

Abstract
In current scenario, various heterocycles have come up exhibiting crucial role in various medicinal agents which are valuable for mankind. Out of diverse range of heterocycle, quinoline scaffold have been proved to play an important role in broad range of biological activities. Several drug molecules bearing a quinoline molecule with useful anticancer, antibacterial activities etc have been marketed such as chloroquine, saquinavir etc. Owing to their broad spectrum biological role, various synthetic strategies such as Skraup reaction, Combes reaction etc. has been developed by the researchers all over the world. But still the synthetic methods are associated with various limitations as formation of side products, use of expensive metal catalysts. Thus, several efforts to develop an efficient and cost effective synthetic protocol are still carried out till date. Moreover, quinoline scaffold displays remarkable antiviral activity. Therefore, in this review we have made an attempt to describe recent synthetic protocols developed by various research groups along with giving a complete explanation about the role of quinoline derivatives as antiviral agent. Quinoline derivatives were found potent against various strains of viruses like zika virus, enterovirus, herpes virus, human immunodeficiency virus, ebola virus, hepatitis C virus, SARS virus and MERS virus etc.
Keywords: Quinoline, Skraup reaction, Combes reaction, Antiviral activity, Ebola virus, Hepatitis C virus, COVID-19
Graphical abstract
1. Introduction
Quinoline or benzo[b]pyridine is a nitrogen containing heterocyclic aromatic compound, acting as a weak tertiary base and has the ability to form salts with acids and undergoing electrophilic substitution reactions as well as reactions similar to those of pyridine and benzene [1]. It is one of the most privileged N-containing motifs till date, most commonly found in various natural products, most common being Cinchona alkaloids and pharmacologically active substances thereby displaying a broad range of biological activity. It is known to possess several biological activities such as antimalarial (Quinine, quinidine, chloroquine, mefloquine, amodiaquine etc), antibacterial (fluoroquinolones such as ciprofloxacin, sparfloxacin), antifungal-antiprotozoal (Clioquinol), anthelmintic (oxamniquine), local anesthetic (dibucaine), antiasthmatic (montelukast), anticancer (camptothecin, irinotecan, topotecan), antipsychotic (Aripiprazole, brexpiprazole), antiglaucoma (cartiolol) and cardiotonic (vesnarinone). A broad spectrum biological activity of quinoline is illustrated in Fig. 1 [[2], [3], [4]].
Fig. 1.

Broad spectrum biological activity of quinoline scaffold.
Several antiviral drugs such as saquinavir, indiravir containing quinoline scaffold are marketed these days but still this area is widely unexplored. Drugs acting specifically on virus targets are available but only for some viral infections. Various molecules have been developed but they are usually associated with drug resistance, cytotoxicity thats by there is an urge to develop more efficient antiviral drugs. In addition to the biological applications, it is being utilized in agrochemicals, anti-foaming agents in refineries and as ligands to prepare phosphorescent complexes in sensors. Quinoline containing few antiviral drugs are shown in Fig. 2 [[5], [6], [7], (a), (b)].
Fig. 2.

Marketed antiviral drugs with quinoline skeleton.
Owing to its immense pharmacological activity, extensive research has been done for the synthesis of quinoline molecule and its derivatives over the years. Till date, several established methods exists for the preparation of quinoline skeleton, most widely being employed as the skraup synthesis with anilines and acrolein, friedländer synthesis with o-amino-benzaldehydes and ketones, combes quinoline synthesis with anilines and 1,3-diketones. Although several methods have been developed, they are associated with certain limitations thus there is a need for developing simple and much more efficient synthetic methods. In this report, we have made an attempt to describe various synthetic methodologies being adopted by various research groups these days along with the importance of quinoline skeleton in the development of antiviral drugs with minimal side effects and drug resistance like issues. We have tried to cover up several viruses responsible for causing severe fatal infections in humans, their targets on which they act. There is very limited review on quinolone as antiviral agents, however we focused on recent literature of synthetic and antiviral effect of quinoline and its derivatives.
2. Synthetic strategies of quinoline scaffold
In literature, several classical strategies have been very well established for the synthesis of quinolines as shown in Fig. 3 [[8], (a), (b), (c)]. However, these methods were usually associated with certain limitations such as requirement of high temperature of approximately 200 °C, limited availability of substrates, poor regioselectivity, low yields, tedious multistep procedures which often require isolation of intermediates, hazardous reagents in stoichiometric amounts which adversely affect the atom economy and environment friendly chemistry. Therefore, since years, modifications have been done and till now being done to address such limitations. In this report, we have made an attempt to describe such modifications which were performed in the span of last two years.
Fig. 3.

Various Classical approaches for quinoline synthesis.
2.1. Catalyst mediated
2.1.1. Nickel-catalyzed
Recently, transition metal-catalyzed coupling reactions have been utilized as an efficient tool for the synthesis of quinoline and its derivatives [[9], (a), (b), (c), (d)]. In 2019, Das et al. reported the nickel catalyzed synthesis of polysubsituted quinolines from α-2-aminoaryl alcohols via sequential dehydrogenation and condensation process which lead to low catalyst loading and wider substrate scope [10]. α-2-Aminoaryl alcohols are effectively employed for acceptorless dehydrogenation (AD) which generally involves the generation of more reactive carbonyl compounds from alcohols by the release of dihydrogen as stoichiometric by product. The in situ generated carbonyl further reacts with a suitable coupling partner to form the desired product, thereby acting as convenient tool for the synthesis of heterocyclic compounds. The nickel catalyst 3 used by Das and co workers was first synthesized by Jager and Goedken [11] and this was further prepared in single step by Niewahner [12].
This protocol allowed the use of primary as well as secondary α-2-aminoaryl alcohols 1 in combination with ketone derivatives 2 for the synthesis of substituted quinoline derivatives with upto 93% isolated yields as shown in Scheme 1 .
Scheme 1.

The nickel catalyzed synthesis of polysubsituted quinolines from α-2-aminoaryl alcohols.
Similarly, Chakraborty and group workers reported a biomimetic method for the construction of polysubstituted quinolines and its derivatives through dehydrogenative condensation/coupling of 2-aminobenzyl alcohols 1 with ketones 2 catalyzed by welldefined singlet diradical Ni (II)-complex 5 [13]. The catalyst is asinglet diradical species containing two antiferromagnetically coupled ligand-centered radicals. With the mechanistic studies in hand it was suggested that the transfer of a hydrogen atom from the alcohol to the coordinated ligand take place viaone electron hydrogen atom transfer process forming ketyl radical intermediate [14]. After the successful synthesis of quinolines, 2-aminoquinolines 7 and quinazolines 9 were also synthesized viabase-promoted intermolecular/intramolecular condensation of 2-aminobenzyl alcohols 1 with 2-phenylacetonitrile 6 (for 2-aminoquinolines) and nitriles 8 (quinazolines) under the similar optimized reaction conditions as shown in Scheme 1. The desired products were synthesized in moderate to good yields from readily accessible starting materials under relatively mild reaction temperature (<100 °C) and aerial conditions as described in Scheme 2 .
Scheme 2.

Biomimetic method for the construction of polysubstituted quinolones.
2.1.2. Iridium-catalyzed
In 2018, Xiong et al. illustrated a straightforward synthesis of quinolones 11 from enones 10 and 2-aminobenzyl alcohols 1 using iridium-catalyzed transfer hydrogenative reactions [15]. This protocol employed the use of readily available [IrCp∗Cl2]2/t-BuOK as the efficient catalyst system that enabled the reaction to proceed with the merits of atom efficiency, mild reaction conditions and operational simplicity. The mechanistic insights suggested that the reactions start with transfer hydrogenation, followed by the Friedländer reaction to give the desirable products. Another report based on synthesis of quinoline through NHC-IrI complex 12 has been reported by Arslan group in 2019 [16]. Iridium(I) complexes are known to have an imidazol-2-ylidene ligand with benzylic wingtips which are known to efficiently catalyze the acceptorless dehydrogenative cyclization of 2-aminobenzyl alcohol with ketones through a borrowing hydrogen (BH) pathway. The BH approach is a tandem process in which alcohols are dehydrogenated followed by base-mediated condensation of the resulting carbonyls with an amine or carbon nucleophile and subsequently the unsaturated intermediate product is reduced. Therefore, substituted quinolines 14 were obtained from the combination of 2-aminobenzyl alcohols 1 and ketones 13 by utilizing NHC-IrI complex 12(in ppm) and 5 mol% of KOH as the catalytic system under air atmosphere and liberating H2 as byproduct as shown in Scheme 3 .
Scheme 3.

Synthesis of quinoline through NHC-IrI complex and [IrCp∗Cl2]2/t-BuOK catalytic system.
2.1.3. Rhodium-catalyzed
Jia and co workers have described a novel synthetic scheme for the synthesis of indolo[3,2-c]quinoline or 3-(2-aminophenyl)quinoline through Rh(III)-catalyzed stepwise dimerization of 2-alkynylanilines under aerobic or anerobic conditions in an efficient and tunable manner [17]. Indolo[3,2-c]quinoline also known as γ-carboline,which is an important scaffold found in numerous natural and synthetic compounds having potential biological activities. Although several methods have been developed for the synthesis of indolo[3,2-c]quinoline, but this method ensures high atom economy and good efficiency. Rh(III)complex was found to be effective catalyst as well as highly compatible with different functional groups. Reaction was carried out using 2-(phenylethynyl)aniline 15 with the promotion of Rh(III)/HFIP under aerobic or anaerobic conditions giving 6-phenyl-11H-indolo[3,2-c]quinolone 16a or 2-(4-benzyl-2-phenylquinolin-3-yl)aniline 17a in good yields as illustrated in Scheme 4 .
Scheme 4.

Synthesis of functionalized quinoline using Rh catalyst.
In 2019, Kumar et al. devised synthesis of functionalized quinolines from hydrazones of 2-aminoacetophenone and alkynes using Rh catalyst under base and oxidant free conditions [18]. Reaction of (Z)-2-(1-(2-phenylhydrazono)ethyl)aniline 18 with phenylacetylenes 19 in presence of Rh[Cp∗Cl2]2as catalyst and acetonitrile as solvent afforded product 20 in moderate to good yields. Mono 22a-b and bis quinolines 22c-d were synthesized by carrying out reaction with 18, 1,4- and 1,3 dialkynes 20 and 21 respectively using standard reaction conditions as shown in Scheme 4. However, it was interesting to note that mono-quinolines 22a-b with free alkyne functionality can be synthesized using similar reaction condition and on further decreasing the amount of alkynes up to 0.5 equiv efficiently provided the bisquinolines 22c-d in moderate yields as shown in Scheme 5 .
Scheme 5.

Synthesis of mono- and bis-quinolines.
2.1.4. Copper-catalyzed
Synthesis of pyrimidine fused quinolines has been reported by Pandat et al., in 2019 using ligand free copper catalytic protocol [19]. Pyrimidine ring fused with quinoline are known as deazaflavins or 5-deazaisoalloxazines while the N-5 analogues of these molecules are known as flavins which are present in biomolecules as riboflavin and flavin adenine dinucleotide (FAD)as shown in Fig. 4 .
Fig. 4.

Some examples of pyrimidine fused with quinoline.
Reaction of 6-amino-1,3-dimethyluracils 23 with 2-bromobenzaldehydes 24 or 2-bromobenzyl bromide derivatives 25 using copper(II) catalyzed Ullman type domino reactions lead to the formation of C-C and C-N bonds as pyrimdine fused quinoline 26 in one pot using DMF as reaction medium and without the use of any ligand. It was suggested that Cu(II) catalyst along with coordinating and reducing property of DMF played a synergistic role in the synthesis of fused quinolines. A range of 2-bromobenzaldehyde derivatives containing both electron donating and with drawing substituents at different positions of the aryl ring such as −CH3, −OCH3, −CN, −NO2, −F, −Br, −CF3 were reacted with the initial substrate 23 and thus pyrimidine fused quinoline derivatives were afforded in moderate to good yields. It was observed that 2-bromobenzaldehydes with electron donating substituents afforded better yields compared to electron withdrawing group as shown in Scheme 6 .
Scheme 6.

Synthesis of pyrimidine fused quinolines using ligand free synthetic protocol.
However, when the reaction of 2-bromobenzyl bromide 25 with 6-amino-1,3-dimethyluracil 23 was carried out using similar reflux conditions the product was obtained only in 33% yield. Therefore, further optimization was carried on by changing the reaction conditions from reflux to microwave heating which led to increased yields. Reaction of benzyl bromides 25 with aminouracil derivatives in the presence of CuCl2 (10 mol %) and K2CO3 (2 equiv) and an oxygen balloon in DMF under reflux conditions was considered as an optimized reaction conditions.
Another, one pot synthetic strategy for the synthesis of 2,4-disubstituted quinolines using copper(II) catalysis was described by Wu and coworkers [20]. Reaction of substituted anilines 27 with dimethyl acetylenedicarboxylate 28 and Cu(OTf)2 catalyst (5 mmol %) in CH3CN at 120 °C generated the substituted quinolines 30 in good to excellent yields (58–96%) with complete regio- and site-selectivity. The regioselective cascade annulation was catalyzed by Cu(OTf)2 employing enamines and propargylic imines as the key intermediates. Interestingly, ketones 29 were replaced as the second molecule of alkyne esters acting as two-carbon source and alkyneprecursor for the Cu(II)-catalyzed second-stage nucleophilic addition followed by intramolecular Friedel-Crafts-type addition and dehydration to produce quinoline products 31 in good yields and excellent regioselectivity as shown in Scheme 7 .
Scheme 7.

Synthesis of 2,4-disubstituted quinolines using copper(II) catalyst.
Formation of heteroaryl-ketone substrates is quite challenging in the field of transition-metal-catalyzed synthesis, owing to their potent coordination ability with transition-metal leading to inactivation of transition-metal catalyst. But surprisingly, the reaction with 1-(pyridin-2-yl)ethanone or its analogues proceeded smoothly to deliver the quinoline products 31(e-s) with good yields after isolation with remarkable tolerance for aniline substrates. The above mentioned results not only the reasoning but also broadly explained the versatility of developed Cu(II)-catalytic system.
Xu et al. have reported an efficient process for the synthesis of quinolines through an N-heterocyclic carbene copper catalyzed indirect Friedländer reaction from 2-aminobenzyl alcohol and aryl ketones using DMSO as an oxidant at room temperature [21]. The indirect Friedländer quinoline synthesis has been developed to improve Friedländer synthesis through the oxidative cyclization of 2-aminobenzylic alcohols instead of o-aminobenzaldehydes with ketones or alcohols. Consequently, three synthetic strategies have been developed for the indirect Friedländer quinoline synthesis which involves: (a) use of ketone as the oxidant through Pfitzner-Moffatt oxidation (b) using oxygen or air as the oxidant at high reaction temperature (c) Through catalytic dehydrogenation, usually under the catalysis of unique metal complex.
Synthesis of quinolines from 2-aminobenzyl alcohol 32 and substituted acetophenone 33 in presence of iPrCuCl as N-heterocyclic carbene complex and DMSO as oxidant was carried out under room temperature to afford quinolines 34 with excellent yields (40–95%). Scope of synthesis was investigated which revealed that with the use of acetophenones substituted with electron donating or withdrawing group, reaction proceeded smoothly via cyclization reaction and afforded the corresponding products in moderate to excellent yields. Ortho-substituted acetophenone showed the inferior result compared with meta- or para-substituted ones because of the steric hindrance associated with the group attached to the phenyl ring of acetophenone. Numerous 2-aminobenzylic alcohols were also screened which led to the conclusion that electron-donating groups and halogens on the phenyl ring can easily undergo the transformation thereby producing quinolines in good to excellent yields as illustrated in Scheme 8 .
Scheme 8.

Synthesis of quinolines through an N-heterocyclic carbene copper catalyst.
2.1.5. Cobalt-catalyzed
Cobalt-catalyzed C-H activation has gained interest due to its high abundance, cheap, excellent reactivity, selectivity and broad substrate scope. Therefore, a unique Co (III) catalyzed- and DMSO involved C-H activation/cyclization from simple, cheap, and readily available anilines 35 with alkynes 36 described the direct and highly efficient synthesis of quinolines 37 with complete regioselectivity and broad substrate group in good to excellent yields [22]. DMSO has been employed both as solvent and C1 building block of quinolines whereas AgSbF6, and K2S2O8 acted as catalyst and cocatalyst respectively, in this one-pot sequence. This transformation to quinolines is a result of first undergoing C-H activation process and utilizing 2-vinylbenzenamine species as the active intermediate. Various substituted anilines were reacted with terminal alkynes which revealed that the reactions proceeded smoothly to provide desired products in good yields (upto 91%) as shown in Scheme 9 .
Scheme 9.

Co (III) catalyzed- and DMSO involved C-H activation/cyclization for quinoline synthesis.
Li et al. have described an alternative approach for the synthesis of quinolines using cobalt catalyzed electrophilic amination of anthranils under mild conditions. Anilines were obtained from the reaction of organozinc pivalates and anthranils [23]. Organozinc pivalates are basically a class of zinc organometallics with advanced air and moisture stability and can be easily stored as solid form for months under argon atmosphere. Alkenyl pivalates 38 was allowed to react best with anthranils 39 in the presence of 10 mol % of CoCl2 at 23 °C for 16 h to provide the quinolines 40 in 62–96% yields. It was clearly observed that after electrophilic amination step, cycloisomerisation (occurring via A to B) was carried out in one-pot procedure, thereby leading to the formation of novel heterocycles as shown in Scheme 10 . Scope of quinoline synthesis was improved using different aryl and heterocyclic zinc pivalates, which were prepared using the corresponding (hetero)aryl bromides by magnesium insertion in the presence of LiCl, or by a directed metalation of heterocyclic substrates with TMP-bases (TMP = 2,2,6,6-tetramethylpiperidyl) such as TMPZnOPiv·LiClorTMPMgCl·LiCl), followed by transmetalation with Zn(OPiv)2 [[24], [25], [26]].
Scheme 10.

Cobalt catalyzed electrophilic amination of anthranils.
Furthermore, some of the heterocycles were further functionalized by metalation. Thus, the condensed heterocycles 40 m and 40f were treated with TMPMgCl·LiCl at 23 °C for 16 h to give complete magnesiation (as shown by iodolysis). Zn(OPiv)2 was added followed by a Negishi cross-coupling using XPhos provided the arylated N-heterocycle bond. Buchwald Hartwig amination of 40g was carried out to afford the aminated thieno[2,3-b]quinoline 41 in 72% yield. The zinc pivalate derived from thieno[2,3-b]quinoline 40f was made to react again with anthranil 39 in the presence of CoCl2 (10 mol %) affording the new heterocycle 42 in 34% yield as illustrated in Scheme 11 .
Scheme 11.

Functionalization of heterocycles by metalation.
2.1.6. Scandium-catalyzed
Tepe’s group have described a one pot synthetic sequence of 2,3-disubstituted quinolines 45 by reacting 3,4,5-trimethoxyaniline 43 and styrene oxide 44 in presence of Sc(OTf)3 and TEMPO using THF as solvent [27]. TEMPO acted as radical scavenger in order to prevent irrelevant oxidation by trapping the radicals generated in the reaction vessel. Styrene oxides substituted with electron donating and withdrawing groups were very well tolerated to afford quinolines in 66–96% yield. Whereas the best yield of 96% was produced by electron withdrawing group mainly due to the reduction of oxidative C-2 dealkylation. However, by reducing the electron density on the aromatic ring directly affected the product yield. Removal of one methoxide from the substrate 43 provided yield upto 78% while presence of only single methoxy group substituted at ortho-, meta- and para positions of anilines generated 54–57% of the product yield as shown in Scheme 12 .
Scheme 12.

Synthesis of 2,3-disubstituted quinolines using Sc(OTf)3.
In 2019, qiu and co workers designed AgSCF3/Na2S2O8 promoted cascade cyclization of o-propargyl arylazide 46 leading to successful generation of trifluoromethylthio-substituted quinolone 47 in a single step via direct trifluoromethyl thiolation/cyclization reaction [28]. Trifluoromethylthio (SCF3) is well known for its high metabolic stability, electonegativity, and lipophilicity thereby giving rise to enhanced absorption rate and membrane permeability in bioavailability. This transformation proceeded effortlessly in moderate to excellent yields with one C(sp2)-N bond and C(sp2)-SCF3 bond constructed consecutively. From the control experiment, it was revealed that radical pathway initiated by a carbon carbon triple bond was crucial in the reaction process as shown in Scheme 12.
2.1.7. Gold metal catalyzed
Liu et al. described gold catalyzed annulations of anthranils with aryloxyethynes or aryl propargyl ethers for the construction of useful benzofuro[2,3-b]quinoline and 6H-chrome no[3,4-b]quinolone scaffolds respectively in one pot strategy [29]. Furoquinoline scaffolds are naturally occurring alkaloids belonging to Rutaceae family, enriched with numerous biological activities such as antiviral, antiplatelet aggregation, cytotoxic and anti-acetylcholinesterase activity. Annulation was carried out using phenoxyethyne 48 and anthranil 49 as starting materials in the presence of gold catalysts equipped with silver salts as in P(t-Bu)2(o-biphenyl)AuCl/AgSbF6 generated furoquinoline 50 in 68% yield with DCE as reaction medium at 80 °C. Correspondingly, anthranils 49 bearing various 5-phenyl substituents with phenoxyethyne 48 afforded novel 6H-chrome no [3,4-b]quinoline derivatives 51 with reasonable yields (59–76%) under similar reaction conditions. The authors proposed a reaction mechanism to proceed via sequential cyclizations among the oxyaryl group, gold carbene and benzaldehyde of the α-imino gold carbene intermediates as illustrated in Scheme 13 .
Scheme 13.

Gold catalyzed synthesis ofbenzofuro[2,3-b]quinoline and 6H-chrome no[3,4-b]quinolone scaffolds.
2.2. Boronic acid mediated
In 2019, Chen group developed a novel palladium-catalyzed approach for the synthesis of 2-arylquinolines via tandem reaction of 2-aminostyryl nitriles with arylboronic acids with good functional group tolerance [30]. Literature reports suggest that, dehydrative cyclization of 2-aminostryl ketones was considered as an attractive protocol for the synthesis of 2-arylquinoline but it was observed that the amino group was unable to approach the carbonyl group for condensation due to sterric reasons. Therefore, this novel approach acts as an alternative for the synthesis of 2-arylquinolines compared to classical condensation reaction. The reaction was carried out between substituted-2-aminostyrylnitrile 52 and phenylboronic acid 53 in the presence of Pd(tfa)2 catalyst, bpy as bidentate ligand, and p-toluenesulfonic acid monohydrate as additive using toluene as solvent at 90 °C to gave the products 55 upto 85% yield as depicted in Scheme 14 . The authors conducted certain preliminary experiments which revealed that this transformation is a result of nucleophilic addition of aryl palladium species to the nitrile in order to generate an aryl ketone intermediate followed by an intramolecular cyclization and dehydration to quinoline ring.
Scheme 14.

Palladium-catalyzed and direct arylation reaction for the synthesis of 2-arylquinolines.
Similarly, another strategy for the selective synthesis of 2-arylquinolines was discovered by Ren et al. by utilizing direct arylation reaction of N-methoxyquinoline-1-tetrafluoroborate derivatives and arylboronic acids at room temperature [31]. N-methoxyquinoline-1-tetrafluoroborate 54 was subjected to react with arylboronic acids 53 in the presence of silver nitrate, Na2S2O8 as oxidant while TFA being additive in DCM: H2O (1:1) solvent system at 25 °C to afford 2-arylquinolines 55 in good to moderate yields. Plausible reaction mechanism suggested that a radical was probably involved in this transformation. Persulfate anion was converted to sulfate radical in the presence of silver(I) salts which could induce the arylboronic acid to produce an aryl radical which was most likely to react with protonated heterocycle to give the desired product as explained in Scheme 14.
2.3. Brønsted acid catalyst
Ahmed and co workers successfully obtained quinolines 58 from the Brønsted acid-catalyzed metal and solvent free Cross-Dehydrogenative Coupling (CDC) of N-alkyl anilines with arylacetylenes or arylethylenes using O2 as the oxidant with satisfactory yields [32]. From control experiments, it was suggested that cyclization reaction involved imine-N-oxide. The initial substrate, N-Benzyl aniline 56 was subjected to cyclization with phenylacetylene 57 using TfOH (15 mol%) as Brønsted acid catalyst at 120 °C for 24 h under O2 atmosphere to afford the substituted quinolines with excellent yields as illustrated in Scheme 15 .
Scheme 15.

Brønsted acid-catalyzed metal and solvent free Cross-Dehydrogenative Coupling.
2.4. Ionic liquid mediated
In 2018, Kia’s group discovered an efficient synthesis for an assembly of pyrido[3,2-g or 2,3-g]quinolines facilitated through the reaction of 2-aminoaryl ketones with terminal and internal alkynes 59 in the presence of propylphosphonium tetrachloroindate ionic liquid supported on nano-silica (PPInCl-nSiO2) as a heterogeneous and reusable catalyst under solvent-free conditions [33]. With optimized condition in hands, 2,3,4-trisubstituted quinolines 61 were derived from the reaction between 2-aminoaryl ketones 60 and terminal and internal alkynes with 65–99% yields. However, when compounds containing 2-aminoaryl ketone groups reacted with similar alkenes it was observed that only unit of 2-aminoaryl ketone was utilized in quinoline synthesis while the other remained on the quinoline backbone which can be further undergo transformation 62. Treatment of 4,6-dibenzoyl-1,3-phenylenediamine 60f and alkynes 59a, 59b in the presence of PPInCl-nSiO2 at 110 °C furnished the related pyrido[3,2-g]quinolines 64 in high yields. Similarly, the reaction of 2,5-diaroyl-1,4-phenylenediamines 60g and 60h with alkynes 59(a-d) led to a variety of symmetric pyrido[2,3-g]quinolones 63 as illustrated in Scheme 16 .
Scheme 16.

Synthesis of pyrido[3,2-g or 2,3-g]quinolines using propylphosphonium tetrachloroindate ionic liquid.
2.5. Transition metal free approach
In 2019, Beesu and Mehta described an approach which can be generally applied for the synthesis of diverse polyfunctional quinolines 67 and isoqunolines 68 from substituted o-chloropridyl ynones 65; particularly involving benzoannulation mediated by nitromethane 66 via transition-metal free, domino Michael addition-SNAr as key steps [34]. The reaction protocol is simple, straightforward, one-pot operation and exhibits satisfactory yields thereby acting as an area for exploitation in targeting more potential quinolines and isoquinoline scaffolds. The reaction was carried out between 2-chloroaryl ynone i.e. [1-(2-chloropyridin-3-yl)-3-phenylprop-2-yn-1-one] 65 and nitromethane 66 in the presence of NaOMe as base with MeOH as solvent at 80 °C to generate quinolines 67 as shown in Scheme 17 . While using K2CO3 as a base and DMSO as a solvent at 110 °C afforded isoquinolines 68 in 75% yield. After the successful development of protocol, several elaborations were carried out which furnished the 8-nitro-6,7-diphenylquinoline 71 and8-nitro-7-phenyl-5-(phenylethynyl)quinoline 72 from trifluoromethanesulfonate as initial substrate 70 which is obtained from a hydroxy quinolone derivative 69. This was subjected to Suzuki and Sonogashira coupling with phenylboronic acid and phenylacetylene under standard reaction conditions as illustrated in Scheme 18 .
Scheme 17.

Benzoannulation mediated synthesis for polyfunctional quinolines.
Scheme 18.

Coupling protocol for synthesis of quinolines.
Tiwari and co workers made an attempt to synthesize 4-aryl quinolines 75 from readily available phenylacetylene 73 and p-toluidine 74 in the presence of K2S2O8 and DMSO in one-pot without utilizing any transition metal catalyst [35]. On conducting control experiments, it was revealed that DMSO serves as carbon source in the reaction. Further, reaction was generalized with various substituted alkynes and anilines resulting in the synthesis of numerous 4-substituted quinolone and wide substrate scope was also explored as shown in Scheme 19 .
Scheme 19.

One-pot metal free synthetic protocol for quinolone synthesis.
This methodology was adopted to generate medicinally important 4-aryl-2-morpholinoquinoline 78 and 4-aryl-2-tosylquinoline 81 from the earlier synthesized quinolines 75c and 75i. These were treated with m-CPBA in chloroform at room temperature under air to give quinoline N-oxides 76 and 79 in 90% and 88%yield, respectively. The intermediate 76 was then subjected to amidation reaction with N-morpholine 77 in the presence of catalytic Cu(OAc)2 and Ag2CO3 to afford 78 in 88% yield. On the other hand, the intermediate 79 was made to react with sodium sulfinate 80 in the presence of I2/TBHP to furnish 81 with excellent yield via one-pot deoxygenation and direct sulfonylation as illustrated in Scheme 20 .
Scheme 20.

One-pot deoxygenation and direct sulfonylation to generate quinolone derivatives.
2.6. Iodine promoted reaction
In 2019, Togo and Naruto unveiled the synthesis of 2-arylquinolines 84 by subjecting β-arylpropionitriles 82 with aryllithiums 83 followed by the reaction with water and N-iodosuccinimide (NIS) under irradiation with a tungsten lamp in good to moderate yields [36]. The authors have revealed that NIS act as iodination reagent for imines to form N-iodoimines, followed by homolytic bond cleavage of the formed N-I bond to generate iminyl radical i.e. an imino-nitrogen centered radical. The generated radical cyclizes onto the aromatic rings to form 2-aryldihyroquinoline finally undergoing oxidation to furnish 2-arylquinolines promoted via NIS as illustrated in Scheme 21 .
Scheme 21.

Iodine-promoted reaction for synthesis of 2-arylquinolines.
Wu and coworkers have reported the synthesis of quinoline skeleton bearing 1,4-carbonyl units via Povarov reaction pathway. This reaction mainly focuses upon using electron rich alkene subtrates, therefore authors described the one pot, multicomponent synthesis of 2,3-diaroyl quinolines via a formal [3 + 2+1] cycloaddition using enaminones, aryl methyl ketones and arylamines as starting materials. Enaminones were found to act as substitutes for α, β-unsaturated ketones by eliminating HNMe2 and enacting as carbonyl precursors to quinoline scaffolds with dicarbonyl units. The reaction was carried out using aryl methyl ketone 85, p-toluidine 86, and enaminone 87 in the presence of I2 and HCl as an additive in presence of DMSO as solvent at 110 °C for 24 h to afford (6-methylquinoline-2,3-diyl)bis(phenylmethanone) 88 in 40–65% yields [37]. The obtained products were further functionalized to give pyridazino[4,5-b] quinolines 89 skeleton in one pot using hydrazine hydrate in 40–58% yield. The mechanistic experiments implied that C-acylimine was a key intermediate involved in Povarov reaction and also indicated that iodine played a crucial role in oxidation process as explained in Scheme 22 .
Scheme 22.

Quinoline skeleton synthesis bearing 1,4-carbonyl units via Povarov reaction pathway.
2.7. N-heterocyclic carbene (NHC) catalyzed
N-heterocyclic carbenes are generally employed for the synthesis of important heterocyclic molecules. Thus, Biju and co workers have described the synthesis of 2-aryl-4-difluoromethyl quinolines by NHC-catalyzed umpolung of aldimines which was found to bear an electron poor double bond due to the presence of CF3 group [38]. It was suggested that the reaction proceeded with imino-stetter transformation with simultaneous formation of the aza-Breslow intermediate generated from the bicyclic triazolium salt using DBU as the base. The reaction was initiated by treating aldimine 90 with the carbene generated from bicyclic triazolium salt 92 using DBU as the base at 100 °C in DMF to furnish quinolines 91 in yields ranging from 55 to 88% as depicted in Scheme 23 .
Scheme 23.

NHC-catalyzed umpolung of aldimines.
Functionalization of the synthesized product 91 was done by undergoing reduction using NaBH3CN which provides tetrahydroquinolines 93 in 95% yields. However, oxidation of 91 with m-CPBA provided quinoline-N-oxide 94 which further underwent Rh-catalyzed C-H activation to generate 8-functionalized quinoline derivative 95 via series of steps involving remote C-H bond activation, alkyne insertion, and intramolecular oxygen atom transfer. Moreover, Pd-catalyzed cross-coupling of 91 with an oxirane aided by the quinolinyl moiety as a directing group afforded functionalized quinoline 96 with a secondary alcohol moiety in 72% yield as illustrated in Scheme 24 .
Scheme 24.

Functionalization of the quinolone skeleton.
In 2019, Ke’s group successfully utilized an unusual non bifunctional outer-sphere strategy in generating new C-C bond through an efficient N-heterocyclic based-Mn complex [39]. This simple and tunable bis-NHC-Mn system was responsible for not only carrying out direct alkylation of ketones with alcohols but also Friedlander annulation to form quinoline derivatives using borrowing hydrogen/hydrogen auto transfer (BH/HA). Various aryl ketones 97 with alcohols 98 smoothly underwent annulation reactions using Mn complex to deliver quinolines 99 in yields ranging from 59% to 71%. It was observed that all the functional groups were well tolerated. The authors tested the hydrogenation ability of Mn-H species towards α,β-unsaturated ketones which is the key step involved in BH/HA process. The transition state free energy of this complex was found to be very low (14.9 kcal/mol) which suggests the superiority of the non bifunctional outer mechanism as shown in Scheme 25 .
Scheme 25.

Non bifunctional outer-sphere strategy through an efficient N-heterocyclic based-Mn complex.
2.8. Miscellaneous
Yan et al. have described one-step methodology for the synthesis of multisubstituted quinoline-4-carboxamides 105 by refluxing a mixture of isatins 100 and various kinds of 1,1-enediamines (EDAM) 101–104 catalyzed by NH2SO3H through a cascade reaction mechanism [40]. This methodology involved two important features; first included N1 amide group of isatins can attack at C2 of EDAMs and even other electrophilic sites (various carbenium ion) while second involves the alkyl/aryl amino group N1 of EDAMs can be cleaved to form amide group by attacking the C2 of isatins as depicted in Scheme 26 . Owing to the above features, a diverse range of quinoline-4-carboxamides was constructed with good to excellent yields via one-pot reaction rather than multi step synthesis. Therefore, this strategy can be simply utilized for the synthesis of potential biological molecules or natural products.
Scheme 26.

Synthesis of multisubstituted quinoline-4-carboxamides through a cascade reaction mechanism.
In 2018, Cheong’s group reported the novel and highly efficient reaction strategy for the on-water synthesis of quinolones 109 from 2-aminochalcones 108 using benzylamine as nucleophilic catalyst [41]. The nucleophilic catalyst underwent conjugate addition to the α,β-unsaturated carbonyl group generating saturated ketones which lead to positioning of two groups amino and carbonyl in close proximity for undergoing condensation through conformational change about the Cα-Cβ single bond. It was also observed that by introducing any alkyl group onto the nitrogen atom of benzylamine considerably decreased the catalytic efficiency of benzylamine while secondary benzylamine furnishing low yields and tertiary amines generating no product. Various functional groups were well-tolerated, with substrates bearing acid-sensitive functional groups such as acetals, silyl ethers, ethers and esters were effortlessly utilized in this protocol to furnished the desired quinolones 109 in excellent yields. The practical feasibility of the reaction was confirmed by recycling the catalyst and then reusing in order to obtain the quinolines in good to excellent yields as illustrated in Scheme 27 .
Scheme 27.

Reaction strategy for the on-water synthesis of quinolines using benzylamine as nucleophilic catalyst.
Bhardwaj and coworkers have reported a green synthetic methodology for the synthesis of 8-aryl-7,8-dihyro [1,3]-dioxolo[4,5-g]quinolin-6(5H)-ones from a three component reaction of Meldrum’s acid, 3,4-methylenedioxy aniline and aromatic acid using a catalytic amount of TiO2 nanoparticles under ultrasonic irradiation [42]. These nanoparticles were prepared by direct interaction of titanium (IV) isopropoxide and extracts of Origanum majorana leaves extract as reducing and capping agent under ultrasonic irradiation. It was observed that the catalyst was easily recovered with its catalytic activity remaining intact. A mixture of Meldrum’s acid 110, 3,4-(methylenedioxy)aniline 111, aldehydes 112 and TiO2 NPs (10 mol %) was taken in a flask with water as a solvent and subjected to sonication to obtain the desired product in excellent yields as shown in Scheme 28 .
Scheme 28.

Three-component strategy for quinolone synthesis using a catalytic amount of TiO2 nanoparticles.
In 2019, Patel’s group demonstrated a novel one-pot synthetic procedure for the regioselective formation of functionalized 6,8-dihydro-1′H,5Hspiro[ [1,3]dioxolo[4,5-g]quinoline-7,5′-pyrimidine]-2′,4′,6’ (3′H)-trione scaffold 115 in excellent reaction yields upto 85% [43]. The formation involves the use of three-component 3,4-methylenedioxyaniline 111, N,N-dimethylbarbituric acid 114 and aryl/heteroaryl aldehyde 112 in the presence of TMGB (Trimethyl glycine betaine)-oxalic (20 mol%) as catalytic system furnishing the product with high diastereoselectivity (dr > 50:1) (syn:anti). Trimethyl glycine betaine is a natural by-product of beet sugar, commonly employed in enzyme catalysis. The protocol was able to tolerate several functional groups with good yields and high diastereoselectivity. The catalyst can be regenerated and reused upto 4 times in a run. A cascade strategy for the regioselective convergent synthesis of a series of 1,3-diazaheterocycle-fused [1,2-a] quinoline derivatives using 2-flurobenzaldehyde and heterocyclic ketene aminals (HKA) was developed by Yan and co workers [44]. The reaction proceeded without any aid of transition metal catalyst, low cost solvent, and mild temperature. The reaction was carried out using 2-fluoro-5-nitro benzaldehyde 116 and heterocyclic ketene aminals 117 in the presence of piperidines as catalyst and 1,4-dioxane solvent under reflux conditions to generate 1,3-diazaheterocycle fused [1,2-a] quinoline derivatives 118 upto 90% yield. Further, 2,3,4,5,6-pentafluorobenzaldehyde was allowed to react with the five-membered ring HKAs 2, a new product 119 was synthesized with piperidine as the substituent. It was suggested that electron withdrawing formyl group at C1 position of perfluorobenzaldehyde makes the fluorine atom at the C-4 position to be easily replaced by piperidine. However, the reaction did not proceed with six and seven membered ring HKAs. Detailed reaction sequence is shown in Scheme 29 .
Scheme 29.

Convergent synthesis of 1,3-diazaheterocycle-fused [1,2-a] quinoline derivatives using heterocyclic ketene aminals (HKA).
Ding and coworkers unveiled an efficient synthesis of multisubstituted 1H-imidazo-[4,5-c]quinoline derivatives through a stepwise van leusen, staudinger, aza-wittig and carbodiimide mediated cyclization reactions [45]. Initially, azides 123 were prepared from the reaction of 2-azidobenzaldehydes 120, amines 121, and TosMIC 122 in the presence of K2CO3 in mixed methanol-DME (1,2-dimethoxyethane) solvent at 76 °C in 60–83% yields. Azides so formed underwent Staudinger reaction rapidly with triphenylphosphine at room temperature to generate iminophosphoranes 124 with N2 release. Simultaneously, isocyanates were added to the reaction mixture resulting in intermolecular aza-wittig reactions occurring at room temperature for aromatic isocyanates and at 70 °C for aliphatic isocyanates to furnish carbodimides 125. At last, cyclizaton of carbodiimides was completed using 1,2-dichlorobenzene under reflux conditions to produce 1H-imidazo-[4,5-c]quinolones 126 in moderate to good yields as shown in Scheme 30 .
Scheme 30.

Synthesis of fused multisubstituted 1H-imidazo-[4,5-c]quinoline derivatives.
Vidal et al. demonstrated the application of ring opening and ring-expansion of cylclopropane ring in order to provide access to aziridino [2,3-c]quinolin-2-ones and cyclopropa[c] quinolin-2-ones which generates heterocyclic ring systems such as quinolin-2-ones substituted with an amino group at the C-3 position and polysubstituted 1-benzazepin-2-ones [46]. These cores act as potential targets for numerous biological activities. Synthesis of aziridino [2,3-c]quinolin-2-ones was initiated from the commercially available (E)-3-(2-nitrophenyl)propenoic acid 127 which was subjected to a series of reaction (Fischer esterification, reduction and diazotation/azidation) to produce ethyl (E)-3-(2-azidophenyl)propenoate 128. To the chloroform solution of 128, bromine was added at 0 °C to provide 2,3-dibromopropanoate 129 which was further treated with various benzylamines in ethanol at room temperature to generate a mixture of cis 130 and trans 131 isomers of the 2-(2-azidophenyl)-3-ethoxycarbonylaziridines in 64–91% yield. Finally, the cis isomer 130 was treated with triphenyl phosphene to furnish aziridino [2,3-c]quinolin-2-ones 132 in good yields and finally stirred in DCM solution for two days to produce 3-benzylamino-1H-quinolin-2-ones 133. This conversion was suggested to be a result of isomerization of the aziridine core into anenamino function involving a simultaneous 1,2 hydrogen migration from carbon to nitrogen. The detailed reaction sequence is illustrated in Scheme 31 .
Scheme 31.

Ring opening and ring-expansion of cylclopropane ring.
However, the synthesis of cyclopropa[c]quinolones was started by selecting 2-(2-azidophenyl)-3-nitrocyclopropane-1,1-dicarboxylates 136 as key precursors. This precursor was prepared from the readily available 2-aminobenzyl alcohol 134 which was subjected to a sequence of diazotation/azidation, oxidation, Henry condensation and subsequent dehydration reactions to generate 2-azido-β-nitrostyrene 135. Then the generated 135 substrate underwent one-pot Michael addition and ring-closure to afford trans-2-(2-azidophenyl)-3-nitrocyclopropane-1,1-dicarboxylates 136 in 56–99% yields. The synthesized azides were subjected to intramolecular aza-wittig reaction using trimethylphosphine in toluene solution at room temperature to produce 2-alkoxy cyclopropa[c]quinolones 138 through the formation of phosphazenes 137. Finally, hydrolysis of imino ether 138 was carried out to obtain the desired product cyclopropa[c]quinolin-2-ones 139 in excellent yields which underwent ring opening to yield benzazepin-2-ones 140 under basic conditions with 75–95% yield as depicted in Scheme 32 .
Scheme 32.

Synthesis of cyclopropa[c]quinolones.
3. Biological activity
From the consequent discovery of tobacco mosaic virus in 1892 to foot-and-mouth disease virus in 1898, first ‘filterable agent’ revealed in humans was yellow fever virus in 1901 [47]. However, till now numerous new species of human viruses are recognized covering upto two third of all new human pathogens. Viruses act like a parasite which cannot reproduce by them but once they infect a host cell they have the ability to direct the host cell machinery to produce similar virus like particles called virion. Several targets have been identified on which the viruses act, thus we have tried to cover some of the viruses which can be inhibited by the molecules bearing quinoline skeleton.
3.1. Zika virus (ZKV)
Zika virus (ZIKV) is one of arboviruses belongs to Flaviviridae family and is primarily known to be transmitted via the bite of mosquitoes of the Aedes type. However, it can also be sexually transmitted, or by blood transfusions and further infections from pregnant women can spread to the baby [[48], [49], [50], [51]]. Owing to the insights of its self-limited infection, ZIKV has been associated with a wide range of neurological diseases both in adults as Guillain-Barré syndrome, foetuses and neonates as pediatric microcephaly [52].
Several researches in the prevention and therapy development have been carried out but still there are no signs of any vaccine or specific antiviral agent against ZIKV [[53], [54], [55]]. However, in 2017, Barbosa-Lima et al. reported that 2,8-bis(triflouromethyl)quinoline derivatives inhibited the ZIKV replication in vitro and their potency was compared with mefloquine which has been approved by Food and Drug Administration (FDA) for the treatment against malaria [56]. The authors have developed triflouromethyl substituents containing quinoline nucleus because this scaffold is present in a variety of bioactive nucleus which are active against several diseases such as malaria, cancer, tuberculosis and viral infections [[57], [58], [59], [60], [61], [62], [63]].
The synthesized 2,8-bis(trifluoromethyl)quinoline derivatives were screened for inhibiting the ZIKV replication. Some of the derivatives showed antiviral activity similar to that of mefloquine. Molecules inhibiting approximately 75% of virus replication or greater, were further examined and compared to mefloquine. Thus, compounds 141a, 141b, 142 and 143 were found to reduce ZIKV RNA production. The structure of these potent molecule is shown in Fig. 5 . On further analyzing the pharmacological activity against ZIKV, it was revealed that mefloquine is approximately three times more potent than chloroquine as shown in Table 1 . Moreover, compounds 141a and 142 exhibited EC50 values of 0.8 μM and had potencies approximately five times higher than mefloquine against ZIKV replication. It was also observed that compounds 141b and 142 were approximately 2 and 3 times more potent than the reference molecule respectively but were found to be less potent than 141a and 142. The selective index (SI) is known as the ratio between the CC50 and EC50 values, and thus 141a was found to be 243 which is four times higher than the SI of mefloquine by carrying out in vitro, study. Conclusively, results indicate that novel 2,8-bis(trifluoromethyl)quinoline derivatives, 141a and 142, have stronger anti-ZIKV activity than mefloquine at the sub-micromolar range.
Fig. 5.

Compounds with reduced ZIKV RNA production.
Table 1.
Potency and cytotoxicity of mefloquine and 2,8-bis(trifluoromethyl)quinoline derivatives against ZIKV replication.
| Compound No. | EC50 (μM) | CC50 (μM) | SIa |
|---|---|---|---|
| Chloroquine | 12.0 ± 3.2 | 412 ± 24 | 34 |
| Mefloquine | 3.6 ± 0.3 | 212 ± 14 | 58 |
| 141a | 0.8 ± 0.06 | 195 ± 8.9 | 243 |
| 141b | 2.0 ± 0.1 | 287 ± 21 | 143 |
| 142 | 0.8 ± 0.03 | 189 ± 10 | 236 |
| 143 | 1.4 ± 0.09 | 316 ± 27 | 225 |
SI, selective index: determined as the ratio between CC50 and EC50.
On the other hand, Li et al., in 2019 described the discovery of andrographolide derivatives against ZIKV infection. Andrographolide 144 is a bicyclic diterpenoid lactone and is one of the major components isolated from Andrographis paniculata. Initially, some active derivatives of andrographolide have been used to treat bacterial and viral infections in China [[64], [65], [66], [67]]. Recently, andrographolide was described as an anti-viral agent against DENV [68,69] and (CHIKV) [70] but does not have a direct antiviral activity [68,69]. Eight derivatives of andrographolide with 14-quinolinyloxy group and related 14-pyridinyloxy group, were tested for anti-Zika activity. These derivatives have been previously described as antibacterial agents [71]. Further modification of 14-(8′-quinolyloxy) group was carried out in a way that sixteen modified derivatives by the introduction of methyl group at 2-position or chloro group at 5,7-positions into 8-quinolinyloxy moiety were subsequently designed and later on synthesized and screened for anti-Zika activity.
ZIKV titer assay (in Vero cells) was carried out to measure ZIKV production using human glioblastoma cells (SNB-19 cell line) infected by PRVABC59 strain at multiplicity of infection (MOI) of 1 [72,73]. Niclosamide has been previously identified as active compounds against Zikavirus infection. Therefore, it was used as a positive and quality control in each antiviral assay. The cytotoxicity of each compound was examined using SNB-19 cells for 24 h and Vero cells for 48 h corresponding to the incubation time in anti-Zika screening.
At first, previously reported derivatives (145 to 152) were screened and it was revealed that 14β-(8′-quinolyloxy) 3,19-diol analog 146 was very effective against Zika virus infection with EC50 of 1.3 μM and selectivity index (SI) > 16, with cytotoxicity level being inappropriate. However, compound 145 also showed similar values against Vero and SNB-19 cell lines, but compound 147 and 148 were completely inactive. These results suggested that more polar 14β-(8′-quinolyloxy) andrographolide derivative 146 is much more active and selective against ZIKV infection than hydrophobic derivatives 145, 147 and 148. Out of two 14-(4′-quinolyloxy)-3,19-diol andrographolide derivatives 149 and 150, only compound 150 showed mild activity (EC50 = 11.0 μM, CC50 = 60.1 μM in SNB-19 cells and CC50 = 22.3 μM in Vero cells) while compound 149 remained inactive. Similarly, 14α-(2′-nitro-pyridinyl-3′-oxy)-3,19-diol andrographolide derivative 9 (EC50 = 12.5 μM, CC50 = 67.7 μM in SNB-19 cells and CC50 = 37.7 μM in Vero cells) displayed mild antiviral activity while the other derivative 151 described no signs of inhibition as summarized in Table 2 . Thus, it was concluded that both of 14β- and 14β-isomers, and modifications at 3-, 19-, or 3, 19-positions are crucial for the anti-Zika activity. The structure of potent molecule is sketched in Fig. 6 .
Table 2.
Reported derivatives (145–152) screened against ZIKV.
| Compound No | EC50 (μM) | CC50 (μM) |
SI |
CC50 (μM) |
SI |
|---|---|---|---|---|---|
| SNB-19 | Vero cells | ||||
| 146 | 1.3 ± 0.1 | 22.7 ± 1.1 | 17.5 | 20.8 ± 0.5 | 16.1 |
| 150 | 11.0 ± 0.2 | 60.1 ± 1.1 | 5.5 | 22.8 ± 0.2 | 2.1 |
| 152 | 12.5 ± 0.4 | 67.7 ± 1.1 | 5.4 | 37.7 ± 0.1 | 3.0 |
| 154a | 8.5 ± 0.4 | 78.4 ± 1.0 | 9.2 | 55.9 ± 0.2 | 6.6 |
| 154b | 16.6 ± 0.3 | 66.5 ± 1.0 | 4.0 | 65.3 ± 0.3 | 3.9 |
Fig. 6.

Natural product based derivatives active against Zika virus.
The potential of compound 146, as lead compound was clearly seen but became insignificant due to its cyctoxicity. It was considered that cytoxicity originates from 1′-N and 8′-O 14-(8′-quinolyloxy) group. Therefore, several modifications were done to improve antiviral efficacy and to reduce cytotoxicity. It was predicted that on introduction of sterically hindered or electrostatic group/s at 2′-position or 7′-position can possibly block some side interactions with 1′-N or 8′-O thereby reducing the cytotoxicity and improving anti-Zika activity.
Thus, eight 14-(2′-methyl 8′-quinolyloxy) derivatives (four 14β-isomers of 154a to 157a and four 14α-siomers of 154b–157b) and eight 14-(5′,7′-dichloro 8′-quinolyloxy) derivatives (four 14β-siomers of 158a to 161a and four 14α-siomers of 158b–161b) were designed, synthesized and tested against Zika virus infection as shown in Fig. 7 . As predicted, eight 14-(2′-methyl 8′-quinolyloxy) andrographolide derivatives were found to be less cytotoxic than 14-(8′-quinolyloxy) andrographolide derivatives. Both 154a (14β) and 154b (14α) were found to be less cytoxic (CC50 values are 78.4 and 66.5 μM in SNB-19 cell line, 55.9 and 65.3 μM in Vero cell line, respectively) and less efficiently active against ZIKV ((EC50 values are 8.5 and 16.6 μM, respectively) as compared to compound 146. Compound 155a (14β) was much less active than compound 146, 154a and 154b, while compound 156a (14β), 157a (14β), and 155b (14α) to 157b (14α) remained completely inactive. It was concluded that 14-(2′-methyl-8′-quinolyloxy) group can only reduce cytotoxicity but cannot improve the antiviral activity whereas modifications at 3- or 19-position can affect both the cytotoxicity and antiviral activity.
Fig. 7.

Structure of mild active quinolone compounds.
At last, 5′,7′-dichloro-8′-quinolinol derivatives were screened. It was observed that 3,19-acetonylidene-protected 5′,7′-dichloro-8′-quinolinol andrographolide derivatives 158a (14β) and 158b (14α) do not show any anti-Zika activity while their corresponding 3,19-acetonylidene-protected 2′-methyl-8′-quinolinol derivatives of 154a and 154b were completely active as shown in Fig. 8 . Both of 3,19-diols of 159a (14β) and 159b (14α) are active against Zikavirus infection with 159a being less active (EC50 = 13.3 μM) and toxic than 159b (EC50 = 7.8 μM). This clearly indicates that the stereochemistry of 14α and 14β affects the inhibitory activity along with the cytotoxicity. 19-Acetylated 14β-derivative 160a was found to be inactive but 19-acetylated-14α-derivative 160b was highly potent anti-Zika agent with EC50 value of 4.5 μM and CC50values of 88.7 μM in SNB-19 cells and 85.0 μM in Vero cells respectively as depicted in Table 3 . On the contrary, 19-acetylated 3-keto-14β-derivative 161a exhibited weak inhibition against Zika virus infection while 19-acetylated 3-keto-14α-derivative 161b was completely inactive.
Fig. 8.

5′,7′-dichloro-8′-quinolinol derivatives active against ZIKV.
Table 3.
Anti-Zika active derivatives of andrographolide.
| Compound No | EC50 (μM) | CC50 (μM) |
SI |
CC50 (μM) |
SI |
|---|---|---|---|---|---|
| SNB-19 | Vero cells | ||||
| 155a | 25.8 ± 1.1 | >100 | >3.9 | 99.8 ± 0.7 | 3.9 |
| 159a | 13.3 ± 0.5 | >100 | >7.5 | >100 | >7.5 |
| 159b | 7.8 ± 0.4 | 85.2 ± 1.0 | 10.9 | 82.5 ± 2.2 | 7.5 |
| 160b | 4.5 ± 0.2 | 88.7 ± 1.1 | 19.7 | 85.0 ± 1.6 | 18.9 |
| 161a | 24.6 ± 0.9 | 72.0 ± 1.0 | 2.9 | 57.4 ± 0.3 | 2.3 |
Conclusively, 14-(2′-methyl 8′-quinolyloxy) and 14-(5′,7′-dicloro 8′-quinolyloxy)derivatives are less cytotoxic than 14-(8′-quinolyloxy) derivatives; and in general, 14-(5′,7′-dichloro-8′-quinolyloxy) derivatives are more potent against Zika infection than 14-(2′-methyl-8′-quinolyloxy) derivatives. Two configurations of 14α and 14β can make significant contribution to the anti-Zika activity, and optimal combination of modifications at 2′- or 5′,7′-positions of 14-(8′-quinolyloxy) and 3- or/and 19-positions improves the antiviral activity and the selectivity.
3.2. Enterovirus (EV-D68)
EV-D68 is an atypical nonpolio enterovirus known for particularly infecting respiratory system of humans, thereby causing moderate-to-severe respiratory infections. It behaves more of like a rhinovirus and is found to replicate more efficiently at 33 °C. Virus is much more suspectible to humans with pre-existing respiratory complications such as asthma or chronic obstructive pulmonary disease. However, several reports discuss about spreading of EV-D68 to central nervous system (CNS) such as spinal cord and cerebrospinal fluid through viremia causing paralysis and viral complications such as AFM. It is important to note that there is currently neither any vaccine nor any antiviral drug have been developed against EV-D68 infection, thus patients tends to depend only upon the supportive care [74,75].
Recently, Wang and coworkers have made an attempt to develop EV-D68 antivirals mainly targeting the viral 2C protein and also improving the antiviral potency of already reported EV-D68 inhibitor dubicaine through structure-activity relationship studies (SAR) as shown in Fig. 9 . 2C protein is a multifunctional protein, exhibiting crucial roles in viral uncoating, RNA binding and replication, membrane rearrangement, encapsidation of the viral genome all of which are essential for viral replication. Thereby, act as an attractive target against anti EV-D68 drug development [76].
Fig. 9.

Established EV-D68 inhibitor and designed compound for EV-D68.
SAR study was carried out on dubicane by varying three positions of the quinoline skeleton. First variation at the 2-postion substituent with an alkoxy or an aromatic group; second one involves the 4-position substituted with different amides while the third variation being the 6-position substitution with F, Cl, or methoxy group. In total, 60 compounds were synthesized and tested against EV-D68 (US/KY/14–18953) virus in the primary viral cytopathic effect (CPE) assay with dubicaine as positive control. From the results, it was observed that compound 162 series with substitution at 2-position had higher potency and selectivity index with most potent being compound 163e with 2-isopropoxy substituent (EC50 = 2.5 ± 0.5 μM, CC50 = 111.2 ± 15.4 μM, SI = 44.5). Therefore, this was selected as reference compound for the next series of compound. Further, compound 164 series with aromatic substitution at 2-position including 164d, 164f, 164g and 164j had improved activity and higher SI compared to 163e while compound 164e, 164l, 164m, 164r and 164s exhibited improved potency but lower value of SI. Rest of the compound of 164 series remained completely ineffective as shown in Table 4 .
Table 4.
Evaluation of anti-EV-D68 activity for the designed compounds.
| Compound No | R2 | EC50 (μM)a | CC50 (μM)b | SI |
|---|---|---|---|---|
| 163e | 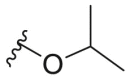 |
2.5 ± 0.5 | 11.2 ± 15.4 | 44.5 |
| 164d |  |
0.6 ± 0.5 | 37.4 ± 6.3 | 62.3 |
| 164e |  |
0.8 ± 0.6 | 6.1 ± 2.2 | 7.6 |
| 164f | 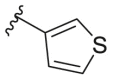 |
1.0 ± 0.4 | 46.9 ± 5.2 | 46.9 |
| 164g | 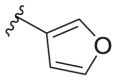 |
1.5 ± 0.9 | 122.5 ± 30.8 | 81.7 |
| 164j | 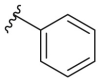 |
0.8 ± 0.3 | 45.1 ± 13.7 | 56.4 |
| 164l | 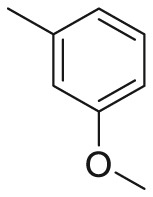 |
1.6 ± 1.8 | 36.5 ± 3.1 | 22.8 |
| 164m |  |
2.4 ± 0.2 | 31.6 ± 4.2 | 13.2 |
| 164r |  |
0.8 ± 0.2 | 11.5 ± 6.7 | 14.4 |
| 164s |  |
1.6 ± 0.1 | 29.5 ± 1.6 | 18.4 |
cN.A. = not applicable. The results are the mean ± standard deviation of three repeats. SI = selectivity index (CC50/EC50).
Antiviral efficacy was determined in the CPE assay with EV-D68 US/KY/14–18953 virus and RD cells.
Cytotoxicity was determined using the neutral red method.
For compound 165 series with 2-position substituted as isopropoxyl being fixed and 4-position being varied. It was observed that compounds 165a and 165e had improved antiviral activity and lower selectivity index compared with 163e while other compounds (165b-d and 165f-h) had decreased antiviral activity. Compound 166 and 167 series with 2-position being benzyl and thienyl, respectively, were also screened suggesting that compound 166a tend to possess significantly high antiviral activity and selectivity index (EC50 = 0.4 ± 0.2 μM, CC50 = 73.7 ± 19. μM, SI = 184.3). However, similar potency (EC50 ≤ 0.1 μM) and SI (SI > 600) was observed with compound 167a and 167c. It was confirmed that 6-position of quinoline should be left unsubstituted as compounds 169. The compound 168 did not show any signs of improved potency and selectivity index compared.
From SAR studies, compound 166, 167a, and 167c were found to be highly potent which were further evaluated against four additional human EV-D68 strains. The results confirmed their similar potent antiviral activity and high selectivity index thereby validating that 2C protein is a potential antiviral drug target. The lead compounds were found to be active against two cell lines A549 and Hela. Since EV-D68 is known to infect CNS, lead compounds were found to inhibit EV-D68 viral replication in neuronal cell lines A172 and SH-SY5Y. But they were less cytotoxic to A172 and SH-SY5Y cells than RD cell lines. On conducting immunofluorescence imaging, western blot and viral RNA levels by RT-qPCR, it was proved that dibucaine and lead compounds shared a similar mechanism of action i.e. inhibition of viral 2C protein which is further inhibiting viral RNA and protein synthesis. Various potent molecules against EV-D68 are shown in Fig. 10 .
Fig. 10.

Potent molecules for EV-D68.
3.3. Dihydroorotate dehydrogenase (DHOD)inhibitor
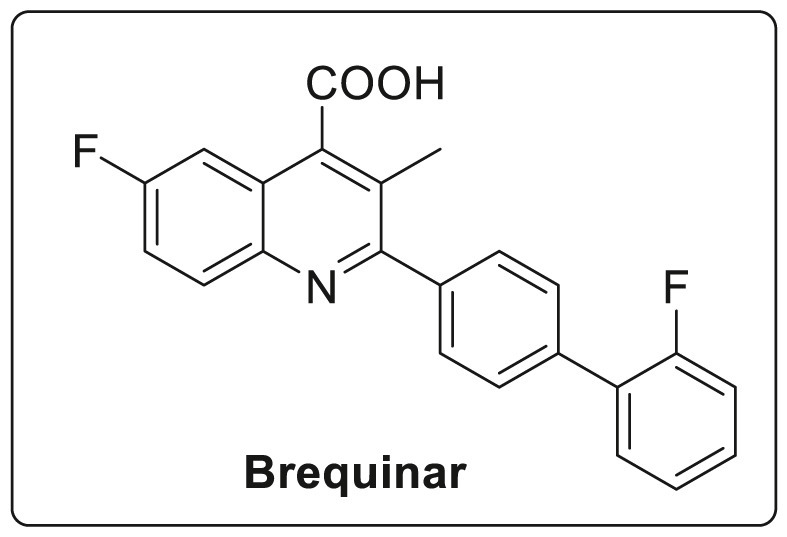
Viruses are known for mutating rapidly which leads to drug resistance and therefore haults the usage of drugs molecules specifically targeting the virus. Resistance to viral inhibitors is a major concern especially for viruses with genomes encoding only 10–12 genes out of which only few are known to function as enzymes [77]. Therefore, it is difficult to find drug molecules that target small molecules. Dihydroorotate dehydrogenase (DHOD) is one of the fourth enzymes involved in the pyrimidine de novo synthesis which catalyses the conversion of dihydroorotate to orotate. Its inhibition causes reduced levels of pyrimidine nucleotides which trigger various activities such as anticancer, immunosuppressive, antimalarial and antifungal. However, Brequinar and leflunomide have evolved as essential inhibitors against DHOD due to their high potency. The structure of brequinar is shown in Fig. 11 [[78], [79], [80]].
Fig. 11.
Structure of brequinar.
Brabender et al. carried out an extensive SAR study for 4-quinolinebearing carboxylic acid group that tend to target DHOD for antiviral activity [81]. From the structural insights of brequinar, it was recognized that there are several pharmacophoric regions where appropriate functional groups are necessary such as presence of free carboxylic acid, hydrophobic moiety at C-2, and an electron withdrawing group at C-7 position.
Thus, structural optimization study of 170 was done by keeping chloroquinoline part fixed and synthesized compounds 171–180. It was observed that replacing the propyl ether of 170 by methyl, ethyl, or butyl (171–173) ethers did not showed any signs of improved activity against vesicular stomatitis virus (VSV) by conducting viral replication assay as shown in Fig. 12 . But replacement of propoxy group by electron withdrawing groups, fluorine 175, bromine 176, or trifluoromethoxy 174 resulted in significant higher activity. Further, 177–179 analogues were prepared by replacing the alkoxy groups of 171, 172 and 174 with their respective alky chain counterparts in order to elucidate the importance of ether linkage. Owing to the beneficial effects of an additional aromatic ring (2-F-Ph) connected to the C(2)-phenyl ring in brequinar, diaryl ether analogue 180 were further explored. This revealed that 180 showed improved activity as compared to other analogues which open up the chances to expand SAR studies by developing a range of diaryl ethers. Then the focus was shifted on 180 by exploring functional group tolerance in 4-quinoline carboxylic acid derivatives. Significant increase in activity (0.11 μM) was seen by replacing C(7)-chlorine with fluorine 181 whereas bromine containing 182 and nitro containing 185 functionality were moderately tolerated (EC50 = 3.10 and1.98 μM, respectively). On the contrary, trifluoromethoxy 183, trifluoromethyl 186, and methoxy 184 functionality were found to be inactive (EC50 = 95.64, 16.58, and 2611 μM, respectively). However, unsubstituted analogue 187 revealed increased activity (EC50 = 0.96 μM) which suggest that small groups like fluorine and hydrogen are well tolerated. In vitro assay was done on 188 and 190 to demonstrate the importance of position of fluorine atom. The results showed that 188 and 190 were 10 times less potent than 197 while 189 with fluorine atom present at C(8) position was active against VSV virus as depicted in Table 5 .
Fig. 12.

Active and inactive compounds of 4-quinoline bearing carboxylic acid derivatives.
Table 5.
SAR studies using different substitutions.
| Compound No. | R1 | R2 | R3 | R4 | EC50 (μM)a |
|---|---|---|---|---|---|
| 180 | Cl | H | H | Ph | 1.3 |
| 181 | F | H | H | Ph | 0.1 |
| 182 | Br | H | H | Ph | 3.1 |
| 183 | OCF3 | H | H | Ph | 95.6 |
| 184 | OCH3 | H | H | Ph | 2611 |
| 185 | NO2 | H | H | Ph | 2.0 |
| 186 | CF3 | H | H | Ph | 16.6 |
| 187 | H | H | H | Ph | 1.0 |
| 188 | H | H | H | Ph | 1.2 |
| 189 | H | F | H | Ph | 158.8 |
| 190 | F | F | H | Ph | 11.6 |
| 191 | F | H | H | CH3 | 0.1 |
| 192 | F | H | H | CH2CH3 | 6.4 |
| 193 | F | H | H | (CH2)2CH3 | 19.0 |
| 194 | F | H | H | (CH2)3CH3 | 2.1 |
| 195 | F | H | H | CH2(C3H5) | 6.8 |
| 196 | F | H | H | CF3 | 1.0 |
| 197 | F | H | H | 4-Cl-Ph | 2.0 |
| 198 | F | H | H | 4-NO2-Ph | 4.9 |
| 199 | F | H | H | 3,4-(OCH2O)-Ph | 0.9 |
| 200 | F | H | H | 2-F-Ph | 0.3 |
| 201 | F | H | H | 3-F-Ph | 0.1 |
| 202 | F | H | H | 4-F-Ph | 1.0 |
| 203 | F | H | H | 2-pyridyl | 22.9 |
| 204 | F | H | H | 3-pyridyl | 2.5 |
| 205 | F | H | H | 2-thiazolyl | 14.6 |
Inhibition of VSV replication in MDCK epithelial cells.
Thus, 197 being the best analogue, several molecules 191–205 were synthesized by modifying the C(2) diaryl ether. It was observed that the diaryl ether 181 was more potent than the corresponding aryl-alkyl ethers 191–196. The 3,4-(methylenedioxy)phenoxy, 4-chlorophenoxy, and 4-nitrophenoxy analogues were approximately 9-to 45-fold less active than the nonsubstituted analogue 181. Out of the three fluorophenyl analogues, 200 and 202 found with inferior activity whereas the 3-F-Ph analogue 201 displayed an activity similar to lead compound 181. However C(3)-methyl substituted analogue 206 was found to less potent than 181 which suggest that introduction of substituent at C(3) and restricting conformational freedom by planarizing fusion of quinoline 207 was not beneficial. A set of diaryl ether analogue 208–210 were synthesized and evaluated for inhibition of VSV replication in MDCK epithelial cells. It was confirmed that activities of 208–210 were inferior than most potent 181. It was seen that the t-butyl substituted analogue 211 and sterically hindered diaryl ether 212 showed signs of improved potency. Based on these results authors made a hypothesis that restriction of rotation across the diaryl ether linkage coupled with the increase in hydrophobicity might be crucial for activity. This hypothesis proved to be fruitful as final analogue 213 showed a significant 50-fold increase in potency (2 nM) versus 181 in the VSV in vitro assay as shown in Fig. 13 .
Fig. 13.

VSV Inhibitor with diaryl ether analogues.
Finally, several analogues in assays measuring inhibition of human DHODH enzyme activity and in vitro influenza-WSN viral replication were compared with the data for inhibition of VSV viral replication. Thus, starting from compound 170 with modest activity, a sub-micromolar analogue 181 was developed and further optimized to the low nanomolar biaryl ether analogue 213. In the VSV assay, compound 213 was 2 times more potent than brequinar at nontoxic concentrations (cytotoxicity in human bronchial epithelial and Caco-2 cells: IC50 ≥ 10 μM). Therefore, analogue 213 was identified as a promising candidate that showed broad spectrum antiviral activity.
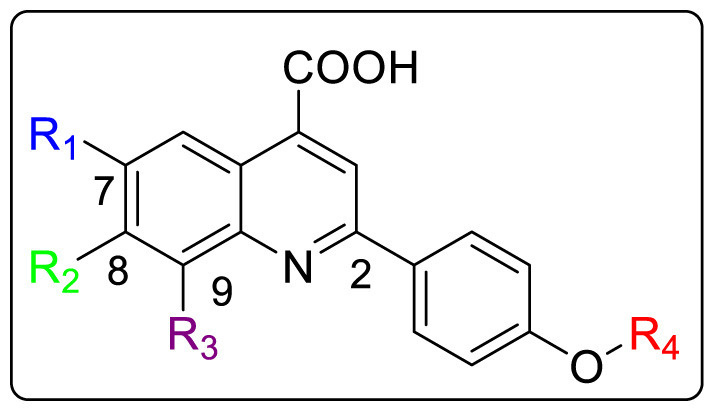
3.4. Human respiratory syncytial virus (HRSV)
RSV is a negative single stranded RNA virus which belongs to the family Paramyxoviridae. It is known for causing acute upper and lower respiratory tract infections in infants, young children and immunocompromised patients. RSV Fusion (F) protein is a surface glycoprotein present on the viral envelope which on combining with G surface glycoprotein mediates the viral entry into host cell. F protein generally triggers the fusion process between viral and host cellular membranes and aids in syncytia formation. Therefore, inhibition of viral entry into the host cell and targeting RSV F protein, act as potential targets for the development of RSV inhibitors. Although, ribavirin has been approved against viral infection but is often associated with potential side effects and low efficacy [82,83].
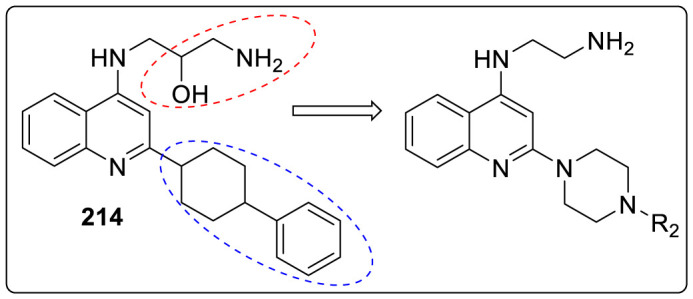
Several attempts have been made to discover new anti-RSV agents out of which Yun and coworkers utilized similarity-based virtual screening approach for developing a novel class of piperazinylquinolines as RSV fusion inhibitors [84]. MOS and ROCS were used in parallel to screen the Roche Smart library (more than one million small molecules) for potential RSV fusion inhibitors. The ligands JNJ-2408068, TMC-353121 and BMS-433771 were selected as reference compounds for the virtual screening. Each method selected the best 1,000 similar compounds to every reference compound in the library based on similarity scores, which resulted in total 6000 similar hits for 3 reference compounds. By removing the duplicate structures, less than 3000 compounds were finally selected for anti-RSV activity. The antiviral activity was examined by carrying out cytopathic effect assay (CPE) which was induced by the RSV long strain of virus replication in HEp-2 human lung epithelial carcinoma cells. Among all the molecules, 1-amino-3-[[2-(4-phenyl-1-piperidyl)-4-quinolyl]amino]propan-2-ol 214, was identified asa potent anti-RSV hit with an EC50 of 0.759 μM, thus used as a starting point for further studies. It was suggested that the piperidyl linker is a deciding factor for conformational control. Thus, piperazinyl analogues, such as N′-[2-(4-phenylpiperazin-1-yl)-4-quinolyl]ethane-1,2-diamine 215, was designed whose conformation helped in retaining antiviral activity, improving metabolic stability, introducing substituents (R2). No significant decrease in activity was seen by replacing the aminoalcohol head part with simple ethyl amine.
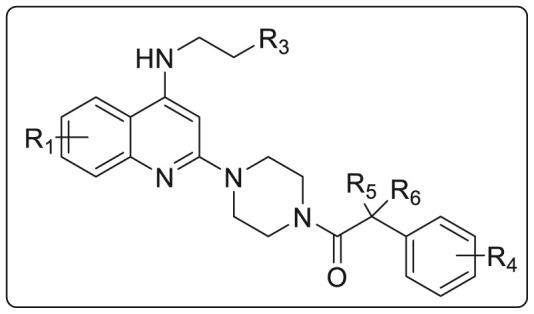
It was observed that when N-piperazine was substituted with meta-chloro phenyl group 217, the potency was maintained but had high cytotoxicity while ortho-chloro 216 and para-chloro 218 exhibited decreased potency. However, by introducing other groups such as cyclo-hexyl 223, methanesulfonamide 227 and benzenesulfonamide 228 showed inferior activity as depicted in Table 6 . 2-phenyl acetamide substituted group was found to have significantly high activity against RSV thus a number of substituted phenylacetamide analogues 229–245 were synthesized. Chloro and fluoro groups at different positions of phenyl ring had similar potency (229–234) while 2-methyl substituted led to increased activity. However, the methyl substitutions at meta- and para positions exhibited decreased activity while some of the electron withdrawing group such as cyano and -CF3 led to slight loss in activity. Further SAR studies were conducted on additional analogues 246–259 by introducing substitutions at quinoline (R1), the head portion (R3), and the benzylic position (R5 and R6). Small alkyl groups 246–252 at the benzylic position were found to block the metabolic hot spot. Gem-dimethyl substituent proved to be 3-times more potent as compared to 225 and the single substituted molecule 246. However, diethyl analogues showed weaker activity that 225. Cyclic substituent 250 was found to be most potent (EC50 = 0.017 μM) but on further enlarging the cyclic ring size, led to significant loss of activity. Reducing basicity of amine at head position by acylation led to 550 times decrease in anti-RSV activity (EC50 = 15.58 μM) compared to 248. N-Methylation of 248 was done to obtain 253 which showed similar activity (EC50 = 0.033 μM) as 248 but bulkier terminal tertiary amine 254 was not well tolerated. Presence of chloro group on the 5, 6, or 7-position of quinoline showed an order of 6-Cl>7-Cl>5-Cl RSV inhibition as shown in Table 7 .
Table 6.
Anti-RSV activity for the synthesized compounds.
| Compound No. | R2 | EC50 (μM) | CC50 (μM) |
|---|---|---|---|
| 214 | 0.759 | 20.80 | |
| 215 | Ph | 0.865 | 20.80 |
| 216 | 2-Cl-Ph | 2.014 | 20.80 |
| 217 | 3-Cl-Ph | 0.977 | 6.40 |
| 218 | 4-Cl-Ph | >100 | 6.60 |
| 219 | COCH3 | 0.317 | >100 |
| 220 | COCH2CH3 | 3.090 | >100 |
| 221 | COCH(CH3)2 | 0.847 | >100 |
| 222 | COCH2CH(CH3)2 | 1.884 | >100 |
| 223 | CO(c-Hexyl) | 9.240 | >100 |
| 224 | COPh | 0.570 | >100 |
| 225 | COCH2Ph | 0.079 | >100 |
| 226 | COCH2CH2Ph | 0.464 | >100 |
| 227 | SO2CH3 | 2.272 | >100 |
| 228 | SO2Ph | 1.166 | 20.50 |
aEC50: the concentration of compound that reduced 50% of the cytopathic effect of RSV. Long strain infected HEp-2 cells. CC50: the concentration of compound that manifests cytotoxicity toward 50% of uninfected HEp-2 cells. Values are means of at least two experiments performed in consecutive weeks.
Table 7.
Anti-RSV activity for the synthesized compounds.
| Compound No. | R1 | R3 | R4 | R5 | R6 | EC50 (μM) | CC50 (μM) |
|---|---|---|---|---|---|---|---|
| 229 | H | H | 2-Cl | H | H | 0.077 | >100 |
| 230 | H | H | 3-Cl | H | H | 0.066 | >100 |
| 231 | H | H | 4-Cl | H | H | 0.058 | >100 |
| 232 | H | H | 2-F | H | H | 0.089 | >100 |
| 233 | H | H | 3-F | H | H | 0.080 | >100 |
| 234 | H | H | 4-F | H | H | 0.095 | >100 |
| 235 | H | H | 2-CH3 | H | H | 0.029 | >100 |
| 236 | H | H | 3-CH3 | H | H | 0.059 | >100 |
| 237 | H | H | 4-CH3 | H | H | 0.142 | >100 |
| 238 | H | H | 2-OCH3 | H | H | 0.056 | >100 |
| 239 | H | H | 3-OCH3 | H | H | 0.098 | >100 |
| 240 | H | H | 4-OCH3 | H | H | 0.679 | >100 |
| 241 | H | H | 3-CN | H | H | 0.225 | >100 |
| 242 | H | H | 3-CF3 | H | H | 0.563 | >100 |
| 243 | H | H | 3-F-4-F | H | H | 0.255 | >100 |
| 244 | H | H | 3-Cl-4-Cl | H | H | 0.220 | 6.68 |
| 245 | H | H | 3-F-5-F | H | H | 0.264 | >100 |
| 246 | H | NH2 | CH3 | H | 0.759 | 20.80 | |
| 247 | H | NH2 | Et | H | 0.865 | 20.80 | |
| 248 | H | NH2 | CH3 | CH3 | 2.014 | 20.80 | |
| 249 | H | NH2 | Et | Et | 0.977 | 6.40 | |
| 250 | H | NH2 | (CH2)2 | >100 | 6.60 | ||
| 251 | H | NH2 | (CH2)3 | 0.317 | >100 | ||
| 252 | H | NH2 | (CH2)4 | 3.090 | >100 | ||
| 253 | H | NHMe | CH3 | CH3 | 0.847 | >100 | |
| 254 | H | NMe2 | CH3 | CH3 | 1.884 | >100 | |
| 255 | H | NHAc | CH3 | CH3 | 9.240 | >100 | |
| 256 | H | OH | CH3 | CH3 | 0.570 | >100 | |
| 257 | 5-Cl | NH2 | CH3 | CH3 | 0.079 | >100 | |
| 258 | 6-Cl | NH2 | CH3 | CH3 | 0.464 | >100 | |
| 259 | 7-Cl | NH2 | CH3 | CH3 | 2.272 | >100 |
In order to determine the direct anti-RSV effect, plaque reduction assay was performed. Compound 248 inhibits RSV replication with IC50 at lower than 100 nM concentration. Interestingly, compound 253 was able to directly inhibit RSV replication but with less potency compared to 248. Therefore, compound 248 and 253 were proven to be RSV fusion inhibitors, which can prevent the fusion process mediated by RSV fusion protein in RSV-F expressing cell lines. DMPK properties for the most potent compound 248 and 253 was examined which revealed that both of them had good oral bioavailability of >40% owing to their good solubility (LYSA > 400 μg/mL) and permeability (PAMPA values of 248 and 253 are 0.74 × 10−6 cm/s and 1.37 × 10−6 cm/s). On carrying docking studies with compound 214 (hit) and 248 described that the quinoline ring and the piperidine ring of compound 214 or the piperazine ring of compound 248 could form the hydrophobic interaction with the residues of Phe140 and Phe488, respectively. Furthermore, a salt bridge formed by the positively charged terminal amine with Asp486, are critical for the binding.
Severson et al. developed a series of enantiomerically pure, sulfonylpyrrolidine-based compounds which were optimized and validated through a high throughput cell-based screening that measures the respiratory syncytial virus (RSV)-induced cytopathic effect (CPE) in HEp-2 cells [85]. A total of 51 hits were selected from a library of 313816 compounds for carrying out an in vitro titer reduction assay to assess their effect on the production of infectious virus. The hit compound 261 (prepared from L- or d-proline) was tested as a racemic mixture out of which S-enantiomer was found to be more active component with a selectivity index of 11.8. Acyclic variants of the linker region which involves methylation at amide nitrogen was found to possess EC50 values > 50 μM as shown in Table 8 .
Table 8.
hRSV CPE Assay potency, cytotoxicity, selectivity index, and logarithmic reduction in viral plaques for analogues with structural variation in the proline linker region of the hit sulfonylpyrrolidine scaffold.
| Compound No. |  |
EC50 (μM)a | CC50 (μM)b | SI | Virus titer log reduction |
|---|---|---|---|---|---|
| 260, ribavirin | NA | 28.4 ± 3.8 | 113.9 ± 38.5 | 4.0 | 2.5 |
| 261 | 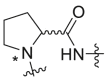 |
5.0 ± 1.4 | 31.5 ± 5.7 | 6.3 | 2.1 |
| 262a | 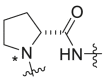 |
>50 | 38.1 ± 2.3 | 0.8 | NT |
| 262b | 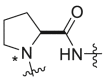 |
2.7 ± 0.9 | 31.8 ± 7.7 | 11.8 | 1.0 |
| 262c | 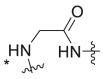 |
>50 | >50 | 1.0 | NT |
| 262d | 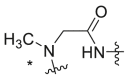 |
>50 | >50 | 1.0 | NT |
| 262e | 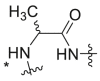 |
>50 | >50 | 1.0 | NT |
Data are an average of ≥3 experiments.
Data are an average of ≥2 experiments. cNT = not tested; NA = not applicable.
On replacing quinoline with a 4-linked benzooxadiazole, a phenyl ring, or a simple methyl group proved to be disadvantageous. Substitution of the sulfonyl group (SO2) with carbonyl functionality or its replacement with CH2 led to complete loss of potency. It was also observed that by increasing steric bulk at C2 marginally improved potency as compared to the monomethyl substitution at the same position. Since 2-alkyl substituent appeared to be crucial in combination with other substituents to retain potency, this dynamic was further explored in order to suggest that the 2,5-dimethylphenyl moiety of analogue 262oproved to be more beneficial in terms of potency and maintaining the cytotoxicity potential. Detailed results are shown in Table 9 .
Table 9.
hRSV CPE Assay potency, cytotoxicity, selectivity index, and logarithmic reduction in viral plaques for analogues with structural variation in the aryl amide region of the sulfonylpyrrolidine scaffold.
| Compound No. |
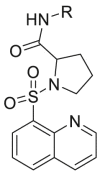 R R |
EC50 (μM)a | CC50 (μM)b | SI | Virus titer log reduction |
|---|---|---|---|---|---|
| 260, ribavirin | NA | 28.4 ± 3.8 | 113.9 ± 38.5 | 3.6 | 2.5 |
| 262b | 2,4-dimethylphenyl | 2.7 ± 0.9 | 31.8 ± 7.7 | 13.1 | 1.0 |
| 262f | 2-methylphenyl | 23.8 ± 10.2 | >50 | 2.1 | NT |
| 262g | 3-methylphenyl | >50 | 25.5 ± 4.1 | 0.5 | NT |
| 262h | 4-methylphenyl | >50 | 31.7 ± 2.9 | 0.6 | NT |
| 262i | phenyl | >50 | 35.6 ± 2.3 | 0.7 | NT |
| 262j | 2-ethylphenyl | 21.2 ± 5.1 | >50 | 2.6 | NT |
| 262k | 2-i-propylphenyl | 17.6 ± 0.9 | 29.9 ± 3.5 | 1.7 | NT |
| 262l | 2,4-dimetyl-3-pyridine | >50 | >50 | 1.0 | NT |
| 262m | 3,5-dimethylphenyl | >50 | 31.3 ± 3.5 | 0.6 | NT |
| 262n | 2,4-dichlorophenyl | >50 | >50 | 1.0 | NT |
| 262o | 2,5-dimethylphenyl | 2.3 ± 0.8 | 30.9 ± 1.1 | 13.4 | 2.0 |
| 262p | 2,6-dimethylphenyl | >50 | >50 | 1.0 | NT |
| 262r | 2-methyl,5-chlorophenyl | 2.2 0.6 | 10 1.3 | 4.3 | NT |
| 262s | 2-methyl,5-trifluoromethylphenyl | >50 | 4.8 1.7 | 0.1 | NT |
| 262t | 2-methyl,5-methoxyphenyl | 4.2 ± 1.3 | 44.2 ± 5.7 | 10.4 | 1.0 |
| 262u | 2-methyl,5-i-propylphenyl | >50 | 9.7 ± 1.2 | 0.2 | NT |
| 262v | 2-methyl,5-i-butylphenyl | >50 | 6.5 ± 0.6 | 0.1 | NT |
| 262w | 2-chloro,5-methylphenyl | >50 | >50 | 1.0 | NT |
| 262x | 2-methoxy,5-methylphenyl | >50 | >50 | 1.0 | NT |
| 262y | 1-(t-butyl)-3-methyl-1H-pyrazol-5-yl | >50 | >50 | 1.0 | NT |
cNT = not tested; NA = not applicable.
Data are an average of ≥3 experiments.
Data are an average of ≥2 experiments.
Further mechanism of action of sulfonylpyrrolidines was examined by checking the inhibitory activity in the cell-based assay in HEp-2 cells. Cells were infected with hRSV in the presence of 25 μM concentration of test compounds 262b, 262t, 262o and ribavarin. Compounds 262b and 262t were found to possess 1 log reduction in virus titer, or 10-fold, as compared to ribavirin which reduced viral titer by 2.5 log units, or 300-fold. Analogue 262o was found to have 100-fold, or 2 log, reduction. Improvements in cell protection against hRSV did not translate to significant improvement in the plaque assay as was seen with ribavirin. Consequently, the titer reduction assay was not used to drive SAR efforts.
3.5. Middle east respiratory syndrome coronavirus (MERS-CoV)
MERS-CoV is fatal coronavirus that has the ability to spread from person to person causing severe respiratory symptoms such as high fever, cough, shortness of breath and acute pneumonia [86,87]. It is a single-stranded, positive-sense RNA virus which tend to utilize host cell components to accomplish their physiological process which includes internalization of the virion, genome replication, packaging and budding of virions. Therefore, each of the above steps can be considered as potential target for the development of novel antiviral agents. Yoon et al. identified 3-acetyl-6-chloro-2-(isopropylamino)-8-(trifluoromethyl)quinolin-4(1H)-one 263 as the lead compound to be active against MERS-CoV infections. So, a series of 3-acyl-2-amino-1,4-dihydroquinolin-4(1H)-one analogues were synthesized and then anti-MERS-CoV activity was evaluated using Vero cells infected with MERS-CoV isolate [88].
SAR studies were conducted by varying the substituents positions of 5–8 of quinoline ring of compound 263. It was observed that electron withdrawing substituents such as 2,4-difluoroaniline, NO2 and CF3 groups were found to cause significant inhibition while the electron donating groups showed no signs of inhibition. Varying the substituents at 2 position of 1,4-dihydroquinoline suggested that 6,8-difluorosubstituent 264g can cause marked changes in inhibition activity. Further, presence of electron withdrawing group at the 2 position shows high binding affinities (0.53–1.44 μM). It was observed that 6,8-difluoro compound 264u and 264v, including 2,3,4-trifluoroaniline and 2,4-difluoro aniline group at 2 position had the potential to cause remarkable inhibition than its corresponding compounds with acetyl group at 3 position (IC50 = 0.086 and 0.79 μM, respectively) as illustrated in Fig. 14 . Thus, compound 264u with isobutyryl group at 3-position and 6,8-difluorophenyl group was found to be potent against MERS-CoV inhibition, displayed good metabolic stability in human, rats and mouse liver microsomes and good oral bioavailability. It was found to have low hERG binding affinity and zero cytotoxicity toward VERO, HFL-1, L929, NIH 3T3, and CHO-K1 cell lines.
Fig. 14.

MERS-CoV inhibitory activity of 3-acyl-2-amino-1,4-dihydroquinolin-4(1H)-one derivatives.
3.6. Dengue virus
Dengue fever, usually a disease is caused by dengue virus which results in an uncontrolled infection transmitted by mosquitoes of the Aedes genus [89]. The virus belongs to the Flavivirus genus belonging to Flaviviridae family and is circulated as four major serotypes which do not induce long-term cross-protection in secondary infections. Currently, there is no availability of specific antiviral therapy against dengue virus except symptomatic treatment. Lleonart’ group have demonstrated the synthesis, screening and characterization of antiviral activity of two quinoline derivatives against dengue virus serotype 2 in vitro as shown in Fig. 15 [90,91].
Fig. 15.

Potent quinoline derivatives against dengue virus serotype.
Out of total 29 compounds, only three compounds were selected for confirmatory tests using viral yield reduction assay which suggested that compound 265 and 266 had shown signs of consistent inhibitory activity against DEN2 in vitro with IC50 of 3.03 μM and 0.59 μM respectively. It was also observed that both the compounds 265 and 266 possessed a marked, dose-dependent effect on the production of the viral envelope glycoprotein which was found to be in harmony with the antiviral activity observed in virus yield reduction assay. The data obtained by authors suggest 2 key points, first, antiviral activity is not due to direct virucidal activity, second, compound tend to act during the early phases of virus life cycle.
In 2017, Gray’s group carried out an extensive SAR study of QL47 (267) which have been confirmed as a potent and covalent inhibitor of BTK and other Tec-family kinases [92,93]. During the SAR study, compound YKL-04–085 was found to be the potent antiviral agent, which lacks the crucial quinoline hydrogen responsible for hydrogen bonding to the kinase hinge. QL47 (267) was found to be the most potent antiviral agent with IC50 value of 0.343 μM against DENV2. However, several BTK/BMX kinase inhibitors such as CG1-1746, Ibrutinib and BMX-IN-1 were evaluated in order to determine the relevance of BTK/BMX kinase as potential targets for viral replication. But from the results none of the compounds showed activity comparable to QL47 (267), thereby suggesting that BTK/BMX are not likely to be targets of QL47(267).
Moreover, it was observed that it possessed broad-spectrum activity against other Flaviviruses such as West Nile Virus (WNV), enteroviruses, vesiculoviruses, pneumoviruses but was associated with poor pharmacokinetic profile. From the structure-based drug designing, QL47 (267) was earlier developed as BTK inhibitor from the highly potent mTOR inhibitor, Torin 2. The tricyclic quinolines of QL47 (267) were found to form a hydrogen bond with the kinase hinge segment, while the two side chains extend toward the back and front of the ATP binding site. The acrylamide arm extends towards Cys 481 of BTK located at the entrance of ATP-binding site. When acrylamide moiety was replaced with unreactive nonpropyl amide to give QL47R (268), no signs of inhibition against DENV2 at concentrations below 10 μM were observed. The structures of BTK/BMX kinase inhibitors are shown in Fig. 16 .
Fig. 16.

Potent BTK/BMX kinase inhibitors.
Methypyrazole substituted analogue 277 described modest improvements in microsomal stability with significant antiviral activity. This analogue was explored because it is key determinant involved in kinase selectivity. It was observed that extended conjugate ring systems (from methylpyrazole to acrylamide) were essential for preserving antiviral activity. Positioning of ortho-substituents to the acrylamide, compound 270 has similar potency to QL47 (267) with potent in cellulo translation inhibition and antiviral activity nut half-life improved only upto 4.5 min. It was presumed due to the intramolecular hydrogen-bonding of the ortho-fluoro group forming a pseudo-five membered ring. However, tricyclic quinolinyl core of QL47 (267) was modified with a five or six membered urea moiety increased antiviral activity and inhibition of DENV reporter replicon translation was observed but didn’t show any improved substantial signs of metabolic stability. The anti-DENV activity and translation effects of DENV reporter replicon translation are correlated because the translation effects of viral genome are crucial for antiviral activity of these compounds. Compound 271, without the N-quinoline nitrogen and methyl pyrazole substituted showed similar activity to QL47 (267) but half life was improved to 4 min. Interestingly, when dimethylamine tail was installed at the acrylamide position, YKL-04–085 and 272 exhibited improved antiviral activity at 2 μM concentration with YKL-04–085 showing marked improvement in half-life of 15.6 min. After the successful discovery of YKL-04–085, it was screened against a panel of kinases out of which only few kinases such as PIM and DDR has 80% of binding affinity. The anti-DENV2 IC90 value was found to be 0.55 μM and exhibited viral inhibition and cytotoxicity at 35-times lower concentrations relative to host-cell proliferation inhibition. YKL-04–085, also described good translation inhibition of DENV2 reporter in cellulo and good pharmacokinetic profile.
3.7. Herpes simplex virus (HSV)
Herpes simplex virus is associated as a common contagious disease, belonging to Herpesviridae family. HSV 1 and HSV 2 tend to affect the human population. HSV 1 causes infections around the mouth pharynx, face and lip area while HSV 2 is associated with infection of the anogenital region [94]. The virus seems to be most contagious when it begins to shed from the human body. Acyclovir, famiclovir and valacyclovir have been approved as antiviral drug for patients suffering from HSV infections. But still there is an urge to develop new molecules against HSV. Therefore, Zhang et al. designed and synthesized novel Pt(II) complexes (TFPPy)PtPic (273) and (TFPQ)PtPic (274) comprising fluorinated phenylpyridine/phenylquinoline scaffolded with pyridine-2-carboxylate as chelating ligands [95]. Both the complexes were investigated for the treatment against HSV-1 infection. The results revealed satisfactory suppression of HSV-1 replication at concentrations of 5–10 μM on conducting plaque assay. The compound (TFPQ)PtPic (273) was found to be more dependent on treatment concentration because of the more significant suppression difference. IC50 of the two complexes against healthy Vero cells were found to be above 28 μM, suggesting that both the complexes have selective inhibition against HSV-1 infection rather than causing inhibition on the un-infected hosting cells. Structure of these complexes is shown in Fig. 17 .
Fig. 17.

Pt(II) complexes with inhibitory activity against HSV-1.
The authors also suggested the inhibitory mechanism which involves the direct ability of the complexes to bind and interfere in the virus DNA genome. Molecular docking studies revealed that the complexes were able to form stable hydrogen bond and hydrophobic interactions to the minor groove of targeted DNA double helix.
3.8. Ebola virus disease (EVD)
Ebola virus disease also known as Ebola haemorrhagic fever caused by pathogen Ebolaviruses, is a severe disease causing fatal illness in humans. It can be rapidly transmitted through direct contacts with infected patients and contaminated materials. Several antiviral drugs have been clinically approved but none of them had shown potential benefits in patients thus, it generates a room for the development of novel antiviral drugs against ebola virus. It was observed that amodiaquine which is an antimalarial drug was found to decrease the mortality rate among the ebola infected patients [96]. Therefore, keeping in mind its beneficial effects against Ebola virus, Sakurai, in 2018, made an attempt to synthesize a series of novel amodiaquine derivatives and evaluated them against EBOV infection [97]. Compounds bearing 4-aminoquinoline scaffold and structurally similar to amodiaquine were synthesized. These were tested using replication component EBOV encoding GFP for antiviral activity with Huh 7 cells for carrying out in vivo studies. A total of 14 compounds were found with improved potency while compounds 275, 276, 277 and 278 were highly potent with IC50 = 0.73, 0.64, 0.29 and 0.72 μM respectively as shown in Fig. 18 . Amodiaquine was found to have IC50 = 2.13 μM against EBOV infection. Cytotoxic profile was evaluated for the 14 compounds by performing infections assays, which revealed that compound 276 and 278 provided SI > 130 describing its low cytotoxicity as compared to amodiaquine which have SI = 37.
Fig. 18.

Antiviral activity of amodiaquine derivatives against EBOV-GFP.
SAR study was conducted by first carrying out modification of the alkyl chain extending from the aminomethyl group (R1). This modification was supported by observing increased potency among the compounds. When halogens were substituted at the 7th position of the quinoline ring (R2), it was seen that most electronegative atoms (-F, -Cl) exhibited lower potency than amodiaquine while least electronegative atom (-Br and -I), increased the potency by 1.4 and 3.3 (compound 276) times respectively as shown in Fig. 19 . Presence of phenol ring adducts at R3 and phenyl ring at R4 were found to have improved activity. Second modification involved varying the alkyl chain length with iodine substitution at the 7th position of the quinoline ring.
Fig. 19.

SAR study result by using compound 276.
Promising results were obtained which included 2 times higher potency than the compound 276 with same alkyl chain as amodiaquine. Compound 279, having the longest chain length was found to have maximal positive effects against viral infection. Cytotoxic values were also higher (SI > 100) than the initial set of derivatives having the alkyl chain extending from the aminomethyl group (R1). Further, compound 276 and 278 were found to inhibit the entry of virus into the host cell more efficiently than amodiaquine which were consistent with the outcomes of EBOV-GFP infection.
3.9. Hepatitis C virus (HCV)
Hepatitis C virus is a positive-strand RNA virus belonging to Flaviviridae family, associated with acute and chronic liver disease [98]. It is a blood borne virus and the most common mode of infection is a result of exposure to small quantities of blood. Triple combination therapy is utilized for curing HCV genotype-1 disease which involves pegylated interferon α (Peg-IFN), ribavirin, and protease inhibitor (telaprevir and boceprevir), but interferons are associated with side effects which limits their use. Therefore, researcher’s main goal is to develop direct acting antiviral therapy (DAA) with high sustainable virological response. The catalytic fragment involved in the HCV RNA replication is the HCV nonstructural 5B protein (NS5B) that contains RNA-dependent RNA polymerase. This enzyme is involved in the life cycle of virus thereby acting as potential therapeutic target to develop novel antiviral agents.
Talamas et al. reported stilbene fragment 281 that is a potent inhibitor of NS5B but still several efforts were made to develop a highly potent inhibitor against the replication of HCV genotype 1a and 1b [99]. In this study, stilbene core was replaced with bicyclic core which was synthesized and further evaluated for antiviral activity. Compound 282 with a bicyclic quinoline core was found to be equipotent against GT-1a and GT-1b strains with better activity against GT-1a when compared with 281. Further, 5-fluoropyridone part was replaced with uracil heterocycle while keeping the methoxy, tert-butyl, and N-phenylmethanesulfonamide substituents intact. Uracil 283 had decreased activity against GT-1a but same potency against GT-1b in replicon assay. Compound bearing naphthalene core exhibited 2 to 3 times increase in potency against both the genotypes. But still 3,5,6,8-tetrasubstituted quinoline core with different heterocyclic head groups was selected for further evaluation owing to its flexibility, convenient synthesis and excellent potency. It was observed that compound 285 had excellent potency and equipotency against both genotypes as compared to other molecules bearing 2(1H)-pyridone, dihydrouracil and cyclic urea head groups. C-linked uracil 286 had improved potency against both genotypes with respect to N-linked uracil 283 as sketched in Fig. 20 .
Fig. 20.

Bicyclic quinoline core possessing molecule with inhibitory activity against HCV.
Molecular docking studies were carried out for compound 285 which suggested that the lower ring of quinoline template makes edge-to-face interactions with the side chain of Tyr448 and tert-buty group fills the lower lipophilic pocket. The above interactions were similar to those observed NS5B fragment 280. It was suggested that quinoline ring tends to hold the methanesulfonamide linker in the correct position. The phenyl ring linked to the quinoline with the methanesulfonamide forms an edge-to-face interaction with Phe193, while sulfonamide group forms hydrogen bond with a distance from the residues Asp318, Asn 291, and Ser288 (2.7, 2.8, and 3.2 Å, respectively). The sulfonamide functional group was essential for the high potency of the template along with the oxygen of the methoxy group on C-6 of the 2(1H)-pyridone is in proximity (3.3 Å) to the carbon of the backbone carbonyl Gly410. Most of the compounds with quinoline core were highly potent and exhibited physicochemical and ADME in vitro properties within the desirable range except for solubility. Thus, in order to improve solubility, planarity of compounds was reduced by introducing replacement of phenyl linker between the quinoline and the methane sulfonamide.
Alkyl and alkyne linker proved to be less active because of the formation of possible conformations which prevents the methanesulfonamide group from forming favorable interactions with Asp318, Asn291, and Ser288. But compound with pyrrolidine linker 287 displayed similar activity as corresponding phenyl linker 284 and also led to increase aqueous solubility as shown in Fig. 21 . Finally, pyridine 288 and pyrrolidone analogue 287 were found to possess best combination of overall properties but 288 had much weaker inhibition compared to 285. 6-fold improvement was observed in the 40% HuS replicon assay. Compounds 285 and 288 underwent pharmacokinetic studies in rat and dog in order to determine their potential as clinical candidates. Ultimately, compound 285 was chosen for clinical development as it met with the expected criteria.
Fig. 21.

Replacements of the phenyl linker with NS5B.
3.10. Human immunodeficiency virus (HIV)
Human immunodeficiency virus infection and acquired immune deficiency syndrome (HIV/AIDS) is a range conditions caused by HIV virus and spreads primarily by unprotected sexual contact, contaminated blood transfusions and from pregnant mothers to their children. Several therapies such as virus-associated reverse transcriptase, protease, or fusion peptides have been developed to inhibit the viral replication. But such therapies are often associated with drug resistance and various side effects thereby limiting their use for treatment of AIDS. Thus, there is an emergent need for developing new class of drugs that can easily replace the existing drugs and act on new targets involving novel molecular mechanisms [100].
Bedoya et al. tested 18 quinoline-based compounds for their antiviral activity against human immunodeficiency syndrome (HIV) [101]. The compounds were found to contain quinoline or tetrahydroquinoline rings and were divided into groups: group 1 comprises of 4-(2-oxopyrrolidinyl-1)-1,2,3,4-tetrahydroquinolineswith 2-(3-nitrophenyl) substituent (N-series) or 2-(3-aminophenyl) moiety (H-series), and group 2 includes 2-(3-nitrophenyl)- or 2-(3-aminophenyl)-substituted quinolines (S-series). Recombinant virus assay (RVA) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay were performed to check anti-HIV activity of compounds. From the results it was confirmed that Compounds belonging to S-series were more potent as compared to Compound of N-series or H-series. In N-series, none of the compounds showed antiviral activity when tested with MTT assay but two tetrahydroquinolines 293 and 294 were found to possess moderate activity when RVA assay was perfomed. Compound 296, 299 and 300 of H-series were active in both the assay with IC50 ranging from 50 to 100 μM and non-specific mechanism of action was observed due to the toxicity. Compound 297 was found to be active against VSV-pseudotyped HIV infections. From S-series compound 301, 303 and 305 were highly potent in both the assays with compound 305 (IC50 = 18.48 μM) being the most potent in MTT assay. Also these three compounds had similar IC50 values against both the viruses (HIV and VSV-pseudotyped) which suggest that they are likely to act when the virus enters the host cell. Detailed structures of potent molecules against HIV are shown in Fig. 22 .
Fig. 22.

Synthesized quinolines with anti-HIV activity.
PCR-based retrotranscriptional assay was performed to analyze the potential action of the 3 most potent compounds of S-series. The results confirmed that the total viral DNA amount was not reduced which suggests that a later step is the probable target for these molecules. HIV transcription is a complex process involving lot of viral and cellular factors thus these compounds were studied as transcription inhibitors. It was observed that all the three compounds were able to decrease HIC transcription through NF-ĸB or specificity protein-1 (Sp1) transcription factors. Compound 297 efficiently abolished the expression of p65 which confirms that quinolines act by blocking nuclear translocation of p65 and thus inhibiting NF-ĸB while IĸB is less expressed.
Bénard et al. have developed a series of compound active against HIV-1 integrase and were further evaluated in both in vitro and ex vivo assays [102]. These inhibitors involve drugs of styrylquinoline family which engages quinoline subunit and an ancillary aromatic ring connected by an ethylenic linker in between. The studies were conducted by replacing the central ethylenic linker with functionalized spacer such as amide, hyradzide, urea, 1-hydroxyprop-1-en-3-one moieties.
MTT assay were perfomed and subsequent IC50 and EC50 values were calculated. It was observed that when ethylenic bridge was replaced by amide group, none of the compound showed any significant inhibition and cytotoxicity. While compound 318, in which the ancillary phenyl ring is unsubstituted had cytotoxicity comparable to compound 307. Compound in which nitrogen atom is attached to the quinoline subunit and in which ancillary nucleus has two hydroxyl groups at C-3′ and C-5′ possessed no in vitro activity but on the contrary corresponding styrylquinolines displayed significant in vitro activity. A micromolar level of antiviral activity was observed with amides 308, 309 and 311 which is comparable to the reference compounds 307 and 317. Styrylquinolines when replaced with hydrazides 311, 312 and urea moiety 313–315, none of them were found to be active but 2,4-dinitrophenylhydrazide 312 was found to restore substantial anti-HIV intergrase activity. However, ketoenol linker 316 which is regarded as potential pharmacopher in the design of HIV-integrase inhibitors remained completely inactive because of the absence of hydroxyl groups on the ancillary phenyl ring as illustrated in Fig. 23 . Di Santo et al. have reported new bifunctional β-diketo acids (BDKA) with enhanced activity against integrase (IN) and HIV-1 infected cells [103]. IN enzyme is known for catalyzing the insertion of viral DNA into the host genome which is derived from reverse transcription of HIV RNA. Integration mainly occurs through a sequence of multiple reactions which includes (i) 3′-processing: it involves the cleavage of a dinucleotide pair from the 3′-end of the viral DNA (ii) strand transfer (ST): insertion of the resulting shortened strands into the host-cell chromosome and (iii) removal of two unpaired nucleotides at the 5′-end of the viral DNA and gap filling process. The authors have utilized DKAs scaffold as they are known for selectively inhibiting the ST reaction of IN and exhibit antiviral activity against HIV-1 infected cells. Thus, inhibitors 327-322 belonging to 4-(4(1H)-quinolinon-3-yl)-2,4-dioxobutanoic acid skeleton was selected because of two reasons, first, alkylating the 4(1H)-quinoline at 1-position with benzyl group to obtain 1,3-disubstituted compound that is able to meet the geometric requirements for IN inhibitory activity, while second involves the introduction of diketoacid at position 6 of aromatic ring via an acetyl intermediate. The structure of compound 330 with inhibitory activity is shown in Fig. 24 .
Fig. 23.

Quinoline derivatives active against HIV-1 integrase.
Fig. 24.
Bifunctional β-diketo acid active against integrase (IN) and HIV-1 infected cells.
The synthesized compounds 319–322 were evaluated for ST inhibition in the presence of magnesium (Mg2+) using a novel electrochemiluminescent assay in vitro as well as examined for ST and3′-P using gel-based assays in the presence of Mg2+ or Mn2+. All the compounds were found to possess potent inhibitory activity against IN for both ST and 3′-P steps. It was also observed that acid derivatives (320, 322) were more potent than the corresponding esters (319, 321) and 1-p-F-benzyl-substituted quinolines (321, 322) were more active. Compound 322 was the most potent among all other compounds with IC50 around 15 nM. It shows selective ST inhibition as well as inhibits integrase with similar potency in the presence of Mg2+ or Mn2+. It revealed promising antiviral efficacy in HIV-1-infected H9/HTLVIIIB cells ((EC50 = 4.29 μM, EC90 = 40 μM) and low cytotoxicity (CC50 > 200 μM, SI > 46.6) along with high potency in inhibiting the replication cycle of HIV-1 in cell based assays. Docking study for compound 322 was performed using AutoDock 3.0.5 program. The results indicated that the carboxylate group of one diketo acid chain of the ligand chelates with the Mg2+ metal ion, whereas the other one inserts between residues K156 and K159, forming H-bonds with both side chains and with N155 CO backbone. The p-F-benzyl group points toward a hydrophobic pocket formed by the catalytic loop residues Y143, P142, I141, and G140 and by residues I60, Q62, V77, V79, H114, G149, V150, I151, E152, S153, and M154. A favorable electrostatic interaction was observed between the fluorine atom on the benzyl ring and the amide group of Q62 residue. A stacked amide-aromatic interaction with the N155 side chain was formed with the quinoline ring of the molecule, while carbonyl oxygen of the quinoline forms a hydrogen bond with the T66 side chain. T66, S153, and M154, side chains were found to be in close proximity of the ligand whose mutations were found to confer resistance to DKAs.
Interestingly, one of the most promising targets for inhibition of HIV-1 replication is the process of Tat/TAR RNA interaction. This Tat-mediated trans-activation process requires structural integrity of TAR RNA and the cooperative interaction of human hsot cell proteins such as positive transcription elongation factor b (P-TEFb), a complex composed of cyclin T1 (CycT1) and cyclin-dependent kinase 9 (CDK9) [[104], [105], [106], [107], [108]]. CycT1 is responsible for remodelling the structure of Tat in order to enhance its affinity for the TAR RNA, whereas TAR RNA inturn enhances the interaction between Tat and CycT1. Yang’s group has proposed a molecular model for the generation of novel Tat-TAR inhibitors which includes an anchor, linker and a positively charged activator. This activator reinforces the binding affinity between the molecule and the negatively charged TAR RNA thereby leading to the disruption of Tat/TAR complex. The compound library was screened for inhibitory activity of Tat-TAR interaction which resulted in emergence of CS3 (323) possessing mild inhibitory activity and antiviral potency. CS3 (323) consisted of a planar 4-chloroquinoline ring with two side chains connected with the aromatic ring by an amide linker. Several modifications to CS3 (323) were done which included modification of two side chains, introduction of substitution in the 4-chloroquinoline ring, substituting nitrogen atom of the quinoline and replacing the chloro group of quinoline with hydroxyl or carbonyl group. Therefore, compounds were designed, synthesized and investigated for antiviral activity using SIV-induced syncytium in CEM cells [109,110]. From the results, it was observed that compound bearing hydroxyl alkane side chain and methyl ester side chain (319n and 319p) both displayed efficient anti SIV-activities and low cytotoxicities especially 319p (EC50 = 1.1 μM) had higher value than 319n probably due to hindrance of hydrogen bond interaction with methyl group. Presence of electron withdrawing or electron releasing group on the quinoline ring proved to be less fruitful for both anti SIV-activity and cytotoxicity. In order to testify the steric hindrance effect of methyl group with hydrogen bond 319z (EC50 = 13.0 μM) was developed which was found to possess higher EC50 than 319s (EC50 = 4.7 μM). Further, introduction of substituent group on nitrogen of quinoline or changing the 4-position into carbonyl (320a-c and 322a-d) did not resulted in any significant changes in anti SIV-activity and cytotoxicity.
Nine compounds with low cytoxicity (CS3 (323), 319d, 319f, 319n, 319p, 319s, 319z, 322a and 322b) were evaluated by Tat dependent HIV-1 LTR-driven CAT gene expression colorimetric enzyme assay in order to ascertain their potential to inhibit HIV-Tat-TAR interaction as shown in Fig. 25 . High inhibitory activity is a result of depressed CAT expression. Thus, 44.2–92.7% range of inhibited CAT expression was observed with compound 322a (44.2%) displaying the lowest CAT expression. Compound 319p (59.6%) and 319c (82.4%) (bearing hydroxylmethyl chain) had lower CAT expression as compared to 319n (53.3%) and 319z (92.7%) (bearing 2-OH ethyl group). This decreased CAT activity suggests that the compounds had the potenty to block the interaction of Tat-TAR in cell based assay. Finally, molecular modeling experiments were performed for 319p, 322a and 322b to study the interaction between these compound and HIV-1 TAR RNA. The free energies turned out to be above-5 kcal/mol, which reflected the binding affinity of the compounds and the TAR RNA leading to conclusion that these compounds may not inhibit Tat-TAR interaction by just binding to TAR RNA. The bound conformations of all these compounds occupied a pocket surrounded by amino acid residues M36, H81, P82,Q35, C34 with fairly low free energies below −10 kcal/mol. H-bond interactions were observed between the side chains of 319p, 322a and 322b and the amide backbone of M36, H81, and P82, describing that the H-bond interaction plays an important role in enhancing the binding preferences of the compounds. Thus from the above suggestion it was confirmed that it was the H-bond interaction being hindered by methyl which resulted 319n to have higher EC50 value than 319p. The aromatic ring moiety of all the three compounds was found to be out of the pocket thereby giving reason to insignificant activity observed while carrying out modifications on the quinoline ring.
Fig. 25.

Quinoline derivatives evaluated for Tat dependent HIV-1 LTR-driven CAT gene.
HIV-1 enzyme reverse transcriptase (RT) is a well known target utilized by Nucleoside (NRTI) and Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTI) as they have the ability to block RT and convert single-stranded RNA genome to double-stranded DNA in HIV-1 replication cycle. Several anti-retroviral agents are associated with resistant thus there is need for development of novel drugs.
Thus, a series of 4-oxoquinoline ribonucleoside derivatives were developed by molecular hybridization and evaluated against human immunodeficiency virus type 1 reverse transcriptase (HIV-1 RT) with azidothymidine (FDA approved drug) being used as reference compound [111,112]. All the compounds displayed good anti-HIV activity ranging from 1.4 to 2.9 μM with compounds 324a and 324d being most potent with IC50 values of 1.4 and 1.6 μM, respectively as illustrated in Fig. 26 . It was observed that absence of halogen group at R1 position is responsible for improved activity. Furthermore, all the compounds were assayed using human primary cells and peripheral blood mononuclear cells (PBMCs) for cytotoxicity. Compounds were found to be 10 times less cytotoxic than azidothymidine with compound 324a and 324d displayed CC50 values of 1486 and 1394 μM respectively. Conformational analysis revealed that greater flexibility in compounds leads to formation of multiple conformations which supports a better fit at the binding site and generation increased inhibition potency. This feature is more favorable for mutant strains with a resilient profile which allows the flexible molecule to be easily accommodated at the RT binding site. Also larger contact surface area provides a room for effective vanderwaal interaction thereby enhancing the activity. Electronic parameters did not show any direct correlation with activity while volume and flexibility were key features for the most active compounds 324a and 324d. The volume of allosteric site HIV-1 RT was calculated to be 822.01 Å which was able to accommodate bulky molecule 324a and 324d. Molecular docking studies revealed that the most active compounds bind to the allosteric site of the enzyme, interacting with important residue such as Tyr318, which is a conserved residue with no mutations. This indicated that 324a as an inhibitor would be less susceptible to viral resistance. Finally, the in silico ADMET results showed that 324a and 324d are similar to AZT.
Fig. 26.

4-oxoquinoline ribonucleoside derivatives against HIV-1 RT.
Overacker et al., in 2019, described the biological evaluation of azaBINOL class of molecules for inhibition against HIV-1RNase H activity [113]. The azaBINOLs are nitrogenous analogues of BINOL containing isostructural 8-(naphth-1-yl)quinoline (8-azaBINOL) and 8,8′-biquinolyl(8,8́-diazaBINOL) motifs. Unique azaBINOLs and two BINOLs were evaluated using pseudo-typed viral particle and single-round infectivity assay resulting in the identification of three promising (325, 326 and 330) antiviral compounds out of which 7-isopropoxy-8-(naphtha-1-yl)quinoline 325 exhibited efficient antiviral activity at micromolar concentration (4–9 μM) with low cytotoxicity as shown in Fig. 27 . However, on closer analysis three more compounds with similar structural to the active compounds features such as naphthol-type regioisomer of quinol-type ether 329 as well as 2-deoxy-8,8′-diazaBINOL congeners of isopropyl ether and carbamates 327 and 328 were also explored. Unfortunately, they possessed inferior antiviral activity compared to 325, 326 and 330. Compound 329 was able to show minor antiviral activity at high concentrations but its IC50 values were higher compared to 325 which is due to the reduced hydrophobic surface and increased steric interaction, respectively. This difference between these two regioisomers suggests that a selective and specific mode of inhibition exists. EASY-HIT was done for 325 which also displayed similar inhibition with low micromolar activity in vitro. Binding affinity (KD) of 38 μM using BLI reveals that 325 binds to HIV RT via 1:1 binding mechanism and recombinant enzyme assay was employed which suggest that it specifically inhibits RNase H. Additive effect was observed by utilizing a combination treatment of 325 with NNRTI efavirenz in a single round infective assay. Interestingly, 325 was able to retain activity against a multi-drug resistant isolate including the mutations induced by NNRTIs in the reverse transcriptase region. Therefore, it is suggested that 325 might be involving a binding site different form that of NNRTI binding site.
Fig. 27.

Active azaBINOL derivatives against HIV-1RNase H activity.
Polyhydroxylated styrylquinolines 331 are potent HIV-1 integrase inhibitors capable of blocking the replication of HIV-1 in cell culture. It has been proposed that these drugs might act prior to integration by preventing viral DNA-integrase binding. Presence of salicylic acid moiety at C-7, C-8 of the quinoline ring and 4′-OH on the ancillary aromatic nucleus are crucial for antiviral activity [114]. Thus, novel styrylquinoline derivatives 332a, b with a carboxyl group either directly bound to the quinoline ring or through an ethylenic spacer were assayed for in vitro inhibitory activity against HIV-1 integrase and for antiviral activity against HIV-1 replication in HeLa P4 cells. Diacids 332a and 332b were found to exhibit high potency against integrase on both 3′-processing and strand transfer assay. But a marked decrease in activity was observed for compound 332b when compared with 331 while activity was retained with 332a with slight increase in cytotoxicity as illustrated in Fig. 28 .
Fig. 28.

Polyhydroxylated styrylquinolines as potent HIV-1 integrase inhibitors.
Ibrahim et al., in 2020, have demonstrated a series of 5-(substituted quinolin-3-yl or napthyl)methylene)-3-substituted imidazoline-2,4-dione 333–350 were designed and evaluated for HIV-1IIIB replication in MT-2 cells [115]. Motivation behind the design of this was from the compounds IV and V which belongs to a series of 4-thiazolidinone-based derivatives that target the inhibition of 6-HB (helix bundle) formation in HIV-1 gp41 [116].
These compounds were found to have high lipophilicity (ClogP>5), high molecular weight (>500), low drug-likeness score and 1–2 violations from Lipnski, Ghose, Egan and Muegge rules. Several modifications in compound IV were done to create a scaffold A which includes the introduction of polar groups (replacing 2 sulfur atoms), quinoline nucleus with hydrophobic substitutents (CH3, OCH3, OCH(CH3)2), 2-position of quinoline substituted with electron donating or electron withdrawing group. The spacer length was increased upto two carbon atoms to enhance hydrophobicity and increase molecular volume thereby allowing them to occupy larger volume in the hydrophobic pocket.
The inhibitory effect of 333–350 was evaluated using p24 ELISA assay on HIV-1 IIIB replication in MT-2 cells with I and II compounds being selected as reference compounds. It was observed that 333–348 derivatives showed superior activity compared to naphthyl derivatives 349 and 350 with compounds 337, 342, 343, 344, 346 and 347 being the best compounds with EC50 ranging from 0.148 to 0.50 μM as shown in Fig. 29 . Compound 337 was found to be most effective against MT-2 cell with an EC50 = 0.148 μM. Reduction in EC50 value upto 5 times was observed by replacing 8-Me group in 337 by 6-Me/OMe/7-Me. Inhibitory effects using primary HIV-1 isolate, 92US657 (class B, R5) on compound 333–350 were determined which revealed that all the compounds inhibited the isolate with EC50 between 0.520 and 11.857 μM with compound 339 being the best inhibitor (EC50 = 0.50 μM). Moreover, two of the best HIV-1 IIIB inhibitors 337 and 343 (EC50 = 1.48 μM and EC50 = 0.33 μM) compared to compound IV with SI = 484 were detected using colorimetric XTT assay on MT-2 cells. From the SAR studies, several conclusions were drawn, which includes the reduction in inhibitory activity and selectivity against HIV by removing the 8-methyl group of compound 337. Introduction of naphthyl moiety by replacing quinoline leads to 10 times decrease in activity and 2 times decrease in selectivity. Further decrease in activity was observed by increasing the length of spacer between the phenyl and imidazole by one carbon atom. 2-position substituted by Cl group instead of OCH3 group led to increase in activity. 7-methoxy group substitution on the quinoline resulted in significant increase in activity. Docking study of the new compounds revealed nice fitting into the hydrophobic pocket of HIV-1 gp41. All the new compounds 333–350 displayed higher affinities than NB-64 due to their extended hydrophobic scaffold. Compound 337 being the most active inhibitor adopted a similar orientation to compound IV with a comparable affinity. It also revealed hydrogen bonding interactions between imidazolidine-2,4-dione ring of the new compounds and LYS574 which was not observed most of the weakly active derivatives. A detailed structure activity analysis is depicted in Table 10 .
Fig. 29.

Series of compounds evaluated for HIV-1IIIB replication in MT-2 cells.
Table 10.
Anti-HIV-1 IIIB activity of compounds 333–350.
| Compound No. | R | X | N | EC50 (μM) |
|---|---|---|---|---|
| 333 | H | Cl | 1 | 50.61 ± 0.01 |
| 334 | 6-CH3 | Cl | 1 | 5.54 ± 0.01 |
| 335 | 6-OCH3 | Cl | 1 | 1.96 ± 0.004 |
| 336 | 7-CH3 | Cl | 1 | 0.72 ± 0.007 |
| 337 | 8-CH3 | Cl | 1 | 0.148 ± 0.006 |
| 338 | H | Cl | 2 | 2.92 ± 0.004 |
| 339 | 6-CH3 | Cl | 2 | 22.76 ± 0.002 |
| 340 | 6-OCH3 | Cl | 2 | 1.01 ± 0.01 |
| 341 | 7-CH3 | Cl | 2 | 2.55 ± 0.004 |
| 342 | 7-OCH3 | Cl | 2 | 0.46 ± 0.002 |
| 343 | 7-OCH(CH3)2 | Cl | 2 | 0.33 ± 0.001 |
| 344 | 8-CH3 | Cl | 2 | 0.50 ± 0.004 |
| 345 | H | OCH3 | 1 | 1.41 ± 0.008 |
| 346 | 6-OCH3 | OCH3 | 1 | 0.27 ± 0.003 |
| 347 | H | OCH3 | 2 | 0.42 ± 0.002 |
| 348 | 6-OCH3 | OCH3 | 2 | 1.51 ± 0.002 |
| 349 | – | – | 1 | 1.52 ± 0.004 |
| 350 | – | – | 2 | 1.93 ± 0.005 |
| NB-64 (II) | 2.4 ± 0.008 |
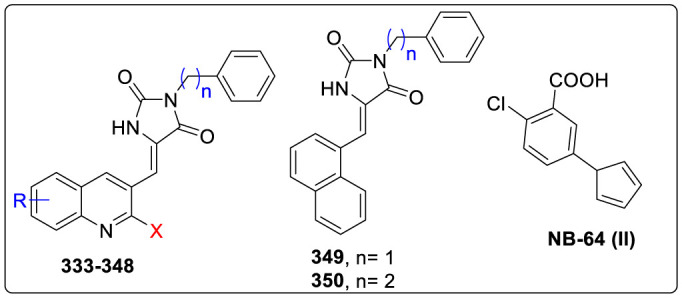
3.11. Severe acute respiratory syndrome-coronavirus infection (SARS-CoV 2)
COVID-19 is serious, deadly and infectious disease caused by severe acute respiratory syndrome coronavirus 2. It has posed a serious threat to global public health and local economies bringing the whole world to a standstill. Very recently, it has been made responsible for causing millions of deaths globally. Currently, there is no vaccine or any specific target available for this disease which is an issue of concern. However, every country’s research scientists are making efforts for the development of a drug molecule against COVID-19 as well as repurposing the already available drugs to be effective in the viral treatment. Thus, Wang and coworkers have reported two drugs, remdesivir (GS-5734) and chloroquine (CQ) to be active against causing inhibition to SARS-CoV-2 infection in vitro [117]. Recently, remdesivir was able to improve the clinical condition of the first patient infected by COVID-19 in USA. CQ, (N4-(7-Chloro-4-quinolinyl)-N1,N1-diethyl-1,4-pentanediamine) has been employed for treating malaria and amoebiasis but is associated with drug resistance. Thus, hydroxychloroquine (HCQ) sulfate was developed and was found to less toxic than CQ in animals. Owing to their structural similarity and mechanism of action as a weak base and immunomodulator, it is better to utilize HCQ for treating SARS-CoV-2 infection as shown in Fig. 30 .
Fig. 30.

Active molecules against SARS-CoV-2 infection.
The antiviral effect of HCQ and CQ was investigated against SARS-CoV-2 infection in vitro using Vero E6 cells and cytotoxcity was measured by standard CCK8 assay. The results were found to be similar for both the agents with CC50 values of 273.20 and 249.50 μM respectively. The MOIs (multiplicity of infection) at 50% maximal concentration was found to be lower for CQ than that of HCQ. On the contrary when immunofluorescence microscopy was done at certain MOIs, HCQ seemed to be less potent than CQ. Time-of-addition experiment revealed that HCQ was able to effectively inhibit the entry as well as post entry stages of SAR-CoV-2 which was also similar to CQ. For better understanding the mechanism of action of CQ and HCQ, immunofluorescence analysis (IFA) and confocal microscopy was conducted, thereby suggesting that both CQ and HCQ were able to block the transports of SAR-CoV-2 from EEs (early endosomes) to ELs (endolysosomes) which is crucial requirement for releasing the viral genome in SAR-CoV-2. Thus, from several studies, it was concluded that HCQ can efficiently inhibit the viral infection in vitro however; it should undergo clinical trials before any further confirmation.
4. Natural products bearing quinoline core
Several natural products consisting of quinoline skeleton are found to bioactive against numerous viral infections. The versatility of quinoline and its derivatives have gained much needed attention in the area of drug development [118,119]. Naturally derived quinolone alkaloid, Uranidine 351 is known to inhibit the RNA-directed DNA synthesis of the reverse transcriptase (RTs) of the HIV-1 and HIV-2 viruses, with the 3-hydroxy-4-oxo system being a crucial element for the inhibitory activity [120]. A gram-negative marine bacterial strain of Pseudomonas sp., 2-undecyl-4(1H)-quinolone has been found to be active against HIV-1 infection [121]. However, furoquinoline alkaloids, γ-fagarine 352, haplopine 353, and (+)-platydesmine 354, as well as 4-methoxy-1-methylquinolin-2-one 355 are known to inhibit HIV-1 replication in H9 lymphocyte cells at low concentrations (EC50 < 5.85 μM) without significantly affecting the growth of uninfected H9 cells [122]. In addition, compound 352 showed the best therapeutic index, while 353, 354, and 355 were less effective. Buchapine [356, 3-(1,1-dimethylallyl)-3-(3-methylbut-2-enyl)-1H-quinoline-2,4-dione] and 3-prenyl-4-prenyloxy-1H-quinolin-2-one 357 obtained from E. roxburghiana were found to be active against infectious HIV-1 (EC50 = 0.940 and 1.64 μM, respectively) in human lymphoblastoid host cells (cell growth IC50 = 29 and 26.9 μM, respectively). They also possessed inhibitory activity in an HIV-1 RT assay (IC50 12 and 8 μM, respectively) [123]. Furthermore, quinoline-containing decadepsipeptides significantly inhibit HIV-1 RT, alongwith a noticeable cytotoxic activity against tumor cell lines. Various modified derivatives of sandramycin 358 (HIV RT IC50 = 0.13 nM) retained their HIV potency, but exhibited 150–1000 fold less cytotoxic activity [124]. In addition to inhibitors of HIV RT, three other decadepsipeptides luzopeptins A-C have been identified for the emerging clinical resistance to recently introduced RT inhibitors [125,126].
Virantmycin 359 inhibited various RNA and DNA viruses, including the Indiana strain of vesicular stomatitis virus, EgyptAr 339 strain of Sindbis virus, MCMILLAN strain of Western equine encephalitis virus, MIYADERA strain of Newcastle disease virus, DIE strain of vaccinia virus, IHD strain of vaccinia virus, HF strain of herpes simplex virus type 1, and UW strain of herpes simplex virus type 2 at very low concentrations [127,128]. The compounds were known to affect the cell membranes, including specific virus receptor sites, and suppressed viral replication at a very early stage. Additionally, compound 359 showed excellent growth inhibition of influenza virus [129], 2-(3,4-Methylenedioxyphenethyl)quinoline 124, chimanine D 122, 2-pentylquinoline 120, and 2-nproplyquinoline 119 from G. longiflora inhibited the growth of cells infected with human T-lymphotropic virus type 1 (HLTV-1) [[130], [131], [132]]. Evolitrine 36 and dictamnine 40 inhibited activation of Epstein-Barr virus early antigen in Raji cells [133]. Various quinoline based natural products are shown in Fig. 31 .
Fig. 31.

Natural quinoline alkaloids with antiviral activity.
5. Conclusion
Based on our interest on medicinal [[134], (a), (b), (c)] and organic Chemistry [(l), [135], (a), (b), (c), (d), (e), (f), (g), (h), (i), (j), (k)], we summarized different quinoline derivatives exhibiting broad spectrum anti-viral activity although quinolines are versatile molecules emerging out to be potent for different diseases. Various classical methods have been developed for synthesis of quinolines and related scaffolds such as Skraup synthesis, Combes reaction but still there is a need to develop better, efficient and versatile synthetic protocol. Based on activity against various viral infections of quinolines compiled in the review, it can be ensured that it will emerge as a potent skeleton against several viruses. Finally, we expect that this scaffold will be a promising lead for the development of drug molecules which will be beneficial for human society especially for viral infection.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We acknowledge the faculty members and management of SVKM’s NMIMS, University for their guidance and support.
References
- 1.Marella A., Tanwar O.P., Saha R., Ali M.R., Srivastava S., Akhter M., Shaquiquzzaman M., Alam M.M. Quinoline: a versatile heterocyclic. Saudi Pharmaceut. J. 2013;21:1–12. doi: 10.1016/j.jsps.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Afzal O., Kumar S., Haider M.R., Ali M.R., Kumar R., Jaggi M., Bawa S. A review on anticancer potential of bioactive heterocycle quinoline, Eur. J. Med. Chem. 2015;97:871–910. doi: 10.1016/j.ejmech.2014.07.044. [DOI] [PubMed] [Google Scholar]
- 3.Kumar S., Bawa S., Gupta H. Biological activities of quinoline derivatives. Mini Rev. Med. Chem. 2009;9:1648–1654. doi: 10.2174/138955709791012247. [DOI] [PubMed] [Google Scholar]
- 4.Kaur K., Jain M., Reddy R.P., Jain R. Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem. 2010;45:3245–3264. doi: 10.1016/j.ejmech.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 5.Aribi F., Panossian A., Vors J.-P., Pazenok S., Leroux F.R. 2,4-Bis(fluoroalkyl)quinoline-3-carboxylates as tools for the development of potential agrochemical ingredients. Eur. J. Org Chem. 2018;2018:3792–3802. [Google Scholar]
- 6.Caeiro G., Lopes J., Magnoux P., Ayrault P., Ramoaribeiro F. A FT-IR study of deactivation phenomena during methylcyclohexane transformation on H-USY zeolites: nitrogen poisoning, coke formation, and acidity-activity correlations. J. Catal. 2007;249:234–243. [Google Scholar]
- 7.(a) Jiang B., Ning X., Gong S., Jiang N., Zhong C., Lu Z.-H., Yang C. Highly efficient red iridium(III)complexes cyclometalated by 4-phenylthieno[3,2-c]quinoline ligands for phosphorescent OLEDs with external quantum efficiencies over 20% J. Mater. Chem. 2017;C5:10220–10224. [Google Scholar]; (b) Mandapati P., Braun J.D., Killeen C., Davis R.L., Williams J.A.G., Herbert D.E. Luminescent platinum(II) complexes of N’N’N amino ligands with benzoannulated N-heterocyclic donor arms: quinolines offer unexpectedly deeper red phosphorescence than phenanthridines. Inorg. Chem. 2019;58:14808–14817. doi: 10.1021/acs.inorgchem.9b02480. [DOI] [PubMed] [Google Scholar]
- 8.(a) Bharate J.B., Vishwakarma R.A., Bharate S.B. Metal-free domino one-pot protocols for quinoline synthesis. RSC Adv. 2015;5:42020–42053. [Google Scholar]; (b) Ramann G., Cowen B. Recent advances in metal-free quinoline synthesis. Molecules. 2016;21:986–1009. doi: 10.3390/molecules21080986. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sharma R., Kour P., Kumar A. A review on transition-metal mediated synthesis of quinolines, J. Chem. Sci. 2018;130:73–98. [Google Scholar]
- 9.(a) Prajapati S.M., Patel K.D., Vekariya R.H., Panchal S.N., Patel H.D. Recent advances in the synthesis of quinolines: a review. RSC Adv. 2014;4:24463–24476. [Google Scholar]; (b) Yamamoto Y. Synthesis of heterocycles via transition-metal-catalyzed hydroarylation of alkynes. Chem. Soc. Rev. 2014;43:1575–1600. doi: 10.1039/c3cs60369e. [DOI] [PubMed] [Google Scholar]; (c) Barluenga J., Rodriguez F., Fananas F.J. Recent advances in the synthesis of indole and quinoline derivatives through cascade reactions. Chem. Asian J. 2009;4:1036–1048. doi: 10.1002/asia.200900018. [DOI] [PubMed] [Google Scholar]; (d) D’Souza D.M., Mueller T.J. Multi-component syntheses of heterocycles by transition-metal catalysts. Chem. Soc. Rev. 2007;36:1095–1108. doi: 10.1039/b608235c. [DOI] [PubMed] [Google Scholar]
- 10.Das S., Maiti D., De Sarkar S. Synthesis of polysubstituted quinolines from α-2-aminoarylalcohols via nickel-catalyzed dehydrogenative coupling. J. Org. Chem. 2018;83:2309–2316. doi: 10.1021/acs.joc.7b03198. [DOI] [PubMed] [Google Scholar]
- 11.Das S., Sinha S., Jash U., Sikari R., Saha A., Barman S.K., Brandao P., Paul N.D. Redox-induced interconversion and ligand-centered hemilability in NiIIcomplexes of redox-noninnocent azo-aromatic pincers. Inorg. Chem. 2018;57:5830–5841. doi: 10.1021/acs.inorgchem.8b00231. [DOI] [PubMed] [Google Scholar]
- 12.Niewahner J.H., Walters K.A., Wagner A. Improved synthesis of geodken’s macrocycle through the synthesis of the dichloride salt. J. Chem. Educ. 2007;84:477–479. [Google Scholar]
- 13.Chakraborty G., Sikari R., Das S., Mondal R., Sinha S., Banerjee S., Paul N.D. Dehydrogenative synthesis of quinolines, 2-aminoquinolines and quinazolines using singlet diradical Ni(II)-catalysts. J. Org. Chem. 2019;84:2626–2641. doi: 10.1021/acs.joc.8b03070. [DOI] [PubMed] [Google Scholar]
- 14.Sikari R., Sinha S., Jash U., Das S., Brandao P., deBruin B., Paul N.D. Deprotonation induced ligan oxidation in a NiII complex of a redox noninnocent N1-(2-aminophneyl)benzene-1,2-diamine and its use in catalytic alcohol oxidation. Inorg. Chem. 2016;55:6114–6123. doi: 10.1021/acs.inorgchem.6b00646. [DOI] [PubMed] [Google Scholar]
- 15.Xiong B., Wang Y., Liu Y., Bao Y., Liu Z., Zhang Y., Ling Y. Straightforward synthesis of quinolines from enones and 2-aminobenzyl alcohols using an iridium-catalyzed transfer hydrogenative strategy. Org. Biomol. Chem. 2018;16:5707–5711. doi: 10.1039/c8ob01321g. [DOI] [PubMed] [Google Scholar]
- 16.Genc S., Arslan B., Gulcemal S., Gunnaz S., Cetinkaya B., Gulcemal D. Iridium(I)-catalyzed C-C and C-N bond formation reactions via the borrowing hydrogen strategy. J. Org. Chem. 2019;84:6286–6297. doi: 10.1021/acs.joc.9b00632. [DOI] [PubMed] [Google Scholar]
- 17.Jia R., Li B., Liang R., Zhang X., Fan X. Tunable synthesis of indolo[3,2-c]quinolines or 3-(2-aminophenyl)quinolines via aerobic/anaerobic dimerization of 2-alkynylanilines. Org. Lett. 2019;21:4996–5001. doi: 10.1021/acs.orglett.9b01534. [DOI] [PubMed] [Google Scholar]
- 18.Kumar P., Garg V., Kumar M., Verma A.K. Rh(III)-catalyzed alkynylation: synthesis of functionalized quinolines from aminohydrazones. Chem. Commun. 2019;55:12168–12171. doi: 10.1039/c9cc06205j. [DOI] [PubMed] [Google Scholar]
- 19.Panday A.K., Mishra R., Jana A., Parvin T., Choudhury L.H. Synthesis of pyrimidine fused quinolines by ligand-free copper-catalyzed domino reactions. J. Org. Chem. 2018;83:3624–3632. doi: 10.1021/acs.joc.7b03272. [DOI] [PubMed] [Google Scholar]
- 20.Wu W., Guo Y., Xu X., Zhou Z., Zhang X., Wua Bo, Yia W. One-pot regioselective synthesis of 2,4-disubstituted quinolines via copper(II)-catalyzed cascade annulations. Org. Chem. Front. 2018;5:1713–1718. [Google Scholar]
- 21.Xu J., Chen Q., Luo Z., Tang X., Zhao J. N-heterocyclic carbene copper catalyzed quinoline synthesis from 2-aminobenzyl alcohols and ketones using DMSO as an oxidant at room temperature. RSC Adv. 2019;9:28764–28767. doi: 10.1039/c9ra04926f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu X., Yang Y., Zhang X., Yi W. Direct synthesis of quinolines via Co(III)-catalyzed and DMSO-involved C-H activation/cyclization of anilines with alkynes. Org. Lett. 2018;20:566–569. doi: 10.1021/acs.orglett.7b03673. [DOI] [PubMed] [Google Scholar]
- 23.Li J., Tan E., Keller N., Chen Y.-H., Zehetmaier P.M., Jakowetz A.C., Bein T., Knochel P. Cobalt-catalyzed electrophilic aminations with anthranils: an expedient route to condensed quinolines. J. Am. Chem. Soc. 2019;141:98–103. doi: 10.1021/jacs.8b11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piller F.M., Appukkuttan P., Gavryushin A., Helm M., Knochel P. Convenient preparation of polyfunctional aryl magnesium reagents by a direct magnesium insertion in the presence of LiCl. Angew. Chem. Int. Ed. 2008;47:6802–6806. doi: 10.1002/anie.200801968. [DOI] [PubMed] [Google Scholar]
- 25.Uzelac M., Kennedy A.R., Hevia E., Mulvey R.E. Transforming LiTMP lithiation of challenging diazines through gallium alkyl trans-metal-trapping. Angew. Chem. Int. Ed. 2016;55:13147–13150. doi: 10.1002/anie.201607284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stathakis C.I., Manolikakes S.M., Knochel P. TMPZnOPiv.LiCl: a new base for the preparation of air-stable solid zinc pivalates of sensitive aromatics and heteroaromatics. Org. Lett. 2013;15:1302–1305. doi: 10.1021/ol400242r. [DOI] [PubMed] [Google Scholar]
- 27.Mehedi M.S.A., Tepe J.J. Sc(OTf)3-Mediated one-pot synthesis of 2,3-disubstituted quinolines from anilines and epoxides. J. Org. Chem. 2020;85:6741–6746. doi: 10.1021/acs.joc.0c00803. [DOI] [PubMed] [Google Scholar]
- 28.Qiu Y.-F., Niu Y.-J., Wei X., Cao B.-Q., Wang X.-C., Quan Z.-J. AgSCF3/Na2S2O8-Promoted trifluoromethylthiolation/cyclization of o-propargyl arylazides/o-alkynyl benzylazides: synthesis of SCF3-substituted quinolines and isoquinolines. J. Org. Chem. 2019;84:4165–4178. doi: 10.1021/acs.joc.9b00181. [DOI] [PubMed] [Google Scholar]
- 29.Patil M.D., Liu R.-S. Direct access to benzofuro[2,3-b] quinoline and 6H-chrome no[3,4-b]quinoline cores through gold-catalyzed annulations of anthranils with arenoxyethynes and aryl propargyl ethers. Org. Biomol. Chem. 2019;17:4452–4455. doi: 10.1039/c9ob00468h. [DOI] [PubMed] [Google Scholar]
- 30.Xu T., Shao Y., Dai L., Yu S., Cheng T., Chen J. Pd-catalyzed tandem reactions of 2-aminostyryl nitriles with arylboronic acids: synthesis of 2-arylquinolines. J. Org. Chem. 2019;84:13604–13614. doi: 10.1021/acs.joc.9b01875. [DOI] [PubMed] [Google Scholar]
- 31.Ren X., Han S., Gao X., Li J., Zou D., Wu Y., Wu Y. Direct arylation for the synthesis of 2-arylquinolines from N-methoxyquinoline-1-ium tetrafluoroborate salts and arylboronic acids. Tetrahedron Lett. 2018;59:1065–1068. [Google Scholar]
- 32.Ahmed W., Zhang S., Yu X., Yamamoto Y., Bao M. Brnsted acid-catalyzed metal-and solvent-free quinoline synthesis from N-alkylanilines and alkynes or alkenes. Green Chem. 2018;20:261–265. [Google Scholar]
- 33.Azizi M., Esfahani M.N., Baltork I.M., Moghadam M., Mirkhani V., Tangestaninejad S., Kia R. Synthesis of quinolines and pyrido[3,2-g or 2,3-g]quinolines catalyzed by heterogeneous propylphosphonium tetrachlorindate ionic liquid. J. Org. Chem. 2018;83:14743–14750. doi: 10.1021/acs.joc.8b02261. [DOI] [PubMed] [Google Scholar]
- 34.Beesu M., Mehta G. Synthesis of quinolines and isoquinolines via site-selective, domino benzannulation of 2- and 3-chloropyridyl ynones with nitromethane. J. Org. Chem. 2019;84:8731–8742. doi: 10.1021/acs.joc.9b00950. [DOI] [PubMed] [Google Scholar]
- 35.Phanindrudu M., Wakade S.B., Tiwari D.K., Likhar P.R., Tiwari D.K. Transition-metal-free approach for the synthesis of 4-arylquinolines from alkynes and anilines. J. Org. Chem. 2018;83:9137–9143. doi: 10.1021/acs.joc.8b01204. [DOI] [PubMed] [Google Scholar]
- 36.Naruto H., Togo H. Preparation of 2-arylquinolines from β-arylpropionitriles with aryllithiums and NIS through iminyl radical-mediated cyclization. Org. Biomol. Chem. 2019;17:5760–5770. doi: 10.1039/c9ob00944b. [DOI] [PubMed] [Google Scholar]
- 37.Zhao P., Wu X., Zhou Y., Geng X., Wang C., Wu Y.-D., Wu A.-X. Direct synthesis of 2,3-diaroyl quinolines and pyridazino[4,5-b]quinolines and I2-promoted one-pot multicomponent reaction. Org. Lett. 2019;21:2708–2711. doi: 10.1021/acs.orglett.9b00685. [DOI] [PubMed] [Google Scholar]
- 38.Patra A., Gelat F., Pannecoucke X., Poisson T., Besset T., Biju A.T. Synthesis of 4-difluoromethylquinolines by NHC-catalyzed umpolung of imines. Org. Lett. 2018;20:1086–1089. doi: 10.1021/acs.orglett.7b04055. [DOI] [PubMed] [Google Scholar]
- 39.Lan X.-B., Ye Z., Huang M., Liu J., Liu Y., Ke Z. Nonbifunctional outer-sphere strategy achieved highly active α-alkylation of ketones with alcohols by N-heterocyclic carbene manganes (NHC-Mn) Org. Lett. 2019;21:8056–8070. doi: 10.1021/acs.orglett.9b03030. [DOI] [PubMed] [Google Scholar]
- 40.Wang B.-Q., Zhang C.-H., Tian X.-X., Lin J., Yan S.-J. Cascade reactions of isatins with 1,1-enediamines: synthesis of multisubstituted quinoline-4-carboxamides. Org. Lett. 2018;20:660–663. doi: 10.1021/acs.orglett.7b03803. [DOI] [PubMed] [Google Scholar]
- 41.Lee S.Y., Chen C.-H. On-water synthesis of 2-substituted quinolines from 2-aminochalcones using benzylamines as the nucleophilic catalyst. J. Org. Chem. 2018;83:13036–13044. doi: 10.1021/acs.joc.8b01675. [DOI] [PubMed] [Google Scholar]
- 42.D. Bhardwaj, A. Singh, R. Singh, Eco-compatible sonochemical synthesis of 8-aryl-7,8-dihydro[1,3]-dioxolo[4,5-g]quinolin-6(5H)-ones using green TiO2, Heliyon 5 (2019) 1256-1280. [DOI] [PMC free article] [PubMed]
- 43.Patel D.M., Patel H.M. Trimethylglycine-betaine-based-catalyst-promoted novel and ecocompatible pseudo-four-component reaction for regioselective synthesis of functionalized 6,8-dihydro-1’H,5H-spiro[[1,3]dioxolo[4,5-g]quinoline-7,5’-pyrimidine]-2’,4’,6’(3’H)-trione derivatives. ACS Sustain. Chem. Eng. 2019;7:18667–18676. [Google Scholar]
- 44.Chen L., Huang R., Kong L.-B., Lin J., Yan S.-J. Facile route to the synthesis of 1,3-diazahetero-cycle-fused [1,2-a]quinoline derivatives via cascade reactions. ACS Omega. 2018;3:1126–1136. doi: 10.1021/acsomega.7b01856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guan Z.-R., Liu Z.-M., Ding M.-W. New efficient synthesis of 1H-imidazo-[4,5-c] quinolines by a sequential Van Leusen/Staudinger/aza-Wittig/carbodiimide-mediated cyclization. Tetrahedron. 2018;74:7186–7192. [Google Scholar]
- 46.Diaz J., Rodenas D., Ballester F.-J., Alajarin M., Orenes R.-A., Andrada P.S., Vidal A. Unlocking the synthetic potential of aziridine and cyclopropane-fused quinolin-2-ones by regioselective fragmentation of its three membered rings. Arab. J. Chem. 2020;13:2702–2714. [Google Scholar]
- 47.Woolhouse M., Scott F., Hudson Z., Howey R., Topping M.C. Human viruses: discovery and emergence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012;367:2864–2871. doi: 10.1098/rstb.2011.0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dick G.W.A., Kitchen S.F., Haddow A.J. Zika virus. I. isolations and serological specificity. Trans. Roy. Soc. Trop. Med. Hyg. 1952;46:509–520. doi: 10.1016/0035-9203(52)90042-4. [DOI] [PubMed] [Google Scholar]
- 49.Rasmussen S.A., Jamieson D.J., Honein M.A., Petersen L.R. Zika virus and birth defects-reviewing the evidence for casuality. N. Engl. J. Med. 2016;374:1981–1987. doi: 10.1056/NEJMsr1604338. [DOI] [PubMed] [Google Scholar]
- 50.Osuna C.E., Lim S., Deleage C., Griffin B.D., Stein D., Schroeder L.T., Omange R., Best K., Luo M., Hraber P.T., Andersen-Elyard H., Ojeda E.F.C., Huang S., Vanlandingham D.L., Higgs S., Perelson A.S., Estes J.D., Safronetz D., Lewis M.G., Whitney J.B. Zika viral dynamics and shedding in rhesus and cynomolgus macaques. Nat. Med. 2016;22:1448–1455. doi: 10.1038/nm.4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haddow A.D., Schuh A.J., Yasuda C.Y., Kasper M.R., Heang V., Huy R., Guzman H., Tesh R.B., Weaver S.C. Genetic characterization of zika virus strains: geographic expansion of the asian lineage. PLoS Neglected Trop. Dis. 2012;6:e1477–e1484. doi: 10.1371/journal.pntd.0001477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.WHO . WHO; 2016. WHO | WHO Announces a Public Health Emergency of International Concern.http://www.who.int/kobe_centre/mediacentre/ZIKA/en/ accessed September 29, 2016. [Google Scholar]
- 53.Brasil P., Sequeira P.C., Freitas A.D., Zogbi H.E., Calvet G.A., de Souza R.V., Siqueira A.M., de Mendonca M.C.L., Nogueira R.M.R., de Filippis A.M.B., Solomon T. Guillain-Barré syndrome associated with zika virus infection. Lancet. 2016;387 doi: 10.1016/S0140-6736(16)30058-7. 1482–1482. [DOI] [PubMed] [Google Scholar]
- 54.Wahid B., Ali A., Rafique S., Idrees M. Current status of therapeutic and vaccine approaches against zika virus. Eur. J. Intern. Med. 2017;44:12–18. doi: 10.1016/j.ejim.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 55.Saiz J., Martín-Acebes M.A. The race to find antivirals for zika virus, Antimicrob. Agents Chemother. 2017;61:e00411–e00417. doi: 10.1128/AAC.00411-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barbosa-Lima G., Moraes A.M., Araújo A. da S., da Silva E.T., De Freitas C.S., Vieira Y.R., Marttorelli A., Neto J.C., Bozza P.T., de Souza M.V.N., Souza T.M.L. 2,8-bis(trifluoromethyl)quinoline analogs show improved anti-zika virus, compared to mefloquine. Eur. J. Med. Chem. 2017;127:334–340. doi: 10.1016/j.ejmech.2016.12.058. [DOI] [PubMed] [Google Scholar]
- 57.Hamann A.R., de Kock C., Smith P.J., van Otterlo W.A.L., Blackie M.A.L. Synthesis of novel triazole-linked mefloquine derivatives: biological evaluation against plasmodium falciparum. Bioorg. Med. Chem. Lett. 2014;24:5466–5469. doi: 10.1016/j.bmcl.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 58.Sumangala V., Poojary B., Chidananda N., Fernandes J., Kumari N.S. Synthesis and antimicrobial activity of 1,2,3-triazoles containing quinoline moiety. Arch Pharm. Res. (Seoul) 2010;33:1911–1918. doi: 10.1007/s12272-010-1204-3. [DOI] [PubMed] [Google Scholar]
- 59.Jayaprakash S., Iso Y., Wan B., Franzblau S.G., Kozikowski A.P. Design, synthesis and SAR studies of mefloquine-based ligands as potential antituberculosis agents. ChemMedChem. 2006;1:593–597. doi: 10.1002/cmdc.200600010. [DOI] [PubMed] [Google Scholar]
- 60.Kgokong J.L., Wachira J.M. Cytotoxicity of novel trifluoromethylquinoline derivatives on human leukemia cells. Eur. J. Pharmaceut. Sci. 2001;12:369–376. doi: 10.1016/s0928-0987(00)00137-8. [DOI] [PubMed] [Google Scholar]
- 61.Briguglio I., Loddo R., Laurini E., Fermeglia M., Piras S., Corona P., Giunchedi P., Gavini E., Sanna G., Giliberti G., Ibba C., Farci P., La Colla P., Pricl S., Carta A. Synthesis, cytotoxicity and antiviral evaluation of nw series of imidazo[4,5-g]quinoline and pyrido[2,3-g]quinoxaline derivatives. Eur. J. Med. Chem. 2015;105:63–79. doi: 10.1016/j.ejmech.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 62.Shin J.-W., Jung K.-H., Lee S.-T., Moon J., Lim J.-A., Byun J.-I., Park K.-I., Lee S.K., Chu K. Mefloquine improved progressive multifocal leukoencephalopathy in a patient with immunoglobulin A nephropathy. J. Clin. Neurosci. 2014;21:1661–1664. doi: 10.1016/j.jocn.2013.12.031. [DOI] [PubMed] [Google Scholar]
- 63.Barrows N.J., Campos R.K., Powell S.T., Prasanth K.R., Schott-Lerner G., Soto-Acosta R., Galarza-Muñoz G., McGrath E.L., Urrabaz-Garza R., Gao J., Wu P., Menon R., Saade G., Fernandez-Salas I., Rossi S.L., Vasilakis N., Routh A., Bradrick S.S., Garcia-Blanco M.A. A screen of FDA-approved drugs for inhibitors of zika virus infection, Cell Host Microbe. 2016;20:259–270. doi: 10.1016/j.chom.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eswaran S., Adhikari A.V., Chowdhury I.H., Pal N.K., Thomas K.D. New quinoline derivatives: synthesis and investigation of antibacterial and antituberculosis properties. Eur. J. Med. Chem. 2010;45:3374–3383. doi: 10.1016/j.ejmech.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 65.Yan Y.Y., Shi G.X., Shao J., Wang T.M., Wang C.Z. Advance in studies on anti-infection of andrographolide and ts derivatives in past 10 years. Zhongguo Zhongyao Zazhi. 2013:3819–3824. [PubMed] [Google Scholar]
- 66.Chinese Pharmacopoeia Commission Chuanhuning (article in Chinese) Chin. Pharmacopoeia. 2010;Part 2:619–620. [Google Scholar]
- 67.Basak A., Cooper S., Roberge A.G., Banik U.K., Chretien M., Seidah N.G. Inhibition of proprotein convertases-1, -7 and furin by diterpines of andrographis paniculata and their succinoyl esters. Biochem. J. 1999;338:107–113. [PMC free article] [PubMed] [Google Scholar]
- 68.Chang R.S., Ding L., Chen G.Q., Pan Q.C., Zhao Z.L., Smith K.M. Dehydroandrographolide succinic acid monoester as an inhibitor against the human immunodeficiency virus. Proc. Soc. Exp. Biol. Med. 1991;197:59–66. doi: 10.3181/00379727-197-43225. [DOI] [PubMed] [Google Scholar]
- 69.Edwin E., Vasantha-Srinivasan P., Senthil-Nathan S., Thanigaivel A., Ponsankar A., Pradeepa V., Selin-Rani S., Kalaivani K., Hunter W.B., Abdel-Megeed A., Duraipandiyan V., Al-Dhabi N.A. Anti-dengue efficacy of bioactive andrographolide from Andrographis paniculata against the primary dengue vector aedes aegypti. Acta Trop. 2016;163:167–178. doi: 10.1016/j.actatropica.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 70.Panraksa P., Ramphan S., Khongwichit S., Smith D.R. Activity of andrographolide against dengue virus. Antivir. Res. 2017;139:69–78. doi: 10.1016/j.antiviral.2016.12.014. [DOI] [PubMed] [Google Scholar]
- 71.Li F., Li X., Sheng D., Chen S., Nie X., Liu Z., Wang D., Zhao Q., Wang Y., Wang Y., Zhou G. Discovery and preliminary SAR of 14-aryloxy-andrographolide derivatives as antibacterial agents with immunosuppressants activity. RSC Adv. 2018;8:9440–9456. doi: 10.1039/c8ra01063c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee E.M., Titus S.A., Xu M., Tang H., Zheng W. High-throughput zika viral titer assay for rapid screening of antiviral drugs. Assay Drug Dev. Technol. 2019;17:128–139. doi: 10.1089/adt.2018.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu M., Lee E.M., Wen Z., Cheng Y., Huang W., Qian X., TCW J., Kouznetsova J., Ogden S.C., Hammack C., Jacob F., Nguyen H.N., Itkin M., Hanna C., Shinn P., Allen C., Michael S.G., Simeonov A., Huang W., Christian K.M., Goate A., Brennand K.J., Huang R., Xia M., Ming G., Zheng W., Song H., Tang H. Identification of small-molecule inhibitors of zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016;22:1101–1107. doi: 10.1038/nm.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hurst B.L., Evans W.J., Smee D.F., Van Wettere A., Tarbet E.B. Virology. 2019;526:146–154. doi: 10.1016/j.virol.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hixon A.M., Clarke P., Tyler K.L. Evaluating treatment efficacy in a mouse model of enterovirus D68-associated paralytic myelitis. J. Infect. Dis. 2017;216:1245–1253. doi: 10.1093/infdis/jix468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Musharrafieh R., Zhang J., Tuohy P., Kitamura N., Bellampalli S.S., Hu Y., Khanna R., Wang J. Discovery of quinoline analogues as potent antivirals against enterovirus D68. J. Med. Chem. 2019;62:4074–4090. doi: 10.1021/acs.jmedchem.9b00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Magden J., Kaariainen L., Ahola T. Inhibitors of virus replication: recent developments and prospects. Appl. Microbiol. Biotechnol. 2005;66:612–621. doi: 10.1007/s00253-004-1783-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu S., Neidhardt E.A., Grossman T.H., Ocain T., Clardy J. Structure of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure. 2000;8:25–33. doi: 10.1016/s0969-2126(00)00077-0. [DOI] [PubMed] [Google Scholar]
- 79.Phillips M.A., Rathod P.K. Plasmodium dihyroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect. Disord. - Drug Targets. 2010;10:226–239. doi: 10.2174/187152610791163336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gustafson G., Davis G., Waldron C., Smith A., Henry M. Identification of a new antifungal target site through a dual biochemical and molecular genetics approach. Curr. Genet. 1996;30:159–165. doi: 10.1007/s002940050115. [DOI] [PubMed] [Google Scholar]
- 81.Das P., Deng X., Zhang L., Roth M.G., Fontoura B.M.A., Phillips M.A., De Brabander J.K. SAR-based optimization of a 4-quinoline carboxylic acid analogue with potent antiviral activity. ACS Med. Chem. Lett. 2013;4:517–521. doi: 10.1021/ml300464h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Borchers A.T., Chang C., Gershwin M.E., Gershwin L.J. Respiratory syncytial virus-A comprehensive review. Clin. Rev. Allergy Immunol. 2013;45:331–379. doi: 10.1007/s12016-013-8368-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao X., Singh M., Malashkevich V.N., Kim P.S. Structural characterization of the human respiratory syncytial virus fusion protein core. Proc. Natl. Acad. Sci. U. S. A. 2000;97:14172–14177. doi: 10.1073/pnas.260499197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zheng X., Wang L., Wang B., Miao K., Xiang K., Feng S., Gao L., Shen H.C., Yun H. Discovery of piperazinylquinoline derivatives as novel respiratory syncytial virus fusion inhibitors. ACS Med. Chem. Lett. 2016;7:558–562. doi: 10.1021/acsmedchemlett.5b00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moore B.P., Chung D.H., Matharu D.S., Golden J.E., Maddox C., Rasmussen L., Noah J.W., Sosa M.I., Ananthan S., Tower N.A., White E.L., Jia F., Prisinzano T.E., Aubé J., Jonsson C.B., Severson W.E. (S)-N-(2,5-dimethylphenyl)-1-(quinoline-8-ylsulfonyl)pyrrolidine-2-carboxamide as a small molecule inhibitor probe for the study of respiratory synctial virus infection. J. Med. Chem. 2012;55:8582–8587. doi: 10.1021/jm300612z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cotten M., Watson S.J., Zumla AI A.I. Spread, circulation and evolution of the middle east respiratory syndrome coronavirus. mBio. 2014;5 doi: 10.1128/mBio.01062-13. e01062–e01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liang R., Wang L., Zhang N. Development of small-molecules MERS-CoV inhibitors. Viruses. 2018;10:721–745. doi: 10.3390/v10120721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yoona J.H., Leea J.Y., Leeb J., Shina Y.S., Jeonb S., Kimc D.E., Minc J.S., Songa J.H., Kimd S., Kwonc S., Jinc Y.-H., Jange M.S., Kima H.R., Parka C.M. Synthesis and biological evaluation of 3-acyl-2-phenylamino-1,4-dihydroquinolin-4(1H)-one derivatives as potential MERS-CoV inhibitors. Bioorg. Med. Chem. Lett. 2019;29:126727–126731. doi: 10.1016/j.bmcl.2019.126727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bhatt S., Gething P.W., Brady O.J., Messina J.P., Farlow A.W., Moyes C.L., Drake J.M., Brownstein J.S., Hoen A.G., Sankoh O. The global distribution and burden of dengue, Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sangkawibha N., Rojanasuphot S., Ahandrik S., Viriyapongse S., Jatanasen S., Salitul V., Phanthumachinda B., Halstead S.B. Risk factors in dengue shock syndrome: a prospective epidemiologic study in Rayong, Thailand. I. The 1980 outbreak. Am. J. Epidemiol. 1984;120:653–669. doi: 10.1093/oxfordjournals.aje.a113932. [DOI] [PubMed] [Google Scholar]
- 91.de la Guardia C., Stephens D.E., Dang H.T., Quijada M., Larionov O.V., Lleonart R. Antiviral activity of novel quinoline derivatives against dengue virus serotype 2. Molecules. 2018;23:672–683. doi: 10.3390/molecules23030672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu H., Wang W., Liu F., Weisberg E.L., Tian B., Chen Y., Li B., Wang A., Wang B., Zhao Z., McMillin D.W., Hu C., Li H., Wang J., Liang Y., Buhrlage S.J., Liang J., Liu J., Yang G., Brown J.R., Treon S.P., Mitsiades C.S., Griffin J.D., Liu Q., Gray N.S. Discovery of a potent, covalent BTK inhibitor for B-cell lymphoma. ACS Chem. Biol. 2014;9:1086–1091. doi: 10.1021/cb4008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liang Y., de Wispelaere M., Carocci M., Liu Q., Wang J., Yang P.L., Gray N.S. Structure-activity relationship study of QL47: a broad spectrum antiviral agent. ACS Med. Chem. Lett. 2017;8:344–349. doi: 10.1021/acsmedchemlett.7b00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chayavichitsilp P., Buckwalter J.V., Krakowski A.C., Friedlander S.F. Herpes simplex. Pediatr. Rev. 2009;30:119–130. doi: 10.1542/pir.30-4-119. [DOI] [PubMed] [Google Scholar]
- 95.Zhang X., Wang F., Zhang C., Wu S., Zheng X., Gonge T., Ding R., Chenf K., Baia D. Novel fluorinated platinum(II) complexes with pyridine-2-carboxylate ligands as radiosentisizer and antiviral agents. Inorg. Chem. Commun. 2018;94:92–97. [Google Scholar]
- 96.Baseler L., Chertow D.S., Johnson K.M., Feldmann H., Morens D.M. The pathogenesis of ebola virus disease. Annu. Rev. Pathol. 2017;12:387–418. doi: 10.1146/annurev-pathol-052016-100506. [DOI] [PubMed] [Google Scholar]
- 97.Sakuraia Y., Sakakibara N., Toyamad M., Babad M., Daveya R.A. Novel amodiaquine derivatives potently inhibit ebola virus infection. Antivir. Res. 2018;160:175–182. doi: 10.1016/j.antiviral.2018.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.World Health Organization Media centre. Hepatitis C. http://www.who.int/mediacentre/factsheets/fs164/en/ accessed May 17, 2013.
- 99.Talamas F.X., Abbot S.C., Anand S., Brameld K.A., Carter D.S., Chen J., Davis D., de Vicente J., Fung A.D., Gong L., Harris S.F., Inbar P., Labadie S.S., Lee E.K., Lemoine R., Le Pogam S., Leveque V., Li J., McIntosh J., Najera I., Park J., Railkar A., Rajyaguru S., Sangi M., Schoenfeld R.C., Staben L.R., Tan Y., Taygerly J.P., Villasenor A.G., Weller P.E. Discovery of N-[4-[6-tert-Butyl-5-methoxy-8-(6-methoxy-2-oxo-1H-pyridin-3-yl)-3-quinolyl]phenyl] methanesulfonamide (RG7109), a potent inhibitor of the hepatitis C virus NS5B polymerase. J. Med. Chem. 2014;57:1914–1931. doi: 10.1021/jm401329s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Adachi A., Gendelman H.E., Koenig S., Folks T., Willey R., Rabson A., Martin M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bedoyaa L.M., Abadb M.J., Calongea E., Saavedrac L.A., Gutierrez C M., Kouznetsov V.V., Alcamia J., Bermejo P. Quinoline-based compounds as modulators of HIV transcription through NF-ĸB and Sp1 inhibition. Antivir. Res. 2010;87:338–344. doi: 10.1016/j.antiviral.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 102.Bénard C., Zouhiri F., Normand-Bayle M., Danet M., Desmaële D., Leh H., Mouscadet J.-F., Mbemba G., Thomas C.-M., Bonnenfant S., Le Bretc M., d’Angelo J. Linker-modified quinoline derivatives targeting HIV-1 integrase: synthesis and biological activity. Bioorg. Med. Chem. Lett. 2004;14:2473–2476. doi: 10.1016/j.bmcl.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 103.Di Santo R., Costi R., Roux A., Artico M., Lavecchia A., Marinelli L., Novellino E., Palmisano L., Andreotti M., Amici R., Galluzzo C.M., Nencioni L., Palamara A.T., Pommier Y., Marchand C. Novel bifunctional quinolonyl diketo acid derivatives as HIV-1 integrase inhibitors: design, synthesis, biological activities and mechanism of action. J. Med. Chem. 2006;49:1939–1945. doi: 10.1021/jm0511583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Peng J., Zhu Y., Milton J.T., Price D.H. Identification of multiple cyclin subunits of human P-TEFb. Gene Dev. 1998;12:755–762. doi: 10.1101/gad.12.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wei P., Garber M.E., Fang S.M., Fischer W.H., Jones K.A. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 106.Price D.H. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell Biol. 2000;20:2629–2634. doi: 10.1128/mcb.20.8.2629-2634.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Matthew P.R., Lin H.C., Ram R.S., Emmanuel A., Arnold B.R., Ajit K. Inhibition of HIV-1 replication in viral mutants with altered TAR RNA stem structures. Virology. 1996;216:411–417. doi: 10.1006/viro.1996.0077. [DOI] [PubMed] [Google Scholar]
- 108.Baba M. Recent status of HIV-1 gene expression inhibitors. Antivir. Res. 2006;71:301–306. doi: 10.1016/j.antiviral.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 109.Hammy F., Brondani V., Flörsheimer A., Stark W., Blommers M., Klimkait T. A new class of HIV-1 tat antagonist acting through Tat-TAR inhibition. Biochemistry. 1998;37:5086–5095. doi: 10.1021/bi972947s. [DOI] [PubMed] [Google Scholar]
- 110.Chen S., Chen R., He M., Pang R., Tan Z., Yang M. Design, synthesis, and biological evaluation of novel quinoline derivatives as HIV-1 Tat-TAR interaction inhibitors. Bioorg. Med. Chem. 2009;17:1948–1956. doi: 10.1016/j.bmc.2009.01.038. [DOI] [PubMed] [Google Scholar]
- 111.Yaseen M.M., Abuharfeil N.M., Alqudah M.A., Yaseen M.M. Mechanism and factors that drive extensive human immunodeficiency virus type-1 hypervariablity: an overview. Viral Immunol. 2017;30:708–726. doi: 10.1089/vim.2017.0065. [DOI] [PubMed] [Google Scholar]
- 112.Forezi L. da S.M., Ribeiro M.M.J., Marttorelli A., Abrantes J.L., Rodrigues C.R., Castro H.C., Souza T.M.L., Boechat F. da C.S., de Souza A.M.T., Cecília M., de Souza B.V. Design, synthesis, in vitro and in silico studies of novel 4-oxoquinoline ribonucleoside derivatives as HIV-1 reverse transcriptase inhibitors. Eur. J. Med. Chem. 2020;194 doi: 10.1016/j.ejmech.2020.112255. [DOI] [PubMed] [Google Scholar]
- 113.Overackera R.D., Banerjeea S., Neuhausa G.F., Sephtona S.M., Herrmannb A., Strotherc J.A., Brack-Wernerc R., Blakemorea P.R., Loesg S. Biological evaluation of molecules of the azaBINOL class as antiviral agents: inhibition of HIV-1RNase H activity by 7-isopropoxy-8-(naphtha-1-yl)quinoline. Bioorg. Med. Chem. 2019;27:3595–3604. doi: 10.1016/j.bmc.2019.06.044. [DOI] [PubMed] [Google Scholar]
- 114.Zouhiri F., Danet M., Bénard C., Normand-Bayle M., Mouscadet J.-F., Leh H., Thomas C.M., Mbemba G., d’Angelo J., Desma D. HIV-1 replication inhibitors of the styrylquinoline class: introduction of an additional carboxyl group at the C-5 position of the quinoline. Tetrahedron Lett. 2005;46:2201–2205. [Google Scholar]
- 115.Ibrahima T.S., Bokhtia R.M., AL-Mahmoudy A.M.M., Taherd E.S., AlAwadha M.A., Elagawanye M., Abdel-Aalb E.H., Pandac S., Goudaf A.M., Asfourg H.Z., Alhakamyh N.A., Youssifi B.G.M. Design, synthesis and biological evaluation of novel 5-((substituted quinolin-3-yl/1-naphthyl) methylene)-3-substituted imidazolidin-2,4-dione as HIV-1 fusion inhibitors. Bioorg. Chem. 2020;99:103782–103791. doi: 10.1016/j.bioorg.2020.103782. [DOI] [PubMed] [Google Scholar]
- 116.Jiang S., Tala S.R., Lu H., Abo-Dya N.E., Avan I., Gyanda K., Lu L., Katritzky A.R., Debnath A.K. Design, synthesis, and biological activity of novel 5-((arylfuran/1H-pyrrol-2-yl) methylene)-2-thioxo-3-(3-(trifluoromethyl)phenyl)thiazolidin-4-ones as HIV-1 fusion inhibitors targeting gp41. J. Med. Chem. 2011;54:572–579. doi: 10.1021/jm101014v. [DOI] [PubMed] [Google Scholar]
- 117.Liu J., Cao R., Xu M., Wang X., Zhang H., Hu H., Li Y., Hu Z., Zhong W., Wang M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020;6:16–19. doi: 10.1038/s41421-020-0156-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shang X.-F., Morris-Natschke S.L., Liu Y.-Q., Guo X., Xu X.-S., Goto M., Li J.-C., Yang G.-Z., Lee K.-H. Biologicall active quinoline and quinazoline alkaloids part I. Med. Res. Rev. 2017:1–54. doi: 10.1002/med.21466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chung P.-Y., Bian Z.-X., Pun H.-Y., Chan D., Chan A.-C., Chui C.-H., Tang J.C.-O., Lam K.-H. Recent advances in research of natural and synthetic bioactive quinolines. Future Med. Chem. 2015;7:947–967. doi: 10.4155/fmc.15.34. [DOI] [PubMed] [Google Scholar]
- 120.Loya S., Rudi A., Tal R., Kashman Y., Loya Y., Hizi A. 3,5,8-Trihydroxy-4-quinolone, a novel natural inhibitor of the reverse transcriptases of human immunodeficiency viruses type 1 and type 2. Arch. Biochem. Biophys. 1994;309:315–322. doi: 10.1006/abbi.1994.1119. [DOI] [PubMed] [Google Scholar]
- 121.Bultel-Poncé V., Berge J.P., Debitus C., Nicolas J.L., Guyot M. Metabolites from the sponge-associated bacterium Pseudomonas species. Mar. Biotechnol. 1999;1:384–390. doi: 10.1007/pl00011792. [DOI] [PubMed] [Google Scholar]
- 122.Cheng M.J., Lee K.H., Tsai I.L., Chen I.S. Two new sesquiterpenoids and anti-HIV principles from the root bark of Zanthoxylum ailanthoides. Bioorg. Med. Chem. 2005;13:5915–5920. doi: 10.1016/j.bmc.2005.07.050. [DOI] [PubMed] [Google Scholar]
- 123.McCormick J.L., McKee T.C., Cardellina J.H., II, Boy M.R. HIV inhibitory natural products. 26. Quinoline alkaloids from Euodia roxburghiana. J. Nat. Prod. 1996;59:469–471. doi: 10.1021/np960250m. [DOI] [PubMed] [Google Scholar]
- 124.Boger D.L., Chen J.H., Saionz K.W., Jin Q. Synthesis of key sandramycin analogs: systematic examination of the intercalation chromophore. Bioorg. Med. Chem. 1998;6:85–102. doi: 10.1016/s0968-0896(97)10014-1. [DOI] [PubMed] [Google Scholar]
- 125.Take Y., Inouye Y., Nakamura S., Allaudeen H.S., Kubo A. Comparative studies of the inhibitory properties of antibiotics on human immunodeficiency virus and avian myeloblastosis virus reverse transcriptases and cellular DNA polymerases. J. Antibiot. 1989;42:107–115. doi: 10.7164/antibiotics.42.107. [DOI] [PubMed] [Google Scholar]
- 126.Boger D.L., Ledeboer M.W., Kume M. Total synthesis of luzopeptins A-C. J. Am. Chem. Soc. 1999;121:1098–1099. [Google Scholar]
- 127.Omura S., Nakagawa A., Hashimoto H. Virantmycin, a potent antiviral antibiotic produced by a strain of Streptomyces. J. Antibiot. 1980;33:1395–1396. doi: 10.7164/antibiotics.33.1395. [DOI] [PubMed] [Google Scholar]
- 128.Nakagawa A., Iwai Y., Hashimoto H. Virantmycin, a new antiviral antibiotic produced by a strain of Streptomyces. J. Antibiot. 1981;34:1408–1415. doi: 10.7164/antibiotics.34.1408. [DOI] [PubMed] [Google Scholar]
- 129.Morimoto Y., Shirahama H. Synthetic studies on virantmycin. 2. Total synthesis of unnatural (+)-virantmycin and determination of its absolute stereochemistry. Tetrahedron. 1996;52:10631–10652. [Google Scholar]
- 130.Fournet A., Hocquemiller R., Roblot F. Les Chimanines, nouvelles quinoleines substituees en 2, isolees d’Une plante bolivienne antiparasitaire: Galipea longiflora. J. Nat. Prod. 1993;56:1547–1552. [Google Scholar]
- 131.Fournet A., Vagneur B., Richomme P., Bruneton J. Aryl-2 et alkyl-2 quinoléines nouvelles isolées d’une Rutacée bolivienne: Galipealongiflora. Can. J. Chem. 1989;67:2116–2118. [Google Scholar]
- 132.Fournet A., Mahieux R., Fakhfakh M.A., Franck X., Hocquemiller R., Figadère B. Substituted quinolines induce inhibition of proliferation of HTLV-1 infected cells. Bioorg. Med. Chem. Lett. 2003;13:891–894. doi: 10.1016/s0960-894x(02)01085-5. [DOI] [PubMed] [Google Scholar]
- 133.Ito C., Itoigawa M., Otsuka T., Tokuda H., Nishino H., Furukawa H. Constituents of Boronia pinnata. J. Nat. Prod. 2000;63:1344–1348. doi: 10.1021/np0000318. [DOI] [PubMed] [Google Scholar]
- 134.(a) Kapoor Y., Kumar K. Structural and clinical impact of anti-allergy agents: an overview. Bioorg. Chem. 2020;94:103351–103375. doi: 10.1016/j.bioorg.2019.103351. [DOI] [PubMed] [Google Scholar]; (b) Kaur R., Manjal S.K., Rawal R.K., Kumar K. Recent synthetic and medicinal perspective of tryptanthrin. Bioorg. Med. Chem. 2017;25:4533–4552. doi: 10.1016/j.bmc.2017.07.003. [DOI] [PubMed] [Google Scholar]; (c) Manjal S.K., Kaur R., Bhatia R., Kumar K., Singh V., Shankar R., Kaur R., Rawal R.K. Synthetic and medicinal perspective of thiazolidinones: a review. Bioorg. Chem. 2017;75:406–423. doi: 10.1016/j.bioorg.2017.10.014. [DOI] [PubMed] [Google Scholar]; (d) Mittal M., Kumar K., Anghore D., Rawal R.K. ICP-MS: analytical method for identification and detection of elemental impurities. Curr. Drug Discov. Technol. 2017;14:106–120. doi: 10.2174/1570163813666161221141402. [DOI] [PubMed] [Google Scholar]; (e) Kaur R., Choudhry S., Kumar K., Gupta M.K., Rawal R.K. Recent synthetic and medicinal perspectives of dihydropyrimidinones: a review. Eur. J. Med. Chem. 2017;132:108–134. doi: 10.1016/j.ejmech.2017.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kumar B., Singh V., Shankar R., Kumar K., Rawal R.K. Synthetic and medicinal prospective of structurally modified curcumins. Curr. Top. Med. Chem. 2017;17:148–161. doi: 10.2174/1568026616666160605050052. [DOI] [PubMed] [Google Scholar]; (g) Talwan P., Choudhary S., Kumar K., Rawal R.K. Chemical and medicinal versatility of substituted 1,4-dihydropyridines. Curr. Bioact. Compd. 2017;13:109–120. [Google Scholar]; (h) Kumar K. TosMIC: a powerful synthon for cyclization and sulphonylation. ChemistrySelect. 2020;5:10298–10328. [Google Scholar]
- 135.(a) Manjal S.K., Pathania S., Bhatia R., Kaur R., Kumar K., Rawal R.K. Diversified synthetic strategies for pyrroloindoles: an overview. J. Heterocycl. Chem. 2019;56:2318–2332. [Google Scholar]; (b) Kaur R., Kapoor Y., Manjal S.K., Rawal R.K., Kumar K. Diversity-oriented synthetic approaches for furoindoline: a review. Curr. Org. Synth. 2019;16:342–368. doi: 10.2174/1570179416666190328211509. [DOI] [PubMed] [Google Scholar]; (c) Chouhan M., Senwar K.R., Kumar K., Sharma R., Nair V.A. Catalytic C-H activation of arylacetylenes: a fast assembly of 3-hydroxy-3-(arylethynyl)indolin-2-ones using CuI/DBU. Synthesis. 2014;46:195–202. [Google Scholar]; (d) Kumar K., More S.S., Khatik G.L., Rawal R.K., Nair V.A. A highly stereoselective chiral auxiliary-assisted reductive cyclization to furoindoline. J. Heterocycl. Chem. 2017;54:2696–2702. [Google Scholar]; (e) Kumar K., Konar D., Goyal S., Gangar M., Chouhan M., Rawal R.K., Nair V.A. Water promoted regiospecific azidolysis and copper catalysed azide alkyne cycloaddition: one-pot synthesis of 3-hydroxy-1-alkyl-3-[(4-aryl/alkyl-1H-1,2,3-triazol-1-yl)methyl]indolin-2-one derivatives. J. Org. Chem. 2016;81:9757–9764. doi: 10.1021/acs.joc.6b01819. [DOI] [PubMed] [Google Scholar]; (f) Kumar K., Siddique J., Gangar M., Goyal S., Rawal R.K., Nair V.A. ZrCl4 catalysed diastereoselective synthesis of spirocarbocyclicoxindolesvia [4+2] cycloaddition. ChemistrySelect. 2016;1:2409–2412. [Google Scholar]; (g) Kumar K., Konar D., Goyal S., Gangar M., Chouhan M., Rawal R.K., Nair V.A. AlCl3/Cyclohexane mediated electrophilic activation of isothiocyanates: an efficient synthesis of thioamides. ChemistrySelect. 2016;1:3228–3231. [Google Scholar]; (h) Kumar K., More S.S., Goyal S., Gangar M., Khatik G.L., Rawal R.K., Nair V.A. A convenient synthesis of 4-alkyl-3-benzyolpyrroles from α,β-unsaturated ketone and tosylmethylisocyanide. Tetrahedron Lett. 2016;57:2315–2319. [Google Scholar]; (i) Kumar K., Mudshinge S.R., Goyal S., Gangar M., Nair V.A. A catalyst free, one pot approach for the synthesis of quinoxaline derivatives via oxidative cyclization of 1,2-diamines and phenacyl bromides. Tetrahedron Lett. 2015;56:1266–1271. [Google Scholar]; (j) Goyal S., Patel J.K., Gangar M., Kumar K., Nair V.A. Zirconocene dichloride catalysed one-pot synthesis of pyrroles through nitroalkene-enamine assembly. RSC Adv. 2015;5:3187–3195. [Google Scholar]; (k) Goyal S., Patel B.K., Sharma R., Chouhan M., Kumar K., Gangar M., Nair V.A. An efficient strategy for the synthesis of syn 1,3-diols via iterative acetate aldol reactions and synthesis of atorvastatin lactone. Tetrahedron Lett. 2015;56:5409–5412. [Google Scholar]; (l) Kumar K., Rawal R.K. CuI/DBU-mediated MBH reaction of isatins: a convenient synthesis of 3-substituted-3-hydroxy-2-oxindole. Chemistryselect. 2020;5:3048–3051. [Google Scholar]