

Abstract
SARS‐CoV‐2 caused the emerging epidemic of coronavirus disease in 2019 (COVID‐19). To date, there are more than 82.9 million confirmed cases worldwide, there is no clinically effective drug against SARS‐CoV‐2 infection. The conserved properties of the membrane fusion domain of the spike (S) protein across SARS‐CoV‐2 make it a promising target to develop pan‐CoV therapeutics. Herein, two clinically approved drugs, Itraconazole (ITZ) and Estradiol benzoate (EB), are found to inhibit viral entry by targeting the six‐helix (6‐HB) fusion core of SARS‐CoV‐2 S protein. Further studies shed light on the mechanism that ITZ and EB can interact with the heptad repeat 1 (HR1) region of the spike protein, to present anti‐SARS‐CoV‐2 infections in vitro, indicating they are novel potential therapeutic remedies for COVID‐19 treatment. Furthermore, ITZ shows broad‐spectrum activity targeting 6‐HB in the S2 subunit of SARS‐CoV and MERS‐CoV S protein, inspiring that ITZ have the potential for development as a pan‐coronavirus fusion inhibitor.
Keywords: estradiol benzoate, fusion inhibitor, itraconazole, SARS‐CoV‐2, spike
An effective drug against COVID‐19 is urgently needed while the current SARS‐CoV‐2 pandemic. Here Itraconazole and Estradiol benzoate are identified to inhibit SARS‐CoV‐2 infection via blocking the spike protein S2 subunit‐mediated membrane fusion and entry into the host cell by targeting the highly conserved six‐helical bundle (6‐HB) fusion core. Furthermore, Itraconazole is confirmed as a potential broad‐spectrum coronavirus fusion inhibitor.
![]()
1. Introduction
Coronaviruses (CoVs) are enveloped viruses divided into αCoV, βCoV, γCoV, and δCoV groups, and 7 among them are found infectious to humans (hCoVs). The epidemic of COVID‐19 broke out in December 2019 and has subsequently spread worldwide and turned into a global pandemic.[ 1 ] Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), a positive‐sense, enveloped single‐stranded RNA β‐coronavirus, was identified as the causative agent of COVID‐19, which closely related to the virus severe acute respiratory syndrome CoV (SARS‐CoV) and Middle East respiratory syndrome CoV (MERS‐CoV). To date, the case fatality rate (CFR) of SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV are 2.2%, 37% and 5%, respectively.[ 2 ] However, there are no clinically approved prophylactic vaccines or drugs available to treat patients infected with the viruses.
The limited understanding of coronavirus led to growing interest in the exploration of viral epidemiology, mechanisms of transmission‐infection, clinical pathologies, and prevention‐treatment strategies by antiviral vaccines and drugs. However, despite the anti‐hCoVs research has been conducted since the SARS epidemic in 2002,[ 3 ] there are still no effective drugs for a coronavirus therapy. The urgent need for treating infected patients demands accelerated antiviral drug discovery. Numerous studies and clinical trials on COVID‐19 are being launched around the world, and several drugs are urgently approved for clinical use, but no solid evidence from randomized clinical trials support their efficiency on COVID‐19 patients.[ 4 ] Vaccines and new drugs targeting various proteins of SARS‐CoV‐2 are being developed, but it takes a long time for a new drug from being designed to being clinically approved. Therefore, finding a specific therapeutic for the emergency response to the SARS‐CoV‐2 by repurposing clinically approved drugs is a feasible approach.
SARS‐CoV‐2, with a genome of 29 881 bp in length (GenBank no. MN908947), encodes 9860 amino acids which form structural and non‐structural proteins.[ 5 ] The S glycoprotein covered on the surface of coronaviruses is a structural protein involved in receptor recognition and viral entry into the host cells, while this protein is critical for viral life cycle and highly conservative to most hCoVs. It has been confirmed that the SARS‐CoV‐2 initiates its infection by binding to the angiotensin‐converting enzyme 2 (ACE2) cell receptor.[ 6 , 7 ] The S protein includes two distinct functional domains: S1 domain and S2 domaiin, responsible for receptor binding and membrane fusion, respectively.[ 7 , 8 , 9 ]
S1 domain recognizes and binds to the cell receptor ACE2 via the receptor binding domain (RBD) of its trimeric S protein, which makes it a valuable drug target. However, previous studies showed that it lacks antibody‐mediated cross‐protection between SARS‐CoV‐2 and SARS‐CoV infections.[ 10 ] It has previously been proved that replacing aspartic acid with glycine at the position 614 of S1 region (D614G) would promote an open conformation that is more favorable to ACE2 association and increased infectivity of the SARS‐CoV‐2,[ 11 ] suggesting that the highly mutable characteristic of S1 domain prevents it to be an ideal broad‐spectrum drug designing target.
The S2 domain is responsible for viral fusion and entry, composed successively of fusion peptide (FP), heptapeptide repeat sequence 1 (HR1), heptapeptide repeat sequence 2 (HR2), transmembrane domain (TM), and cytoplasmic domain (CP).[ 12 ] HR1 and HR2 are the main structures of S2 subunit as they could form a six‐helical bundle (6‐HB), which is essential for the viral fusion.[ 13 ] Once binding to the host cell, the FP inserts into the target cell membrane, the pre‐hairpin coiled‐coil of HR1 domain is exposed to initiate the interaction between HR1 and HR2 to form a homotrimer assembly. Three highly conserved hydrophobic grooves are exposed on the surface of HR2, while it forms helix and loop to interact with HR1 domain, then the 6‐HB is formed. The viral envelope and cell membrane are brought into close proximity for viral fusion, thus enabling the virus to bind and fuse with the cell membrane.[ 14 ] Unlike the S1 subunit, the S2 subunit showed low variability, low fluctuations in molecular dynamics (MD), and displayed a trimer cavity.[ 15 ] The HR domain has been attracted many interests for discovering a therapeutic agent. In contrast with RBD region, the HR domain and the mode of interaction between HR1 and HR2 are highly conserved among hCoVs, even in other class I fusion proteins.[ 16 ] Previous studies of class I viral fusion proteins from enveloped viruses have reported that peptides derived from HR2 domain could bind to the viral HR1 domain competitively and inhibit the viral infection, elucidating that the HR domain may be an ideal target for the entry inhibitors against SARS‐CoV‐2 infection.
Earlier studies have successfully designed peptides that can effectively inhibit viral entry by binding to the HR region of hCoVs.[ 14 ] In addition to peptides fusion inhibitors, we screened clinically approved drugs targeting to the S2 subunit via virtual screening, each of the drugs in this collection has been previously optimized for safety and bioavailability. We succeeded in finding two drugs, Itraconazole (ITZ) and Estradiol benzoate (EB), which could potentially inhibit the viral fusion to the host cells, further we validated them on pseudotyped and authentic SARS‐CoV‐2 infections in vitro.
ITZ is a highly effective broad‐spectrum antifungal of the triazole group, which also presents a great diversity of non‐antifungal activity and has been used to treat a wide range of diseases. It has been reported that ITZ has potential to be an alternative drug for the treatment of advanced cancer patients, particularly those difficult to treat with conventional therapies.[ 17 ] More studies presented it can be used to regulate inflammation or immune related diseases.[ 18 ] EB is a pro‐drug ester of estradiol, a naturally occurring hormone that circulates endogenously within the human body through binding to the estrogen receptor (ER) including ERα and ERβ subtypes.[ 19 ] EB is typically used in hormone therapy with women, treatment for gynecological disorders,[ 20 ] and cure of prostate cancer.[ 21 ]
A series of experiments were carried out and two drugs were found with potential to inhibit the viral entry by binding to HR1 region and preventing the formation of 6‐HB. ITZ and EB also exhibited promising anti‐SARS‐CoV‐2 activity in cell‐based assays. Subsequent experiments with other hCoV pseudoviruses (PsV) and cell‐cell fusion assays proved that ITZ not only inhibited the entry of SARS‐CoV‐2, but also had inhibitory effects on SARS‐CoV and MERS‐CoV infections, which brings expectations to broadly inhibit coronaviruses of the β family.
2. Results and Discussion
2.1. Virtual Screening Identified ITZ and EB Were Interacted with S2 Domain of Spike Protein
The recent outbreak of COVID‐19 caused by SARS‐CoV‐2 has become a global pandemic, and there is a number of clinically approved drugs that have been repurposed as antiviral candidates at clinical trials currently, however, without sufficient information, suggesting for effective drugs targeting SARS‐CoV‐2 infection.[ 22 ] Various viral proteins responsible for different physiological stages of coronavirus have been shown valuable targets based on our previous studies on coronaviruses and drugs targeting class I fusion proteins. As the S protein is responsible for viral entry and fusion of SARS‐CoV‐2, it is a potential promising drug targeting for virus entry inhibitor.
The S protein is composed of S1 subunit and S2 subunit, on S1 subunit there is an RBD region responsible for virus binding to the host cell receptor ACE2, thus the S1 subunit was previously considered as a suitable virus entry inhibitor target.[ 23 ] However, subsequent studies have revealed that the RBD region is highly mutable in the evolutionary process, which may lead to significant differences in receptor recognition regions among different coronaviruses, making RBD unsuitable as a target for broad‐spectrum anti‐coronavirus drug screening.[ 24 ]
The HR1 and HR2 from S2 subunit could form a 6‐HB, a structure core essential for the viral fusion. The HR regions and the modes of action between HR1 and HR2 are conserved in various coronaviruses, and it has been demonstrated in previous studies that peptides derived from the HR2 region of class I fusion proteins could compete for binding to viral HR1, thus effectively inhibit viral infection. Therefore, in previous studies, peptides derived from coronaviruses have also been considered as an option for broad‐spectrum anti‐coronavirus drugs,[ 16 , 25 ] many drugs targeting at other viral 6‐HB have been developed and showed promising interests.[ 26 ] Recent studies also showed this fusion domain of SARS‐CoV‐2 can produce broad cross‐neutralization antibody, which make the fusion domain as a promising drug target.[ 27 ] However, it is challenging to quickly overcome the existing barriers to form effective drugs because of the unstable structure of peptides in solution, the same is true for other small molecular drugs, where the formation of a new drug requires a lengthy animal and clinical trials before clinical usage, which makes it improbable to address the current notorious issue of COVID‐19. Therefore, screening the clinically approved drugs and validation the drug effects through in vitro is one effective way to solve the current dilemma.[ 28 ] As a novel choice, we chose the S2 subunit as the target for our anti‐coronavirus drug study, using the pre‐fusion and post‐fusion conformations of S protein close to HR1 region as the binding site, which are the hot sites to interfere with the formation of 6‐HB, screened the clinically approved drugs by virtual screening.
In order to rapidly identify potential drugs against the ongoing SARS‐CoV‐2 infection, we facilitated drug repurposing from clinically approved drug library (DrugBank) which carried out by other study to repurposing anti‐SARS‐CoV‐2 drugs by selected metallo‐compounds recently.[ 28 ] Initially, virtual screening was carried out targeting HR1 domain with pre‐fusion or post‐fusion conformation of spike protein, respectively. In the pre‐fusion S protein structure, there isn't any interaction information between HR1 domain and HR2 domain because of the lacking HR2 residues from the structure, but a visible channel is identified between two interwoven protomers closed to HR1 domain (Figure 1a), this channel was defined as the pre‐fusion drug target. While in the post‐fusion conformation structure, three HR1 domain form into a 3‐helical bundle face‐to‐face, and HR1 domain bind to HR2 domain via its back side (Figure 1d). The interface between HR1 domain and HR2 domain is defined as potential post‐fusion drug target site. This stragtegy can potentially identify inhibitors to interfere with the formation of 6‐HB, which was validated by Ebola fusion inhibitors identification.[ 29 ] Parallelly virtual screening targeting to these pre‐fusion conformation and post‐fusion conformations were performed from clinically approved drug library (Figure 1a,d). Two clinically approved drugs, ITZ and EB, presented feasible binding affinities both in pre‐fusion and post‐fusion conformations targeting HR1 domain. ITZ has −9.4 and −8.6 kcal mol−1 binding affinities to pre‐fusion and post‐fusion conformations, respectively (Figure 1b,c). In addition, EB has −8.6 and −8.3 kcal mol−1 binding affinities to pre‐fusion and post‐fusion conformations, respectively (Figure 1e,f). ITZ interacted with 16 and 15 residues with pre‐fusion and post‐fusion S structures, respectively (Figure S1b,c, Supporting Information), however, EB only interacted with 10 and 11 residues with pre‐fusion and post‐fusion S structures, respectively (Figure S1d,e, Supporting Information). Most of those residues are located in the S protein HR1 region (Figure S1a, Supporting Information).
Figure 1.
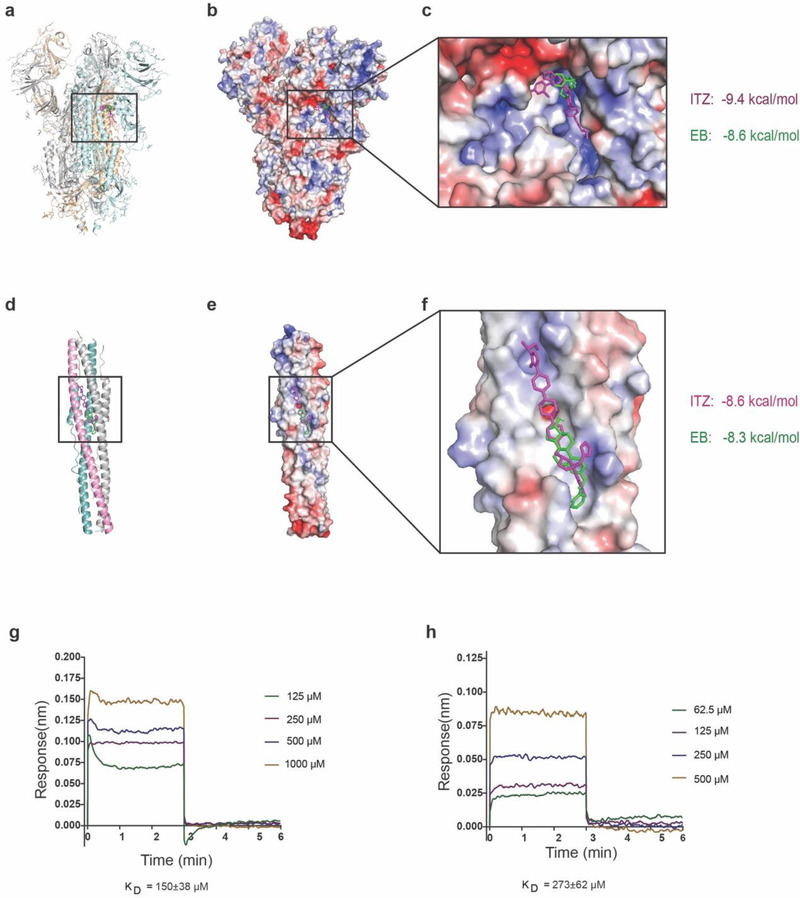
ITZ and EB interacted with S2 domain of spike protein via virtual screening. a) Potential drug binding sites of S protein in pre‐fusion conformation, b) ITZ colored in magenta and EB colored in green were bound to the pre‐fusion conformation of S protein from virtual screening, c) binding details of ITZ and EB to pre‐fusion of S protein. d) Potential drug binding sites of S protein in post‐fusion conformation, e) ITZ colored in magenta and EB colored in green bound to the post‐fusion conformation of S protein from virtual screening, f) binding details and binding affinities of ITZ and EB to post‐fusion of S protein, g) binding between ITZ and SARS‐CoV‐2 S protein by BLI, h) binding between EB and SARS‐CoV‐2 S protein by BLI.
To verify the candidate drugs from virtual screening, we performed the direct binding assay between ITZ/EB and SARS‐CoV‐2 S protein by biolayer interferometry (BLI) technology. We observed the direct binding with binding affinities ≈ 150 µm within ITZ and S protein interaction, and 273 µm within EB and S protein interaction (Figure 1g,h). Other attempts to detect the binding affinities between ITZ/EB and S2 protein were failed because of lacking expressed and purified S2 protein. This direct binding data gave the strongly evidences to support the interaction between ITZ/EB and SARS‐CoV‐2 S protein.
2.2. ITZ and EB Blocked Spike Protein‐Mediated Membrane Fusion
To determine whether these two drugs inhibited the entry of SARS‐CoV‐2 into host cell, we first developed an SARS‐CoV‐2 S protein‐mediated cell‐cell fusion assay in which the 293T cells transiently expressed SARS‐CoV‐2 S protein and EGFP (293T//SARS‐CoV‐2‐S/EGFP) as effecter cells, while Vero cells as host cells. The fusion syncytium between the two cells were calculated, and the percentage of S protein‐mediated viral‐cell fusion inhibited by drug was analyzed. It was found that no more syncytium were observed under bright‐field or fluorescence‐field microscopy after the co‐incubation with one of these two drugs at the screening concentration, ITZ and EB (Figure 2a). After treatment with ITZ or EB, we further found ITZ and EB inhibited the formation of syncytium in a dose‐dependent manner (Figure S2a,b, Supporting Information). The half maximal inhibitory concentration (IC50) values of ITZ and EB for inhibiting SARS‐CoV‐2 S protein‐mediated intercellular fusion in cell experiments were 0.45 µM and 1.02 µM, respectively (Figure 2b,c).
Figure 2.
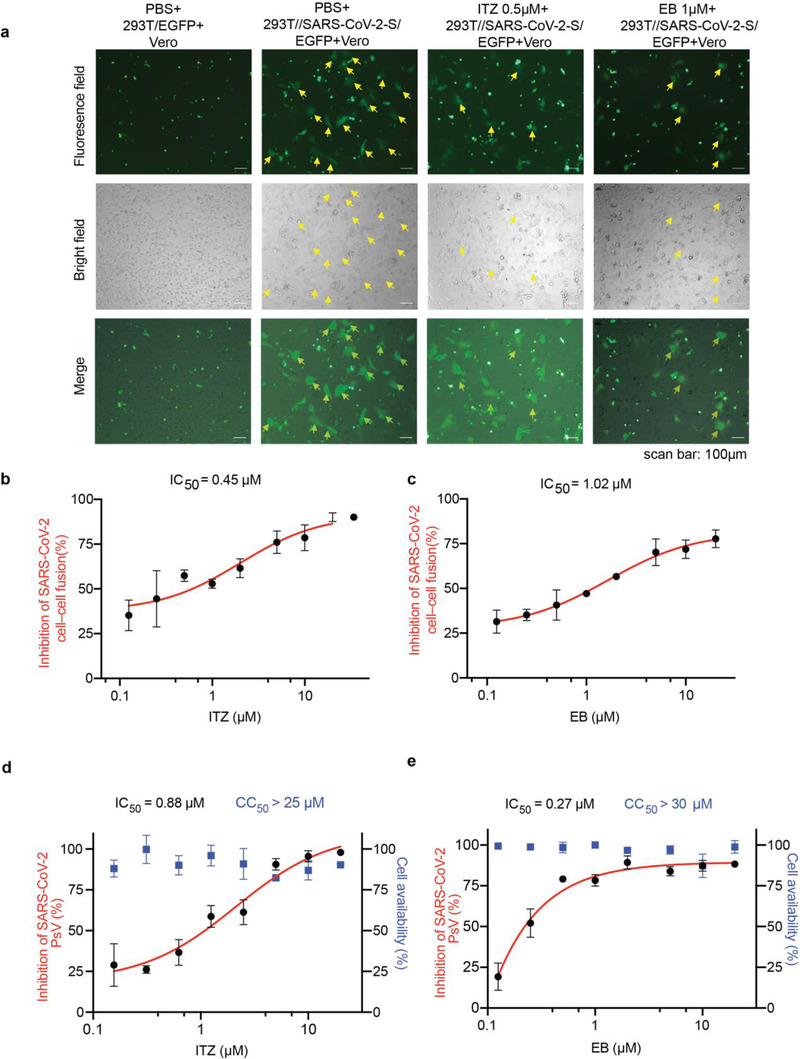
ITZ and EB inhibited SARS‐CoV‐2 infection via blocking spike protein‐mediated membrane fusion. a) Cell‐cell fusion mediated by SARS‐CoV‐2 S protein. ITZ and EB inhibited the SARS‐CoV‐2 S‐mediated syncytium formation on Vero cells at 24 h. 293T/EGFP, HEK‐293T cells transfected with vector (pAAV‐IRES‐EGFP), scale bars, 100 µm. b,c) Quantification of GFP‐positive syncytia. Three images per condition were acquired and processed. Data were normalized to PBS controls and depicted as fold changes in mean GFP expression. Dose‐response curves and IC50s of b) ITZ and c) EB on inhibiting SARS‐CoV‐2 S‐mediated syncytium formation. d,e) Inhibition of entry of SARS‐CoV‐2 PsV by d) ITZ and e) EB. SARS‐CoV‐2 PsV were pretreated with gradient concentrations of ITZ or EB, then inoculated with 293T‐ACE2 cells. The luciferase activity were measured 48 h post transduction. The blue squares indicate the percent cytotoxicity of the ITZ or EB. Results are representative of n = 3. All experiments were done in triplicates and repeated three times, data are expressed as means ± S.D.(error bar).
Additionally, SARS‐CoV‐2 S protein‐mediated PsV assays were developed to evaluate the inhibitory activity of ITZ and EB on the SARS‐CoV‐2 infection as previously.[ 13 ] PsV is a classical tool that can mimic the entry of virus into cells. It is widely used to evaluate the inhibitory activity of antiviral drugs against packaged virus infection via the surface envelope protein. From the PsV assays results, it indicated that ITZ and EB could successfully inhibit the invasion of SARS‐CoV‐2 PsV into cells stably expressing human ACE2 (293T‐ACE2), with IC50 values of 0.88 and 0.27 µm, respectively, which exhibiting consistent inhibitory activity to SARS‐CoV‐2 infection comparing to cell‐cell fusion assays. And the cell availability assay indicated ITZ and EB had low cell toxicity with half cytotoxic concentrations (CC50) values more than 25 and 30 µm respectively (Figure 2d,e).
From the SARS‐CoV‐2 cell‐cell fusion assays and pseudotyped virus assays, ITZ and EB were found to target the spike protein to inhibit the membrane fusion of SARS‐CoV‐2. Since ITZ and EB both have reasonable inhibitory ability to SARS‐CoV‐2, they are worthy to be investigated further.
2.3. ITZ and EB Blocked the Formation of 6‐HB between HR1 and HR2
To decipher the mechanism of ITZ and EB in inhibiting SARS‐CoV‐2 infection, flow cytometry analysis was carried out to detect the binding of SARS‐CoV‐2 RBD to ACE2 receptor. ITZ (10 µm) showed no effect on RBD‐Fc protein binding to the ACE2 receptor, while the binding between SARS‐CoV‐2 RBD and 293T‐ACE2 cells was slightly weakened after EB (10 µm) treatment (Figure S1f–i, Supporting Information). These data provide evidence that ITZ may blocks SARS‐CoV‐2 S protein‐mediated cell entry by targeting to the regions in S2 subunit. We then further developed a peptides‐based assay to explore the interaction between ITZ/EB and spike protein. Since peptides derived from HR1 and HR2 regions of type I fusion proteins could simulate the fusion core structure of S proteins in vitro,[ 25 ] the HR1 domain can interact with HR2 domain from post‐fusion structure of spike protein. Based on the HR1/HR2 interaction information, we synthesized peptide HR1 (HR1P) and peptide HR2 (HR2P) to elucidate the inhibitory mechanism of ITZ and EB.[ 30 ] At the same time, we performed biophysical analysis of ITZ and EB inhibiting SARS‐CoV‐2 PsV infection by non‐denatured polyacrylamide gel electrophoresis (N‐PAGE) and circular dichroism (CD) spectroscopy.
N‐PAGE is a polyacrylamide gel electrophoresis method for proteins that remain active without addition of denatured agents, which has been successfully applied to test the formation of 6‐HB structures of other type I fusion proteins.[ 25 , 31 ] We used N‐PAGE to test whether ITZ and EB can interact with HR1 or HR2 to inhibit the formation of 6‐HB. HR1P was incubated with ITZ or EB at indicated concentrations for the first 30 min. HR2P was then added for another 30 min co‐incubation. In the gel, the individual HR1P is positively charged and moves upwards under the negatively charged electrophoresis condition, with no bands shown in the gel (lane 2, Figure 3a,b), while negatively charged HR2P alone showed a band at lower position in the gel (lane 3, Figure 3a,b). The HR1P/HR2P mixture showed a band on the upper part of the gel at about 26 kDa (lane 4, Figure 3a,b) and this band represented 6‐HB formed by the interaction between HR1P and HR2P, according to the related studies.[ 32 , 33 ] After adding ITZ (lane 5–8, Figure 3a) or EB (lane 5–7, Figure 3b), the bands representing 6‐HB showed a weakening trend on the gel according to the concentration increase of drugs. Troglitazone, a drug with no effect on cell‐cell fusion assay, was selected as negative control and showed no effect on 6‐HB formation (Figures S2c, S3g, Supporting Information).
Figure 3.
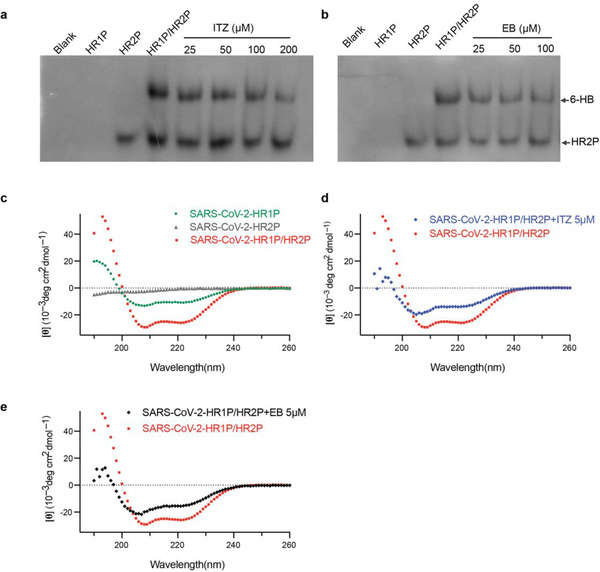
Molecular mechanism of ITZ and EB for inhibiting SARS‐CoV‐2 infection. a,b) Dose‐dependent inhibition of 6‐HB formation by ITZ (a) and EB (b), as assesed by N‐PAGE. HR1P (lane 2) were prtreated with indicated concentration of ITZ or EB, followed by the addition of HR2P (lane 3). ITZ (a, lane 4–8) or EB (b, lane 4–7) inhibit the 6‐HB (upper part) formation between HR1P and HR2P. c‐e) ITZ and EB interfered the formation of 6‐HB, as analysed using CD spectroscopy. The CD profile of HR1P/HR2P mixture (10µM) showed the characteristic α‐helical spectrum with double minima peak at 208 and 222 nm (c). With the addition of ITZ (d) or EB (e), the secondary structures of 6‐HB in HR1P/HR2P mixture were affected, showing a lower α‐helicity.
To examine the structural characteristics of HR1 and HR2 regions of the SARS‐CoV‐2 S protein, peptides corresponding to various concentration of ITZ or EB were analyzed by CD spectroscopy, a technique that monitor the secondary structural and disordered content of peptides. HR1P was incubated with 50 mm phosphate buffer (pH 7.2) or ITZ/EB at room temperature for 30 min, then HR2P was added for a 30 min co‐incubation, CD wave scanning was measured from 190 m to 260 nm at 4 °C.
As shown in Figure 3c, the CD profile of HR1P/HR2P mixture showed the characteristic α‐helical spectrum with double minima peak at 208 and 222 nm, while HR1P alone displayed a relatively low α‐helicity and HR2P alone showed a random coil structure. With the addition of ITZ or EB, the secondary structure of 6‐HB in HR1P/HR2P mixture was affected, showing a lower α‐helicity (Figure 3d,e and Figure S3a–f, Supporting Information), suggesting that ITZ and EB inhibit the SARS‐CoV‐2 infection by reducing the formation of 6‐HB. It was reported that chloroquine (CQ) was an effective antiviral drug but not target to SARS‐CoV‐2 S2 or 6‐HB.[ 34 ] So it was selected as a negative control in this assay. CQ showed no effects on 6‐HB formation (Figure S3f, Supporting Information).
2.4. ITZ and EB Inhibited Authentic SARS‐CoV‐2 Virus In Vitro
To validate the efficiency for SARS‐CoV‐2 inhibiting, we tested the inhibiting activity with authentic SARS‐CoV‐2 infection in vitro. As above mentioned, our N‐PAGE and CD assays identified ITZ and EB as potent inhibitors of SARS‐CoV‐2 S‐mediated membrane fusion. These inhibitions were most likely a results of inhibition of the formation of 6‐HB fusion core between HR1 and HR2 regions. Subsequently, we evaluated the inhibitory activity of ITZ and EB against authentic SARS‐CoV‐2 infection by using in vitro virus infection assays.[ 35 ] We measured the inhibition of SARS‐CoV‐2 and the production of progeny viruses by ITZ and EB.
Vero‐E6 cells infected with SARS‐CoV‐2 at 0.05 MOI were incubated with ITZ or EB at different dilution concentrations, then the copies of SARS‐CoV‐2 in cellular supernatant were calculated by monitoring the viral RNA via real‐time quantitative reverse transcription PCR (RT‐qPCR). As a result, ITZ and EB exhibited antiviral activity against SARS‐CoV‐2 with median effective concentration (EC50) values of 3.25 and 6.72 µm, respectively (Figure 4a,b). Meanwhile, both ITZ and EB had no toxicity on Vero‐E6 cells at 200 µm. Corresponding to that, the infection of progeny viruses from diluted cellular supernatant after the treatment of ITZ or EB was also detected by plaque formation assays, CQ was made as a control. As shown in Figure 4c–f, plaques in Vero‐E6 cells reduced with the concentration increase of ITZ or EB, which further verified the inhibitory effect of ITZ and EB on SARS‐CoV‐2 infection. Moreover, an immunofluorescence assay was conducted to detect SARS‐CoV‐2 N expression which reflected the infection of SARS‐CoV‐2 in Vero‐E6 cells. In Figure 4g,h, a dose‐dependent inhibitory effect on SAR‐CoV‐2 infection was observed after the treatment of ITZ or EB. Both drugs also showed lower cytotoxic effects on different cell lines (Figure S5, Supporting information). All the evidences revealed that ITZ and EB can potentially be used as treatment to against SARS‐CoV‐2 infection.
Figure 4.
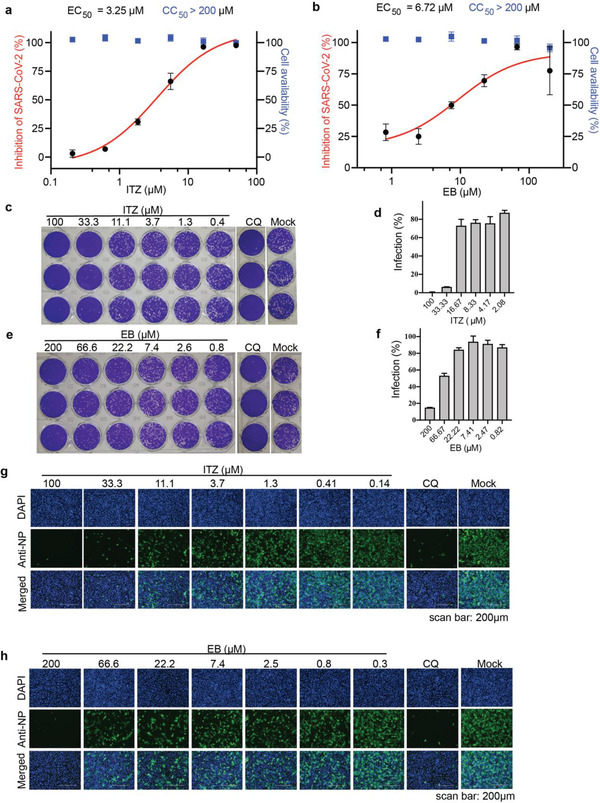
ITZ and EB inhibited authentic SARS‐CoV‐2 virus entry in vitro. a,b) Inhibition of SARS‐CoV‐2 infection by a) ITZ and b) EB. Vero‐E6 cells were pretreated with gradient concentrations of ITZ or EB for 1 hour and infected by SARS‐CoV‐2 at MOI = 0.05. Viral infection (red) and cell viability (bule) were determined by using RT‐qPCR and CCK‐8 assays, respectively. c‐f), There images per concentration of ITZ (c,d) or EB (e,f) were acquired, as detected by using plaque assay. CQ (10 µM) was made as a positive control. (Data are represent as mean ± SD of the percentage infection relative to Mock, n = 3), Immunofluorescence images of g) ITZ or h) EB dose‐dependently inhibiting SARS‐CoV‐2 N protein expression (blue: DAPI; green: SARS‐CoV‐2 N protein). CQ (10 µm) was made as a positive control. Scale bars: 200 µm.
2.5. ITZ Is a Potential Pan‐Coronavirus Countermeasure
To expand our knowledge to understand the broad‐spectrum inhibition of coronaviruses targeting spike membrane fusion domain, we selected ITZ to evaluate the pan‐coronavirus inhibitory activity with pseudoviral assay.
As previous studies investigated, the HR regions and the modes of interaction between HR1 and HR2 were highly conserved among different coronaviruses using class I fusion proteins, the broad‐spectrum anti‐coronavirus activity of fusion inhibitors are expected. To further evaluate the breadth of fusion inhibitory activity, we investigated ITZ activity with PsV and cell‐cell fusion assays mediated by the S protein of SARS‐CoV or MERS‐CoV. As expected, ITZ inhibited spike protein mediated entry of SARS‐CoV PsV and MERS‐CoV PsV into host cells with IC50s values of 1.2 and 1.66 µm, respectively, at the meantime, we determined the cell availability for ITZ and EB on host cell, the results present low cell toxicity (Figure 5a,d,b). In the meantime, ITZ (10, 20 µm) significantly inhibited SARS‐CoV or MERS‐CoV S‐mediated syncytium formation (Figure 5c,e and Figure S4a,b, Supporting Information). And the similar IC50 values on the spike protein mediated cell fusion assays (Figure 5d,f) were found. CD assays also showed ITZ can disrupt 6‐HB formation of SARS‐CoV and MERS‐CoV (Figure 5g–h). However, using the similar strategies, we could not detect significantly antiviral ability for EB to inhibit SARS‐CoV or MERS‐CoV S protein‐mediated cell fusion and PsV infection (Figure S4c–f, Supporting Information). N‐PAGE and CD assays also showed no effects for EB inhibiting 6‐HB formation of SARS‐CoV or MERS‐CoV (Figure S4g–n, Supporting Information). Those data indicated ITZ and EB have different binding modes to various coronaviruses, and ITZ may interact with more key residues of S2 subunit of S protein, which consistent with the docking data (Figure S1, Supporting Information). Meanwhile, we found that ITZ and EB showed no cytotoxic effect on normal human bronchial epithelial cells (BEAS‐2B) (Figure S5c,g, Supporting Information), implying an acceptable toxicity of ITZ and EB.
Figure 5.
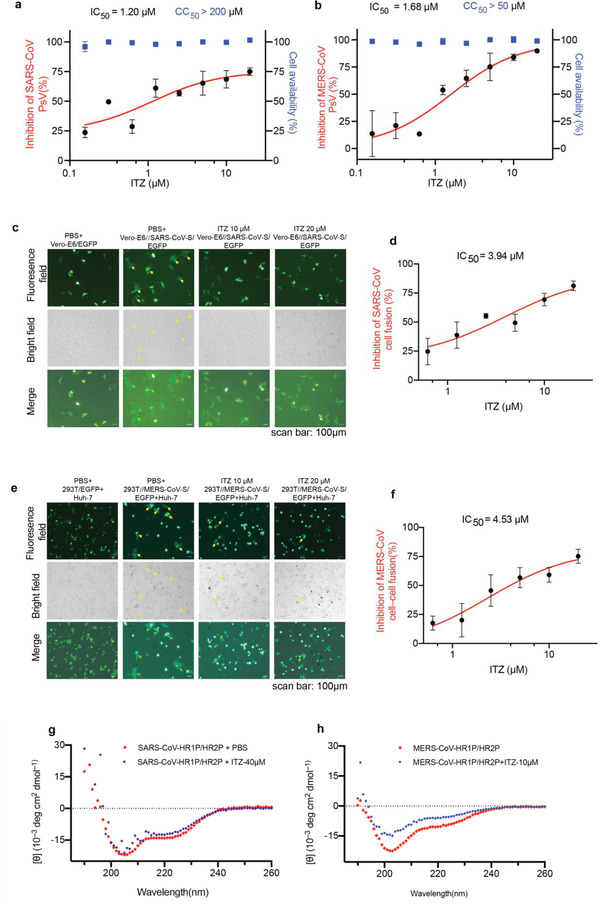
The inbition of ITZ on SARS‐CoV and MERS‐CoV PsV infection. a,b) Pseudovirus infection (red) and cell viability (bule) were determined by using luciferase and CCK‐8 assays, respectively. Dose‐response curves and IC50s of ITZ on inhibiting the entry of a) SARS‐CoV PsV and b) MERS‐CoV PsV. Each data point represents the mean of there technical replicates (n = 3), data are expressed as means ± SD (error bar). c) Cell‐cell fusion mediated by SARS‐CoV S protein. Vero‐E6 cells transiently expressed SARS‐CoV S protein and EGFP (293T//SARS‐CoV‐S/EGFP). ITZ (10, 20µm) significantly inhibited SARS‐CoV S‐mediated syncytium formation. Vero‐E6/EGFP, Vero‐E6 cells transfected with vector (pAAV‐IRES‐EGFP), scale bars, 100 µm. d) Dose‐response curve and IC50 of ITZ on inhibiting SARS‐CoV S‐mediated syncytium formation. e) ITZ (10, 20µm) inhibited the MERS‐CoV S‐mediated syncytium formation on Huh‐7 cells at 24 h. 293T/EGFP, HEK‐293T cells transfected with vector (pAAV‐IRES‐EGFP). Scale bars, 100 µm. f) Dose‐response curve and IC50 of ITZ on inhibiting MERS‐CoV S‐mediated syncytium formation and quantification of GFP‐positive syncytia. Three images per condition were acquired and processed. Data were normalized to PBS controls and were depicted as fold changes in mean GFP expression. g,h) ITZ interfered with the formation of 6‐HB of g) SARS‐CoV or h) MERS‐CoV, as analysed using CD spectroscopy. The CD profiles of SARS‐CoV HR1P/HR2P (15 µm) and MERS‐CoV HR1P/HR2P mixture (10 µm) showed the characteristic α‐helical spectrum with double minima peak at 208 and 222 nm. With the addition of ITZ (40 µm for SARS‐CoV and 10 µm for MERS‐CoV), the secondary structures of 6‐HB in HR1P/HR2P mixture were affected, showing a lower α‐helicity.
SARS‐CoV‐2 poses a great threat to human life and economic development. In fact, it has the potential to coexist with humans for a long time. Therefore, the emergency of specific anti‐hCoV drugs with broad‐spectrum inhibitory activity is of great significance for solving the current dilemma of COVID‐19 and preventing possible coronavirus diseases in the future. In our studies, ITZ and EB, viral fusion inhibitors, are drugs repurposed from clinically approved drugs, they bind to the S protein HR1 region to inhibit the SARS‐CoV‐2 entry, presenting broading‐spectrum inhibitory activity. It provides the new choice to fight future hCoV pandemic.
3. Conclusion
We identified two drugs, ITZ and EB, can bind to pre‐fusion and post‐fusion conformation of spike protein fusion domain to prevent S protein‐mediated viral invasion. Moreover, ITZ presents anti‐SARS‐CoV and anti‐MERS‐CoV activities at the several micromolar level, suggesting its broad‐spectrum anti‐coronavirus agents against multiple hCoV infections. Together, ITZ has the potential to become a specific drug against SARS‐CoV‐2 infection and broad‐spectrum coronavirus inhibitor to fight future coronavirus infections. Since many of these clinically approved molecules have been advanced into the clinic, the known pharmacological and human safety profiles of these drugs will accelerate their preclinical and clinical evaluation for COVID‐19 treatment.
4. Experimental Section
Materials
HEK‐293T (Human, embryonic kidney), Vero (African green monkey, kidney), Vero‐E6 (African green monkey, kidney), and Huh‐7 (Human, liver) cells were cultivated in Dulbecco's Modified Eagle Medium (Gibco) supplemented with 10% fetal bovine serum (Capricorn Scientific), 100 U mL−1 of penicillin and 0.1 µg mL−1 of streptomycin (Gibco). HEK‐293T cells that stably express human ACE2 (293T‐ACE2) have been described previously[ 13 ] and were cultivated in the presence of 2 µg mL−1 puromycin (Invivogen). BEAS‐2B (Human, epithelial cells were isolated from normal bronchial epithelium) was cultured in Lonza BEGM BulletKit (Lonza). All cell lines were obtained from ATCC (Manassas, VA) and were incubated at 37 °C and 5% CO2 in a humidified atmosphere. ITZ and EB were purchased from TargetMol (China). The peptides, including SARS‐CoV‐2 HR1P (residues 924–965), SARS‐CoV‐2 HR2P (residues 1168‐1203),[ 36 ] SARS‐CoV HR1P (residues 892–931), SARS‐CoV HR2P (residues 1153‐1189),[ 32 ] MERS‐CoV HR1P (residues 998‐1039), and MERS‐CoV HR2P (residues 1251‐1286)[ 30 ] were synthesized by GL Biochem Ltd (China).
Plasmids encoding SARS‐CoV S glycoprotein (pcDNA3.1‐SARS‐CoV S), SARS‐CoV‐2 S glycoprotein (pcDNA3.1‐SARS‐CoV‐2 S and pAAV‐IRES‐EGFP‐SARS‐CoV‐2 S) and MERS‐CoV S glycoprotein (pcDNA3.1‐MERS‐CoV S and pAAV‐IRES‐EGFP‐MERS‐CoV S) have been described previously.[ 37 ] Empty luciferase reporter vector (pNL4‐3.Luc.R‐E‐) was maintained in our laboratory. Plasmid pAAV‐IRES‐EGFP was purchased from Hedgehogbio Science and Technology Ltd.
Structure‐Based Virtual Screening
Virtual screening strategies were modified from previous studies in screening Ebola virus fusion inhibitors.[ 29 ] Pre‐fusion structure (pdb code: 6vsb) and post‐fusion core of 6‐HB structure (pdb code: 6LXT) were selected as the docking structure. In pre‐post structure, a channel between two protomers close to HR1 region at S2 fusion domain of the spike protein was defined as the docking site. In the post‐fusion structure, one of the HR2 residues from 1163 to 1197 were removed and the channel occupied by this HR2 was defined as the docking grid, clinically approved drugs from DrugBank were prepared in MGLTools1.5.4 and virtual screening was performed via program autodock vina. For each drug docking, ten binding modes were analyzed and the pose with top score was selected according to the binding affinity from computation, and further manually checked the interaction.
Cell–Cell Fusion Assay
To prepare effector cells (293T//SARS‐CoV‐2‐S/EGFP), HEK‐293T cells were transfected with pAAV‐IRES‐GFP‐SARS‐CoV‐2 S or vehicle pAAV‐IRES‐GFP for 48 h with PolyJet (SignaGen) transfection. Targeted cells (Vero) were seeded in 96‐well plates (104 cells for each well) 4 h prior to cell‐cell fusion assay. 293T//SARS‐CoV‐2‐S/EGFP cells were incubated with ITZ or EB at room temperature for 30 min and were overlaid on Vero cells with 2 × 104 cells well−1. 293T/EGFP cells were used as a negative control. After 24 h incubation, the samples were visualized by bright‐field and fluorescence‐field microscopy using a Zeiss scanner and the ZEN imaging software.
To prepare Vero‐E6//SARS‐CoV‐S/EGFP cells as described before,[ 38 ] Vero‐E6 cells were grown on coverslips in 24‐well plates and transfected with pAAV‐IRES‐GFP‐SARS‐CoV S or pAAV‐IRES‐GFP with PolyJet (SignaGen) transfection. After 6 h, Vero‐E6//SARS‐CoV‐S/EGFP cells were treated with gradient concentrations of ITZ for 24 h. Cells were incubated for 1 h in serum‐free medium containing of 2 µg mL−1 trypsin to induce fusion. The medium were replaced with fresh DMEM containing 10% FBS, and the cells were incubated for 6 h. Vero‐E6/EGFP cells were used as a negative control. The samples were visualized by bright‐field and fluorescence‐field microscopy using a Zeiss scanner.
To prepare effector cells (293T//MERS‐CoV‐S/EGFP), HEK‐293T cells were transfected with pAAV‐IRES‐GFP‐MERS‐CoV S or pAAV‐IRES‐GFP for 48 h with PolyJet (SignaGen) transfection. After Huh‐7 cells were co‐cultured with 293T//MERS‐CoV‐S/EGFP or 293T/EGFP cells in the absence or presence of ITZ or EB for 48 h, cell‐cell fusion were photographed under a Zeiss microscope with fluorescence or bright light.
Pseudovirus (PsV) Assay
Pseudovirus was produced by co‐transfection HEK‐293T cells with pNL4‐3.Luc.R‐E‐ and plasmids encoding either SARS‐CoV‐2 S, SARS‐CoV S or empty vector by using PolyJet (SignaGen). MERS‐CoV PsV were produced by co‐transfection 293T cells with psPAX2, pLenti‐GFP, and plasmid encoding MERS‐CoV S by using PolyJet (SignaGen). The PsV supernatants were harvested at 48 h post transfection, centrifuged at 2000 × g for 10 min and passed through 0.45 µm filter to remove cell debris. For infection, target cells (293T‐ACE2, Vero‐E6 or Huh‐7) were grown in 96‐well plates until they reached 60–80% confluency. PsV supernatant were mixed with gradient concentrations of ITZ or EB at room temperature for 30 min. Then 100 µL well−1 of the respective mixture were added to target cells (triplicate samples). At 48 or 72 h post‐transduction, culture supernatants were aspirated, and cells were lysed with 1× cell lysis reagent (Promega) for 10–15 min at room temperature. The cells lysates were then transferred to white, opaque‐walled 96‐well plates, and luciferase activity was quantified by measuring luminescence upon addition of luciferase assay substrate (Promega) using a Synergy HTX (Bio Tek).
Native‐PAGE (N‐PAGE)
N‐PAGE was used to detect the inhibitory activity of ITZ or EB on formation of 6‐HB between HR1P and HR2P as described previously. SARS‐CoV‐2 HR1P was dissolved in ddH2O, and SARS‐CoV‐2 HR2P was dissolved in phosphate buffer (pH 7.4). HR1P (30 µm) with or without indicated concentration of ITZ or EB were incubated at room temperature for 30 min, followed by the addition of HR2P (30 µm). The mixtures were incubated for another 30 min and separated by 18% Tris‐glycine gel with constant 125 V at room temperature for 2 h. The gel was stained with coomassie blue staining (HaoMa Biotechnology) and imaged with a Tanon 2500‐B scanner (Tanon Science and Technology).
Circular Dichroism Spectroscopy
CD spectra were monitored on a Chirascan plus ACD (Applied Photophysics Ltd). HR1P and HR2P (from SARS‐CoV‐2/ SARS‐CoV / MARS‐CoV for ITZ) were dissolved in buffer (0.1 m KCl, 0.05 m PO4, pH 7.2) at a final concentration of 10 µm. Briefly, HR1P were incubated with PBS or drugs at 25 °C for 30 min, followed by addition of HR2P (10 µm). After further incubation for 30 min, the CD wave scans were measured from 190 to 260 nm at 4 °C with the bandwidth of 2 nm and the step size of 1nm. The [θ]222 value of −33 000 deg cm2 dmol−1 was taken to correspond to 100% α‐helical content as described previously.[ 39 ]
Assay of Antiviral Activities of ITZ and EB with authentic SARS‐CoV‐2 In Vitro
Authentic SARS‐CoV‐2 (isolate Wuhan‐Hu‐1) was preserved at Wuhan institute of virology, Chinese Academy of Sciences. Its associated operations were all performed in a biosafety level 3 (BSL‐3) facility. The target Vero‐E6 cells were pre‐treated with gradient‐diluted ITZ or EB for 1 hour, and SARS‐CoV‐2 virus were then added at a multiplicity of infection (MOI) of 0.05. The viruses and cells were incubated for 1 h at 37 °C. After that, fresh medium with corresponding concentrations of ITZ or EB were added. Supernatants were lysed using MiniBEST Viral RNA/DNA Extraction Kit (Takara) 24 h later, and the viral RNAs were reversed transcription with PrimeScriptTM RT reagent Kit and gDNA Eraser (Takara), viral copies in cell supernatants were quantified from viral cDNA by a standard curve method on ABI 7500 (Takara TB Green Premix Ex Taq II, Japan) with a pair of primers targeting S gene. The forward primer 5’‐GCTCCCTCTCATCAGTTCCA‐3’ and the reverse primer: 5’‐CTCAAGTGTCTGTGGATCACG‐3’.
Indirect Immunofluorescence Assay
Vero‐E6 cells collected from the authentic SARS‐CoV‐2 inhibition assay as described above were fixed by 4% paraformaldehyde (Bio‐Rad), permeabilized with 0.2% Triton X‐100 (Sigma) for 30min. Non‐specific binding sites were blocked with 2% skim milk and incubated Anti‐NP polyantibodies. Subsequently, cells were incubated with goat‐anti‐Rabbit IgG H&L (Alexa Fluor 488) (Abcam) for 1 h followed counterstained with DAPI staining solution. Fluorescence images were acquired using Axio Observer microscope (Zeiss).
Plaque Reduction Assay
Vero‐E6 cells were seeded in 24‐well plates overnight. Cellular supernatants collected from the authentic SAR‐CoV‐2 inhibition assay were first diluted 1000 folds with 2% FBS DMEM, and then 200 µL liquor were added to each well and incubated with the cells for 1 h at 37 °C. After that, inoculum was absolutely removed, cells were washed twice with PBS, and the monolayer cells were overlaid with 2% FBS DMEM containing 0.9% methylcellulose. Four days later, the methylcellulose medium were aspirated, and cells were washed with PBS. After fixing by 4% paraformaldehyde overnight (Bio‐Rad), cells were followed by staining with 1% crystal violet. The plaques were manually counted and the pictures were captured with a Leica Camera.
Flow Cytometry Assay
SARS‐CoV‐2 RBD‐Fc protein was expressed and purified according to previous study.[ 40 ] Flow cytometry analysis was carried out as described previously.[ 41 ] Briefly, the 293T‐ACE2 cells were plated in 12‐well plates overnight. RBD‐Fc protein (2 µg mL−1) were pre‐incubated with different concentration of ITZ or EB at room temperature for 30 min. The mixture was added to 293T‐ACE2 cells and incubation at 37 °C for 20 min. After staining with goat anti‐human FITC‐conjugated IgG antibody (1:500, Sigma), the cells were analyzed by flow cytometer (BD FACSCanto II) and Flowjo software.
Biolayer Interferometry
The SARS‐CoV‐2 S protein was expressed and purified from 293F cells according to previous studies.[ 9 , 42 ] S protein was immobilized on the streptavidin (SA) biosensors via reagent NHS‐PEG12‐Biotin (Thermo Fisher, USA). Biotin‐S protein was diluted in PBS to a concentration of 20 µg mL−1 and loaded for 300 s. BLI assays were performed on the FortBio Octet K2 instrument and the affinity constant (K D), association rate constant (K on) and dissociation rate constant (K off) were calculated using 1:1 binding model on GraphPad Prism software version 8.0.
Cytotoxicity Assay
Cytotoxicity of drugs to cells (Vero‐E6, Vero, Huh‐7, 293T‐ACE2, BEAS‐2B) was detected by Cell Counting Kit‐8 (CCK‐8) (Topscience). Briefly, cells were seeded into 96‐well plates (104 per well) and cultured at 37 °C overnight. A total 100 µL of DMEM containing indicated concentrations of ITZ or EB was added to cells. Following 24, 48, or 72 h incubation, 10 µL of CCK‐8 solution was dropped to each well 4 h prior to the measurement of absorbance at 450 nm.
Statistical Analysis
All statistical data analyses were performed in Prism version 8.0. EC50 and IC50 values were calculated using a one‐way analysis of variance (ANOVA), p values < 0.05 were considered significant. For details regarding the statistical tests applied, please refer to the figure captions.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting information
Acknowledgements
C.Y. and X.Y.P. contributed equally to this work. This project granted by National Natural Science Foundation of China (32041005 to R.Z.), Major scientific and technological projects of Guangdong Province (2019B020202002 to S.L.) and China Academy of Traditional Chinese Medicine (ZZ13‐035‐02, 2019XZZX‐LG04 to S.L.); Fund of Natural Science Foundation of Guangdong Province (2018A030313056 and 202020012611500005 to W.X.). Guangzhou Science and Technology Program (201904010477 to W.X. and 2020B111110001 to S.L.).
Contributor Information
Shuwen Liu, Email: liusw@smu.edu.cn.
Gengfu Xiao, Email: xiaogf@wh.iov.cn.
Wei Xu, Email: xuwei3322@smu.edu.cn.
References
- 1. Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., Tan W., China Novel Coronavirus I., Research T., N. Engl. J. Med. 2020, 382, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Peiris J. S. M., Lai S. T., Poon L. L. M., Guan Y., Yam L. Y. C., Lim W., Nicholls J., Yee W. K. S., Yan W. W., Cheung M. T., Cheng V. C. C., Chan K. H., Tsang D. N. C., Yung R. W. H., Ng T. K., Yuen K. Y., Lancet 2003, 361, 1319; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B., Lancet 2020, 395, 497; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yeung M. L., Yao Y., Jia L., Chan J. F., Chan K. H., Cheung K. F., Chen H., Poon V. K., Tsang A. K., To K. K., Yiu M. K., Teng J. L., Chu H., Zhou J., Zhang Q., Deng W., Lau S. K., Lau J. Y., Woo P. C., Chan T. M., Yung S., Zheng B. J., Jin D. Y., Mathieson P. W., Qin C., Yuen K. Y., Nat. Microbiol. 2016, 1, 16004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nkengasong J., Nat. Med. 2020, 26, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanders J. M., Monogue M. L., Jodlowski T. Z., Cutrell J. B., JAMA, J. Am. Med. Assoc. 2020, 323, 1824. [DOI] [PubMed] [Google Scholar]
- 5.a) Chen L., Liu W., Zhang Q., Xu K., Ye G., Wu W., Sun Z., Liu F., Wu K., Zhong B., Mei Y., Zhang W., Chen Y., Li Y., Shi M., Lan K., Liu Y., Emerg. Microbes Infect. 2020, 9, 313;. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chan J. F., Kok K. H., Zhu Z., Chu H., To K. K., Yuan S., Yuen K. Y., Emerg. Microbes Infect. 2020, 9, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoffmann M., Kleine‐Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N. H., Nitsche A., Muller M. A., Drosten C., Pohlmann S., Cell 2020, 181, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cai Y., Zhang J., Xiao T., Peng H., Sterling S. M., R. M. Walsh, Jr. , Rawson S., Rits‐Volloch S., Chen B., Science 2020, 369, 1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Zhou P., Yang X. L., Wang X. G., Hu B., Zhang L., Zhang W., Si H. R., Zhu Y., Li B., Huang C. L., Chen H. D., Chen J., Luo Y., Guo H., Jiang R. D., Liu M. Q., Chen Y., Shen X. R., Wang X., Zheng X. S., Zhao K., Chen Q. J., Deng F., Liu L. L., Yan B., Zhan F. X., Wang Y. Y., Xiao G. F., Shi Z. L., Nature 2020, 579, 270;. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Walls A. C., Park Y. J., Tortorici M. A., Wall A., McGuire A. T., Veesler D., Cell 2020, 181, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wrapp D., Wang N., Corbett K. S., Goldsmith J. A., Hsieh C. L., Abiona O., Graham B. S., McLellan J. S., Science 2020, 367, 1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Yang R., Lan J., Huang B., A R., Lu M., Wang W., Wang W., Li W., Deng Y., Wong G., Tan W., EBioMedicine 2020, 58, 102890;. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lv H., Wu N. C., Tsang O. T., Yuan M., Perera R., Leung W. S., So R. T. Y., Chan J. M. C., Yip G. K., Chik T. S. H., Wang Y., Choi C. Y. C., Lin Y., Ng W. W., Zhao J., Poon L. L. M., Peiris J. S. M., Wilson I. A., Mok C. K. P., Cell Rep. 2020, 31, 107725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Korber B., Fischer W. M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E. E., Bhattacharya T., Foley B., Hastie K. M., Parker M. D., Partridge D. G., Evans C. M., Freeman T. M., de Silva T. I., McDanal C., Perez L. G., Tang H., Moon‐Walker A., Whelan S. P., LaBranche C. C., Saphire E. O., Montefiori D. C., Cell 2020, 182, 812;. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang L., Jackson C. B., Mou H., Ojha A., Rangarajan E. S., Izard T., Farzan M., Choe H., Nat. Commun. 2020, 11, 6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu C., Feng Y., Gao F., Zhang Q., Wang M., Biochem. Biophys. Res. Commun. 2006, 345, 1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xia S., Zhu Y., Liu M., Lan Q., Xu W., Wu Y., Ying T., Liu S., Shi Z., Jiang S., Lu L., Cell Mol. Immunol. 2020, 17, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xia S., Xu W., Wang Q., Wang C., Hua C., Li W., Lu L., Jiang S., Int. J. Mol. Sci. 2018, 19, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kalathiya U., Padariya M., Mayordomo M., Lisowska M., Nicholson J., Singh A., Baginski M., Fahraeus R., Carragher N., Ball K., Haas J., Daniels A., Hupp T. R., Alfaro J. A., J. Clin. Med. 2020, 9, 1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu S. W., Xiao G. F., Chen Y. B., He Y. X., Niu J. K., Escalante C. R., Xiong H. B., Farmar J., Debnath A. K., Tien P., Jiang S. B., Lancet 2004, 363, 938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aftab B. T., Dobromilskaya I., Liu J. O., Rudin C. M., Cancer Res. 2011, 71, 6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Tsai Y. C., Tsai T. F., Dermatol. Ther. 2019, 9, 271;. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Aftab B. T., Dobromilskaya I., Liu J. O., Rudin C. M., Cancer Res. 2011, 71, 6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Refaie M. M. M., El‐Hussieny M., Biomed. Pharmacother. 2017, 95, 223. [DOI] [PubMed] [Google Scholar]
- 20. Israel G. E., Tarver D. E., Transgender Care: Recommended Guidelines, Practical Information, and Personal Accounts, Temple University Press, Philadelphia, PA: 2001. [Google Scholar]
- 21. Di Donato M., Cernera G., Giovannelli P., Galasso G., Bilancio A., Migliaccio A., Castoria G., Mol. Cell. Endocrinol. 2017, 457, 35. [DOI] [PubMed] [Google Scholar]
- 22. Ludwig S., Zarbock A., Anesth. Analg. 2020, 131, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Ying T., Du L., Ju T. W., Prabakaran P., Lau C. C., Lu L., Liu Q., Wang L., Feng Y., Wang Y., Zheng B. J., Yuen K. Y., Jiang S., Dimitrov D. S., J. Virol. 2014, 88, 7796;. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tai W., Wang Y., Fett C. A., Zhao G., Li F., Perlman S., Jiang S., Zhou Y., Du L., J. Virol. 2017, 91, e01651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Li F., J. Virol. 2015, 89, 1954;. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang Y., Yang C., Xu X. F., Xu W., Liu S. W., Acta Pharmacol. Sin. 2020, 41, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu L., Liu Q., Zhu Y., Chan K. H., Qin L. L., Li Y., Wang Q., Chan J. F. W., Du L. Y., Yu F., Ma C. Q., Ye S., Yuen K. Y., Zhang R. G., Jiang S. B., Nat. Commun. 2014, 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pattnaik G. P., Chakraborty H., J. Membr. Biol. 2020, 253, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a) Liu B., Shi Y., Zhang W., Li R., He Z., Yang X., Pan Y., Deng X., Tan M., Zhao L., Zou F., Zhang Y., Pan T., Zhang J., Zhang X., Xiao F., Li F., Deng K., Zhang H., Cell Mol. Immunol. 2020, 17, 1098;. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ma X., Zou F., Yu F., Li R., Yuan Y., Zhang Y., Zhang X., Deng J., Chen T., Song Z., Qiao Y., Zhan Y., Liu J., Zhang J., Zhang X., Peng Z., Li Y., Lin Y., Liang L., Wang G., Chen Y., Chen Q., Pan T., He X., Zhang H., Immunity 2020, 53, 1315;. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Song G., He W.‐t., Callaghan S., Anzanello F., Huang D., Ricketts J., Torres J. L., Beutler N., Peng L., Vargas S., Cassell J., Parren M., Yang L., Ignacio C., Smith D. M., Voss J. E., Nemazee D., Ward A. B., Rogers T., Burton D. R., Andrabi R., bioRxiv 2020, 2020.09.22.308965. [Google Scholar]
- 28. Yuan S., Wang R., Chan J. F., Zhang A. J., Cheng T., Chik K. K., Ye Z. W., Wang S., Lee A. C., Jin L., Li H., Jin D. Y., Yuen K. Y., Sun H., Nat. Microbiol. 2020, 5, 1439. [DOI] [PubMed] [Google Scholar]
- 29. Singleton C. D., Humby M. S., Yi H. A., Rizzo R. C., Jacobs A., J. Virol. 2019, 93, e00676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu L., Liu Q., Zhu Y., Chan K. H., Qin L., Li Y., Wang Q., Chan J. F., Du L., Yu F., Ma C., Ye S., Yuen K. Y., Zhang R., Jiang S., Nat. Commun. 2014, 5, 3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leslie G. J., Wang J. B., Richardson M. W., Haggarty B. S., Hua K. L., Duong J., Secreto A. J., Jordon A. P. O., Romano J., Kumar K. E., DeClercq J. J., Gregory P. D., June C. H., Root M. J., Riley J. L., Holmes M. C., Hoxie J. A., PLoS Pathog. 2016, 12, e1005983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu S., Xiao G., Chen Y., He Y., Niu J., Escalante C. R., Xiong H., Farmar J., Debnath A. K., Tien P., Jiang S., Lancet 2004, 363, 938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu Q., Xia S., Sun Z., Wang Q., Du L., Lu L., Jiang S., Antimicrob. Agents Chemother. 2015, 59, 742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Celı K. I., Onay‐Besı Kcı A., Ayhan‐Kilcigı L. G., J. Biomol. Struct. Dyn. 2020, 1. [Google Scholar]
- 35. Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G., Cell Res. 2020, 30, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xia S., Zhu Y., Liu M., Lan Q., Xu W., Wu Y., Ying T., Liu S., Shi Z., Jiang S., Lu L., Cell Mol. Immunol. 2020, 17, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Romeo A., Iacovelli F., Falconi M., Virus Res. 2020, 286, 198068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Belouzard S., Chu V. C., Whittaker G. R., Proc. Natl. Acad. Sci. USA 2009, 106, 5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xia S., Yan L., Xu W., Agrawal A. S., Algaissi A., Tseng C. K., Wang Q., Du L., Tan W., Wilson I. A., Jiang S., Yang B., Lu L., Sci. Adv. 2019, 5, eaav4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang C., Pan X., Xu X., Cheng C., Huang Y., Li L., Jiang S., Xu W., Xiao G., Liu S., Signal Transduct. Target Ther. 2020, 5, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gu H., Chen Q., Yang G., He L., Fan H., Deng Y. Q., Wang Y., Teng Y., Zhao Z., Cui Y., Li Y., Li X. F., Li J., Zhang N. N., Yang X., Chen S., Guo Y., Zhao G., Wang X., Luo D. Y., Wang H., Yang X., Li Y., Han G., He Y., Zhou X., Geng S., Sheng X., Jiang S., Sun S., Qin C. F., Zhou Y., Science 2020, 369, 1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hsieh C. L., Goldsmith J. A., Schaub J. M., DiVenere A. M., Kuo H. C., Javanmardi K., Le K. C., Wrapp D., Lee A. G., Liu Y., Chou C. W., Byrne P. O., Hjorth C. K., Johnson N. V., Ludes‐Meyers J., Nguyen A. W., Park J., Wang N., Amengor D., Lavinder J. J., Ippolito G. C., Maynard J. A., Finkelstein I. J., McLellan J. S., Science 2020, 369, 1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information