

Abstract
Undifferentiated endometrial carcinoma is an aggressive type of endometrial carcinoma that typically presents with advanced stage disease and rapid clinical progression. In contrast to dedifferentiated endometrial carcinoma, undifferentiated carcinoma lacks a concurrent differentiated (typically low-grade endometrioid) carcinoma component, though the undifferentiated component of dedifferentiated carcinoma is similar histologically and immunophenotypically to pure undifferentiated carcinoma. We recently identified three mutually exclusive mechanisms of switch/sucrose non-fermentable (SWI/SNF) complex inactivation (BRG1 inactivation, INI1 inactivation or ARID1A/ARID1B co-inactivation) that are associated with histologic dedifferentiation in the majority of dedifferentiated endometrial carcinoma. In the current study, we aimed to determine by immunohistochemistry whether these patterns of SWI/SNF inactivation also occur in undifferentiated endometrial carcinomas. Of the 34 undifferentiated carcinomas examined, 17 (50%) exhibited SWI/SNF complex inactivation, with 11 tumors showing complete loss of both ARID1A and ARID1B, 5 showing complete loss of BRG1 and 1 showing complete loss of INI1. Ten of the remaining 17 undifferentiated carcinomas showed the following alterations: 5 tumors (15%) showed loss of ARID1A only with intact ARID1B, BRG1 and INI1 expression, 4 tumors (12%) showed mutated patterns of p53 staining with intact SWI/SNF protein expression, and 1 tumor (3%) harbored a POLE exonuclease domain mutation (P286R). SWI/SNF complex-inactivated tumors presented more frequently with extrauterine disease spread than those with intact expression (88% versus 41%, respectively). In addition, patients with SWI/SNF complex-inactivated tumors had a significantly worse disease-specific survival (p=0.02). The findings here demonstrate frequent SWI/SNF complex inactivation in undifferentiated endometrial carcinomas, which has future implications regarding therapies that target chromatin remodelling and epigenetic control.
Keywords: Endometrial cancer, undifferentiated carcinoma, ARID1A, ARID1B, SMARCA4, SMARCB1
Introduction
Undifferentiated endometrial carcinoma is a highly aggressive subtype of endometrial cancer. About half of the patients present with high stage disease (FIGO stage 3–4) and patients with surgically non-debulkable disease typically experience a rapidly progressive clinical course that cannot be controlled with conventional chemotherapies.1–3 Histologically, undifferentiated endometrial carcinoma is composed of a sheet-like pattern-less proliferation of monotonous tumor cells. The tumor cells typically possess scant to moderate amount of cytoplasm, which can impart a rhabdoid appearance in less cohesive areas. These tumors lack any evidence of glandular formation or squamous differentiation. Because of this undifferentiated appearance, a definitive diagnosis may be difficult on endometrial biopsy/curettage specimens. Ancillary immunohistochemical analysis demonstrating the presence of focal keratin or epithelial membrane antigen (EMA) expression in the absence of other demonstrable cellular differentiation can be used to support its diagnosis. Mismatch repair (MMR) protein immunohistochemistry can be useful as about half of undifferentiated endometrial carcinomas are MMR protein-deficient, 3, 4 whereas MMR protein deficiency is uncommon in other entities that may enter into the differential diagnosis such as undifferentiated uterine sarcoma or high-grade endometrial stromal sarcoma.5
Dedifferentiated endometrial carcinoma contains a component of undifferentiated carcinoma in addition to a component of typically low-grade endometrioid-type carcinoma.6 The undifferentiated component of dedifferentiated endometrial carcinoma exhibits similar histologic and immunophenotypic features to those seen in pure undifferentiated endometrial carcinomas. We recently identified inactivation of core components of the switch/sucrose non-fermentable (SWI/SNF) chromatin remodelling complex proteins, specifically ARID1A/ARID1B (co-inactivation), BRG1 (encoded by SMARCA4) and INI1 (encoded by SMARCB1) as the likely molecular triggers underlying the apparent “dedifferentiation” of low-grade endometrioid carcinoma in about 70% of dedifferentiated endometrial carcinomas.7–9 The loss of these core SWI/SNF complex proteins was hypothesized to prevent müllerian epithelial differentiation, thereby arresting tumor cells in an undifferentiated state with significantly reduced to absent PAX8 and estrogen receptor (ER) expression, in contrast to the differentiated endometrioid carcinoma component that expresses PAX8, ER and other epithelial markers.4, 10 Given the histologic and immunophenotypic similarities between pure undifferentiated endometrial carcinoma and the undifferentiated component of dedifferentiated endometrial carcinoma, we hypothesized that the same spectrum of SWI/SNF complex inactivation also occurs in undifferentiated endometrial carcinoma. While there is evidence supporting this notion, as demonstrated in a recent study by Ramalingam et al. that reported the loss of BRG1 or INI1 expression in about 25% of undifferentiated endometrial carcinomas,11 the full range of SWI/SNF complex inactivation mechanisms including ARID1A/ARID1B co-inactivation has not been comprehensively evaluated in undifferentiated endometrial carcinoma. In addition to SWI/SNF complex inactivation, we and others have also identified POLE exonuclease domain mutations in a small subset of dedifferentiated endometrial carcinomas.7, 8, 12, 13 In this study, we examined ARID1A, ARID1B, BRG1, INI1 and p53 expression, and POLE mutation status, in a series of undifferentiated endometrial carcinomas.
Materials and methods
Study samples
The study included 34 undifferentiated endometrial carcinomas (Table 1). These cases were obtained from Calgary Laboratory Services (Calgary, Canada), Royal Alexandra Hospital (Edmonton, Canada), Vancouver General Hospital (Vancouver, Canada), Memorial Sloan Kettering Cancer Centre (New York, United States) and King Edward Memorial Hospital (Perth, Australia). All of the endometrial carcinomas included in this study were from hysterectomy specimens and all cases were centrally reviewed (M.K, C.H.L and C.J.R.S), fulfilling the morphologic features described by Silva et al.1, 2 The study was approved by the Institutional Review Board (University of Alberta, Pro00042667).
Table 1:
Clinicopathologic features of the 34 undifferentiated endometrial carcinomas.
| Case | Age (year) | FIGO 2009 stage | Follow-up period (year) | Status at last follow-up | p53 | PAX8 | ER | MLH1 | PMS2 | MSH2 | MSH6 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 55 | 3B | 0.25 | DOD | wt | (+)(<10%) | (−) | loss | loss | intact | intact |
| 2 | 55 | 4B | 0.67 | AWD | wt | (−) | (−) | loss | loss | intact | intact |
| 3 | 54 | 4B | 0.33 | AWD | wt | (−) | (−) | loss | loss | intact | intact |
| 4 | 66 | 3A | 1.00 | DOD | wt | (−) | (−) | loss | loss | intact | intact |
| 5 | 69 | 4A | 3.58 | NED | wt | (−) | (−) | loss | loss | intact | intact |
| 6 | 56 | 4A | 0.25 | DOD | wt | (−) | (−) | loss | loss | intact | intact |
| 7 | 52 | 3C | 0.08 | DOD | wt | (+) (50%) | (−) | intact | intact | intact | intact |
| 8 | 48 | 4B | 0.21 | DOD | wt | (−) | (−) | intact | intact | intact | intact |
| 9 | 48 | 4B | NA | NA | wt | (−) | (−) | intact | intact | intact | intact |
| 10 | 57 | 1B | 5.17 | NED | wt | (−) | (−) | intact | intact | intact | intact |
| 11 | 65 | 4B | 0.13 | DOD | wt | (−) | (−) | intact | intact | intact | intact |
| 12 | 56 | 2A | 1.67 | NED | wt | (−) | (−) | intact | intact | intact | loss |
| 13 | 54 | 3C | 0.67 | DOD | wt | (−) | (−) | loss | loss | intact | intact |
| 14 | 59 | 4B | 0.42 | DOD | wt | (−) | (−) | intact | intact | intact | intact |
| 15 | 65 | 4A | 0.13 | DOD | mutated | (−) | (−) | intact | intact | intact | intact |
| 16 | 26 | 4B | 0.42 | DOD | wt | (−) | (−) | intact | intact | intact | intact |
| 17 | 62 | 3B | 0.67 | DOD | wt | (−) | (−) | loss | loss | intact | intact |
| 18 | 62 | 4B | 1.50 | NED | wt | (+)(100%) | (+) | intact | intact | intact | intact |
| 19 | 58 | 3B | 0.50 | DOD | mutated | (−) | (−) | intact | loss | intact | intact |
| 20 | 63 | 3C | 0.25 | DOD | wt | (+)(<10%) | (−) | loss | loss | intact | intact |
| 21 | 75 | 3C | 0.60 | AWD | wt | (−) | (−) | intact | loss | intact | intact |
| 22 | 56 | 2B | 6.50 | NED | wt | (−) | (−) | intact | intact | loss | loss |
| 23 | 41 | 1B | 5.00 | NED | wt | (−) | (−) | intact | intact | intact | intact |
| 24 | 56 | 1B | NA | NA | wt | (−) | (−) | loss | loss | intact | intact |
| 25 | 59 | 1A | 3.00 | NED | wt | (−) | (−) | loss | loss | intact | intact |
| 26 | 57 | 4B | 2.33 | DOD | wt | (−) | (−) | loss | loss | intact | intact |
| 27 | 66 | 4B | 0.83 | DOD | wt | (−) | (−) | intact | intact | intact | intact |
| 28 | 74 | 2A | 2.17 | NED | wt | (−) | (−) | intact | intact | intact | intact |
| 29 | 74 | 4A | 3.00 | DOD | wt | (−) | (−) | intact | intact | intact | intact |
| 30 | 54 | 2B | 10.00 | DOUC | wt | (−) | (−) | intact | intact | intact | intact |
| 31 | 93 | 1B | 0.40 | DOD | mutated | (−) | (−) | loss | loss | intact | intact |
| 32 | 67 | 1A | 3.00 | DOUC | mutated | (−) | (−) | intact | intact | intact | intact |
| 33 | 63 | 1A | 3.42 | NED | mutated | (−) | (−) | intact | intact | intact | intact |
| 34 | 78 | 1B | 3.00 | DOD | mutated | (−) | (−) | intact | intact | intact | intact |
wt: wild-type pattern; FIGO: The International Federation of Gynecology and Obstetrics; ER: estrogen receptor; DOD: died of disease; AWD: alive with disease; NED: no evidence of disease; DOUC: died of other cause; NA: follow-up information not available.
Immunohistochemistry and interpretation
Immunohistochemistry was performed on whole tissue sections. The slides were incubated with antibodies to ARID1A (1:200, HPA005456, Sigma), ARID1B (1:100, clone 2D2, H00057492-M01, Abnova), BRG1 (1:25, clone EPNCIR111A, ab110641, Abcam) and INI1 (1:50, 25/BAF47, 612110, BD Biosciences); BRG1 and INI1 were processed using Ventana Benchmark XT (Ventana Medical Systems, Tucson, AZ, USA), while ARID1A and ARID1B were processed using Dako Omnis Autostainer (Dako Canada ULC, Mississauga, Ontario, Canada). The detection system used was the Bond polymer refine (Leica Microsystems, Wetzlar, Germany). For mismatch repair proteins (MLH1, MSH2, MSH6 and PMS2), the primary antibodies used and the staining methods are as reported previously.8 The slides were incubated with MLH1 (DAKO clone ES05 1:100), MSH2 (NCL clone 25D12 prediluted), MSH6 (BD Bioscience 44/MSH6 1:2000), PMS2 (BD Bioscience A16–4 1:100) and processed using the Leica Bond Max platform (Leica Microsystems, Wetzlar, Germany) as per manufacturer’s protocol with proprietary reagents.
PAX8, ER and p53 immunohistochemistry was performed as previously described.10 The mouse monoclonal PAX8 antibody, clone BC12 (catalog number: ACI 438) was obtained from Biocare Medical (Concord, California, United States); the rabbit monoclonal ER antibody, clone SP1 (catalog number: RM-9101) was obtained from Thermo Fisher Scientific (Ottawa, Ontario, Canada); the mouse monoclonal p53 antibody, clone DO-7 (catalog number: M7001) was obtained from Dako (Burlington, Ontario, Canada). Primary incubations were performed for 60 min at 1:100 dilution for PAX8, 60 min at 1:25 dilution for ER and 32 min at 1:800 dilution for p53, all at 37C using Ventana antibody diluents. The Ventana Universal Secondary Antibody was used for 32 min at 37°C. The detection system used was the Ventana DABMap kit (ER) and Ventana OptiView DAB kit (p53).
For ARID1A, ARID1B, BRG1, INI1 and MMR proteins, cases were scored as showing intact expression if any tumor cells showed nuclear staining and deficient if the nuclei were unstained in the presence of internal positive control immunoreactivity. For PAX8, ER and p53, only nuclear staining was considered and evaluated. PAX8 and ER immunostains were scored as positive if any tumor cells exhibited moderate to strong positive (definite) nuclear staining; tumors were considered PAX8-negative or ER-negative only if there was adequate nuclear staining of internal positive control tissue. p53 immunostain was considered to be abnormal (mutated pattern) if there was either diffuse moderate to strong uniform nuclear staining in ≥ 70% of tumor cells (overexpression), or complete absence of nuclear staining in the presence of focal stromal cell staining (null pattern ). p53 immunostain was considered normal (wild type pattern) if any degree of non-diffuse nuclear staining (<70%) of the tumor cells was present.14
POLE exonuclease domain sequencing
All tumor blocks underwent pathology review to identify regions with high (>50%) tumor cellularity. DNA was extracted from tissue cores; the purity and yield were determined using a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Primer sets that cover the exonuclease domain regions of POLE in which mutations were previously identified in endometrial carcinomas were used to amplify exonuclease domain genomic regions – exon 9 (forward: 5’-TGTTCAGGGAGGCCTAATGG-3’; reverse: 5’-AACAAATACTAACAGTGGGG-3’), exon 10 (forward: 5’-GCTGCAATTCTGATCTGACG-3’; reverse: 5’-CAGCCTCTGACTTGTGCTGA-3’), exon 11 (forward: 5’- CTTCTGAACTTTGGGAGAGG-3’; reverse: 5’- CACCTCCTAAGTCGACATGG-3’), exon 12 (forward: 5’- GCATTAGAGCCTGACCTGC-3’; reverse: 5’- ACAGCACAGTCTGCAAGAGG-3’), exon 13 (forward: 5’- CGGGATGTGGCTTACGTGC-3’; reverse: 5’- TTGCATCTGTCTGTGTGGTG-3’), exon 14 (forward: 5’- TCTGTGCTTCACACTTGACC-3’; reverse: 5’- GACATCCACCTCCATTCAGC-3’) as previously described.15 PCR amplifications were performed as previously described using 50ng genomic DNA and the primer sets using High-Fidelity Tag DNA polymerase (Invitrogen, Carlsbad, CA, USA) 16. Prior to sequencing, PCR amplicons were electrophoresed and visualized to confirm the presence of desired amplicons and absence of off-target amplification products. PCR amplicons were then purified using Axygen™ AxyPrep Mag™ PCR Clean-up Kits according to manufacturer’s protocol (Axygen Biosciences, Union City, CA, USA) and resuspended in 30ul double-distilled water. Direct bi-directional sequencing was performed on a 3730xl DNA Analyzer (Applied Biosystems, Carlsbad, CA) with 96 capillaries. Sequencing reactions were performed as previously described and all mutation positive samples were re-sequenced to confirm mutational status.15
Statistical analysis
Univariate disease-specific survival analysis was performed by generating Kaplan-Meier curves and log-rank statistics were applied. A p-value < 0.05 was considered statistically significant.
Results
Clinicopathologic features
The clinicopathologic features of the 34 undifferentiated endometrial carcinomas are summarized in Table 1. The average age was 60 years (range 26 to 93 years). The majority of patients presented with stage 3 or 4 disease (22/34, 65%), and approximately half of the patients died from their disease within a year of initial presentation.
Histologically, all tumors demonstrated a cellular sheet-like growth of monomorphic cells that possessed scanty to moderate amount of cytoplasm (Figures 1 and 2). There was no differentiated element/architecture such as squamous differentiation or glandular/papillary formation. Mitotic activity was high in all cases (> 20 MF/10 HPF) and necrosis was present in all cases, with prominent geographic necrosis noted in 5 cases (Figure 2A). The majority of cases (28 of 34 cases) exhibited areas of cellular discohesion and rhabdoid cytologic features. Scattered tumor giant cells were seen in 3 cases. Immunophenotypically, keratin stains (AE1/3, CAM5.2) and EMA showed at least focal positivity in all cases. One case expressed ER and PAX8 staining was positive in 4 cases (focal in 2 cases with <10% tumor cell staining). MMR protein staining showed deficient expression in 17 cases (13 with MLH1 and PMS2 loss, 1 with MSH2 and MSH6 loss, 2 with isolated PMS2 loss and 1 with only MSH6 loss). P53 immunohistochemistry showed a wild-type pattern in 28 cases, while 6 showed mutated staining patterns (4 with diffuse strong nuclear staining and 2 with null pattern staining).
Figure 1: Representative cases of undifferentiated endometrial carcinomas showing ARID1A/ARID1B co-deficiency.
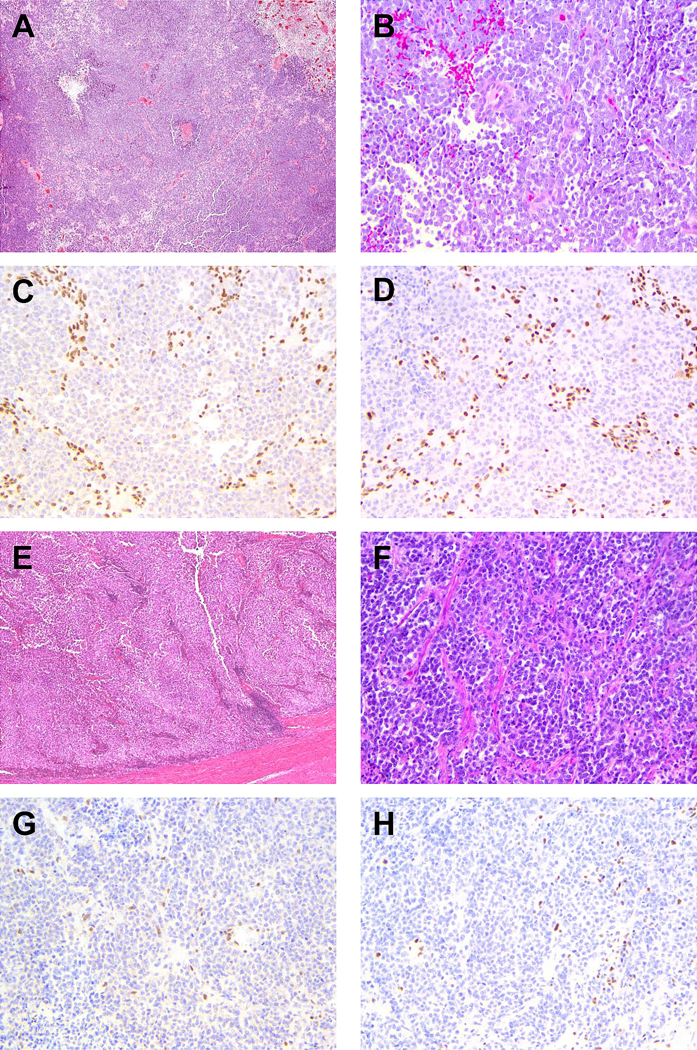
A and B) low magnification and high magnification H&E images of case 3. C and D) case 3 showing loss of ARID1A and ARID1B nuclear staining respectively in the presence of stromal internal positive control. E and F) low magnification and high magnification H&E images of case 10, which shows significant tumor necrosis. G and H) case 10 showing loss of ARID1A and ARID1B nuclear staining respectively in the presence of stromal internal positive control.
Figure 2: Representative cases of undifferentiated endometrial carcinomas showing BRG1 deficiency or isolated ARID1A deficiency.
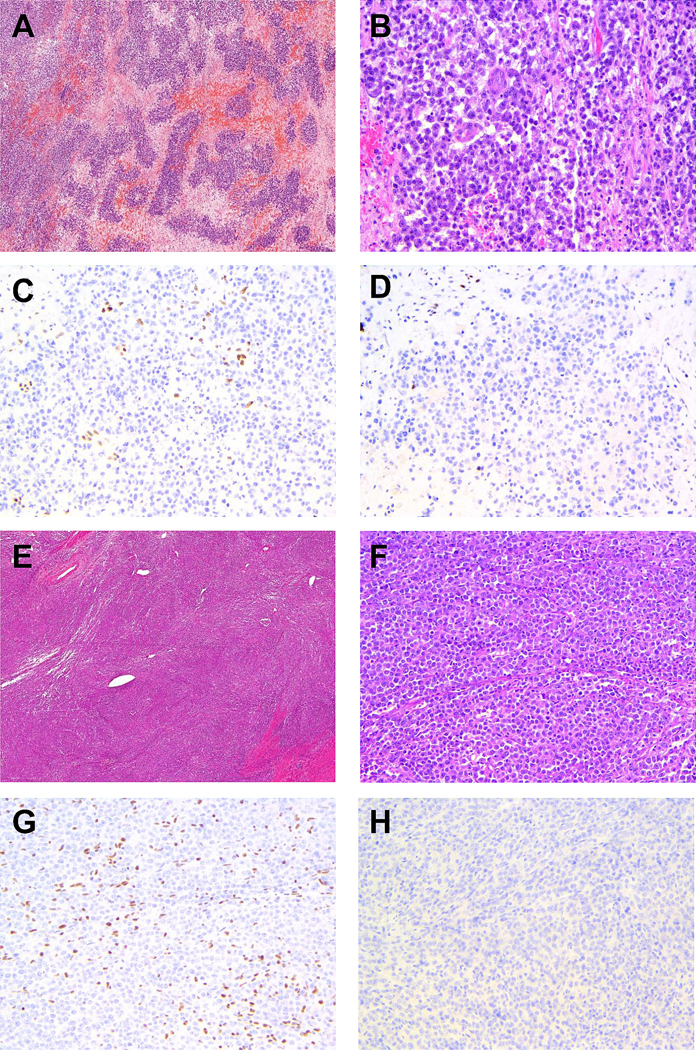
A and B) low magnification and high magnification H&E images of case 12. C) case 12 showing loss of BRG1nuclear staining in the presence of stromal internal positive control. D) case 12 showing absent MSH6 nuclear staining in the presence of stromal internal positive control. E and F) low magnification and high magnification H&E images of case 22. G) case 22 showing loss of ARID1A in the presence of internal positive control. H) case 22 showing absent PAX8 nuclear staining.
SWI/SNF complex protein immunohistochemistry and POLE mutation status
Of the 34 cases, 17 (50%) exhibited one of the three patterns of SWI/SNF complex inactivation that were associated with dedifferentiation in endometrial cancer,7, 8 with 11 tumors showing complete loss of both ARID1A and ARID1B (Figure 1), 5 showing complete loss of BRG1 (Figure 2A–D) and 1 showing complete loss of INI1 (Table 2). Among these 17 SWI/SNF complex-inactivated tumors, 16 showed wild-type p53 staining with 1 BRG1-deficient tumor (case 15) showing a mutated staining pattern. PAX8 expression was absent in 15 of 17 SWI/SNF complex-inactivated tumors, with one case (case 1, ARID1A/1B co-deficient) showing very focal and weak staining (<10% cells positive) and another case (case 7, ARID1A/1B co-deficient) showing PAX8 expression in 50% of tumor cells. ER was negative in all 17 SWI/SNF complex-inactivated tumors, and MMR protein-deficiency was observed in 9 of these cases (53%).
Table 2:
The results of BRG1, INI1, ARID1A and ARID1B immunohistochemistry, and POLE exonuclease domain mutation analysis in 34 undifferentiated endometrial carcinomas.
| Case | BRG1 | INI1 | ARID1A | ARID1B | POLE status |
|---|---|---|---|---|---|
| 1 | Intact | Intact | Loss | Loss | wt |
| 2 | Intact | Intact | Loss | Loss | wt |
| 3 | Intact | Intact | Loss | Loss | wt |
| 4 | Intact | Intact | Loss | Loss | wt |
| 5 | Intact | Intact | Loss | Loss | wt |
| 6 | Intact | Intact | Loss | Loss | wt |
| 7 | Intact | Intact | Loss | Loss | wt |
| 8 | Intact | Intact | Loss | Loss | wt |
| 9 | Intact | Intact | Loss | Loss | wt |
| 10 | Intact | Intact | Loss | Loss | wt |
| 11 | Intact | Intact | Loss | Loss | wt |
| 12 | Loss | Intact | Intact | Intact | wt |
| 13 | Loss | Intact | Intact | Intact | wt |
| 14 | Loss | Intact | Intact | Intact | wt |
| 15 | Loss | Intact | Intact | Intact | wt |
| 16 | Loss | Intact | Intact | Intact | wt |
| 17 | Intact | Loss | Loss | Intact | wt |
| 18 | Intact | Intact | Loss | Intact | mutated (P286R) |
| 19 | Intact | Intact | Loss | Intact | wt |
| 20 | Intact | Intact | Loss | Intact | wt |
| 21 | Intact | Intact | Loss | Intact | wt |
| 22 | Intact | Intact | Loss | Intact | wt |
| 23 | Intact | Intact | Loss | Intact | wt |
| 24 | Intact | Intact | Intact | Intact | wt |
| 25 | Intact | Intact | Intact | Intact | wt |
| 26 | Intact | Intact | Intact | Intact | wt |
| 27 | Intact | Intact | Intact | Intact | wt |
| 28 | Intact | Intact | Intact | Intact | wt |
| 29 | Intact | Intact | Intact | Intact | wt |
| 30 | Intact | Intact | Intact | Intact | wt |
| 31 | Intact | Intact | Intact | Intact | wt |
| 32 | Intact | Intact | Intact | Intact | wt |
| 33 | Intact | Intact | Intact | Intact | wt |
| 34 | Intact | Intact | Intact | Intact | wt |
wt: wild-type.
Among the remaining 17 undifferentiated carcinomas, 5 (cases 18 to 23) (15%) showed loss of ARID1A only (with intact ARID1B, BRG1 and INI1 expression) and wild-type POLE status (Figure 2E–H); one of the 5 ARID1A-deficient tumors showed a mutated p53 staining pattern. Four tumors (12%) showed mutated p53 staining pattern with intact SWI/SNF protein expression and wild-type POLE status and one (3%) harbored a POLE exonuclease domain mutation (P286R). The remaining seven tumors (21%) showed no alterations, displaying intact SWI/SNF protein expression, wild-type p53 staining and a lack of POLE exonuclease domain mutation. The single POLE-mutated tumor was the only case in this cohort that showed both PAX8 and ER immunopositivity.
Clinical significance of SWI/SNF complex inactivation
Clinical follow-up was available in 32 of 34 cases. Patients with tumors showing ARID1A/B co-deficiency, BRG1 deficiency or INI1 deficiency more frequently presented with extrauterine disease (88% versus 41%), and these SWI/SNF complex-inactivated cases had a significantly worse disease-specific survival (Figure 3) (p=0.02).
Figure 3:

Kaplan-Meier disease specific survival analysis comparing SWI/SNF complex-inactivated undifferentiated endometrial carcinomas (BRG1-deficient, INI1-deficient or ARDI1A/1B co-deficient; indicated by solid black line) versus tumors lacking these patterns of SWI/SNF complex deficiencies (indicated by solid gray line).
Discussion
We examined here the expression of selected SWI/SNF complex, MMR and p53 proteins as well as POLE exonuclease domain mutation status in a series of undifferentiated endometrial carcinomas. Similar to earlier observations in dedifferentiated endometrial carcinoma,7, 8 we identified a spectrum of SWI/SNF complex inactivation in about half of the cases examined, with the most common pattern of inactivation being a combined loss of ARID1A and ARID1B expression, followed by loss of BRG1 expression and less frequently loss of INI1 expression. This demonstrates shared oncogenic mechanisms between undifferentiated carcinomas and dedifferentiated carcinomas, and raises the possibility that at least a subset of undifferentiated endometrial carcinomas may have arisen through dedifferentiation with the precursor differentiated carcinoma component either completely overgrown or not sampled. An additional subset of undifferentiated carcinomas showed only loss of ARID1A with intact expression of other SWI/SNF complex proteins surveyed. The significance of isolated loss of ARID1A expression is unclear in the context of undifferentiated carcinoma as this occurs not infrequently in well differentiated endometrioid carcinomas and it was not associated with histologic dedifferentiation in dedifferentiated endometrial carcinomas.8, 17 Like dedifferentiated endometrial carcinomas, we also observed frequent MMR protein deficiency in undifferentiated endometrial carcinoma, with about half of the cases being MMR protein-deficient irrespective of SWI/SNF complex status. 3, 4 This suggests a tendency for undifferentiated carcinoma to occur in a MMR protein-deficient molecular background.
The SWI/SNF chromatin remodelling complex regulates transcription through repositioning of nucleosomes.18–20 In the SWI/SNF A complex (ubiquitously expressed in mammals), BRG1 is a core catalytic subunit whereas ARID1A and ARID1B serve as the DNA binding/targeting components. Loss of either BRG1 or ARID1A/1B therefore results in inactivation of the SWI/SNF A complex. At the cellular level, SWI/SNF inactivation can disrupt cell-lineage specific differentiation,21 thereby arresting the cells in an undifferentiated state with increased ability for expansion/self-renewal. In this study, about half of undifferentiated endometrial carcinomas showed SWI/SNF complex inactivation, with mutually exclusive ARID1A/B co-deficiency, BRG1 deficiency or INI1-deficiency. In keeping with our understanding of the importance of SWI/SNF complex in maintaining cellular differentiation, the great majority of these SWI/SNF complex-inactivated tumors exhibit an undifferentiated immunophenotype, with absent PAX8 and ER expression.
Immunophenotypically, all BRG1- or INI1-deficient tumors showed a complete absence of PAX8 and ER immunostaining, consistent with our previous observations in the undifferentiated component of dedifferentiated endometrial carcinomas.10 In comparison, 2 of 11 ARID1A/1B co-deficient tumors showed PAX8 immunopositivity in <10% and 50% of tumor cells, similar to our reported findings in ARID1A/1B co-deficient dedifferentiated endometrial carcinoma. A potential reason for the occasional focal/patchy PAX8 expression may be related to the different mechanisms of SWI/SNF complex inactivation involved. There are several mammalian SWI/SNF complexes, including SWI/SNF A and SWI/SNF B that are expressed ubiquitously. While BRG1 and INI1 are obligatory core components for both SWI/SNF A and SWI/SNF B complexes, the former utilizes ARID1A or ARID1B, and the latter ARID2 and PBRM1, as their DNA binding domains. As such, loss of ARID1A and ARID1B is expected only to inactivate the SWI/SNF A complex, whereas loss of either BRG1 or INI1 is expected to inactivate both the SWI/SNF A and the SWI/SNF B complexes. This difference in the extent of SWI/SNF complex inactivation may account for the occasional expression of transcriptional factors such as PAX8 that facilitate müllerian differentiation. In comparison, in the 17 SWI/SNF complex-intact tumors, PAX8 expression was absent in all except 2 cases, and ER was absent in all except 1 case. Interestingly, it was the single POLE mutated (P286R) tumor that was positive for both PAX8 (diffuse immunopositivity) and ER. Overall, 12% of undifferentiated carcinomas in our current series were PAX8-positive. This is comparable to what Ramalingam et al reported in their series, where 3 of 15 (20%) undifferentiated carcinomas were PAX8-positive.22
Our findings also demonstrate significant molecular heterogeneity among undifferentiated endometrial carcinomas as defined histologically, and support the earlier observations made by Rosa-Rosa et al.12 In the context of the proposed molecular subtypes of endometrial cancer by The Cancer Genome Atlas (TCGA),23 about half of undifferentiated endometrial carcinomas arise in a MMR-deficient molecular context, which belongs to the high microsatellite instability (MSI-H) hypermutated molecular subtype, while 15% of cases exhibit mutated p53 staining patterns, intact MMR protein expression and wild-type POLE status, which would be classified under the copy number high serous carcinoma-like molecular subtype. A single case (3%) in our cohort harbored a hotspot POLE exonuclease domain mutation, therefore belonging to the POLE ultramutated molecular subtype. The remaining one-third of undifferentiated endometrial carcinoma show a wild-type p53 staining pattern, wild-type POLE status and intact MMR protein expression, suggesting that these belong to the copy number low, endometrioid molecular subtype. Among SWI/SNF complex-inactivated tumors, slightly more than half arose in a MMR protein-deficient/MSI-H molecular context while 40% arose in a copy number low, endometrioid molecular context. Only one SWI/SNF complex-inactivated tumor in our cohort arose in a copy number high, serous-like molecular context. These findings highlight the molecular heterogeneity that exists within undifferentiated endometrial carcinoma
Clinically, SWI/SNF complex deficiency defines a highly aggressive subset of undifferentiated endometrial carcinomas in which one-year disease specific survival is 26% in the SWI/SNF complex-inactivated tumors compared to 75% in SWI/SNF complex-intact tumors. It therefore appears that there are important biological differences between SWI/SNF complex-inactivated and intact undifferentiated endometrial carcinomas. Moreover, this distinction may become clinically relevant as investigational drugs such as EZH2 inhibitors have shown preclinical efficacy in targeting BRG1-deficient ovarian cancer,24, 25 INI1-deficient malignant rhabdoid tumors,26, 27 and INI1-deficient atypical rhabdoid/teratoid tumors.28 It is possible that these chromatin remodelling/epigenetic targeting therapies will prove to be more effective treatment for SWI/SNF complex-inactivated undifferentiated or dedifferentiated endometrial carcinomas.
In summary, we identified frequent inactivation of core SWI/SNF complex proteins in undifferentiated endometrial carcinomas. Moreover, SWI/SNF complex deficiency defines a highly aggressive subset of undifferentiated endometrial carcinomas with rapid disease progression. This may have important therapeutic implications in the near future with the emergence of drugs that specifically target the chromatin remodelling/epigenetic landscape.
Acknowledgement
We thank Taryn Rutherford for study coordination as well as Shuhong Liu and Young Ou (APRL) for immunohistochemical stains.
Sources of support/funding: This study is supported in part by research funds from Cancer Research Society of Canada (20313), Royal Alexandra Hospital foundation, Alberta Cancer Foundation, Calgary Laboratory Services Internal Research Competition (RS14-513). Dr. Soslow is supported in part by the MSK Cancer Center Support Grant P30 CA008748.
Footnotes
Disclosure: The study authors have no conflicts of interest to declare.
References
- 1.Silva EG, Deavers MT, Malpica A. Undifferentiated carcinoma of the endometrium: a review. Pathology. 2007;39:134–138. [DOI] [PubMed] [Google Scholar]
- 2.Altrabulsi B, Malpica A, Deavers MT, et al. Undifferentiated carcinoma of the endometrium. Am J Surg Pathol. 2005;29:1316–1321. [DOI] [PubMed] [Google Scholar]
- 3.Tafe LJ, Garg K, Chew I, Tornos C, Soslow RA. Endometrial and ovarian carcinomas with undifferentiated components: clinically aggressive and frequently underrecognized neoplasms. Mod Pathol. 2010;23:781–789. [DOI] [PubMed] [Google Scholar]
- 4.Ramalingam P, Masand RP, Euscher ED, Malpica A. Undifferentiated Carcinoma of the Endometrium: An Expanded Immunohistochemical Analysis Including PAX-8 and Basal-Like Carcinoma Surrogate Markers. Int J Gynecol Pathol. 2015. [DOI] [PubMed] [Google Scholar]
- 5.Hoang LN, Ali RH, Lau S, Gilks CB, Lee CH. Immunohistochemical survey of mismatch repair protein expression in uterine sarcomas and carcinosarcomas. Int J Gynecol Pathol. 2014;33:483–491. [DOI] [PubMed] [Google Scholar]
- 6.Silva EG, Deavers MT, Bodurka DC, Malpica A. Association of low-grade endometrioid carcinoma of the uterus and ovary with undifferentiated carcinoma: a new type of dedifferentiated carcinoma? Int J Gynecol Pathol. 2006;25:52–58. [DOI] [PubMed] [Google Scholar]
- 7.Karnezis AN, Hoang LN, Coatham M, et al. Loss of switch/sucrose non-fermenting complex protein expression is associated with dedifferentiation in endometrial carcinomas. Mod Pathol. 2016;29:302–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coatham M, Li X, Karnezis AN, et al. Concurrent ARID1A and ARID1B inactivation in endometrial and ovarian dedifferentiated carcinomas. Mod Pathol. 2016;29:1586–1593. [DOI] [PubMed] [Google Scholar]
- 9.Stewart CJ, Crook ML. SWI/SNF complex deficiency and mismatch repair protein expression in undifferentiated and dedifferentiated endometrial carcinoma. Pathology. 2015;47:439–445. [DOI] [PubMed] [Google Scholar]
- 10.Hoang LN, Lee YS, Karnezis AN, et al. Immunophenotypic features of dedifferentiated endometrial carcinoma Insights from BRG1/INI1-deficient tumors. Histopathology. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramalingam P, Croce S, McCluggage WG. Loss of expression of SMARCA4 (BRG1), SMARCA2 (BRM) and SMARCB1 (INI1) in undifferentiated carcinoma of the endometrium is not uncommon and is not always associated with rhabdoid morphology. Histopathology. 2016;70:359–366. [DOI] [PubMed] [Google Scholar]
- 12.Rosa-Rosa JM, Leskela S, Cristobal-Lana E, et al. Molecular genetic heterogeneity in undifferentiated endometrial carcinomas. Mod Pathol. 2016;29:1390–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Espinosa I, Lee CH, D’Angelo E, Palacios J, Prat J. Undifferentiated and Dedifferentiated Endometrial Carcinomas With POLE Exonuclease Domain Mutations Have a Favorable Prognosis. Am J Surg Pathol. 2017. [DOI] [PubMed] [Google Scholar]
- 14.Kobel M, Piskorz AM, Lee S, et al. Optimized p53 immunohistochemistry is an accurate predictor of TP53 mutation in ovarian carcinoma. J Pathol Clin Res. 2016;2:247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meng B, Hoang LN, McIntyre JB, et al. POLE exonuclease domain mutation predicts long progression-free survival in grade 3 endometrioid carcinoma of the endometrium. Gynecol Oncol. 2014;134:15–19. [DOI] [PubMed] [Google Scholar]
- 16.Isphording A, Ali RH, Irving J, et al. YWHAE-FAM22 endometrial stromal sarcoma: diagnosis by reverse transcription-polymerase chain reaction in formalin-fixed, paraffin-embedded tumor. Hum Pathol. 2013;44:837–843. [DOI] [PubMed] [Google Scholar]
- 17.Guan B, Mao TL, Panuganti PK, et al. Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am J Surg Pathol. 2011;35:625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masliah-Planchon J, Bieche I, Guinebretiere JM, Bourdeaut F, Delattre O. SWI/SNF chromatin remodeling and human malignancies. Annu Rev Pathol. 2015;10:145–171. [DOI] [PubMed] [Google Scholar]
- 19.Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv. 2015;1:e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Haswell JR, Roberts CW. Molecular pathways: SWI/SNF (BAF) complexes are frequently mutated in cancer--mechanisms and potential therapeutic insights. Clin Cancer Res. 2014;20:21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alver BH, Kim KH, Lu P, et al. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun. 2017;8:14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramalingam P, Masand RP, Euscher ED, Malpica A. Undifferentiated Carcinoma of the Endometrium: An Expanded Immunohistochemical Analysis Including PAX-8 and Basal-Like Carcinoma Surrogate Markers. Int J Gynecol Pathol. 2016;35:410–418. [DOI] [PubMed] [Google Scholar]
- 23.Kandoth C, Schultz N, Cherniack AD, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Chen SY, Karnezis AN, et al. The histone methyltransferase EZH2 is a therapeutic target in small cell carcinoma of the ovary, hypercalcemic type. J Pathol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan-Penebre E, Armstrong K, Drew A, et al. Selective Killing of SMARCA2- and SMARCA4-deficient Small Cell Carcinoma of the Ovary, Hypercalcemic Type Cells by Inhibition of EZH2: In Vitro and In Vivo Preclinical Models. Mol Cancer Ther. 2017;16:850–860. [DOI] [PubMed] [Google Scholar]
- 26.Moreno N, Kerl K. Preclinical Evaluation of Combined Targeted Approaches in Malignant Rhabdoid Tumors. Anticancer Res. 2016;36:3883–3887. [PubMed] [Google Scholar]
- 27.Knutson SK, Warholic NM, Wigle TJ, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110:7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alimova I, Birks DK, Harris PS, et al. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro Oncol. 2013;15:149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]