

Abstract
Background
Ubiquitin‐specific protease 7 (USP7) belongs to the group of deubiquitinating enzymes (DUBs), which remove ubiquitin which controls various cellular processes such as chromosome segregation, DNA repair, gene expression, protein localization, kinase activity, protein degradation, cell cycle progression, and apoptosis. It is critical for several important functions in the cell, and therefore dysregulation of USP7 can contribute to tumorigenesis.
Objectives
Alterations in the USP7 protein have been identified in various malignancies of humans. Our aim was to examine whether USP7 could be a potential therapeutic target in hematopoietic cancers of dogs.
Methods
The expression level of USP7 in lymphocytes from healthy dogs and canine lymphoma cells was determined, and the effect of USP7 inhibition on the vital functions of canine cancer cells was examined.
Results
We showed that USP7 was overexpressed in lymphomas in dogs. The USP7 inhibitor P5091 has selective cytotoxic activity in canine lymphoma and leukemia cell lines. Our results indicate that inhibition of USP7 leads to a disruption of cell cycle progression, and triggers DNA damage and apoptosis. The observed proapoptotic effect of the USP7 inhibitor most likely is not dependent on the p53 pathway.
Conclusions and Clinical Importance
Our results suggest that USP7 could be explored as a potential therapeutic target in dogs with lymphoma. The effectiveness of USP7 inhibition in malignant cells is predicted to be independent of their p53 status.
Keywords: anticancer activity, canine hematopoietic cancers, deubiquitylases, deubiquitylases inhibitors
1. INTRODUCTION
Ubiquitin‐specific protease 7 (USP7), also called Herpes virus‐associated ubiquitin‐specific protease (HAUSP), is a 135‐kDa cellular protein discovered in the late 1990s that interacts with herpes virus regulatory protein. 1 It is widely expressed in all tissue types, and in the cell it is mainly found in the nucleus but also in the cytoplasm and mitochondria. 2 , 3 Ubiquitin‐specific protease 7 belongs to the group of deubiquitinating enzymes (DUBs) that remove ubiquitin and control various cellular processes, such as chromosome segregation, DNA repair, gene expression, protein localization, kinase activation, protein degradation, cell cycle progression, and apoptosis. 4 , 5 Ubiquitin‐specific protease 7 is evolutionarily conserved in humans, mice and rats with approximately 98.6% amino acid sequence homology. 6 By regulating the expression and function of various proteins, USP7 plays a critical role in cancers, neurological disorders, metabolic disorders, immune dysfunctions and many more disease processes. The first substrate identified for USP7‐mediated deubiquitination was the tumor suppressor protein TP53 (p53). 7 Stabilization of p53 in cancer cells by inhibition of USP7 protein leads to cell cycle arrest in these tumor cells and triggers apoptosis. 8 , 9 However, in some cell types, USP7 inhibition produces cytotoxic effects that are not caused by direct p53 stabilization. 10 , 11 , 12 On the basis of this observation, several other USP7 substrates have been identified that potentially could affect cell growth and survival, including mouse double minute 2 homolog, transcription factor Forkhead box protein O4, phosphatase and tensin homolog, β‐catenin, and checkpoint with forkhead and ring finger domain protein (CHFR). 10 , 13 , 14 The identification of these substrates indicates that USP7 controls additional vital cellular functions beyond those mediated by p53 stability. In addition to the aforementioned substrates, USP7 regulates the stability of other proteins in the DNA damage response, thereby contributing to maintenance of genome stability. Examples are mediator of DNA damage checkpoint protein 1 (MDC1) and Claspin, important for the detection and signaling of DNA damage, checkpoint kinase Chk1, repair and replication protein Rad18 and DNA polymerase eta, required for DNA damage tolerance. 15 , 16 , 17 , 18 , 19 In addition, accumulating evidence suggests that USP7 controls DNA replication. 20 , 21 Finally, inhibition of USP7 was shown to trigger apoptosis in cancer cells by causing oxidative and endoplasmic reticulum (ER) stress. 22
Because some of the better characterized USP7 substrates play crucial roles in tumor suppression, DNA repair, immune responses and epigenetic control, it is not surprising that USP7 has been linked to cancer. 13 Its altered expression was found in various types of cancers in humans (eg, bladder, prostate, colon, lung, liver, ovary, breast and nervous system cancers, and leukemias). 23 Therefore, attempts to inhibit USP7 activity from a therapeutic perspective have yielded a number of different synthetic inhibitors mainly with cytotoxic and proapoptotic activity. 8 Both newly developed small molecule inhibitors, such as HBX 41108, HBX 19818, HBX 28258, P5091, P22077, FT671, FT827, GNE‐6640, and GNE‐6776 as well as previously known compounds with antitumor activity for which inhibition of USP7 is an additional activity (imatinib, arsenic trioxide, metformin) currently are used in ongoing studies. 23 In recent years, an increasing number of studies has indicated the antitumor effect when USP7 was lost or inhibited in various types of cancer (eg, prostate, 24 colorectal, 10 ovarian, 12 , 25 colon, 26 breast, 27 hematopoietic cancers, gliomas, 28 neuroblastomas 9 ). The P5091 inhibitor, in addition to in vitro experimental studies, has been used in in vivo studies, mainly in mouse xenograft models of multiple myeloma. These studies indicated that USP7 inhibition is tolerable at doses that are effective in tumor suppression, causing no significant weight loss or cachexia. 29 , 30
Interestingly, despite the growing number of studies on this topic, the mechanisms by which USP7 inhibitors affect the functions of cancer cells remain to be explained and therefore further research is still needed to fully exploit the therapeutic potential of USP7 inhibition. Because the inhibition of USP7 function is still a novel and promising direction in the development of new, molecularly targeted treatments in human oncology, we investigated whether USP7 also can be a target for drug development in veterinary oncology. Consequently, we investigated expression levels of USP7 and cytotoxic effect of its inhibition in a model of the most common hematopoietic cancer in dogs, non‐Hodgkin lymphoma.
2. MATERIALS AND METHODS
2.1. Cell lines and cell culture
The following canine cancer cell lines were used in our study: CLBL‐1 (B‐cell lymphoma), GL‐1 (B‐cell leukemia), CL‐1 (primitive αβ T‐cell leukemia), CLB70 (B‐cell chronic lymphocytic leukemia), CNK‐89 (Natural killer‐cell lymphoma), P114 (mammary cancer), D‐17 (osteosarcoma) and 1 human cell line U2OS (osteosarcoma), which served as a control. The CLBL‐1 cell line was obtained from Barbara C. Ruetgen, Institute of Immunology, Department of Pathobiology, University of Veterinary Medicine, Vienna, Austria, 31 GL‐1 and CL‐1 cells were obtained from Yasuhito Fujino and Hajime Tsujimoto from the University of Tokyo, Department of Veterinary Internal Medicine 32 , 33 and CLB70 34 and CNK‐89 were established in our laboratory. The CNK‐89 cell line represents fully functional canine NK cells. These cells can be activated by IL‐12 alone, or in combination with several other interleukins, and also can exert a cytotoxic effect on target cells. The CNK‐89 cell line expresses CD5, CD8, CD45, CD56, CD79a, and NKp46.
The P114 cell line was obtained from Dr Rutteman (Utrecht University, the Netherlands), 35 and D‐17 and U2OS were purchased from the ATCC collection (ATCC.CCL‐183 and ATCC.HTB‐96, respectively).
After examining the level of USP7 protein expression in various cell lines, for additional tests investigating the effect of USP7 inhibition on cell cycle progression, apoptosis and DNA damage, CLBL‐1, CLB70, CNK‐89, and GL‐1 cell lines were selected.
The cell lines were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium, CLBL‐1 and GL‐1 (Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Wrocław, Poland), or Advanced RPMI (Gibco, Grand Island, New York, CLB70 and CNK‐89) culture medium supplemented with 2 mM L‐glutamine (Sigma Aldrich, Steinheim, Germany), 100 U/mL penicillin, 100 μg/mL streptomycin (Sigma Aldrich), and 10% to 20% heat‐inactivated fetal bovine serum (FBS, Gibco, Grand Island, New York).
2.2. Isolation of lymphoma samples
Lymphoma samples were obtained from dogs diagnosed with lymphoma, between the years 2018 and 2019 at the Veterinary Clinic “NeoVet” S.C. in Wrocław (Poland) during standard diagnostic procedures. The research therefore did not require Institutional Animal Care and Use Committee (IACUC) approval. In total, 8 lymph node fine‐needle aspirates from the popliteal lymph node were collected from dogs of different breeds, age, and sex. The samples came from 4 females and 4 males ranging in age from 5 to 13 years (mean, 9 years) representing different breeds (3 mixed breed, 2 miniature schnauzers, 1 Chinese crested dog, 1 golden retriever, and 1 boxer). In 7 dogs, neoplastic growth was derived from B cells (7 high‐grade B‐cell lymphomas) whereas 1 dog was diagnosed with T‐cell lymphoblastic lymphoma (boxer). The diagnosis was made on the basis of clinical examination and evaluation of cytological preparations. Cell phenotype was determined by flow cytometry using antibodies directed against canine CD3, CD4, CD8, CD14, CD21, CD34, CD45, MHCII, and CD79α according to a previously reported procedure in our laboratory. 36
Cell suspensions were obtained after 3 cycles of washing with phosphate‐buffered saline (PBS) and erythrocyte lysis buffer (0.84% ammonium chloride, Sigma Aldrich). The samples were taken from dogs that had not been previously treated with any chemotherapeutic agents.
2.3. Isolation of peripheral blood mononuclear cells from healthy donors
Peripheral blood samples from 8 dogs of different breeds, age, and sex, remaining after routine diagnostic procedures (Veterinary Clinic “NeoVet” S.C. in Wrocław, Poland) were used for the isolation of peripheral blood mononuclear cells (PBMCs). The PBMCs were obtained by density gradient centrifugation (Histopaque 1077, Sigma Aldrich), erythrocyte lysis (0.84% ammonium chloride) and 3 cycles of washing with PBS.
2.4. Cell proliferation assay
The proliferation of canine cells was determined using the MTT test (Sigma Aldrich). In brief, 1 × 105 cells per well were seeded in a 96‐well plate (Thermo Fisher Scientific, Roskilde, Denmark), and USP7 inhibitor P5091 (Selleckchem, Texas) was added in increasing concentrations (0.6, 1.25, 2.5, 5, 10, and 20 μM). Cells incubated in culture medium alone were used as controls. After incubation for 24, 48, 72, and 96 hours, 20 μL of MTT solution (5 mg/mL) were added to each well. After dissolving the content, the optical density of wells was measured using a spectrophotometric microplate reader (Elx800, BioTek, Winooski, VT) at a reference wavelength of 570 nm. The results were means of 3 independent experiments (different plates, different days) with 3 wells each.
2.5. Western blotting
A total of 5 × 106 cells were rinsed with cold PBS, suspended in lysis buffer (50 mM Tris‐HCl pH 7.5, 100 mM NaCl, 1% NP‐40, and protease inhibitors set) and incubated for 20 minutes on ice. The suspensions were centrifuged at 10000 rpm at 4°C for 12 minutes. Then, sodium dodecyl sulfate (SDS) sample buffer was added to clear supernatants and the samples were boiled at 95°C for 5 minutes and subjected to SDS‐polyacrylamide gel electrophoresis on 10‐15% gel.
For USP7 and γH2AX Western blot analysis, transfer was performed using a Trans Blot Turbo, Biorad semi‐dry chamber and nitrocellulose membranes (Sigma Aldrich). Then, the membranes were incubated with Pierce Western Blot Signal Enhancer (Thermo Scientific, Mississippi) and blocked with Western Dot blocking buffer (Invitrogen, California; blocking conditions: 180 minutes, 37°C). After blocking, the membranes were incubated with primary antibodies: goat polyclonal antibodies anti‐HAUSP/USP7 (ab 157 132; Abcam); murine monoclonal antibodies anti‐γH2A.X clone 9F3 (ab26350; Abcam) and murine monoclonal anti‐β‐actin antibody clone C4 (sc‐47 778; Santa Cruz, California). Membranes were incubated with the primary antibody (dilution 1:2000) overnight at 4°C. The membranes were incubated with the secondary antibody for 90 minutes at room temperature. The reaction was developed using Clarity Western ECL (BioRad) as a substrate. Membrane visualization was performed using ChemiDoc Touch Instruments (exposure: first image, 1 second; last image, 60 seconds; images, 20; BioRad). For USP7 expression quantification, Western blot normalization using a single protein (housekeeping protein, β‐actin) was performed using Image LabTM software (version 5.2.1; BioRad).
2.6. Apoptosis assays by flow cytometry
After seeding at a density of 1 × 105/mL in 96‐well plates (TPP, Trasadingen, Switzerland), the cells were incubated for 24 hours and 48 hours with increasing concentrations of P5091 (2, 4, and 8 μM). Cells then were collected, suspended in a binding buffer, and stained with Annexin V‐FITC and propidium iodide (PI, final PI concentration, 1 μg/mL). Flow cytometric analysis was performed immediately using a flow cytometer (FACS Calibur; Becton Dickinson, Biosciences, San Jose, California). CellQuest 3.lf. software (Becton Dickinson) was used for data analysis.
2.7. Cell cycle analysis
For this test, the same concentrations of P5091 were used as in the apoptosis studies (2, 4, and 8 μM), but because live cells are needed to analyze changes in the course of the cell cycle, concentrations of 2 and 4 μM were chosen for additional studies.
For cell cycle analysis (bromodeoxyuridine [BrdU]/PI staining), the cells were incubated with 10 μM BrdU for 15 minutes. After fixation in 70% ethanol, the cells were washed (0.5% Tween‐20 in PBS) and incubated in a denaturing solution (0.5% Triton X‐100, 2 M HCl) for 30 minutes at 37°C. Then, the cells were neutralized with 1 M Tris‐HCl pH 7.5. After washing with PBS, cells were incubated with anti‐BrdU antibody (GenScript) in bovine serum albumin (BSA)‐T‐PBS (1% BSA, 0.5% Tween‐20 in PBS) for 16 hours at 4°C. After washing with BSA‐T‐PBS, the cells were incubated with Alexa 488‐conjugated anti‐mouse secondary antibody (Life Technologies, Carlsbad, California) and incubated with 25 μg/mL PI and 100 μg/mL RNase (Sigma Aldrich). The samples were analyzed by flow cytometry using a MacsQuant Analyzer and Macsquantify software (Miltenyi Biotec, San Diego, California).
2.8. Statistical analysis
All data are shown as means with SDs. Statistical differences were analyzed using 1‐way analysis of variance followed by Tukey's multiple comparison test. Statistical analysis was performed using STATISTICA version 13.3 software (TIBCO Software Inc., Palo Alto, California) and GraphPad Prism Version 8.0.2 (GraphPad Software, Inc, California). Results were considered significant at P < .05.
3. RESULTS
3.1. USP7 overexpression in canine lymphoma cells
To determine whether USP7 may be a potential therapeutic target in hematopoietic malignancies of dogs, we analyzed USP7 expression levels in lymphomas in dogs. For this purpose, we first compared the expression level of USP7 in selected established canine cancer cell lines (CLBL‐1, CLB70, CNK‐89, GL‐1, CL‐1, D‐17, and P114) with expression in human osteosarcoma cell line U2OS, characterized by high USP7 protein expression level. 37 Figure 1A shows that in all tested cell lines the level of USP7 protein expression was comparable to the level in U2OS cells, which means that USP7 expression level is increased in the tested canine cell lines. Next, we compared the expression level of USP7 in PBMCs obtained from healthy donors (n = 8) to that in U2OS cells. The comparison indicated lower protein expression of USP7 in healthy donor PBMCs than in the U2OS cell line (Figure 1B). Subsequently, the expression level of USP7 in the lymphocytes from dogs with lymphoma (n = 8) was analyzed using the same experimental conditions. Different from healthy donors, the expression levels of USP7 were high in at least 4 of 8 samples (Figure 1C) and, when results were quantified and normalized with respect to the U2OS control, a clear increase was observed (Figure 1D).
FIGURE 1.
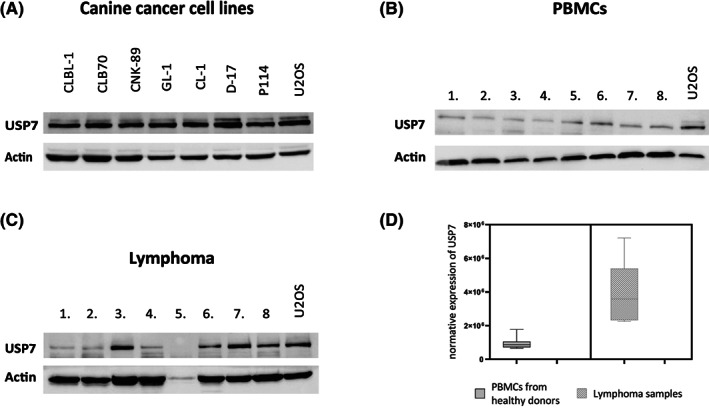
Representative graphical results showing expression of ubiquitin‐specific protease 7 (USP7) protein in canine cancer cell lines, A, peripheral blood mononuclear cells (PBMCs), B, and cells from lymphoma samples, C. Statistical analysis of USP7 expression level in healthy donor cells (PBMCs) and lymphoma samples compared to U2OS cells (arbitrary units), D
3.2. The USP7 inhibitor P5091 inhibits proliferation of canine lymphoma cell lines
To investigate the hypothesis of the potential use of USP7 as a new therapeutic target in dogs with lymphoma, we investigated the effect of a USP7 inhibitor (USP7i, P5091) on cell proliferation using the MTT test which, by determining the metabolic activity of the cells, showed changes in the rate of cell proliferation. The assay indicated that USP7i acutely inhibited proliferation of all cell lines used in our study and the effect was both concentration‐ and time‐dependent (Table 1). The number of actively dividing cells decreased between 24 and 72 hours of incubation, but the difference between 72 and 96 hours was minimal. The CLBL‐1 and CNK‐89 cell lines were most sensitive to the antiproliferative activity of P5091, with a half maximal inhibitory concentrations (IC50) of approximately 8 μM for 24 hours of incubation and up to 1 μM for 96 hours. The GL‐1 cells were more resistant, with an IC50 of between 13 μM for 24 hours treatment and 6 μM for 96 hours.
TABLE 1.
Comparison of IC50 values (μM) for all tested cell lines after 24, 48, 72, and 96 hours of incubation with USP7 inhibitor P5091 evaluated with MTT assay
| Time/Cell line | CLBL‐1 | CLB70 | CNK‐89 | GL‐1 |
|---|---|---|---|---|
| 24 h | 8.04a± 0.53 | 10.19a ± 3.11 | 8.21a ± 1.63 | 12.77b ± 3.08 |
| 48 h | 6.27a ± 1.07 | 6.41a ± 0.78 | 3.48b ± 0.43 | 11.30c ± 1.50 |
| 72 h | 3.58a ± 0.61 | 4.46a ± 1.47 | 0.97b ± 0.29 | 5.63a ± 0.53 |
| 96 h | 3.4a ± 0.30 | 3.83a ± 0.65 | 0.95b ± 0.51 | 5.75c ± 0.91 |
Note: The concentration range used was 0.6, 1.25, 2.5, 5, 10, and 20 μM. For each cell line, values without common letters (a, b, c) in the superscript differ statistically (P < .05). Results are presented as average ± the SD of three independent experiments (three wells each).
3.3. Effect of USP7 inhibition on cell cycle progression
The observed antiproliferative effect of P5091 on canine lymphoma and leukemia cell lines could be explained by either an effect of inhibition of USP7 on cell cycle progression or by triggering cell death. Because previous studies reported that treatment with P5091 affected the cell cycle progression of human cells, 9 , 10 we started by analyzing possible changes in cell cycle progression after treatment with USP7i (Figure 2). Contrary to our expectations, the effect of USP7i on the cell cycle was ambiguous. A slight increase in the percentage of cells in the G1 phase was observed in CLB70 and CNK‐89 cell lines, accompanied by a decrease in the percentage of cells in the phase of DNA synthesis (Figure 2C,D and E,F, respectively). In any case, no clear cell cycle arrest was identified in any phase of the cycle, both after 24 and 48 hours of incubation (Figure 2).
FIGURE 2.
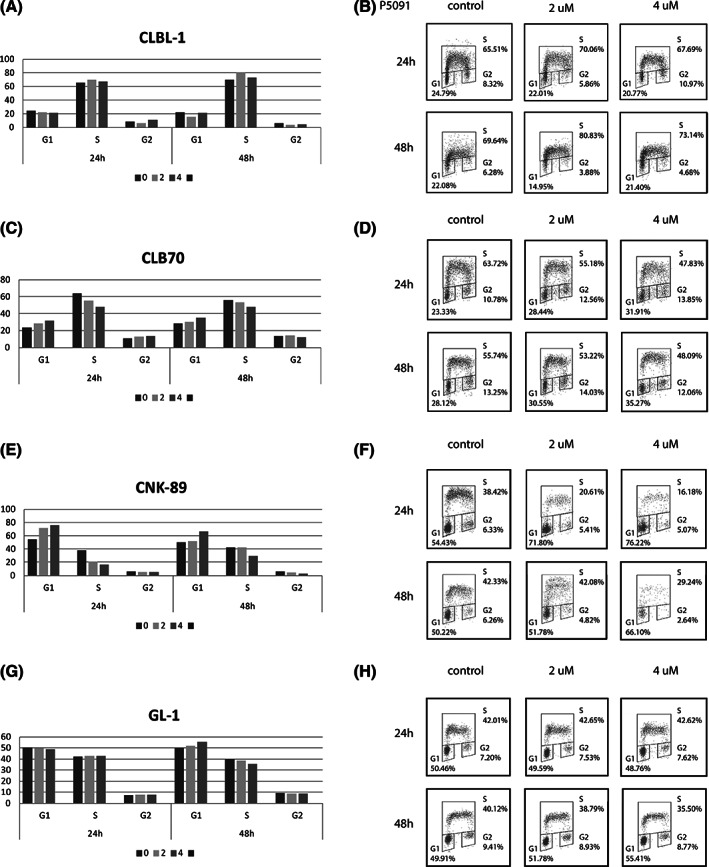
Effect of ubiquitin‐specific protease 7 (USP7) inhibition with P5091 on cell cycle progression. Cell cycle distribution as determined by bromodeoxyuridine/propidium iodide (BrdU/PI) staining, presented as percentage of cells (y‐axis) in different phases of the cell cycle for the CLBL‐1, A, CLB70, C, CNK‐89, E, and GL‐1, G, cell lines with the corresponding representative dotplots, B,D,F,H
3.4. The USP7 inhibitor P5091 induces apoptosis in canine lymphoma/leukemia cell lines
We subsequently examined the possibility that USP7 inhibition might trigger cell death in canine lymphoma/leukemia cells. The most common types of cell death, apoptosis and necrosis, were analyzed and our results showed that the cytotoxic effect of USP7i on canine lymphoma/leukemia cells was at least partly caused by induction of cell death. After as early as 24 hours of incubation with USP7i, a distinct population of annexin V and PI double‐positive cells (annexin V+/PI+) was identified in all cell lines, representing cells in late apoptosis. In contrast, P5091 treatment did not have significant pro‐apoptotic effects in the primary culture of PBMCs obtained from healthy donors, which indicates that the observed effect is limited to canine lymphoma and leukemia cells (Figure 3A,B). Induction of apoptosis was highest in CLBL‐1 and CNK‐89 cells, where the percentages of late apoptotic cells, after incubation with 8 μM P5091, were 75.4% ± 8.2% and 64.4% ± 11.3%, respectively. The CLB70 cell line was less susceptible to the pro‐apoptotic effect of USP7 inhibition, but 27.6% ± 2.7% apoptotic cells still were detected after 24 hours of incubation with 8 μM of the tested inhibitor. In the most resistant cell line GL‐1, most of the cells were alive under these conditions, and apoptotic cells accounted for 13.2% ± 6.8%. Longer incubation with P5091 (48 hours) only slightly increased the percentage of apoptotic cells in each investigated cell line (Figure 3C‐F).
FIGURE 3.
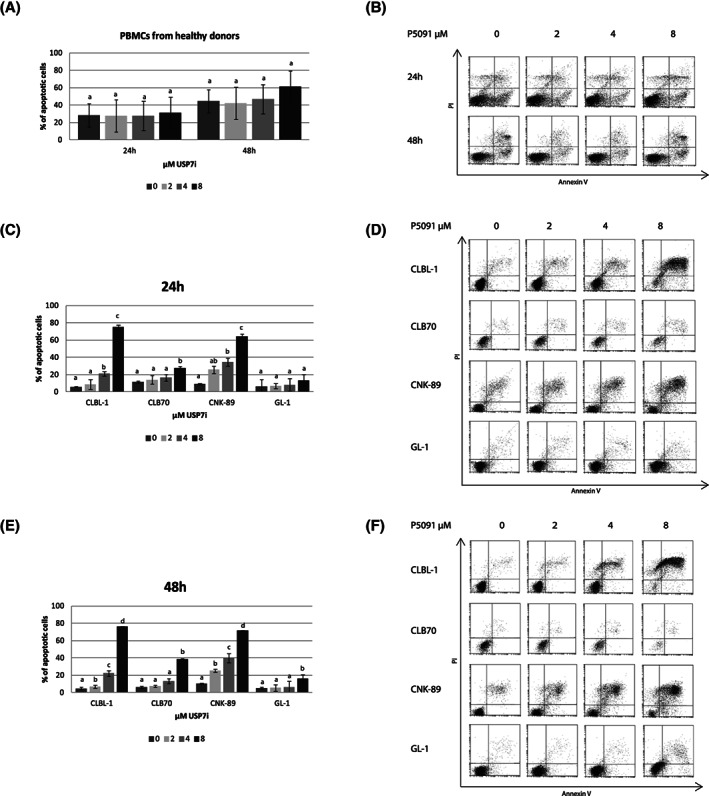
Effect of the ubiquitin‐specific protease 7 (USP7) inhibition on the induction of apoptosis. Results of annexin V/propidium iodide (PI) staining after treatment for 24 and 48 hours with P5091 for canine lymphocytes obtained from healthy donors (n = 4), A, with representative dotplots, B. Analysis as in A, but for CLBL‐1, CLB70, CNK‐89 and GL‐1 cell lines after 24 hours, C, and 48 hours, E, of treatment with P5091, with representative dotplots, D, F, respectively. For each cells, values without common letters (a, b, c) in the superscript differ statistically (P < .05)
3.5. Inhibiting USP7 triggers DNA damage in canine lymphoma/leukemia cell lines
After establishing that P5091 triggers apoptosis in lymphoma/leukemia cells, we examined if the induced apoptosis resulted from DNA damage. Therefore, phosphorylation of H2AX on serine 139, named γH2AX and generally considered as a marker of DNA damage, 38 , 39 was studied by Western blot under identical conditions as during the apoptosis assessment. Figure 4A‐D shows that treatment of all used canine lymphoma/leukemia cells with P5091 increased the amount of γH2AX at both time points. To ensure that DNA damage was the cause and increased phosphorylation of H2AX was not the effect of cell apoptosis, the test also was performed after a shorter incubation with P5091 (2, 4, 6, and 8 hours). Because the study substantially shortened the incubation time, it was decided to use higher concentrations of the inhibitor so as to visualize the changes taking place in the cell so early (up to 10 μM). Inhibition of USP7 triggered H2AX phosphorylation after a 2‐ to 4‐hour treatment, a time at which no apoptosis was expected (Figure 4E‐H).
FIGURE 4.
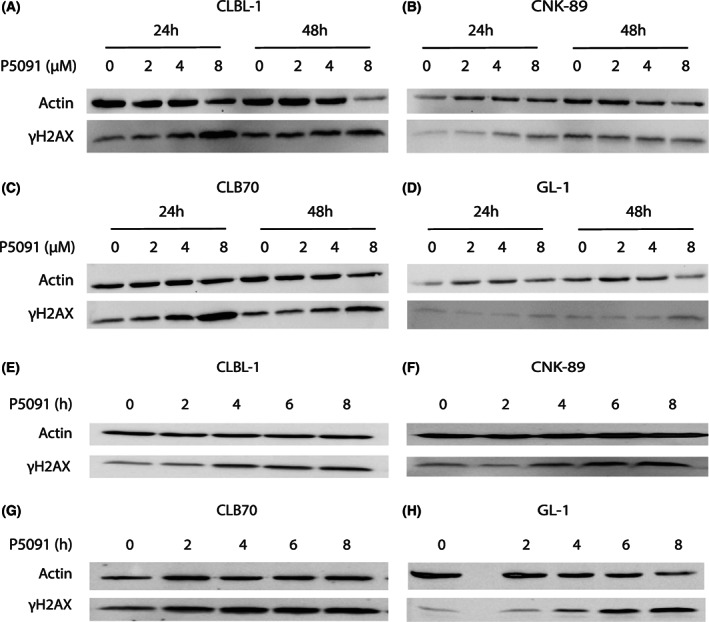
Ubiquitin‐specific protease 7 (USP7) triggers DNA damage in a concentration‐ and time‐dependent manner. Western blot analysis for phosphorylated H2AX (γH2AX) of CLBL‐1, CNK‐89, CLB70, and GL‐1 cells after 24 and 48 hours of incubation with different concentrations (2, 4, and 8 μM) of P5091, A‐D. Western blot analysis as in A‐D, in CLBL‐1, CNK‐89, CLB70, and GL‐1 cells after incubation with 10 μM of P5091 for various incubation times (2, 4, 6, and 8 hours), E‐H
4. DISCUSSION
Our aim was to determine whether USP7 could be a potential therapeutic target in hematopoietic cancers in dogs. Because USP7 plays a role in various physiological processes in different cells, numerous studies indicate that its altered expression and function underlie many diseases, including cancers. 23
The most common anomaly associated with USP7 in cancer is its overexpression. 13 Our study indicated that USP7 expression is higher in hematopoietic cancers in dogs than in normal lymphocytes from healthy donors. Because ours is the first study performed in dogs and we lacked a positive control overexpressing USP7, we assessed USP7 expression in canine cells by comparing it with USP7 expression in the U2OS human osteosarcoma cell line, characterized by increased USP7 expression. 37 Despite the relatively low number of analyzed samples (8 from healthy donors and 8 from dogs with lymphoma), statistical analysis showed significant differences (Figure 1D) in USP7 expression between these 2 groups. Considering the limited number of samples, we did not draw conclusions on the type of lymphoma and the level of USP7 expression. However, the investigated panel of cell lines had increased USP7 expression in other types of canine cancers as well, such as osteosarcoma and mammary tumors. As in research in humans, where enhanced expression indicated susceptibility of cancer cells to pharmacological inhibition of USP7, 16 , 26 , 28 , 40 our results suggested that USP7 may be a potential therapeutic target in lymphomas in dogs.
After confirming USP7 overexpression in canine cells, our next step was to determine the sensitivity of these cells to the pharmacological inhibition of this enzyme. Numerous studies indicate that USP7 inhibitors restrain the proliferation of cancer cells, as demonstrated in studies using chronic human leukemia cells, 41 in colorectal carcinoma cell lines and tissues 10 or human melanoma cells. 22 Knowing this action of USP7i, we first evaluated the cytotoxic effect of P5091 using the MTT test. The assay identified significant sensitivity of the investigated canine lymphoma/leukemia cell lines to pharmacological inhibition of USP7 activity. Comparing the IC50 of P5091 for human cell lines of colon carcinoma, 10 multiple myeloma, 30 and melanoma, 22 it can be concluded that in general canine lymphoma and leukemia cell lines are much more sensitive to the toxic effects of inhibition of USP7 than are some human cell lines. The IC50 for canine lymphomas/leukemias cell lines is an order of magnitude lower than that for the mentioned human cell lines. In summary, our study identified the investigated canine lymphoma/leukemia cell lines as being highly sensitive to the antiproliferative effect of P5091.
Ubiquitin‐specific protease 7 controls many proteins involved in cell cycle progression such as Chfr, Chk1, Claspin, and p53. 21 As a result of USP7i treatment, many types of cancer cells arrest in the G1 phase of the cell cycle, most likely because of stabilization of the p53 protein. 8 , 9 , 42 Therefore, we evaluated the changes in the cell cycle progression of canine cancer cells after P5091 treatment. Contrary to expectations, we did not find a strong effect of USP7i on the course of cell cycle progression. Although we found that cell proliferation was slower, no effect on a specific cell cycle phase was observed. Such a result may indicate that the mechanism of action of the P5091 inhibitor on canine lymphoma/ leukemia cells is not a consequence of the effect of inhibition of USP7 on p53 protein stabilization. These observations confirm the results of Western blot analysis, in which no activation of the p53 pathway was observed after treatment with P5091 (A. Pawlak, unpublished data).
Having analyzed cell cycle progression, we investigated if USP7 inhibition causes any particular type of cell death. We showed that inhibition of USP7 in canine lymphoma/leukemia cells may cause apoptosis (Figure 3). This result was much weaker than the antiproliferative effect but at least 2 features of apoptosis were confirmed for all cell lines: externalization of phosphatidylserine (PS) and poly (ADP‐ribose) polymerase (PARP) fragmentation. Induction of apoptosis is the second best known mechanism of USP7i anti‐tumor activity, and also caused by the previously mentioned activation of the p53 pathway. 3 , 7 , 9 , 42 , 43 Induction of apoptosis as a result of this pathway activation is the best known and documented mechanism of USP7i action, but apparently such was not the case in the tested canine lymphoma and leukemia cell lines. This finding is consistent with our observations that apoptosis begins rapidly after incubation with USP7i (after 24 hours of treatment the cells were already in late apoptosis), which suggests the presence of another, p53‐independent, mechanism of apoptosis. Such a p53‐independent mechanism of USP7 inhibition was reported for different cell lines. 23 One of these mechanisms is based on the fact that USP7 inhibition may alter homologous recombination repair (HRR) of DNA double strand breaks (DSBs) in some types of cancer cells. For example, USP7 is upregulated in chronic lymphocytic leukemia (CLL) cells and its loss or inhibition disrupts HRR by modulating the stability of the DNA damage‐responsive E3 ubiquitin ligase RAD18. 16 Targeting USP7 in CLL patients thus may have therapeutic potential even in tumors with defective p53 or those resistant to ibrutinib (Bruton's tyrosine kinase inhibitor). Another proof for a p53‐independent role of USP7 in regulating DSB repair is a previous study, 17 that determined that depletion of USP7 in cervical cancer cells impaired engagement of the MRN‐MDC1 complex and consequent recruitment of downstream factors p53‐binding protein 1 (53BP1) and breast cancer protein 1 (BRCA1), critical factors in the early response after DNA damage at sites of DNA lesions. Destabilization of MDC1 disrupts cervical cancer cell survival and cellular resistance to genotoxic insults, supporting the pursuit of USP7 as a potential therapeutic target for MDC1‐proficient cancers. 17 A different p53‐independent mechanism of USP7 inhibition also leading to DNA damage was found in human melanoma cells. Inhibition of DUB activity by USP7i caused ER stress by causing accumulation of polyubiquitinated proteins in cancer cells and increasing concentrations of intracellular reactive oxygen species (ROS). 22 Although moderate concentrations of intracellular ROS can promote cell proliferation, survival, and transformation, excessive ROS concentrations can induce apoptosis by disrupting intracellular macromolecules such as proteins, lipids, and nucleic acids. 44 Because no activation of the p53 pathway was identified in the examined canine hematopoietic tumor cells, the examples of USP7i modes of action presented above are feasible. This hypothesis is supported by the fact that DNA damage was found to be an effect of USP7i in canine lymphoma/leukemia cells. Initially, we observed phosphorylated H2AX, a marker of DNA damage, after 24 and 48 hours of incubation with P5091. After analyzing the results, when it became clear that p53 likely was not fully responsible for the cytotoxic effect of the tested inhibitor, we investigated if DNA damage was not the cause of the observed cell death. The positive staining for phosphorylated H2AX already was detected after 2 hours of incubation with USP7i, which led to the hypothesis that it is the actual cause of cell death, and this mechanism is in fact independent of the p53 pathway. Particularly interesting results were obtained for the GL‐1 cell line, where DNA damage clearly was visible after treatment with the USP7 inhibitor (Figure 4H), whereas only slight changes were observed after 24 to 48 hours incubation. One possible interpretation of this observation is that the DNA repair mechanisms are working more efficiently in GL‐1 cells than in the other cell lines used in our study. The GL‐1 cell line, compared to the other lymphoma/leukemia cell lines used, is less sensitive to the effects of various substances and drugs with antitumor activity, 45 which may be related to an efficient DNA repair system. This observation substantiates the proposed hypothesis. The observed DNA damage after USP7 inhibition in canine lymphoma/leukemia cells requires further in‐depth research.
Despite great interest in the possible use of USP7 inhibition as a therapeutic target and many years of research, none of the USP7 inhibitors has entered the clinical trial phase yet, probably because of the very extensive effects of USP7 protein inhibition itself as well as additional effects of individual inhibitors. The potential of the antitumor inhibition of USP7 appears to be beyond doubt and we therefore believe that use of a canine model (first in vitro and then in vivo) may provide valuable information and influence the pace of research in trials of human patients. Designing research in the field of veterinary oncology, where studies on dogs precede and support discoveries in human medicine, is becoming an important direction in contemporary comparative oncology.
5. CONCLUSIONS
Having established that USP7 expression differs in canine lymphoma cells and normal lymphocytes, we hypothesized that this protein is involved in tumor development or the course of this type of malignancy in dogs or both. In addition, the selective cytotoxic activity of P5091i on canine lymphoma cells indicated USP7 as a potentially interesting therapeutic target. Additional research is needed to clarify the mechanism of USP7i action in canine lymphoma/leukemia cells and to optimize the therapeutic potential of USP7 inhibition in cancer treatment in dogs. At this point, it can be stated that inhibition of USP7 may be an interesting therapeutic target in hematopoietic cancers in dogs. Knowing the role of non‐Hodgkin's lymphoma in dogs as a model for the same disease in humans, clinical tests in dogs could become the basis for the introduction of similar treatments in human medicine.
CONFLICT OF INTEREST DECLARATION
Authors declare no conflict of interest.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
Authors declare no IACUC or other approval was needed.
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
ACKNOWLEDGMENTS
We are grateful to B.C. Ruetgen (Institute of Immunology, Department of Pathobiology, University of Veterinary Medicine, Vienna) for providing CLBL‐1 cell line, Y. Fujino and H. Tsujimoto (Department of Veterinary Internal Medicine, University of Tokyo) for providing the GL‐1 and CL‐1 cell lines and Dr Rutteman (Utrecht University, the Netherlands) for providing the P114 cell line. The publication is financed under the Leading Research Groups support project from the subsidy increased for the period 2020–2025 in the amount of 2% of the subsidy referred to Art. 387 (3) of the Law of 20 July 2018 on Higher Education and Science, obtained in 2019. This work was supported by the Ministerio de Ciencia, Innovación y Universidades (SAF2016‐80626‐R to RF and VAJS), co‐funded by EU‐ERDF.
Pawlak A, Bajzert J, Bugiel K, et al. Ubiquitin‐specific protease 7 as a potential therapeutic target in dogs with hematopoietic malignancies. J Vet Intern Med. 2021;35:1041–1051. 10.1111/jvim.16082
Funding information Leading Research Groups; Ministerio de Ciencia, Innovación y Universidades, Grant/Award Number: SAF2016‐80626‐R
REFERENCES
- 1. Everett RD, Meredith M, Orr A, Cross A, Kathoria M, Parkinson J. A novel ubiquitin‐specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 1997;16:1519‐1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fernandez‐Montalvan A, Bouwmeester T, Joberty G, et al. Biochemical characterization of USP7 reveals post‐translational modification sites and structural requirements for substrate processing and subcellular localization. FEBS J. 2007;274:4256‐4270. [DOI] [PubMed] [Google Scholar]
- 3. Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007;26:923‐934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ramakrishna S, Suresh B, Baek KH. The role of deubiquitinating enzymes in apoptosis. Cell Mol Life Sci. 2011;68:15‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reyes‐Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin‐specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheon KW, Baek KH. HAUSP as a therapeutic target for hematopoietic tumors (review). Int J Oncol. 2006;28:1209‐1215. [PubMed] [Google Scholar]
- 7. Li M, Chen D, Shiloh A, et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416:648‐653. [DOI] [PubMed] [Google Scholar]
- 8. Colland F, Formstecher E, Jacq X, et al. Small‐molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther. 2009;8:2286‐2295. [DOI] [PubMed] [Google Scholar]
- 9. Fan YH, Cheng J, Vasudevan SA, et al. USP7 inhibitor P22077 inhibits neuroblastoma growth via inducing p53‐mediated apoptosis. Cell Death Dis. 2013;17:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. An T, Gong Y, Li X, et al. USP7 inhibitor P5091 inhibits Wnt signaling and colorectal tumor growth. Biochem Pharmacol. 2017;131:29‐39. [DOI] [PubMed] [Google Scholar]
- 11. Bernardi R, Ghia P. Reactivating nuclear PTEN to treat CLL. Oncotarget. 2017;8(22):35486‐35487. 10.18632/oncotarget.17543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang M, Zhang Y, Wang T, et al. The USP7 inhibitor P5091 induces cell death in ovarian cancers with different P53 status. Cell Physiol Biochem. 2017;43:1755‐1766. [DOI] [PubMed] [Google Scholar]
- 13. Nicholson B, Suresh Kumar KG. The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem Biophys. 2011;60:61‐68. [DOI] [PubMed] [Google Scholar]
- 14. Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53‐Mdm2 pathway. Mol Cell. 2004;13:879‐886. [DOI] [PubMed] [Google Scholar]
- 15. Alonso‐de Vega I, Martin Y, Smits VA. USP7 controls Chk1 protein stability by direct deubiquitination. Cell Cycle. 2014;13:3921‐3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Agathanggelou A, Smith E, Davies NJ, et al. USP7 inhibition alters homologous recombination repair and targets CLL cells independently of ATM/p53 functional status. Blood J Am Soc Hematol. 2017;130:156‐166. [DOI] [PubMed] [Google Scholar]
- 17. Su D, Ma S, Shan L, et al. Ubiquitin‐specific protease 7 sustains DNA damage response and promotes cervical carcinogenesis. J Clin Invest. 2018;128:4280‐4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qian J, Pentz K, Zhu Q, et al. USP7 modulates UV‐induced PCNA monoubiquitination by regulating DNA polymerase eta stability. Oncogene. 2015;34:4791‐4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Faustrup H, Bekker‐Jensen S, Bartek J, Lukas J, Mailand N. USP7 counteracts SCFbetaTrCP‐ but not APCCdh1‐mediated proteolysis of Claspin. J Cell Biol. 2009;184:13‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jagannathan M, Nguyen T, Gallo D, et al. A role for USP7 in DNA replication. Mol Cell Biol. 2014;34:132‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smits VA, Freire R. USP7/HAUSP: a SUMO deubiquitinase at the heart of DNA replication. Bioessays. 2016;38:863‐868. [DOI] [PubMed] [Google Scholar]
- 22. Lee G, Oh T‐I, Um KB, et al. Small‐molecule inhibitors of USP7 induce apoptosis through oxidative and endoplasmic reticulum stress in cancer cells. Biochem Biophys Res Commun. 2016;470:181‐186. [DOI] [PubMed] [Google Scholar]
- 23. Bhattacharya S, Chakraborty D, Basu M, Ghosh MK. Emerging insights into HAUSP (USP7) in physiology, cancer and other diseases. Signal Transduct Target Ther. 2018;3:018‐0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morra F, Merolla F, Napolitano V, et al. The combined effect of USP7 inhibitors and PARP inhibitors in hormone‐sensitive and castration‐resistant prostate cancer cells. Oncotarget. 2017;22:16463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bastola P, Neums L, Schoenen FJ, Chien J. VCP inhibitors induce endoplasmic reticulum stress, cause cell cycle arrest, trigger caspase‐mediated cell death and synergistically kill ovarian cancer cells in combination with Salubrinal. Mol Oncol. 2016;10:1559‐1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Becker K, Marchenko ND, Palacios G, Moll UM. A role of HAUSP in tumor suppression in a human colon carcinoma xenograft model. Cell Cycle. 2008;7:1205‐1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hernandez‐Perez S, Cabrera E, Salido E, et al. DUB3 and USP7 de‐ubiquitinating enzymes control replication inhibitor Geminin: molecular characterization and associations with breast cancer. Oncogene. 2017;36:26. [DOI] [PubMed] [Google Scholar]
- 28. Cheng C, Niu C, Yang Y, Wang Y, Lu M. Expression of HAUSP in gliomas correlates with disease progression and survival of patients. Oncol Rep. 2013;29:1730‐1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rawat R, Starczynowski DT, Ntziachristos P. Nuclear deubiquitination in the spotlight: the multifaceted nature of USP7 biology in disease. Curr Opin Cell Biol. 2019;58:85‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chauhan D, Tian Z, Nicholson B, et al. A small molecule inhibitor of ubiquitin‐specific protease‐7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22:345‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rutgen BC, Hammer SE, Gerner W, et al. Establishment and characterization of a novel canine B‐cell line derived from a spontaneously occurring diffuse large cell lymphoma. Leuk Res. 2010;34:932‐938. [DOI] [PubMed] [Google Scholar]
- 32. Nakaichi M, Taura Y, Kanki M, et al. Establishment and characterization of a new canine B‐cell leukemia cell line. J Vet Med Sci. 1996;58:469‐471. [DOI] [PubMed] [Google Scholar]
- 33. Momoi Y, Okai Y, Watari T, Goitsuka R, Tsujimoto H, Hasegawa A. Establishment and characterization of a canine T‐lymphoblastoid cell line derived from malignant lymphoma. Vet Immunol Immunopathol. 1997;59:11‐20. [DOI] [PubMed] [Google Scholar]
- 34. Pawlak A, Ziolo E, Kutkowska J, et al. A novel canine B‐cell leukaemia cell line. Establishment, characterisation and sensitivity to chemotherapeutics. Vet Comp Oncol. 2017;15:1218‐1231. [DOI] [PubMed] [Google Scholar]
- 35. Timmermans‐Sprang EP, Gracanin A, Mol JA. High basal Wnt signaling is further induced by PI3K/mTor inhibition but sensitive to cSRC inhibition in mammary carcinoma cell lines with HER2/3 overexpression. BMC Cancer. 2015;15:015‐1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pawlak A, Rapak A, Drynda A, et al. Immunophenotypic characterization of canine malignant lymphoma: a retrospective study of cases diagnosed in Poland lower Silesia, over the period 2011‐2013. Vet Comp Oncol. 2016;1:52‐60. [DOI] [PubMed] [Google Scholar]
- 37. Turnbull AP, Ioannidis S, Krajewski WW, et al. Molecular basis of USP7 inhibition by selective small‐molecule inhibitors. Nature. 2017;550:481‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fernandez‐Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair. 2004;3:959‐967. [DOI] [PubMed] [Google Scholar]
- 39. Podhorecka M, Skladanowski A, Bozko P. H2AX phosphorylation: its role in DNA damage response and cancer therapy. J Nucleic Acids. 2010;3:920161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carra G, Panuzzo C, Torti D, et al. Therapeutic inhibition of USP7‐PTEN network in chronic lymphocytic leukemia: a strategy to overcome TP53 mutated/deleted clones. Oncotarget. 2017;8:35508‐35522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Agathanggelou A, Smith E, Davies NJ, et al. USP7 inhibition alters homologous recombination repair and targets CLL cells independently of ATM/p53 functional status. Blood. 2017;130:156‐166. [DOI] [PubMed] [Google Scholar]
- 42. Kon N, Kobayashi Y, Li M, Brooks CL, Ludwig T, Gu W. Inactivation of HAUSP in vivo modulates p53 function. Oncogene. 2010;29:1270‐1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shaw PH. The role of p53 in cell cycle regulation. Pathol Res Pract. 1996;192:669‐675. [DOI] [PubMed] [Google Scholar]
- 44. Maharjan S, Oku M, Tsuda M, Hoseki J, Sakai Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci Rep. 2014;4(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pawlak A, Rapak A, Zbyryt I, Obminska‐Mrukowicz B. The effect of common antineoplastic agents on induction of apoptosis in canine lymphoma and leukemia cell lines. In Vivo. 2014;28:843‐850. [PubMed] [Google Scholar]