

Abstract
Protein complexes are key macromolecular machines of the cell, but their description remains incomplete. We and others previously reported an experimental strategy for global characterization of native protein assemblies based on chromatographic fractionation of biological extracts coupled to precision mass spectrometry analysis (CF/MS), but the resulting data is challenging to process and interpret. Here, we describe EPIC (Elution Profile-based Inference of Complexes), a software toolkit for automated scoring of large-scale CF/MS data to define high-confidence multi-component macromolecules from diverse biological specimens. As a case study, we used EPIC to map the global interactome of Caenorhabditis elegans, defining 612 putative worm protein complexes linked to diverse biological processes. These included novel subunits and assemblies unique to nematodes that we validated using orthogonal methods. The EPIC software is freely available as a Jupyter notebook packaged in a Docker container and the open source code is accessible via GitHub.
INTRODUCTION
Systematic mapping of multi-protein complexes formed by protein-protein interactions (PPI) is critical to understand the mechanistic basis of cellular processes. Affinity purification coupled to mass spectrometry (AP/MS)1 is a powerful method for identifying such assemblies and has been applied widely2–9, but is difficult to scale up or apply to non-model organisms. Biochemical co-fractionation coupled to mass spectrometry (CF/MS) is a more efficient and flexible alternate strategy for examining native macromolecules on a global scale10,11. CF/MS is based on biophysical (typically chromatographic) co-purification of stable-associated proteins starting from cell-free mixtures (e.g. tissue lysates). However, sophisticated data processing is needed to define genuine interactions, which can be challenging to implement.
To facilitate such studies, we have developed a simplified, standardized and fully automated CF/MS data analysis software toolkit, EPIC, which enables routine scoring and interpretation of large-scale CF/MS data regardless of sample source. Using supervised machine learning EPIC integrates experimentally derived CF profiles and complementary functional evidence from public databases to create probabilistic PPI networks, which are then clustered to define high-confidence complexes.
We demonstrate the utility and performance of EPIC by applying it to the nematode Caenorhabditis elegans. By analyzing quantitative mass spectrometry generated for whole organism soluble protein extracts resolved by ion-exchange chromatography, we identified 612 putative complexes from a network of 16,098 high-confidence PPIs that encompassed 3,855 worm proteins, most of which have never been reported before. The resulting ‘WormMap’ reveals assemblies with links to disparate lineage-restricted processes, conserved animal systems and human disease. To facilitate community adoption of CF/MS workflows, the EPIC toolkit is freely available as a Jupyter notebook packaged in a Docker container (https://hub.docker.com/r/baderlab/bio-epic/).
RESULTS
Systematic scoring of PPI networks and identification of native multi-protein complexes
CF/MS is based on extensive experimental separation of native macromolecular mixtures under non-denaturing conditions. While there is no universally optimal protocol, ion-exchange high-performance chromatography (IEX-HPLC) is efficient at resolving stable endogenous complexes (Fig. 1a). To maximize coverage, non-ionic detergents can be added to solubilize hydrophobic complexes9, chemical cross-linkers can be used to stabilize labile assemblies12, and organelle compartments can be enriched prior to HPLC. For example, bead-based pre-fractionation (see Online Methods) improves detection of less abundant macromolecules (Supplementary Fig. 1), while concomitantly reducing ‘chance’ co-elution (i.e. co-fractionation of functionally unrelated proteins). The results from a CF/MS experiment can be summarized as a matrix of biochemical fractions versus protein identities containing MS-derived protein amounts for each fraction (e.g. summed precursor ion intensities or spectral counts).
Figure 1: EPIC workflow.
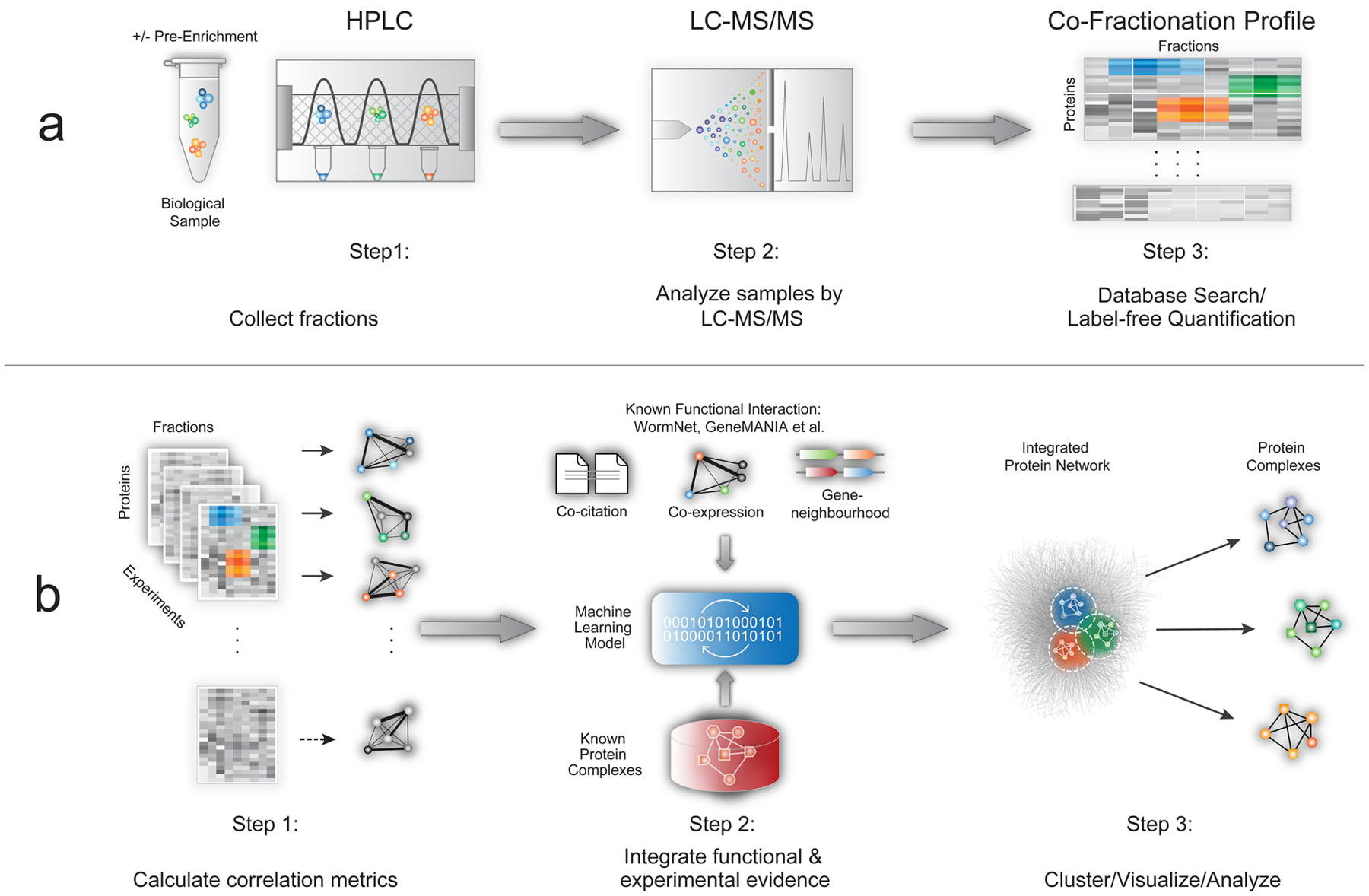
a) CF/MS experiments have three main steps: biochemical fractionation, MS analysis, and protein profile scoring. b) Automated computational analysis using EPIC takes CF/MS data as input and consists of three main steps: (i) calculation of co-elution profile similarity using correlation metrics; (ii) co-complex PPI scoring using machine learning-based integration of experimental and functional evidence; (iii) prediction, clustering, and benchmarking of derived complexes.
EPIC software environment
EPIC employs python scripts to score CF/MS data, with modules to (i) process protein co-elution profiles, (ii) optionally download supporting functional association information from public databases (CORUM13, UniProt14, IntAct15, GO16, GeneMANIA17, STRING18, InParanoid19), (iii) predict and benchmark predicted associations versus curated reference assemblies (CORUM, IntAct and GO, Supplementary Fig. 2), and (iv) cluster and visualize the resulting PPI network using Cytoscape20 (Fig. 1b). Given suitable experimental CF/MS data and a standard taxonomy identifier for the organism under study, the software collects required information from online sources and automates all data processing (scoring through visualization). In addition to convenient automation, EPIC outperforms an existing computational tool21 both in terms of prediction quality and quantity (Supplementary Table 1; see Online Methods) and can process both isotope labelled and label-free CF/MS data as input.
Since stably-associated components within a complex are expected to co-fractionate together, EPIC first computes pairwise protein profile similarity using up to eight correlation metrics (Euclidean, Jaccard, Apex, Pearson, Pearson with Poisson noise, weighted cross correlation, mutual information, and Bayes correlation22) that emphasize different profile features (Supplementary Notes). Positive (known) and negative reference co-complex PPIs display distinct correlation distributions (Supplementary Fig. 3). While it is likely not possible to pre-define a universally optimal combination of correlation metrics for all possible CF/MS experiments, EPIC provides default parameters tuned on comprehensive CF/MS data (described below), and can optimize settings for any given data set. To reduce computational time, proteins observed in only one fraction and protein pairs with co-fractionation correlation scores less than 0.5 are removed (see Online Methods and Supplementary Fig. 4) before generating a scored co-complex PPI vector for each input experiment. Multiple correlation vectors are then combined and input into a supervised machine-learning model that is both trained to predict new PPIs and benchmarked against reference positive (annotated) PPIs (i.e. co-complex relationships curated in the CORUM, IntAct and GO databases) and negatives (i.e. combinations of proteins in distinct complexes).
To generate a comprehensive reference (gold standard) set for both training and benchmarking, EPIC retrieves species-specific complexes from the IntAct and GO complex databases. Since positive examples are limited for certain species, like C. elegans, the benchmark is supplemented by mapping annotated human protein complexes from the CORUM database based on stringent one-to-one orthology (InParanoid). To minimize redundancy and bias, complexes with the majority of subunits in common (overlap score >0.8) are merged, while large assemblies with 50+ members (e.g. ribosome) that could dominate learning are eliminated.
EPIC uses support vector machine (SVM) and random forest (RF) classifiers by default, but other algorithms can be substituted programmatically. Since CF/MS data is often incomplete (e.g. due to proteome under-sampling) or noisy (e.g. chance co-elution of unrelated proteins), EPIC can integrate additional supporting evidence (e.g. functional interactions inferred from co-expression, domain co-occurrence, and co-citation) from public sources such as GeneMANIA or STRING, thereby producing richer and more accurate interaction networks. To avoid circularity, functional interactions based on published PPIs are excluded. To ensure all complexes have CF/MS experimental support, those complexes inferred based solely on functional evidence are removed. Prediction performance is evaluated by 2-fold cross-validation (i.e. against an independent ‘holdout’ set of reference protein complexes; see Online Methods).
Finally, EPIC applies network-partitioning to define complex membership. ClusterONE23 is used by default, though other algorithms can be evaluated to optimize complex definition24. Each cluster is compared to annotated complexes curated in CORUM, GO and IntAct, and overall performance is measured by three complementary evaluation metrics (maximum matching ratio, accuracy, and overlap score; see Supplementary Notes), from which a single summary composite score is calculated to assign prediction quality23.
Optimizing EPIC performance
We optimized EPIC performance using a novel data set of 1,380 IEX-HPLC fractions generated for soluble worm protein extracts from mixed stage C. elegans cultures. Co-eluting proteins were acid-precipitated, alkylated and trypsin digested, and the resulting peptide mixtures analyzed by precision Orbitrap MS. To optimize major EPIC parameters (MS search tool, set of profile correlation metric and machine learning classifier), we compared predicted complexes from each parameter setting (2,040 parameter combinations) against an independent benchmark of known complexes compiled from CORUM, IntAct and GO using composite score as the evaluation measure (Fig. 2a; see Supplementary Notes). Optimized parameters substantially improved the resulting composite score compared to previously used parameters10,11 (Fig. 2b). We evaluated the performance benefit of integrating functional interactions with the CF/MS data, again based on composite score, and found that including GeneMANIA, STRING, or WormNet25 clearly boosted performance (Fig. 2b and Supplementary Fig. 5). Functional evidence was not effective when used alone as input to predict complexes (Supplementary Fig. 5 and Supplementary Fig. 6). Since CF/MS studies consume considerable resources (e.g. LC/MS run time), we used EPIC to explore the ‘cost/benefit’ ratio of repeat biochemical fractionations by evaluating the relationship between prediction accuracy and the number of experiments performed. We calculated the average composite score by randomly sampling different numbers of co-fractionation experiments. Notably, while performance steadily improved as more data was acquired, prediction performance grew fastest over the first 2–4 separations (Fig. 2c; see Online Methods for details), suggesting an efficient lower bound (i.e. ~4 IEX-HPLC experiments) for study design.
Figure 2: EPIC parameter evaluation.
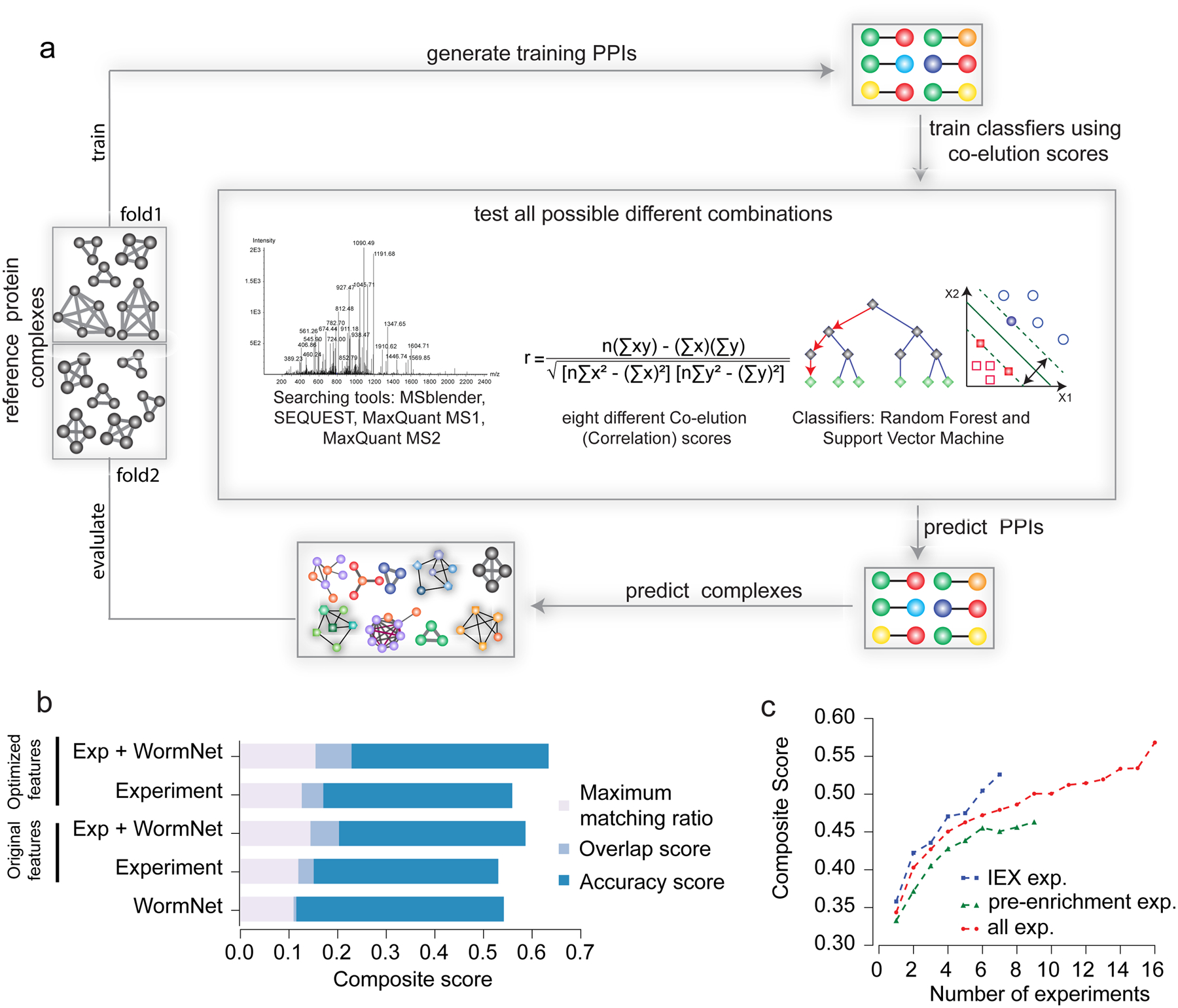
a) Computational procedures for protein interaction and co-complex prediction, driven by global optimization of classifier performance. b) Bar chart shows predicted worm complex scores (maximal matching ratio, overlap and accuracy, the sum of which forms the composite score) using different combinations of experimental (CF/MS) data, functional evidence (WormNet) and correlation scores. “Original features” indicates results from the set of correlation metrics (parameters) used in previous publications, and “optimized features” indicates our newly optimized EPIC parameters. c) Average composite scores for different number of co-fractionation experiments.
WormMap – a comprehensive map of soluble protein complexes in C. elegans
Using all 16 C. elegans co-fractionation experiments with optimized parameter settings and including functional interactions, EPIC predicted 16,098 high-confidence co-complex PPIs among 3,855 worm proteins (~25% of the nematode proteome), each directly supported by CF/MS data (at least one co-elution correlation score >0.5). Most (13,547) of these PPIs have not been reported before (compared to iRefWeb26, BioGRID27 or our previously generated Metazoan Complex Map11) (Supplementary Fig. 7; see Supplementary Table 2 for complete listing). Partitioning the network using ClusterONE predicts 612 complexes (Fig. 3a) of which only 150 map to known assemblies in CORUM, GO, and IntAct. Most of the novel complexes appear to be clade-specific as only 89 are also found in the Metazoan Complex Map (see Supplementary Table 3 for complete listing).
Figure 3: Prediction, benchmarking and analysis of C. elegans protein complexes.
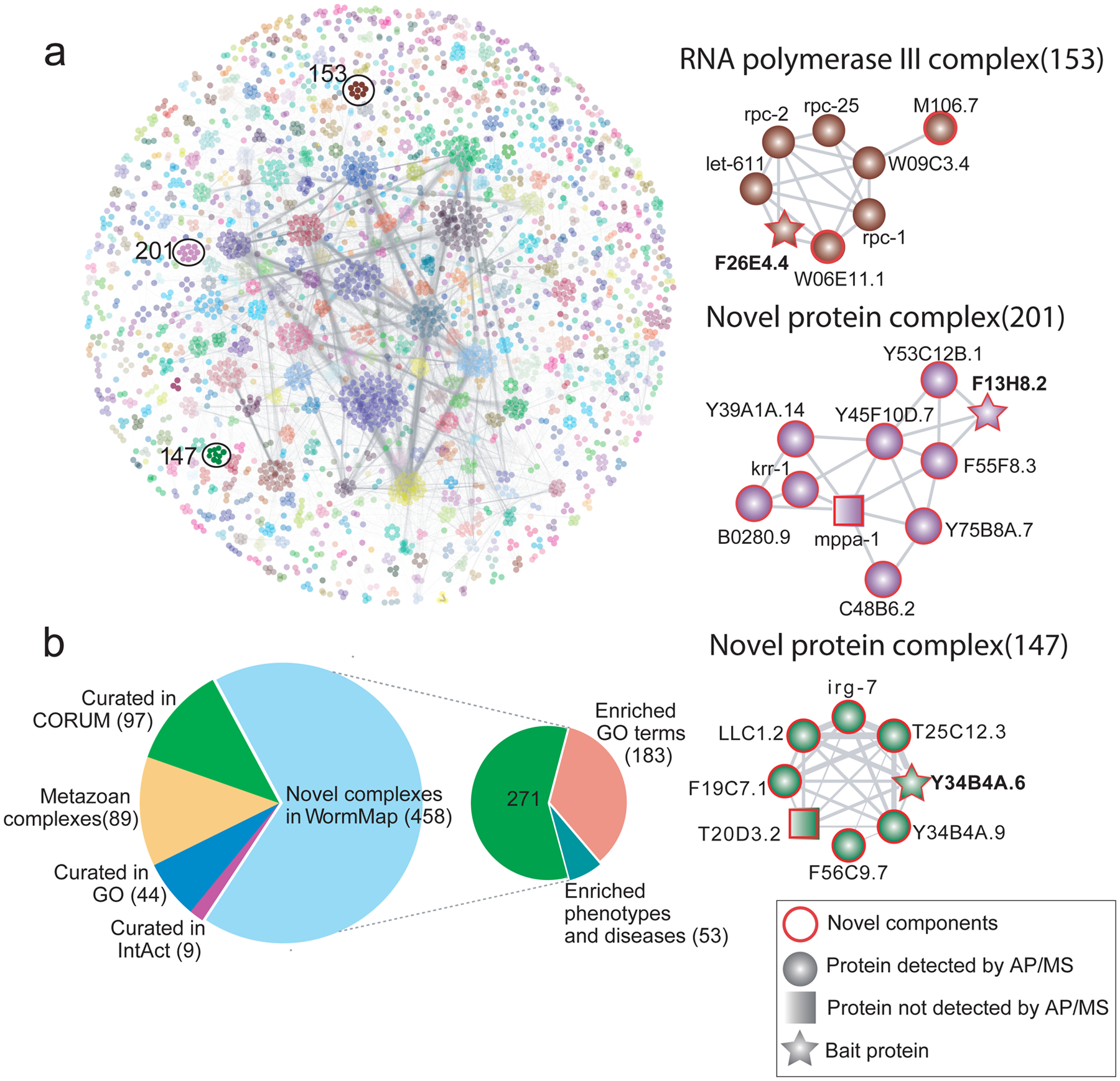
a) EPIC-derived WormMap. Protein nodes are coloured according to complex assignments, with novel assemblies and components highlighted with red circles. Spectral counts recorded across validation AP/MS experiments are summarised in Supplementary Table 4 Interacting proteins identified with low counts, indicating a weak MS signal, were nonetheless consistent with co-elution evidence. Grey lines between proteins indicate the interactions are supported by strong co-elution evidence. b) Pie charts showing the overlap of predicted worm complexes with previously known macromolecules (from CORUM, GO, IntAct, and the metazoan protein complex map11) and enrichment of putative novel assemblies for select biological function (GO terms), phenotype and/or disease associations.
We used multiple independent approaches to assess the accuracy of the predicted worm protein complexes. Experimentally, we used an established, orthogonal biochemical approach (AP/MS; see Online Methods) to validate both entirely novel assemblies as well as previously reported assemblies for which EPIC predicted unexpected new components (Fig. 3a and Supplementary Table 4). For example, we verified three new nematode-specific components (F26E4.4, W06E11.1 and M106.7) of the worm RNA polymerase III machinery, one of which (M106.7) has DNA and nucleotide binding activity28 (Fig. 3a). We also validated unc-15 as part of a large myosin complex, an association not reported in a public database or our training set, but has been observed in previous work29. Likewise, we verified a predicted novel 10-member complex (Fig. 3a), for which most components have limited functional annotation in WormBase30, suggesting an overlooked biological role. Two of the subunits (B0280.9 and krr-1) are orthologs of human small-subunit processome components involved in ribosomal biogenesis, suggesting a related function in nematodes. Intriguingly, another subunit, Y45F10D.7, is an ortholog of human WDR36, which is linked to primary open-angle glaucoma type 1G (GLC1G)31, potentially providing a mechanistic connection. We also confirmed another putative novel complex with eight protein components (Fig. 3a) containing mostly uncharacterized components according to UniProt14 and WormBase30. Irg-7 is the only annotated subunit, with links to innate immunity and expression in the intestine32, suggesting a potential role in the host response to animal pathogens.
To assess the physiological significance of the putative worm assemblies, we analyzed the network of complexes for coherent biological functions (based on GO annotations, Supplementary Table 5), mutant phenotypes (based on information from WormBase30, Supplementary Table 6), or disease associations (based on orthology to human proteins in genetic disorder databases such as OMIM33 and HGMD34, Supplementary Table 7). Strikingly, almost half of the novel complexes in WormMap were enriched for associations to essential processes, phenotypes or diseases (Fig. 3b). For example, knockdown of components of dozens of complexes either cause embryonic lethality or sterility, and have links to cancer in humans, reinforcing the utility of EPIC for gaining fundamental mechanistic insight into large CF/MS data.
DISCUSSION
Current knowledge of the physical networks of cells and tissues remains limited for many species, particularly non-traditional animal models. The majority of known/curated protein assemblies are annotated to mammals, whereas inference based on homology may not be the ideal for more distant organisms. CF/MS is an ideal experimental technology to address this, as it can be applied directly to any biological sample. However, CF/MS data is complex and challenging to process. We have developed the EPIC software to facilitate routine CF/MS analysis of native macromolecular assemblies in diverse contexts. EPIC provides optimized computational workflows, does not require expert computational skills to run, automates the entire data analysis process, and is applicable to diverse model systems. We used EPIC to map protein complexes in C. elegans, which has classically been studied using genetic methods, thereby revealing nematode-specific biochemical network adaptations. In practice, EPIC enables users to process their own data and supply their own manually curated reference protein complexes to optimize classifier training.
We have shown that EPIC predicts complexes with high accuracy, particularly if four or more biochemical separations are available. While transient or unstable macromolecules may not be efficiently detected by CF/MS, chemical cross-linking can potentially be beneficial12, while other gentle separation techniques, such as isoelectric focusing (Pourhaghighi et al., submitted) and size-exclusion chromatography35, can provide complementary data. Regardless, to mitigate the false discovery rate, EPIC implements customizable data filtering procedures and can optionally integrate supporting independent functional evidence. Integrating functional evidence will reduce false negative PPIs, but may introduce bias towards well-studied proteins36. While it is difficult to evaluate this bias, we note that many WormMap complexes, including those validated by AP/MS, contain uncharacterized proteins or proteins with diverse functional annotations, which suggests that EPIC is not strongly affected by this bias.
EPIC is both open source (https://github.com/BaderLab/EPIC) and compatible with disparate proteomic sampling techniques, including ‘top-down’ analysis of intact proteins37 and sample multiplexing (isotopic labelling)38 to map differential networks across conditions39. To facilitate broader uptake, we provide an automatically executable Jupyter-based notebook along with a Docker container (https://hub.docker.com/r/baderlab/bio-epic/) encompassing all necessary scripts and packages, enabling easy installation, deployment and optimization on any operating system. The distributed version of EPIC has step-by-step instructions and a user-friendly interface that enables uploading of local user defined CF/MS data files and the graphical display of results.
ONLINE METHODS:
Protein Extract Preparation.
Mixed-staged N2 strain C. elegans were collected in M9 buffer (standard recipe40), and re-suspended into lysis buffer (50 mM HEPES pH7.4, 1 mM MgCl2, 1 mM EGTA, 100 mM KCl) plus protease inhibitor cocktail (Roche). Worms were lysed by 3 rounds of 10 sec sonication on ice (Branson Sonifer 450, output 6.0, duty cycle 60%). Soluble protein lysate [~2 mg/ml] was collected by filter centrifugation (Ultrafree®-MC-HV, 0.45 μm). Bradford assay was used to determine protein concentration.
Pre-enrichment before HPLC fractionation
Differential affinity capture beads (NuGel PROspector; BSG) were used to pre-enrich the worm lysate according to the manufacturer’s protocol. After removal of lipids and insoluble biomass, extract incubated with different reagent beads (PRO-A, PRO-B, PRO-C, PRO-L, PRO-N, PRO-R). The suspensions were mixed for 10 min at 4 °C, centrifuged using Spin-X filters, and the filtrate was collected as ‘flow-through’ fractions. Bound proteins were eluted with 200 ul elution buffer (0.2 M Tris, 0.5 M NaCl, pH 9.0). The buffer was exchanged for HPLC loading buffer by Zeba desalt spin column (Thermo) before HPLC fractionation.
HPLC separations
C. elegans lysate and affinity enriched eluates (plus flow-through fractions) were individually fractionated by ion-exchange liquid chromatography using a quaternary pump 1100 HPLC system (Agilent Technologies). Whole proteome lysate was resolved into 120 fractions on a PolyCATWAX mixed-bed ion exchange column (200 × 4.6 mm id, 12 μm, 1500 A) over a 240 min salt gradient (0.15 to 1.5 M NaCl). Enriched eluates were separated on a PolyCATWAX mixed-bed ion exchange column (200 × 4.6mm id, 5 μm, 1000A) into 60 fractions using a 120 min salt gradient (0.15 to 1.5 M NaCl). The detailed protocol has been described previously10.
LC-MS/MS analysis
Proteins from the HPLC fractions were acid precipitated, re-dissolved and digested by sequencing grade trypsin overnight at 37 °C. The resulting peptides were dried and solubilized in 5% formic acid. Data-dependent LC-MS/MS was performed using a nano-flow HPLC System (EASY-nLC, Proxeon, Odense, Denmark) coupled to an LTQ Orbitrap Velos Mass Spectrometer (Thermo Fisher). After loading onto a 2.5 cm C18 trap column (75 mm inner diameter) packed with 100A Luna 5u C18 beads (Phenomenex) using an auto-sampler, peptides were separated on a 10 cm analytical column (75 mm i.d.) packed with 2 mm Zorbax 80XDB C18 reverse phase beads (Agilent). A 60 min gradient consisting from 5% to 35% ACN in water (with 1% formic acid) was used to elute peptides. Electro-spray ionization was performed using at 2.5kV spray voltage, and the instrument was operated in a data dependent mode (one full MS1 ion survey scan directing consecutive MS2 acquisition scans on the top 10 most prominent precursor ions). Collison induced dissociation (CID) directed peptide fragmentation was performed by 35% normalized collision energy.
Protein identification and label free quantification.
Raw spectral files were converted into mzXML format using the ReAdW software. A canonical FASTA file for protein searching was downloaded from the UniProt database and appended with common contaminants and reverse decoy sequences to assess the false-discovery rate (FDR). The peptide-spectrum matches from three different searching engines (comet, MSGF+ and X!Tandem) were integrated probabilistically using MSblender41, setting the FDR to less than 1% for peptide and protein identifications. Parameter settings and detailed search protocols are available online (http://www.marcottelab.org/index.php/MSblender). MaxQuant42 (Version 1.6.0.16) search was performed at a fragment ion mass tolerance of 20 pp., maximum missed cleavage of 2 and a 1% false discovery level (controlled by target/decoy approach). SEQUEST (Version 2.7) search was performed at 20 pp. fragment ion mass tolerance and one missed cleavage allowance. The STATQUEST43 model was used to assign confidence scores to all putative matches of peptides and proteins and a false discovery rate was controlled at 1% for all identifications.
Generating GFP tagged worm strains for AP/MS
To create GFP-tagged proteins for AP/MS experiments, C. elegans strains were grown and maintained at 20 °C on nematode growth media (NGM) plates seeded with E. coli strain OP50. Some strains (wild-type N2 and RW1596: myo-3 (st386) stEx30 [myo-3p::GFP::myo-3 + rol-6(su1006)44) were ordered from the CGC (https://cgc.umn.edu/). Extra-chromosomal array strains containing a C-terminal GFP translational fusion construct of F26E4.4, Y34B4A.6 and F13H8.2 were also generated in this study. For instance, the open reading frame and 617 bp promoter region of F26E4.445 were amplified and cloned into the pPD95.75 vector (Fire Lab Vector Kit). The construct was then injected at 20 ng/μl along with pRF4 as a co-injection marker. Roller positive F2 animals were isolated and imaged to confirm the GFP expression (rol-6 was used as a co-injection marker). Mixed stage worms were harvested for AP/MS validation studies. All other GFP-tagged strains (Y34B4A.6 and F13H8.2) are generated in a similar fashion.
Affinity purification mass spectrometry validation
Affinity purification was performed essentially as described46 with minor modifications. Briefly, frozen cell pellets were re-suspended in high-salt NP-40 lysis buffer (10 mM Tris-HCl pH 8.0, 420ml NaCl, 0.1% NP-40) with protease and phosphatase inhibitors (Roche). After 3 freeze-thaw cycles, each lysate was briefly sonicated, treated with nuclease (Thermo Scientific Cat #88700), followed by centrifugation at 14,000 rpm. The resulting soluble protein extract was split for technical replicate purifications. Each lysate was incubated at 4 °C on a rotator with 1 μg of rabbit anti-GFP antibody (Thermo Scientific Cat #G10362) for 2 hrs, followed by incubation with 25 μl of Protein-G Dynabeads slurry for 1 hr. The beads were washed twice with low-salt buffer (10mM Tris-HCl pH 8.0, 100 mM NaCl) and bound proteins subsequently eluted (X 4) with 1% ammonium hydroxide pH 11. Recovered protein samples were dried, re-suspended in 50 mM ammonium bicarbonate, reduced with 5 mM DTT at 56 °C for 45 min and alkylated with 10 mM iodoacetamide at room temperature for 45 min in the dark. Trypsin digestion was performed overnight at 37 °C. Peptide samples were de-salted and re-suspended in 1% formic acid and then analyzed by data dependent (top-15 MS2) acquisition on a Q Exactive HF mass spectrometer (Thermo Scientific) using a 90-minute gradient on the same HPLC system described above. The resulting MS spectra were searched with MSblender.
Computational workflow
In the following sections, we describe the computational components of the EPIC workflow that use machine learning methods with the goal of identifying as many interacting proteins as possible, while minimizing the ‘chance co-elution’ problem, based on MS-based protein profiles (Supplementary Fig. 8). For each fractionation experiment, search results are summarized into a single data matrix, in which, each row represents an identified protein while each column value refers to an estimated relative protein amount (spectral count or summed MS1 ion intensity) for a corresponding fraction. Under the assumption that proteins not detected by mass spectrometry are likely to be low abundant or simply not expressed, the values of missing proteins are set to zero. In the case of multiple co-fractionation experiments, a single unified matrix is created. For classification, EPIC has two major steps: first, a training set of co-complex PPIs is derived from reference protein complex datasets (e.g. CORUM) that map onto the experimental data. Second, one of two built-in machine-learning algorithms (SVM, RF) is used to define a probabilistic interaction network from which protein complexes are inferred using a network-partitioning algorithm.
Data processing
Several steps are required to pre-process the raw mass spectrometry co-elution table in order to improve the quality of the predicted network, similar to those performed in prior work11. However, we have added new features to improve prediction quality and to reduce the computational runtime.
Removing ‘one-hit-wonders’
The central principle of EPIC is based on the guilt-by-association approach, which posits that proteins that are physically associated tend to elute at the same time. However, to meaningfully evaluate fractionation data, EPIC requires the proteins to be present across multiple biochemical fractions within the same experiment. Thus, proteins measured in exactly only one fraction are deemed ‘one-hit-wonders’ and removed from further analysis. The reason for discarding such proteins is not because we assume they were falsely measured, but rather that EPIC measures co-elution profile similarities based on correlation metrics that evaluate similarity over the entire elution profile, which is not effective for singletons. Some proteins may be identified in only one fraction in multiple experiments. However, if we predict PPIs in this way, overall performance is markedly decreased (data not shown). Hence, each experiment is processed individually in EPIC, followed by merging/concatenating all the resulting co-elution correlation metric scores into a single unified matrix for machine learning. From the initial raw MS data, we observed that MSblender is highly sensitive and identifies the largest amount of peptides of which many are one-hit-wonders. However, even after removing one-hit-wonders, MSblender still has the largest amount of identified peptides compared with single search engines, resulting in the highest predicted quality protein complexes (Supplementary Fig. 9; see main text).
Elution data normalization
Before calculating correlation coefficient metrics, the protein elution profile matrix is normalized column-wise to correct for slight sample injection variation. The protein elution profile matrix for each co-fractionation experiment consists of MS1 ion intensity or MS2 spectral counts for M proteins across N fractions. Thus, before calculating protein elution profile similarities, the raw data of each protein in each fraction is normalized by dividing the amount of the particular protein (either MS1 ion intensity or MS2 spectral counts) by the total amount of proteins in corresponding fractions. So given a protein elution matrix A of the size M × N, where each Ai,j denotes the value of MS1 intensity or MS2 spectral counts of a particular protein i in fraction j, the column-wise normalized protein elution profile matrix Bi,j is calculated as:
Some similarity score metrics (i.e. Euclidean distance score) require row-wise normalization after column-wise normalization to make sure the sum of each row equal 1. So the final normalized protein elution profile matrix Ci,j is calculated as:
Creating candidate protein pairs
In previous work11, we first created all possible pairs of proteins for each experiment, followed by calculating their corresponding co-elution scores and then removed all protein pairs without co-elution correlation scores equal or more than 0.5. However, this approach is computationally demanding and requires high-performance computational resources to perform all calculations in a reasonable amount of time. Thus, we decided to apply a pre-filtering step: instead of calculating all possible protein pairs for each experiment we first generate a super-set of all possible protein pairs across all experiments and remove those pairs for which the two proteins do not overlap (never occur in same fraction across all experiments). Usually, this filtering-step removes a substantial (up to 60%) of possible candidate pairs, significantly reducing computational time. In the subsequent step, we calculate co-elution scores for each candidate protein pair across each experiment and then summarize the results into matrices, and then we remove all protein pairs whose co-elution score is below 0.5 across all experiments.
Machine learning prediction
At the end of the data processing, EPIC generates a co-elution matrix, which contains rows for each protein pair and columns for each co-elution score across all co-fractionation experiments. In cases where a protein pair was not present in one of the experiments, we set all of its co-elution scores for the given experiment to zero. In the subsequent section, we describe how EPIC creates co-elution protein-protein interaction network and the set of protein complexes:
Reference data set
Our goal is to make EPIC a generic tool for surveying protein complexes in different species. To facilitate standardization, we decided to use CORUM database13 as the source of the gold standard set, as it is the largest manually curated protein complex database available. EPIC utilizes human protein complexes for generating the necessary reference data, since protein complex information is typically sparse for the majority of species and as CORUM itself mainly curates human protein complex information. EPIC automatically downloads the current CORUM version and retains only those complexes that are annotated for human or mammals. Further, only protein complexes defined based on biochemical approaches are retained in the reference dataset, as protein complexes defined based on non-biochemical methods might not be expected to co-elute by chromatographic separation.
As an added set, EPIC downloads all human protein complexes from the IntAct database, for which again only complexes detected by biochemical methods are retained.
Additionally, EPIC automatically downloads a set of curated protein complexes in the Gene Ontology (GO) database, annotated based on biochemical evidence for relevant target species (e.g. C. elegans).
We then generate an extracted set of positive and negative protein-protein interactions for both the training and holdout protein complexes, respectively. PPIs are defined as positive if they are observed in the same protein complex. If proteins exist in the protein complex dataset but never appear in the same protein complex, then these two proteins are defined as negative PPIs.
For mapping human proteins to the input species (test sample), we integrated the InParanoid database, which is also automatically downloaded for each EPIC run. We only consider one-to-one orthologous protein mappings between human and the test species with an InParanoid confidence score of 100%. In this manner, curated human protein complexes are projected on to corresponding orthologous protein complexes in a target species of interest. To avoid bias, protein complexes with less than three members and large assemblies with more than 50 proteins are removed, because these would dominate the machine learning process. Further, to remove redundancy in our data set, highly overlapping protein complexes (high fraction of shared components) are merged. We evaluate the overlap of two complexes A and B as follows, where |A| denotes the number of proteins in A:
Protein complexes are merged if they have an overlap score of at least 0.8. This automatic process for generation of reference data set currently only supports UniProt identifiers because they are used by GO, IntAct, InParanoid and CORUM.
Train machine-learning classifier and predict protein-protein interactions
The machine learning classifier is trained on the sets of positive and negative PPIs as we defined before based on CORUM, IntAct, and GO. We create the union of training set by merging the training set obtained from the above three databases, in which only the protein pairs have at least one elution profile similarity score larger than 0.5 (among all co-fractionation experiment and among all correlation metrics) are retained. We then train the classifier on this reduced set of negative and positive interactions with correlation metrics scores from different co-fractionation experiments as input features. Because the classifier is trained to distinguish true-positive co-complex membership with high co-elution score from non-interacting protein pairs including false-positive chance co-elution associations that also have high co-elution scores, we decided to additionally integrate functional evidence data (i.e. GeneMANIA, STRING and WormNet) into the machine learning method. However, to reduce circular reasoning in the machine learning step, functional evidence derived from “physical interaction”, “protein complexes” and “predicted interactions” are excluded from input features.
EPIC generates a set of protein-protein interactions using the classifier trained on experimental data with an option to include functional evidence. Then a set of protein-protein interactions are predicted by the classifier trained on experimental data or optionally experimental data integrated with functional data. A protein elution profile correlation score cut-off was applied to ensure all PPIs have experimental evidence support. EPIC then applies a clustering algorithm (see below) to predict protein complexes based on the PPIs from the combined set of data. Novel protein complexes are identified by comparing the predicted set of complexes from above and the curated protein complexes from the major databases (CORUM, IntAct and GO) by setting a liberal overlap score cut-off at 0.25.
Predict protein complexes from the protein-protein interaction network
In the final step, EPIC generates a set of putative complexes from the predicted protein interaction network. As with our previous work, we use the ClusterONE clustering method, since it has been shown to provide excellent performance among several different clustering algorithms for predicting protein complexes from protein-protein interaction networks, thus we do not investigate clustering methods here.
Benchmarking
We extensively benchmarked EPIC and optimized parameters for each step of the EPIC pipeline on WormMap data. In an ideal scenario, we would evaluate the complete space of all possible parameters, however the space for searching the optimal parameter configuration grows exponentially (2|parameters|) with the number of parameters we want to configure. Thus, to make the benchmarking of EPIC feasible, we investigated only one parameter at a time while keeping the remaining parameters fixed. First we will describe benchmarking statistics and evaluation criteria followed by the results of benchmarking.
Feature parameters
In this part, we evaluate the optimal parameter settings for co-elution scores (if any parameter setting is involved). From the total of eight correlation features, two of them have parameters to optimize: the prior used for the Bayes correlation and the number of noise iterations for Pearson correlation plus noise (PCCN). We evaluated those parameters based on how well they can predict PPIs (i.e. precision, recall, F1, auROC, auPR). To be consistent, all the evaluations were performed using elution data generated by the MSblender search engine, as it is the search engine used in our previous publication that generated the largest data set with the most identified proteins. The results for number of noise iterations can be found in Supplementary Fig. 10 and we observed optimal scores obtained for five noise iterations. After analyzing the three possible Bayes priors, we observed no significant differences between the three different priors based on ROC and PR curves (Supplementary Fig. 11). However, if we analyze the evaluation metrics for predicted protein complexes we see the best composite score for the zero-count prior (Bayes3) (Supplementary Table 9). Thus, we use the zero-count prior for EPIC.
EPIC parameter optimization by nested cross validation
It is not possible to provide globally optimal parameters for all data sets. In EPIC, we developed a nested cross validation strategy to optimize parameters for our worm data and used the optimized set of parameters to generate our WormMap. As described in Fig. 2a, we first collected and merged all worm protein complexes from CORUM, GO and IntAct. We first used k-means clustering and an overlap score as the measurement metric to divide the whole set of reference protein complexes set into two distinct sets of complexes. We then balanced the two sets while minimizing the overlap by pruning. The first half is used for training (based on our co-fractionation data) while the second half is used as the ‘holdout’ set for evaluation (2-fold cross validation at the protein complex level). In our study and in the EPIC software, we implement two machine-learning classifiers, support for four protein searching/quantification tools and eight different correlation scores, which gives us 2,040 total parameter combinations. We trained machine-learning classifiers with our worm co-fractionation data to predict protein-protein interactions and protein complexes for each of the 2,040 different parameters combinations. The resulting 2,040 predicted protein complex sets were then benchmarked with the held out “test” half of the curated protein complexes using composite score (see main text and above) as the evaluation metric. Random forest in general outperformed support vector machine for predicting protein complexes (Supplementary Fig. 12a). MSblender gives the best composite score compared with other protein search/quantification tools (Supplementary Fig. 12b). To get a relatively good prediction, at least three different correlation scores are required (Supplementary Fig. 12c). The optimized set of parameters (machine-learning classifier: random forest, protein searching/quantification tool: MSblender, correlation scores: mutual information, Bayes correlation, Euclidean distance, weighted cross correlation and apex score) for generating WormMap is the combination that gives the highest composite score. Functional evidence data was then added to the matrix formed by the optimal set of correlation scores for predicting protein-protein interactions. Since extensive computational resources are required for this optimization, we performed this analysis on the SciNet supercomputing platform (https://www.scinethpc.ca/). We provide a parameter optimization function in the EPIC software and encourage users to optimize their parameters using their own data if a super computing resource is available, but otherwise, we recommend using the default EPIC parameters, which are the ones that were found optimal for WormMap using the above procedure.
Exploring the value of the additional experiments
After nested cross validation, the selected optimal correlation score combination and random forest machine learning classifier was used for evaluating if a pre-enrichment step improves protein complex prediction and what is the most economic way to perform experiments. We performed the analysis using data collected from pre-enrichment, non-pre-enrichment (IEX) and the combination of both (all experiments), individually. Similar to the step of nested cross validation, we benchmarked the predicted protein complexes using composite score, based on our 2-fold cross-validation strategy. For each specific number of experiments, we considered all combinations and reported the average of the evaluating metrics. For example, for the first point in the plot indicating use of one experiment, we analyzed each of our seven IEX experiments individually to predict complexes, evaluated the composite score and then calculated the average of number predicted complexes and composite scores over the seven experiments. We observed a positive correlation between composite score and the number of experiments (Supplementary Fig. 13a). After five experiments, using IEX alone performed much better than using all experiments. Similarly, a sharp increase is observed for the last point of the “all experiments” line (red line). We then asked if the sharp increase of IEX performance is the result of sacrificing the coverage of predicted protein complexes. To balance the coverage of predicted protein complexes and composite score, we then plotted “composite score × the number of predicted complexes” vs. “number of experiments” (Supplementary Fig. 13b). In this plot, we noticed the “all experiments” line reached its stationary phase at nine experiments. We also noticed a dramatic decrease of the “IEX” line at seven experiments, which shows that the sharp increase of composite score for “IEX” is due to a decrease in the number of predicted protein complexes. Also, when using all 16 experiments, the composite score is maximized. Thus, the general guideline would be to use as many experiments as possible and that pre-enrichment will help protein complex prediction in terms of both composite score and coverage, however, if mass spectrometry time is limited, a reasonable lower bound is to run four IEX experiments.
Cut-off for correlation coefficient
We plotted the histogram of maximal correlation scores for all positive protein-protein interactions among all seven different correlation coefficients (apex score is not included, since it is either 0 or 1) across all experiments performed (Supplementary Fig. 3a). We noticed there is a clear cut-off at 0.5, which suggests we could retain protein pairs with a co-elution correlation score over 0.5 for machine learning prediction, as pairs without any co-elution score over 0.5 are not likely to be positive interactions.
Comparison of EPIC with PrInCE
To objectively compare the performance of the two tools, we downloaded the example SILAC co-fractionation data available from the PrInCE website (condition1.csv and condition2.csv) and used this as input data to predict protein complexes using both PrInCE and EPIC. We then compared the results (predicted complexes) with a benchmark set of reference assemblies (i.e. CORUM) using the multifactor composite score as the stringent evaluation metric. The resulting set of protein complexes predicted by EPIC with the SILAC data alone produced a substantially higher composite score than PrInCE achieved (Supplementary Table 1) and that EPIC also predicted up to five times as many complexes (with comparable or higher reliability) than PrInCE (Supplementary Table 1).
Disease and phenotype enrichment analysis
Since there is a lack of information available for Worm gene disease associations, we combined several human resources and mapped human gene names to worm gene names via 1:1 orthology using InParanoid. Gene disease associations were retrieved from the Online Mendelian Inheritance in Man (OMIM), UniProt, and ClinVar databases. However, OMIM only provides gene-disease associations, and thus we retrieved a mapping from gene name to UniProt identifier via the UniProt identifier mapping web service. Moreover, OMIM does not provide a classification system for their diseases and different OMIM IDs might describe the same disease (e.g. Alzheimer has multiple identifiers depending on the types). Thus, we mapped each OMIM disease identifier to their corresponding disease ontology identifier (DOID). In the final step, we combined the resulting data set with a set of DOID annotations for Worm genes from the WormBase database. For phenotype analysis, we annotated our protein complexes with phenotype information taken from WormBase. Statistical enrichment for both phenotype and disease was determined by Fisher exact test, and Benjamini-Hochberg procedure was applied for multiple testing correction.
GO Enrichment
The Gene Ontology (GO) is a controlled vocabulary that describes genes by using three categories: molecular function, cellular component and biological process. We inferred enriched GO terms using the g:Profiler R package50. To ensure we only get significant hits we only considered GO terms with less than 500 proteins annotated to it, and the p-value was corrected by the conservative Bonferroni correction procedure.
Supplementary Material
ACKNOWLEDGMENTS:
This study was supported by a Foundation Grant (FDN #148399) from the Canadian Institute of Health Research (CIHR) to A. Emili, and U.S. National Institutes of Health grants (P41 GM103504, GM070743) to G. D. Bader. L.Z Hu was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) Mass Spectrometry-Enabled Science and Engineering (MS-ESE) program.
Footnotes
DATA AVAILABILITY and ACCESSION:
The supporting co-fractionation data is available via ProteomeXchange with the identifier PXD011182. The entire WormMap network (Cytoscape format) is available on GitHub (https://github.com/BaderLab/EPIC/tree/master/WormMap). The EPIC software code is publicly available on GitHub (https://github.com/BaderLab/EPIC).
COMPETING INTERESTS:
The authors declare no competing interests.
REFERENCES
- 1.Rigaut G et al. A generic protein purification method for protein complex characterization and proteome exploration. Nature biotechnology 17, 1030–1032, doi: 10.1038/13732 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Krogan NJ et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Gavin AC et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147, doi: 10.1038/415141a (2002). [DOI] [PubMed] [Google Scholar]
- 4.Ho Y et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415, 180–183, doi: 10.1038/415180a415180a [pii] (2002). [DOI] [PubMed] [Google Scholar]
- 5.Gavin AC et al. Proteome survey reveals modularity of the yeast cell machinery. Nature 440, 631–636 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Hu P et al. Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS biology 7, e96, doi: 10.1371/journal.pbio.1000096 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huttlin EL et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 162, 425–440, doi: 10.1016/j.cell.2015.06.043 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hein MY et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163, 712–723, doi: 10.1016/j.cell.2015.09.053 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Babu M et al. Interaction landscape of membrane-protein complexes in Saccharomyces cerevisiae. Nature 489, 585–589, doi: 10.1038/nature11354 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Havugimana PC et al. A census of human soluble protein complexes. Cell 150, 1068–1081, doi: 10.1016/j.cell.2012.08.011 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wan C et al. Panorama of ancient metazoan macromolecular complexes. Nature 525, 339–344, doi: 10.1038/nature14877 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu F, Rijkers DT, Post H & Heck AJ Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nature methods 12, 1179–1184, doi: 10.1038/nmeth.3603 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Ruepp A et al. CORUM: the comprehensive resource of mammalian protein complexes--2009. Nucleic acids research 38, D497–501, doi: 10.1093/nar/gkp914 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.UniProt C UniProt: a hub for protein information. Nucleic acids research 43, D204–212, doi: 10.1093/nar/gku989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orchard S et al. The MIntAct project-IntAct as a common curation platform for 11 molecular interaction databases. Nucleic acids research 42, D358–D363, doi: 10.1093/nar/gkt1115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The Gene Ontology, C. Expansion of the Gene Ontology knowledgebase and resources. Nucleic acids research 45, D331–D338, doi: 10.1093/nar/gkw1108 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuberi K et al. GeneMANIA prediction server 2013 update. Nucleic acids research 41, W115–122, doi: 10.1093/nar/gkt533 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szklarczyk D et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic acids research 45, D362–D368, doi: 10.1093/nar/gkw937 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sonnhammer EL & Ostlund G InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic acids research 43, D234–239, doi: 10.1093/nar/gku1203 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504, doi: 10.1101/gr.1239303 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stacey RG, Skinnider MA, Scott NE & Foster LJ A rapid and accurate approach for prediction of interactomes from co-elution data (PrInCE). Bmc Bioinformatics 18, 457, doi: 10.1186/s12859-017-1865-8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez-Taltavull D, Ramachandran P, Lau N & Perkins TJ Bayesian Correlation Analysis for Sequence Count Data. Plos One 11, e0163595, doi: 10.1371/journal.pone.0163595 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nepusz T, Yu H & Paccanaro A Detecting overlapping protein complexes in protein-protein interaction networks. Nature methods 9, 471–472, doi: 10.1038/nmeth.1938 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiwie C, Baumbach J & Rottger R Comparing the performance of biomedical clustering methods. Nature methods 12, 1033–1038, doi: 10.1038/nmeth.3583 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Cho A et al. WormNet v3: a network-assisted hypothesis-generating server for Caenorhabditis elegans. Nucleic acids research 42, W76–82, doi: 10.1093/nar/gku367 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner B et al. iRefWeb: interactive analysis of consolidated protein interaction data and their supporting evidence. Database (Oxford) 2010, baq023, doi:baq023 [pii] 10.1093/database/baq023 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chatr-Aryamontri A et al. The BioGRID interaction database: 2017 update. Nucleic acids research 45, D369–D379, doi: 10.1093/nar/gkw1102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mulder NJ et al. The InterPro Database, 2003 brings increased coverage and new features. Nucleic acids research 31, 315–318 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kagawa H, Gengyo K, McLachlan AD, Brenner S & Karn J Paramyosin gene (unc-15) of Caenorhabditis elegans. Molecular cloning, nucleotide sequence and models for thick filament structure. J Mol Biol 207, 311–333 (1989). [DOI] [PubMed] [Google Scholar]
- 30.Harris TW et al. WormBase: a comprehensive resource for nematode research. Nucleic acids research 38, D463–467, doi: 10.1093/nar/gkp952 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monemi S et al. Identification of a novel adult-onset primary open-angle glaucoma (POAG) gene on 5q22.1. Hum Mol Genet 14, 725–733, doi: 10.1093/hmg/ddi068 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Yunger E, Safra M, Levi-Ferber M, Haviv-Chesner A & Henis-Korenblit S Innate immunity mediated longevity and longevity induced by germ cell removal converge on the C-type lectin domain protein IRG-7. PLoS genetics 13, e1006577, doi: 10.1371/journal.pgen.1006577 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amberger JS, Bocchini CA, Schiettecatte F, Scott AF & Hamosh A OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic acids research 43, D789–798, doi: 10.1093/nar/gku1205 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stenson PD et al. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 133, 1–9, doi: 10.1007/s00439-013-1358-4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olinares PD, Ponnala L & van Wijk KJ Megadalton complexes in the chloroplast stroma of Arabidopsis thaliana characterized by size exclusion chromatography, mass spectrometry, and hierarchical clustering. Molecular & cellular proteomics : MCP 9, 1594–1615, doi: 10.1074/mcp.M000038-MCP201 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skinnider MA, Stacey RG & Foster LJ Genomic data integration systematically biases interactome mapping. PLoS Comput Biol 14, e1006474, doi: 10.1371/journal.pcbi.1006474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tran JC et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 480, 254–258, doi: 10.1038/nature10575 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Werner T et al. Ion coalescence of neutron encoded TMT 10-plex reporter ions. Analytical chemistry 86, 3594–3601, doi: 10.1021/ac500140s (2014). [DOI] [PubMed] [Google Scholar]
- 39.Ideker T & Krogan NJ Differential network biology. Molecular systems biology 8, 565, doi: 10.1038/msb.2011.99 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stiernagle T in WormBook : the online review of C. elegans biology (ed The C. elegans Research Community; ) (WormBook). [Google Scholar]
- 41.Kwon T, Choi H, Vogel C, Nesvizhskii AI & Marcotte EM MSblender: A probabilistic approach for integrating peptide identifications from multiple database search engines. Journal of proteome research 10, 2949–2958, doi: 10.1021/pr2002116 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cox J & Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature biotechnology 26, 1367–1372, doi: 10.1038/nbt.1511 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Kislinger T et al. PRISM, a generic large scale proteomic investigation strategy for mammals. Molecular & cellular proteomics : MCP 2, 96–106, doi: 10.1074/mcp.M200074-MCP200 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Campagnola PJ et al. Three-dimensional high-resolution second-harmonic generation imaging of endogenous structural proteins in biological tissues. Biophys J 82, 493–508, doi: 10.1016/S0006-3495(02)75414-3 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dupuy D et al. A first version of the Caenorhabditis elegans Promoterome. Genome Res 14, 2169–2175, doi: 10.1101/gr.2497604 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kwan J et al. DLG5 connects cell polarity and Hippo signaling protein networks by linking PAR-1 with MST1/2. Genes Dev 30, 2696–2709, doi: 10.1101/gad.284539.116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wehrens R, Melssen W, Buydens L & de Gelder R Representing structural databases in a self-organizing map. Acta Crystallogr B 61, 548–557, doi: 10.1107/S0108768105020331 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Brohee S & van Helden J Evaluation of clustering algorithms for protein-protein interaction networks. Bmc Bioinformatics 7, 488, doi: 10.1186/1471-2105-7-488 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scrucca L, Fop M, Murphy TB & Raftery AE mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J 8, 289–317 (2016). [PMC free article] [PubMed] [Google Scholar]
- 50.Reimand J et al. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic acids research 44, W83–89, doi: 10.1093/nar/gkw199 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.