

Abstract
Somatic mutations are a large category of genetic variations, which play an essential role in tumorigenesis. Detection of somatic single nucleotide variants (SNVs) could facilitate downstream analysis of tumorigenesis. Many computational methods have been developed to detect SNVs, but most require normal matched samples to differentiate somatic SNVs from the normal state, which can be difficult to obtain. Therefore, developing new approaches for detecting somatic SNVs without matched samples are crucial. In this work, we detected somatic mutations from individual tumor samples based on a novel machine learning approach, svmSomatic, using next-generation sequencing (NGS) data. In addition, as somatic SNV detection can be impacted by multiple mutations, with germline mutations and co-occurrence of copy number variations (CNVs) common in organisms, we used the novel approach to distinguish somatic and germline mutations based on the NGS data from individual tumor samples. In summary, svmSomatic: (1) considers the influence of CNV co-occurrence in detecting somatic mutations; and (2) trains a support vector machine algorithm to distinguish between somatic and germline mutations, without requiring normal matched samples. We further tested and compared svmSomatic with other common methods. Results showed that svmSomatic performance, as measured by F1-score, was significantly better than that of others using both simulation and real NGS data.
Keywords: Somatic mutation, Germline mutation, Next-generation sequencing, Single nucleotide variations, Copy number variants, Support vector machine
DEAR EDITOR,
Somatic mutations are a large category of genetic variations, which play an essential role in tumorigenesis. Detection of somatic single nucleotide variants (SNVs) could facilitate downstream analysis of tumorigenesis. Many computational methods have been developed to detect SNVs, but most require normal matched samples to differentiate somatic SNVs from the normal state, which can be difficult to obtain. Therefore, developing new approaches for detecting somatic SNVs without matched samples are crucial. In this work, we detected somatic mutations from individual tumor samples based on a novel machine learning approach, svmSomatic, using next-generation sequencing (NGS) data. In addition, as somatic SNV detection can be impacted by multiple mutations, with germline mutations and co-occurrence of copy number variations (CNVs) common in organisms, we used the novel approach to distinguish somatic and germline mutations based on the NGS data from individual tumor samples. In summary, svmSomatic: (1) considers the influence of CNV co-occurrence in detecting somatic mutations; and (2) trains a support vector machine algorithm to distinguish between somatic and germline mutations, without requiring normal matched samples. We further tested and compared svmSomatic with other common methods. Results showed that svmSomatic performance, as measured by F1-score, was significantly better than that of others using both simulation and real NGS data.
In recent years, many developed tools have achieved good results in somatic mutation detection. These approaches can be classified into two categories: i.e., those using paired tumor-normal samples to distinguish somatic mutations from uncommon germline polymorphisms, e.g., VarDict (Lai et al., 2015), Muse (Fan et al., 2016), and FaSD-somatic (Wang et al., 2014), and those using tumor samples without normal matched samples, e.g., SomVarIUS (Smith et al., 2016), SNVer (Wei et al., 2011), and ISOWN (Kalatskaya et al., 2017). The first detection category has the advantage of excluding germline mutations with allele frequencies ≥1% in global populations (Sherry et al., 2001). However, rare germline mutations specific to an individual can affect the detection of somatic mutations. Furthermore, obtaining matched normal samples in clinical practice can be difficult. The second detection category can save on sequencing costs and is favored in clinical practice. However, some novel single nucleotide variants (SNVs) found in individuals will severely influence somatic mutation detection accuracy, resulting in higher false positives (Liu et al., 2016). In general, existing methods achieve relatively good detection results, but these tools only consider one type of variation in the genome.
With the above considerations, we propose a new machine learning-based method, named svmSomatic, to distinguish somatic and germline mutations without normal matched samples using next-generation sequencing (NGS) cancer genome data. The svmSomatic approach incorporates copy number variation (CNV) analysis in somatic mutation detection, extracts a set of somatic-relevant features at each site, and trains the support vector machine (SVM) classifier. We applied svmSomatic using real and simulation sequencing data. Results showed that this method is superior to others with consideration of the influence of CNVs.
The svmSomatic procedure workflow is shown in Figure 1A. The process starts with input of a tumor sample without normal matched samples and a human reference genome, followed by short-read alignment. As svmSomatic is focused on distinguishing somatic SNVs from germline SNVs and considers the influence of CNVs, we used existing methods to first detect SNVs and CNVs. Therefore, svmSomatic follows a four-step process for task learning. In the first step, five somatic SNV-related features are extracted: i.e., read depth, allele frequency (AF), mapping quality, mismatched reads, and copy number of each site. In the second step, the SVM is employed to complete classification (Hastie & Tibshirani, 1998). In the third step, the SMV classifier is trained with the labeled samples. In the final step, the trained SVM classifier is used to distinguish between germline and somatic mutations.
Figure 1.

Overview of svmSomatic method and performance comparison among five methods
A: Overview of svmSomatic. Input is a tumor-only sample aligned to a human reference genome. Based on STIC and FREEC, five features related to somatic SNVs were selected for SVM training. Trained classifier was then used to distinguish between germline and somatic mutations; B: Performance comparisons of five methods based on F1-score using simulation datasets with tumor purity ranging from 0.2 to 0.8 and coverage of 30X. C: Overlap between methods in terms of total number of detected somatic SNVs using real dataset.
The detection of CNVs and SNVs is the first step before running the svmSomatic algorithm. Currently, many existing methods can detect CNVs and SNVs. We chose our previously proposed method STIC (Yuan et al, 2020b) for the detection of SNVs and the classic method FREEC (Boeva et al., 2012) for the detection of CNVs. Both methods can work on single tumor samples without normal matched samples and exhibit reasonable performance, even when tumor purity (fraction of tumor cells in tumor tissue mixture) is relatively low. We also conducted a simulation experiment to demonstrate the performance of the two methods, with results presented in Supplementary Text 1. It should be noted that this preprocess step is relatively independent from the implementation of the svmSomatic algorithm and users can choose other methods for the detection of CNVs and SNVs according to their requirements.
Genomic data were extracted using BWA (Li & Durbin, 2009) and SAM tools (Li et al., 2009). Four features were extracted from the Pileup file, including read depth, number of mismatched reads, AF, and average mapping quality. Finally, according to the FREEC results, copy number information was added to each SNV site as the fifth feature. These five features are associated with SNVs (Yuan et al., 2020b). Read depth denotes the number of reads aligned on some sites and provides important information for the deduction of copy number and number of variant alleles. AF can distinguish germline and somatic mutations. Due to the influence of tumor purity and copy number, the number of mismatched reads will vary, and the AF value will deviate from the ideal. Average mapping quality also considers sequencing errors. These five features show good separability and reliability, allowing the classifier to easily distinguish between somatic and germline mutations. Table 1 shows the features and their corresponding definitions.
Table 1. Description of five extracted features.
| Feature | Description |
| Read depth | Number of reads mapped to each site |
| Mismatched reads | Number of mismatched reads |
| Allele frequency | Ratio of a particular allele to total number of alleles |
| Ave. mapping quality | Average mapping quality of reads matched to each site |
| Copy number | Copy number of reads mapped to each site |
Distinction between somatic and germline mutations is primarily achieved through AF. Studies have shown that for heterozygous and homozygous genotypes, the AF of germline SNVs is 0.5 and 1, respectively (Xu, 2018). However, when germline AF is involved in somatic copy number change events, it may deviate from 0.5 or 1. Similarly, AF with somatic mutations can fluctuate due to CNV, normal tissue mixing, and subcloning (Cun et al., 2018; Xi et al., 2020). Therefore, it is necessary to add copy number as a feature to the classifier.
Here, the SVM was selected as the algorithm classifier as it shows outstanding performance in classification problems. The design of the SVM classifier considers the distance between different categories to determine the optimal classification boundary by maximizing the distance between classes (Guyon et al., 1993; Lappalainen et al., 2015). We used the SVM as a binary classifier. Further details can be found in Supplementary Text 2.
Crucially, the SVM classifier must be trained before performing classification. We trained the SVM classifier using simulation datasets. In brief, 10-fold cross-validation was used to assess algorithm performance and chose the best classification strategy. We generated 100 000 SNVs, containing 50 000 somatic mutations and 50 000 germline mutations. The training dataset contain 45 000 randomly selected somatic mutations and 45 000 randomly selected germline mutations. The training dataset contained only two data types, labeled 1 and 0, representing germline and somatic mutations, respectively. The best parameter combination was chosen using 10-fold cross-validation based on the highest F1-score. Further details can be found in Supplementary Text 3.
The new approach consists of two parameters, i.e., C and γ. The best method to determine the optimal parameter values in space was C=
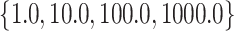 and γ=
and γ=
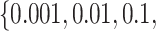
 , with the parameter combination C=1 000.0 and γ=0.1. However, due to hyperparameter distribution characteristics (Liu et al., 2006), the best combination was not unique. Here, we only present an optimal combination.
, with the parameter combination C=1 000.0 and γ=0.1. However, due to hyperparameter distribution characteristics (Liu et al., 2006), the best combination was not unique. Here, we only present an optimal combination.
To evaluate performance, we applied the newly proposed method using the simulation datasets. As the simulation data showed a clear pattern, we calculated sensitivity and precision of the simulation experiment results and then used the F1-scores for comprehensive evaluation (Yuan et al., 2012, 2017). In addition, we compared the new approach to four classic methods (i.e., STIC (Yuan et al, 2020b), FaSD-somatic (Wang et al., 2014), SNVSniffer (Liu et al., 2016), and VarScan2 (Koboldt et al., 2012)) using their default parameters for reasonable and fair comparison.
SInC (Pattnaik et al., 2014) was used to generate sequencing reads of chromosome 21. A total of 100 000 somatic SNVs and 100 000 germline SNVs were simulated. Half of the SNVs were heterozygous and the other half homozygous. We also simulated 226 CNVs in chromosome 21 ranging in length from 10 000 to 100 000. The simulated CNV types included gain and loss with copy numbers of 0, 1, 3, 4, 5, and 6. To simulate different tumor purity levels, a pair of tumor-normal matched genomes was prepared. The tumor genome contained 200 000 SNVs and the normal genome contained only germline SNVs. FASTQ files from mixed samples with tumor purity ranging from 0.2 to 0.8 were generated. The sequencing coverage depths were 10X, 20X, 30X, 40X, and 50X. To reduce the influence of noise from instruments and equipment, 10 simulation experiments for each coverage were carried out. The results presented are the average of the 10 replicates. Comparisons of the svmSomatic approach and four other methods were performed with the above data. Results are shown in Figure 1B, with coverage of 30X, and Supplementary Text 4. The recall and precision results of the five methods are presented in Supplementary Text 5.
As shown in Figure 1B, the prediction of somatic SNVs improved with the increase in tumor purity; when tumor purity remained constant, prediction of somatic SNVs increased with the increase in coverage. In contrast, for STIC, the overall performance fluctuated with the increase in tumor purity. Somatic SNV prediction by STIC was dependent on AF, and thus was impacted by the increase in copy number. SNVSniffer and VarScan2 achieved satisfactory results at various coverages. However, FaSD-somatic was greatly affected by coverage, and only achieved good results when coverage was high. Overall, the performance of svmSomatic showed advantage over the other methods.
The svmSomatic method was also applied to real data. As several of the methods (FaSD-somatic (Wang et al., 2014), VarScan2 (Koboldt et al., 2012), and SNVSniffer (Liu et al., 2016)) require matched samples for comparison, we collected paired tumor-normal samples (EGAR00001008630 and EGAR00001008681) for this experiment. Figure 1C shows the results of svmSomatic and other methods for chromosome 21. The blacked numbers in the table represents the number of somatic SNVs detected. SvmSomatic predicted the largest number of somatic SNVs, followed by STIC (Yuan et al, 2020b), FaSD-somatic (Wang et al., 2014), SNVSniffer (Liu et al., 2016), and VarScan2 (Koboldt et al., 2012). For sample data, the F1-score could not be calculated. Thus, to evaluate method performance using real data, overlap among the five methods was analyzed using the overlapping density score (ODS), which developed by Yuan (Yuan et al, 2020a) as expressed in Equation (1).
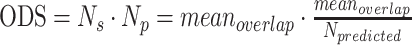 |
1 |
where
 is the mean number of overlaps of one method with other methods and
is the mean number of overlaps of one method with other methods and
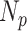 is the mean number of overlaps divided by the total predictions by the method. Here, we assumed that the overlaps between different methods were true positives. Thus,
is the mean number of overlaps divided by the total predictions by the method. Here, we assumed that the overlaps between different methods were true positives. Thus,
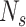 could be defined as sensitivity and
could be defined as sensitivity and
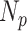 could be defined as precision. The product of
could be defined as precision. The product of
 and
and
 is similar to the area under an ROC curve (AUC), but the greater the value, the higher the performance. ODS(FaSD-somatic)=137.7, ODS(VarScan2)=101.8, ODS(SNVSniffer2)=45.2, ODS(STIC)=887.5, ODS(svmSomatic)=899.3, svmSomatic had the highest ODS value, followed by STIC, FaSD-somatic, VarScan2, and SNVSniffer. These results indicate that svmSomatic has a higher
is similar to the area under an ROC curve (AUC), but the greater the value, the higher the performance. ODS(FaSD-somatic)=137.7, ODS(VarScan2)=101.8, ODS(SNVSniffer2)=45.2, ODS(STIC)=887.5, ODS(svmSomatic)=899.3, svmSomatic had the highest ODS value, followed by STIC, FaSD-somatic, VarScan2, and SNVSniffer. These results indicate that svmSomatic has a higher
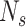 , higher mean number of overlaps with other methods, and higher sensitivity. Overall, svmSomatic showed slightly better results compared to the simulation data when applied to real data.
, higher mean number of overlaps with other methods, and higher sensitivity. Overall, svmSomatic showed slightly better results compared to the simulation data when applied to real data.
In this paper, we developed a new open-source method (svmSomatic) to distinguish somatic SNVs from germline SNVs in tumor-only NGS data. SvmSomatic considers the influence of copy number variation when distinguishing SNVs. Furthermore, it is a single-sample-based method that does not require normal matched samples. The approach can be applied for individual chromosomes as well as whole exome and genome data. The detection of somatic SNVs should facilitate downstream research on tumors, including gene annotation and targeted drug therapy. SvmSomatic is written in Python language and implemented on the Linux system. The source code and manual documents are freely available at https://github.com/BDanalysis/svmSomatic.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
AUTHORS’ CONTRIBUTIONS
Y.F.M. and X.G.Y. participated in the design of algorithms and experiments. Y.F.M. and Y.P.C. participated in analysis of the performance of the proposed method. Y.P.C. and X.G.Y. directed and conceived the study and helped edited the manuscript. All authors read and approved the final version of the manuscript.
Funding Statement
This study was supported by the CAS Pioneer Hundred Talents Program and National Natural Science Foundation of China (32070683) to Y.P.C
Contributor Information
Xi-Guo Yuan, Email: xiguoyuan@mail.xidian.edu.cn.
Yu-Peng Cun, Email: cunyupeng@mail.kib.ac.cn.
References
- 1.Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, et al Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012;28(3):423–425. doi: 10.1093/bioinformatics/btr670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cun YP, Yang TP, Achter V, Lang U, Peifer M Copy-number analysis and inference of subclonal populations in cancer genomes using Sclust. Nature Protocols. 2018;13(6):1488–1501. doi: 10.1038/nprot.2018.033. [DOI] [PubMed] [Google Scholar]
- 3.Fan Y, Xi L, Hughes DST, Zhang JJ, Zhang JH, Futreal PA, et al MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biology. 2016;17(1):178. doi: 10.1186/s13059-016-1029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guyon I, Boser BE, Vapnik V. 1993. Automatic capacity tuning of very large VC-dimension classifiers. In: Proceedings of Advances in Neural Information Processing Systems 5. Denver: NIPS, 147–155.
- 5.Hastie T, Tibshirani R Classification by pairwise coupling. The Annals of Statistics. 1998;26(2):451–471. doi: 10.1214/aos/1028144844. [DOI] [Google Scholar]
- 6.Kalatskaya I, Trinh QM, Spears M, Mcpherson JD, Bartlett JMS, Stein L ISOWN: accurate somatic mutation identification in the absence of normal tissue controls. Genome Medicine. 2017;9(1):59. doi: 10.1186/s13073-017-0446-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koboldt DC, Zhang QY, Larson DE, Shen D, McLellan MD, et al VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Research. 2012;22(3):568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lai ZW, Markovets A, Ahdesmaki M, Johnson J Abstract 4864: VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Cancer Research. 2015;75(15):4864–4864. doi: 10.1093/nar/gkw227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lappalainen I, Almeida-King J, Kumanduri V, Senf A, Spalding JD, Ur-Rehman S, et al The European Genome-phenome Archive of human data consented for biomedical research. Nature Genetics. 2015;47(7):692–695. doi: 10.1038/ng.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu RM, Liu EQ, Yang J, Li M, Wang FL. 2006. Optimizing the hyper-parameters for SVM by combining evolution strategies with a grid search. In: Proceedings of International Conference on Intelligent Computing. Kunming, China: Springer, 712–721.
- 13.Liu YC, Loewer M, Aluru S, Schmidt B SNVSniffer: an integrated caller for germline and somatic single-nucleotide and indel mutations. BMC Systems Biology. 2016;10(S2):47. doi: 10.1186/s12918-016-0300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pattnaik S, Gupta S, Rao AA, Panda B SInC: an accurate and fast error-model based simulator for SNPs, Indels and CNVs coupled with a read generator for short-read sequence data. BMC Bioinformatics. 2014;15:40. doi: 10.1186/1471-2105-15-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al dbSNP: the NCBI database of genetic variation. Nucleic Acids Research. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith KS, Yadav VK, Pei SS, Pollyea DA, Jordan CT, De S SomVarIUS: somatic variant identification from unpaired tissue samples. Bioinformatics. 2016;32(6):808–813. doi: 10.1093/bioinformatics/btv685. [DOI] [PubMed] [Google Scholar]
- 17.Wang WX, Wang PW, Xu F, Luo RB, Wong MP, Lam TW, et al FaSD-somatic: a fast and accurate somatic SNV detection algorithm for cancer genome sequencing data. Bioinformatics. 2014;30(17):2498–2500. doi: 10.1093/bioinformatics/btu338. [DOI] [PubMed] [Google Scholar]
- 18.Wei Z, Wang W, Hu PZ, Lyon GJ, Hakonarson H SNVer: a statistical tool for variant calling in analysis of pooled or individual next-generation sequencing data. Nucleic Acids Research. 2011;39(19):e132. doi: 10.1093/nar/gkr599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xi JN, Yuan XG, Wang MH, Li A, Li XL, Huang Q Inferring subgroup-specific driver genes from heterogeneous cancer samples via subspace learning with subgroup indication. Bioinformatics. 2020;36(6):1855–1863. doi: 10.1093/bioinformatics/btz793. [DOI] [PubMed] [Google Scholar]
- 20.Xu C A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Computational and Structural Biotechnology Journal. 2018;16:15–24. doi: 10.1016/j.csbj.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan XG, Miller DJ, Zhang JY, Herrington D, Wang Y An overview of population genetic data simulation. Journal of Computational Biology. 2012;19(1):42–54. doi: 10.1089/cmb.2010.0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan XG, Zhang JY, Yang LY IntSIM: an integrated simulator of next-generation sequencing data. IEEE Transactions on Biomedical Engineering. 2017;64(2):441–451. doi: 10.1109/TBME.2016.2560939. [DOI] [PubMed] [Google Scholar]
- 23.Yuan X, Bai J, Zhang J, Yang L, Duan J, Li Y, et al CONDEL: Detecting copy number variation and genotyping deletion zygosity from single tumor samples using sequence data. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2020a;17(4):1141–1153. doi: 10.1109/TCBB.2018.2883333. [DOI] [PubMed] [Google Scholar]
- 24.Yuan X, Ma C, Zhao H, Yang L, Wang S, Xi J STIC: Predicting single nucleotide variants and tumor purity in cancer genome. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2020b doi: 10.1109/TCBB.2020.2975181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data to this article can be found online.