

Abstract
The tumorigenicity and toxicity of induced pluripotent stem cells (iPSCs) and their derivatives are major safety concerns in their clinical application. Recently, we developed granulocyte-macrophage colony-stimulating factor (GM-CSF)-producing proliferating myeloid cells (GM-pMCs) from mouse iPSCs as a source of unlimited antigen-presenting cells for use in cancer immunotherapy. As GM-pMCs are generated by introducing c-Myc and Csf2 into iPSC-derived MCs and are dependent on self-produced GM-CSF for proliferation, methods to control their proliferation after administration should be introduced to improve safety. In this study, we compared the efficacy of two promising suicide gene systems, herpes simplex virus-thymidine kinase (HSV-TK)/ganciclovir (GCV) and inducible caspase-9 (iCasp9)/AP1903, for safeguarding GM-pMCs in cancer immunotherapy. The expression of HSV-TK or iCasp9 did not impair the fundamental properties of GM-pMCs. Both of these suicide gene-expressing cells selectively underwent apoptosis after treatment with the corresponding apoptosis-inducing drug, and they were promptly eliminated in vivo. iCasp9/AP1903 induced apoptosis more efficiently than HSV-TK/GCV. Furthermore, high concentrations of GCV were toxic to cells not expressing HSV-TK, whereas AP1903 was bioinert. These results suggest that iCasp9/AP1903 is superior to HSV-TK/GCV in terms of both safety and efficacy when controlling the fate of GM-pMCs after priming antitumor immunity.
Keywords: cancer immunotherapy, induced pluripotent stem cell, antigen-presenting cell, granulocyte-macrophage colony-stimulating factor, c-Myc, suicide gene, iCasp9, HSV-TK
Graphical abstract
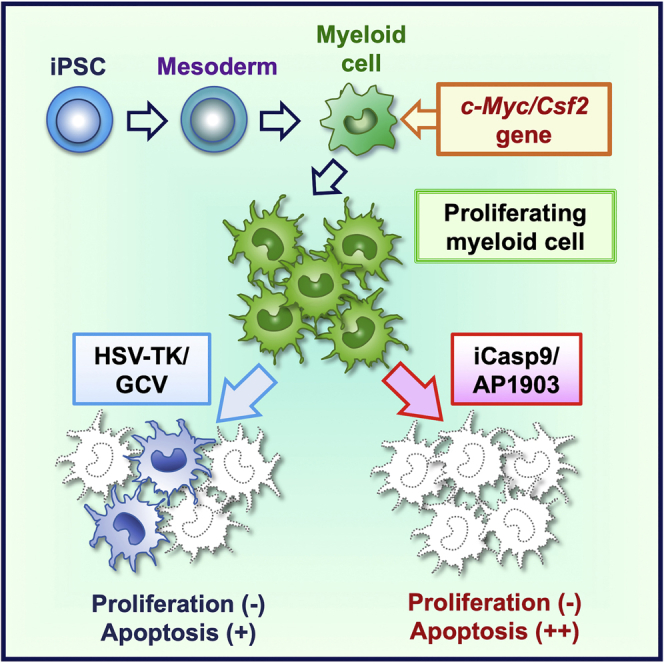
GM-CSF-producing, proliferating myeloid cells (GM-pMCs) from iPSCs have been developed as potent antigen-presenting cells for cancer immunotherapy. However, their potential tumorigenicity and toxicity are major safety concerns for their clinical applications. Uemura and colleagues demonstrated that the inducible caspase-9 suicide system can efficiently control the fates of GM-pMCs.
Introduction
The recent success in cancer treatments using immune checkpoint inhibitors and multiple types of immune cells has garnered the attention of researchers studying the immunological mechanisms by which cancer cells can be eliminated.1,2 Dendritic cell (DC)-based vaccines, which activate endogenous tumor-reactive T cells through the administration of tumor antigen-loaded DCs, are some of the most promising approaches in cancer therapy.3, 4, 5 Conventional DC-based vaccines typically utilize autologous peripheral blood DC precursors derived from patients with cancer. However, their capacity to differentiate and function appropriately after differentiation is often limited due to disease progression or immunosuppressive cancer treatment, leading to modest clinical efficacy.6
To overcome such issues, we established a technology for generating DC-like antigen-presenting cells (APCs) derived from induced pluripotent stem cells (iPSCs), which possess an unrestricted potential to proliferate and differentiate.7, 8, 9, 10, 11, 12, 13 Myeloid lineage cells were initially induced from mouse embryonic stem cells (ESCs) or iPSCs, and then transfected with the pro-proliferative c-Myc gene to generate cytokine-dependent proliferating myeloid cells (ESC- or iPSC-pMCs).10 By genetically engineering these iPSC-pMCs, we further expressed granulocyte macrophage colony-stimulating factor (GM-CSF), a growth factor for iPSC-pMCs, to establish GM-pMCs.13 GM-pMCs express major histocompatibility complex (MHC) class I/II and costimulatory (CD80 and CD86) molecules and proliferate in a manner that depends on the self-produced GM-CSF. These cells have the capacity to capture, process, and present extracellular antigen proteins in the context of MHC class I molecules, and the process is known as cross-presentation.13,14 Moreover, the administration of GM-pMCs loaded with an MHC class I-restricted cancer antigen peptide or a cancer antigen protein stimulates antigen-specific cytotoxic T lymphocytes to inhibit tumor growth; this is comparable with the effect of functionally complete bone marrow-derived DCs.13 These findings suggest that this GM-pMC system, using human leukocyte antigen (HLA)-matched iPSCs, enable a stable supply of functional APCs on a large scale without the need for repeated invasive blood samplings, thereby serving as an alternative to autologous DCs. Although GM-pMCs have a high proliferative capacity, they are not associated with either tumorigenicity or autoimmune-related organ dysfunction.13 Moreover, even when irradiated before administration to suppress proliferation, GM-pMCs efficiently induce the proliferation of cancer-reactive T cells in vivo.13 However, as all possible adverse events cannot be predicted, another safety net that is applicable even after administration might be needed.
Recently, suicide gene systems that enable the conditional elimination of administered gene-transduced cells and their progenies have been developed.15,16 Herpes simplex virus-thymidine kinase (HSV-TK)/ganciclovir (GCV) and inducible caspase-9 (iCasp9)/AP1903 are well-known examples of such systems and have been validated in clinical settings, including cancer gene therapy for solid tumors and safeguarding adoptive T cell therapy following hematopoietic stem-cell transfer for hematologic malignancies.17,18 Moreover, they have been used as safety switches in PSC-based regenerative medicine and cell-based immunotherapy.16,19, 20, 21 Introducing these suicide gene systems should increase the safety of GM-pMC-based vaccines. However, proliferation signals are consistently activated in GM-pMCs, and the suicide gene that is more suitable for this system is not clear.
In this study, we established safeguarding systems controlling the c-Myc/Csf2-mediated proliferative potential of iPSC-derived antigen-presenting GM-pMCs through inducible expression of HSV-TK or iCasp9.
Results
Suicide gene expression does not alter the basic characteristics and antitumor effects of GM-pMCs
HSV-TK or iCasp9 were introduced into GM-pMCs using lentiviral vectors to generate GM-pMC-HSV-TK and GM-pMC-iCasp9, respectively. The resulting cells were sorted based on the expression of a co-introduced, truncated CD19, and further isolated using the limiting dilution method to obtain monoclonal cell lines that highly express the transduced genes (Figures 1A and 1B; Figure S1A). In the iPSC-pMC system, we previously found that gene expression mediated by the EF1α promoter was less likely to decrease after long-term culture compared with that mediated by the cytomegalovirus (CMV) promoter (unpublished observation); thus, the EF1α promoter was used in this study. Although the expression of GM-CSF did not change after 30 days of culture, a slight decrease in the expression of these two suicide genes was observed (Figure S1B). These observations are conceivably due to the difference between the genes whose products are used as growth factors (GM-CSF) and genes that are not used for proliferation (HSV-TK and iCasp9). GM-pMCs express myeloid lineage markers (CD11b+, CD11cint, F4/80high, DEC205high, Gr-1low, and CD33+), antigen-presenting molecules (MHC class I/II), and costimulatory molecules (CD40, CD80, and CD86). Suicide gene transfer did not alter the expression of these markers (Figure 1C), GM-CSF production (Figure 1D), or GM-CSF-dependent proliferation (Figure 1E). In our prophylactic cancer vaccine study, the administration of OVA257–264 peptide-loaded GM-pMC-HSV-TK or GM-pMC-iCasp9 cells inhibited the growth of OVA-expressing melanoma cells (MO4) as effectively as GM-pMCs (Figures 2A and 2D). These findings suggest that the introduction of HSV-TK or iCasp9 does not adversely affect the fundamental properties and antitumor efficacy of GM-pMCs.
Figure 1.

Suicide gene transfer does not alter the fundamental properties of GM-pMCs
(A) Schematic of viral vectors expressing herpes simplex virus-thymidine kinase (HSV-TK) or inducible caspase 9 (iCasp9), co-expressed with truncated CD19 as a marker and a selection gene. (B) Expression of GM-CSF and transduced suicide gene. (C) Expression of differentiation markers, antigen-presenting molecules, and co-stimulatory molecules. Left panels, representative flow cytometry profiles of the indicated surface molecules. Right panels, expression of surface molecules associated with T cell stimulation. (D) GM-CSF production at 24 h. GM-CSF levels in the culture supernatants were evaluated using ELISA. (E) Cell proliferation. Cells were cultured in the presence of the indicated cytokines. Proliferation was measured using the MTT assay. The culture medium served as a control. (D and E) Data are shown as mean ± SD of triplicate cultures and are representative of two (D) or three (E) independent experiments.
Figure 2.

Suicide gene transfer does not impair the cancer vaccine effects of GM-pMCs
(A) Schematic illustration of the experiment. C57BL/6 mice were vaccinated twice with the OVA257–264 peptide-loaded GM-pMC, GM-pMC-HSV-TK, or GM-pMC-iCasp9 cells, followed by inoculation with MO4 tumor cells. (B) The tumor size in individual mice from one experiment is shown (n = 8 mice). (C) The median tumor size for each group in (B) is shown (n = 8 mice). (D) Kaplan-Meier survival curves were evaluated using the log-rank test (n = 8 mice). ∗∗∗p < 0.001.
Suicide gene-expressing GM-pMCs are selectively eradicated following the induction of apoptosis
First, we compared the apoptosis-induction efficiency of HSV-TK/GCV and iCasp9/AP1903 in vitro. Both induced apoptosis selectively in gene-expressing cells in the presence of the corresponding drug (Figure S2A). The proliferation of GM-pMC-HSV-TK cells was effectively inhibited by 1–10 μM GCV. However, treatment with >100 μM GCV considerably inhibited proliferation of cells, including HSV-TK non-expressing cells, suggesting nonspecific cytotoxicity (Figure 3A). In contrast, the proliferation of GM-pMC-iCasp9 cells was considerably inhibited at low doses of AP1903 (0.1–1 nM), whereas non-expressing cells were not affected, even when exposed to 1,000- to 10,000-fold higher doses (1,000 nM; Figure 3B). When treated with 10 μM GCV, the viability of GM-pMC-HSV-TK cells gradually decreased (day 1, 70.0%; day 2, 20.0%; day 3 or later, <0.1% relative to day 0; Figure 3C). On the contrary, GM-pMC-iCasp9 cells were promptly eradicated in the presence of 10 nM AP1903 (day 1, 3.7%; day 2, 0.4%; day 3 or later, <0.1% relative to day 0; Figure 3D). During the 7 days of observation, re-expansion, which suggests the emergence of apoptosis-resistant cells, was not observed for either of the suicide gene systems. Neither drug affected the viability or GM-CSF production of cells not expressing the suicide gene (Figures 3C–3E; Figure S2A). In contrast, GM-CSF production by both GM-pMC-iCasp9 and GM-pMC-HSV-TK cells was significantly reduced as a result of the induction of apoptosis (24 h). The amount of GM-CSF was considerably lower in GM-pMC-iCasp9 cells than in GM-pMC-HSV-TK cells (Figure 3E). These findings suggest that the iCasp9/AP1903 system induces the apoptosis of GM-pMCs more quickly and effectively than does the HSV-TK/GCV system in vitro.
Figure 3.

iCasp9/AP1903 induces apoptosis in GM-pMCs more efficiently than does HSV-TK/GCV
(A and B) Cell proliferation. Cells were cultured for 72 h in 96-well culture plates in the presence of increasing doses of GCV (A) or AP1903 (B). The culture medium served as a control. Proliferation was determined using the MTT assay. (C and D) Number of cells after suicide induction. Cells (1.0 × 105/well) were seeded in 24-well culture plates in the presence of 10 μM GCV (C) or 10 nM AP1903 (D). The culture medium served as a control. Viable cells were enumerated by direct counting of trypan-blue-stained-cells at the indicated time points. See also Figure S2. (E) GM-CSF production. Cells were cultured for 24 h in the presence of 10 μM GCV or 10 nM AP1903. GM-CSF levels in the culture supernatants were evaluated using ELISA. The medium served as a control. (A–E) Data are shown as mean ± SD of triplicate cultures and are representative of two independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001.
iCasp9/AP1903 controls the fate of transferred cells more efficiently than HSV-TK/GCV
We evaluated the in vivo efficacy of both suicide gene systems using luciferase (Luc)-transfected cells. Mice were subcutaneously (s.c.) inoculated with suicide gene-expressing GM-pMCs and intraperitoneally (i.p.) injected with the corresponding apoptosis-inducing drug for 5 consecutive days. While the exogenous cell numbers gradually diminished without any treatment, the administration of GCV or AP1903 did not affect the suicide gene non-expressing cells (Figure 4A). When treated with 100 mg/kg GCV, the luminescence of injected GM-pMC-HSV-TK-Luc cells decreased by 60.7% on day 1, 75.1% on day 2, 84.6% on day 3, 88.7% on day 4, and 97.5% on day 7 from the initial luminescence on day 0 (Figure 4B). Meanwhile, 2.5 mg/kg AP1903 reduced the luminescence of GM-pMC-iCasp9-Luc by 65.5% on day 1, 92.5% on day 2, 92.5% on day 3, 94.2% on day 4, and 98.2% on day 7 from the initial luminescence on day 0 (Figure 4C). These findings revealed that both suicide systems were effective in vivo, with similar efficacies on day 7. However, the iCasp9/AP1903 system eliminated the gene-expressing cells more promptly than did the HSV-TK/GCV system, consistent with the in vitro data. An analysis of the long-term effectiveness of these systems in severely immunocompromised mice (BABL/c Rag2−/−Jak3−/−)22 did not reveal cells expressing the suicide gene after 35 days of suicide induction, suggesting no evidence of apoptosis-resistant cells (Figure S2B).
Figure 4.

iCasp9/AP1903 eradicates the transferred cells more efficiently than does HSV-TK/GCV
(A–C) In vivo suicide control of GM-pMCs. C57BL/6 mice were inoculated with luciferase (Luc)-expressing GM-pMCs (A), GM-pMC-HSV-TK cells (B), or GM-pMC-iCasp9 cells (C) (1.0 × 106 cells) and treated with 100 mg/kg GCV or 2.5 mg/kg AP1903 for 5 consecutive days (days 0–4). In vivo cell survival was monitored by bioluminescence imaging. Left panels, bioluminescence images of representative mice from two independent experiments are shown (n = 6 mice). Right panels, total flux (photons) for the indicated treatment groups are shown as mean ± SEM (n = 6 mice). ∗p < 0.05, ∗∗p < 0.01.
Survivin inhibitor YM-155 does not enhance iCasp9-mediated apoptosis
Survivin (also known as baculoviral inhibitor of apoptosis [IAP] repeat-containing protein 5 [BIRC5]) functions as a key regulator of mitosis and programmed cell death. It forms a complex with X-linked inhibitor of apoptosis (XIAP, also known as BIRC4) and directly inhibits cell death mediated by the caspase cascade pathway.23,24 Therefore, it is conceivable that the combination of iCasp9/AP1903 with a survivin inhibitor might enhance iCasp9-mediated apoptosis. To examine this possibility, we used a survivin inhibitor YM-155, which selectively eliminates survivin-overexpressing cells. Although GM-CSF is known to increase survivin expression,25,26 there was no increase in survivin gene expression after GM-CSF transfer (Figure 5A). In addition, YM-155 had no effect on the survival of either iCasp9-expressing or non-expressing GM-pMCs (Figure S3A). Consistently, the presence of YM-155 did not promote AP1903-induced GM-pMC-iCasp9 apoptosis in vitro and in vivo (Figures 5A and 5B; Figure S3B). These results suggest that YM-155 does not enhance iCasp9-mediated apoptosis in the GM-pMC system.
Figure 5.

Survivin is not associated with apoptosis induction in GM-pMCs
(A) Birc5 mRNA expression. Cells were cultured without adding GM-CSF to the medium. Birc5 mRNA was quantified using qRT-PCR. iPSC-pMCs cultured in the presence of recombinant GM-CSF (30 ng/mL) served as a reference. (B) Number of cells after suicide induction. Cells (1.0 × 105/well) were cultured in the presence of 10 nM AP1903 and/or 10 nM YM-155. (A and B) Data are shown as mean ± SD of triplicate cultures and are representative of two independent experiments. See also Figure S3A. (C) In vivo effect of YM155 on suicide induction. C57BL/6 mice were inoculated with Luc-expressing GM-pMC-iCasp9 cells (1.0 × 106 cells) and treated with 2.5 mg/kg AP1903 and/or 5 mg/kg YM-155 for 5 consecutive days (days 0–4). Upper panels, bioluminescence images of representative mice from two independent experiments are shown (n = 6). Lower left, total flux (photons) for the indicated treatment groups are shown as mean ± SEM (n = 6 mice). Lower right, reduction rate of total flux at the indicated time points compared with that on day 0. Data are shown as mean ± SEM (n = 6 mice). See also Figure S3B.
Discussion
The development of iPSCs with unrestricted proliferation potential and differentiation capacity has made a breakthrough in regenerative medicine and cell-based therapies.27, 28, 29 Recent advances in iPSC engineering might enable a stable, large-scale supply of functional immune cells of uniform quality, leading to improved cancer immunotherapy.30 However, considering their actual clinical application, the cells should be equipped with safety switches that enable clinicians to immediately control the fates of cells in case of an unexpected adverse event, while retaining their optimal antitumor efficacy. Although increased serum GM-CSF level, tumorigenesis, and systemic adverse effects were not observed in our GM-pMC-based cancer vaccine experiments,13 some concerns, such as tumor formation and unwanted immune-modulating effects, remain because GM-pMCs have a high proliferative potential and the GM-CSF they produce might cause unanticipated events. To address these issues, we attempted to further control the fate of GM-pMCs using two promising suicide gene systems, by comparing their efficacies.
The HSV-TK/GCV system has been extensively studied in the fields of cancer gene therapy, regenerative medicine, and immune cell therapy.17,21,31 This system is highly cell cycle-dependent; it induces apoptosis only in cells in the S and G2 phases by inhibiting DNA synthesis. Therefore, its killing effect is limited to rapidly dividing cells, whereas slowly or non-dividing cells evade death.32,33 As cancer cells are highly proliferative, gene therapy that directly introduces HSV-TK into tumor tissues and induces apoptosis seems to be reasonable. HSV-TK exerts a bystander effect that ablates not only the transduced cells, but also the adjacent non-transduced cells through gap junctions; thus, it could be a good therapeutic strategy, particularly for solid tumors.31,34,35 This cell cycle dependency has been exploited in the treatment of spinal cord injuries using HSV-TK-expressing, iPSC-derived, neural stem/progenitor cells (iPSC-NS/PCs), resulting in the selective eradication of proliferative tumorigenic cells while sparing mature post-mitotic neuronal cells, thus preserving reconstructed motor function.36 As GM-pMCs are highly proliferative, we first considered that HSV-TK/GCV might be effective in inducing apoptosis. However, it is possible that HSV-TK-expressing cells could be eliminated by the host’s immune system due to the immunogenicity of HSV-TK, thereby impairing therapeutic efficacy.37 However, this does not seem to occur because GM-pMC-HSV-TK cells showed antitumor efficacy comparable with that of the parental GM-pMCs in this study. The ablation of HSV-TK-expressing cells by GCV is relatively slow, requiring approximately 3 days; therefore, it appears that iCasp9 might be superior to HSV-TK in situations where more rapid cell elimination is needed, such as during severe adverse events related to cell-based immunotherapy.17 In addition, although GCV is a safe and well-tolerated drug, its long-term and high-dose administration introduces a risk of serious toxicity, including renal dysfunction, liver dysfunction, and myelosuppression, necessitating careful management.38 Moreover, GCV is one of the essential antiviral drugs used to treat herpes virus infections, but its utility might be restricted in patients transplanted with HSV-TK-expressing cells.
Apoptosis induction by iCasp9/AP1903 is cell cycle-independent and rapidly eliminates all iCasp9-expressing cells.39 Therefore, it seems that the iCasp9/AP1903 system is suitable for safeguarding cell-based immunotherapies rather than preventing teratoma formation in iPSC-derived organs, which is expected after long-term engraftment. Recently, iCasp9 has been utilized as a safety switch for chimeric antigen receptor (CAR)-T cell therapy.15,16 In GM-pMCs, iCasp9/AP1903 induced apoptosis more effectively than did HSV-TK/GCV both in vitro and in vivo. Murine caspase-9 shares 72% homology and 87% similarity with human caspase-9 at the protein level. A previous report demonstrated the effectiveness of iCasp9 in murine cells, showing a rapid elimination of iCasp9-expressing murine iPSCs in the presence of AP1903.40 These observations strongly suggest that human iCasp9 cleaves murine caspase-3, although this should be clarified directly in further experiments.
Some reports have suggested that altered expression of HSV-TK or iCasp9 due to methylation, mutation, or silencing can result in apoptosis-resistant or -escaping cells.38,41 One of the limitations of our study is that these phenomena have not been fully investigated. Various cancer-related pro-inflammatory cytokines, which reflect the tumor microenvironment, should be elevated in tumor-bearing mouse. Accordingly, it is possible that GM-pMCs might acquire tumorigenicity or apoptosis resistance in response to these altered cytokine milieus. To clarify these points, the findings herein should be further elucidated in a tumor-bearing mouse model. We previously found that irradiation prior to administration prevented the in vivo proliferation of GM-pMCs while maintaining their anti-tumor efficacy.13 Therefore, combining irradiation with a suicide gene system may overcome such a limitation and improve the safety of GM-pMCs. To establish GM-pMCs, iPSC-derived MCs were initially transfected with c-Myc, and then with Csf2, using lentiviral vectors. To further incorporate suicide genes, a third round of gene transfer is required. There is a concern that repeated gene transfer may negatively affect cell function or stability. Therefore, when producing human GM-pMCs, other approaches that enable targeted gene delivery and multiple gene transfer, such as the use of bicistronic expression vector systems, may be needed.42, 43, 44
The survivin inhibitor YM-155 selectively eliminates survivin-overexpressing cells, such as cancer cells or undifferentiated PSCs; therefore, it has potential as a novel anti-cancer drug or preventive agent of PSC-derived teratoma formation.45, 46, 47 This small molecule does not exert cytotoxic effects on normal, differentiated cells and is well tolerated in clinical use.48,49 YM-155 also has the ability to inhibit murine survivin.50 Although survivin inhibition is known to enhance caspase-dependent apoptosis,46 we did not observe a similar effect in GM-pMCs, which may be due to the lack of elevated levels of survivin.
In conclusion, iCasp9/AP1903 is superior to HSV-TK in terms of both safety and effectiveness as a safeguard against GM-pMC-based immunotherapy. By ensuring the safety of GM-pMCs using this system, GM-pMC-based cancer vaccines might become more suitable for clinical applications.
Materials and methods
Mice
Female C57BL/6 mice were purchased from Charles River Laboratories (Yokohama, Japan). BALB/c Rag2−/−Jak3−/− mice were provided by Dr. S. Okada (Kumamoto University, Japan).22 In all experiments, animals were randomly assigned to the experimental groups. All animals were humanely sacrificed at the end of the study. All mice were maintained under specific pathogen-free conditions and used at 6–12 weeks of age. All animal studies were performed in accordance with the procedures approved by the Animal Research Committee of the National Cancer Center (Tokyo, Japan).
Cells and reagents
MO4, an OVA-expressing B16-F10 melanoma cell line of C57BL/6 origin, was cultured in Roswell Park Memorial Institute 1640 medium (Sigma-Aldrich) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco). Murine iPSC-pMCs and GM-pMCs were established as described previously10,13 and cultured in alpha-modified Eagle’s minimum essential medium (Invitrogen) supplemented with 30 ng/mL GM-CSF (Miltenyi Biotec), 50 ng/mL M-CSF (BioLegend), and 20% FBS. HSV-TK- and iCasp9-expressing GM-pMCs or iPSC-pMCs were generated by transducing GM-pMCs with a lentiviral vector encoding a synthetic HSV-TK gene (psetz-hsv1tk, InvivoGen) or iCasp951 co-expressed with truncated CD19 (GenBank: NM001770) in the presence of Polybrene (Sigma-Aldrich). All cell lines were tested and found to be free of mycoplasma contamination. GCV was purchased from Hoffmann-La Roche. AP1903 was purchased from MedChemExpress, and YM-155 was purchased from Cayman Chemical.
Generation of recombinant lentivirus
HIV-1-based lentiviral vectors pseudotyped with the vesicular stomatitis virus G glycoprotein (VSV-G) were generated by transient transfections with pCAG-HIVgp, pCMV-VSV-G-RSV-Rev, and CSII-EF-MCS in 293T cells using Lipofectamine 2000 (Thermo Fisher Scientific). After 48 h, the vector-containing supernatant was harvested and passed through 0.45-μm filter. The lentivirus vector was concentrated using the Lenti-X concentrator (Clontech) according to the manufacturer’s instructions.
In vivo tumor models
GM-pMCs were incubated with 10 μM OVA257–264 peptide (SIINFEKL, Eurofins Genomics) for 8–12 h at 37°C. The mice were i.p. treated with these peptide-loaded GM-pMCs (1.0 × 105 cells) twice at a 7-day interval, followed by a s.c. inoculation of 2.0 × 105 MO4 tumor cells into the right flank, 7 days later. The mice were monitored for tumor growth and survival. Tumor size was measured twice a week until the mice either died or were sacrificed when the tumors exceeded 20 mm in diameter. Tumor volume was calculated from its length (a) and width (b) as follows: ab2/2.
Flow cytometry
Single-cell suspensions were incubated with Fc receptor blocking reagent (Miltenyi Biotec), and then stained with the fluorophore-conjugated monoclonal antibodies following the manufacturer’s instructions (Table S1). Cell viability was determined using an annexin V apoptosis detection kit (BioLegend) according to the manufacturer’s instructions. Cells were analyzed using a flow cytometer (FACSCanto II or Accuri C6; BD Biosciences); data were analyzed using FlowJo software v10.7 (Tree Star).
Cytokine measurements
Cytokine levels in culture supernatants were measured using an enzyme-linked immunosorbent assay kit (BioLegend) according to the manufacturer’s instructions.
In vitro cell proliferation assay
Cell proliferation was evaluated using the standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) assay. The cells were seeded in 96-well culture plates and labeled with MTT at each time point. After 4 h of labeling, the plate was centrifuged to sediment the cells, and the supernatant was removed by tapping. Subsequently, 200 μL of dimethyl sulfoxide was added to each well and mixed thoroughly. After 5 min, the absorbance of the sample was measured at 570 nm.
In vivo bioluminescence imaging
The mice were i.p. injected with 200 μL of d-luciferin (15 mg/mL; VivoGlo luciferin; Promega) under 2% inhaled isoflurane anesthesia. Bioluminescence images were obtained using in vivo imaging system (IVIS) Lumina II with Living Image software version 3.2 (PerkinElmer).
Real-time PCR
The total RNA was extracted using the RNeasy Mini kit plus (QIAGEN), and cDNA was synthesized using the PrimeScript II first-strand cDNA synthesis kit (Takara Bio). Transcripts were quantified by quantitative real-time PCR on an ABI Prism 7500 sequence detector using the TaqMan Gene Expression Assay (Applied Biosystems). The following probes purchased from Applied Biosystems were used: Birc5 (Mm00599749_m1) and glyceraldehyde-3-phosphate dehydrogenase (Gapdh; Mm99999915_g1). mRNA expression levels were calculated using the change-in-cycling threshold (Ct) method, and the results were normalized to levels of the control gene Gapdh.
Statistical analysis
Statistical analyses were conducted using JMP Genomics software version 14 (SAS Institute, Cary, NC, USA). Unpaired two-tailed Student’s t tests were used for comparisons between the groups. One-way ANOVA with Tukey’s test was used for multiple-group comparisons. Kaplan-Meier survival analysis was based on the endpoint reached (when mice died or were sacrificed when tumors exceeded 20 mm in diameter). Differences between survival curves were evaluated using log-rank tests. Results were considered significant at p <0.05.
Acknowledgments
We thank Dr. H. Miyoshi (RIKEN) for providing the plasmid vectors pCSII-EF, pCMV-VSV-G-RSV-Rev, and pCAG-HIVgp. We thank M. Ozaki (National Cancer Center) for secretarial support. This work was partially supported by JSPS KAKENHI grants JP17K16877, JP20K09186, and JP20H03759, and by AMED grants JP19bm0404054h0001 and JP20bm0404054h0002.
Author contributions
Conceptualization, R.Z. and Y.U.; methodology, R.Z.; investigation, H.M., R.Z., Y.H., T.I., M.Y., and Y.U.; resources, R.Z., T.L., T.I. and S.O.; writing – original draft, H.M. and Y.U.; writing – review & editing, H.M., R.Z., T.K., H.T., S.F., S.K., and Y.U.; administrative support, A.I., H.O., and T.N.; supervision: Y.U.; funding acquisition, R.Z., T.I., T.N., and Y.U.
Declaration of interests
Y.U. received research funding from AGC, Inc., Japan. The remaining authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.03.002.
Supplemental information
References
- 1.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg S.A., Restifo N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anguille S., Smits E.L., Lion E., van Tendeloo V.F., Berneman Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014;15:e257–e267. doi: 10.1016/S1470-2045(13)70585-0. [DOI] [PubMed] [Google Scholar]
- 4.Garg A.D., Coulie P.G., Van den Eynde B.J., Agostinis P. Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol. 2017;38:577–593. doi: 10.1016/j.it.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Palucka K., Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 2013;39:38–48. doi: 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bol K.F., Schreibelt G., Gerritsen W.R., de Vries I.J., Figdor C.G. Dendritic cell-based immunotherapy: State of the art and beyond. Clin. Cancer Res. 2016;22:1897–1906. doi: 10.1158/1078-0432.CCR-15-1399. [DOI] [PubMed] [Google Scholar]
- 7.Senju S., Hirata S., Matsuyoshi H., Masuda M., Uemura Y., Araki K., Yamamura K., Nishimura Y. Generation and genetic modification of dendritic cells derived from mouse embryonic stem cells. Blood. 2003;101:3501–3508. doi: 10.1182/blood-2002-07-2254. [DOI] [PubMed] [Google Scholar]
- 8.Matsuyoshi H., Senju S., Hirata S., Yoshitake Y., Uemura Y., Nishimura Y. Enhanced priming of antigen-specific CTLs in vivo by embryonic stem cell-derived dendritic cells expressing chemokine along with antigenic protein: Application to antitumor vaccination. J. Immunol. 2004;172:776–786. doi: 10.4049/jimmunol.172.2.776. [DOI] [PubMed] [Google Scholar]
- 9.Senju S., Suemori H., Zembutsu H., Uemura Y., Hirata S., Fukuma D., Matsuyoshi H., Shimomura M., Haruta M., Fukushima S. Genetically manipulated human embryonic stem cell-derived dendritic cells with immune regulatory function. Stem Cells. 2007;25:2720–2729. doi: 10.1634/stemcells.2007-0321. [DOI] [PubMed] [Google Scholar]
- 10.Zhang R., Liu T.Y., Senju S., Haruta M., Hirosawa N., Suzuki M., Tatsumi M., Ueda N., Maki H., Nakatsuka R. Generation of mouse pluripotent stem cell-derived proliferating myeloid cells as an unlimited source of functional antigen-presenting cells. Cancer Immunol. Res. 2015;3:668–677. doi: 10.1158/2326-6066.CIR-14-0117. [DOI] [PubMed] [Google Scholar]
- 11.Liu H., Chen L., Liu J., Meng H., Zhang R., Ma L., Wu L., Yu S., Shi F., Li Y. Co-delivery of tumor-derived exosomes with alpha-galactosylceramide on dendritic cell-based immunotherapy for glioblastoma. Cancer Lett. 2017;411:182–190. doi: 10.1016/j.canlet.2017.09.022. [DOI] [PubMed] [Google Scholar]
- 12.Tsuchiya N., Zhang R., Iwama T., Ueda N., Liu T., Tatsumi M., Sasaki Y., Shimoda R., Osako Y., Sawada Y. Type I interferon delivery by iPSC-derived myeloid cells elicits antitumor immunity via XCR1+ dendritic cells. Cell Rep. 2019;29:162–175.e9. doi: 10.1016/j.celrep.2019.08.086. [DOI] [PubMed] [Google Scholar]
- 13.Mashima H., Zhang R., Kobayashi T., Hagiya Y., Tsukamoto H., Liu T., Iwama T., Yamamoto M., Lin C., Nakatsuka R. Generation of GM-CSF-producing antigen-presenting cells that induce a cytotoxic T cell-mediated antitumor response. OncoImmunology. 2020;9:1814620. doi: 10.1080/2162402X.2020.1814620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joffre O.P., Segura E., Savina A., Amigorena S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012;12:557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 15.Bonifant C.L., Jackson H.J., Brentjens R.J., Curran K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics. 2016;3:16011. doi: 10.1038/mto.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu S., Yi M., Qin S., Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol. Cancer. 2019;18:125. doi: 10.1186/s12943-019-1057-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciceri F., Bonini C., Stanghellini M.T., Bondanza A., Traversari C., Salomoni M., Turchetto L., Colombi S., Bernardi M., Peccatori J. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): A non-randomised phase I-II study. Lancet Oncol. 2009;10:489–500. doi: 10.1016/S1470-2045(09)70074-9. [DOI] [PubMed] [Google Scholar]
- 18.Di Stasi A., Tey S.K., Dotti G., Fujita Y., Kennedy-Nasser A., Martinez C., Straathof K., Liu E., Durett A.G., Grilley B. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y., Chang T., Long Y., Huang H., Kandeel F., Yee J.K. Using gene editing to establish a safeguard system for pluripotent stem-cell-based therapies. iScience. 2019;22:409–422. doi: 10.1016/j.isci.2019.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rossignoli F., Grisendi G., Spano C., Golinelli G., Recchia A., Rovesti G., Orsi G., Veronesi E., Horwitz E.M., Dominici M. Inducible caspase9-mediated suicide gene for MSC-based cancer gene therapy. Cancer Gene Ther. 2019;26:11–16. doi: 10.1038/s41417-018-0034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiuru M., Boyer J.L., O’Connor T.P., Crystal R.G. Genetic control of wayward pluripotent stem cells and their progeny after transplantation. Cell Stem Cell. 2009;4:289–300. doi: 10.1016/j.stem.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ono A., Hattori S., Kariya R., Iwanaga S., Taura M., Harada H., Suzu S., Okada S. Comparative study of human hematopoietic cell engraftment into BALB/c and C57BL/6 strain of Rag-2/Jak3 double-deficient mice. J. Biomed. Biotechnol. 2011;2011:539748. doi: 10.1155/2011/539748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mita A.C., Mita M.M., Nawrocki S.T., Giles F.J. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res. 2008;14:5000–5005. doi: 10.1158/1078-0432.CCR-08-0746. [DOI] [PubMed] [Google Scholar]
- 24.Altieri D.C. Survivin, cancer networks and pathway-directed drug discovery. Nat. Rev. Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 25.Gu L., Chiang K.Y., Zhu N., Findley H.W., Zhou M. Contribution of STAT3 to the activation of survivin by GM-CSF in CD34+ cell lines. Exp. Hematol. 2007;35:957–966. doi: 10.1016/j.exphem.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 26.Hu H., Shikama Y., Matsuoka I., Kimura J. Terminally differentiated neutrophils predominantly express Survivin-2α, a dominant-negative isoform of survivin. J. Leukoc. Biol. 2008;83:393–400. doi: 10.1189/jlb.0507282. [DOI] [PubMed] [Google Scholar]
- 27.Choi K.D., Vodyanik M., Slukvin I.I. Hematopoietic differentiation and production of mature myeloid cells from human pluripotent stem cells. Nat. Protoc. 2011;6:296–313. doi: 10.1038/nprot.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minagawa A., Yoshikawa T., Yasukawa M., Hotta A., Kunitomo M., Iriguchi S., Takiguchi M., Kassai Y., Imai E., Yasui Y. Enhancing T cell receptor stability in rejuvenated iPSC-derived T cells improves their use in cancer immunotherapy. Cell Stem Cell. 2018;23:850–858.e4. doi: 10.1016/j.stem.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Shi Y., Inoue H., Wu J.C., Yamanaka S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017;16:115–130. doi: 10.1038/nrd.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sachamitr P., Leishman A.J., Davies T.J., Fairchild P.J. Directed differentiation of human induced pluripotent stem cells into dendritic cells displaying tolerogenic properties and resembling the CD141+ subset. Front. Immunol. 2018;8:1935. doi: 10.3389/fimmu.2017.01935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Westphal M., Ylä-Herttuala S., Martin J., Warnke P., Menei P., Eckland D., Kinley J., Kay R., Ram Z., ASPECT Study Group Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade glioma (ASPECT): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14:823–833. doi: 10.1016/S1470-2045(13)70274-2. [DOI] [PubMed] [Google Scholar]
- 32.Brenner M.K., Gottschalk S., Leen A.M., Vera J.F. Is cancer gene therapy an empty suit? Lancet Oncol. 2013;14:e447–e456. doi: 10.1016/S1470-2045(13)70173-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leen A.M., Heslop H.E., Brenner M.K. Antiviral T-cell therapy. Immunol. Rev. 2014;258:12–29. doi: 10.1111/imr.12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCormick F. Cancer gene therapy: Fringe or cutting edge? Nat. Rev. Cancer. 2001;1:130–141. doi: 10.1038/35101008. [DOI] [PubMed] [Google Scholar]
- 35.Nasu Y., Saika T., Ebara S., Kusaka N., Kaku H., Abarzua F., Manabe D., Thompson T.C., Kumon H. Suicide gene therapy with adenoviral delivery of HSV-tK gene for patients with local recurrence of prostate cancer after hormonal therapy. Mol. Ther. 2007;15:834–840. doi: 10.1038/sj.mt.6300096. [DOI] [PubMed] [Google Scholar]
- 36.Kojima K., Miyoshi H., Nagoshi N., Kohyama J., Itakura G., Kawabata S., Ozaki M., Iida T., Sugai K., Ito S. Selective ablation of tumorigenic cells following human induced pluripotent stem cell-derived neural stem/progenitor cell transplantation in spinal cord injury. Stem Cells Transl. Med. 2019;8:260–270. doi: 10.1002/sctm.18-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Traversari C., Marktel S., Magnani Z., Mangia P., Russo V., Ciceri F., Bonini C., Bordignon C. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 2007;109:4708–4715. doi: 10.1182/blood-2006-04-015230. [DOI] [PubMed] [Google Scholar]
- 38.Zhao Q., Lu B., George S.K., Yoo J.J., Atala A. Safeguarding pluripotent stem cells for cell therapy with a non-viral, non-integrating episomal suicide construct. Biomaterials. 2012;33:7261–7271. doi: 10.1016/j.biomaterials.2012.06.038. [DOI] [PubMed] [Google Scholar]
- 39.Yagyu S., Hoyos V., Del Bufalo F., Brenner M.K. An inducible caspase-9 suicide gene to improve the safety of therapy using human induced pluripotent stem cells. Mol. Ther. 2015;23:1475–1485. doi: 10.1038/mt.2015.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu C., Hong S.G., Winkler T., Spencer D.M., Jares A., Ichwan B., Nicolae A., Guo V., Larochelle A., Dunbar C.E. Development of an inducible caspase-9 safety switch for pluripotent stem cell-based therapies. Mol. Ther. Methods Clin. Dev. 2014;1:14053. doi: 10.1038/mtm.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yagyu S., Hoyos V., Del Bufalo F., Brenner M.K. Multiple mechanisms determine the sensitivity of human-induced pluripotent stem cells to the inducible caspase-9 safety switch. Mol. Ther. Methods Clin. Dev. 2016;3:16003. doi: 10.1038/mtm.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lipus A., Janosz E., Ackermann M., Hetzel M., Dahlke J., Buchegger T., Wunderlich S., Martin U., Cathomen T., Schambach A. Targeted integration of inducible caspase-9 in human iPSCs allows efficient in vitro clearance of iPSCs and iPSC-macrophages. Int. J. Mol. Sci. 2020;21:2481. doi: 10.3390/ijms21072481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Z.D., Tchao J., Wu L., Carman A.J. Precision installation of a highly efficient suicide gene safety switch in human induced pluripotent stem cells. Stem Cells Transl. Med. 2020;9:1378–1388. doi: 10.1002/sctm.20-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang S., Cohen C.J., Peng P.D., Zhao Y., Cassard L., Yu Z., Zheng Z., Jones S., Restifo N.P., Rosenberg S.A., Morgan R.A. Development of optimal bicistronic lentiviral vectors facilitates high-level TCR gene expression and robust tumor cell recognition. Gene Ther. 2008;15:1411–1423. doi: 10.1038/gt.2008.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee M.O., Moon S.H., Jeong H.C., Yi J.Y., Lee T.H., Shim S.H., Rhee Y.H., Lee S.H., Oh S.J., Lee M.Y. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl. Acad. Sci. USA. 2013;110:E3281–E3290. doi: 10.1073/pnas.1303669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelly R.J., Lopez-Chavez A., Citrin D., Janik J.E., Morris J.C. Impacting tumor cell-fate by targeting the inhibitor of apoptosis protein survivin. Mol. Cancer. 2011;10:35. doi: 10.1186/1476-4598-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bedel A., Beliveau F., Lamrissi-Garcia I., Rousseau B., Moranvillier I., Rucheton B., Guyonnet-Dupérat V., Cardinaud B., de Verneuil H., Moreau-Gaudry F., Dabernat S. Preventing pluripotent cell teratoma in regenerative medicine applied to hematology disorders. Stem Cells Transl. Med. 2017;6:382–393. doi: 10.5966/sctm.2016-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tolcher A.W., Mita A., Lewis L.D., Garrett C.R., Till E., Daud A.I., Patnaik A., Papadopoulos K., Takimoto C., Bartels P. Phase I and pharmacokinetic study of YM155, a small-molecule inhibitor of survivin. J. Clin. Oncol. 2008;26:5198–5203. doi: 10.1200/JCO.2008.17.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Giaccone G., Zatloukal P., Roubec J., Floor K., Musil J., Kuta M., van Klaveren R.J., Chaudhary S., Gunther A., Shamsili S. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J. Clin. Oncol. 2009;27:4481–4486. doi: 10.1200/JCO.2008.21.1862. [DOI] [PubMed] [Google Scholar]
- 50.Guo K., Huang P., Xu N., Xu P., Kaku H., Zheng S., Xu A., Matsuura E., Liu C., Kumon H. A combination of YM-155, a small molecule survivin inhibitor, and IL-2 potently suppresses renal cell carcinoma in murine model. Oncotarget. 2015;6:21137–21147. doi: 10.18632/oncotarget.4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tey S.K., Dotti G., Rooney C.M., Heslop H.E., Brenner M.K. Inducible caspase 9 suicide gene to improve the safety of allodepleted T cells after haploidentical stem cell transplantation. Biol. Blood Marrow Transplant. 2007;13:913–924. doi: 10.1016/j.bbmt.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.