

Abstract
The ongoing pandemic of the coronavirus disease (Covid‐19), caused by the spread of the severe acute respiratory syndrome coronavirus 2 (SARS CoV‐2), highlights the need for broad‐spectrum antiviral drugs. In this Essay, it is argued that such agents already exist and are readily available while highlighting the challenges that remain to translate them into the clinic. Multivalent inhibitors of viral infectivity based on polymers or supramolecular agents and nanoparticles are shown to be broadly acting against diverse pathogens in vitro as well as in vivo. Furthermore, uniquely, such agents can be virucidal. Polymers and nanoparticles are stable, do not require cold chain of transportation and storage, and can be obtained on large scale. Specifically, for the treatment of respiratory viruses and pulmonary diseases, these agents can be administered via inhalation/nebulization, as is currently investigated in clinical trials as a treatment against SARS CoV‐2/Covid‐19. It is believed that with due optimization and clinical validation, multivalent inhibitors of viral infectivity can claim their rightful position as broad‐spectrum antiviral agents.
Keywords: antivirals, microbicides, multivalency
Ongoing pandemic highlights the urgent need to identify broad‐spectrum antiviral agents. In this Essay it is argued that such agents are already known (polymers, nanoparticles) and available for translation to clinic, with a particular promise of drug administration via inhalation.
![]()
The current coronavirus (SARS‐CoV‐2/COVID‐19) pandemic has highlighted the world's need for broad spectrum antiviral agents. We need drugs to buy time for vaccine development or to manage viral outbreaks when the development of a vaccine is elusive. These drugs should be available at the start of the pandemic. Their role would be to stop or at least to slow down the contagion by decreasing the average number of infected people by a single sick patient. Medicinal chemistry of antiviral agents has been a story of incredible success,[ 1 ] but each virus has been, and is being treated individually, via the so‐called “one bug–one drug” approach.[ 2 ] For a drug to be ready even before a new virus emerges, it has to be a broad‐spectrum one,[ 2 ] an antiviral counterpart of penicillin (the first broad‐spectrum antibiotic).[ 3 ] “Broad spectrum” of activity means that one drug would be suited as an antiviral measure against diverse viruses, from different virus families. Ideally, broad spectrum antiviral drug would be effective against nonenveloped and enveloped viruses, with dense glycoprotein corona or sparse, with RNA and/or DNA based genome, and diverse, dissimilar viral proteins. The aim of this Essay is to illustrate that promising candidates for such agents in fact are known and are ready to start the translational path, namely multivalent, macro‐ or supramolecular inhibitors of virus cell entry.
Polymers represent a historically important class of multivalent antiviral agents. In fact, the history of polymers and antiviral agents goes back by almost a century, to the times almost as far back as the origin of polymers as chemical entities: observation of antiviral effects for polymers can be traced back at least to the 1930s.[ 4 ] Initial experiments were inspired by the observations that proteins can inhibit infectivity of tobacco mosaic virus (plant virus) and later vaccinia virus (a classic “enveloped” virus). These proteins were of protamine family and were positively charged. In parallel, in 1942, Cohen[ 5 ] reported isolation and crystallization of plant viruses by the negatively charged polymers, namely heparin, hyaluronic acid, and chondroitin sulfate. In those early days, it was known that heparin can bind proteins and Cohen aimed to see if heparin can replace the nucleic acid within the viral capsid. Instead, result of this study was crystallization of several plant viruses and influenza, in the presence of polyanions. Subsequent development of antiviral polymers was fairly rapid and productive, both for polycations and polyanions, natural and synthetic by origin.[ 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 ]
In 1968, De Somer et al. presented what can be considered the first attempted illustration that polyanions are in fact broad‐spectrum antiviral agents, in vitro.[ 14 ] This study also provided an early indication of antiviral effects of polymers being a fairly universal phenomenon, observed for many polymers rather than for a select few. The same team and in the same year also presented what is, to our knowledge, the first illustration of antiviral effects elicited by polymers in vivo.[ 15 ] Subsequent decades of research on inhibitors of virus cell entry were fruitful in finding many new polymers with lower inhibitory concentrations and also in enlarging the chemical toolbox to other macromolecules.[ 16 ]
With regards to the mechanism of antiviral activity of polymers, in 1951 Stahmann co‐workers made an observation that inhibition of virus infectivity by poly‐Lysine correlates with the ionization of lysine amine (is pH dependent) and occurs very fast, but is also quickly reversed.[ 17 ] Independently, Vaheri[ 18 ] documented that antiviral effects of heparin are reversed by simple dilution of the culture medium. The hypothesis that was put forward based on these results was that there is a direct contact between the viral particles and the infectivity inhibitor, and that the main driving force for this interaction is electrostatics (ionic bonds). De Somer et al. postulated that in vivo, another mechanism contributes to the overall antiviral activity of polyanions, namely stimulation of production of interferon,[ 15 ] an ultrapotent cytokine that activates immunity. Nevertheless, it is now accepted that multivalent inhibitors (polymers, dendrimers, and nanoparticles) form direct contact with the viral particles and in doing so neutralize infectivity (Figure 1 ) acting as entry inhibitors.[ 16 , 19 ] Sulfated, sulfonated, and carboxyl‐containing agents are able to inhibit a large number of viruses, especially the ones whose viral attachment ligands seeks heparan sulfate proteoglycans (HSPG).[ 20 , 21 , 22 ] Macromolecules containing sialic acids mimics bind to and inhibit viruses whose viral attachment ligand binds to sialic acids (e.g., influenza).[ 19 , 23 , 24 , 25 ]
Figure 1.
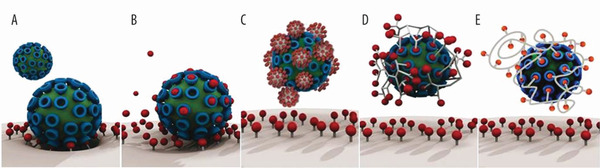
A) Interaction between a virus particle and a cell surface can be inhibited using B) small molecule drugs or—more effectively—by multivalent inhibitors of virus cell entry such as C) nanoparticles, D) hyperbranched polymers, and E) linear polymers. Adapted with permission.[ 19 ] Copyright 2016, American Chemical Society.
As was the case with small molecule antivirals, development of macromolecular inhibitors was also spurred by the discovery of the human Immunodefficiency virus (HIV) as a causative agent of the acquired immunodeficiency syndrome (AIDS), and the realization that polymers are potent and efficacious inhibitors of infectivity of this virus. Regretfully, clinical trials have failed.[ 26 ] One of the reasons of failure is that systemic antiviral effects require continuous and maintained presence of polymers at a relatively high concentration, which can be toxic. The second reason is that virus inhibition by polymers is a reversible process, which means that the pathogen can escape from the inhibitor.[ 26 ] By far and large, this led to the demise of interest in polymers as antivirals.
There are two notable exceptions. First one is illustrated by the nucleic acid polymers (phosphothioate backbone), negatively charged macromolecules per se, that bind hepatitis B viral particle surface antigen.[ 27 ] Hepatitis B virus (HBV) is rather unique in that infected cells produce new viral particles but also produce a tremendously higher number of empty pseudoparticles that carry HBV surface antigen. The sheer amount of the latter distracts and exhausts the natural immune response and the body fails to clear the viral load. Administration of nucleic acid polymers effectively neutralizes the HBV pseudoparticles and this effect is currently being investigated in clinical trials as a measure towards curing HBV viremia as well as the coinfection with hepatitis B and D viruses.[ 27 ] Second success of polymers as antiviral agents is that of the Starpharma product Vivagel. In this case, dendritic polyanion polymers are used as ingredients in condom lubricants such as to counter infectivity of HIV, the herpes simplex virus (HSV), and possibly other sexually transmitted viruses.
Ionic, electrostatic interactions between polymers and viruses are the common denominator in the observed antiviral activity of the compounds described so far,[ 16 ] regardless of the virus particle isoelectric point.[ 28 ] However, electrostatic interactions alone are insufficient to create a persistent binding. Indeed, electrostatic polymer‐virus interaction is reversible, and so is the antiviral effect. Game‐changing opportunity was proposed through the combined use of two weak interactions, namely electrostatics and hydrophobic interactions. Typically, this combination leads to lower inhibition concentrations and in a number of cases it also leads to a damage to the viral envelop and an irreversible antiviral effect.[ 29 , 30 ] This is best‐illustrated by the example of sulfated polysaccharides. While effect of sulfated polysaccharides is reversed by dilution, modification of these molecules with hydrophobic groups (aliphatic or aromatic) affords unique, virucidal materials.[ 29 , 30 ] Importance of combining electrostatics with hydrophobic interactions to make broad spectrum antivirals was also highlighted in our own recent work.[ 22 ] Specifically, Zelikin lab analyzed a panel of polyanions as broad spectrum antiviral agents and observed that lead candidates were united by combining in their structure anionic charge and well‐defined hydrophobicity of the backbone.[ 22 ] Furthermore, importance of hydrophobic–hydrophilic balance is also highlighted in the structure of the nucleic acid inhibitors in clinical trials;[ 31 ] and the dendrimer molecule in the Vivagel preparation is also a sulfonate derivative of a hydrophobic aromatic group.
We strongly argue that reversibility in the interaction remains the biggest challenge in the development of broad‐spectrum antivirals (as long as it is engineered in the molecules while keeping the toxicity profile as low as possible). Stellacci lab showed that multivalent, supramolecular inhibitors of viral infectivity based on the round‐shaped gold nanoparticles, which act as an anchoring surface for a mixture of octanethiols and mercapto‐undecane sulfonic acids (MUS), were able to bind to HSPG seeking viruses basically in the same way (and with very similar inhibitory concentration) as heparin.[ 32 ] Yet, differently than heparin, upon binding these particles were able to damage the virus (see Figure 2 ), rendering the interaction irreversible (i.e., virucidal mechanism). It was postulated that the key for the change in mechanism from reversible binding (virustatic) to irreversible inhibition (virucidal) is the addition of multivalent hydrophobic interactions to the established electrostatic ones. Indeed, a direct comparison between MUS coated nanoparticles with nanoparticles known in the literature[ 33 ] coated with mercaptoethane sulfonic acid (MES) was performed. The particles differed only in the length of the hydrophobic linkers. The inhibitory concentration found through standard assays was very similar. Standard dilution assays were then performed (virucidal assays). These tests led to results that were significantly different. The more hydrophobic particles (MUS coated) showed irreversible inhibition, while the less hydrophobic ones (and heparin) showed an effect that could be easily reversed with dilution. The interpretation of the experiments has been strengthened recently because the MUS linker was used to chemically modify the primary face of beta‐cyclodextrins.[ 34 ] Similarly to previous results, these modified cyclodextrins showed irreversible inhibition in virucidal assays. Furthermore, another assay showed that when inhibiting HSV‐2 these cyclodextrins elicited DNA release, confirming a damage on the envelope as well as on the capsid of the virus. Also for these MUS modified cyclodextrins a comparison with similar sulfonated cyclodextrins, with shorter ligands, showed that only the former lead to irreversible inhibition. The key role of the hydrophobic linkers was further confirmed by comparing undecanesulfonic acids with oligoethyleneglycol sulfonic acid moieties on cyclodextins. It was shown that changing from an aliphatic to an ethyleneglycol chain had minimal effect on the inhibitory concentration but made the interaction reversible. Importantly, ex vivo only the irreversible inhibitors showed sustained protection from a viral infection.
Figure 2.
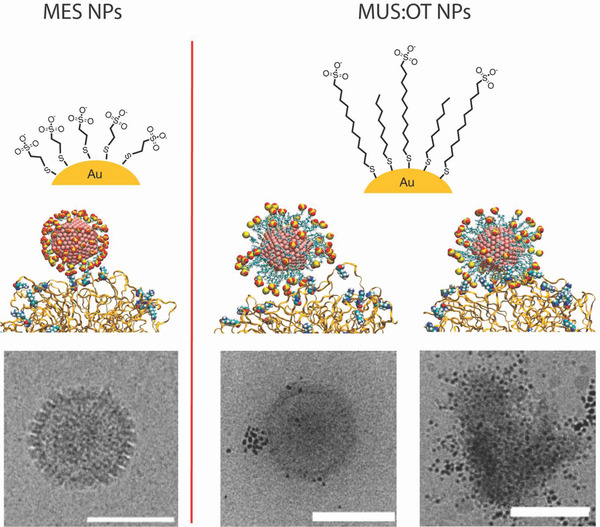
Schematic drawing of the working hypothesis for the mechanism proposed for the antiviral nanoparticles described in refs. [33] and [24]. Left) A schematic of virustatic effect that is described in the literature for MES‐coated nanoparticles (ref. [33]). Right) The proposed virucidal mechanism (ref. [24]). Images present chemical schemes for the particles (top), molecular dynamics calculation results (middle), and cryo‐transmission electron microscopy (TEM) images of herpes simplex virus 2 interacting with the nanoparticles. The main difference between the two types of particles is that while both attach to the virus only the virucidal are able to affect the structural integrity of the virus.
It should be noted that Stellacci's multivalent hydrophobic/sulfonated macromolecules do not solely damage the envelope of viruses but they are capable of damaging the capsid also, eventually eliciting the release of viral genetic materials. They do so while having no evident effect on mammalian cell membranes. It is not obvious that these molecules are the only ones to have these effects on viruses. Unfortunately, the use of dilution assays and of DNA/RNA release assays as a means of characterizing the antiviral mechanism of multivalent inhibitors is scarce, hence there are many open questions whether many of the published materials are in fact virustatic or virucidal. We encourage the field to add virucidal assays as standard characterization test, given how important the result is to the translate in vitro into in vivo effects.
In many aspects, we believe that synthetic multivalent synthetic inhibitors of viral infectivity as broad spectrum antiviral agents compare favorably to the biological neutralizing agents (Table 1 ). Macromolecular/supramolecular inhibition relies on nonspecific interactions and can therefore be optimized into a truly broad‐spectrum antiviral effect, whereas antibodies are designed to be highly specific to each and every (re)emerging pathogen. Polymers and nanoparticles can be manufactured on large scale and are stable at room temperature—and thus can be stockpiled, whereas antibodies are expensive, require cold chain of transportation, and have limited shelf‐life. The one aspect that speaks not in favor of multivalent synthetic inhibitors is that these are at the very beginning of the translational path.
Table 1.
Comparative analysis of synthetic mutltivalent inhibitors of virus cell entry and neutralizing antibodies as perspective broad‐spectrum antiviral agents
| Synthetic multivalent inhibitors (polymers, nanoparticles, dendrimers) | Antibodies | |
|---|---|---|
| Interaction with pathogens | Nonspecific | Specific |
| Spectrum of activity against pathogens | Broad, across the species of viridae | Specific to the individual pathogen |
| Mutation Resistance | Potentially very good | Limited |
| Toxicity | To be investigated case by case | Often ideal |
| Production scale | Large | Limited |
| Stability in storage | High, even at room temperature | Requires cold chain transportation and storage |
| Price | Low | High |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
There is also ample space for improvement for macro/supramolecular inhibitors. Specifically, we need to improve the potency of these agents and minimize toxicity and side effects. Indeed, polyanions exhibit anticoagulant and anti‐inflammatory activity, and interact with serum albumin (causing albumin aggregation).[ 35 ] Together with the failure of polyanions as injectable inhibitors of infectivity of HIV, this signifies that systemic administration of polyanions is not a viable measure.
Instead, it may be more prudent to develop antiviral formulations for inhalation. Drug repurposing, specifically for administration via inhalation and localized activity in the respiratory tract, has proven to be highly successful and hold translational potential.[ 36a ] Good case in point, there is an on‐going clinical trial for nebulized heparin as a treatment of Covid19 (NCT04397510), carried out in part due to direct antiviral effects of heparin.[ 21 , 36 ] Another sulfated polysaccharide, iota‐carrageenan, also showed promise in clinical trials as an inhalable antiviral.[ 37 , 38 ] We strongly believe that formulation of multivalent (polymer, nanoparticles) inhibitors of viral infectivity for inhalation and/or pulmonary administration has a significant promise to deliver the highly sought‐after broad‐spectrum antiviral agents, specifically as a preventative measure against respiratory viral infections.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
A.N.Z. wishes to acknowledge financial support from the Horizon 2020 project Fight‐nCoV and the Independent Research Fund Denmark (DFF‐FTP Project No. 4184‐00177).
Biographies
Alexander N. Zelikin finished his education in polymer chemistry in Moscow State University (Russia) in 2003 and subsequently held research positions at MIT, Cornell University, and the University of Melbourne. In 2009, he joined Aarhus University (Aarhus, Denmark) where he lectures in medicinal chemistry. His research interests include polymer chemistry, materials science, medicinal chemistry (prodrug design), and artificial biology.

Francesco Stellacci graduated in materials engineering in Politecnico di Milano (Italy) in 1998. After a postdoctoral experience in the Department of Chemistry at the University of Arizona (USA), he became an assistant professor in the Department of Materials Science and Engineering at the Massachusetts Institute of Technology (MIT, USA) in 2002. In 2010 he moved as a Full Professor to the Ecole Polytechnique Fédérale de Lausanne (EPFL, Switzerland) where he has appointments in the Institute of Materials and in the Bioengineering Institute. His research interests are in solid–liquid interfaces, nanoparticles characterization, and the development of broad‐spectrum antivirals.

Zelikin A. N., Stellacci F., Broad‐Spectrum Antiviral Agents Based on Multivalent Inhibitors of Viral Infectivity. Adv. Healthcare Mater. 2021, 10, 2001433. 10.1002/adhm.202001433
Contributor Information
Alexander N. Zelikin, Email: zelikin@chem.au.dk.
Francesco Stellacci, Email: francesco.stellacci@epfl.ch.
References
- 1. De Clercq E., Li G., Clin. Microbiol. Rev. 2016, 29, 695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bekerman E., Einav S., Science 2015, 348, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clardy J., Fischbach M. A., Currie C. R., Curr. Biol. 2009, 19, R437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McClean D., J. Pathol. Bacteriol. 1931, 34, 459. [Google Scholar]
- 5. Cohen S. S., J. Biol. Chem. 1942, 144, 353. [Google Scholar]
- 6. Ginsberg H. S., Goebel W. F., Horsfall F. L., Proc. Soc. Exp. Biol. Med. 1947, 66, 99. [DOI] [PubMed] [Google Scholar]
- 7. Green M., Stahmann M. A., Proc. Soc. Exp. Biol. Med. 1953, 83, 852. [DOI] [PubMed] [Google Scholar]
- 8. Green M., Stahmann M. A., Proc. Soc. Exp. Biol. Med. 1954, 87, 507. [DOI] [PubMed] [Google Scholar]
- 9. Green M., Stahmann M. A., Rasmussen A. F., Proc. Soc. Exp. Biol. Med. 1953, 83, 641. [DOI] [PubMed] [Google Scholar]
- 10. Nahmias A. J., Kibrick S., J. Bacteriol. 1964, 87, 1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nahmias A. J., Kibrick S., Bernfeld P., Proc. Soc. Exp. Biol. Med. 1964, 115, 993. [DOI] [PubMed] [Google Scholar]
- 12. Burger W. C., Stahmann M. A., J. Biol. Chem. 1951, 193, 13. [PubMed] [Google Scholar]
- 13. Rubini J. R., Rasmussen A. F., Stahmann M. A., Proc. Soc. Exp. Biol. Med. 1951, 76, 662. [DOI] [PubMed] [Google Scholar]
- 14. De Somer P., De Clercq E., Billiau A., Schonne E., Claesen M., J. Virol. 1968, 2, 878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Somer P., De Clercq E., Billiau A., Schonne E., Claesen M., J. Virol. 1968, 2, 886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith A. A. A., Kryger M. B. L., Wohl B. M., Ruiz‐Sanchis P., Zuwala K., Tolstrup M., Zelikin A. N., Polym. Chem. 2014, 5, 6407. [Google Scholar]
- 17. Stahmann M. A., Graf L. H., Patterson E. L., Walker J. C., Watson D. W., J. Biol. Chem. 1951, 189, 45. [PubMed] [Google Scholar]
- 18. Vaheri A., Acta Pathol. Microbiol. Scand. Suppl. 1964, 171, 1. [PubMed] [Google Scholar]
- 19. Bhatia S., Camacho L. C., Haag R., J. Am. Chem. Soc. 2016, 138, 8654. [DOI] [PubMed] [Google Scholar]
- 20. Rusnati M., Vicenzi E., Donalisio M., Oreste P., Landolfo S., Lembo D., Pharmacol. Ther. 2009, 123, 310. [DOI] [PubMed] [Google Scholar]
- 21. Mycroft‐West C. J., Su D., Pagani I., Rudd T. R., Elli S., Guimond S. E., Miller G., Meneghetti M. C. Z., Nader H. B., Li Y., Nunes Q. M., Procter P., Mancini N., Clementi M., Forsyth N. R., Turnbull J. E., Guerrini M., Fernig D. G., Vicenzi E., Yates E. A., Lima M. A., Skidmore M. A., bioRxiv 2020, 10.1101/2020.04.28.0667612020.04.28.066761. [DOI] [PMC free article] [PubMed]
- 22. Schandock F., Riber C. F., Rocker A., Muller J. A., Harms M., Gajda P., Zuwala K., Andersen A. H. F., Lovschall K. B., Tolstrup M., Kreppel F., Munch J., Zelikin A. N., Adv. Healthcare Mater. 2017, 6, 1700748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Papp I., Sieben C., Sisson A. L., Kostka J., Böttcher C., Ludwig K., Herrmann A., Haag R., Chembiochem 2011, 12, 887. [DOI] [PubMed] [Google Scholar]
- 24. Strauch E. M., Bernard S. M., La D., Bohn A. J., Lee P. S., Anderson C. E., Nieusma T., Holstein C. A., Garcia N. K., Hooper K. A., Ravichandran R., Nelson J. W., Sheffler W., Bloom J. D., Nat. Biotechnol. 2017, 35, 667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reuter J. D., Myc A., Hayes M. M., Gan Z., Roy R., Qin D., Yin R., Piehler L. T., Esfand R., Tomalia D. A., Baker J. R., Bioconjugate Chem. 1999, 10, 271. [DOI] [PubMed] [Google Scholar]
- 26. Pirrone V., Wigdahl B., Krebs F. C., Antiviral Res. 2011, 90, 168. [DOI] [PubMed] [Google Scholar]
- 27. Bazinet M., Pântea V., Cebotarescu V., Cojuhari L., Jimbei P., Albrecht J., Schmid P., Gal F. L., Gordien E., Krawczyk A., Mijočević H., Karimzadeh H., Roggendorf M., Vaillant A., Lancet Gastroenterol. Hepatol. 2017, 2, 877. [DOI] [PubMed] [Google Scholar]
- 28. Michen B., Graule T., J. Appl. Microbiol. 2010, 109, 388. [DOI] [PubMed] [Google Scholar]
- 29. Said J., Trybala E., Andersson E., Johnstone K., Liu L., Wimmer N., Ferro V., Bergström T., Antiviral Res. 2010, 86, 286. [DOI] [PubMed] [Google Scholar]
- 30. Lundin A., Bergström T., Andrighetti‐Fröhner C. R., Bendrioua L., Ferro V., Trybala E., Antiviral Res. 2012, 93, 101. [DOI] [PubMed] [Google Scholar]
- 31. Vaillant A., Antiviral Res. 2016, 133, 32. [DOI] [PubMed] [Google Scholar]
- 32. Cagno V., Andreozzi P., D'Alicarnasso M., Silva P. J., Mueller M., Galloux M., Goffic R. L., Jones S. T., Vallino M., Hodek J., Weber J., Sen S., Janeček E.‐R., Bekdemir A., Sanavio B., Martinelli C., Donalisio M., Welti M.‐A. R., Eleouet J.‐F., Han Y., Kaiser L., Vukovic L., Tapparel C., Král P., Krol S., Lembo D., Stellacci F., Nat. Mater. 2018, 17, 195. [DOI] [PubMed] [Google Scholar]
- 33. Baram‐Pinto D., Shukla S., Gedanken A., Sarid R., Small 2010, 6, 1044. [DOI] [PubMed] [Google Scholar]
- 34. Jones S. T., Cagno V., Janeček M., Ortiz D., Gasilova N., Piret J., Gasbarri M., Constant D. A., Han Y., Vuković L., Král P., Kaiser L., Huang S., Constant S., Kirkegaard K., Boivin G., Stellacci F., Tapparel C., Sci. Adv. 2020, 6, eaax9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zuwala K., Riber C. F., Lovschall K. B., Andersen A. H. F., Sorensen L., Gajda P., Tolstrup M., Zelikin A. N., J. Controlled Release 2018, 275, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.a) Newman S. P., Adv. Drug Delivery Rev. 2018, 133, 5; [DOI] [PubMed] [Google Scholar]; b) Conzelmann C., Müller J. A., Perkhofer L., Sparrer K. M., Zelikin A. N., Münch J., Kleger A., Clin. Med. 2020, 20, e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eccles R., Meier C., Jawad M., Weinmüllner R., Grassauer A., Prieschl‐Grassauer E., Respir. Res. 2010, 11, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eccles R., Winther B., Johnston S. L., Robinson P., Trampisch M., Koelsch S., Respir. Res. 2015, 16, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]