

Abstract
Mitochondria share extensive evolutionary conservation across nearly all living species. This homology allows robust insights to be gained into pathophysiologic mechanisms and therapeutic targets for the heterogeneous class of primary mitochondrial diseases through the study of diverse in vitro cellular and in vivo animal models. Dramatic advances in genetic technologies, ranging from RNA interference to achieve graded knock-down of gene expression to CRISPR/Cas-based gene editing that yields a stable gene knock-out or targeted mutation knock-in, have enabled the ready establishment of mitochondrial disease models for a plethora of individual nuclear gene disorders. These models are complemented and extended by the use of pharmacologic inhibitor-based stressors to characterize variable degrees, onset, duration, and combinations of acute on chronic mitochondrial dysfunction in individual respiratory chain enzyme complexes or distinct biochemical pathways in mitochondria. Herein is described the rationale for, and progress made in, “therapeutic cross-training”, a novel approach meant to improve the validity and rigor of experimental conclusions when testing therapies by studying treatment effects in multiple, evolutionarily-distinct species, including C. elegans (invertebrate, worm), D. rerio (vertebrate, zebrafish), M. musculus (mammal, mouse), and/or human patient primary fibroblast cell line models of primary mitochondrial disease. The goal of these pre-clinical studies is to identify lead therapies from candidate molecules or library screens that consistently demonstrate efficacy, with minimal toxicity, in specific subtypes of mitochondrial disease. Conservation of in vitro and in vivo therapeutic effects of lead molecules across species has proven extensive, where molar concentrations found to be toxic or efficacious in one species are often consistent with therapeutic effects at similar doses seen in other mitochondrial disease models. Phenotypic outcome studies in all models are prioritized at the level of survival and function, to reflect the ultimate goal of developing highly potent therapies for human mitochondrial disease. Lead compounds that demonstrate significant benefit on gross phenotypes may be further scrutinized in these same models to decipher their cellular targets, mechanism(s), and detailed biochemical effects. High-throughput, automated technologic advances will be discussed that enable efficient, parallel screening in a diverse mitochondrial disease disorders and overarching subclasses of compounds, concentrations, libraries, and combinations. Overall, this therapeutic cross-training approach has proven valuable to identify compounds with optimal potency and safety profiles among major biochemical subtypes or specific genetic etiologies of mitochondrial disease. This approach further supports rational prioritization of lead compounds, target concentrations, and specific disease phenotypes, outcomes, and subgroups to optimally inform the design of clinical trials that test their efficacy in human mitochondrial disease subjects.
INTRODUCTION.
Primary mitochondrial disease (PMD) is a highly heterogeneous class of inherited diseases, both in causal genetic etiologies and in manifesting symptoms, which shares an impaired capacity to generate cellular energy through aerobic respiration (Rahman and Rahman 2018). The continually expanding number to now hundreds of unique genetic etiologies across both nuclear and mitochondrial genomes being identified (Frazier et al 2019; Falk 2020) has greatly improved understanding that impaired mitochondrial respiratory capacity is a common final pathway resulting from disruption in a wide array of upstream mitochondrial and cellular pathways (Zhang et al 2013; Zhang and Falk 2014). These core learnings imply that therapies may potentially target a range of physiologic levels (Gorman et al 2016), including genetic (Setten et al 2019; Doudna 2020; Mok et al 2020) or cell based strategies (Hsu et al 2016; Naeem and Sondheimer 2019) to correct individual genetic deficiencies (Jurkute et al 2019), cellular signaling network enhancers that boost endogenous mitochondrial content or functions (Zhang and Falk 2014; McCormack et al 2015), vitamin cofactors and supplements that enhance residual mitochondrial enzymatic functions (Parikh et al 2009; Barcelos et al 2020), antioxidants to mitigate oxidative stress through a variety of mechanisms (Polyak et al 2018), signaling or organelle modulators to target cytosolic translation and/or mitophagy to mitigate cellular proteotoxic stress (Peng et al 2015), and nutrients to enhance alternative energy production pathways (Falk 2018; Kuszak et al 2018). However, there has previously been no objective methodology to readily compare these approaches or pre-determine which therapeutic strategies or combinations are likely to be effective and tolerated in different individuals, phenotypes, biochemical subclasses, or specific genetic etiologies of mitochondrial disease. This challenge has directly contributed to the limited success of clinical trials to test candidate interventions in human mitochondrial disease patients (Pfeffer et al 2012), with no pre-clinical means broadly applicable beyond a single etiology to improve the focus of clinical trial designs to match specific therapeutic candidates with likely responders and prioritized outcomes.
As mitochondria share extensive evolutionary conservation in structure and function across nearly all living species, improved understanding of pathophysiologic mechanisms and therapeutic targets for the heterogeneous class of primary mitochondrial diseases may be obtained using a range of cellular (in vitro) and animal (in vivo) models (Kuszak et al 2018). Dramatic advances in genetic technologies across diverse species, ranging from RNA interference to achieve graded knock-down of gene expression (Kurreck 2009) to CRISPR/Cas-based gene editing that yields a stable gene knock-out or targeted mutation knock-in (Doudna and Charpentier 2014), have enabled the ability to readily establish mitochondrial disease animal and cell models for a plethora of individual nuclear gene disorders (Vergano et al 2014; Kuszak et al 2018). These models require careful phenotypic assessment to readily use for therapeutic screening, as they may show widely variable phenotypes under basal conditions. However, as human mitochondrial diseases are often characterized by sensitivity to stressors (nutritional, infectious, etc.) that causes acute decompensation and multi-organ progression, the utility of genetic-based mitochondrial disease models is complemented and extended by pharmacologic inhibitor-based stressor models to characterize variable degrees, onset, duration, and combinations of acute on chronic mitochondrial dysfunction in distinct respiratory chain complexes or biochemical pathways (Rea et al 2010; Byrnes et al 2018).
“Therapeutic cross-training” is a useful way to conceptualize the novel, pre-clinical research approach we have developed to enhance the validity and rigor of experimental conclusions when testing therapies for mitochondrial diease. Specifically, we seek to identify lead therapies from candidate molecule analyses, or broad-based drug or genetic library screens, that consistently demonstrate efficacy with minimal toxicity in specific mitochondrial disease subtypes and prioritized outcomes using multiple evolutionarily-distinct species that have complementary experimental strengths and physiologic relevance. Models frequently used by our research group include Caenorhabditis elegans (invertebrate, worm), Danio rerio (vertebrate, zebrafish), Mus musculus (mammal, mouse), and human patient primary fibroblast cell line models of primary mitochondrial disease (Rea et al 2010; Kuszak et al 2018). Of course, other models may also be considered that hold unique value, such as Escherichia coli (bacteria), Saccharomyces cerevisiae (yeast), and Drosophila melanogaster (invertebrate, fruit fly) (Rea et al 2010; Sen and Cox 2017; Malina et al 2018). Experimental conservation of in vitro and in vivo therapeutic effects for lead molecules across species has proven to be high, where molar concentrations found to be toxic or efficacious in one species are often consistent with therapeutic effects seen in other mitochondrial disease models (Peng et al 2015; Kwon et al 2017; Polyak et al 2018; Guha et al 2019). Phenotypic outcomes at the level of survival and function are prioritized in all models to reflect the ultimate goal of developing potent therapies for human mitochondrial disease that are aligned with regulatory agency requirements such as the Food and Drug Administration (Center for Drug Evaluation and Research 2015). Compounds showing significant phenotypic benefit may then warrant deeper characterization in these same models to decipher their cellular targets, mechanism(s), pharmacologic properties, and detailed biochemical effects (MacRae and Peterson 2015; Carretero et al 2017). High-throughput, automated technologic advances are reviewed that enable more efficient, parallel screening of multiple compounds, concentrations, libraries, and combinations in a wide range of distinct mitochondrial disease disorders and overarching subclasses. Overall, a therapeutic cross-training approach in pre-clinical animal and cellular models has proven valuable to identify lead compounds with optimal potency and safety profiles in different mitochondrial disease subtypes. It further supports rational prioritization of lead compounds, target concentrations, and specific disease phenotypes and subgroups to optimize the design of clinical trials, which ultimately represent the essential level of evidence necessary to demonstrate to determine their safety and efficacy for a target indication in human mitochondrial disease subjects.
Animal Model Considerations for Therapeutic Development in Mitochondrial Disease.
The classical therapeutic development pipeline for human disease commonly entails large-scale clinical trials in hundreds to thousands of homogenous patients to test a candidate intervention on a postulated clinically-meaningful and responsive outcome in a defined study population (Figure 1A). Unfortunately, this approach is highly costly, in the order of tens to hundreds of millions of dollars to design and perform Food and Drug Administration (FDA)-approved clinical trials and frequently unsuccessful, with fewer than 5% of candidate therapies obtaining regulatory approval based on demonstrated safety and efficacy on their primary outcome (Falk 2017). Mitochondrial diseases face additional challenges to this pipeline, with typically insufficient patient numbers to apply this classical paradigm, and extensive heterogeneity complicating assessment of validated outcome measures or biomarkers that are generalizable to all or even most patients (Karaa et al 2017). As an alternative approach to empiric design of clinical trials, pre-clinical models offer an objective means in which to first evaluate the therapeutic effects in a desired disease population (such as all molecularly-confirmed mitochondrial disease patients or a defined subset that matches anticipated therapeutic mechanisms), on specific gross phenotypic outcomes that may best inform subsequent impact on patient-important outcomes (such as survival, activity, or organ-level effect), and potentially identify a biomarker that may be informative in a clinical trial as a companion diagnostic (Figure 1A). Therapeutic mechanisms can also be further elucidated to guide a therapeutic development program, if desired, although typically regulatory agencies require evidence of safety rather than mechanism. Modeling a candidate therapy in multiple genetic models in a given species and across diverse species may add a further measure of utility in trial design, where showing benefit in one population but either harm or lack of efficacy in another subpopulation would directly inform specific clinical trial populations to include or exclude. Confirming therapeutic benefits of a specific therapy across diverse mitochondrial disease species adds weight to the likelihood that the restoration of cellular physiology achieved with that therapeutic approach is meaningful and worth pursuing further in human mitochondrial disease patients. Of course, it is critically important to recognize that unique medicinal chemistry challenges exist when making the leap of translating therapeutic approaches from animal models to humans, such as drug metabolism, delivery, and adverse effects, all of which must be carefully considered beyond therapeutic target alone. However, matching the right therapeutic target to the right patient for the right disease indication is the definition of precision medicine (Falk 2017; Ginsburg and Phillips 2018), represents an important goal to enable design of smaller trials with improved dosing consistency and less heterogeneity between patients in achieving the primary outcome (Peck 2018). This approach also improves safety and reduces drug development costs, by limiting the trial design to the most likely responders.
Figure 1. Mitochondrial disease therapeutic pipeline development.
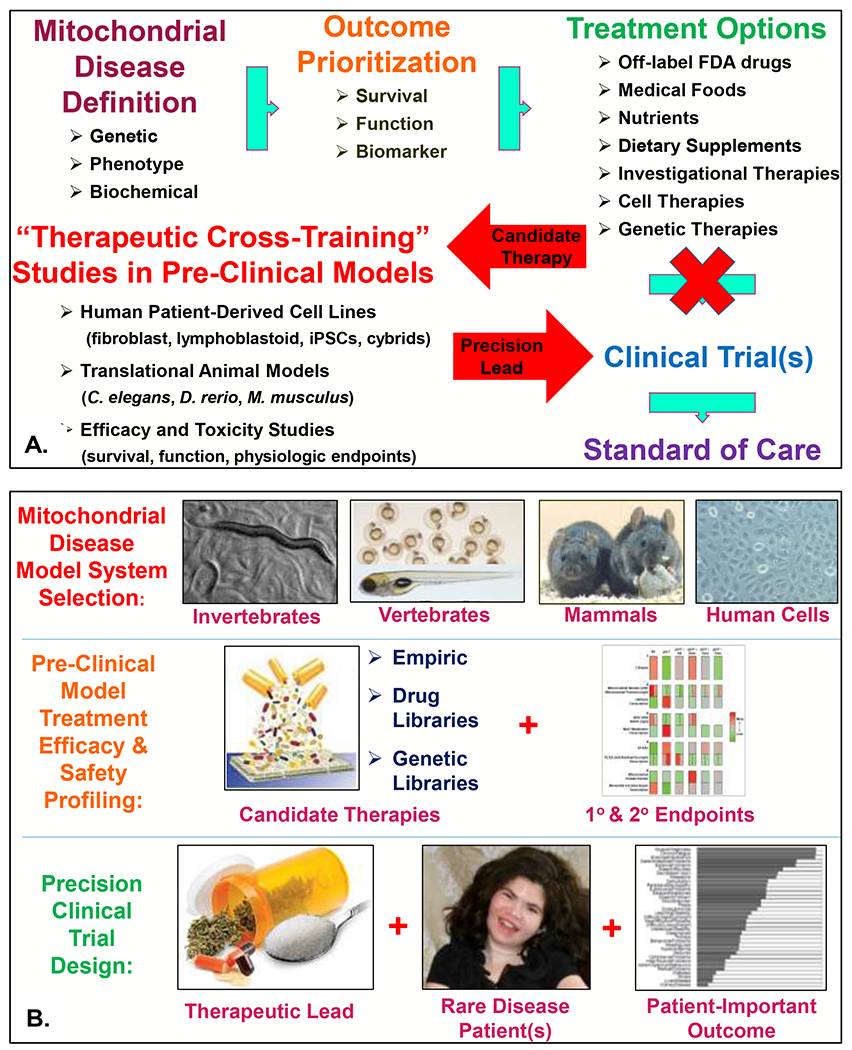
(A) The role of pre-clinical, translational research investigations in mitochondrial disease animal and cellular models to guide improved clinical trial design (modified from original publication) (Falk 2017). IPSCs, induced pluripotent stem cells. Cybrids, transmitochondrial hybrid lines that are the gold-standard for establishing the functional pathogenicity of mitochondrial DNA heteroplasmic variants (Giles et al 1980). (B) Schematic of ‘therapeutic cross-training” approach to support development of precision mitochondrial medicine therapies, utilizing C. elegans (worm), D. rerio (zebrafish), M. musculus (mouse), and H. sapiens (patient cells) to provide rigorous pre-clinical data in different species to optimize lead therapies and outcomes that support improved clinical trial design. The schematic in the middle right panel showing summarized interpretation of primary and secondary outcome measures assayed in a complex I C. elegans model summarizes data was originally published by our research group (McCormack et al 2015), as was the original data summarized in the bottom right panel indicating mitochondrial disease patient-prioritized outcomes (Zolkipli-Cunningham et al 2018). The patient displayed in the bottom center panel is a 16-year-old girl with FBXL4-based primary mitochondrial disease (Gai et al 2013), with photo shared per family consent per CHOP Institutional Review Board-approved study #08-6177 (Falk, PI).
In an effort to create a standard pipeline for pre-clinical therapeutic development in cell and animal models of mitochondrial disease, we have developed and utilize a “therapeutic cross-training” approach (Figure 1B). This model draws on genetic and pharmacologic models of diverse mitochondrial diseases that we and others have developed and extensively characterized over time, particularly in genes or processes that directly impair mitochondrial respiratory chain complex activities. While the details of all of the specific models and outcomes studied extends beyond the scope of this review (although an overview of general experimental outcomes is discussed below), the general approach employed to generate and study these models is highly similar. First, public repositories are screened for existing mutants to readily obtain, such as through the online Wormbase resource (Harris et al 2020) to identify worm strains that are available as frozen strains in Caenorhabditis genomics center (CGC) or other repositories for C. elegans, and the online Zfin resource (The Zebrafish Information Network) (Ruzicka et al 2019) to identify zebrafish strains that may be obtained in the form of frozen sperm from the Sanger repository for D. rerio. If these are not readily available or have been found to be embryonic lethal, then a range of other genetic approaches may be used to study a specific gene or pathway. In C. elegans, feeding RNA interference can be readily used to knock-down gene expression to variable degrees to generate large numbers of animals to study. More recently, CRISPR/Cas9 low-cost approaches have become commercially available to generate stable knock-out or knock-in alleles for a nuclear gene in several months’ time. For zebrafish, a range of genetic approaches may be used, where CRISPR/Cas9 is readily applied to generate knock-out alleles with high efficiency for nuclear genes over the course of 6-12 months, although generating targeted knock-in alleles can be more challenging. Mouse models of mitochondrial disease due to either nuclear gene or mitochondrial DNA gene mutations have been challenging to establish but are increasingly possible to generate and permit targeted analysis of individual candidate therapies (Inoue et al 2000; Tyynismaa and Suomalainen 2009; Wallace and Fan 2009; Ahola-Erkkila et al 2010; Yatsuga and Suomalainen 2012; Ruzzenente et al 2016). In addition, a large repository of primary human fibroblast cell lines and Epstein Bar Virus (EBV)-transformed lymphoblastoid cell lines from definite primary mitochondrial disease patients with molecularly-confirmed etiologies, as well as their unaffected family members, has been created at our Children’s Hospital of Philadelphia Mitochondrial Medicine Frontier Program through an approved institutional review board (IRB) study, and may also be accessible through a virtual repository organized by the North American Mitochondrial Disease Consortium (Rosales et al 2020). Collectively, these animal and cellular models across different species that can now be readily generated using a range of robust experimental methodologies have highly complementary utility to better understand the mechanisms and therapeutic targets in primary mitochondrial disease.
While regulatory agencies do not require evidence of therapeutic development in pre-clinical models to allow clinical trials to proceed, it may be highly informative to clinical trial design and outcomes to rigorously evaluate therapies first in relevant cellular and animal models. Although mice have long been considered an informative model when they exist to enable candidate drug modeling, it is widely acknowledged that many therapies shown promising in mice do not show similar benefit in humans both in general (Henderson et al 2019) and specifically in the case of mitochondrial disease (Ahola-Erkkila et al 2010; Yatsuga and Suomalainen 2012; Ahola et al 2016; Steele et al 2020). The same limitation may well hold true in other individual models, including worms, zebrafish, and human cells. However, the extensive homology of mitochondrial genomics, structure, and function across species supports the value of pursuing a “therapeutic cross-training” approach. Rather than studying treatment effects in only one disease model, this alternative approach sets a higher threshold for pre-clinical evidence by requiring efficacy and tolerability be demonstrated in at least one other species. Thus, identifying a therapy with a consistently promising therapeutic profile in multiple distinct species at specific dosing levels and on objectively measured outcomes can add rigor to prioritize it’s further evaluation in subsequent human clinical trials.
No single gold-standard model exists in which to test all mitochondrial disease therapies, given there are a range of different biochemical and genetic etiologies for which different therapeutic approaches may be indicated. When using any animal model, it is important to assure there is good homology in the given species for the desired therapeutic target in humans. However, it is highly important that any established models in which therapies are tested and published be accurately documented and/or deposited for general access in public repositories such as Wormbase for C. elegans, Zfin for zebrafish, and biobanks such as Coriell or the NIH North American Mitochondrial Disease Consortium (NAMDC) virtual fibroblast repository for human cell lines. This practice lowers the burden of individual investigators having to maintain mutant lines, strains and cells long-term, while making sure the models remain accessible for future comparative studies of additional therapies that may be under development by others to guide improved community understanding of optimal therapies for specific outcomes and types of mitochondrial disease. It would also be valuable when developing a new therapy in an established mitochondrial disease animal model to include previously published therapeutics as controls to allow replication and/or side-by-side comparison with novel therapeutic leads on specific outcomes that may guide their prioritization for further evaluation in human clinical trials
Candidate drug therapies for mitochondrial disease.
While mitochondrial biochemistry is complex (Gorman et al 2016), one simplified framework to consider for therapeutic target development in mitochondrial disease or dysfunction is to replenish the myriad of products that are directly generated by the respiratory chain (Figure 2A). In this model, major respiratory chain products that may become deficient in mitochondrial disease to the point of causing clinical symptoms and requiring therapeutic correction include (1) adenosine triphosphate (ATP), which requires alternative nutritional approaches that provide sufficient glucose to generate anaerobically through glycolysis; (2) oxidants in the form of superoxide and hydrogen peroxide, which disperse throughout the mitochondria and cell to cause oxidative damage of DNA, lipids and proteins, and require antioxidant therapies to mitigate their effect; (3) nicotinamide adenine dinucleotide in both reduced (NADH) and oxidized (NAD+) forms, where the reduction:oxidation (redox) ratio dictates the flux of hundreds of downstream cellular equilibrium reactions. Indeed, complex I is enzymatically named NADH dehydrogenase, conveying its central role to generate intra-mitochondrial NAD+, which when deficient may be replenished through NAD+ donating therapies in the nicotinic acid class (Zhang et al 2013; McCormack et al 2015); and (4) nucleic acids, including the pyrimidine, uridine (King and Attardi 1989) as well as the impaired mitochondrial salvage pathway for purines and pyrimidines that may be replaced with targeted nucleoside therapies (Wang 2016).
Figure 2. Candidate therapeutic approaches in primary mitochondrial disease.
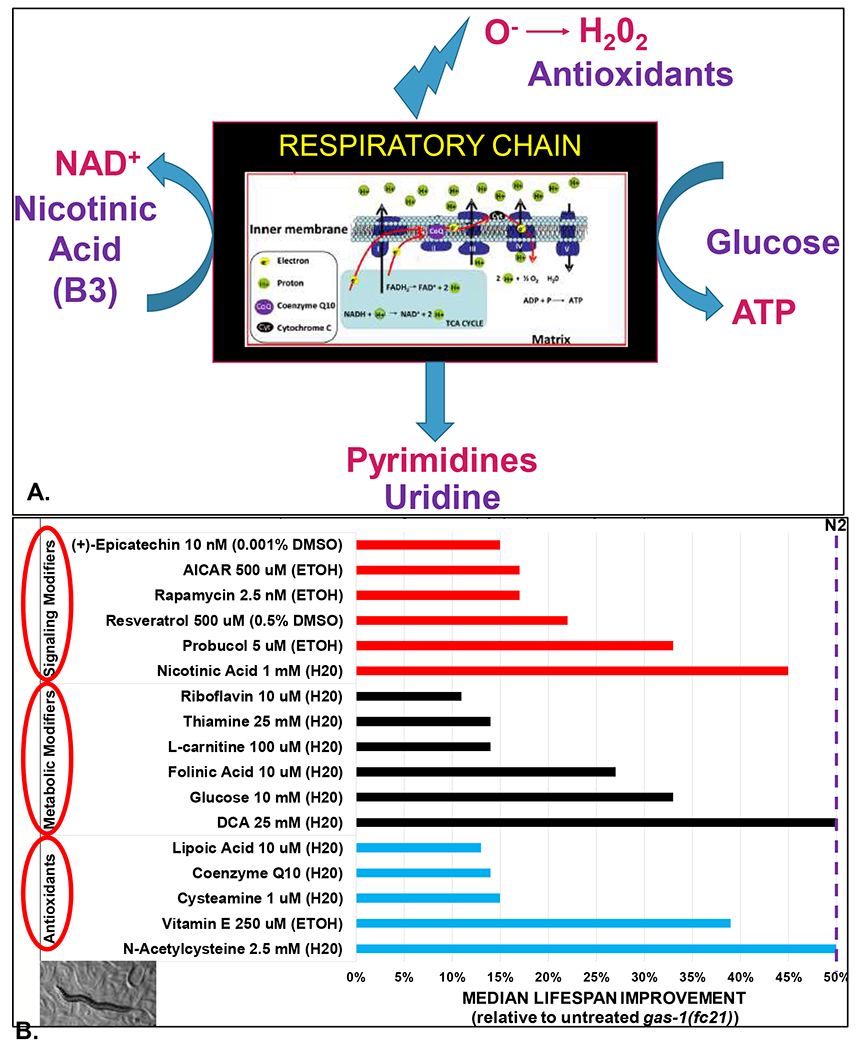
(A) Schematic of “black box product” therapies for mitochondrial disease. Major products generated by the mitochondrial respiratory chain include ATP, pyrimidines, NAD+, and oxidants (superoxide and hydrogen peroxide). To balance deficiencies of these products that occur in mitochondrial respiratory chain dysfunction, therapeutic classes to consider include nutrients (glucose) to optimize anaerobic glycolytic ATP generation in the cytosol, nucleic acids, NAD+ precursors, and antioxidants. (B) Lead drug classes and candidates identified through individual therapy modeling in the gas-1(fc21) C. elegans model of mitochondrial respiratory chain complex I disease that harbors a stable, homozygous missense mutation replacing a conserved arginine with a lysine in the NDUFS2 complex I subunit orthologue (Kayser et al 1999). Effective drugs were grouped into three general categories, namely antioxidants, metabolic modifiers, and signaling modifiers. The degree to which each drug restored shortened gas-1(fc21) animal lifespan toward that of wild-type (N2 Bristol) worms is shown, along with the maximally tolerated (or in some cases optimized) dose and buffer used for each compound. These C. elegans lifespan studies were performed manually and analyzed by a range of research scientists in the Falk Research laboratory at The Children’s Hospital of Philadelphia over many years, with results published in previous publications and/or performed by Julian Ostrovsky, MS (McCormack et al 2015; Polyak et al 2018; Guha et al 2019). Cysteamine bitartrate was provided by Raptor Therapeutics in an investigator-initiated therapy (Guha et al 2019). Dichloroacetate was provided for study by Dr. Peter Stacpoole at University of Florida and also independently obtained from Sigma. Epicatechin was provided by Dr. George Schreiner at Cardero Therapeutics (Falk et al, abstract presentation at 2014 Society of Inherited Metabolic Disease (SIMD) annual meeting). All other compounds were obtained from commercial sources, as previously described (McCormack et al 2015; Peng et al 2015; Kwon et al 2018; Polyak et al 2018; Guha et al 2019). Inset at bottom left depicts variable stage worms on a nematode growth media plate and tracks showing through the E. coli lawn, with a central adult work surrounded by visible eggs and larvae. H20, water. ETOH, ethanol. DMSO, dimethyl sulfoxide.
A wide range of therapies targeting these deficiencies as well as secondary biochemical and signaling changes throughout the cell have been postulated to have benefit in mitochondrial disease. While none have received FDA regulatory approval to date or been demonstrated to have individual benefit in randomized, double blind clinical trials of human mitochondrial disease patients, empiric use of several therapies with predicted benefit and generally good tolerability is commonly used in the care of mitochondrial disease patients (Parikh et al 2009; Barcelos et al 2020). Our research group has systematically tested dozens of these empirically-rationalized vitamin, supplement, cofactor, and repurposed therapies using a well-established C. elegans model of mitochondrial complex I disease, gas-1(fc21), which harbors a homozygous missense mutation in the NDUFS2 subunit of complex I (Kayser et al 1999). This mutant worm strain has 30% of wild-type complex I activity, reduced mitochondrial content and membrane potential, increased mitochondrial oxidative burden, reduced fecundity, delayed development, and significantly reduced lifespan (Kayser et al 2001; Falk et al 2006; Falk et al 2008; Dingley et al 2010; Kwon et al 2018). Therapeutic modeling of individual therapies in this model has demonstrated that at least 17 individual therapies significantly rescue, to variable extent, the short lifespan of these complex I disease worms (Figure 2B). These therapies are grossly classified into three major categories, namely antioxidants, metabolic modifiers, and signaling modifiers. Interestingly, therapeutic modeling of several agents has revealed there is extensive conservation of in vitro and in vivo therapeutic effects of lead molecules across species. Indeed, molar concentrations found to be toxic or efficacious in one species are often consistent with therapeutic effects at similar doses seen in other mitochondrial disease models. Indeed, previous work from our group has demonstrated this principle when comparing therapeutic effects on gas-1(fc21) worms’ lifespan with complex I disease model effects in other species. This has included survival under stress conditions and/or diverse aspects of mitochondrial of physiology or cellular transcriptome adaptations in human complex I disease patient fibroblast cells, and/or prevention of brain death and improvement of neuromuscular function, swimming activity, and/or survival in zebrafish models of complex I dysfunction for diverse candidate therapies including probucol, rapamycin, cysteamine bitartrate, N-acetylcysteine, vitamin E, and glucose (Falk et al 2011; Zhang et al 2013; McCormack et al 2015; Peng et al 2015; Byrnes et al 2018; Kwon et al 2018; Polyak et al 2018; Guha et al 2019). Additional mitochondrial disease therapies in these and other treatment classes are now under development by a range of biopharmaceutical companies, reflecting the growing recognition that mitochondrial disease will likely be amenable to a range of therapeutic approaches and an arsenal of therapies will be required to reach effective therapies for the diversity of mitochondrial etiologies and phenotypes.
High Throughput Drug Screen: Rationale and approach.
Therapeutic modeling approaches may vary with specific drug development goals, and the conservation of a specific pathway or gene in the relevant species. Phenotypic outcome studies in all models are prioritized at the level of survival and function, to reflect the ultimate goal of developing highly potent therapies for human mitochondrial disease that improve clinically meaningful outcomes at the level of survival, function, or feeling (Center for Drug Evaluation and Research 2015). Lead compounds that demonstrate significant benefit on gross phenotypes may be scrutinized in these same models to decipher their cellular targets, mechanism(s), pharmacologic properties, and detailed biochemical effects (MacRae and Peterson 2015; Carretero et al 2017). To facilitate high-throughput screening of multiple molecules in a drug class or large-scale library, developing and automating experimental analyses is important to enable efficient, parallel screening in a wide range of distinct mitochondrial disease disorders and overarching subclasses of multiple compounds, concentrations, libraries, and combinations (Figure 3).
Figure 3. Overview of high-throughput screening approach in C. elegans, zebrafish, and human cell models of primary mitochondrial disease.
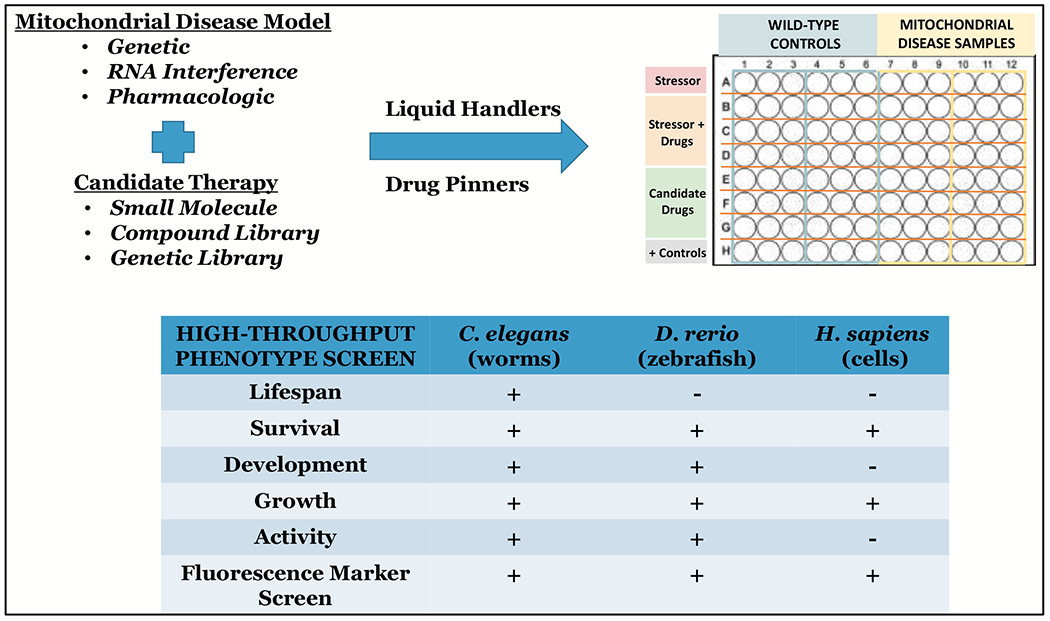
Precision mitochondrial medicine development is facilitated by matching a carefully-phenotyped primary mitochondrial disease model generated by any of multiple cutting-edge genetic or pharmacologic techniques with a candidate therapy or therapeutic library. Once conditions are established to evaluate a pathologic phenotype present in the model, high-throughput liquid handling and drug pinning systems can be efficiently harnessed to perform massively parallel evaluation in multi-well plate format of candidate therapies in cell and/or animal models of mitochondrial disease, with automated analyses to improve the efficiency and throughput of screens. In vivo phenotypes are prioritized for high-throughput screening in C. elegans and zebrafish that are indicative of lifespan, survival, development, growth, activity, and any of multiple different fluorescence-based indicators of mitochondrial or broader cellular pathophysiology. A range of human cells may also be amenable to high-throughput screens to assess in vitro survival, growth and proliferation, and fluorescence indicators of mitochondrial or cellular pathophysiology, with careful consideration given to growth conditions, cell passage, confluence, and expression of the relevant causal gene.
Although mitochondrial disease is defined by genetic and clinical heterogeneity, common outcomes prioritized for therapeutic targeting by mitochondrial disease patients include fatigue, exercise intolerance, muscle weakness, gastrointestinal disturbances, balance problems, and developmental delays (Zolkipli-Cunningham et al 2018). Therefore, we have prioritized development of high-throughput assays that start with rapid interrogation of gross phenotypic abnormalities in C. elegans and zebrafish models that include survival, growth and development, activity (at rest and when provoked), neuromuscular function, and growth and development. Each model requires careful assessment of its gross phenotypes and functions, such that consistent abnormalities can then be used to screen for therapeutic effect. A comparison of major assay types amenable to study in C. elegans, zebrafish, and human cells is shown in Figure 3. In C. elegans, the 2-week median lifespan of wild-type worms represents a particularly strong indicator of integrated physiologic effects, although classical lifespan measurement assays that require daily physical touching of each worm limits its experimental utility. This challenge has been addressed through the use of automated lifespan quantitation systems, which harness blue light technology in 24- to 240-well plate formats to stimulate worms to move with robotic systems to quantify the presence and degree of their response (Churgin et al 2017). This automated lifespan system improves experimental capacity by 10-fold to 100-fold in terms of the number of conditions whose impact on both lifespan and healthspan can be interrogated in parallel with minimal technician time. As wild-type zebrafish may survive on the order of 2-3 years, lifespan assays are not as readily performed in zebrafish as in C. elegans. However, survival assays can be an important and relatively rapid outcome for models that are shown to be short-lived. More generally useful in zebrafish are swimming activity assays, which correlate well with the prioritized mitochondrial disease patient outcomes of fatigue and exercise capacity. Semi-automated commercial systems are available that quantify fish swimming patterns and distance traveled over minutes to hours under basal or stressed conditions in a 96-well plate format that allows diverse therapies to be compared in zebrafish larvae (Graham et al 2016; Shen et al 2020).
In addition, some disorders have focused symptom burden in specific organs, such as metabolic strokes in the brain, cardiomyopathy or arrhythmias in the heart, and vision loss from retinal or optic nerve problems (Haas et al 2007; 2020). While C. elegans are useful for interrogation of integrated physiology at the level of development, fecundity, neuromuscular activity, and survival (O’Reilly et al 2014), zebrafish provide the advantage of being vertebrate animals with discrete organs including brain, eyes, otic vesicle, heart, liver, intestinal tract, skeletal muscle, and swim bladder. Targeted assays can be used to efficiently screen for dysfunction in all of these organs (Stewart et al 2014; Goessling and Sadler 2015; Scott et al 2016; Gut et al 2017; Byrnes et al 2018; Fontana et al 2018; Guha et al 2019; Shen et al 2020). Both models share the benefit of having large brood sizes that lend themselves to higher-throughput analyses. C. elegans are hermaphrodites (and can also sexually reproduce with males to enable targeted mating) that have a 3.5-day developmental cycle from egg through four larval developmental stages to reach adulthood and each adult yields approximately 300 offspring within the first 3 to 5 days of adulthood. Zebrafish develop externally follow egg fertilization through sexual reproduction, yield a fully formed larvae in 24 hours that lives off of a yolk-sac for 1 week before requiring feeding, and after reaching sexual maturity at 3-4 months post fertilization can be mated weekly to generate 100-200 offspring per mating pair. Such large brood sizes that can be rapidly generated greatly exceed that achieved with mammalian models, and represent a major benefit for high-throughput drug screening with large sample sizes and ease of generating replication datasets.
All three models discussed here also have the benefit of being transparent, allowing for a wide range of fluorescence analyses to be performed to interrogate targeted aspects of mitochondrial physiology, protein expression, and cellular biology. C. elegans are transparent throughout their lifecycle where they can ingest fluorescent dyes that target specific organelles or processes. They are also easily manipulated by genetic crossing to generate fluorescent marker lines that are stable and heritable between generations. Examples of C. elegans markers relevant for mitochondrial disease research include in vivo indicators of relative mitochondrial mass, membrane potential, oxidant burden, stress, and mitophagy induction, as well as markers for a host of other organelle, cell-type, and protein function that may be useful in specific scenarios for screening purposes (Kayser et al 2001; Silverman et al 2009; Dingley et al 2010; Luke and O’Reilly 2015; Kwon et al 2018; Thomas et al 2019). Zebrafish embryos and larvae are transparent when given a chemical to inhibit their pigment formation for the first week of life, allowing fluorescent dyes to be injected in the yolk sac that target different mitochondrial or cellular components, and are also readily amenable to a range of heritable fluorescent marker lines that convey similar in vivo aspects of mitochondrial and cellular physiology as described for C. elegans (Byrnes et al 2018; Liptak et al 2019). Human mitochondrial disease patient fibroblast and lymphoblastoid cells have been extensively used to functionally validate novel genetic causes and model therapies of mitochondrial disease, when the causal gene is expressed in the available cells. Outcomes readily assessed in human patient cells include survival assays that may use fluorescent dyes, and a range of mitochondrial-targeted fluorescence analyses to interrogate in vitro aspects of mitochondrial and broader cellular function (Dingley et al 2012; Saada 2014; Kong et al 2018). Fluorescence analyses in each of these models may all be done at the individual cell or animal level for assay development and validation purposes, but then are readily amenable to high-throughput image analysis using 96-well plate readers, flow-sorting methodologies, and/or high-content imaging systems (Silverman et al 2009; Dingley et al 2012; Kwon et al 2018). Further, multiple fluorescent markers can be combined, to facilitate screen of a given compound set or drug library on simultaneous outcomes that may inform optimal selection, such as mitochondrial content and membrane potential or mitophagy and oxidative stress.
For any drug screen, careful attention is required to include appropriate positive and negative controls, a wide dynamic range, and a validated assay that will reliably inform the intended outcome (Ding et al 2017). Additional analyses can be performed to validate therapeutic leads that are prioritized at different doses and exposure time courses based on results of one or more phenotypic or in vivo fluorescence screen across the different mitochondrial disease species models. Common read-outs used to better characterize therapeutic effects on specific aspects of mitochondrial dysfunction following high-throughput screens include ex vivo targeted analyses in whole animals, organs, tissues or cells. These may include biochemical read-outs of mitochondrial respiratory chain oxidative phosphorylation function by extracellular flux or high-resolution respirometry methodologies, spectrophotometric analyses of respiratory chain enzymatic capacities, blue native gel analyses of respiratory chain supercomplexes, high performance liquid chromatography (HPLC) methods to quantify specific mitochondrial or cellular metabolites, genomic analyses of transcriptome expression, proteomic analyses, metabolomic analyses, stable isotope-based analyses of flux through specific intermediary metabolic pathways, as well as a variety of histologic and imaging analyses of mitochondrial structure, function, and dynamics (Falk et al 2009; Zhang et al 2013; O’Reilly et al 2014; Vergano et al 2014; McCormack et al 2015; Peng et al 2015; Enns and Cowan 2017; Byrnes et al 2018; Connolly et al 2018; Kong et al 2018; Polyak et al 2018; Guha et al 2019).
CONCLUSION.
In summary, “therapeutic cross-training” across diverse mitochondrial disease animal and cell model species that have complementary experimental strengths and physiologic relevance offers a useful pre-clinical research approach to improve the rigor and efficiencies of developing precision mitochondrial medicines. The goal of these studies is to identify lead therapies from candidate molecule analyses, or broad-based drug or genetic library screens, that consistently demonstrate efficacy with minimal toxicity in specific mitochondrial disease subtypes and prioritized outcomes across well characterized and evolutionarily-distinct mitochondrial disease models that lend themselves to high-throughput discoveries. The high conservation of mitochondrial genomics, structure, and function serves as strong rationale for pursuit of this translational modeling approach, with the goal to identify compounds having optimal potency and safety profiles among major biochemical subtypes or specific genetic etiologies of mitochondrial disease consistently in more than one species. Validation of therapeutic leads in multiple species and disease subtypes adds multiple layers of objective evidence to identify and derisk therapeutic leads and support their rational prioritization, target concentration selection, and specific disease phenotypes, outcomes, companion biomarkers, and subgroups to focus further clinical drug development. Ultimately, the long-term goal of this work is to accelerate the pace and potency of therapeutic lead discovery to optimally inform precision clinical trial design that will test their efficacy in targeted small cohorts of human subjects with primary mitochondrial disease.
Informed Consent.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study. Additional informed consent was obtained from all patients for which identifying information is included in this article.
Animal Studies.
This article does not contain any studies with human or animal subjects performed by the author.
ACKNOWLEDGEMENTS.
I am deeply grateful to the many mitochondrial disease patients and families, research scientists, and lifescience companies who partner with us to provide inspiration, guide research priorities, develop therapies, and work tirelessly to perform research that advances our collective goal to develop safe and effective therapies to improve the lives of patients living with mitochondrial disease.
FUNDING.
This work was funded in part by generous philanthropic donations from mitochondrial disease patient families, the Mitochondrial Medicine Frontier Program at The Children’s Hospital of Philadelphia, Raptor Pharmaceuticals (to study cysteamine bitartrate in pre-clinical animal models), and the National Institutes of Health (R01-HD065858, R01-GM120762, and R35-GM134863). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
CONFLICTS OF INTEREST.
Marni J. Falk, MD (MJF) is co-inventor on International Patent Application No. PCT/US19/39631 based on U.S. Serial No. 62/690,718 filed on 6/27/2018 and U.S. Serial No. 62/830,850 filed on 4/8/2019 Entitled, “Compositions and Methods for Treatment of Mitochondrial Respiratory Chain Dysfunction and Other Mitochondrial Disorders and Methods for Identifying Efficacious Agents for the Alleviating Symptoms of the Same,” filed in the Name of The Children’s Hospital of Philadelphia on 6/27/2019. MJF is a co-founder of MitoCUREia, Inc, scientific advisory board member with equity interest in RiboNova, Inc., and scientific board member as paid consultant with Khondrion (active) and Larimar Therapeutics (pending). MJF has previously been or is currently engaged with several companies involved in mitochondrial disease therapeutic pre-clinical and/or clinical stage development as a paid consultant (Astellas (formerly Mitobridge) Pharma Inc, Cyclerion Therapeutics, Imel Therapeutics, NeuroVive, Reneo Therapeutics, Stealth BioTherapeutics) and/or a sponsored research collaborator (AADI Therapeutics, Cardero Therapeutics, Cyclerion Therapeutics, Minovia Therapeutics Inc, Mission Therapeutics, NeuroVive, Raptor Therapeutics, REATA Inc., RiboNova Inc, Standigm Therapeutics, Stealth BioTherapeutics).
REFERENCES.
- Ahola-Erkkila S, Carroll CJ, Peltola-Mjosund K, et al. (2010) Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum Mol Genet 19: 1974–1984. [DOI] [PubMed] [Google Scholar]
- Ahola S, Auranen M, Isohanni P, et al. (2016) Modified Atkins diet induces subacute selective ragged-red-fiber lysis in mitochondrial myopathy patients. EMBO Mol Med 8: 1234–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcelos I, Shadiack E, Ganetzky R, Falk MJ (2020) Mitochondrial Medicine Therapies: Rationale, Evidence, and Dosing Guidelines. Curr Opin Pediatr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes J, Ganetzky R, Lightfoot R, et al. (2018) Pharmacologic modeling of primary mitochondrial respiratory chain dysfunction in zebrafish. Neurochem Int 117: 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero M, Solis GM, Petrascheck M (2017) C. elegans as Model for Drug Discovery. Curr Top Med Chem 17: 2067–2076. [DOI] [PubMed] [Google Scholar]
- Center for Drug Evaluation and Research OoTS, Food and Drug Administration (2015) FDA Memorandum: Critical Path Innovation Meeting Regarding Drug Development for Mitochondrial Diseases. In Editor ed.^eds. Book FDA Memorandum: Critical Path Innovation Meeting Regarding Drug Development for Mitochondrial Diseases Office of Dietary Supplements. [Google Scholar]
- Churgin MA, Jung SK, Yu CC, Chen X, Raizen DM, Fang-Yen C (2017) Longitudinal imaging of Caenorhabditis elegans in a microfabricated device reveals variation in behavioral decline during aging. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly NMC, Theurey P, Adam-Vizi V, et al. (2018) Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ 25: 542–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M, Kaspersson K, Murray D, Bardelle C (2017) High-throughput flow cytometry for drug discovery: principles, applications, and case studies. Drug Discov Today 22: 1844–1850. [DOI] [PubMed] [Google Scholar]
- Dingley S, Chapman KA, Falk MJ (2012) Fluorescence-activated cell sorting analysis of mitochondrial content, membrane potential, and matrix oxidant burden in human lymphoblastoid cell lines. Methods Mol Biol 837: 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingley S, Polyak E, Lightfoot R, et al. (2010) Mitochondrial respiratory chain dysfunction variably increases oxidant stress in Caenorhabditis elegans. Mitochondrion 10: 125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA (2020) The promise and challenge of therapeutic genome editing. Nature 578: 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346: 1258096. [DOI] [PubMed] [Google Scholar]
- Enns GM, Cowan TM (2017) Glutathione as a Redox Biomarker in Mitochondrial Disease-Implications for Therapy. J Clin Med 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MJ (2017) Chapter 3. Case Studies in Precision Drug Development: Using Genetics in Clinical Trials for Mitochondrial Disease. Book Chapter 3. Case Studies in Precision Drug Development: Using Genetics in Clinical Trials for Mitochondrial Disease. Washington, D.C.: The National Academies Press, 37–42. [Google Scholar]
- Falk MJ (2018) Nutritional Inadequacies In Mitochondrial-associated Metabolic Disorders. In Editor ed.êds. Book Nutritional Inadequacies In Mitochondrial-associated Metabolic Disorders Washington, D.C.: The National Academies Press, 24–30. [Google Scholar]
- Falk MJ (2020) Mitochondrial Disease Genes Compendium: From Genes to Clinical Manifestations, London, UK: Elsevier Academic Press. [Google Scholar]
- Falk MJ, Kayser EB, Morgan PG, Sedensky MM (2006) Mitochondrial complex I function modulates volatile anesthetic sensitivity in C. elegans. Curr Biol 16: 1641–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MJ, Polyak E, Zhang Z, et al. (2011) Probucol ameliorates renal and metabolic sequelae of primary CoQ deficiency in Pdss2 mutant mice. EMBO Mol Med 3: 410–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MJ, Rosenjack JR, Polyak E, et al. (2009) Subcomplex Ilambda specifically controls integrated mitochondrial functions in Caenorhabditis elegans. PLoS One 4: e6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MJ, Zhang Z, Rosenjack JR, et al. (2008) Metabolic pathway profiling of mitochondrial respiratory chain mutants in C. elegans. Mol Genet Metab 93: 388–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana BD, Mezzomo NJ, Kalueff AV, Rosemberg DB (2018) The developing utility of zebrafish models of neurological and neuropsychiatric disorders: A critical review. Exp Neurol 299: 157–171. [DOI] [PubMed] [Google Scholar]
- Frazier AE, Thorburn DR, Compton AG (2019) Mitochondrial energy generation disorders: genes, mechanisms, and clues to pathology. J Biol Chem 294: 5386–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai X, Ghezzi D, Johnson MA, et al. (2013) Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am J Hum Genet 93: 482–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles RE, Stroynowski I, Wallace DC (1980) Characterization of mitochondrial DNA in chloramphenicol-resistant interspecific hybrids and a cybrid. Somatic Cell Genet 6: 543–554. [DOI] [PubMed] [Google Scholar]
- Ginsburg GS, Phillips KA (2018) Precision Medicine: From Science To Value. Health Aff (Millwood) 37: 694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goessling W, Sadler KC (2015) Zebrafish: an important tool for liver disease research. Gastroenterology 149: 1361–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman GS, Chinnery PF, DiMauro S, et al. (2016) Mitochondrial diseases. Nat Rev Dis Primers 2: 16080. [DOI] [PubMed] [Google Scholar]
- Graham S, Rogers RP, Alper RH (2016) An automated method to assay locomotor activity in third instar Drosophila melanogaster larvae. J Pharmacol Toxicol Methods 77: 76–80. [DOI] [PubMed] [Google Scholar]
- Guha S, Konkwo C, Lavorato M, et al. (2019) Pre-clinical evaluation of cysteamine bitartrate as a therapeutic agent for mitochondrial respiratory chain disease. Hum Mol Genet 28: 1837–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut P, Reischauer S, Stainier DYR, Arnaout R (2017) Little Fish, Big Data: Zebrafish as a Model for Cardiovascular and Metabolic Disease. Physiol Rev 97: 889–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas RH, Parikh S, Falk MJ, et al. (2007) Mitochondrial disease: a practical approach for primary care physicians. Pediatrics 120: 1326–1333. [DOI] [PubMed] [Google Scholar]
- Harris TW, Arnaboldi V, Cain S, et al. (2020) WormBase: a modern Model Organism Information Resource. Nucleic Acids Res 48: D762–D767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson CJ, Kapelyukh Y, Scheer N, et al. (2019) An Extensively Humanized Mouse Model to Predict Pathways of Drug Disposition and Drug/Drug Interactions, and to Facilitate Design of Clinical Trials. Drug Metab Dispos 47: 601–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YC, Wu YT, Yu TH, Wei YH (2016) Mitochondria in mesenchymal stem cell biology and cell therapy: From cellular differentiation to mitochondrial transfer. Semin Cell Dev Biol 52: 119–131. [DOI] [PubMed] [Google Scholar]
- Inoue K, Nakada K, Ogura A, et al. (2000) Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat Genet 26: 176–181. [DOI] [PubMed] [Google Scholar]
- Jurkute N, Harvey J, Yu-Wai-Man P (2019) Treatment strategies for Leber hereditary optic neuropathy. Curr Opin Neurol 32: 99–104. [DOI] [PubMed] [Google Scholar]
- Karaa A, Rahman S, Lombes A, et al. (2017) Common data elements for clinical research in mitochondrial disease: a National Institute for Neurological Disorders and Stroke project. J Inherit Metab Dis 40: 403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser EB, Morgan PG, Hoppel CL, Sedensky MM (2001) Mitochondrial expression and function of GAS-1 in Caenorhabditis elegans. J Biol Chem 276: 20551–20558. [DOI] [PubMed] [Google Scholar]
- Kayser EB, Morgan PG, Sedensky MM (1999) GAS-1: a mitochondrial protein controls sensitivity to volatile anesthetics in the nematode Caenorhabditis elegans. Anesthesiology 90: 545–554. [DOI] [PubMed] [Google Scholar]
- King MP, Attardi G (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246: 500–503. [DOI] [PubMed] [Google Scholar]
- Kong J, Peng M, Ostrovsky J, et al. (2018) Mitochondrial function requires NGLY1. Mitochondrion 38: 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurreck J (2009) RNA interference: from basic research to therapeutic applications. Angew Chem Int Ed Engl 48: 1378–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszak AJ, Espey MG, Falk MJ, et al. (2018) Nutritional Interventions for Mitochondrial OXPHOS Deficiencies: Mechanisms and Model Systems. Annu Rev Pathol 13: 163–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YJ, Falk MJ, Bennett MJ (2017) Flunarizine rescues reduced lifespan in CLN3 triple knock-out Caenorhabditis elegans model of batten disease. J Inherit Metab Dis 40: 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YJ, Guha S, Tuluc F, Falk MJ (2018) High-throughput BioSorter quantification of relative mitochondrial content and membrane potential in living Caenorhabditis elegans. Mitochondrion 40: 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liptak N, Bosze Z, Hiripi L (2019) GFP transgenic animals in biomedical research: a review of potential disadvantages. Physiol Res 68: 525–530. [DOI] [PubMed] [Google Scholar]
- Luke CJ, O’Reilly LP (2015) Microscopic Investigation of Protein Function in C. elegans Using Fluorescent Imaging. Curr Protoc Cytom 74: 12 41 11–12 41 17. [DOI] [PubMed] [Google Scholar]
- MacRae CA, Peterson RT (2015) Zebrafish as tools for drug discovery. Nat Rev Drug Discov 14: 721–731. [DOI] [PubMed] [Google Scholar]
- Malina C, Larsson C, Nielsen J (2018) Yeast mitochondria: an overview of mitochondrial biology and the potential of mitochondrial systems biology. FEMS Yeast Res 18. [DOI] [PubMed] [Google Scholar]
- McCormack S, Polyak E, Ostrovsky J, et al. (2015) Pharmacologic targeting of sirtuin and PPAR signaling improves longevity and mitochondrial physiology in respiratory chain complex I mutant Caenorhabditis elegans. Mitochondrion 22: 45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok BY, de Moraes MH, Zeng J, et al. (2020) A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583: 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naeem MM, Sondheimer N (2019) Heteroplasmy Shifting as Therapy for Mitochondrial Disorders. Adv Exp Med Biol 1158: 257–267. [DOI] [PubMed] [Google Scholar]
- O’Reilly LP, Benson JA, Cummings EE, Perlmutter DH, Silverman GA, Pak SC (2014) Worming our way to novel drug discovery with the Caenorhabditis elegans proteostasis network, stress response and insulin-signaling pathways. Expert Opin Drug Discov 9: 1021–1032. [DOI] [PubMed] [Google Scholar]
- O’Reilly LP, Luke CJ, Perlmutter DH, Silverman GA, Pak SC (2014) C. elegans in high-throughput drug discovery. Adv Drug Deliv Rev 69–70: 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh S, Saneto R, Falk MJ, et al. (2009) A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol 11: 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck RW (2018) Precision Medicine Is Not Just Genomics: The Right Dose for Every Patient. Annu Rev Pharmacol Toxicol 58: 105–122. [DOI] [PubMed] [Google Scholar]
- Peng M, Ostrovsky J, Kwon YJ, et al. (2015) Inhibiting cytosolic translation and autophagy improves health in mitochondrial disease. Hum Mol Genet 24: 4829–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF (2012) Treatment for mitochondrial disorders. Cochrane Database Syst Rev: CD004426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak E, Ostrovsky J, Peng M, et al. (2018) N-acetylcysteine and vitamin E rescue animal longevity and cellular oxidative stress in pre-clinical models of mitochondrial complex I disease. Mol Genet Metab 123: 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman J, Rahman S (2018) Mitochondrial medicine in the omics era. Lancet 391: 2560–2574. [DOI] [PubMed] [Google Scholar]
- Rea SL, Graham BH, Nakamaru-Ogiso E, Kar A, Falk MJ (2010) Bacteria, yeast, worms, and flies: exploiting simple model organisms to investigate human mitochondrial diseases. Dev Disabil Res Rev 16: 200–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales XQ, Thompson JLP, Haas R, et al. (2020) The North American mitochondrial disease registry. J Transl Genet Genom 4: 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzicka L, Howe DG, Ramachandran S, et al. (2019) The Zebrafish Information Network: new support for non-coding genes, richer Gene Ontology annotations and the Alliance of Genome Resources. Nucleic Acids Res 47: D867–D873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzenente B, Rotig A, Metodiev MD (2016) Mouse models for mitochondrial diseases. Hum Mol Genet 25: R115–R122. [DOI] [PubMed] [Google Scholar]
- Saada A (2014) Mitochondria: mitochondrial OXPHOS (dys) function ex vivo--the use of primary fibroblasts. Int J Biochem Cell Biol 48: 60–65. [DOI] [PubMed] [Google Scholar]
- Scott CA, Marsden AN, Slusarski DC (2016) Automated, high-throughput, in vivo analysis of visual function using the zebrafish. Dev Dyn 245: 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen A, Cox RT (2017) Fly Models of Human Diseases: Drosophila as a Model for Understanding Human Mitochondrial Mutations and Disease. Curr Top Dev Biol 121: 1–27. [DOI] [PubMed] [Google Scholar]
- Setten RL, Rossi JJ, Han SP (2019) The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov 18: 421–446. [DOI] [PubMed] [Google Scholar]
- Shen Q, Truong L, Simonich MT, Huang C, Tanguay RL, Dong Q (2020) Rapid well-plate assays for motor and social behaviors in larval zebrafish. Behav Brain Res 391: 112625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman GA, Luke CJ, Bhatia SR, et al. (2009) Modeling molecular and cellular aspects of human disease using the nematode Caenorhabditis elegans. Pediatr Res 65: 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele H, Gomez-Duran A, Pyle A, et al. (2020) Metabolic effects of bezafibrate in mitochondrial disease. EMBO Mol Med 12: e11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart AM, Braubach O, Spitsbergen J, Gerlai R, Kalueff AV (2014) Zebrafish models for translational neuroscience research: from tank to bedside. Trends Neurosci 37: 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BJ, Wight IE, Chou WYY, et al. (2019) CemOrange2 fusions facilitate multifluorophore subcellular imaging in C. elegans. PLoS One 14: e0214257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyynismaa H, Suomalainen A (2009) Mouse models of mitochondrial DNA defects and their relevance for human disease. EMBO Rep 10: 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergano SS, Rao M, McCormack S, et al. (2014) In vivo metabolic flux profiling with stable isotopes discriminates sites and quantifies effects of mitochondrial dysfunction in C. elegans. Mol Genet Metab 111: 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Fan W (2009) The pathophysiology of mitochondrial disease as modeled in the mouse. Genes Dev 23: 1714–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L (2016) Mitochondrial purine and pyrimidine metabolism and beyond. Nucleosides Nucleotides Nucleic Acids 35: 578–594. [DOI] [PubMed] [Google Scholar]
- Yatsuga S, Suomalainen A (2012) Effect of bezafibrate treatment on late-onset mitochondrial myopathy in mice. Hum Mol Genet 21: 526–535. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Falk MJ (2014) Integrated transcriptome analysis across mitochondrial disease etiologies and tissues improves understanding of common cellular adaptations to respiratory chain dysfunction. Int J Biochem Cell Biol 50: 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Tsukikawa M, Peng M, et al. (2013) Primary respiratory chain disease causes tissue-specific dysregulation of the global transcriptome and nutrient-sensing signaling network. PLoS One 8: e69282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolkipli-Cunningham Z, Xiao R, Stoddart A, et al. (2018) Mitochondrial disease patient motivations and barriers to participate in clinical trials. PLoS One 13: e0197513. [DOI] [PMC free article] [PubMed] [Google Scholar]