

Abstract
The interaction of positively charged N-terminal histone tails with nucleosomal DNA plays an important role in chromatin assembly and regulation, modulating their susceptibility to post-translational modifications and recognition by chromatin-binding proteins. Here, we report residue-specific 15N NMR relaxation rates for histone H4 tails in reconstituted nucleosomes. These data indicate that H4 tails are strongly dynamically disordered, albeit with reduced conformational flexibility compared to a free peptide with the same sequence. Remarkably, the NMR observables were successfully reproduced in a 2-μs MD trajectory of the nucleosome. This is an important step toward resolving an apparent inconsistency where prior simulations were generally at odds with experimental evidence on conformational dynamics of histone tails. Our findings indicate that histone H4 tails engage in a fuzzy interaction with nucleosomal DNA, underpinned by a variable pattern of short-lived salt bridges and hydrogen bonds, which persists at low ionic strength (0–100 mM NaCl).
Keywords: fuzzy protein–DNA interactions, histone tails, molecular dynamics, NMR spectroscopy, nucleosome
Graphical Abstract
Histone H4 Tails in Nucleosomes: a Fuzzy Interaction with DNA 15N relaxation data suggest that amino-terminal histone H4 tails in reconstituted nucleosome are flexible. This result is rationalized by suitably designed MD simulations, showing that H4 tails are involved in a fuzzy interaction with nucleosomal DNA while retaining their disordered character.
Introduction
In eukaryotic cells, gene expression, replication and repair are controlled by an extremely complex network of mechanisms centered on chromatin. The fundamental building block of chromatin is the nucleosome core particle (NCP), which consists of histone octamer (assembled from two copies each of H2A, H2B, H3 and H4 histones) and ca. 147 bp double-stranded DNA wrapped around the histone core. All histones contain N-terminal tail segments, ranging from a dozen to several dozen residues. The histone tails are rich in positively charged amino acids, particularly lysines; some also include multiple glycine residues. It has been discovered early on that histone tails are disordered,[1] which is consistent with their amino-acid composition.[2] It has also been found that histone tails can weakly bind to the negatively charged DNA; the interaction is electrostatic in nature and can be scaled down by neutralizing modifications of Lys and Arg residues (or simply by raising the ionic strength of the solution).[3] Multiple lines of evidence indicate that histone tails remain disordered within the NCP and maintain tenuous contact with nucleosomal DNA (nDNA).[4]
More recent studies shed further light on the status of histone tails in nucleosomes. The rapidly increasing number of X-ray structures[5] show a lack of definable electron density from the tails, suggesting that the tails remain disordered even in the highly constrained crystalline environment at low temperature. Several NMR studies took advantage of NCP samples reconstituted with isotopically labeled recombinant histones. The HSQC spectra of such samples consist of a small number of relatively sharp peaks, corresponding to the dynamic tails.[6] In the case of the H3 tail, these resonances are slightly shifted relative to the free peptide of the same sequence[7] and show several-fold reduced mobility compared to the free peptide.[7a] Of note, H3, H4 and H2A tails all remain dynamic in the condensed nucleosomal arrays,[8] as well as in the dense NCP precipitate environment.[9] Solid-state NMR experiments employing dipolar or, alternatively, J-coupling-based transfer help to delineate the boundaries of histone tails. For example, in the case of H4 tail residues 1–15 are moving rapidly, whereas residues 16–24 appear to be moving more slowly.[9b]
Collectively, this experimental evidence has informed our understanding of the histone tails status in nucleosomes. It has been hypothesized that the tails tend to interact with nDNA (or histone core) via nonspecific, transient interactions that are mainly electrostatic in origin.[7a] Despite the tenuous character of the interaction between the histone tails and nDNA, this interaction is not inconsequential. A number of experimental studies have shown that NCPs with deleted histone tails are characterized by increased exposure of nDNA,[10] moderately lower stability[11] and compromised compaction properties.[12] Furthermore, the interaction of histone tails with nDNA makes these tails somewhat less available for binding of transcription regulatory factors. Posttranslational modifications of the tails tend to partially offset this effect, weakening tail interactions with the body of NCP.[6,7, 13]
Given the physiological significance of “fuzzy” interactions between the histone tails and nDNA, it is desirable to characterize these interactions with atomic level detail. This is, however, a challenging problem—the interactions are highly dynamic and therefore difficult to capture experimentally. In this situation, valuable insights can be obtained from Molecular Dynamics (MD) simulations. All-atom MD simulations of NCPs in explicit solvent became feasible ca. 10 years ago, and a number of such studies have been undertaken to date. Regarding the disordered N-terminal histone tails, these studies unanimously report that the tails “collapse” onto the surface of the nucleosome and become tightly bound to nDNA.[7b,13b, 14] In other words, the tails are largely immobilized on the surface of the nucleosome, remaining conformationally constrained on the time scale of hundreds of nanoseconds. This description, however, is inconsistent with the experimental evidence, especially NMR data, which are indicative of robust conformational dynamics of the histone tails on the time scale of ca. 10 ns (see Stützer et al.[7a] and other references above).
This contradiction has been noted previously, leading to speculation that MD captures those species that are trapped in local free-energy minima. These species, featuring immobilized histone tails, are assumed to be in slow exchange with other species, featuring disordered tails.[7b, 14d] It remains unclear, however, why MD simulations fail to observe these latter states. The discrepancy between the modeling results and the experimental findings remains an outstanding paradox, which hinders further progress in this important area. Here we seek to resolve this paradox. Specifically, we report residue-specific NMR chemical shifts and 15N spin relaxation rates for histone H4 tails in nucleosomes as well as an extended, microsecond MD trajectory of the nucleosome core particle that is consistent with these (and other) experimental data.
Results
For the purpose of this study, we focused on the mobile N-terminal tails of histone H4 proteins (hereafter referred to as N-H4 tails). We prepared several NCP samples reconstituted with recombinant 15N- or 15N,13C-labeled histone H4 from Xenopus Laevis and the 147-bp Widom 601 DNA motif (see Figures S1 and S2 for details).[15] As expected, the 1H,15N-HSQC spectra of these samples contained a handful of observable peaks; they were successfully assigned to residues 3–15 from the N-H4 tail, see Figure 1A. The positions of the peaks are near identical to those in the spectrum of the free peptide, which replicates the sequence of the H4 tail,[16] see Figure S3. This immediately suggests that the first fifteen N-H4 residues remain strongly disordered also in the nucleosome core particle.
Figure 1.

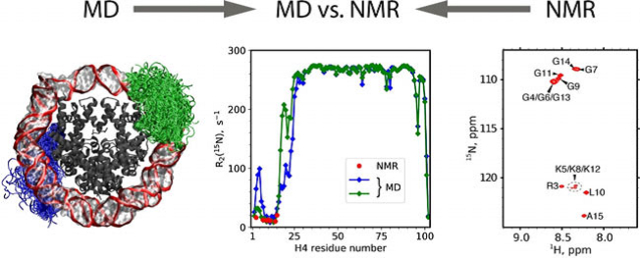
(A) 1H,15N-HSQC spectrum of mononucleosomes containing 15N-labeled H4. (B) Dynamics of N-H4 tails (pictured as blue tubes) in mononucleosome according to the MD simulation data. To generate this plot, a 2-μs trajectory of NCP in TIP4P-D water was sampled with the step of 10 ns; the extracted frames were overlaid onto the reference structure 3LZ0. The DNA backbone is shown as a red band, bases and sugars are shown as a semi-transparent surface, the bodies of histone proteins are dark grey (with tails of H3, H2A, and H2B histones not shown). The visible asymmetry in spatial distribution of the N-H4–1 and N-H4–2 tails reflects a limited convergence of the simulation.
The NCP sample with 50 mM concentration was further used to measure site-specific backbone amide 15N longitudinal (R1) and transverse (R2) relaxation rates for those N-H4 sites that give rise to well-resolved spectral resonances. The measurements were conducted using recently reported high-accuracy experiments.[17] The obtained R2 values fall in the range 10–20 s−1 (see Table S1), which is an order of magnitude higher than in the free N-H4 peptide. Furthermore, the R2 vs. the residue number profile is different from what one may expect for a freely moving protein tail. Specifically, residue R3 with R2=16.4 s−1 is followed by a stretch of residues with R2 from 9.9 to 12.5 s−1 and then A15 with R2=20.3 s−1. Clearly, the motion is partially constrained, which particularly affects R3 at the extreme N-terminus of the H4 tail.
In an attempt to rationalize these data, we have turned to MD simulations. The starting coordinates of the NCP were taken from the most relevant crystallographic structure 3LZ0.[18] The histone tails that are missing from the crystallographic coordinates were rebuilt with extended conformations (see Supporting Information). The nucleosome particle was placed in a large truncated octahedron box with TIP3P[19] water. For this system, the MD trajectory was recorded using the Amber 18[20] program with Amber ff14SB[21] force field.
To assess the validity of the obtained MD model, we have calculated per-residue 15N relaxation rates in the two H4 proteins within the NCP. The relaxation rate constants were computed with an established approach that uses MD-derived temporal correlation functions.[16] Of note, these calculations require the NCP tumbling time, which was obtained by performing HYDROPRO[22] calculations on the MD frames (sampled at 1 ns intervals) and subsequently averaging the results, τrot=146 ns. This value is in line with the earlier estimates based on NMR linewidths and fluorescence anisotropy decay rates.[23] Given that histone tails in our model are tightly coordinated to the body of the NCP (rather than extended into solvent), the application of HYDROPRO appears justified.[24] In addition to the 15N relaxation rates, we have also calculated secondary chemical shifts δsec =δ–δrc[25] for the two H4 histones. For this purpose, the NCP trajectory was processed using SHIFTX2[26] and SHIFTS[27] programs (see Supporting Information).
The comparison of the MD-based predictions and the experimental results is illustrated in Figure S4. Both experiment and computations yield near-zero δsec values for backbone 13C spins in the N-H4 tails, confirming the lack of any appreciable secondary structure in the tail region. However, the calculated relaxation rates are clearly at odds with the experiment. Specifically, 15N R2 rates for residues 3–15 in N-H4 are predicted to range from 39 to 151 s−1, overestimated on average by eight-fold relative to the available experimental values. The corresponding R1 rates are underestimated on average by three-fold. This immediately suggests that N-H4 tails are overstabilized in the MD trajectory, experiencing only limited and/or slow conformational dynamics. From the spin-relaxation perspective, parts of the simulated tails resemble the rigid NCP body, where the calculated 15N R2 rates are ca. 240 s−1 (see Figure S4). These findings are in agreement with the previous observations from MD modeling studies[7b,13b, 14] (and, in fact, add a useful quantitative dimension to these observations).
Given the major discrepancies between the experimental and simulated NMR relaxation rates, the question naturally arises of what is the potential fault in the MD simulations. We believe that the problem lies with the traditional MD setup that uses the standard TIP3P water model, which has recently been shown to produce overly compact (collapsed) representations of disordered proteins.[28] One can expect that this problem would also affect the disordered histone tails in the NCP. Recent efforts to resolve this problem have been mainly focused on water-water and water-protein interactions.[28b,c,29] Another complication that can adversely affect the MD model stems from the imperfect parameterization of salt bridges.[30] This issue is relevant for the interaction between positively charged N-H4 tails and the negatively charged nDNA. Likewise, there have been recent attempts to improve this particular aspect of MD simulations.[31]
To explore the more advanced MD modeling options, we have conducted a series of NCP simulations using the following MD setups: Amber ff15ipq + SPC/Eb,[31a] CHARMM c36m + CHARMM-modified TIP3P,[29b] CHARMM c36m with CUFIX corrections[32] + CHARMM-modified TIP3P, Amber ff14SB with the latest version of CUFIX correction[31b] (including re-parameterized interactions of arginines and lysines with the DNA phosphate groups) + TIP3P or, alternatively, TIP4P/Ew.[33] The lengths of the individual trajectories ranged from 0.5 to 1.0 μs. While these more advanced models tend to improve the situation, the results still fall short of quantitative agreement with experiment. The predicted 15N R2 rates for residues at the extremity of the N-H4 tails remain significantly overestimated, whereas the R1 rates are significantly underestimated. The only MD simulation that was able to predict spin relaxation rates in reasonable agreement with experiment was one that used the TIP4P-D[28b] water model. This model includes a corrected parameter for dispersion interactions originating at water molecules (which are underestimated in conventional water models). Consequently, it promotes the solvation of peptide chains and thus tilts the balance toward disordered protein states. The success of the TIP4P-D water model in reproducing various experimental characteristics of intrinsically disordered proteins, as well as proteins containing disordered regions, has been well documented.[28b, 34]
The N-H4 conformational ensemble from (Amber ff14SB / TIP4P-D) trajectory is illustrated in Figure 1B. The H4 tails still tend to coalesce on the surface of nucleosomal DNA and, similarly, the other histone tails are also positioned over the nDNA surface. We have used the CRYSOL[35] and GNOM[36] programs to determine the radius of gyration for the simulated NCP. The calculated value, 43.0±0.9 Å, is in agreement with the experimental result of 43.7 Å.[37] Furthermore, processing the trajectory with HYDROPRO reproduces the experimentally determined translational diffusion coefficient, 3.6 × 10−7 cm2 s−1[38] (the same calculation predicts τrot=163 ns). Hence, the experimental data appear to be consistent with the MD model featuring a relatively compact arrangement of the histone tails over the surface of nDNA.
However, in spite of the apparent attraction to nDNA, the N-H4 tails remain highly mobile in the MD model employing TIP4P-D water. This is evident from the calculated 15N spin relaxation rates (Figures 2A,B). One of the two tails in the simulation (green symbols in the plot) produces R2 rates in good agreement with experimental data, along with R1 rates that are only moderately underestimated. The other tail (blue symbols in the plot) produces overestimated R2 values for residues R3 and G7 (and presumably for the entire stretch of residues in between), but aside from that also shows reasonable agreement with the experimental results. Given the magnitude of the R2 rates, it can be immediately suggested that N-H4 tails (residues 1–15) are an order of magnitude more mobile than the histone core, but an order of magnitude less mobile than the corresponding free peptide.[16]
Figure 2.

(A,B) 15N relaxation rate constants R1, R2 and (C,D) 13Cα/β secondary chemical shifts δsec for H4 histone proteins in NCP. Experimental values are shown with red circles, the results of MD-based calculations—with blue and green diamonds (first and second copy of H4, as enumerated in the structure 3LZ0). The MD trajectory is that of NCP in TIP4P-D water, with a net length of 2 μs. Additionally, 1HN and 15N δsec data are summarized in Figure S5.
What are the specific interactions underpinning this dynamic behavior of N-H4? To answer this question, we have tracked the formation/dissolution of salt bridges and hydrogen bonds throughout the course of the (Amber ff14SB / TIP4P-D) trajectory. The results for the two N-H4 tails are summarized in Figure 3.
Figure 3.

Traces of the interactions between N-terminal tails of H4 histones (two copies in the NCP, residues 1–24) and the remaining part of the NCP. Each horizontal band represents a specific interaction involving one of the tail residues (indicated on the y-axis). For a given residue, interactions are sorted according to the time of the first appearance; the ordered list that identifies all interactions can be found in Table S2. Hydrogen bonds and salt bridges are coded blue and red, respectively. The definition of salt bridges between DNA phosphate group and arginine or lysine side chains is illustrated in Figure S6.
Considering the results in this graph, we first focus on the most flexible portion of the N-H4 tail, residues 1–15. We observe that charged residues within this segment form salt bridges with phosphate groups in the DNA backbone (red bands in the plot), but these are mostly transient, fleeting interactions. One good example is residue R3 in the second tail (N-H4–2, right half of the plot in Figure 3). This residue is found to form a salt bridge in 49% of all frames. Most of these interactions are short-lived and go away after several nanoseconds to several tens of nanoseconds. Others are more persistent, existing in on-off mode for hundreds of nanoseconds. Interestingly, such long-lasting salt bridges are invariably stabilized by hydrogen bonds (blue bands in the plot) in the proximal residues. In this particular case, the more persistent salt bridges involving R3 occur in concert with hydrogen bonds involving S1, G4 or K5. Similar pattern is also observed elsewhere in the plot. These observations are consistent with what is known about the dynamic nature of salt bridges.[39] Considering other positively charged residues in the N-H4–2 (1–15) segment, K5 and K8 have been engaged in salt-bridge interactions in 15% of all frames, while for K12 this proportion was only 9%. This is ca. 3-fold lower compared to the R3 propensity to form salt bridges. Such an outcome is expected since lysine-phosphate interactions are known to be weaker than arginine-phosphate interactions.[40] In summary, it appears that salt bridges alone are insufficient to produce a stable connection between N-H4 and nDNA. However, a sequence of rapidly formed and dissolved salt-bridge interactions apparently succeeds in keeping N-H4 in the vicinity of nDNA. Once hydrogen bonds are formed between N-H4 and nDNA, a more stable configuration ensues, where salt bridges also tend to become more permanent.
Generally, the behavior of the N-H4–2 segment (residues 1–15), as illustrated in Figure 3, is consistent with a concept of fuzzy complex.[41] While loosely associated with nDNA, this segment maintains a high degree of mobility. The intermittent salt bridges and hydrogen bonds provide temporary “anchors”, while the stretches of the peptide chain in between such anchors remain conformationally mobile (akin to floppy loops). In particular, R3 is an important anchoring residue, as confirmed by the elevated R2 rate (both predicted and found experimentally at this site). The fuzzy behavior of N-H4–2 (1–15) is visualized in Movie S1A.
The scenario observed for the other tail (N-H4–1, left portion of the plot in Figure 3) is somewhat different. The N-terminal segment exhibits similar fuzzy dynamics up to 710 ns in the trajectory. At this point, it forms a local contact with nDNA, which remains present (subject to several rearrangements) until the end of the simulation. Three distinct long-lived states observed in this timeframe all feature hydrogen bonds between the R3 side chain, positioned in the DNA minor groove, and adenine-thymine base pair dA-31(strand I) / dT31(strand J). It is, in fact, common to find arginine side chains inserted into minor grooves, where guanidinium group interacts particularly strongly with the adenine-thymine pair.[42] However, despite the relatively stable anchoring through R3, a large stretch of N-H4–1 remains highly mobile, resembling a floppy loop. This is evidenced by the low R2 rates calculated for residues 8–14 (blue symbols in Figure 2B).
Of interest, residue R3 shows average separation of 2.6 Å with the body of nucleosome in both N-H4 tails, see Figure S7A. Apparently, this residue maintains close contact with nDNA irrespective of whether it forms relatively stable hydrogen bonds with nDNA (such as the case with N-H4–1) or becomes engaged in a series of short-lived salt-bridge interactions (such as the case with N-H4–2). At the same time, the average separation between cationic residue K12 and the body of the NCP is significantly higher: 5.1 Å and 6.5 Å for N-H4–1 and N-H4–2, respectively. As already noted, this residue is a part of a flexible loop-like segment, which hovers over the surface of NCP, but at some distance from nDNA. It is relatively rare that K12 becomes directly engaged in salt bridges, making this residue amenable to posttranslational modifications and recognition by reader proteins.[43]
As for the remaining portion of the tails, 16–24, these residues become increasingly restrained toward the structured part of H4. According to the MD data, this region is characterized by conformational heterogeneity, as well as a reduced level of conformational mobility (compared to 1–15). This is consistent with the absence of electron density from residues 16–24 in the crystallographic coordinates 3LZ0 and the absence of the corresponding spectral signals in the HSQC spectrum of NCP (see Figure 1A).
In addition to structural parameters, we are also interested in temporal characteristics of the conformational dynamics in N-H4. Those can be conveniently assessed using reorientational correlation functions of the backbone N-HN vectors,[44] illustrated in Figure S8. For the most mobile residues, such as G9 and K12, the correlation functions are determined solely by conformational dynamics of the tail, with no appreciable contribution from the overall tumbling of the NCP. The characteristic time constants span the range from a picosecond to tens of nanoseconds, reflecting a range of motions such as ultrafast librations, axial fluctuations of peptide planes, local rotameric jumps and concerted jumps associated with major conformational rearrangements of the tail.[16] Residue R3, which is engaged in fuzzy interactions, experiences somewhat slower dynamics; accordingly, its correlation function is impacted to some degree by the overall NCP tumbling. As a point of contrast, residue A38, which belongs to the rigid body of the H4 histone integrated into the structure of NCP, senses only two motional modes: ultrafast librations and the overall tumbling with characteristic time of 163 ns, see Figure S8 (panel O).
Considering the significance of electrostatic interactions in the fuzzy dynamics of N-H4, one may expect to obtain interesting insights from relaxation measurements in NCP samples with different solvent ionic strength. In addition to our standard conditions of 100 mM NaCl, we have also measured site-specific 15N relaxation rates at 0 and 10 mM NaCl (summarized in Figure S9). Aside from residue R3, which appears to be insensitive to salt concentration in this range, the other residues show the expected decreases in dynamics under low-salt conditions, as evidenced by the increasing 15N R2 values. The effect is, however, moderate, meaning that the tail residues 1–15 retain a high degree of conformational flexibility even at 0 mM NaCl.
This observation is consistent with the MD data, where we cannot discern any significant differences between N-H4 dynamics at 100 and 0 mM salt (not shown). To probe the effect of electrostatics further, we have recorded a 1.35-μs trajectory of NCP in solution with 800 mM NaCl. Although nucleosome particles are known to dissociate under high-salt conditions, on a microsecond time scale this trajectory provides a reasonable in silico model to characterize the effect of salt on histone tail dynamics (see discussion following Figure S10). The N-H4 (1–15) dynamics observed in this trajectory is distinctly different from the low-salt simulations. In particular, the shape of the simulated R2 profile suggests that N-H4 behaves as a random-coil tail, with R3 no longer acting as an anchor residue (Figure S10). This result supports our notion that the fuzzy interaction between N-H4 tail and nDNA is largely electrostatic in nature.
Concluding Remarks
The above analysis exposes the limitations of the (Amber ff14SB / TIP4P-D) model. The results from the two tails point toward the lack of convergence in the simulation (e.g., compare blue and green symbols in Figures 2A,B). It is known that TIP4P-D can have a certain destabilizing effect on protein structure.[28b] In our case, the histone core of the nucleosome has remained stable, with the secondary-structure Cα rmsd fluctuating around the average value of 1.50 Å (see Figure S11A). At the same time, the nDNA rmsd increased progressively, reaching at the end of the simulation the level of 4.8 Å (average value over the final 0.5 μs). As one may expect, the increased mobility of nDNA involves mainly the outer turn (nucleotides −72 to −39 and 39 to 72), which shows weaker interactions with the histone core.[45] Specifically, we observe local displacements of the DNA turns (i.e. local contraction or stretching of the spiral), plus occasional disengagement of the first few base pairs from the histone core. Fortunately, the N-H4 tails interact almost exclusively with the inner turn (nucleotides −38 to 38) and thus remain relatively unaffected, see Figure S12. In spite of the described shortcomings, however, the (Amber ff14SB / TIP4P-D) model provides a consistent picture of the N-H4 dynamics that is in reasonable agreement with experimental data. Generally, MD simulations of large biomolecular systems often push the limits in terms of convergence and/or stability, but nonetheless produce valuable structural and functional insights.[46]
As a point of comparison, let us briefly survey the results from the (Amber ff14SB / TIP3P) simulation that are documented in the Supporting Information (see Figures S4, S10C,D and S12–S15, as well as Movie S1B). In this trajectory, N-H4 tails become coordinated to nDNA and largely immobilized. Characteristically, the entire length of the segment 3–15 is in contact with nDNA, as multiple residues, including G4, G6, G7, G11 and G13, make hydrogen bonds to nucleobases, phosphate groups or ribose rings. Consequently, this model fails to correctly predict the 15N relaxation rates in the N-H4 tails. In contrast, the (Amber ff14SB / TIP4P-D) simulation paints a different picture. While N-H4 tails maintain contact with nDNA in this latter trajectory, they also retain a great deal of mobility, reminiscent of a fuzzy complex. The transient anchoring of N-H4 to nDNA occurs mainly through arginine side chains, while lysines are mostly projected into solvent, which makes them amenable to acetylation, methylation and other modifications. These modifications control nucleosome packing, as well as recognition by reader proteins, with far-reaching consequences for cell status and disease.[47] Of note, the fuzzy state of histone tails is apparently preserved in nucleosome arrays, including large and highly compacted arrays, as reported by us previously.[8] Clearly there is only a limited amount of bulk solvent contained within the condensed array. Nevertheless, the histone tails presumably retain their ability to glide over the surface of nDNA, belonging either to their own nucleosome or to the other nearby nucleosomes.
The presented (Amber ff14SB / TIP4P-D) model predicts 15N spin relaxation rates that are in good agreement with our experimental results. Furthermore, it successfully reproduces other measurable parameters, such as chemical shifts, radius of gyration and coefficient of translational diffusion of the NCP. It is also consistent with the X-ray diffraction data, which lack the definable electron density from the histone tails. Hence this model reconciles MD and experimental perspectives on the dynamics of histone tails in nucleosomes, thereby resolving the existing controversy. Generally, MD models are of major importance for characterization of histone tails in nucleosomes (and other conformationally dynamic elements of biomolecular systems), effectively replacing conventional structural data such as crystallographic or NMR coordinates. In the context of chromatin biology, such accurate modeling of native and post-translationally modified histone tails in conjunction with quantitative experimental data is expected to lead to detailed atomistic view of nucleosome complexes and assemblies and an enhanced understanding of fundamental cellular processes.
Supplementary Material
Acknowledgements
The research was supported by NIH grants R01GM118664 to C.P.J., R01GM123743 to C.P.J. and M.G.P., S10OD012303 to C.P.J., NSF grant MCB-1715174 to C.P.J. (experimental component) and RSF grant 15-14-20038 to N.R.S. (modeling component). N.R.S. acknowledges grant 51142660 from St. Petersburg State University that provided funds to purchase GPU-powered workstations. Exploratory computations were performed at the Computer Center of St. Petersburg State University. We thank Prof. Catherine Musselman for providing the pJ201 plasmid.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.202012046.
Contributor Information
Sevastyan O. Rabdano, Laboratory of Biomolecular NMR, St. Petersburg State University, St. Petersburg 199034 (Russian Federation)
Matthew D. Shannon, Department of Chemistry and Biochemistry, The Ohio State University, Columbus, OH 43210 (USA)
Sergei A. Izmailov, Laboratory of Biomolecular NMR, St. Petersburg State University, St. Petersburg 199034 (Russian Federation)
Nicole Gonzalez Salguero, Department of Chemistry and Biochemistry, The Ohio State University, Columbus, OH 43210 (USA).
Mohamad Zandian, Department of Chemistry and Biochemistry, The Ohio State University, Columbus, OH 43210 (USA).
Rudra N. Purusottam, Department of Chemistry and Biochemistry, The Ohio State University, Columbus, OH 43210 (USA)
Michael G. Poirier, Department of Physics, The Ohio State University, Columbus, OH 43210 (USA)
Nikolai R. Skrynnikov, Laboratory of Biomolecular NMR, St. Petersburg State University, St. Petersburg 199034 (Russian Federation); Department of Chemistry, Purdue University, West Lafayette, IN 47906 (USA).
Christopher P. Jaroniec, Department of Chemistry and Biochemistry, The Ohio State University, Columbus, OH 43210 (USA).
References
- [1] a).Boublik M, Bradbury EM, Crane-Robinson C, Johns EW, Eur. J. Biochem 1970, 17, 151–159; [DOI] [PubMed] [Google Scholar]; b) Lilley DMJ, Howarth OW, Clark VM, Pardon JF, Richards BM, FEBS Lett 1976, 62, 7–10. [DOI] [PubMed] [Google Scholar]
- [2].Drozdetskiy A, Cole C, Procter J, Barton GJ, Nucleic Acids Res 2015, 43, W389–W394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cary PD, Crane-Robinson C, Bradbury EM, Dixon GH, Eur. J. Biochem 1982, 127, 137–143. [DOI] [PubMed] [Google Scholar]
- [4] a).Weintraub H, Vanlente F, Proc. Natl. Acad. Sci. USA 1974, 71, 4249–4253; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Walker IO, Biochemistry 1984, 23, 5622–5628; [DOI] [PubMed] [Google Scholar]; c) Hilliard PR, Smith RM, Rill RL, J. Biol. Chem 1986, 261, 5992–5998; [PubMed] [Google Scholar]; d) Stefanovsky VY, Dimitrov SI, Russanova VR, Angelov D, Pashev IG, Nucleic Acids Res 1989, 17, 10069–10081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kale S, Goncearenco A, Markov Y, Landsman D, Panchenko AR, Curr. Opin. Struct. Biol 2019, 56, 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhou BR, Feng HQ, Ghirlando R, Kato H, Gruschus J, Bai YW, J. Mol. Biol 2012, 421, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7] a).Stützer A, Liokatis S, Kiesel A, Schwarzer D, Sprangers R, Soding J, Selenko P, Fischle W, Mol. Cell 2016, 61, 247–259; [DOI] [PubMed] [Google Scholar]; b) Morrison EA, Bowerman S, Sylvers KL, Wereszczynski J, Musselman CA, eLife 2018, 7, e31481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gao M, Nadaud PS, Bernier MW, North JA, Hammel PC, Poirier MG, Jaroniec CP, J. Am. Chem. Soc 2013, 135, 15278–15281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9] a).Xiang SQ, le Paige UB, Horn V, Houben K, Baldus M, van Ingen H, Angew. Chem. Int. Ed 2018, 57, 4571–4575; Angew. Chem. 2018, 130, 4661 – 4665; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shi XY, Prasanna C, Nagashima T, Yamazaki T, Pervushin K, Nordenskiold L, Angew. Chem. Int. Ed 2018, 57, 9734–9738; Angew. Chem. 2018, 130, 9882–9886. [DOI] [PubMed] [Google Scholar]
- [10].Polach KJ, Lowary PT, Widom J, J. Mol. Biol 2000, 298, 211–223. [DOI] [PubMed] [Google Scholar]
- [11] a).Iwasaki W, Miya Y, Horikoshi N, Osakabe A, Taguchi H, Tachiwana H, Shibata T, Kagawa W, Kurumizaka H, FEBS Open Bio 2013, 3, 363–369; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Widlund HR, Vitolo JM, Thiriet C, Hayes JJ, Biochemistry 2000, 39, 3835–3841. [DOI] [PubMed] [Google Scholar]
- [12] a).Robinson PJJ, An W, Routh A, Martino F, Chapman L, Roeder RG, Rhodes D, J. Mol. Biol 2008, 381, 816–825; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dorigo B, Schalch T, Bystricky K, Richmond TJ, J. Mol. Biol 2003, 327, 85–96. [DOI] [PubMed] [Google Scholar]
- [13] a).Wakamori M, Fujii Y, Suka N, Shirouzu M, Sakamoto K, Umehara T, Yokoyama S, Sci. Rep 2015, 5, 17204; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gatchalian J, Wang XD, Ikebe J, Cox KL, Tencer AH, Zhang Y, Burge NL, Di L, Gibson MD, Musselman CA, et al. , Nat. Commun 2017, 8, 1489; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Munari F, Soeroes S, Zenn HM, Schomburg A, Kost N, Schroder S, Klingberg R, Rezaei-Ghaleh N, Stutzer A, Gelato KA, et al. , J. Biol. Chem 2012, 287, 33756–33765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14] a).Roccatano D, Barthel A, Zacharias M, Biopolymers 2007, 85, 407–421; [DOI] [PubMed] [Google Scholar]; b) Erler J, Zhang RH, Petridis L, Cheng XL, Smith JC, Langowski J, Biophys. J 2014, 107, 2911–2922; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li ZH, Kono H, Sci. Rep 2016, 6, 31437; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Shaytan AK, Armeev GA, Goncearenco A, Zhurkin VB, Landsman D, Panchenko AR, J. Mol. Biol 2016, 428, 221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lowary PT, Widom J, J. Mol. Biol 1998, 276, 19–42. [DOI] [PubMed] [Google Scholar]
- [16].Kämpf K, Izmailov SA, Rabdano SO, Groves AT, Podkorytov IS, Skrynnikov NR, Biophys. J 2018, 115, 2348–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17] a).Lakomek NA, Ying JF, Bax A, J. Biomol. NMR 2012, 53, 209–221; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gairí M, Dyachenko A, González MT, Feliz M, Pons M, Giralt E, J. Biomol. NMR 2015, 62, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Vasudevan D, Chua EYD, Davey CA, J. Mol. Biol 2010, 403, 1–10. [DOI] [PubMed] [Google Scholar]
- [19].Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- [20].Case DA, Ben-Shalom IY, Brozell DS, Cerutti DS, Cheatham TE, Cruzeiro VWD, Darden TA, Duke RE, Ghoreishi D, Gilson MK, et al. , AMBER 18, University of California, San-Francisco, 2018. [Google Scholar]
- [21].Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C, J. Chem. Theory Comput. 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].García de la Torre J, Huertas ML, Carrasco B, Biophys. J 2000, 78, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23] a).Feigon J, Kearns DR, Nucleic Acids Res 1979, 6, 2327–2337; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Brown DW, Libertini LJ, Small EW, Biochemistry 1991, 30, 5293–5303. [DOI] [PubMed] [Google Scholar]
- [24].Amorós D, Ortega A, García de la Torre J, J. Chem. Theory Comput 2013, 9, 1678–1685. [DOI] [PubMed] [Google Scholar]
- [25].Spera S, Bax A, J. Am. Chem. Soc 1991, 113, 5490–5492. [Google Scholar]
- [26].Han B, Liu YF, Ginzinger SW, Wishart DS, J. Biomol. NMR 2011, 50, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xu XP, Case DA, Biomol J. NMR 2001, 21, 321–333. [DOI] [PubMed] [Google Scholar]
- [28] a).Best RB, Mittal J, Proteins Struct. Funct. Bioinf 2011, 79, 1318–1328; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Piana S, Donchev AG, Robustelli P, Shaw DE, J. Phys. Chem. B 2015, 119, 5113–5123; [DOI] [PubMed] [Google Scholar]; c) Shabane PS, Izadi S, Onufriev AV, J. Chem. Theory Comput. 2019, 15, 2620–2634. [DOI] [PubMed] [Google Scholar]
- [29] a).Best RB, Zheng WW, Mittal J, Chem J. Theory Comput 2014, 10, 5113–5124; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmuller H, MacKerell AD, Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30] a).Debiec KT, Gronenborn AM, Chong LT, J. Phys. Chem. B 2014, 118, 6561–6569; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xue Y, Yuwen TR, Zhu FQ, Skrynnikov NR, Biochemistry 2014, 53, 6473–6495. [DOI] [PubMed] [Google Scholar]
- [31] a).Debiec KT, Cerutti DS, Baker LR, Gronenborn AM, Case DA, Chong LT, J. Chem. Theory Comput 2016, 12, 3926–3947; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) You S, Lee H, Kim K, Yoo J, Chem J. Theory Comput. 2020, 16, 4006–4013. [DOI] [PubMed] [Google Scholar]
- [32].Yoo J, Aksimentiev A, Phys. Chem. Chem. Phys 2018, 20, 8432–8449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Horn HW, Swope WC, Pitera JW, Madura JD, Dick TJ, Hura GL, Head-Gordon T, J. Chem. Phys 2004, 120, 9665–9678. [DOI] [PubMed] [Google Scholar]
- [34] a).Henriques J, Skepo M, J. Chem. Theory Comput 2016, 12, 3407–3415; [DOI] [PubMed] [Google Scholar]; b) Zapletal V, Mladek A, Melkova K, Lousa P, Nomilner E, Jasenakova Z, Kuban V, Makovicka M, Lanikova A, Zidek L, et al. , Biophys. J 2020, 118, 1621–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Svergun D, Barberato C, Koch MHJ, J. Appl. Crystallogr 1995, 28, 768–773. [Google Scholar]
- [36].Svergun DI, J. Appl. Crystallogr 1992, 25, 495–503. [Google Scholar]
- [37].Chen YJ, Tokuda JM, Topping T, Sutton JL, Meisburger SP, Pabit SA, Gloss LM, Pollack L, Nucleic Acids Res 2014, 42, 8767–8776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kitevski-LeBlanc JL, Yuwen T, Dyer PN, Rudolph J, Luger K, Kay LE, J. Am. Chem. Soc 2018, 140, 4774–4777. [DOI] [PubMed] [Google Scholar]
- [39] a).Tomlinson JH, Ullah S, Hansen PE, Williamson MP, J. Am. Chem. Soc 2009, 131, 4674–4684; [DOI] [PubMed] [Google Scholar]; b) Esadze A, Chen CY, Zandarashvili L, Roy S, Pettitt BM, Iwahara J, Nucleic Acids Res 2016, 44, 6961–6970; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Baird-Titus JM, Thapa M, Doerdelmann T, Combs KA, Rance M, Biochemistry 2018, 57, 2796–2813. [DOI] [PubMed] [Google Scholar]
- [40] a).Su YC, Doherty T, Waring AJ, Puchala P, Hong M, Biochemistry 2009, 48, 4587–4595; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) DeRouchey J, Hoover B, Rau DC, Biochemistry 2013, 52, 3000–3009; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li LB, Vorobyov I, Allen TW, J. Phys. Chem. B 2013, 117, 11906–11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tompa P, Fuxreiter M, Trends Biochem. Sci 2008, 33, 2–8. [DOI] [PubMed] [Google Scholar]
- [42] a).Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B, Nature 2009, 461, 1248–1253; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chiu TP, Rao S, Mann RS, Honig B, Rohs R, Nucleic Acids Res 2017, 45, 12565–12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lang DN, Schumann M, Gelato K, Fischle W, Schwarzer D, Krause E, Proteomics 2013, 13, 2989–2997. [DOI] [PubMed] [Google Scholar]
- [44].Chandrasekhar I, Clore GM, Szabo A, Gronenborn AM, Brooks BR, J. Mol. Biol 1992, 226, 239–250. [DOI] [PubMed] [Google Scholar]
- [45].Mihardja S, Spakowitz AJ, Zhang YL, Bustamante C, Proc. Natl. Acad. Sci. USA 2006, 103, 15871–15876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46] a).Salvi N, Abyzov A, Blackledge M, Angew. Chem. Int. Ed 2017, 56, 14020–14024; Angew. Chem. 2017, 129, 14208 – 14212; [DOI] [PubMed] [Google Scholar]; b) Kurauskas V, Izmailov SA, Rogacheva ON, Hessel A, Ayala I, Woodhouse J, Shilova A, Xue Y, Yuwen T, Coquelle N, et al. , Nat. Commun 2017, 8, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47] a).Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL, Science 2006, 311, 844–847; [DOI] [PubMed] [Google Scholar]; b) Goudarzi A, Zhang D, Huang H, Barral S, Kwon OK, Qi SK, Tang ZY, Buchou T, Vitte AL, He TM, et al. , Mol. Cell. 2016, 62, 169–180; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Price BD, D’Andrea AD, Cell 2013, 152, 1344–1354; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Shang WH, Hori T, Westhorpe FG, Godek KM, Toyoda A, Misu S, Monma N, Ikeo K, Carroll CW, Takami Y, et al. , Nat. Commun 2016, 7, 13465; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Nativio R, Donahue G, Berson A, Lan YM, Amlie-Wolf A, Tuzer F, Toledo JB, Gosai SJ, Gregory BD, Torres C, et al. , Nat. Neurosci 2018, 21, 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.