differences between type I and type II probe distributions with BMIQ method [23].
After normalization, we removed the batch effect caused by sample source. ChAMP called the differentially methylated probes (DMPs) using the corrected matrix of expression values with gene-wise linear models. A total of 10,264 DMPs were identified with FDR<0.05, and 1,459 DMPs with FDR<0.01 were selected as the input for further analysis. To estimate leukocyte subpopulations, we used ChAMP 450k reference databases for whole blood and performed regression method by Houseman et.al [24] to deconvolute cell populations for each blood cell type.
Then we used 5-fold cross-validated random forest model to identify the methylation signature that associates with GS. Training set was determined randomly as 80% of total dataset for each fold. Random forest trees were not pruned, and the number of trees was set as 400 to increase model power and also to decrease the FDR. After the first model was trained, probe importance (Decrease Gini) was ranked for further model selection. We decided the best probe number for the random forest model based on AUC of training and testing set and prediction accuracy.
Results
Patient characteristics
We performed whole genome CpG methylation profiling in leukocyte DNA from 287 PCa patients with GS=6 and GS≥8. All patients were Caucasians. Most patients (85.3%) were 55 years and older. The mean ages (SD) of GS=6 and GS≥8 patients were 63.49 (SD: 5.46) and 63.68 (7.29), respectively. Only 7.8% were current smokers. About half were GS=6 (N=140) and half GS≥8 (N=147) patients. The patients had predominantly T1 (68.2%) tumors and had PSA<10 ng/ml (72.4%).
Leukocyte DNA methylation pattern of GS≥8 and GS=6 PCa patients
After normalization among all patients, a total of 464,867 cytosine positions in CpG dinucleotides on Human Methylation 450k BeadArray were analyzed. We first compared the global methylation level between GS≥8 and GS=6 patients. Although there was no significant differences in the overall global methylation level (mean β values of all the measured CpG sites) between GS≥8 and GS=6 patients, there were distinct methylation patterns among different functional regions (Figure 1A). The mean β value was the lowest in the core promoter region (TSS-200, within 200 base pairs of the transcription start site [TSS]), followed by Exon 1, 5’ untranslated region (UTR), and TSS-1500 (within 1,500 base pairs of the TSS), all of which had mean β values between 0.18 and 0.40; whereas the mean β values of CpG sites located in gene body, 3’ UTR, and intergenic region (IGR) were much higher (0.63 to 0.78). More importantly, the mean β values of CpG sites in TSS-200 and Exon 1 were significantly higher in GS≥8 patients than in GS=6 patients (P=0.013 and 0.017, respectively), whereas the methylation levels in gene body, 3’ UTR, IGR and overall methylation level (all) were higher in GS=6 than GS≥8 patients, although the difference did not reach statistical significance (Figure 1A).
Figure 1.
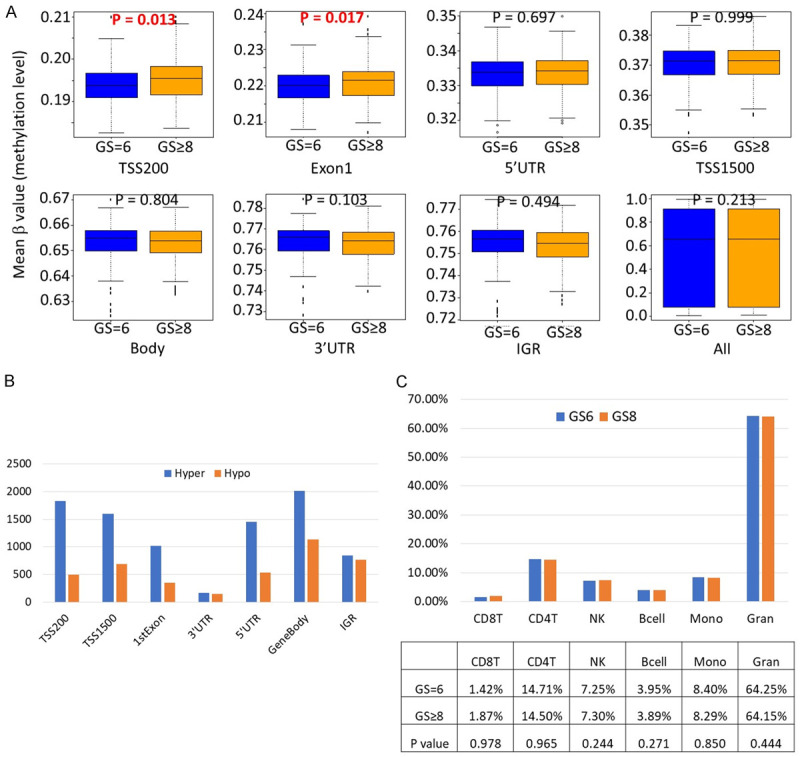
Overall leukocyte DNA hypermethylation in transcriptionally active regions in GS≥8 patients compared to GS=6 patients. A. Comparisons of mean β value of CpG sites by locations of CpG sites relative to gene structure; B. Comparisons of the total numbers of significantly hypermethylated and hypomethylated CpG sites by locations of CpG sites relative to gene structure; C. Comparisons of the frequencies of major leukocyte subpopulations between GS≥8 and GS=6 patients. Abbreviations: TSS200: within 200 bp of the transcription start site (TSS); TSS1500: within 1500 bp of the TSS; UTR: untranslated region; IGR: intergenic regions.
There were 10,264 differentially methylated CpG probes (DMPs) between GS≥8 and GS=6 patients with FDR<0.05, among which 6,876 were hypermethylated and 3,389 were hypomethylated in GS≥8 compared to GS=6 patients. In a breakdown of significant hypermethylated and hypomethylated CpG sites by CpG locations, there were significantly more hypermethylated than hypomethylated CpG sites in transcriptionally active regions, in particular, TSS200, Exon 1, 5’ UTR, and TSS1500, whereas the numbers of significantly hypermethylated and hypomethylated CpG sites were similar in 3’ UTR and IGR (Figure 1B).
Among 6,876 hypermethylated CpG sites in GS≥8 patients, 3,771 were located in CpG islands. Since hypermethylation in CpG islands is more likely to affect host gene expression, we performed gene set enrich analysis (GSEA) using host genes of these 3,771 DMPs. The top enriched pathways included RNA-binding, enzyme-biding, ribonucleotide binding, and regulation of gene expression.
Leukocyte DNA methylation can be used to quantify different leukocyte subproportions [24,25]. We estimated the frequencies of B cell, CD8+ and CD4+ T cell, natural killer cell, granulocytes, and monocytes using methylation profiles (Figure 1C). The frequencies of major leukocyte subpopulations were similar between GS≥8 and GS=6 patients, which indicates the leukocyte methylation differences between GS≥8 and GS=6 are not likely due to different immune cell compositions.
A leukocyte CpG methylation signature for predicting aggressive PCa
To identify a CpG methylation signature that distinguish GS≥8 from GS=6 patients, we used the normalized β value of 1,459 CpG sites with FDR<0.01 as input to train the 5-fold cross validated random forest model. The testing set AUC was 0.836 and prediction accuracy was 0.757. After ranking the probes with their contribution to the model (decreasing Gini), we improved the model by training the model with fewer top ranked DMPs. When we used top 10 differentially methylated DMPs, the prediction reached 80% and additional DMPs only modestly increased the prediction accuracy, up to 85% (Figure 2A). For the final model with the top 97 DMPs, the testing AUC was 0.920, and predicting accuracy was 0.847 (Figure 2B). The Multidimensional Scaling (MDS) plot indicated a strong ability of our model to cluster patients (Figure 2C). Figure 2D shows the heatmap of using those 97 CpG sites to group GS≥8 from GS=6 patients and there was a clear separation of these two groups. The characteristics of the top 97 differentially methylated CpG sites between GS=6 and GS≥8 patients are shown in Table 1.
Figure 2.
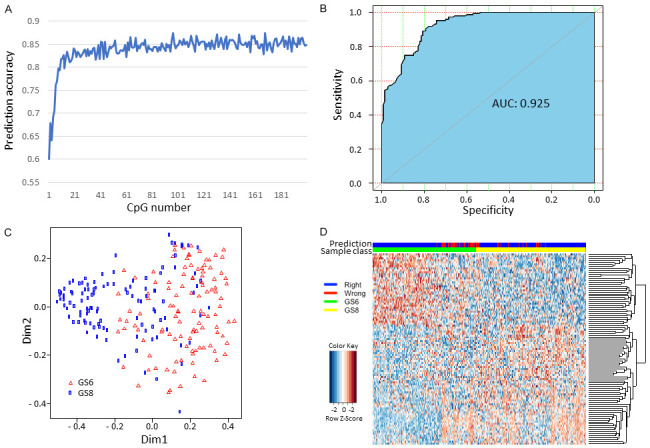
Leukocyte DNA methylation signature that differentiates GS≥8 patients from GS=6 patients. A. Prediction accuracy based on the number of differentially methylated CpG probes (DMPs); B. The ROC and AUC of prediction model using top 97 DMPs; C. Multidimensional Scaling (MDS) plot indicating the ability of the model to cluster patients; D. Supervised clustering of GS≥8 and GS=6 patients.
Table 1.
Top 97 differentially methylated CpG sites between GS=6 and GS≥8 patients
| CpG ID | β value | P value | Chromosome | Position | Gene | CpG Location | |
|---|---|---|---|---|---|---|---|
|
| |||||||
| GS=6 | GS≥8 | ||||||
| cg00111102 | 0.9175 | 0.9328 | 1.73E-05 | 20 | 60509975 | CDH4 | Body-shore |
| cg00216361 | 0.0491 | 0.0543 | 1.96E-05 | 3 | 115342527 | GAP43 | 1st Exon-open sea |
| cg00419564 | 0.0223 | 0.0266 | 4.21E-07 | 1 | 153508860 | S100A6 | TSS200-shore |
| cg00567696 | 0.0492 | 0.0564 | 2.37E-06 | 6 | 46097521 | ENPP4 | TSS200-shore |
| cg00619978 | 0.5270 | 0.5650 | 4.13E-06 | 5 | 180046052 | FLT4 | Body-island |
| cg00843795 | 0.6578 | 0.7677 | 1.32E-05 | 7 | 105163736 | PUS7 | TSS1500-shore |
| cg00850868 | 0.6365 | 0.6548 | 9.03E-09 | 10 | 64437920 | IGR | NA |
| cg01071346 | 0.0294 | 0.0335 | 5.61E-06 | 1 | 2480431 | IGR | chr1:2477563-2478363 |
| cg01077623 | 0.7255 | 0.7055 | 2.16E-05 | 7 | 55757733 | FKBP9L | TSS1500-open sea |
| cg01466348 | 0.9389 | 0.9245 | 5.40E-07 | 2 | 161503843 | IGR | NA |
| cg01890546 | 0.9143 | 0.9227 | 1.64E-06 | 7 | 884588 | UNC84A | Body-shelf |
| cg02005490 | 0.9271 | 0.9150 | 6.24E-06 | 5 | 1959850 | IGR | NA |
| cg02048674 | 0.0437 | 0.0505 | 2.24E-06 | 19 | 49991517 | RPL13AP5 | Body-island |
| cg02383160 | 0.0280 | 0.0331 | 1.23E-07 | 11 | 62496393 | TTC9C | 1st Exon-shore |
| cg02895995 | 0.0641 | 0.0741 | 4.94E-08 | 19 | 7554069 | PEX11G | TSS200-shore |
| cg03014008 | 0.5970 | 0.6156 | 1.93E-07 | 20 | 57463767 | GNAS | 3’ UTR-island |
| cg03354554 | 0.2366 | 0.2169 | 2.08E-05 | 11 | 9781412 | IGR | chr11:9779592-9780470 |
| cg03414732 | 0.0713 | 0.0597 | 3.06E-09 | 18 | 32870301 | ZNF271 | Body-island |
| cg04208114 | 0.0454 | 0.0554 | 4.16E-08 | 1 | 59012469 | OMA1 | TSS200-island |
| cg04250904 | 0.6706 | 0.6485 | 2.25E-07 | 19 | 12623422 | ZNF709 | 5’ UTR-shore |
| cg04442328 | 0.1526 | 0.1675 | 1.34E-05 | 3 | 185304136 | SENP2 | 1st Exon-island |
| cg04913913 | 0.1189 | 0.1070 | 7.40E-07 | 6 | 31126599 | CCHCR1 | TSS1500-shore |
| cg05176964 | 0.9753 | 0.9701 | 3.67E-06 | 22 | 42910177 | RRP7A | Body-island |
| cg06295548 | 0.8495 | 0.8741 | 3.28E-08 | 4 | 146296778 | IGR | NA |
| cg06434972 | 0.0371 | 0.0426 | 3.48E-07 | 7 | 44122219 | POLM | TSS200-island |
| cg06834240 | 0.1333 | 0.1507 | 4.07E-08 | 16 | 79632625 | MAF | 3’ UTR-island |
| cg07374247 | 0.0157 | 0.0187 | 3.00E-08 | 6 | 27860935 | HIST1H2AM | 1st Exon-shore |
| cg07872947 | 0.9402 | 0.9481 | 1.47E-06 | 2 | 1732172 | PXDN | Body-open sea |
| cg08005992 | 0.1080 | 0.1219 | 7.13E-06 | 11 | 31832959 | PAX6 | TSS200-island |
| cg08907257 | 0.8877 | 0.8974 | 1.34E-05 | 16 | 2223188 | TRAF7 | Body-shelf |
| cg09618381 | 0.8791 | 0.8566 | 6.12E-07 | 6 | 150379479 | IGR | chr6:150378838-150379048 |
| cg09910998 | 0.5714 | 0.5810 | 1.18E-06 | 7 | 94285942 | SGCE | TSS1500-island |
| cg10149161 | 0.0831 | 0.0680 | 3.18E-10 | 11 | 64578067 | MEN1 | TSS200-island |
| cg10438391 | 0.2942 | 0.2522 | 2.42E-05 | 8 | 144631915 | IGR | chr8:144631767-144631971 |
| cg10446143 | 0.0558 | 0.0626 | 2.16E-05 | 21 | 44394730 | PKNOX1 | 5’ UTR-island |
| cg10632144 | 0.9276 | 0.8967 | 1.27E-05 | 13 | 50252564 | EBPL | Body-open sea |
| cg10797195 | 0.0457 | 0.0514 | 4.70E-06 | 1 | 45805338 | MUTYH | 5’ UTR-shore |
| cg10919522 | 0.2547 | 0.2330 | 1.84E-05 | 14 | 74227441 | C14orf43 | 5’ UTR-shore |
| cg11214243 | 0.0373 | 0.0424 | 2.36E-07 | 11 | 65405388 | SIPA1 | TSS200-shelf |
| cg11678250 | 0.4900 | 0.4546 | 3.31E-06 | 7 | 136362483 | IGR | NA |
| cg11956953 | 0.0881 | 0.0726 | 1.85E-05 | 17 | 27347092 | IGR | chr17:27346853-27347222 |
| cg12791243 | 0.0682 | 0.0609 | 1.01E-06 | 4 | 79698201 | BMP2K | Body-shore |
| cg13038108 | 0.0267 | 0.0317 | 4.34E-07 | 4 | 39461155 | LIAS | Body-shore |
| cg13785223 | 0.4713 | 0.4286 | 4.20E-06 | 13 | 114905788 | IGR | NA |
| cg14235800 | 0.8887 | 0.8982 | 2.01E-05 | 9 | 104238593 | C9orf125 | Body-open sea |
| cg14323199 | 0.0517 | 0.0572 | 3.99E-06 | 17 | 60705839 | MRC2 | Body-island |
| cg14416269 | 0.2198 | 0.1914 | 4.71E-06 | 4 | 6271139 | WFS1 | TSS1500-shore |
| cg14420670 | 0.0328 | 0.0382 | 8.18E-08 | 6 | 29617961 | IGR | chr6:29617765-29617974 |
| cg14951488 | 0.1623 | 0.1538 | 1.73E-05 | 10 | 95256188 | CEP55 | TSS200-island |
| cg15248835 | 0.0413 | 0.0485 | 1.76E-05 | 8 | 9761171 | LOC157627 | TSS1500-island |
| cg15354625 | 0.9302 | 0.9372 | 4.06E-06 | 11 | 78381223 | ODZ4 | Body-open sea |
| cg15404375 | 0.9397 | 0.9458 | 2.02E-05 | 4 | 111866546 | IGR | NA |
| cg15731816 | 0.0361 | 0.0408 | 6.70E-07 | 14 | 75230414 | YLPM1 | 1st Exon-island |
| cg15896939 | 0.9280 | 0.9352 | 1.35E-05 | 1 | 156030809 | RAB25 | TSS200-opensea |
| cg15935247 | 0.7404 | 0.7189 | 2.31E-06 | 17 | 56606842 | 4-Sep | TSS200-shelf |
| cg16311364 | 0.5388 | 0.4808 | 1.07E-06 | 10 | 46912902 | FAM35B | Body-shore |
| cg16374753 | 0.9620 | 0.9682 | 2.27E-06 | X | 79279642 | TBX22 | Body-open sea |
| cg16513883 | 0.8804 | 0.8923 | 2.90E-05 | 5 | 9295286 | SEMA5A | Body-open sea |
| cg16619899 | 0.8632 | 0.8492 | 6.18E-06 | 8 | 915860 | IGR | chr8:914817-915894 |
| cg16925090 | 0.0692 | 0.0790 | 4.46E-06 | 11 | 101785516 | KIAA1377 | TSS1500-shore |
| cg17098965 | 0.3396 | 0.3067 | 3.26E-07 | 20 | 52199520 | ZNF217 | 5’ UTR-shore |
| cg17329834 | 0.8211 | 0.7991 | 4.98E-06 | 6 | 131380543 | EPB41L2 | 5’ UTR-shelf |
| cg17392909 | 0.2192 | 0.2510 | 4.35E-09 | 10 | 135187035 | ECHS1 | TSS200-island |
| cg17524854 | 0.0458 | 0.0515 | 8.21E-08 | 12 | 67663046 | CAND1 | TSS200-island |
| cg18050997 | 0.8858 | 0.8970 | 6.34E-08 | 8 | 8176225 | PRAGMIN | Body-island |
| cg18483322 | 0.0772 | 0.0845 | 3.01E-06 | 2 | 97523826 | ANKRD39 | TSS200-island |
| cg18651347 | 0.8273 | 0.8092 | 2.18E-05 | 7 | 70102632 | AUTS2 | Body-open sea |
| cg18725195 | 0.6238 | 0.6550 | 5.79E-07 | 5 | 976058 | IGR | NA |
| cg18943383 | 0.0329 | 0.0402 | 6.49E-09 | 6 | 27777858 | HIST1H3H | 1st Exon-island |
| cg19239278 | 0.7810 | 0.7583 | 2.98E-06 | 19 | 19513162 | GATAD2A | 5’ UTR-shelf |
| cg19242459 | 0.9213 | 0.9302 | 1.99E-08 | 2 | 239006511 | SCLY | Body-shelf |
| cg19757631 | 0.8720 | 0.8529 | 6.65E-06 | 1 | 11118889 | SRM | Body-shore |
| cg19864851 | 0.0275 | 0.0333 | 1.48E-08 | 10 | 75503847 | SEC24C | TSS1500-shore |
| cg20153768 | 0.0334 | 0.0376 | 2.42E-05 | 6 | 26123228 | HIST1H2AC | TSS1500-shore |
| cg20390613 | 0.0432 | 0.0513 | 1.86E-07 | 1 | 12678355 | DHRS3 | TSS1500-island |
| cg20539816 | 0.9396 | 0.9334 | 4.13E-07 | 17 | 5988249 | WSCD1 | Body-open sea |
| cg21636841 | 0.8416 | 0.8540 | 1.43E-05 | 11 | 968731 | AP2A2 | Body-open sea |
| cg22028624 | 0.8741 | 0.8513 | 1.01E-07 | 11 | 70281091 | CTTN | Body-open sea |
| cg22110517 | 0.9086 | 0.9168 | 4.36E-06 | 17 | 4800583 | MINK1 | 3’ UTR-shore |
| cg22407822 | 0.6570 | 0.6790 | 7.54E-08 | 20 | 57463658 | GNAS | 3’ UTR-island |
| cg22716488 | 0.0387 | 0.0437 | 2.15E-06 | 6 | 35995431 | MAPK14 | TSS200-island |
| cg22826071 | 0.0364 | 0.0452 | 2.91E-08 | 19 | 344165 | MIER2 | Body-island |
| cg22961241 | 0.8822 | 0.8999 | 1.56E-06 | 6 | 32917502 | HLA-DMA | Body-open sea |
| cg23496597 | 0.6552 | 0.6715 | 1.23E-06 | 20 | 57463725 | GNAS | 3’ UTR-island |
| cg23983453 | 0.4253 | 0.4789 | 8.55E-06 | 5 | 92925524 | NR2F1 | Body-shore |
| cg24337701 | 0.6228 | 0.5930 | 1.87E-05 | 8 | 141275191 | TRAPPC9 | Body-open sea |
| cg24751378 | 0.7014 | 0.7205 | 2.26E-06 | 21 | 30396349 | USP16 | TSS1500-shore |
| cg25079743 | 0.0262 | 0.0311 | 7.23E-07 | 16 | 30441674 | DCTPP1 | TSS1500-island |
| cg25198967 | 0.8953 | 0.9066 | 7.74E-08 | 3 | 52325846 | GLYCTK | Body-shelf |
| cg25554036 | 0.2521 | 0.2127 | 7.34E-07 | 4 | 6271136 | WFS1 | TSS1500-shore |
| cg25696807 | 0.7097 | 0.6766 | 6.39E-07 | X | 145109374 | MIR891A | Body-open sea |
| cg25697492 | 0.1212 | 0.1110 | 1.54E-05 | 19 | 2950919 | IGR | chr19:2950359-2950962 |
| cg25748441 | 0.0329 | 0.0383 | 2.47E-05 | 2 | 202122587 | CASP8 | 5’ UTR-open sea |
| cg25806190 | 0.8185 | 0.7973 | 3.24E-06 | 2 | 232878174 | DIS3L2 | 5’ UTR-open sea |
| cg26127025 | 0.9343 | 0.9201 | 1.11E-06 | 5 | 2703138 | IGR | NA |
| cg26683137 | 0.3639 | 0.3234 | 7.98E-06 | 17 | 33447208 | FNDC8 | TSS1500-shore |
| cg27482619 | 0.0574 | 0.0663 | 7.74E-11 | 10 | 30818479 | IGR | NA |
Comparison of leukocyte DNA methylation between PCa patients and healthy controls
We compared our data with a publically available leukocyte 450K methylation dataset of healthy controls (GSE85210). There were 172 healthy men in the dataset. The mean β values of all the CpG sites were significantly lower in PCa patients than in healthy controls (P=0.011), indicating global hypomethylation of PCa patients. However, the mean β values of CpG sites in TSS-200 and Exon 1 regions were significantly higher in PCa patients than in healthy controls (P<0.001 for both) (Figure 3).
Figure 3.
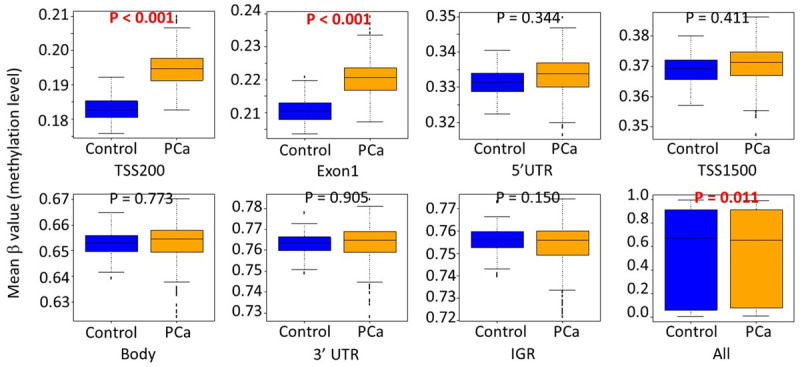
Comparisons of mean β value of CpG sites by locations of CpG sites relative to gene structure between PCa patients and healthy controls. Abbreviations: TSS200: within 200 bp of the transcription start site (TSS); TSS1500: within 1500 bp of the TSS; UTR: untranslated region; IGR: intergenic regions.
Discussion
The main purpose of this study was to identify intrinsic biological differences between clinically defined non-aggressive (GS=6) and aggressive (GS≥8) that may serve as predictors of aggressive PCa. We performed a genome-wide CpG methylation profiling of leukocyte DNA from 287 PCa patients with GS=6 and GS≥8. We found leukocyte DNA hypermethylation in transcriptionally active regions in aggressive PCa patients and identified a 97-CpG signature that could distinguish aggressive from non-aggressive PCa. To our knowledge, this is the first study to report leukocyte CpG methylation signature for the prediction of aggressive PCa.
We found the mean methylation level was the lowest in the core promoter region (TSS-200), followed by Exon 1, 5’ UTR, and TSS-1500, but considerably higher in gene body, 3’ UTR, and intergenic regions, consistent with literature reports of low methylation in the transcriptionally active regions, indicating open chromatin structure [26]. More importantly, we observed hypermethylation of leukocyte DNA in GS≥8 patients compared to GS=6 patients in the most transcriptionally active regions (TSS200 and Exon 1). In gene set enrichment analysis, the top enriched pathways included RNA-binding, enzyme-binding, ribonucleotide binding, and regulation of gene expression. These findings indicate an overall down-regulation of gene expression in leukocytes of GS≥8 patients, likely affecting inflammatory response and immune function and contributing to the aggressive phenotypes. Likewise, when we used a publically available dataset of leukocyte DNA methylation in healthy men and compared to the data in our PCa patients, we observed hypermethylation of leukocyte DNA in PCa patients in the most transcriptionally active regions (TSS200 and Exon 1), supporting an overall down-regulation of gene expression in leukocytes of PCa patients, particularly aggressive PCa, that affects inflammatory response and immune function and contributes to PCa development and progression. We also observed an overall lower methylation of leukocyte DNA in PCa patients compared to healthy individuals, and in GS≥8 than in GS=6 patients. Global hypomethylation in tumor tissues is a well-established cancer promoting event [5,6,27]. It has also been hypothesized that global DNA hypomethylation in leukocytes may be a cancer risk factor due to increased genomic instability [7,8]. There are some supporting evidence for this notion, but the data were not consistent [7,8]. Previous studies evaluating global DNA methylation and cancer risks mostly used methylation of short repetitive DNA sequences (e.g., LINE-1 and Alu) as surrogates to represent global DNA methylation level. In our study, we used the mean β value of all the assayed CpG sites, which provides a more accurate estimate of global DNA methylation level. Our data support the notion that global hypomethylation in leukocyte DNA contributes to the development and progression of PCa likely through general genomic instability.
Leukocyte DNA methylation is at the interphase between genetics and environment. It is under strong influence of genetics and also has been linked to immune cell subpopulation, aging, and smoking. We did not observe significant differences in the immune cell subpopulations between GS=6 and GS≥8 patients, indicating that there were minimal immune cell turnovers between aggressive and non-aggressive PCa patients and the methylation level differences between GS=6 and GS≥8 patients were consistent across all immune cell types. The absolute methylation level difference (β value difference) of each individual CpG site between GS=6 and GS≥8 patients was modest, and the prediction accuracy of our model reached a plateau of 85%. This limitation of predicting aggressive PCa using leukocyte DNA methylation is not surprising given the predominant background of normal immune cells.
We only included GS=6 and GS≥8 patients in this analysis because they have distinct clinical phenotypes. This study design is intended to identify biological features that differentiate clinically defined aggressive diseases from non-aggressive diseases. GS=7 patients, on the other hand, have intermediate risks of progression and their outcomes are more heterogeneous and more difficult to predict. Future studies are needed to determine whether leukocyte DNA methylation can predict more aggressive clinical behavior in GS=7 patients.
In summary, we performed a large scale DNA methylation profiling of leukocyte DNA in clinically defined aggressive and non-aggressive PC patients. We observed hypermethylation in transcriptionally active regions of aggressive PCa patients compared to non-aggressive PCa patients and global hypomethylation in PCa patients. We identified a 97-CpG methylation signature in leukocytes that is associated with aggressive PCa at diagnosis. Our study also provides biological insights into the modulation of immune system by aggressive PCa.
Acknowledgements
We thank the Population Genetics Core of MD Anderson Cancer Center for performing methylation arrays. This study was supported by a Cancer Prevention and Research Institute of Texas (CPRIT) grant (RP140556), a National Cancer Institute Specialized Program of Research Excellence (SPORE) grant (CA140388), and MD Anderson Cancer Center start-up fund to Jian Gu.
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Mettlin C, Lee F, Drago J, Murphy GP. The American Cancer Society National Prostate Cancer Detection Project. Findings on the detection of early prostate cancer in 2425 men. Cancer. 1991;67:2949–2958. doi: 10.1002/1097-0142(19910615)67:12<2949::aid-cncr2820671202>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 3.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae SS, Boysen G, Auclair D, Onofrio RC, Park K, Kitabayashi N, MacDonald TY, Sheikh K, Vuong T, Guiducci C, Cibulskis K, Sivachenko A, Carter SL, Saksena G, Voet D, Hussain WM, Ramos AH, Winckler W, Redman MC, Ardlie K, Tewari AK, Mosquera JM, Rupp N, Wild PJ, Moch H, Morrissey C, Nelson PS, Kantoff PW, Gabriel SB, Golub TR, Meyerson M, Lander ES, Getz G, Rubin MA, Garraway LA. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao S, Geybels MS, Leonardson A, Rubicz R, Kolb S, Yan Q, Klotzle B, Bibikova M, Hurtado-Coll A, Troyer D, Lance R, Lin DW, Wright JL, Ostrander EA, Fan JB, Feng Z, Stanford JL. Epigenome-wide tumor DNA methylation profiling identifies novel prognostic biomarkers of metastatic-lethal progression in men diagnosed with clinically localized prostate cancer. Clin Cancer Res. 2017;23:311–319. doi: 10.1158/1078-0432.CCR-16-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455. doi: 10.1126/science.1083557. [DOI] [PubMed] [Google Scholar]
- 6.Breivik J, Gaudernack G. Genomic instability, DNA methylation, and natural selection in colorectal carcinogenesis. Semin Cancer Biol. 1999;9:245–254. doi: 10.1006/scbi.1999.0123. [DOI] [PubMed] [Google Scholar]
- 7.Woo HD, Kim J. Global DNA hypomethylation in peripheral blood leukocytes as a biomarker for cancer risk: a meta-analysis. PLoS One. 2012;7:e34615. doi: 10.1371/journal.pone.0034615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barchitta M, Quattrocchi A, Maugeri A, Vinciguerra M, Agodi A. LINE-1 hypomethylation in blood and tissue samples as an epigenetic marker for cancer risk: a systematic review and meta-analysis. PLoS One. 2014;9:e109478. doi: 10.1371/journal.pone.0109478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flanagan JM, Wilson A, Koo C, Masrour N, Gallon J, Loomis E, Flower K, Wilhelm-Benartzi C, Hergovich A, Cunnea P, Gabra H, Braicu EI, Sehouli J, Darb-Esfahani S, Vanderstichele A, Vergote I, Kreuzinger C, Castillo-Tong DC, Wisman GBA, Berns EM, Siddiqui N, Paul J, Brown R. Platinum-based chemotherapy induces methylation changes in blood DNA associated with overall survival in patients with ovarian cancer. Clin Cancer Res. 2017;23:2213–2222. doi: 10.1158/1078-0432.CCR-16-1754. [DOI] [PubMed] [Google Scholar]
- 10.Dugue PA, Bassett JK, Joo JE, Jung CH, Ming Wong E, Moreno-Betancur M, Schmidt D, Makalic E, Li S, Severi G, Hodge AM, Buchanan DD, English DR, Hopper JL, Southey MC, Giles GG, Milne RL. DNA methylation-based biological aging and cancer risk and survival: Pooled analysis of seven prospective studies. Int J Cancer. 2018;142:1611–1619. doi: 10.1002/ijc.31189. [DOI] [PubMed] [Google Scholar]
- 11.Dong L, Ren H. Blood-based DNA methylation biomarkers for early detection of colorectal cancer. J Proteomics Bioinform. 2018;11:120–126. doi: 10.4172/jpb.1000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ambatipudi S, Langdon R, Richmond RC, Suderman M, Koestler DC, Kelsey KT, Kazmi N, Penfold C, Ho KM, McArdle W, Ring SM, Pring M, Waterboer T, Pawlita M, Gaunt TR, Davey Smith G, Thomas S, Ness AR, Relton CL. DNA methylation derived systemic inflammation indices are associated with head and neck cancer development and survival. Oral Oncol. 2018;85:87–94. doi: 10.1016/j.oraloncology.2018.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joo JE, Dowty JG, Milne RL, Wong EM, Dugué PA, English D, Hopper JL, Goldgar DE, Giles GG, Southey MC kConFab. Heritable DNA methylation marks associated with susceptibility to breast cancer. Nat Commun. 2018;9:867. doi: 10.1038/s41467-018-03058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joyce BT, Zheng Y, Nannini D, Zhang Z, Liu L, Gao T, Kocherginsky M, Murphy R, Yang H, Achenbach CJ, Roberts LR, Hoxha M, Shen J, Vokonas P, Schwartz J, Baccarelli A, Hou L. DNA methylation of telomere-related genes and cancer risk. Cancer Prev Res (Phila) 2018;11:511–522. doi: 10.1158/1940-6207.CAPR-17-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dugue PA, Dowty JG, Joo JE, Wong EM, Makalic E, Schmidt DF, English DR, Hopper JL, Pedersen J, Severi G, MacInnis RJ, Milne RL, Giles GG, Southey MC. Heritable methylation marks associated with breast and prostate cancer risk. Prostate. 2018;78:962–969. doi: 10.1002/pros.23654. [DOI] [PubMed] [Google Scholar]
- 16.FitzGerald LM, Naeem H, Makalic E, Schmidt DF, Dowty JG, Joo JE, Jung CH, Bassett JK, Dugue PA, Chung J, Lonie A, Milne RL, Wong EM, Hopper JL, English DR, Severi G, Baglietto L, Pedersen J, Giles GG, Southey MC. Genome-wide measures of peripheral blood dna methylation and prostate cancer risk in a prospective nested case-control study. Prostate. 2017;77:471–478. doi: 10.1002/pros.23289. [DOI] [PubMed] [Google Scholar]
- 17.Barry KH, Moore LE, Sampson JN, Koutros S, Yan L, Meyer A, Reddy M, Oler AJ, Cook MB, Fraumeni JF Jr, Yeager M, Amundadottir LT, Berndt SI. Prospective study of DNA methylation at chromosome 8q24 in peripheral blood and prostate cancer risk. Br J Cancer. 2017;116:1470–1479. doi: 10.1038/bjc.2017.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han Y, Xu J, Kim J, Wu X, Gu J. Methylation of subtelomeric repeat D4Z4 in peripheral blood leukocytes is associated with biochemical recurrence in localized prostate cancer patients. Carcinogenesis. 2017;38:821–826. doi: 10.1093/carcin/bgx064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han Y, Xu J, Kim J, Wu X, Gu J. LINE-1 methylation in peripheral blood leukocytes and clinical characteristics and prognosis of prostate cancer patients. Oncotarget. 2017;8:94020–94027. doi: 10.18632/oncotarget.21511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011;6:692–702. doi: 10.4161/epi.6.6.16196. [DOI] [PubMed] [Google Scholar]
- 21.Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, Beck S. ChAMP: 450k chip analysis methylation pipeline. Bioinformatics. 2014;30:428–430. doi: 10.1093/bioinformatics/btt684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baron U, Werner J, Schildknecht K, Schulze JJ, Mulu A, Liebert UG, Sack U, Speckmann C, Gossen M, Wong RJ, Stevenson DK, Babel N, Schurmann D, Baldinger T, Bacchetta R, Grutzkau A, Borte S, Olek S. Epigenetic immune cell counting in human blood samples for immunodiagnostics. Sci Transl Med. 2018;10:eaan3508. doi: 10.1126/scitranslmed.aan3508. [DOI] [PubMed] [Google Scholar]
- 26.Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. 2016;17:551–565. doi: 10.1038/nrg.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zelic R, Fiano V, Grasso C, Zugna D, Pettersson A, Gillio-Tos A, Merletti F, Richiardi L. Global DNA hypomethylation in prostate cancer development and progression: a systematic review. Prostate Cancer Prostatic Dis. 2015;18:1–12. doi: 10.1038/pcan.2014.45. [DOI] [PubMed] [Google Scholar]