

Abstract
There is a growing demand for sustainable methods in research and development, where instead of hazardous chemicals, an aqueous medium is chosen to perform biological reactions. In this Perspective, we examine the history and current methodology of using enzymes to generate artificial single-stranded DNA. By using traditional solid-phase phosphoramidite chemistry as a metric, we also explore criteria for the method of template-independent enzymatic oligonucleotide synthesis (TiEOS). As its key component, we delve into the biology of one of the most enigmatic enzymes, terminal deoxynucleotidyl transferase (TdT). As TdT is found to exponentially increase antigen receptor diversity in the vertebrate immune system by adding nucleotides in a template-free manner, researchers have exploited this function as an alternative to the phosphoramidite synthesis method. Though TdT is currently the preferred enzyme for TiEOS, its random nucleotide incorporation presents a barrier in synthesis automation. Taking a closer look at the TiEOS cycle, particularly the coupling step, we find it is comprised of additions > n+1 and deletions. By tapping into the physical and biochemical properties of TdT, we strive to further elucidate its mercurial behavior and offer ways to better optimize TiEOS for production-grade oligonucleotide synthesis.
Graphical Abstract
In the mid-20th century, there were several key breakthroughs in the fields of genetics and biochemistry that have led us to current day medicine. This was a cascade of events that started with X-ray-induced gene knockout studies in 1941, which made the connection that genes were directly involved in enzyme function.1 It soon followed that genes themselves were comprised of nucleic acids (DNA),2 and a double helix was an orderly structure of nucleic acids that stored genetic information3 and could be precisely replicated by a DNA polymerase.4
It was then understood the power to manipulate such biological systems was harnessed in the form of single-stranded (ss) DNA with a defined sequence.5 As such, a great deal of work in the 1950s went toward developing methods for chemically synthesizing polynucleotides from nucleic acid monomers.6–8 At about the same time, the potential to synthesize polynucleotides enzymatically was also being realized.9 However, nucleotide (nt) addition was limited to homopolymeric tract formation, and efforts were eclipsed by the chemical method, which allowed synthesis in a controlled, stepwise manner.8 Its development continued over the next 30 years,10,11 which culminated in the traditionally accepted solid-phase phosphoramidite method used on all major synthesis platforms to date (Figure 1).
Figure 1.
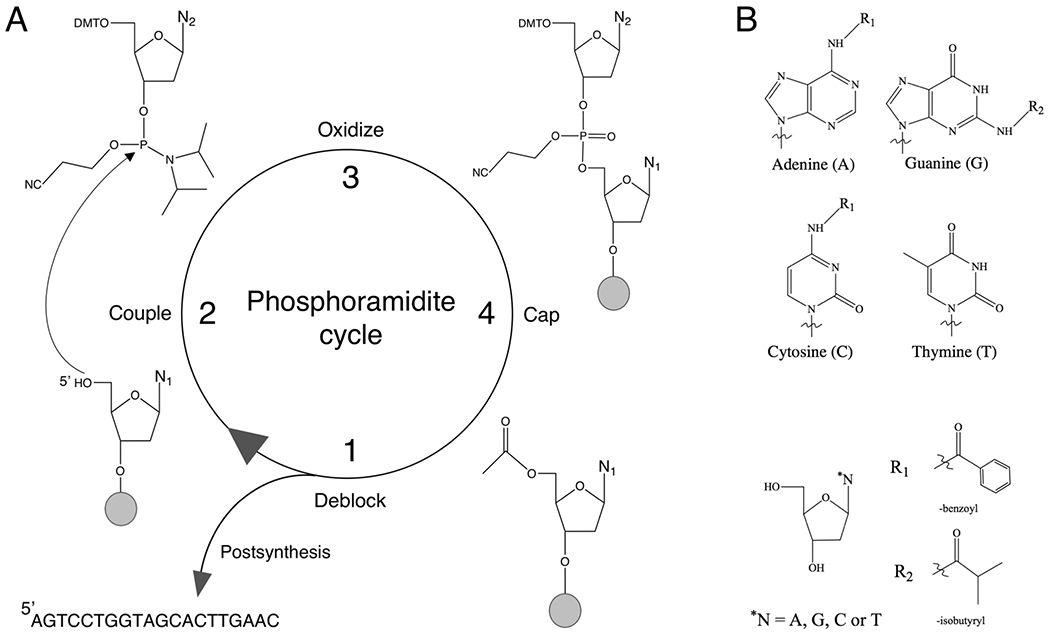
(A) Traditional 3′ → 5′ solid-phase phosphoramidite chemical synthesis cycle. The cycle begins with the first nucleoside tethered to a solid substrate by a cleavable succinate linker (alkaline-labile); the 5′ region, protected by dimethoxytrityl (DMT), is deblocked with acid (e.g., trichloroacetic acid/dichloromethane) to yield a hydroxyl group (step 1). In the coupling reaction (step 2), the phosphorus atom of an incoming nucleoside phosphoramidite is activated (e.g., with ethylthio-1H-tetrazole) for nucleophilic attack by the 5′ oxygen of the N1 nucleoside. Here, exocyclic amines for adenine, guanine, and cytosine are also reactive and must be blocked to prevent branching (panel B, R1 and R2). Once the nucleoside phosphoramidite is added, the phosphite bond (also protected with cyanoethyl) is oxidized with iodine/pyridine to generate a phosphate linkage to stabilize the sugar backbone (step 3). In step 4, oligonucleotide DNA strands (n) that fail to couple (n−1) are capped (acetylated) at the 5′ end to prevent any further reactions in successive cycles; n − 1 strands that remain uncapped will present with internal deletions. Wash steps are introduced using acetonitrile (petroleum byproduct) to clear the reaction well/column of waste. The cycle is repeated until the full-length product is generated, and then it is released from the solid support with ammonium hydroxide (methylamine); incubation at an elevated temperature (for ≤16 h) will remove all protecting groups.12 N1 and N2 denote nucleobases; n is the full-length oligonucleotide. (B) Nucleobase structures and exocyclic amine protecting groups. R1 and R2 denote benzoyl and isobutyryl, respectively.
One of the major drawbacks with the phosphoramidite method is the use of hazardous chemicals described in each step of Figure 1. These are listed by the U.S. Environmental Protection Agency for community awareness and emergency planning.13 Since the 1990s, there has been a growing trend toward “green” technology development in terms of sustainable chemistry.14 Possible advantages of using an enzyme for ssDNA synthesis also include (i) longer strand generation,15 (ii) a lower error rate,16 (iii) a faster cycle time, and (iv) a lower cost of production. These have very important implications in many fields of research, particularly in the areas of synthetic biology17 and DNA data storage.18,19 Another advantage of oligonucleotide production in an aqueous medium is portability. This comes into play especially in diagnostics and point-of-care devices,20,21 as well as for infield applications from forensics22 to synthesis in space (stanfordssi.org).
Until recently, the use of enzymes to make ssDNA continued a very slow and unproductive journey alongside the chemical method. In this Perspective, we first evaluate the origins of enzymatic oligonucleotide synthesis; and with the knowledge gained from prior research, we explore the method defined as template-independent enzymatic oligonucleotide synthesis (TiEOS). We also observe parallels with the solid-phase phosphoramidite cycle as a metric for quality assessment to gain a better appreciation for the prospects and challenges inherent with using an enzyme to make ssDNA. From a biological point of view, we also pay particular attention to how additions > n+1 and deletions might be generated and controlled during the TiEOS cycle.
ENZYMES AND METHODOLOGIES USED FOR OLIGONUCLEOTIDE SYNTHESIS: A BRIEF HISTORY
In 1955, Severo Ochoa discovered polynucleotide phosphorylase (PNPase) isolated from the microorganism Azotobacter vinelandii.9 Believed to be a component of RNA metabolism, PNPase was used in vitro to synthesize polyribonucleotides (Figure 2).
Figure 2.
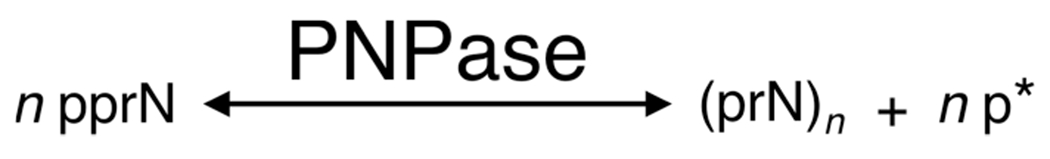
Proposed pathway of PNPase RNA synthesis and degradation. From left to right, 5′ diphosphate ribonucleosides (pprN) drive the reaction toward PNPase catalysis of RNA (n monomers long), with the release of orthophosphate. At high concentrations, phosphate byproduct (p*) drives the reaction (right to left) toward phosphorolysis, degrading RNA into its 5′ diphosphate ribonucleoside monomers.23
In 1959, F. J. Bollum described the first ssDNA polymerase, terminal deoxynucleotidyl transferase (TdT), capable of template-independent (primer-free) synthesis24,25 (Figure 3). Before characterization of this enzyme, we understood DNA polymerization to be constrained to primer–template duplex-driven synthesis.
Figure 3.
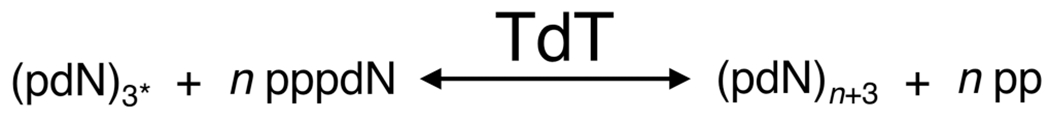
TdT polymerization of ssDNA. Here, a 3′-hydroxylated initiator strand (also termed an acceptor) is required [in the presence of a divalent cation (e.g., Mg2+)] for TdT to catalyze ssDNA using a deoxyribonucleoside triphosphate monomer (dNTP, also termed a donor). Here dNTP (pppdN) monomers are the substrate for addition of TdT to the 3′ end of an initiator strand (pdN)3* to generate ssDNA (pdN)n+3 with the release of pyrophosphate (pp). 3* indicates the length must be at least 3 nt. There are two primary isoforms of the TdT gene, TdTL (3′ exonuclease activity only, arrow right to left) and TdTS (3′ terminal transferase only, arrow left to right).
In 1962, Bollum confirmed dNTPs were being added to the initiator at the 3′ oxygen, because blocking it with an acetyl group prevented further nucleotide addition. As a consequence, Bollum proposed TdT could be used for ssDNA polymerization with monomers blocked at the 3′ position for stepwise synthesis of oligonucleotides with a defined sequence.26 In 1965, Letsinger and Mahadevan reported the first chemical DNA synthesis (dCpT) on a polymer support.27 Now with the potential to anchor the starting material (initiator) to a solid substrate, single monomer additions could be repeated on a per cycle basis, and unincorporated substrate could be washed from the reaction mixture between steps without loss of initiator.
In 1971, Mackey and Gilham used PNPase to synthesize RNA of a defined sequence; via the introduction of nucleotides blocked at the 2′ (3′) end, it was theorized RNA monomers could also be added to an initiator in a controlled stepwise manner.28 Here, they proposed a blocking group should (i) be chemically stable, (ii) have conditions for removal that are compatible with the enzymatic reaction, (iii) be small enough for the enzyme to be fully incorporated, and (iv) completely block any further enzymatic nucleotide additions. To demonstrate this, they successfully coupled 5′ diphosphate 2′-O-(α-methoxyethyl)-uridine to the 3′ end of an adenosine trinucleotide initiator in the presence of PNPase (Figure 4). A year later, Gilham and Smith would apply the same principle to generate ssDNA.29
Figure 4.
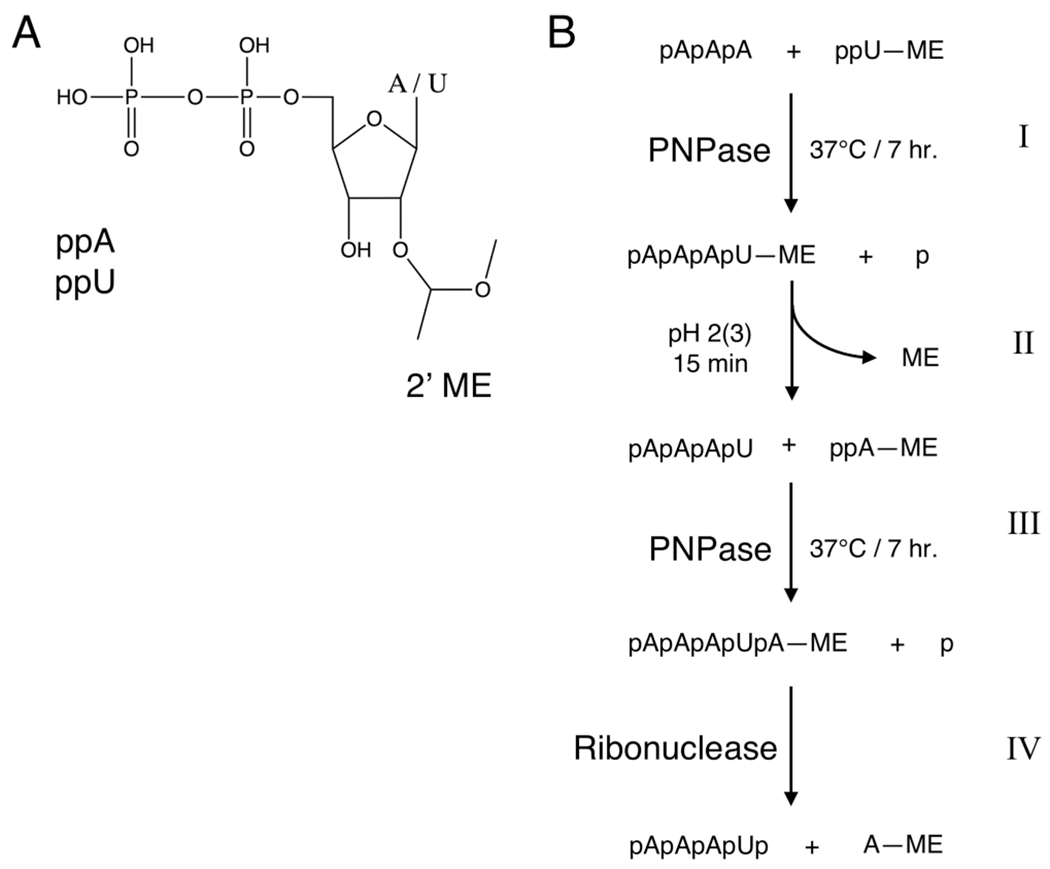
PNPase synthesis of polyribonucleotides of defined sequence. (A) Ribonucleotide representation, either adenosine (A) or uridine (U), diphosphate (pp), with 2′ (3′)-O-(α-methoxyethyl) blocking group (ME). (B) Solution-phase enzymatic RNA synthesis with 2′-protected ribonucleotides. (I) Adenosine trinucleotide (initiator) is coupled to 5′ diphosphate uridine protected at the 2′ position with α-methoxyethyl in the presence of PNPase at 37 °C for 7 h to generate 2′-protected tetranucleotide. (II) The α-methoxyethyl protecting group is removed with acid [pH 2 (3) for 15 min]. (III) The next nucleotide, 2′-protected 5′ ADP, is coupled to the tetranucleotide initiator (conditions are the same as those in step I). (IV) The structure of a blocked pentanucleotide is then confirmed by hydrolysis with pancreatic ribonuclease.
In 1978, England and Uhlenbeck introduced the first ligation method by which oligoribonucleotide synthesis was performed with T4 RNA ligase using 5′, 3′ ribonucleoside bisphosphates (prNp).30 In 1984, Schott and Schrade reported single-step oligonucleotide synthesis with TdT,31 during which they added unprotected dNTPs to initiator strands of variable length, and in 1999, the first report of solid-phase enzymatic DNA synthesis was made using T4 RNA ligase (Figure 5).32 This demonstrated the basic repeating synthesis cycle whereby protected monomers could be added in a stepwise manner and then deblocked at the 3′ end for further additions of defined sequence; as the initiator strand remains tethered without loss during wash steps, it is finally released by enzymatic cleavage postsynthesis.
Figure 5.
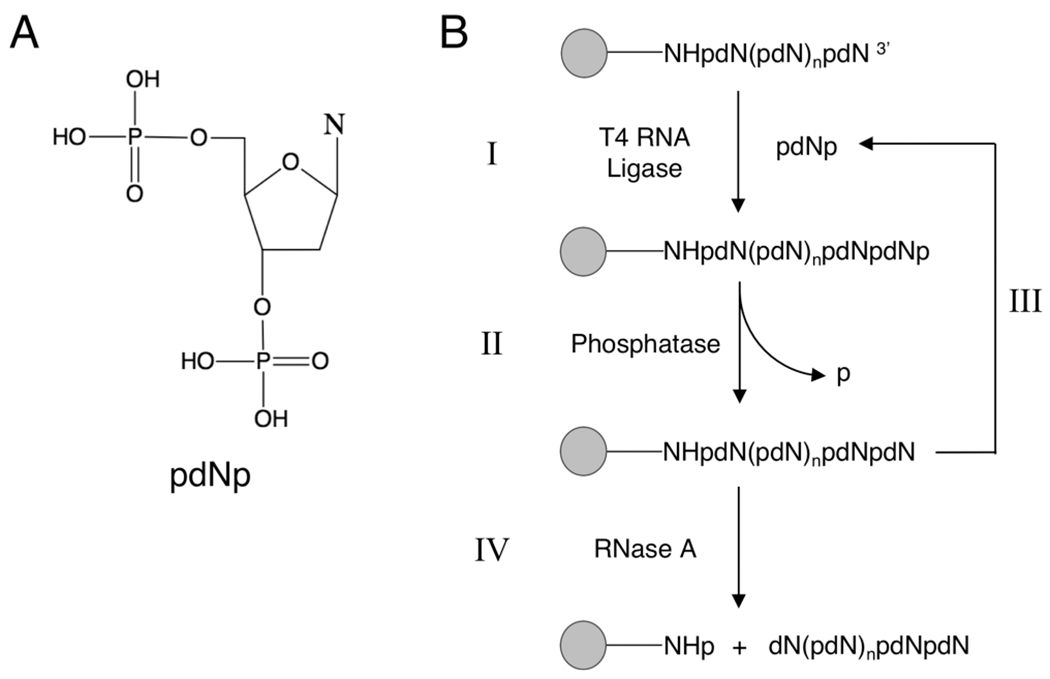
Solid-phase T4 RNA ligation of bisphosphate monomers to generate ssDNA. (A) Structure of 5′, 3′ bisphosphate-2′-deoxynucleoside (pdNp), where dN is any 2′ deoxynucleoside (A, G, C, or T). (B) Enzymatic synthesis performed in the solid phase, where 5′-phosphorylated initiator is covalently attached to Tentagel. (I) pdNp is ligated to the initiator at the 3′ hydroxyl in the presence of T4 RNA ligase. (II) The 3′ phosphate (p) is removed with alkaline phosphatase. (III) The process is repeated until the full-length product is established. (IV) The target strand is enzymatically released from the support with RNase A, yielding 5′, 3′ dephosphorylated product.
Despite the capability of PNPase to synthesize both ribo- and deoxyribonucleotides, limitations outweigh its benefits. (i) Purification is an extremely difficult, multistep process. (ii) PNPase has a preference for purine-rich initiators, which may limit sequence design. (iii) As the primer molecules become longer, accumulation of orthophosphate drives the reaction backward in phosphorolysis (strand degradation) (Figure 233).
Advantages of using T4 RNA ligase as a method of oligonucleotide production are also eclipsed by its limitations. (i) Initiator strands with uracil are less reactive. (ii) Ligation times are very long (≤144 h).32 (iii) The rate of T4 RNA ligase activity decreases after incubation at 37 °C.30 (iv) If free unblocked nucleotides are present in the reaction mixture, monomers will react with themselves and/or add to the initiator to generate homopolymeric tracts;34 this can substantially reduce the target product yield if reactions favor synthesis of unblocked donor generation. (v) If the unblocked donor is ≥8 nt, intramolecular cyclization may occur,35 thus competing with acceptor (initiator) polymerization.
Though terminal transferase would prove to be a far better candidate for enzymatic oligonucleotide synthesis compared to its predecessors, it is still less than perfect. The remainder of this Perspective delves into its role in the synthesis cycle, with a particular focus on types and causes of failure strand generation.
TERMINAL DEOXYNUCLEOTIDYL TRANSFERASE (TDT)
TdT is part of the X Family of low-fidelity DNA polymerases that includes, Pol β, λ, μ, σ1, and σ2.36 Pol β is involved in base excision repair (BER) and nonhomologous end joining (NHEJ), with Pol λ, μ, and TdT being active in NHEJ and V(D)J recombination in eukaryotes.37 The primary function of TdT is to increase antigen receptor diversity through random nucleotide incorporation in the vertebrate adaptive immune system. This creates a vast catalog of immunoglobulins (1014) and T-cell receptors (1018) that recognize and target most foreign pathogens encountered during an organism’s life span.38 In general, the terminal transferase gene (e.g., human) has two splice variants, TdTL (long amino acid sequence, associated with only 3′ → 5′ exonuclease activity) and TdTS (short amino acid sequence, acts solely as a terminal transferase).39–41 It is understood in vivo nucleotide additions and deletions during antigen receptor gene rearrangement are modulated by the co-expression of these two variants. For the remainder of this Perspective, we will be referring to the isoform, TdTS, which is typically isolated (e.g., from calf thymus42) for use in a number of biological applications, including TUNEL, RACE, modified end labeling, and homopolymer tailing.43,44
As TdT is the only enzyme fully dedicated to template-independent synthesis, both Pol λ and μ partially share these properties but are not suitable for synthesis automation. While processivity is most active with dsDNA binding and extension, Pol λ and μ can accommodate single-strand addition to some degree. For example, Pol λ requires a 3′ overhang (≥9 nt) to perform template-independent synthesis, and polymerization occurs with only certain sequences.45 Pol μ, which shares 41% amino acid identity with TdT, is a dual-mode DNA polymerase where it acts either by polymerization extension (template-dependent) or by randomly adding nucleotides across from an abasic site in a sequence-independent nucleotidyl transferase manner.46
TEMPLATE-INDEPENDENT ENZYMATIC OLIGONUCLEOTIDE SYNTHESIS (TIEOS)
To develop a cycle of repeating steps for enzymatic ssDNA synthesis in an automated capacity (Figure 6), we first need to consider criteria gleaned from past history exploring various enzymes and substrates used to generate polynucleotides. These necessary components include (i) a solid substrate for retaining the polynucleotide during synthesis, (ii) an initiator strand (>3 nt) for enzyme binding and polymerization, (iii) an enzyme that is template-independent, (iv) protected nucleotides for controlled, stepwise addition, and (v) a means of final product release from the support. In doing this, we define the method as template-independent enzymatic oligonucleotide synthesis (TiEOS) (Figure 6).
Figure 6.
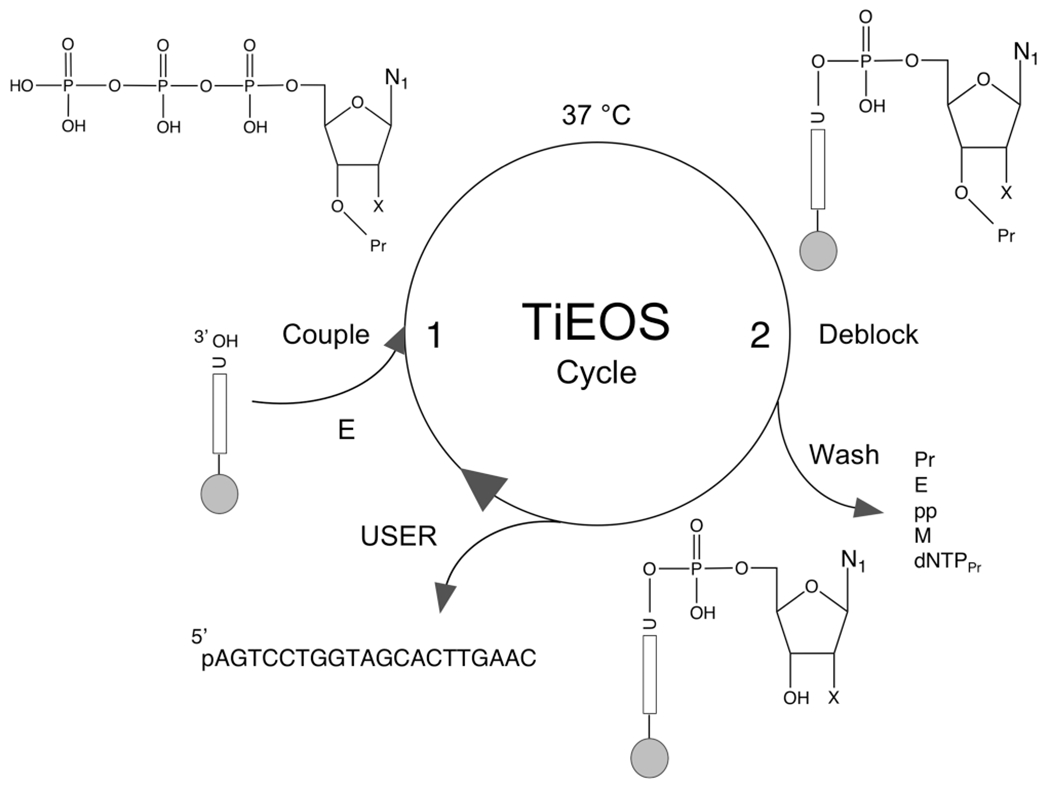
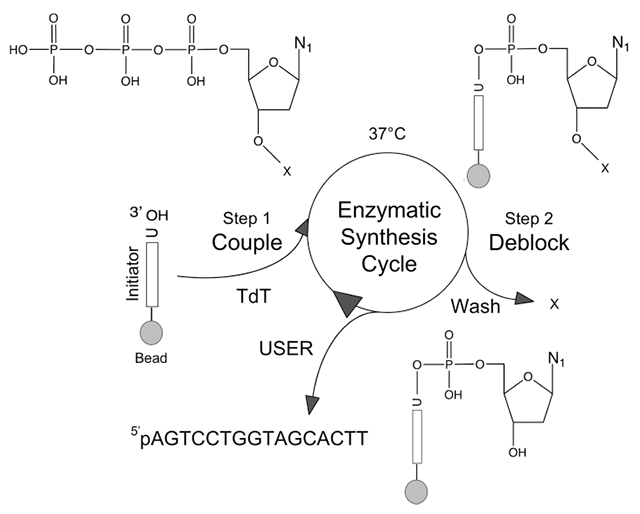
Proposed cycle for template-independent enzymatic oligonucleotide synthesis (TiEOS). In step 1, an incoming 3′-protected (Pr) dNTPPr (or NTPPr) is coupled to a 20 nt initiator (e.g., 37 °C, 30 s), which allows for enzyme (TdT) attachment and polymerization. Here the initiator is chemically presynthesized and covalently tethered {e.g., carbodiimide chemistry to a solid substrate [e.g., superparamagnetic beads (SPMB), silicon, or glass]}. A 20 nt long initiator was chosen to prevent steric hindrance at the surface of the beads during polymerization. In step 2, newly added dNTPPr is deblocked at the 3′ to hydroxyl (dNTP), followed by a wash step to remove protecting groups, enzyme, pyrophosphate, metal ions, and unincorporated dNTPPr. The cycle is repeated until the FLP is completed. For the purposes of enzymatic target strand release, uracil is placed at the 3′ end of the initiator for a point of cleavage; uracil DNA glycosylase and endonuclease VIII (USER) cleave the target strand [now 5′-phosphorylated (p)] from the support.47 Legend: gray spheres, solid support; vertical bars, initiator strands (>20 nt); E, enzyme (e.g., terminal transferase); U, uridine; N, nucleobase (see Figure 1B); p, phosphate; O, oxygen; pp, pyrophosphate; M, metal (divalent cation); X, either H (dNTP), OH (NTP for RNA synthesis), or a protecting group (Figure 8).
ADDITIONS > N+1
In living polymerization,48,49 where “chain ends remain active until killed”, TdT can generate a homopolymer tract of up to 8000 nt.43 It was determined that the length of the product generated from the 3′ end of the initiator is proportional to the monomer to initiator ratio (M/I).50 As such, the extent of polymerization can be controlled by combining a higher M/I ratio and a longer incubation period so that the initiator is fully extended. For our purposes, we are primarily interested in a controlled, stepwise nucleotide addition in the TiEOS cycle. Once added to the initiator strand, the 3′ region of a single dNTPPr [Pr is a protecting group (see Figure 8)] is protected from further extension until it is selectively converted to the reactive hydroxyl group (dNTPPr → dNTP); as such, dNTPPr is also termed the reversible terminator (RT). Because RT stability, efficiency of TdT incorporation, and completeness of deprotection are all major factors affecting TiEOS product quality and final yield, we next focus on how additions > n+1 are generated in the TiEOS cycle.
Figure 8.
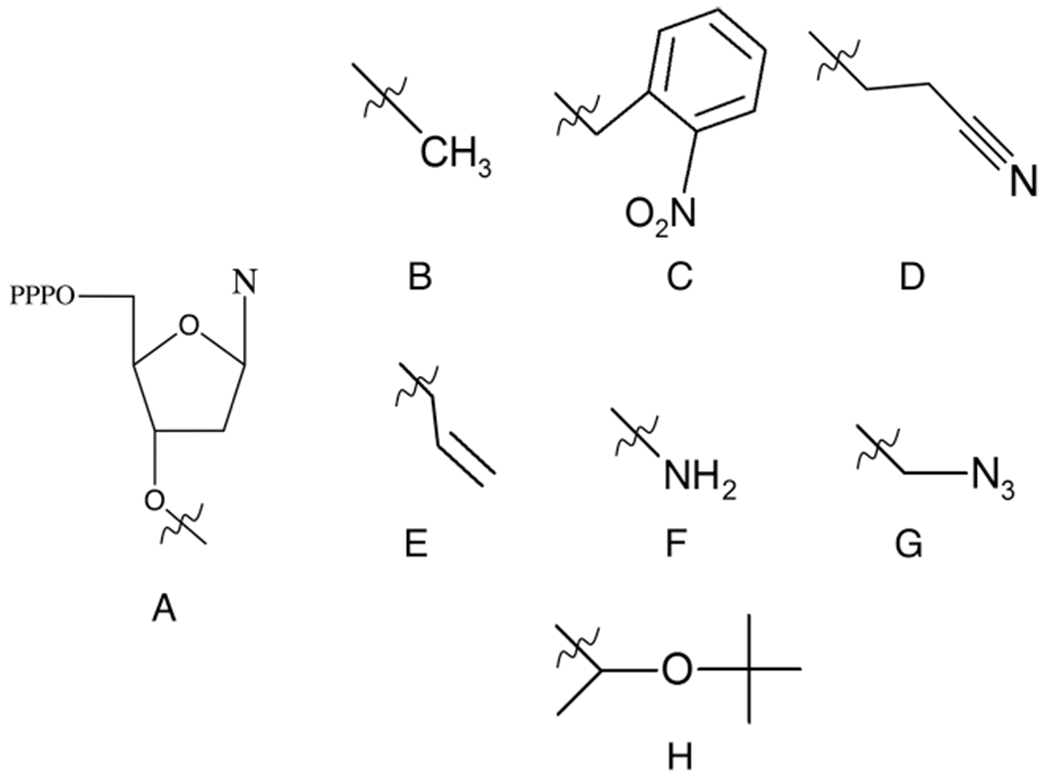
3′ reversible terminator protecting groups. (A) General structure for a 3′ O-triphosphate nucleotide, where N is any one of the nucleobases shown in Figure 1B. (B) Methyl, (C) 2-nitrobenzyl, (D) 3′-O-(2-cyanoethyl), (E) allyl, (F) amine, (G) azidomethyl, and (H) tert-butoxy ethoxy (TBE).
As a metric of quality assessment, it is important to compare TiEOS with the chemical method in terms of stepwise synthesis. Using step 2 in Figure 1 as a reference, the incoming nucleoside phosphoramidite is coupled (in the presence of an activator) to the first base preattached to the solid support. Here, DMT at the 5′ position of each phosphoramidite monomer prevents homopolymeric tract formation. If, however, premature detritylation occurs due to residual acid leftover from step 1 (inadequate washing) or an acidic activator [5-ethylthio-1H-tetrazole pKa (5.2) > 4, 5-dicyanoimidazole pKa (4.28)51], product > n+1 maybe generated. Side reactions can also occur during synthesis at the internucleotide linkage and nucleobases themselves. To prevent this, the phosphorus oxygen and nucleobase exocyclic amines are blocked by cyanoethyl, and isobutyryl or benzoyl protecting groups, respectively (Figure 1A,B).
In TiEOS, strands > n+1 can be generated by unblocked nucleotides during the couple step. Figure 7 demonstrates the pattern observed for uncontrolled homopolymeric tract formation and may be representative of a TiEOS cycle reaction contaminated by unblocked nucleotides.34
Figure 7.
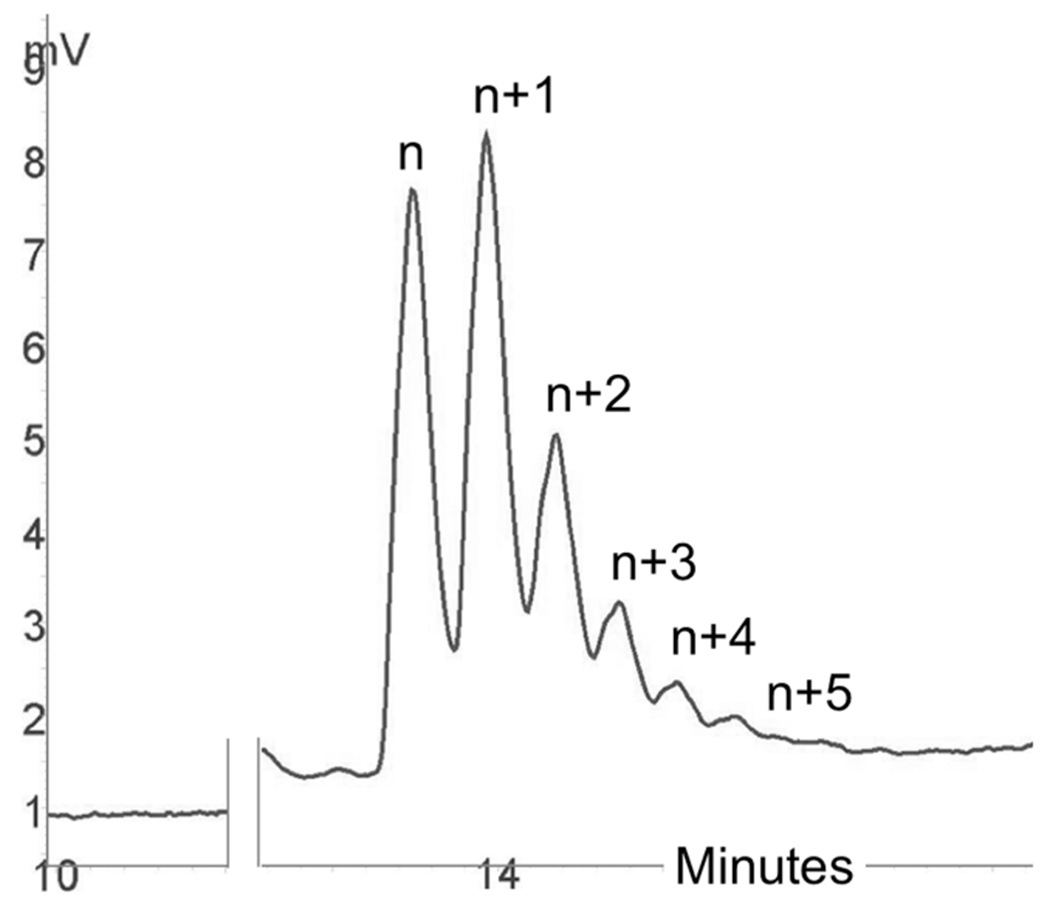
Homopolymeric tract formation due to 3′ unblocked dNTP contamination during TiEOS. Here, n represents the initiator strand; n + 1, n+2, etc., represent single-thymidine nucleotide additions, where anything greater than n+1 is considered a failure. This sample was generated using 60 units of TdT (M0315, NEB), 2 μL of buffer, 2 μL of CoCl2, 1 μL of MgCl2 (50 mM), 500 μM dTTP (3′ unblocked), and 50 pmol of T20 initiator (water added to a final volume of 20 μL), for 10 min at 37 °C. The reaction was terminated using 5% 0.4 M EDTA. The chromatogram for this figure was generated using reverse-phase high-performance liquid chromatography (RP-HPLC); conditions included a DNASep column (C-18) using reverse-phase buffers (ADS Biotec). Samples were processed at 80 °C with ultraviolet detection at 260 nm.
If the dNTPPr reservoir contains hydroxylated material, multiple dNTPs could be added consecutively onto the unblocked initiator (n). This can be a very complicated problem in terms of (i) percent homopolymeric tract formation (n+2, n+3, n+4, etc.), (ii) percent initiator left unreacted (n), and (iii) percent initiator correctly extended (n+1). Therefore, in addition to the criteria set by Mackey and Gilham for the protected monomer, we suggest reservoir purity and completeness of deprotection are also major considerations for the TiEOS coupling step.28 Figure 8 provides several possible protected dNTP analogues.
In 1994, Metzker and colleagues developed a set of reversible terminators as a gel-free alternative to the Sanger sequencing method, which includes 3′-O-methyl-dTTP (dATP) and 3′-O-(2-nitrobenzyl)-dATP (Figure 8B,C).52 Here they demonstrated these RTs could arrest template-dependent polymerase activity of Bst and AmpliTaq. Knapp et al. also reported fluorescently labeled RTs blocked with 2-cyanoethyl (Figure 8D) prevented further nucleotide addition by TdT.53 Ju et al. also discussed development of fluorescently labeled RT, allyl (Figure 8E), for use in sequencing by synthesis (SBS) reactions.54 Chen et al. also offered 3′-O-allyl, hydroxyamine, and azidomethyl (Figure 8E–G, respectively) as alternative RTs for SBS.55
With respect to Figure 8, there are two RT options that may be of particular interest for the TiEOS application. The first is photocleavable 2-nitrobenzyl (Figure 8C), which has been shown to cause TdT arrest after its incorporation onto blunt-ended, duplex DNA.56 This could be very beneficial in terms of high-throughput synthesis automation; however, at the time of this writing, only the dATP monomer with 2-nitrobenzyl is available for purchase (TriLink Biotechnologies). The second RT with strong potential in TiEOS is 3′ hydroxyamine (Figure 8F),57 which presents with mild reagent deblocking conditions using aqueous sodium nitrite and is currently available for all dNTPs (Firebird Biomolecular Sciences).
Moreover, the native form of TdT appears to allow some incorporation of these 3′-blocked monomers (Figure 8) as shown with 2-nitrobenzyl.56 Boulé et al. also observed TdT had an only weak preference for dNTP over 2′ OH NTP (RNA) polymer growth of the dA10 initiator.58 Winz et al. also used TdT to label DNA through copper-catalyzed azide–alkyne cycloaddition.59 Even in our own lab, we demonstrated that TdT incorporates 3′-tert-butoxy ethoxy-dTTP (dTTPTBE)60 (Figure 9).
Figure 9.
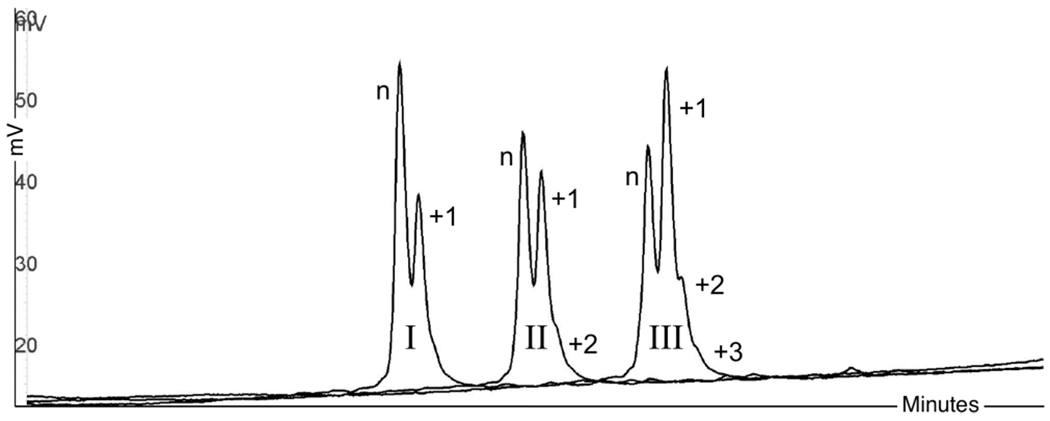
Incorporation of dTTPTBE by TdT over time. n → n+1 conversion after (I) 5, (II) 10, and (III) 15 min. Reactions were stopped immediately after each time point with 5% 0.4 M EDTA. Reaction conditions included 3 μL (60 units) of TdT (M0315, NEB), 2 μL of buffer, 2 μL of CoCl2, 1 μL of MgCl2 (50 mM), 1 μL of initiator (20 μM T20), 1 μL of dTTPTBE (TriLink Biotechnologies, 50 mM), and 10 μL of water (all stock reagents from NEB). See Figure 7 for HPLC conditions.
Figure 9 shows possible distributive properties of TdT over time (5, 10, and 15 min).41 Percent conversion n → n+1 for each sample was calculated at 22% (I), 52% (II), and 55% (III) (based on the percent area for each target peak using chromatogram analysis reports). Reactions were stopped with ethylenediaminetetraacetic acid (EDTA), which is a cation chelating agent and, therefore, shuttles away metal ions necessary for polymerase activity. As indicated in sample II, the shoulder peak may represent a second dTTP addition, same as III with +2 and +3 additions. Because heat-labile dNTP monomers (e.g., dNTPTBE) were specifically developed for use in hot-start polymerase chain reaction (PCR) protocols,60 TBE will be lost at high temperatures; also, dNTPTBE → dNTP conversion is on a sliding scale, and even at 37 °C, TBE protection is compromised, especially over longer periods of incubation (Figure 9, II and III at 10 and 15 min). This is also a good example of possible unblocked dNTP contamination during the TiEOS coupling step (compare with the pattern in Figure 7).
As for any modified stock dNTP product, trace contamination may be present, which cannot be filtered out 100% by analytical methods such as HPLC purification. For reversible terminators, in particular hydroxylated dNTPs must be removed prior to TiEOS application to prevent homopolymeric tract formation during synthesis. To achieve this, we suggest exhausting unblocked dNTPs through a simple polymerase extension reaction. For example, chemically synthesize two complementary primers (top and bottom, 20 and 35 nt, respectively), where the top primer is 5′-biotinylated and the bottom sequence contains a homopolymeric tract at the 5′ end (e.g., A15, G15, C15, or T15); with the top strand immobilized to streptavidin-coated superparamagnetic beads (SPMBs), hybridize the bottom strand to introduce a 3′ recess. To each set add the respective dNTP sample (dATPPr + T15) with a standard polymerase (e.g., AmpliTaq, 72 °C for 10 min); only unblocked, hydroxylated nucleotides will be added to the 3′ OH of the top strand forming duplex DNA. Using an external magnet, SPMBs can be separated from the purified dNTPPr sample, which is now ready for use in the TiEOS application.
Though several dNTP analogues (Figure 8) are readily incorporated by the native form of TdT (e.g., bovine), addition and removal of these 3′ blocking structures are rate-limiting in stepwise synthesis. As such, considerations must be made with respect to (i) gaining access to the 3′ moiety for its complete and efficient removal, (ii) the fate of TdT at this critical step [the enzyme may require inactivation by heating (75 °C, 20 min) or treatment with EDTA (0.2 M)61 to force its dissociation from the initiator–dNTPPr complex], and (iii) whether residue modifications in the 3′ binding pocket are necessary to improve dNTP analogue uptake to match the TdT rate of standard nucleotide incorporation in living polymerization. There is also the added time to chemically and/or enzymatically remove the 3′ blocking moiety and thoroughly wash it from the reaction well before beginning the next cycle.
DELETIONS
While homopolymeric tract formation can be managed to a certain degree, a much more complicated issue arises with deletions in TiEOS. To put this in perspective, we must first understand conditions affecting coupling efficiency in the chemical method (Figure 1), and how product ≤ n−1 is minimized. The coupling reaction time for standard phosphoramidite monomers is typically 20–40 s and is largely dependent on the activator used and scale of synthesis. To avoid unreacted oligonucleotides (attached to the solid substrate), reagents are typically delivered in excess to ensure maximum coverage of the substrate-bound product. The general rule is 1 of 100 oligonucleotide molecules will fail to react during the coupling step. This is restated as a 99% coupling efficiency (CE), which is defined by the equation full-length product (FLP) = (CE)n, where n is the polymer length (n−1, if the first nucleoside is preattached to the support); the purity of FLP generated during chemical synthesis is directly proportional to the total number of monomers added.62 For example, synthesis of a 20 nt strand with a CE of 99% will generate 81.7% FLP, while synthesis of 40 nt will yield 66.9% FLP with the same CE. Depurination is also another factor that contributes to synthesis failures generated via the phosphoramidite method.63 This may be caused by residual acid leftover from the deblocking step, where adenosine is particularly susceptible to cleavage at the glycosidic bond, generating an apurinic site.
Though the process by which failures are generated in TiEOS differs from the chemical method, contamination by product ≤ n−1 is still a problem. As such, these deletions may occur when the initiator is not fully consumed by the end of the cycle (n → n+1 conversion is <100%, and unreacted material is allowed to carry over into consecutive cycles).34 To emphasize a statement made earlier, the primary function of TdT is to increase antigen receptor diversity through random nucleotide incorporation. Because this adaptive immunity arose in the vertebrate system ~500 million years ago,64 it makes use of terminal transferase for nucleotide addition in a controlled, stepwise manner particularly challenging. While deletions may be due in part to an inadequate nucleotide:TdT:initiator ratio, we believe the main cause is random enzyme–nucleotide polymerization efficiency. This is where TdT appears to have a preference for adding one specific nucleobase over another, thus affecting the order and degree of nucleotide incorporation.40,50 Table 1 below provides examples of this mercurial behavior.
Table 1.
Examples of Random TdT Nucleotide Incorporation/Polymerization
| solution-phase polymerization | G = A > T > C42 | A > G > T > C58 | G > C > T > A65 | A > T > C > G66 | (C > A > G > T)a66 |
| solid-phase polymerizationb | T > C > A > G67 | A > T >> C = G68 | |||
| influence of initiator 3′ nucleotide | T25 > C25 > A25 (dCTP)68 | A25 > C25 = T25 (dATP)68 | A25 = C25 > T25 (dTTP)68 | C25 > T25 = A25 (dGTP)68 | (dTTP > dATP)c43 |
| influence of nearest neighbor nucleotide | 31.9% AA, 24.4% CA, 24.4% AC, and 19.3% CC (dATP, dCTP)d69 | 9.7% AA, 10.1% CA, 12.9% GA, 10.9% CC, 9.2% AC, 10.1% GC, 13.9% GG, 13.8% AG, and 9.2% CG (dATP, dCTP, dGTP)d69 | |||
| influence of metal ions | G > A > C > T (Mg2+)41 | T > C > G > A (Co2+)41 | Zn2+ 70,71 | ||
| mixed-base polymerization | AATCACGGGCGGCGCATCAGGAGAGACGCAAAGATGGCCCCCGGCCTTATCCAGCCATTTCGGCATAAGTGAAAAAAAAAAGCCGGGAAATGGGCACATTTCCAAGGGCGCTTTTACAGGGAGCGGCGCGGCCCTGAACGAGCTTGGCGGCTGAGCCGGCTCGCGCCCCGGAGTCAC (3′) (G > C > A > T) | ||||
Steady-state kinetics employed for single-nucleotide incorporation efficiencies at C > A > G > T.
Surface-initiated enzymatic polymerization (SIEP) on a gold substrate.
Where initiators were terminated with A, G, C, and T, dTTP had an incorporation efficiency that was higher than that of dATP.43
Mixed nucleotides (50% dATP and dCTP and 33% each dATP, dCTP, and dGTP). The efficiency of nucleotide incorporation by TdT (excluding influence of nearest neighbor nucleotide) is based on the extent of polymerization as measured via polyacrylamide gel electrophoresis. All are homopolymers except where otherwise indicated. Mixed-base polymerization (performed in house) showed heteropolymeric ssDNA generation with nucleotide percent incorporation at G > C > A > T; experimental conditions include initiator (20 nt) 40 pm, 20 units of TdT (bovine, NEB), 2 mM dNTP mix (equimolar A, G, C, and T), 1× CoCl2, 1× TdT buffer, water added to a final volume of 20 μL, 37 °C/60 min. Samples were sequenced using the Sanger method.
Physiological conditions in vivo, particularly nucleotide imbalance, may also greatly affect the TdT order of nucleotide incorporation.72,73 For example, in patients with adenosine deaminase deficiency where there is an accumulation of dATP, N-region insertions during V(D)J recombination were 49% AT compared with 24% in normal B cells.73
In our own lab, we demonstrated TdT randomly incorporates dNTPs (G > C > A > T) onto an initiator (Table 1, mixed-base polymerization), even from a standard dNTP mix (each nucleobase represented in equimolar amounts). The initiator strand we used, TTTAGTCCTGGTAGTACTTGAAC (3′), also served as a forward primer to amplify FLP for sequencing; we added a poly A tail at the 3′ end for annealing T20 as a reverse primer. Also, the initiator nucleotide composition can affect TdT initiation efficiencies. For example, Tjong et al. found that dT10 was preferred over dC10, which resulted in an only 70% initiation efficiency.43
Short of manipulating the amino acid sequence of the terminal transferase itself, how might the information presented in Table 1 be beneficial in terms of generating ssDNA with higher yield and purity via synthesis automation? Assuming the species of TdT used in TiEOS (e.g., bovine) is consistent in its nucleobase preference, one could predict how efficient TdT might be at incorporating, for example, dATPPr onto an initiator with T at the 3′ end. If this coupling event is expected to be only 95% successful (compared with dTTPPr at 100%, same conditions), synthesis parameters could be optimized by either (i) delivering a larger volume of the dATP stock solution or (ii) increasing the reaction time. Alternatively, each dNTPPr could be delivered in excess, and the reaction time could be adjusted to exceed the rate-limiting dNTPPr addition.
Despite any preference TdT may have for one particular nucleobase over another, a finely tuned ratio of enzyme to initiator is critical for addressing as many starting molecules as possible. Tang et al. found that by increasing this ratio from 0.1 (0.05 unit of TdT/μL) to 2 (1 unit/μL), the polydispersity index decreased from 1.31 to 1; here, the fraction of unextended initiator remained below 20% for each nucleotide added.48
Guanosine tracts of ≥4 nt may also be problematic in TiEOS. Because of the increased level of hydrogen bonding at the N7 ring position, G-quadruplexes are formed through intra-, bi-, or tetramolecular strand folding.74 The effects are exhibited in downstream applications such as PCR where polymerase arrest sites manifest as hairpin structures causing premature double-stranded product termination.75 It has been noted in several cases that guanosine homopolymer tracts either completely failed or showed TdT polymerization efficiencies significantly lower than that of dATP, dCTP, or dTTP.66,68 In addition to the effects of temperature and pH, the presence of metal cations can further stabilize G-quadruplexes generally in the following order: K+ > Ca2+ > Na+ > Mg2+ > Li+ and K+ > Rb+ > Cs+.76 Other reports suggest divalent cations may actually destabilize the G-quadruplex (e.g., Zn2+, Mg2+, Co2+, Mn2+, Ca2+, and Ni2+74). There are several options for minimizing secondary structure formation in G-rich sequences during TiEOS. For example, 7-deaza-dGTP disrupts hydrogen bonding between neighboring guanosines by displacing the ring nitrogen from position 7 to 8. Also, a library of reagent additives is available, which includes betaine, dimethyl sulfoxide, formamide, glycerol, NP-40, Tween 20, trehalose, and EcoSSB.77–81
Even with the phosphoramidite method, deletions may cause problems in application if not blocked from further reacting in downstream cycles. As previously mentioned, the 5′ oxygen of uncoupled molecules is acetylated during the capping step (Figure 1). These truncated species can then be removed enzymatically82 or by gel/column-based methods of filtration postsynthesis to purify the final product.83–85 In TiEOS, then, how might failures be minimized and/or removed? One possibility is by introducing an enzyme with 3′ exonuclease activity such as Exo I, which catalyzes the complete degradation of 3′-hydroxylated ssDNA;86 this may prove to be beneficial in eliminating the unreacted initiator product. Whether the 3′ blocking group is impervious to specific exonuclease digestion, this must be tested empirically (see Figure 8 for protecting group examples), but as with any oligonucleotide purification method, the final FLP yield may be compromised.
TDT KINETICS AND FACTORS THAT CONTRIBUTE TO RANDOM NUCLEOBASE INCORPORATION
It should be clear by this point the effects of random nt incorporation by terminal transferase can be quite extensive (Table 1). This is most apparent when factored into oligonucleotide synthesis, where quality and product yield may be negatively impacted. A closer look at the physical properties and behaviors of TdT through its kinetic pathway could shed more light on this unpredictable behavior. Because of its kinship with terminal transferase, we compare central elements of the Pol β pathway (Figure 10) with that of TdT (Figure 11).
Figure 10.
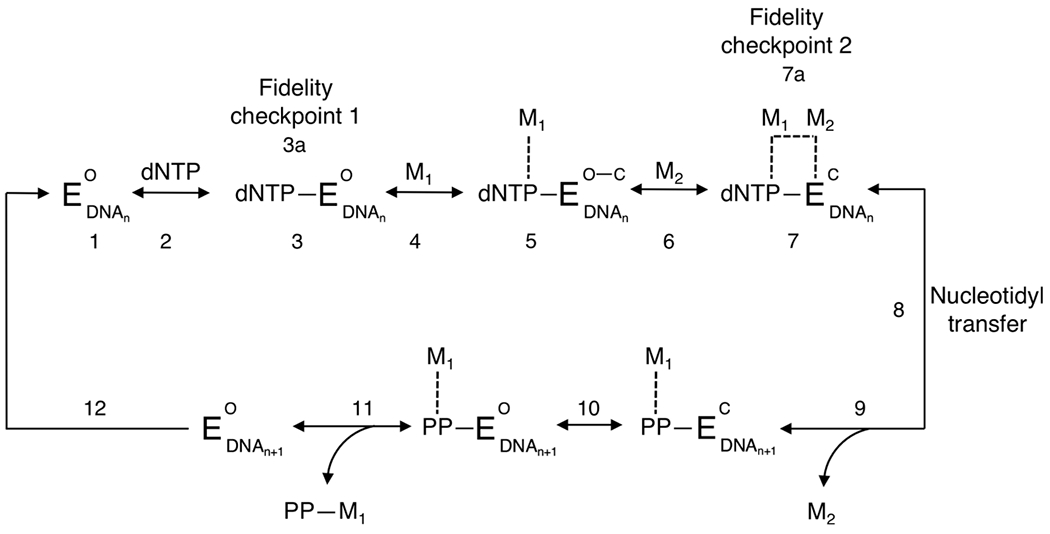
Kinetic pathway of Pol β processive (template-dependent) enzymatic synthesis. Components of the pathway: EO, enzyme (Pol β) in the open binary conformation; EC, closed ternary conformation; dNTP, any deoxynucleotide triphosphate; M1 and M2, metal ions [e.g., magnesium (Mg2+)]; DNAn, dsDNA strand ≥10 (n) nt (template and primer); DNAn+1, primer extended by 1 nt; PP, pyrophosphate. Dashed lines indicate interaction of the metal ion with the nucleotide triphosphate, enzyme binding site, and pyrophosphate. Pol β is in an open binary complex bound to the template and primer strand (step 1). The first incoming nucleotide (dNTP) binds to the active site, converting the polymerase complex into the ternary conformation (step 2). The triphosphate, of which the charge is neutralized by binding pocket side chain residues, is in the extended orientation and stabilized by hydrogen bonding via surrounding water molecules (step 3). dNTP is now paired with the template nucleotide in a buckled conformation.87 The first metal ion (M1) binds to the active site (steps 4 and 5).88 M1 coordinates the geometry of α-, β-, and γ-phosphate oxygens, including carboxylates, Asp190, and Asp192, partially closing the ternary complex (step 5).89 The catalytic ion, M2, then binds (steps 6 and 7) and coordinates the α-phosphate of the incoming nucleotide and 3′ oxygen of the primer strand (step 7) at a distance of 3.4 Å.90 The ternary complex is now in the fully closed conformation. With the correct nucleotide incorporated, the closed enzyme complex is now poised for nucleotidyl transfer (step 8). This slow, rate-limiting chemical step involves 3′ OH proton abstraction from the primer followed by 3′ O-nucleophilic attack on the dNTP α-phosphate (DNAn → DNAn+1).89 The catalytic ion (M2) is next expelled (step 9), and Pol β undergoes a rapid transition to the open conformation (step 10). Finally, the pyrophosphate–M1 complex dissociates from the enzyme active site (step 11), and with the Pol β enzyme still bound to the template–primer DNAn+1, it continues the processive addition of the second incoming dNTP through the translocation step (step 12).
Figure 11.
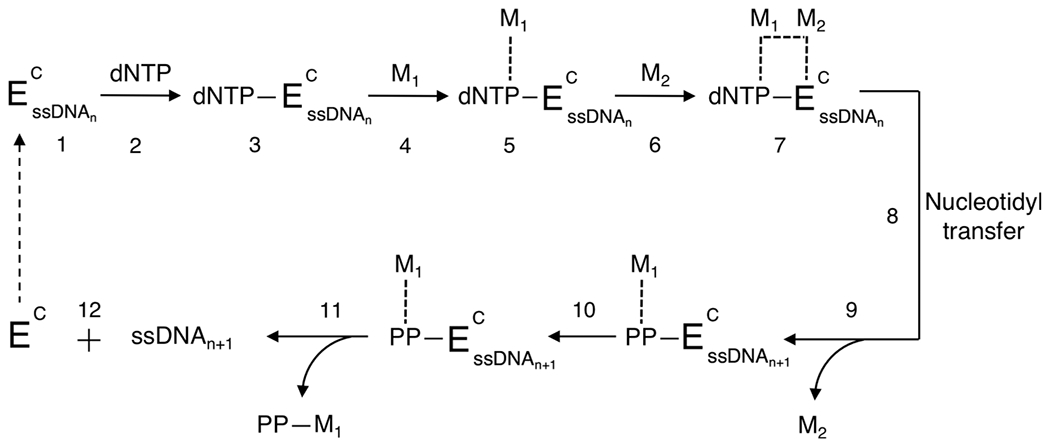
Proposed kinetic pathway of TdT distributive (template-independent) enzymatic synthesis. See Figure 10 for nomenclature. Here “n” is any strand ≥3 nt in length. Also, M1 and M2 can be either divalent cation, Mg2+ or Co2+, whereas Zn2+ may act as an allosteric cofactor. This pathway represents transferase catalytic activity for the TdTS variant of the TdT gene. The dashed arrow following step 12 indicates TdT either dissociates from the initiator and binds to another strand or remains bound to the original initiator during polymerization.
Unlike Pol β, TdT assumes a permanently closed ternary conformational complex; here, Arg258 in binding motif A of Pol β, which is essential for maintaining an open conformation, is absent in TdT. Instead, TdT contains loop 1, a Lariat-like conformation, which clamps down on the initiator via hydrogen bonding and precludes dsDNA from the active site.91 Upon entering the binding pocket, the triphosphate of the incoming dNTP is held in place by water molecules, where the sugar and nucleobase are loosely seated (steps 1–3).87 At this first possible checkpoint shown in Figure 10 (step 3a), Pol β determines whether dNTP pairing is a match with the template strand before metal ions commit the nucleotide to its geometry. Mismatched nucleotides that might otherwise be rejected at this stage in a template-dependent configuration are more blindly accepted in the kinetic pathway of terminal transferase. At the second fidelity checkpoint (Figure 10, step 7a), a major conformational shift occurs after both metal ions bind in the active site and properly align the nucleotide through an induced-fit mechanism; the enzyme goes from an open to closed ternary conformation.92–94 If an improperly seated nucleotide has escaped the first fidelity checkpoint (Figure 10, step 3a), Pol β closure will sandwich the mismatch into the active site, increasing the distance from 3.4 Å (between the α-phosphate of the incoming nucleotide and the 3′ oxygen of the primer strand) to 3.9 Å.95 This in turn destabilizes the closed enzyme conformation of the active site. Under ideal conditions, the mismatched nucleotide is then expelled from the complex before the chemical step, and the polymerase returns to an open configuration. For TdT, there is no template for comparison; therefore, no fidelity checkpoints exist to dictate nucleotide specificity. As mentioned earlier, TdTL and TdTS act in concert in vivo; therefore, nucleotide additions and deletions are moderated. For TiEOS, we consider only the isoform with transferase activity (GenBank entry AAA36726.1) (Figure 11).
As determined by crystal structures of terminal transferase, a lack of specific contact between the incoming nucleobase and surrounding residues may explain indiscriminant dNTP allowance.91 This argument is strengthened by the fact TdT permits considerable flexibility toward various nucleobase modifications. For example, (i) Jarchow-Choy et al. demonstrated that their extended nucleobase dNTP analogues (xDNA) were incorporated by TdT with kinetic efficiencies comparable to those of the natural dNTP controls;66 (ii) similarly, Berdis and McCutcheon incorporated the dNTP analogues 5-(nitro, phenyl, naphthyl, and cyclohexyl)-indolyl-2′-deoxyriboside triphosphate;65 (iii) Sørensen et al. used TdT to incorporate dNTP-coupled proteins and other macromolecules (e.g., polyethylene glycol, dendrimer, and streptavidin96); and (iv) as mentioned earlier, Winz showed use of azide-functionalized pyrimidine adducts with TdT for click chemistry applications in DNA labeling.59
Unlike Pol β, which preferentially binds Mg2+, TdT can accept multiple nucleotide binding and catalytic divalent cations (Figure 11, steps 4–7); these include Mg2+, Co2+, and Zn2+ (allosteric cofactor). The specificity of nucleobase incorporation by TdT appears to be largely ion-dependent as demonstrated by Fowler and Suo: G > A > C > T and T > C > G > A, respectively [Mg2+ (purines) and Co2+ (pyrimidines)].41,97 It has been shown, too, that micromolar quantities of Zn2+ increase the efficiency of nucleotide incorporation for both Mg2+ and Co2+ (generally at millimolar concentrations per reaction).70,71 Chang and Bollum identify Zn2+ as a nonessential allosteric cofactor for terminal transferase, which loosely interacts with the initiator and TdT binding site to induce conformational changes that increase the rate of catalysis.71 Whether Mg2+ or Co2+ competes for the active site when present in the same reaction, tailoring the TiEOS coupling reaction buffers separately for purines (Mg2+ and Zn2+) and pyrimidines (Co2+ and Zn2+) may be advisable for improving dNTP incorporation efficiency.
It has been postulated, too, because the TdT-bound initiator is not in an α-helix configuration, translocation is discouraged, and the enzyme completely dissociates from the initiator strand (ssDNAn+1) (Figure 11, step 12).41 Efficiency of nucleotide incorporation may also be influenced by the probability of TdT moving from one strand to another between single-nucleotide additions.58 TdT as a distributive enzyme is further explained by the observation that complete dNTP incorporation appears to be time-dependent.43 Also, the transition from the open to closed conformation in template-dependent polymerases (e.g., Pol β and Thermus aquations Pol I) is associated with the translocation step and, therefore, may be unnecessary for terminal transferase due to its distributive property.91
CONCLUDING REMARKS
As its key purpose is to increase antigen receptor diversity, terminal transferase has inherent contrivances that resist normal, predictable behavior observed with most replicative, high-fidelity polymerases. Therefore, these represent many challenges to its use in the oligonucleotide synthesis cycle for high-throughput automation. For TiEOS to work using TdT on a production level, it must be at least comparable to, if not exceed the synthesis output and efficiency of, the solid-phase phosphoramidite method.
To minimize additions > n+1 and deletions during synthesis, TiEOS might be improved by (i) determining the ideal TdT:dNTPPr:initiator ratio, (ii) tailoring dNTPPr buffers98 for the specific nucleotide being incorporated (Mg2+ for purines and Co2+ for pyrimidines) with Zn2+ as an enhancer, (iii) filtering the dNTPPr stock mix from unblocked monomers, (iv) digesting unreacted initiators with an exonuclease that targets 3′-hydroxylated ssDNA, (v) introducing additives to prevent secondary structure formation for G-rich polymers, and (vi) adjusting coupling times based on TdT-specific nucleotide incorporation. If TdT nucleotide incorporation cannot be controlled by the synthesis environment alone, there are options to modify the enzyme’s amino acid structure.99,100 To touch on this, the field of protein engineering maintains a rapidly growing toolbox of methodologies for altering protein structure and function (e.g., rational design and directed evolution).101 Helpful tools, including reconstructed evolutionary adaptive path (REAP) analysis, combine a protein’s evolutionary and functional history to best predict which amino acids may be replaced to achieve a target outcome.102 Artificially modified noncanonical amino acids (Ncas) can also expand the genetic code to yield greater protein structural and functional diversity.101 Currently, there are ~70 Ncas available (e.g., with methyl, glycosyl, and phosphoryl groups). Another area of protein engineering specifically focuses on designing and evolving polymerases for the purpose of propagating genetic information using noncognate substrates such as XNA103 This has many implications in the study of diversifying molecular heredity. Several tools are available as well for screening and detecting polymerase activity such as phage display and compartmentalized self-replication.104 While codon-optimizing the binding pocket of TdT to decrease steric hindrance through site-directed mutagenesis is one technique that has been employed, care must be taken to avoid introducing undesired results (e.g., replacing Arg336 with either glutamine or alanine significantly decreases activity for TdT incorporation of dATP).105 Attention may also be focused on altering the nucleobase binding region to more tightly regulate dNTP incorporation. For example, as shown in the Protein Data Bank (PDB entry 4I27, ternary complex of mouse TdT with ssDNA and incoming nucleotide), there is a vast open space opposite the incoming nucleobase. If loop 1, which is in the proximity, can be extended to moderate nucleobase binding, this may allow for a more uniform incorporation of all four standard dNTPs (A, G, C, and T).
Finally, we consider the prospective cost of synthesizing oligonucleotides with an enzyme compared with that of the phosphoramidite method. While there are several factors that contribute to the total cost such as the scale of synthesis, strand length, synthesis platform, and throughput (e.g., total number of samples generated either by column, titer plate, or array),106 we base our estimates on synthesis of 1000 1000-nucleotide samples at 1 fm each (see Table 2).
Table 2.
Prospective Cost Comparison between Enzymatic and Chemical Methods of Oligonucleotide Synthesisa
| method | cost (USD) per dNTPPr incorporationb | cost (USD) of TdT | cost (USD) of dNTPPr | total cost (USD) of TdT + dNTPPrc |
|---|---|---|---|---|
| TiEOS with TdTd | 1.30 × 10−7 | 1.36 × 105 | 1.30 × 10−1 | 1.36 × 105 |
| TiEOS without TdTe | 1.30 × 10−7 | 1.36 × 102 | 1.30 × 10−1 | 1.36 × 102 |
| phosphoramidite | 2.70 × 10−3 | – | – | 2.70 × 103 |
Values in this table represent total costs for 1 fm-scale production of 1000 samples, at 1000 nt/strand, where the rate of nucleotide incorporation is estimated to be 250 fm s−1 (unit of TdT)−1.61
Includes cost for nucleoside phosphoramidite additions.
Total cost for phosphoramidite method also included.
Fresh TdT .
recycled TdT (attached to solid substrate). All costs reflect current market pricing.
Table 2 shows TiEOS can be significantly more cost-efficient than the phosphoramidite method if TdT is recycled ($136 and $2700, compared with $136000 if a fresh stock of TdT is introduced every cycle). Recycling TdT can be done by covalently attaching the enzyme to a solid substrate;107,108 as such, TdT has been shown to yield homopolymers up to 8000 nt during living polymerization,43 which is a testament to the enzyme’s longevity in a single reaction.
Because enzymatic oligonucleotide synthesis has not yet been reduced to standard practice, many unknowns still exist. For example, how much will TdT’s random nucleobase incorporation actually contribute to deletions via the unreacted initiator? Also, can dNTP analogues with 3′ blocking moieties be accepted into the binding pocket efficiently enough to match TdT nucleotide incorporation rates during living polymerization? Whether these concerns may be resolved or controlled by optimizing the buffers and/or reaction conditions alone is uncertain, but the option of protein engineering TdT for maximum performance holds even greater potential. Our objective for this Perspective was to shed light on such key issues, using the phosphoramidite cycle as a metric for improving stepwise, enzymatic oligonucleotide synthesis. Therefore, a sustainable method for producing artificial ssDNA promises to transform many fields of research and development, particularly in the area of synthetic biology. This in turn will greatly benefit both the environment and healthcare system.
ACKNOWLEDGMENTS
The authors thank Dr. Mohsen Nemat-Gorgani for his feedback regarding the manuscript.
Funding
This work was funded by National Institutes of Health Grant 2P01HG000205-24 to R.W.D.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Beadle GW, and Tatum EL (1941) Genetic Control of Biochemical Reactions in Neurospora. Proc. Natl. Acad. Sci. U. S. A 27, 499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hershey AD, and Chase M (1952) Independent functions of viral protein and nucleic acid in growth of bacteriophage. J. Gen. Physiol 36, 39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Watson JD, and Crick FH (1953) Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 171, 737–738. [DOI] [PubMed] [Google Scholar]
- (4).Lehman IR, Bessman MJ, Simms ES, and Kornberg A (1958) Enzymatic synthesis of deoxyribonucleic acid. I. Preparation of substrates and partial purification of an enzyme from Escherichia coli. J. Biol. Chem 233, 163–170. [PubMed] [Google Scholar]
- (5).Khorana HG (1968) Synthetic nucleic acids and the genetic code. JAMA, J. Am. Med. Assoc 206, 1978–1982. [PubMed] [Google Scholar]
- (6).Michelson AM, and Todd AR (1955) Nucleotides 0.32. Synthesis of a Dithymidine Dinucleotide Containing a 3′-5′-Internucleotidic Linkage. J. Chem. Soc 0, 2632–2638. [Google Scholar]
- (7).Todd A (1958) Synthesis in the study of nucleotides; basic work on phosphorylation opens the way to an attack on nucleic acids and nucleotide coenzymes. Science 127, 787–792. [DOI] [PubMed] [Google Scholar]
- (8).Khorana HG (1968) Synthesis in the study of nucleic acids. The Fourth Jubilee Lecture. Biochem. J 109, 709–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Grunberg-Manago M, Ortiz PJ, and Ochoa S (1956) Enzymic synthesis of polynucleotides. I. Polynucleotide phosphorylase of azotobacter vinelandii. Biochim. Biophys. Acta 20, 269–285. [DOI] [PubMed] [Google Scholar]
- (10).Brown DM (1993) A brief history of oligonucleotide synthesis. Methods Mol. Biol 20, 1–17. [DOI] [PubMed] [Google Scholar]
- (11).Reese CB (2005) Oligo- and poly-nucleotides: 50 years of chemical synthesis. Org. Biomol. Chem 3, 3851–3868. [DOI] [PubMed] [Google Scholar]
- (12).Behlke MA, and Devor EJ (2005) Chemical Synthesis of Oligonucleotides, Integrated DNA Technologies, Coralville, IA. [Google Scholar]
- (13). https://www.epa.gov/sites/production/files/2015-03/documents/list_of_lists.pdf.
- (14).Anastas PT (2009) The transformative innovations needed by green chemistry for sustainability. ChemSusChem 2, 391–392. [DOI] [PubMed] [Google Scholar]
- (15).Tang L, Navarro LA Jr., Chilkoti A, and Zauscher S (2017) High-Molecular-Weight Polynucleotides by Transferase-Catalyzed Living Chain-Growth Polycondensation. Angew. Chem., Int. Ed 56, 6778–6782. [DOI] [PubMed] [Google Scholar]
- (16).Tindall KR, and Kunkel TA (1988) Fidelity of DNA synthesis by the Thermus aquaticus DNA polymerase. Biochemistry 27, 6008–6013. [DOI] [PubMed] [Google Scholar]
- (17).Mueller S, Coleman JR, and Wimmer E (2009) Putting synthesis into biology: a viral view of genetic engineering through de novo gene and genome synthesis. Chem. Biol 16, 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Church GM, Gao Y, and Kosuri S (2012) Next-generation digital information storage in DNA. Science 337, 1628. [DOI] [PubMed] [Google Scholar]
- (19).Goldman N, Bertone P, Chen S, Dessimoz C, LeProust EM, Sipos B, and Birney E (2013) Towards practical, high-capacity, low-maintenance information storage in synthesized DNA. Nature 494, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hamon M, and Hong JW (2013) New tools and new biology: recent miniaturized systems for molecular and cellular biology. Mol. Cells 36, 485–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Cui F, Rhee M, Singh A, and Tripathi A (2015) Microfluidic Sample Preparation for Medical Diagnostics. Annu. Rev. Biomed. Eng 17, 267–286. [DOI] [PubMed] [Google Scholar]
- (22).Halamek J (2014) New age of quick and onsite bioassays for forensics: where are we now? Bioanalysis 6, 429–431. [DOI] [PubMed] [Google Scholar]
- (23).Mohanty BK, and Kushner SR (2000) Polynucleotide phosphorylase functions both as a 3′ right-arrow 5′ exonuclease and a poly(A) polymerase in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A 97, 11966–11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bollum FJ (1959) Thermal conversion of nonpriming deoxyribonucleic acid to primer. J. Biol. Chem 234, 2733–2734. [PubMed] [Google Scholar]
- (25).Bollum FJ (1960) Calf thymus polymerase. J. Biol. Chem 235, 2399–2403. [PubMed] [Google Scholar]
- (26).Bollum FJ (1962) Oligodeoxyribonucleotide-primed reactions catalyzed by calf thymus polymerase. J. Biol. Chem 237, 1945–1949. [PubMed] [Google Scholar]
- (27).Letsinger RL, and Mahadevan V (1965) Oligonucleotide Synthesis on a Polymer Support. J. Am. Chem. Soc 87, 3526–3527. [DOI] [PubMed] [Google Scholar]
- (28).Mackey JK, and Gilham PT (1971) New approach to the synthesis of polyribonucleotides of defined sequence. Nature 233, 551–553. [DOI] [PubMed] [Google Scholar]
- (29).Gilham S, and Smith M (1972) Enzymatic synthesis of deoxyribo-oligonucleotides of defined sequence. Nat. New Biol 238, 233–234. [DOI] [PubMed] [Google Scholar]
- (30).England TE, and Uhlenbeck OC (1978) Enzymatic oligoribonucleotide synthesis with T4 RNA ligase. Biochemistry 17, 2069–2076. [DOI] [PubMed] [Google Scholar]
- (31).Schott H, and Schrade H (1984) Single-step elongation of oligodeoxynucleotides using terminal deoxynucleotidyl transferase. Eur. J. Biochem 143, 613–620. [DOI] [PubMed] [Google Scholar]
- (32).Schmitz C, and Reetz MT (1999) Solid-phase enzymatic synthesis of oligonucleotides. Org. Lett 1, 1729–1731. [DOI] [PubMed] [Google Scholar]
- (33).Kimhi Y, and Littauer UZ (1968) Purification and properties of polynucleotide phosphorylase from Escherichia coli. J. Biol. Chem 243, 231–240. [PubMed] [Google Scholar]
- (34).Minhaz Ud-Dean SM (2008) A theoretical model for template-free synthesis of long DNA sequence. Syst. Synth Biol 2, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kaufmann G, and Kallenbach NR (1975) Determination of recognition sites of T4 RNA ligase on the 3′-OH and 5′ -P termini of polyribonucleotide chains. Nature 254, 452–454. [DOI] [PubMed] [Google Scholar]
- (36).Ramadan K, Shevelev I, and Hubscher U (2004) The DNA-polymerase-X family: controllers of DNA quality? Nat. Rev. Mol. Cell Biol 5, 1038–1043. [DOI] [PubMed] [Google Scholar]
- (37).Yamtich J, and Sweasy JB (2010) DNA polymerase family X: function, structure, and cellular roles. Biochim. Biophys. Acta, Proteins Proteomics 1804, 1136–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sadofsky MJ (2001) The RAG proteins in V(D)J recombination: more than just a nuclease. Nucleic Acids Res. 29, 1399–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Thai TH, Purugganan MM, Roth DB, and Kearney JF (2002) Distinct and opposite diversifying activities of terminal transferase splice variants. Nat. Immunol 3, 457–462. [DOI] [PubMed] [Google Scholar]
- (40).Motea EA, and Berdis AJ (2010) Terminal deoxynucleotidyl transferase: the story of a misguided DNA polymerase. Biochim. Biophys. Acta, Proteins Proteomics 1804, 1151–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fowler JD, and Suo Z (2006) Biochemical, structural, and physiological characterization of terminal deoxynucleotidyl transferase. Chem. Rev 106, 2092–2110. [DOI] [PubMed] [Google Scholar]
- (42).Kato KI, Goncalves JM, Houts GE, and Bollum FJ (1967) Deoxynucleotide-polymerizing enzymes of calf thymus gland. II. Properties of the terminal deoxynucleotidyltransferase. J. Biol. Chem 242, 2780–2789. [PubMed] [Google Scholar]
- (43).Tjong V, Yu H, Hucknall A, Rangarajan S, and Chilkoti A (2011) Amplified on-chip fluorescence detection of DNA hybridization by surface-initiated enzymatic polymerization. Anal. Chem 83, 5153–5159. [DOI] [PubMed] [Google Scholar]
- (44). https://www.neb.com/-/media/catalog/datacards-or-manuals/m0315datasheet-lot0101204.pdf.
- (45).Maga G, Ramadan K, Locatelli GA, Shevelev I, Spadari S, and Hubscher U (2005) DNA elongation by the human DNA polymerase lambda polymerase and terminal transferase activities are differentially coordinated by proliferating cell nuclear antigen and replication protein A. J. Biol. Chem 280, 1971–1981. [DOI] [PubMed] [Google Scholar]
- (46).Covo S, Blanco L, and Livneh Z (2004) Lesion bypass by human DNA polymerase mu reveals a template-dependent, sequence-independent nucleotidyl transferase activity. J. Biol. Chem 279, 859–865. [DOI] [PubMed] [Google Scholar]
- (47).Nair SV, Witek MA, Jackson JM, Lindell MA, Hunsucker SA, Sapp T, Perry CE, Hupert ML, Bae-Jump V, Gehrig PA, Wysham WZ, Armistead PM, Voorhees P, and Soper SA (2015) Enzymatic cleavage of uracil-containing single-stranded DNA linkers for the efficient release of affinity-selected circulating tumor cells. Chem. Commun. (Cambridge, U. K.) 51, 3266–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Tang L, Navarro LA Jr., Chilkoti A, and Zauscher S (2017) High-Molecular-Weight Polynucleotides by Transferase-Catalyzed Living Chain-Growth Polycondensation. Angew. Chem. Int. Ed 56, 6778–6782. [DOI] [PubMed] [Google Scholar]
- (49).Webster OW (1991) Living polymerization methods. Science 251, 887–893. [DOI] [PubMed] [Google Scholar]
- (50).Bollum FJ (1974) in The Enzymes (Boyer PD., Ed.) Vol. 10, p 145, Academic Press, New York. [Google Scholar]
- (51).Kates SA, and Albericio F (2000) Solid-Phase Synthesis: A Practical Guide, Marcel Dekker, New York. [Google Scholar]
- (52).Metzker ML, Raghavachari R, Richards S, Jacutin SE, Civitello A, Burgess K, and Gibbs RA (1994) Termination of DNA synthesis by novel 3/-modified-deoxyribonucleoside 5′-triphosphates. Nucleic Acids Res. 22, 4259–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Knapp DC, Serva S, D’Onofrio J, Keller A, Lubys A, Kurg A, Remm M, and Engels JW (2011) Fluoride-cleavable, fluorescently labelled reversible terminators: synthesis and use in primer extension. Chem. - Eur. J 17, 2903–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Ju J, Kim DH, Bi L, Meng Q, Bai X, Li Z, Li X, Marma MS, Shi S, Wu J, Edwards JR, Romu A, and Turro NJ (2006) Four-color DNA sequencing by synthesis using cleavable fluorescent nucleotide reversible terminators. Proc. Natl. Acad. Sci. U. S. A 103, 19635–19640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Chen F, Dong M, Ge M, Zhu L, Ren L, Liu G, and Mu R (2013) The history and advances of reversible terminators used in new generations of sequencing technology. Genomics, Proteomics Bioinf. 11, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Mathews AS, Yang H, and Montemagno C (2016) Photo-cleavable nucleotides for primer free enzyme mediated DNA synthesis. Org. Biomol. Chem 14, 8278–8288. [DOI] [PubMed] [Google Scholar]
- (57).Hutter D, Kim MJ, Karalkar N, Leal NA, Chen F, Guggenheim E, Visalakshi V, Olejnik J, Gordon S, and Benner SA (2010) Labeled nucleoside triphosphates with reversibly terminating aminoalkoxyl groups. Nucleosides, Nucleotides Nucleic Acids 29, 879–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Boule JB, Rougeon F, and Papanicolaou C (2001) Terminal deoxynucleotidyl transferase indiscriminately incorporates ribonucleotides and deoxyribonucleotides. J. Biol. Chem 276, 31388–31393. [DOI] [PubMed] [Google Scholar]
- (59).Winz ML, Linder EC, Andre T, Becker J, and Jaschke A (2015) Nucleotidyl transferase assisted DNA labeling with different click chemistries. Nucleic Acids Res. 43, e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Koukhareva I, and Lebedev A (2009) 3′-Protected 2′-deoxynucleoside 5′-triphosphates as a tool for heat-triggered activation of polymerase chain reaction. Anal. Chem 81, 4955–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61). https://www.neb.com/-/media/catalog/datacards-or-manuals/m0315datasheet-lot0101204.pdf.
- (62).Temsamani J, Kubert M, and Agrawal S (1995) Sequence identity of the n-1 product of a synthetic oligonucleotide. Nucleic Acids Res. 23, 1841–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Krotz AH, Cole DL, and Ravikumar VT (2003) Synthesis of antisense oligonucleotides with minimum depurination. Nucleosides, Nucleotides Nucleic Acids 22, 129–134. [DOI] [PubMed] [Google Scholar]
- (64).Hirano M, Das S, Guo P, and Cooper MD (2011) The evolution of adaptive immunity in vertebrates. Adv. Immunol 109, 125–157. [DOI] [PubMed] [Google Scholar]
- (65).Berdis AJ, and McCutcheon D (2007) The use of non-natural nucleotides to probe template-independent DNA synthesis. ChemBioChem 8, 1399–1408. [DOI] [PubMed] [Google Scholar]
- (66).Jarchow-Choy SK, Krueger AT, Liu H, Gao J, and Kool ET (2011) Fluorescent xDNA nucleotides as efficient substrates for a template-independent polymerase. Nucleic Acids Res. 39, 1586–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Chow DC, Lee WK, Zauscher S, and Chilkoti A (2005) Enzymatic fabrication of DNA nanostructures: extension of a self-assembled oligonucleotide monolayer on gold arrays. J. Am. Chem. Soc 127, 14122–14123. [DOI] [PubMed] [Google Scholar]
- (68).Chow DC, and Chilkoti A (2007) Surface-initiated enzymatic polymerization of DNA. Langmuir 23, 11712–11717. [DOI] [PubMed] [Google Scholar]
- (69).Ratliff RL, Schwartz AW, Kerr VN, Williams DL, Ott DG, and Hayes FN (1968) Heteropolydeoxynucleotides synthesized with terminal deoxyribonucleotidyltransferase. II. Nearest neighbor frequencies and extent of digestion by micrococcal deoxyribonuclease. Biochemistry 7, 412–418. [DOI] [PubMed] [Google Scholar]
- (70).Chang LM, and Bollum FJ (1990) Multiple roles of divalent cation in the terminal deoxynucleotidyltransferase reaction. J. Biol. Chem 265, 17436–17440. [PubMed] [Google Scholar]
- (71).Deibel MR Jr., and Coleman MS (1980) Biochemical properties of purified human terminal deoxynucleotidyltransferase. J. Biol. Chem 255, 4206–4212. [PubMed] [Google Scholar]
- (72).Traut TW (1994) Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem 140, 1–22. [DOI] [PubMed] [Google Scholar]
- (73).Gangi-Peterson L, Sorscher DH, Reynolds JW, Kepler TB, and Mitchell BS (1999) Nucleotide pool imbalance and adenosine deaminase deficiency induce alterations of N-region insertions during V(D)J recombination. J. Clin. Invest 103, 833–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Bhattacharyya D, Mirihana Arachchilage G, and Basu S (2016) Metal Cations in G-Quadruplex Folding and Stability. Front. Chem 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Jensen MA, Fukushima M, and Davis RW (2010) DMSO and betaine greatly improve amplification of GC-rich constructs in de novo synthesis. PLoS One 5, e11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Hardin CC, Watson T, Corregan M, and Bailey C (1992) Cation-dependent transition between the quadruplex and Watson-Crick hairpin forms of d(CGCG3GCG). Biochemistry 31, 833–841. [DOI] [PubMed] [Google Scholar]
- (77).Rees WA, Yager TD, Korte J, and von Hippel PH (1993) Betaine can eliminate the base pair composition dependence of DNA melting. Biochemistry 32, 137–144. [DOI] [PubMed] [Google Scholar]
- (78).Varadaraj K, and Skinner DM (1994) Denaturants or cosolvents improve the specificity of PCR amplification of a G + C-rich DNA using genetically engineered DNA polymerases. Gene 140, 1–5. [DOI] [PubMed] [Google Scholar]
- (79).Winship PR (1989) An improved method for directly sequencing PCR amplified material using dimethyl sulfoxide. Nucleic Acids Res. 17, 1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).McConlogue L, Brow MA, and Innis MA (1988) Structure-independent DNA amplification by PCR using 7-deaza-2′-deoxyguanosine. Nucleic Acids Res. 16, 9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Spiess A-N, Mueller N, and Ivell R (2004) Trehalose is a potent PCR enhancer: lowering of DNA melting temperature and thermal stabilization of taq polymerase by the disaccharide trehalose. Clin. Chem 50, 1256–1259. [DOI] [PubMed] [Google Scholar]
- (82).Jensen M, and Davis R (2017) RecJ 5′ Exonuclease Digestion of Oligonucleotide Failure Strands: A "Green" Method of Trityl-On Purification. Biochemistry 56, 2417–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Vorndam AV, and Kerschner J (1986) Purification of Small Oligonucleotides by Polyacrylamide-Gel Electrophoresis and Transfer to Diethylaminoethyl Paper. Anal. Biochem 152, 221–225. [DOI] [PubMed] [Google Scholar]
- (84).Warren WJ, and Vella G (1995) Principles and Methods for the Analysis and Purification of Synthetic Deoxyribonucleotides by High-Performance Liquid-Chromatography. Mol. Biotechnol 4, 179–199. [DOI] [PubMed] [Google Scholar]
- (85).Glen-PakTM Cartridges: DNA and RNA Purification. http://www.glenresearch.com/Technical/GlenPak_UserGuide.pdf.
- (86).Brody RS, and Doherty KG (1985) Stereochemical course of hydrolysis of DNA by exonuclease I from Escherichia coli. Biochemistry 24, 2072–2076. [DOI] [PubMed] [Google Scholar]
- (87).Freudenthal BD, Beard WA, and Wilson SH (2012) Structures of dNTP intermediate states during DNA polymerase active site assembly. Structure 20, 1829–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Yang L, Arora K, Beard WA, Wilson SH, and Schlick T (2004) Critical role of magnesium ions in DNA polymerase beta’s closing and active site assembly. J. Am. Chem. Soc 126, 8441–8453. [DOI] [PubMed] [Google Scholar]
- (89).Balbo PB, Wang EC, and Tsai MD (2011) Kinetic mechanism of active site assembly and chemical catalysis of DNA polymerase beta. Biochemistry 50, 9865–9875. [DOI] [PubMed] [Google Scholar]
- (90).Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, and Wilson SH (2006) Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure 14, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Delarue M, Boule JB, Lescar J, Expert-Bezancon N, Jourdan N, Sukumar N, Rougeon F, and Papanicolaou C (2002) Crystal structures of a template-independent DNA polymerase: murine terminal deoxynucleotidyltransferase. EMBO J. 21, 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Arora K, Beard WA, Wilson SH, and Schlick T (2005) Mismatch-induced conformational distortions in polymerase beta support an induced-fit mechanism for fidelity. Biochemistry 44, 13328–13341. [DOI] [PubMed] [Google Scholar]
- (93).Showalter AK, and Tsai MD (2002) A reexamination of the nucleotide incorporation fidelity of DNA polymerases. Biochemistry 41, 10571–10576. [DOI] [PubMed] [Google Scholar]
- (94).Sawaya MR, Prasad R, Wilson SH, Kraut J, and Pelletier H (1997) Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry 36, 11205–11215. [DOI] [PubMed] [Google Scholar]
- (95).Batra VK, Beard WA, Shock DD, Pedersen LC, and Wilson SH (2008) Structures of DNA polymerase beta with active-site mismatches suggest a transient abasic site intermediate during misincorporation. Mol. Cell 30, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Sorensen RS, Okholm AH, Schaffert D, Kodal AL, Gothelf KV, and Kjems J (2013) Enzymatic ligation of large biomolecules to DNA. ACS Nano 7, 8098–8104. [DOI] [PubMed] [Google Scholar]
- (97).Tauraite D, Jakubovska J, Dabuzinskaite J, Bratchikov M, and Meskys R (2017) Modified Nucleotides as Substrates of Terminal Deoxynucleotidyl Transferase. Molecules 22, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Chirpich TP (1977) Factors affecting terminal deoxynucleotidyl transferase activity in cacodylate buffer. Biochem. Biophys. Res. Commun 78, 1219–1226. [DOI] [PubMed] [Google Scholar]
- (99).Modified template-independent enzymes for polydeoxynucleotide (2015) Patent WO2016064880.
- (100).Methods and apparatus for synthesizing nucleic acids (2013) Patent US8808989B1.
- (101).Windle CL, Simmons KJ, Ault JR, Trinh CH, Nelson A, Pearson AR, and Berry A (2017) Extending enzyme molecular recognition with an expanded amino acid alphabet. Proc. Natl. Acad. Sci. U. S. A 114, 2610–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Chen F, Gaucher EA, Leal NA, Hutter D, Havemann SA, Govindarajan S, Ortlund EA, and Benner SA (2010) Reconstructed evolutionary adaptive paths give polymerases accepting reversible terminators for sequencing and SNP detection. Proc. Natl. Acad. Sci. U. S. A 107, 1948–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Pinheiro VB, and Holliger P (2012) The XNA world: progress towards replication and evolution of synthetic genetic polymers. Curr. Opin. Chem. Biol 16, 245–252. [DOI] [PubMed] [Google Scholar]
- (104).Houlihan G, Arangundy-Franklin S, and Holliger P (2017) Exploring the Chemistry of Genetic Information Storage and Propagation through Polymerase Engineering. Acc. Chem. Res 50, 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Yang B, Gathy KN, and Coleman MS (1994) Mutational analysis of residues in the nucleotide binding domain of human terminal deoxynucleotidyl transferase. J. Biol. Chem 269, 11859–11868. [PubMed] [Google Scholar]
- (106).Kosuri S, and Church GM (2014) Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods 11, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107). https://tools.thermofisher.com/content/sfs/brochures/1602163-Crosslinking-Reagents-Handbook.pdf.
- (108).Brena B, Gonzalez-Pombo P, and Batista-Viera F (2013) Immobilization of enzymes: a literature survey. Methods Mol. Biol 1051, 15–31. [DOI] [PubMed] [Google Scholar]