

Abstract
Resisting cell death is one of the hallmarks of cancer. Necroptosis is a form of non-caspase dependent necrotic cell death mediated by receptor-interacting protein kinase-1/3 (RIP1/3), which represents another mode of programmed cell death besides apoptosis. RIP3 also acts as an energy metabolism regulator associated with switching cell death from apoptosis to necroptosis. Trichothecin (TCN) is a sesquiterpenoid originating from endophytic fungi and shows potent anti-tumor bioactivity. Our current findings reveal that RIP3 mediates TCN-induced necroptosis through up-regulating PYGL and PDC-E1α to promote mitochondria energy metabolism and ROS production. RIP3 might be involved in sensitizing tumor cells to chemotherapy induced by TCN. Therefore, activating RIP3 to initiate necroptosis contributes to the bioactivity of TCN. Moreover, TCN could be exploited for therapeutic gain through up-regulating RIP3 to sensitize cancer chemotherapy.
Keywords: Trichothecin, necroptosis, RIP3, mitochondrial metabolism, ROS
Introduction
Resisting cell death is one of the hallmarks of cancer [1,2]. The development of acquired resistance to pro-apoptotic antitumor drugs is a major challenge for cancer chemotherapy [3]. To induce non-apoptotic cell death in tumor cells is considered as a promising approach to circumvent such resistance and would be developed as a supporting factor for chemotherapeutic drugs [4,5].
Cell death can be divided into several main forms in the light of morphological features and biochemical characteristics, such as apoptosis [6], autophagy [7], necrosis [8], necroptosis [9] and ferroptosis [10]. Programmed cell death is defined as a gene-controlled active cell suicide process [11-13]. Necroptosis is a form of caspase (cysteinyl aspartate specific proteinase)-independent programmed cell death. In this case, cells lose their plasma membrane integrity, leading to leakage of intracellular contents and increased permeability [14-17]. Hence, necroptosis shows morphological features of organelle swelling, such as mitochondria swelling, in addition to cytoplasmic swelling and vacuolation. Necroptotic cell death lacks of typical apoptotic features, such as membrane blebbing, chromatin condensation and intranucleosomal DNA fragmentation, but may show TUNEL positivity [18].
Receptor-Interacting Protein Kinase-3 (RIP3) belongs to the RIP family and is a RIP1 binding protein with homology to RIP1 and RIP2. It plays an essential role in tumor necrosis factor (TNF) or Toll-like receptors (TLRs)-mediated cell necrosis signaling pathway [19]. The N-terminus of RIP3 contains an active kinase domain (KD), which is conserved among other RIP kinases. Unlike the N-terminus, each RIP family member encodes a unique C-terminus that determines different signaling molecules would be recruited. RIP3 contains a C-terminal motif called RIP homotype interaction motif (RHIM) [20,21], which also exists in the bridging intermediate domain (ID) of RIP1 and mediates the interaction of RIP1 and RIP3 to induce necroptosis [22]. The RHIM-RHIM interaction between RIPK1 and RIPK3 results in the formation of necrosome, which is an oligomeric cytoplasmic complex. RIPK3 is subsequently activated and further recruit and phosphorylate mixed lineage kinase domain-like (MLKL) [23-26]. Consequently, MLKL oligomers move to the membrane and destroy its integrity through non-specific pore formation or interaction with TRPM7 calcium channel protein to promote calcium and sodium influx, resulting in necroptotic cell death [27,28].
Mitochondria play a critical role in regulating cell energy metabolism, signal transduction and cell death [29]. Among these, the most important function of mitochondria is to generate ATP through oxidative phosphorylation (OXPHOS) to power cells [30,31]. During the process of ATP production, the electrons may leave the electron transfer chain and hit surrounding molecules to generate reactive oxygen species (ROS) eventually [32,33], which is mainly in the form of superoxide radical O2•- and is subsequently converted to hydrogen peroxide [34-36]. With the enhancement of aerobic respiration, mitochondrial ROS level increases concomitantly [37]. Apart from its critical role in necroptotic process, RIP3 is also regarded as an energy metabolism regulator. Several key enzymes of metabolic pathways, such as glycogen phosphorylase (PYGL), glutamine ligase (GLUL), glutamate dehydrogenase 1 (GLUD1) and pyruvate dehydrogenase (PDC) are known targets of RIP3 [37,38]. PYGL catalyzes the degradation of glycogen by releasing glucose-1-phosphate for utilizing glycogen as an energy source [39]. GLUL is responsible for the condensation of glutamate (Glu) and ammonia to generate glutamine (Gln) and GLUD1 converts Glu to a-ketoglutarate. Both of them are essential for the use of amino acid Glu or Gln as substrates for energy production by means of OXPHOS [40,41]. PDC decarboxylates pyruvate into acetyl-CoA, which enters into the TCA cycle as a supply for OXPHOS to promote aerobic respiration [38]. RIP3 activates these downstream targets to increase aerobic respiration and its by-product ROS, which can drive necroptotic cell death [37,38].
Natural products provide a unique source for the discovery of innovative leading compounds and drugs, which exhibits promising pharmacological effects and anticancer activities through inducing cell death and enhancing chemotherapeutic sensitivity [4,42-44]. Trichothecin (TCN) is a natural secondary metabolite extracted from the endophytic fungi of Maytenus confertiflorus and it belongs to the family of trichothecene mycotoxins [45]. Previous studies of our and other groups have showed that TCN exerts a broad-spectrum anti-tumor activity through cell cycle arrest, proliferation inhibition, apoptotic induction and invasion impairment with less toxic side effects [46,47]. In this study, we found for the first time that TCN induces necroptosis in apoptosis-resistant cancer cells. Moreover, we have demonstrated that RIP3 mediates TCN-induced necroptosis through activating mitochondria energy metabolism and ROS production.
Materials and methods
Cell culture
Human colon cancer HT29 cell line (ATCC HTB-38) and Burkitt lymphoma Raji cell line (ATCC CCL-86) were cultured in RPMI-1640 media containing 10% (v/v) heat-inactivated fetal bovine serum (FBS), 1% w/v glutamine and 1% w/v antibiotics. Both the cell lines were cultured at 37°C in a humidified incubator containing 5% CO2.
Reagents and antibodies
z-VAD-fmk, a cell-permeable and irreversible pan-caspase inhibitor and cisplatin were from MedChemExpress (Monmouth Junction, NJ, USA). TCN was obtained from Kunming Institute of Botany, the Chinese Academy of Sciences (purity > 99%, HPLC analysis). Dimethyl sulphoxide (DMSO, Sigma) was used to dissolve TCN. The antibody against β-actin was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-RIP1 was from BD Biosciences (San Jose, CA, USA). Anti-RIP3 and anti-phosphorylated MLKL were from Abcam (Cambridge, MA, USA). The antibody against cleaved-Caspase 3 and Ki67 was obtained from Cell Signaling Technologies (Danvers, MA, USA).
Cell viability assay
The cell proliferation rate was assessed by plating cells into 96-well plates (2 × 103 cells per well) and absorption measurement was performed at 490 nm by a BioTek microplate reader (Beckman, Brea, CA, USA) with CCK-8 kit (Beyotime, China).
Cell permeability assay
Cell permeability assay was performed using the cell-impermeable dye Sytox Green (Invitrogen, USA). Briefly, cells were incubated with 30 nM Sytox Green dead cell stain for 10 min in the dark at room temperature and then imaged with a DMI3000 fluorescence microscope (Leica, Germany).
Transmission electron microscopy (TEM)
TEM was performed to identify the cells undergoing necroptosis. Cells were fixed with ice-cold 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) at 4°C for 4 h, then were post-fixed in 1% osmium tetroxide, dehydrated in graded alcohol and embedded in epoxy resin. The sections were counterstained with uranyl acetate and lead citrate and detected with a HT7700 TEM (Hitachi, Japan) at an accelerating voltage of 100 kV.
Immunofluorescence analysis
The cells were grown on glass coverslips overnight and treated with DMSO or TCN for 24 h, followed by fixation with 4% paraformaldehyde. After permeabilization with 0.3% Triton X-100 in PBS, the cells were blocked with 5% BSA for 1 h and then incubated with the primary antibody overnight. The coverslips were washed with PBS and then incubated with appropriate fluorescent secondary antibody for 1 h, followed by washing and mounting using DAPI. Images were obtained by confocal microscopy (TCS SP8, Leica) and quantified using the NIH Image J software.
Annexin V PE/7-ADD cell death detection
Annexin V PE/7-ADD apoptosis detection kit (4A Biotech, Beijing, China) was used to detect cell death. Cells were collected and successively incubated with Annexin V/PE and 7-AAD in PBS. Finally, samples were applied for flow cytometry (Becton-Dickinson, USA) and analyzed using FlowJo ver.10.0 software. The sum of Q1 and Q2 quadrants represents the rate of necroptotic cell death.
Measurement of the cellular oxygen consumption rate using the XF extracellular flux analyzer
Cells were plated in XF96 cell culture plates (Seahorse Bioscience, North Billerica, MA, USA) and incubated overnight. Cells were equilibrated with Seahorse XF base medium supplemented with glucose, glutamine, pyruvate and incubated into a 37°C non-CO2 incubator for 45 min to 1 h prior to the XF assay. Oligomycin was prepared for final concentrations of 1.5 μM, carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP), rotenone and antimycin A were prepared for a final concentration of 0.5 μM, and they were sequentially injected from the reagent ports automatically to the wells. Real-time measurements of oxygen consumption rate (OCR) in pmole per min for cells in culture medium were plotted over time. The OCR measurements were normalized to protein levels in each well.
Measurement of mitochondrial ROS accumulation
Cells were plated into 6-well plates and stained with 5 μM MitoSOX Red (Molecular Probes, Eugene, OR, USA) for 30 min at 37°C. Mitochondrial ROS content was detected by flow cytometry (Becton Dickinson, USA) and analyzed by flowjo ver.7.6 software. The fluorescence intensity reflected the amount of mitochondrial ROS and an increase in fluorescence intensity represents an enhanced generation of mitochondrial ROS.
Immunohistochemical analysis
The tumor tissue sections were deparaffinized in environmentally friendly dewaxing agent (solarbio, china) and rehydrated with an ethanol-aqueous solutions of decreasing concentrations. For antigen retrieval, tissue sections were incubated in 10 mM sodium citrate buffer (pH = 6.0) for 20 min in a microwave oven. The endogenous peroxidase activity was removed by incubating with 3% hydrogen peroxide for 10 min and was blocked in normal donkey serum for 30 min. The primary antibodies (anti-RIP3, anti-Ki67) were applied at 4°C overnight. Chromogen was developed using DAB (Zsgbbio, China) and counterstained with hematoxylin staining kit. Immunohistochemical staining of these sections was evaluated based on all of the available tumor cells or epithelial cells meeting the typical morphological criteria by 3 pathologists using the qualitative scale that is described in the literature. The number of cells stained was scored as 0 (no staining), 1 (< 1/3 positive cells), 2 (> 1/3 and < 2/3 positive cells) and 3 (> 2/3 positive cells). The intensity of staining ranged from 1 (weak) to 3 (strong). The immune reactive score was calculated by multiplying the percentage of positive cells and staining intensity.
Tumor xenograft studies
Xenograft experiment using HT29 cells was performed by subcutaneous injection of 3 × 106 cells into 5-week-old BALB/c nu/nu female mice. After the tumors grew to a volume of about 80-100 mm3, mice were randomly divided into four groups (n = 4 for each): untreated (vehicle), treatment with TCN (1 mg/kg) or DDP (1 mg/kg), treatment with TCN (0.5 mg/kg) combined with DDP (1 mg/kg) by intraperitoneal injection every day for 13 days, respectively. Tumor volume was calculated according to the formula (V = length × width2/2). At the end of experiments, the mice were euthanized by CO2 inhalation and the tumors were stripped and weighed. Animal care experimental procedures were conducted in accordance with the approval of Xiangya hospital of Central South University (Changsha, China).
Statistical analysis
Statistical analyses were all performed using ANOVA and two-tailed Student’s t test and a p value < 0.05 was considered statistically significant. Statistical calculations were performed using SPSS ver.16.0 software.
Results
TCN induces necroptosis in apoptosis-resistant cancer cells
In the presence of different concentrations of zVAD.fmk, a pan-caspase inhibitor, the protein levels of cleaved form of caspase-3 and PARP were substantially decreased in a dose-dependent manner both in HT29 and Raji cells, which indicate an effective inhibition of apoptotic pathway (Supplementary Figure 1A, 1B). Hence, the dose of 20 μM zVAD.fmk was adopted to pre-treat the cancer cells to mimic a prototypic model of apoptosis-resistance due to caspase pathway inhibition. To gain insight into the potential role of Trichothecin (TCN) in apoptosis-resistant cancer cells, firstly, CCK-8 assay was performed to investigate its effect on cell proliferation. Cells were pretreated with zVAD.fmk (20 μM) for 1 h, followed by TCN treatment for 24 h. The results showed that TCN treatment substantially inhibited the cell proliferation in apoptosis-resistant HT29 and Raji cells (Figure 1A, 1B). Sytox Green Nucleic Acid Stain is a high-affinity nucleic acid fluorescent probe that can easily penetrate cells with compromised plasma membrane, while be unable to across the membrane of live cells. We used sytox green to stain apoptosis-resistant HT29 and Raji cells upon TCN treatment and observed that green fluorescence was substantially intensified in a dose-dependent manner (Figure 1C). This suggests that TCN treatment significantly disrupts the cell membrane integrity. Accordingly, we further used TME to observe the morphology of TCN-induced cell death in apoptotic-resistant HT29 and Raji cells. Compared with the control group, TEM analysis showed necroptotic-like morphological changes in the TCN group such as mitochondrial swelling and plasma membrane rupture (Figure 1D). Taken together, these experiments indicate that TCN treatment might induce necroptotic cell death in apoptosis-resistant cancer cells.
Figure 1.
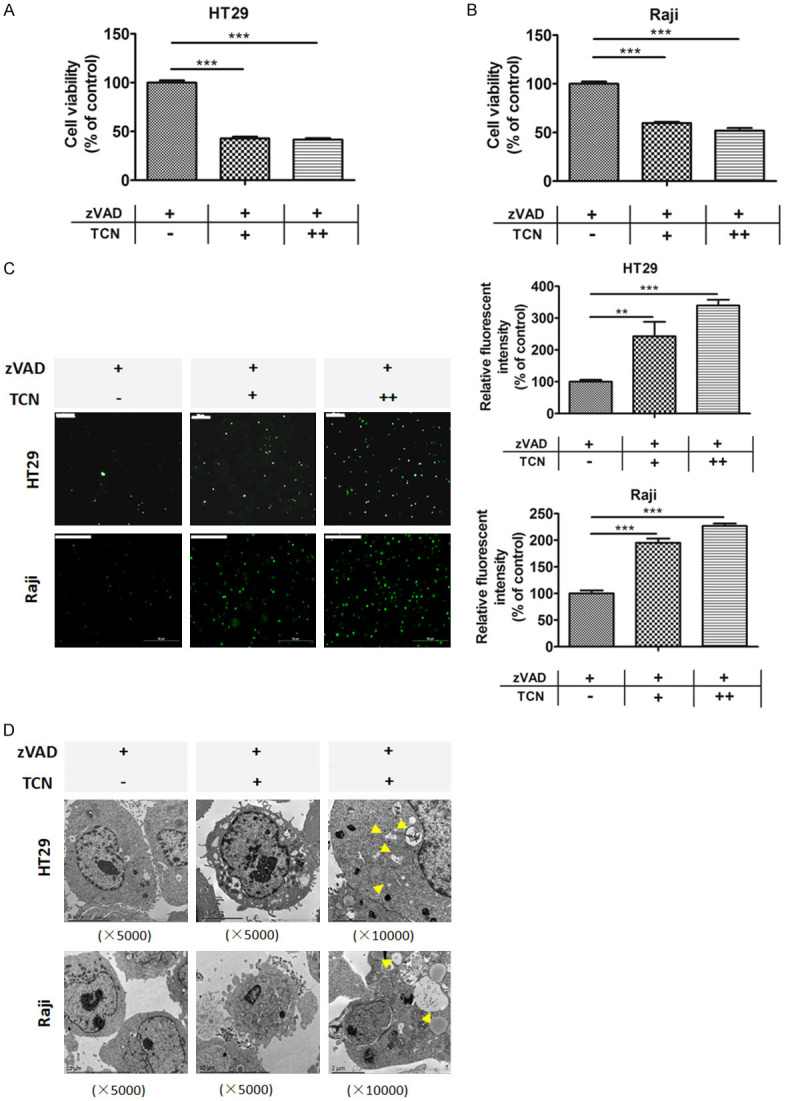
TCN induces necroptosis in apoptosis-resistant cancer cells. Cells were pretreated with zVAD.fmk (20 μM) for 1 h, followed by different dosages of TCN (0, 1, 2 μM) treatment for 24 h. Cell viability of (A) HT29 and (B) Raji cells were analyzed by CCK-8 assay. (C) Cells were stained with SYTOX Green Nucleic Acid Stain and imaged by fluorescent microscopy. The intensity of green fluorescence was analyzed using Image J software and shown in bar graphs. Scale bar, 50 μm. Data are shown as mean values ± S.D. of independent, triplicate experiments. The asterisks (**, ***) indicate significant differences (P < 0.01, P < 0.001, respectively). (D) Representative electron microscope photographs of HT29 and Raji cells pretreated with zVAD.fmk (20 μM) for 1 h, followed by TCN (0, 1 μM) treatment for 24 h. TCN treatment resulted in necroptotic cell death, which was featured by plasma membrane rupture, organelle swelling and vacuolation as indicated by yellow arrows in HT29 and Raji cells.
TCN up-regulates RIP3 upon caspase inhibition
In order to further clarify the underlying mechanism of TCN-induced necroptosis in apoptotic-resistant cancer cells, we examined both the mRNA and protein levels of RIP1 and RIP3 in HT29 and Raji cells. The results demonstrated that TCN treatment augmented the expression of RIP3 rather than RIP1 in a dose-dependent manner (Figure 2A, 2B). Moreover, the fluorescence intensity of RIP3 was enhanced upon TCN treatment (Figure 2C). Although the mRNA level was up-regulated, we did not observe coincident change on protein expression of RIP1 with TCN treatment (Supplementary Figure 2A, 2B). Similar results were confirmed by immunofluorescent assay (Supplementary Figure 2C). We further tested whether TCN treatment facilitated the interaction between RIPK1 and RIPK3. Computerized quantification of fluorescent co-localization between RIPK1 and RIPK3 showed no significant difference in the presence or absent of TCN (Supplementary Figure 2D), which suggests that TCN treatment might have no effect on the interaction between RIP1 and RIP3 as well as the RIP1/3 necrosome formation. These findings indicate that TCN might induce necroptosis through up-regulating RIP3, not RIP1 or the formation of RIP1/3 necrosomes. MLKL can be recruited and further be phosphorylated by RIP3, which is essential for executing necroptosis. We tested p-MLKL expression levels upon TCN treatment and observed that it was substantially augmented in a TCN dose-dependent manner both in HT29 and Raji cells (Figure 2B). Altogether, these data indicate that TCN treatment leads to up-regulation of RIP3, consequently recruits and phosphorylates downstream MLKL to induce necroptosis in apoptosis-resistant cancer cells, while RIP1 might not be involved in this event.
Figure 2.

TCN up-regulates RIP3 upon caspase inhibition. Cells were pretreated with zVAD.fmk (20 μM) for 1 h, followed by different dosages of TCN (0, 1, 2 μM) treatment for 24 h. A. The mRNA levels of RIPK3 were examined using real-time PCR in HT29 and Raji cells. B. The protein levels of RIPK3 and p-MLKL (S358) were detected by western blot assay in HT29 and Raji cells. β-actin was used as a loading control. C. HT29 cells were seeded on coverslips overnight and pretreated with zVAD.fmk (20 μM) for 1 h, followed by TCN (0, 1 μM) treatment for 24 h. The fluorescence of RIPK3 was detected by confocal microscopy and nuclei were stained blue. Quantification of the fluorescent intensity of RIPK3 imaging was shown as bar graphs. Scale bar, 25 μm. Data are shown as mean values ± S.D. of independent, triplicate experiments. The asterisks (*, **, ***) indicate significant differences (P < 0.05, P < 0.01, P < 0.001, respectively).
Silencing of RIP3 rescues cancer cells from TCN-induced necroptosis
RIP3 has emerged as a critical regulator of necroptosis. To further investigate the role of RIPK3 in TCN-induced necroptosis, four non-overlapping short hairpin RNAs targeting RIP3 (shRIP3 3#-6#) were transfected into HT29 cells, respectively. Western blot analysis confirmed that shRIP3 3# and 6# exhibited the best knock down efficiency of RIP3 (Supplementary Figure 3). CCK-8 assay was used to examine the cell proliferation of each designated group (Figure 3A). The results showed that TCN treatment significantly inhibited cell proliferation in apoptotic-resistant HT29 and Raji cells. However, knocking down of RIP3 restored cell viability suppressed by TCN treatment (Figure 3A, 3B). Sytox Green staining assay confirmed that TCN-induced necroptotic cell death was rescued by RIP3 knocking down (Figure 3C). Flow cytometry was further performed. Similarly, as shown in Figure 3D, TCN increased necroptotic cell death both in HT29 and Raji cells, which was reversed by the depletion of RIP3. These data suggest that silencing of RIP3 reduces the sensitivity of HT29 cells upon TCN treatment and protects cells from TCN-induced necroptotic cell death. Moreover, TEM analysis illustrated that TCN treatment induced cell death characterized by disrupt plasma membrane, mitochondrial swelling and vacuolation. Whereas, depletion of RIP3 substantially attenuated TCN-induced morphological changes both in apoptotic-resistant HT29 and Raji cells (Figure 3D). These findings support the notion that RIP3 mediates the TCN-induced necroptosis in apoptotic-resistant cancer cells.
Figure 3.

Silencing of RIP3 rescues cancer cells from TCN-induced necroptosis. Cells were pretreated with zVAD.fmk for 1 h, followed by different dosages of TCN (0 or 1 μM) treatment for 24 h. Cell viability of (A) HT29 shCON/shRIP3 (3# or 6#) and (B) Raji shCON/shRIP3 (3# or 6#) cells as designated in each group were analyzed by CCK-8 assay. (C) Cells were stained with SYTOX Green Nucleic Acid Stain and imaged by fluorescent microscopy. The intensity of green fluorescence was analyzed and shown in bar graphs. Scale bar, 50 μm. (D) Cells were stained using Annexin V PE/7-ADD apoptosis detection kit and applied to flow cytometry analysis. The sum of Q1 and Q2 quadrants was shown as bar graphs, which reflects the rate of necroptotic cell death. Data are shown as mean values ± S.D. of independent, triplicate experiments. The asterisks (**, ***) indicate significant differences (P < 0.01, P < 0.001, respectively). (E) Representative electron microscope photographs of HT29 shCON/shRIP3 and Raji shCON/shRIP3 cells as designated in each group.
RIP3 mediates TCN-induced necroptosis through activating mitochondria energy metabolism and ROS production
RIP3 acts as an energy metabolism regulator associated with switching cell death from apoptosis to necroptosis. In order to explore the downstream effector of RIP3, we screened the mRNA levels of RIP3 targets involved in energy metabolism, including PYGL, GLUL, GLUD1 and pyruvate dehydrogenase complex (PDC-E1α, E1β, E2, E3 subunits) upon TCN treatment in apotosis-resistant cancer cells. All of them are responsible for the use of energy substrates such as glucose, glutamate and glutamine, to boost aerobic respiration. Among them, PYGL and PDC-E1α mRNAs were substantially up-regulated induced by TCN both in HT29 (Figure 4A) and Raji cells (Figure 4B). In addition, the mRNA levels of PDC-E3 in HT29 and GLUL in Raji cells were also augmented with TCN treatment (Figure 4A, 4B). Moreover, knockdown of RIP3 led to substantial reduction of the mRNA levels of both PYGL and PDC-E1α (Figure 4C, 4D). These results indicate that RIP3 mediates the up-regulation of PYGL and PDC-E1α transcriptions induced by TCN in apotosis-resistant cancer cells.
Figure 4.

RIP3 mediates the up-regulation of PYGL and PDC-E1α induced by TCN. Cells were pretreated with zVAD.fmk for 1 h, followed by different dosages of TCN (0, 1 or 2 μM) treatment for 24 h. The mRNA levels of PYGL, GLUL, GLUD1 and PDC-E1α, E1β, E2, E3 genes were detected by real-time PCR both in (A) HT29 and (B) Raji cells. The mRNA levels of PYGL and PDC- E1α genes was examined by real-time PCR in (C) HT29 shCON/shRIP3 (3# or 6#) (D) Raji shCON/shRIP3 (3# or 6#) cells as designated in each group.
Since mitochondria play an important role in regulating cell energy metabolism and the energy substrates are metabolized to acetyl-CoA and enter into the Krebs cycle to boost OXPHOS, we further tested the change of oxygen consumption rate (OCR) in the absence or presence of TCN in apoptotic-resistant cancer cells. As expected, compared to the DMSO control, the basal respiration and maximal respiration were both enhanced upon TCN treatment (Figure 5A). However, knockdown of RIP3 (shRIP3 3#, 6#) reversed the up-regulation of OCRs induced by TCN treatment (Figure 5A). Considering that the mitochondrial respiratory chain consists of the main intracellular source of reactive oxygen species (ROS) in most tissues, the boosted mitochondrial energy metabolism should be accompanied by enhanced ROS production. Accordingly, we further tested mitochondrial ROS levels in the presence or absence of TCN in apoptotic-resistant cancer cells. The results illustrated that mitochondrial ROS was markedly augmented upon TCN treatment. Knock down of RIP3 substantially attenuated ROS production in the mitochondria (Figure 5B, 5C). Moreover, treatment with antioxidant, N-acetylcysteine (NAC) markedly rescued cell death induced by TCN (Figure 5D, 5E). Collectively, these findings support the conclusion that up-regulation of RIP3 to activate mitochondria energy metabolism and its by-product ROS is responsible for TCN to trigger necroptosis, especially when apoptosis is restricted in cancer cells.
Figure 5.

RIP3 mediates TCN-induced necroptosis through activating mitochondria energy metabolism and ROS production. HT29 shCON/shRIP3 cells were pretreated with zVAD.fmk for 1 h, followed by different dosages of TCN (0, 1 μM) treatment for 24 h. (A) Left, seahorse extracellular flux analyzer measurements of OCR metabolic profile using the mito stress cell assay. Traces shown were representative of two independent experiments in which each data point represents replicates of five wells. Data are shown as mean values ± S.D. Right, quantitative determination of basal and maximal OCR values of each designated group. Mitochondrial ROS levels of (B) HT29 and (C) Raji cells were determined by using MitoSOX Red, a specific mitochondrial probe, and detected by flow cytometry. Cell viability of (D) HT29 and (E) Raji cells were analyzed by CCK-8 assay. Data are shown as mean values ± S.D. of independent, triplicate experiments. The asterisks (*, **, ***) indicate significant differences (P < 0.05, P < 0.01, P < 0.001, respectively).
TCN sensitizes cisplatin (DDP) in tumor treatment in vivo
A xenograft tumor model using HT29 cells, which is a representative of the in vitro studies, was adopted to examine whether TCN would promote chemosensitivity in tumor treatment in vivo. Administration with DDP at a dose of 1 mg/kg had no effect on tumor growth (Figure 6A, 6B), which indicated that HT29 xenograft tumor was resistant to DDP treatment. In addition, DDP group showed a severe side effect with obvious weight loss of animals relative to the vehicle group (Figure 6C). However, the combination of DDP with TCN (0.5 mg/kg for each) achieved a significant inhibition of tumor load by 2-fold compared to the vehicle group (Figure 6A, 6B). Moreover, this regimen effectively ameliorated the side effect caused by administration of DDP alone (Figure 6C). Immunohistochemistry (IHC) staining of Ki67 showed a significant inhibition of tumor cell proliferation in DDP/TCN-treated group compared with vehicle or DDP group (Figure 6D). The up-regulation of RIP3 expression as well as phosphorylation level of MLKL in DDP/TCN-treated group further suggested that RIP3-mediated necroptotic cell death should contribute to the increased chemosensitivity of tumor cells (Figure 6D). Therefore, these results indicate that TCN might augment RIP3 expression to sensitize tumor cells to DDP chemotherapy.
Figure 6.
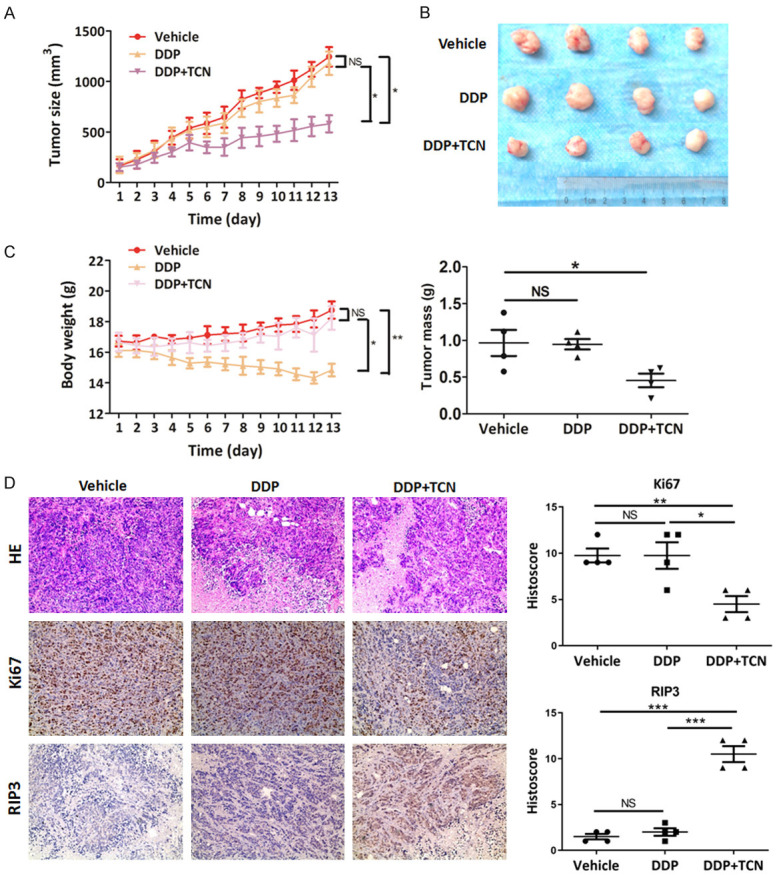
TCN sensitizes cisplatin in tumor treatment in vivo. A. Athymic BALB/c nude mice bearing HT29 cells were randomly separated into 4 groups (n = 4) and intraperitoneal administrated with corn oil (vehicle), TCN (1 mg/kg), DDP (1 mg/kg) or DDP combined with TCN (0.5 mg/kg each) every day for 13 days. Tumor volume was examined every day and shown in the graph. B. Upper, at the end of the experiment, the mice were sacrificed and the tumors were separated. Lower, tumor mass of each group was weighed and shown in the graph. C. During the experiment, body weight of the mice in each group was monitored and shown in the graph. D. Images of tumor sections in each group stained with hematoxylin-eosin (HE) and indicated antibodies. Antibody staining is in brown and nuclear counter staining is in blue. Scatter diagram shows Histoscore for the indicated antibody staining in tumor samples.
Discussion
Necroptosis is typically defined as a form of programmed necrotic cell death mediated by RIP1 and RIP3 kinases. RIPK1 and RIPK3 are regarded as central regulators, which are activated and form the necrosome complex through RHIM domain interaction to initiate necroptosis. In this paper, we have demonstrated that upon TCN treatment, the expression of RIP3 as well as the phosphorylation level of its downstream effector MLKL was substantially augmented in apoptosis-resistant cancer cells. Whereas, the change of RIP1 protein expression was not so evident. Moreover, the interaction between RIPK1 and RIPK3 can’t be enhanced by TCN treatment, which suggests that TCN treatment has no effect on the RIP1/3 necrosome formation. Although RIP1 plays an important regulatory role in the process of necrosis, it is not necessary for the occurrence of necroptosis. RIP3 can be activated by several stimuli to trigger necroptosis [48,49]. Lipopolysaccharide (LPS) or double-stranded RNA interacts with Toll-like receptor 3/4 via TRIF (TIR-domain-containing adapter-inducing interferon-β), which is a RHIM binding protein, to activate RIP3 directly [50]. During virus infection-induced necroptosis, a DNA-dependent activator of interferon-regulatory factors (DAI) interacts with RIP3 via RHIM to mimic the RIP1/3 necrosome complex, resulting cell death [51]. Moreover, RIP3 oligomerization can be enhanced by chemical agents to induce necroptosis in a RHIM domain-independent manner [52]. Thus, we conclude that TCN treatment might up-regulate RIP3 to induce necroptosis of apoptosis-resistant cancer cells in a RIP1-independent manner.
Cancer cells frequently display fundamentally altered energy metabolism, which is featured by increased aerobic glycolysis and reduced OXPHOS [53,54]. Here, we illustrated that TCN addition reactivated OXPHOS level to promote mitochondrial metabolism in apoptotic-resistant cancer cells. As an energy regulator, RIP3 mediates the action of TCN. PYGL and PDC are all downstream targets of RIP3, both of which are responsible for the utilization of carbon sources to boost OXPHOS. Through up-regulation of PYGL and PDC-E1α, RIP3 prompts aerobic respiration.
The enhanced respiration should be accompanied by accumulated ROS production. Growing evidence supports that ROS accumulation exerts the executioner and mediator of necroptosis [55-58]. ROS could change mitochondrial permeability, eventually resulting in necroptosis. Curcumol, a natural sesquiterpenoid hemiketal compound, induces necroptosis in hepatic stellate cells through JNK1/2-ROS signaling [57]. Mitochondrial ROS generation induced by C/EBP homologous protein promoted necroptotic cell death in human lung cancer cells [56]. In this study, we have demonstrated that concomitant ROS generation induced by OXPHOS augmentation facilitates necroptotic cell death and contributes to the anti-tumor activity of TCN. Moreover, RIP3 mediates the augmentation of ROS content induced by TCN.
Necroptosis might act as an alternative pathway to initiate cell death when apoptosis is restrained. Since RIP3 serves as a critical regulator of necroptosis, silencing RIP3 causes cancer cells to develop resistance to 5-FU [59,60], cisplatin [61-63], camptothecin and etoposide and the activation of key regulators in necroptotic pathway can increase the sensitivity of cells to chemotherapy drugs [5]. Here, we demonstrated that administration of TCN combination with cisplatin significantly increased the chemotherapeutic sensitivity of cancer cells in vivo.
Conclusions
Our study supports the view that TCN up-regulates RIP3 expression rather than RIP1 or the interaction between RIP1 and RIP3 forming necrosomes to induce necroptosis. RIP3 mediates this process through reprogramming of mitochondrial energy metabolism and ROS production as well as phosphorylation of MLKL (Figure 7). Thus, activating RIP3 to reprogram mitochondrial energy metabolism and redox homeostasis might be a novel strategy for intervention against cancer, especially in the case of apoptosis being restrained. TCN might facilitate therapeutic gain via single administration or combination with chemotherapy drugs by up-regulation of RIP3 to induce necroptotic cell death and is deserved for further exploration.
Figure 7.

Potential mechanism of RIP3-mediated necroptosis induced by TCN in cancer cells. In the case of apoptosis being restrained, TCN up-regulates RIPK3 to recruit and phosphorylate downstream MLKL; In the meanwhile, RIPK3 activates PYGL, PDC, etc to promote mitochondrial respiration and the accompanied ROS accumulation. These effects facilitate necroptosis induced by TCN and benefit for the chemotherapeutic sensitivity of cancer cells.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81874195, 81702720), the National Natural Science Foundation of Hunan (2020JJ4769), Clinical Medical Technology Innovation Guidance Project of China Hunan Provincial Science & Technology Department (2018SK50903), the Innovative Free Orientation Project of Central South University (No. 1053320190913). The ethics approval statements for animal work were provided by the Animal Care and Use Committee of Xiangya hospital in Central South University.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Baudino TA. Targeted cancer therapy: the next generation of cancer treatment. Curr Drug Discov Technol. 2015;12:3–20. doi: 10.2174/1570163812666150602144310. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Xu B, Xu M, Tian Y, Yu Q, Zhao Y, Chen X, Mi P, Cao H, Zhang B, Song G, Zhan YY, Hu T. Matrine induces RIP3-dependent necroptosis in cholangiocarcinoma cells. Cell Death Discov. 2017;3:16096. doi: 10.1038/cddiscovery.2016.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wattanathamsan O, Hayakawa Y, Pongrakhananon V. Molecular mechanisms of natural compounds in cell death induction and sensitization to chemotherapeutic drugs in lung cancer. Phytother Res. 2019;33:2531–2547. doi: 10.1002/ptr.6422. [DOI] [PubMed] [Google Scholar]
- 5.Moriwaki K, Bertin J, Gough PJ, Orlowski GM, Chan FK. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015;6:e1636. doi: 10.1038/cddis.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D’Orazi G. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY) 2016;8:603–619. doi: 10.18632/aging.100934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antunes F, Erustes AG, Costa AJ, Nascimento AC, Bincoletto C, Ureshino RP, Pereira GJS, Smaili SS. Autophagy and intermittent fasting: the connection for cancer therapy? Clinics (Sao Paulo) 2018;73(Suppl 1):e814s. doi: 10.6061/clinics/2018/e814s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vergara GA, Eugenio GC, Malheiros SMF, Victor EDS, Weinlich R. RIPK3 is a novel prognostic marker for lower grade glioma and further enriches IDH mutational status subgrouping. J Neurooncol. 2020;147:587–594. doi: 10.1007/s11060-020-03473-0. [DOI] [PubMed] [Google Scholar]
- 10.Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, Li B. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34. doi: 10.1186/s13045-019-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lockshin RA, Zakeri Z. Programmed cell death and apoptosis: origins of the theory. Nat Rev Mol Cell Biol. 2001;2:545–550. doi: 10.1038/35080097. [DOI] [PubMed] [Google Scholar]
- 12.Wallach D, Kang TB, Kovalenko A. Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat Rev Immunol. 2014;14:51–59. doi: 10.1038/nri3561. [DOI] [PubMed] [Google Scholar]
- 13.Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: toward identification of the effector molecules. Science. 2016;352:aaf2154. doi: 10.1126/science.aaf2154. [DOI] [PubMed] [Google Scholar]
- 14.Berman A, Winkelmann RK. Seborrheic keratoses: appearance in course of exfoliative erythroderma and regression associated with histologic mononuclear cell inflammation. Arch Dermatol. 1982;118:615–618. doi: 10.1001/archderm.118.8.615. [DOI] [PubMed] [Google Scholar]
- 15.Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, Cheng H, Jin K, Ni Q, Yu X, Liu C. The role of necroptosis in cancer biology and therapy. Mol Cancer. 2019;18:100. doi: 10.1186/s12943-019-1029-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Najafov A, Chen H, Yuan J. Necroptosis and cancer. Trends Cancer. 2017;3:294–301. doi: 10.1016/j.trecan.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14:48. doi: 10.1186/s12943-015-0321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou W, Yuan J. SnapShot: necroptosis. Cell. 2014;158:464–464. e1. doi: 10.1016/j.cell.2014.06.041. [DOI] [PubMed] [Google Scholar]
- 19.Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem. 2002;277:9505–9511. doi: 10.1074/jbc.M109488200. [DOI] [PubMed] [Google Scholar]
- 20.Zhang D, Lin J, Han J. Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol. 2010;7:243–249. doi: 10.1038/cmi.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 2013;27:1640–1649. doi: 10.1101/gad.223321.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Humphries F, Yang S, Wang B, Moynagh PN. RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ. 2015;22:225–236. doi: 10.1038/cdd.2014.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y, Gao H, Hu Y, Fang Y, Qi C, Huang J, Cai X, Wu H, Ding X, Zhang Z. High glucose-induced apoptosis and necroptosis in podocytes is regulated by UCHL1 via RIPK1/RIPK3 pathway. Exp Cell Res. 2019;382:111463. doi: 10.1016/j.yexcr.2019.06.008. [DOI] [PubMed] [Google Scholar]
- 25.Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. 2019;20:19–33. doi: 10.1038/s41583-018-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jun-Long H, Yi L, Bao-Lian Z, Jia-Si L, Ning Z, Zhou-Heng Y, Xue-Jun S, Wen-Wu L. Necroptosis signaling pathways in stroke: from mechanisms to therapies. Curr Neuropharmacol. 2018;16:1327–1339. doi: 10.2174/1570159X16666180416152243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnston A, Wang Z. Necroptosis: MLKL polymerization. J Nat Sci. 2018;4:e513. [PMC free article] [PubMed] [Google Scholar]
- 28.Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity. 2017;47:51–65. e7. doi: 10.1016/j.immuni.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Area-Gomez E, Guardia-Laguarta C, Schon EA, Przedborski S. Mitochondria, OxPhos, and neurodegeneration: cells are not just running out of gas. J Clin Invest. 2019;129:34–45. doi: 10.1172/JCI120848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zdralevic M, Vucetic M, Daher B, Marchiq I, Parks SK, Pouyssegur J. Disrupting the ‘Warburg effect’ re-routes cancer cells to OXPHOS offering a vulnerability point via ‘ferroptosis’-induced cell death. Adv Biol Regul. 2018;68:55–63. doi: 10.1016/j.jbior.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 31.Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem. 2017;292:7189–7207. doi: 10.1074/jbc.M116.774471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Curi R, Newsholme P, Marzuca-Nassr GN, Takahashi HK, Hirabara SM, Cruzat V, Krause M, de Bittencourt PI Jr. Regulatory principles in metabolism-then and now. Biochem J. 2016;473:1845–1857. doi: 10.1042/BCJ20160103. [DOI] [PubMed] [Google Scholar]
- 33.du Plessis SS, Agarwal A, Mohanty G, van der Linde M. Oxidative phosphorylation versus glycolysis: what fuel do spermatozoa use? Asian J Androl. 2015;17:230–235. doi: 10.4103/1008-682X.135123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newsholme P, Cruzat VF, Keane KN, Carlessi R, de Bittencourt PI Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem J. 2016;473:4527–4550. doi: 10.1042/BCJ20160503C. [DOI] [PubMed] [Google Scholar]
- 35.Li G, Qin Y. Mitochondrial translation factor EF4 regulates oxidative phosphorylation complexes and the production of ROS. Free Radic Res. 2018;52:1250–1255. doi: 10.1080/10715762.2018.1479063. [DOI] [PubMed] [Google Scholar]
- 36.Costa A, Scholer-Dahirel A, Mechta-Grigoriou F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin Cancer Biol. 2014;25:23–32. doi: 10.1016/j.semcancer.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 37.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 38.Yang Z, Wang Y, Zhang Y, He X, Zhong CQ, Ni H, Chen X, Liang Y, Wu J, Zhao S, Zhou D, Han J. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat Cell Biol. 2018;20:186–197. doi: 10.1038/s41556-017-0022-y. [DOI] [PubMed] [Google Scholar]
- 39.Favaro E, Bensaad K, Chong MG, Tennant DA, Ferguson DJ, Snell C, Steers G, Turley H, Li JL, Gunther UL, Buffa FM, McIntyre A, Harris AL. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab. 2012;16:751–764. doi: 10.1016/j.cmet.2012.10.017. [DOI] [PubMed] [Google Scholar]
- 40.Palmieri EM, Menga A, Martin-Perez R, Quinto A, Riera-Domingo C, De Tullio G, Hooper DC, Lamers WH, Ghesquiere B, McVicar DW, Guarini A, Mazzone M, Castegna A. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep. 2017;20:1654–1666. doi: 10.1016/j.celrep.2017.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu Z, Boggon TJ, Jin P, Yi H, Wright ER, Duong D, Seyfried NT, Egnatchik R, DeBerardinis RJ, Magliocca KR, He C, Arellano ML, Khoury HJ, Shin DM, Khuri FR, Kang S. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell. 2015;27:257–270. doi: 10.1016/j.ccell.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng C, Li Z, Zhao X, Liao C, Quan J, Bode AM, Cao Y, Luo X. Natural alkaloid and polyphenol compounds targeting lipid metabolism: treatment implications in metabolic diseases. Eur J Pharmacol. 2020;870:172922. doi: 10.1016/j.ejphar.2020.172922. [DOI] [PubMed] [Google Scholar]
- 43.Cheng C, Zhuo S, Zhang B, Zhao X, Liu Y, Liao C, Quan J, Li Z, Bode AM, Cao Y, Luo X. Treatment implications of natural compounds targeting lipid metabolism in nonalcoholic fatty liver disease, obesity and cancer. Int J Biol Sci. 2019;15:1654–1663. doi: 10.7150/ijbs.33837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo X, Yu X, Liu S, Deng Q, Liu X, Peng S, Li H, Liu J, Cao Y. The role of targeting kinase activity by natural products in cancer chemoprevention and chemotherapy (Review) Oncol Rep. 2015;34:547–554. doi: 10.3892/or.2015.4029. [DOI] [PubMed] [Google Scholar]
- 45.Su J, Zhao P, Kong L, Li X, Yan J, Zeng Y, Li Y. Trichothecin induces cell death in NF-kappaB constitutively activated human cancer cells via inhibition of IKKbeta phosphorylation. PLoS One. 2013;8:e71333. doi: 10.1371/journal.pone.0071333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo X, Li N, Zhao X, Liao C, Ye R, Cheng C, Xu Z, Quan J, Liu J, Cao Y. DHRS2 mediates cell growth inhibition induced by Trichothecin in nasopharyngeal carcinoma. J Exp Clin Cancer Res. 2019;38:300. doi: 10.1186/s13046-019-1301-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liao C, Li M, Li X, Li N, Zhao X, Wang X, Song Y, Quan J, Cheng C, Liu J, Bode AM, Cao Y, Luo X. Trichothecin inhibits invasion and metastasis of colon carcinoma associating with SCD-1-mediated metabolite alteration. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865:158540. doi: 10.1016/j.bbalip.2019.158540. [DOI] [PubMed] [Google Scholar]
- 48.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 49.Wang T, Jin Y, Yang W, Zhang L, Jin X, Liu X, He Y, Li X. Necroptosis in cancer: an angel or a demon? Tumour Biol. 2017;39:1010428317711539. doi: 10.1177/1010428317711539. [DOI] [PubMed] [Google Scholar]
- 50.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108:20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2019;26:564. doi: 10.1016/j.chom.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 52.Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 54.Luo X, Hong L, Cheng C, Li N, Zhao X, Shi F, Liu J, Fan J, Zhou J, Bode AM, Cao Y. DNMT1 mediates metabolic reprogramming induced by Epstein-Barr virus latent membrane protein 1 and reversed by grifolin in nasopharyngeal carcinoma. Cell Death Dis. 2018;9:619. doi: 10.1038/s41419-018-0662-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Basit F, van Oppen LM, Schockel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, Grefte S, Kopitz C, Heroult M, Hgm Willems P, Koopman WJ. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8:e2716. doi: 10.1038/cddis.2017.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma YM, Peng YM, Zhu QH, Gao AH, Chao B, He QJ, Li J, Hu YH, Zhou YB. Novel CHOP activator LGH00168 induces necroptosis in A549 human lung cancer cells via ROS-mediated ER stress and NF-kappaB inhibition. Acta Pharmacol Sin. 2016;37:1381–1390. doi: 10.1038/aps.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jia Y, Wang F, Guo Q, Li M, Wang L, Zhang Z, Jiang S, Jin H, Chen A, Tan S, Zhang F, Shao J, Zheng S. Curcumol induces RIPK1/RIPK3 complex-dependent necroptosis via JNK1/2-ROS signaling in hepatic stellate cells. Redox Biol. 2018;19:375–387. doi: 10.1016/j.redox.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu P, Hu S, Jin Q, Li D, Tian F, Toan S, Li Y, Zhou H, Chen Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018;16:157–168. doi: 10.1016/j.redox.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grassilli E, Narloch R, Federzoni E, Ianzano L, Pisano F, Giovannoni R, Romano G, Masiero L, Leone BE, Bonin S, Donada M, Stanta G, Helin K, Lavitrano M. Inhibition of GSK3B bypass drug resistance of p53-null colon carcinomas by enabling necroptosis in response to chemotherapy. Clin Cancer Res. 2013;19:3820–3831. doi: 10.1158/1078-0432.CCR-12-3289. [DOI] [PubMed] [Google Scholar]
- 60.Oliver Metzig M, Fuchs D, Tagscherer KE, Grone HJ, Schirmacher P, Roth W. Inhibition of caspases primes colon cancer cells for 5-fluorouracil-induced TNF-alpha-dependent necroptosis driven by RIP1 kinase and NF-kappaB. Oncogene. 2016;35:3399–3409. doi: 10.1038/onc.2015.398. [DOI] [PubMed] [Google Scholar]
- 61.Wu X, Wu MY, Jiang M, Zhi Q, Bian X, Xu MD, Gong FR, Hou J, Tao M, Shou LM, Duan W, Chen K, Shen M, Li W. TNF-alpha sensitizes chemotherapy and radiotherapy against breast cancer cells. Cancer Cell Int. 2017;17:13. doi: 10.1186/s12935-017-0382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roy A, Sarker S, Upadhyay P, Pal A, Adhikary A, Jana K, Ray M. Methylglyoxal at metronomic doses sensitizes breast cancer cells to doxorubicin and cisplatin causing synergistic induction of programmed cell death and inhibition of stemness. Biochem Pharmacol. 2018;156:322–339. doi: 10.1016/j.bcp.2018.08.041. [DOI] [PubMed] [Google Scholar]
- 63.Engel JB, Martens T, Hahne JC, Hausler SF, Krockenberger M, Segerer S, Djakovic A, Meyer S, Dietl J, Wischhusen J, Honig A. Effects of lobaplatin as a single agent and in combination with TRAIL on the growth of triple-negative p53-mutated breast cancers in vitro. Anticancer Drugs. 2012;23:426–436. doi: 10.1097/CAD.0b013e32834fb8ce. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.