

Abstract
Chemical cross-linking with mass spectrometry (XL-MS) has emerged as a useful tool for the large-scale study of protein structures and interactions from complex biological samples including intact cells and tissues. Quantitative XL-MS (qXL-MS) provides unique information on protein conformational and interaction changes resulting from perturbations such as drug treatment and disease state. Previous qXL-MS studies relied on the incorporation of stable isotopes into the cross-linker (primarily deuterium) or metabolic labeling with SILAC. Here, we introduce isobaric quantitative protein interaction reporter (iqPIR) technology which utilizes stable isotopes selectively incorporated into the cross-linker design, allowing for isobaric cross-linked peptide pairs originating from different samples to display distinct quantitative isotope signatures in tandem mass spectra. This enables improved quantitation of cross-linked peptide levels from proteome-wide samples because of the reduced complexity of tandem mass spectra relative to MS1 spectra. In addition, because of the isotope incorporation in the reporter and the residual components of the cross-linker that remain on released peptides, each fragmentation spectrum can offer multiple independent opportunities and, therefore, improved confidence for quantitative assessment of the cross-linker pair level. Finally, in addition to providing information on solvent accessibility of lysine sites, dead end iqPIR cross-linked products can provide protein abundance and/or lysine site modification level information all from a single in vivo cross-linking experiment.
Graphical Abstract
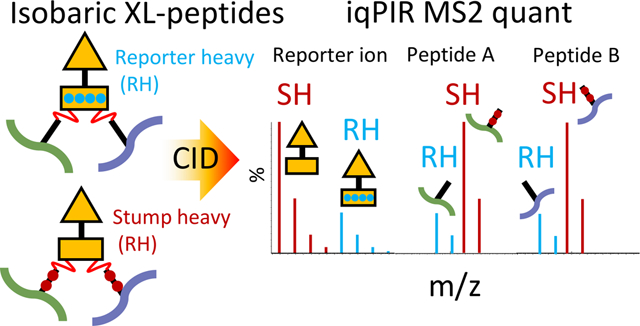
Chemical cross-linking with mass spectrometry (XL-MS) provides spatial information on proximal cross-linker reactive amino acid residues on the surfaces of protein molecules. As such XL-MS has become a useful tool for structural biology providing information that is largely complementary to techniques such as X-ray crystallography, nuclear magnetic resonance spectroscopy, and cryo-EM. XL-MS provides unique advantages over other techniques including being applicable to proteins and protein complexes of any size, providing data on flexible and disordered regions, sampling the complete ensemble of conformations in solution, and being applicable to unpurified proteins and complexes even as they exist in their native cellular environments. In addition to low-resolution structural information on proteins, intermolecular cross-links provide direct physical evidence for protein–protein interactions (PPIs). As such, XL-MS serves as a technique to bridge traditional structural biology techniques and methods to measure PPIs in a large scale, such as affinity purification mass spectrometry (AP-MS) and yeast two-hybrid (Y2H) screening. Protein interaction reporter (PIR) technology was initially introduced in 20051 utilizing selectively cleavable cross-linkers for facile identification of cross-linked peptide pairs by MS analysis. PIR and similar strategies from other research groups enabled large-scale in situ XL-MS studies providing useful structural and PPI information from cultured bacterial cells,2,3 mammalian cells,4,5 isolated organelles,6–8 and tissues.9
Quantifying cross-linked peptides provides unique information on protein conformational and interaction changes resulting from perturbations such as drug treatment, post-translational modifications, and disease state. Previous qXL-MS studies have relied on the incorporation of stable isotopes into the cross-linker (primarily deuterium),10 metabolic labeling using heavy isotope amino acid residues (SILAC),11–13 or incorporation of isobaric mass tags with additional chemical labeling steps.14 Additionally, cross-linked peptide pairs have been quantified by targeted MS2-based quantification with parallel reaction monitoring (PRM).15 PRM offers excellent sensitivity and accuracy at the cost of being relatively low throughput and generally requires separate LC–MS analyses for quantification and identification. Label-free qXL-MS using PRM was successfully used to help elucidate conformational changes in the Hsp90-Aha1 complex.16
Here, we report on the development of isobaric quantitative protein interaction reporter (iqPIR) technology which utilizes stable isotopes selectively incorporated into the cross-linker design, allowing for cross-linked peptide pairs originating from different samples to have exactly the same mass in MS1 measurements, yet display distinct quantitative isotope signatures in tandem MS. Akin to the benefits afforded by isobaric mass tags for traditional proteomics including iTRAQ17 and TMT,18 iqPIR provides multiple benefits over other existing qXL-MS strategies. These include increased signal-to-noise because of additive contributions of MS1 signals, generation of multiple fragment ions carrying quantitative information in a single tandem MS spectrum, avoidance of chromatographic alignment and peak assignment problems, elimination of retention time shifts between isotope partners as commonly observed with deuterium, applicability to systems where metabolic labeling is impractical, and potential for multiplexed quantitation. However, in contrast with iTRAQ and TMT, which primarily rely on quantification based on the relative intensities of reporter ions generated from the released chemical tag, iqPIR MS2 are rich in quantitative information as PIR reporter ions, intact released peptide ions, and many backbone fragment ions of the cross-linked peptides all contain encoded isotope signatures that can be used for quantification. Multiple opportunities for quantitative assessment in each MS2 spectrum make iqPIR quantitation less susceptible to ratio suppression commonly observed with MS2 reporter ion-based methods.19 Herein, we describe the synthesis and characterization of iqPIR reagents, the development of informatics for quantification, and the application to protein model systems as well as drug-treated HeLa cell samples.
EXPERIMENTAL SECTION
Synthesis.
The iqPIR cross-linker, sequence = Gly-Gly-Lys(biotin)-Lys-Pro2-Asp2-succinate2-N-hydroxyphthalimide (NHP)2, was synthesized on Rink amide ProTide resin (CEM) by solid phase peptide synthesis using a CEM Liberty Lite peptide synthesizer. For the stump heavy (SH) version, stable heavy isotopes were incorporated into the stump portion of the iqPIR molecule via 13C2-succinic anhydride. For the reporter heavy (RH) version, 13C2-Gly was used for the incorporation of heavy isotopes into the reporter region of the iqPIR molecule. Activated NHP esters were incorporated by incubating the resin containing the iqPIR molecule with a 12-fold molar excess of N-(trifluoroacetoxy)-pthalimide (TFA-NHP) in pyridine. The reaction was carried out for 20 min at room temperature. The resin was then transferred to a Poly-Prep column (Bio-Rad) and coupled to a vacuum flask. The resin was washed extensively with dimethylformamide (DMF) followed by extensive washes with dichloromethane (DCM). The cross-linker was cleaved from the resin by incubation with 95% TFA, 2.5% DCM, and 2.5% H2O for 3 h at room temperature. The cross-linker was then precipitated in cold diethyl ether and washed extensively with fresh cold diethyl ether. The resulting pellet was dried by vacuum centrifugation. The resulting product was dissolved in DMSO to a concentration of 200 mM, determined by UV–vis absorbance.
Generation of iqPIR Cross-linked Standard Protein Mixtures.
One mg of each of the three purified proteins; serum albumin (BSA, Sigma), myoglobin from horse (MYG, Sigma), and alcohol dehydrogenase from yeast (ADH1, Sigma) were dissolved in separate 1 mL aliquots of 170 mM Na2HPO4 pH 8. Chemical cross-linking with iqPIR was carried out by adding either the SH or RH iqPIR from a concentrated stock in DMSO to the protein containing solution at a final concentration of 1 mM and allowing the reaction to proceed for 30 min at 22 °C with constant mixing at 600 rpm on a Thermomixer. Aliquots of the cross-linked proteins were then mixed at the following SH/RH ratios to generate three different mixed samples each containing 100 μg total of each of the three proteins; mix 1 = BSA 4:1, Myg 1:10, ADH 1:2, mix 2 = BSA 1:10, Myg 2:1, ADH 4:1, mix 3 = BSA 2:1, Myg 1:4, and ADH 10:1. Disulfides in the three mixed samples were reduced by adding TCEP to 5 mM from a 500 mM stock and mixing at room temperature for 30 min. Reduced thiols were alkylated with 10 mM iodoacetamide added from a 1 M stock. Proteins were digested with a 1:200 ratio of trypsin to protein at 37 °C for 16 h. The resulting peptide samples were desalted by solid phase extraction using Waters C18 SepPak cartridges.
HeLa Cell Cross-linking with iqPIR.
HeLa cells were cultured in 15 cm diameter dishes at 37 °C in a humidified atmosphere containing 5% CO2, in DMEM supplemented with 10% Fetalgro bovine growth serum (Rocky Mountain Biologicals) and 100 U/mL penicillin–streptomycin. For drug treatment experiments, when cells reached 80% confluence, 17-AAG (Cayman Chemicals) was added from a 0.5 mM stock in DMSO to the growth media at 500 nM final concentration and the cells were incubated for 18 h. To a separate 15 cm dish of cells, 0.1% DMSO was added as a vehicle control. Prior to cross-linking, the growth media was aspirated away and cells were washed twice with 5 mL PBS. Cells were then released from the dish by incubation for 3 min at 37 °C in 5 mL PBS containing 20 mM EDTA. Detached cells were transferred to 15 mL centrifuge tubes and pelleted at 300 g for 3 min. The cell pellet was washed twice with 10 mL PBS containing 1 mM Ca2+ and Mg2+. The cell pellet was then suspended in 500 μL of 170 mM Na2HPO4 pH 8.0 and iqPIR cross-linker (either SH or RH) was added to a final concentration of 10 mM. The cross-linking reaction was carried out for 30 min at room temperature with constant mixing. Cross-linked cells were mixed at a 1:1 17-AAG:DMSO mixture. After 30 min, the cells were pelleted by centrifugation at 300g for 3 min and the supernatant was removed. The cell pellet was washed three times with 1 mL 0.1 M NH4HCO3, pelleting the cells by centrifugation in between wash steps. Cells were lysed by suspending the cell pellet in 8M urea in 0.1M NH4HCO3 followed by reducing disulfides by adding TCEP to 5 mM from a 500 mM stock and mixing at room temperature for 30 min. Reduced thiols were alkylated with 10 mM iodoacetamide added from a 1 M stock. Proteins were digested with a 1:200 ratio of trypsin to protein at 37 °C for 16 h. Resulting peptide samples were desalted by solid phase extraction using Waters C18 SepPak cartridges. Desalted samples were concentrated by vacuum centrifugation using an EZ2-Plus evaporator. Peptide samples were then adjusted to a volume of 0.5 mL with 7 mM KH2PO4, 30% acetonitrile pH 2.8 before being fractionated by strong cation exchange chromatography (SCX) using an Agilent 1200 series HPLC system equipped with a 250 × 10.0 mm column packed with Luna 5 μm 100A particles (Phenomenex). Peptide separation was accomplished using a binary mobile phase solvent system consisting of solvent A (7 mM KH2PO4, 30% acetonitrile pH 2.8) and solvent B (7 mM KH2PO4, 350 mM KCl, 30% acetonitrile pH 2.8) at a flow rate of 1.5 mL/min using the following gradient program: 0–7.5 min 100% A, 7.5–47.5 min 95% A/5% B to 40% A/60% B, 47.5–67.5 min 40% A/60% B to 100% B, 67.5–77.5 min 100% B, and 77.5–97.5 min 100% A. A total of 15 fractions (5 min time slices, 7.5 mL each) were collected starting after an initial 17.5 min delay. Resulting SCX fractions were concentrated by vacuum centrifugation before their pH was adjusted to 8.0 by the addition of 0.1 M NH4HCO3. Fractions 6 and 7 were combined as well as fractions 11–14. To each of five fractions (6–7, 8, 9, 10, 11–14), 200 μL of monomeric avidin slurry (Thermo) was added and the samples were mixed for 30 min at room temperature. To remove non-biotin containing peptides, the avidin beads were washed 3 x with 3 mL of 100 mM NH4HCO3 pH 8.0 before eluting the cross-linked peptides by incubating the beads for 5 min each with two 500 μL aliquots of 70% acetonitrile, 30% H20 containing 0.5% formic acid. The enriched cross-linked peptide sample was then concentrated by vacuum centrifugation and stored at −80 °C until LC–MS analysis.
LC–MS Analysis.
Cross-linked samples were analyzed in technical triplicate by liquid chromatography mass spectrometry using an Easy-nLC (Thermo Scientific) coupled to a Q Exactive Plus mass spectrometer (Thermo Scientific). Peptides were loaded (3 μL injection volume) onto a 3 cm × 100 μm inner diameter fused silica trap column packed with a stationary phase consisting of 5 μm Reprosil C8 particles with 120 Å pores (Dr. Maisch GmbH) with a flow rate of 2 μL/min of the mobile phase consisting of solvent A (H2O containing 0.1% formic acid) for 10 min. Peptides were then fractionated over a 60 cm × 75 μm inner diameter fused silica analytical column packed with 5 μm Reprosil C8 particles with 120 Å pores by applying a linear gradient from 95% solvent A, 5% solvent B (acetonitrile containing 0.1% formic acid) to 60% solvent A, 40% solvent B over either 120 (standard protein samples) or 240 min (HeLa samples) at a flow rate of 300 nL/min. Eluting peptide ions were ionized by electrospray ionization by applying a positive 2.2 kV potential to a laser pulled spray tip at the end of the analytical column. The mass spectrometer was operated using a top five data-dependent acquisition method with a resolving power setting of 70,000 for MS1 and MS2 scans. Additional settings include an AGC target value of 1e6 with a maximum ion time of 100 ms for the MS1 scans and an AGC value of 5e4 with a maximum ion time of 300 ms for the MS2 scans. Charge state exclusion parameters were set to only allow ions with charge states from 4+ to 7+ to be selected for MS2. Ions selected for MS2 were isolated with a 3 m/z window and fragmented by HCD using a normalized collision energy setting of 30. Ions for which MS2 was performed were then dynamically excluded from further selection for MS2 for 30 s.
Informatics.
Raw files were converted to mzXML format and searched for PIR mass relationships with Mango.20 The resulting MS2 files were searched using Comet4 against the sequence databases, containing both forward and reverse databases. For standard protein samples, the database consisted of 4327 forward and reverse protein sequences, including those for ALBU_BOVIN, ADH1_YEAST, and MYG_HORSE along with a background of known false positive proteins from Bacillus. subtilis (https://doi.org/10.1093/bioinformatics/bty720). For HeLa cell samples, the database consisted of a subset of the UniProt reference proteome database for Homo sapiens containing both forward and reverse sequences for 3515 proteins identified from our previous study of 17-AAG-treated HeLa cells.12 Resulting pepXML files were then analyzed with XLinkProphet21 and filtered to an estimated false discovery rate of less than 1% at the nonredundant peptide pair level.
For quantification, php scripts were developed to extract peak intensities and calculate quantitative ratios from the MS2 spectra. Inputs are the raw files in mzXML format, the corresponding noise files after analyzing RAW files with getNoise, and the XLinkProphet analysis pepXML file. Cross-link results analyzed with XLinkProphet are read in pepXML format, filtered according to the user-specified criteria (e.g., estimated 1% non-redundant cross-link FDR). All cross-links passing the threshold are analyzed for quantitation in their corresponding MS2 scan. The noise for that scan is read from the noise file of that run, so a minimum signal to noise ratio can be imposed. Based on the cross-link identification, masses of the two released peptides with stump modifications, as well as their sequences and site of stump modification, are noted. These are used to calculate the m/z values of both released peptide ions with charges from 1 to 3, in both the light and heavy forms. The m/z values of reporters are fixed and can also be assessed for quantitation if desired. Based on the sequences of the peptides, fragment ions containing the stump modification, those predicted to be present in both light and heavy forms, were noted so their singly charged light and heavy m/z values could be used for quantitation. Quantitation of specified m/z values was achieved by summing the intensities of all m/z values at the MS2 scan within 20 ppm tolerance of the targeted ions. In order to ensure high quality quantitation, several filters were imposed on the data. For all ions, a minimum signal to noise of 5 was imposed.
For the reporter ions which have nonoverlapping light (SH) and heavy (RH) isotope peak groups, the observed isotope peak intensities were required to adequately match theoretical intensities, as computed by Yergey.22 The error was computed for the 3 isotopes as the sum of the squared differences between the relative intensities of the observed and theoretical isotopes, then taking its square root and dividing by the number of isotopes, N. In our analysis, N = 3.
This gives the average deviation of the observed and expected relative isotope intensities. A maximum difference of 0.045 was allowed separately for the light and heavy ions. Quantitation was only accepted from light and heavy reporter ions that both agreed within 0.045 of the expected relative intensities.
For the peptide and fragment ions, the light (RH) and heavy (SH) quantitation required deconvolution because the third isotope of the light form coincides with the monoisotopic peak of the heavy form. Similarly, the fourth light and second heavy ion peaks coincide. In these cases, the theoretical relative intensities were used to help deconvolute the light and heavy ion contributions of coincident peaks. The ratio of the second to monoisotopic light peaks was noted, as well as the theoretical ratio. Both those isotopes are exclusively contributed by light peptides or fragments. If the absolute value of the difference in ratios between the observed and theoretical intensities exceeded 0.4, quantitation was aborted. In a similar manner, the second and third heavy isotope peaks were assumed to be predominantly contributed from the heavy, though in the case of heavy peptides could contain some light contribution as well. The observed and theoretical heavy third over second isotope intensities were compared, their absolute value was not allowed to exceed 0.4.
Light and heavy isotope peak intensities matching sufficiently with the expected values were then deconvoluted to yield a heavy to light ratio, minimizing the error between the observed relative isotope peak intensities and those expected based on the light/heavy ratio and the theoretical relative isotope peak intensities predicted by the ion chemical composition. The theoretical relative intensities for the monoisotopic through fourth isotope offset peaks, normalized to 1, are designated as ξ 0 through ξ 4, while the relative intensities of the observed isotope peaks normalized to 1 are designated O0 through O4 (Figure S1A).
Define the ratio R as the light ion total intensity L divided by the heavy ion total intensity H
Note that the expected L and H contributions for the first five isotope peaks is equal to L(ξ0 + ξ 1 + ξ 2 + ξ 3 + ξ 4) and H(ξ0 + ξ1 + ξ2), respectively. The light contribution is equal to the total L defined over the first five isotopes, whereas the heavy contribution is equal to the total H defined over its first three isotopes, corresponding to isotope offset peaks 2 through 4. Thus, the expected light + heavy contributions to the first five isotope peaks, Ei, i = 0 to 4, based on the ratio R and normalized to sum to 1, are
Now consider the observed ion isotope peak relative intensities O0 through O4 for the first five isotope peaks, normalized to 1. The summed error between the observed ion isotope peak relative intensities and those expected based on ratio R is
We can then derive the value of R that minimizes the error
To simplify the equation for error, we define the following variables
Define the following three terms
Now the equation for error becomes
and its derivative with respect to R
Setting this derivative to zero leads to the following equation for the optimal ratio Ropt that minimizes the error
Only optimal ratios with a coincident error not exceeding 0.05 were accepted.
The second derivative of error with respect to ratio
is calculated for each computed ratio to ensure that it is greater than zero, reflective of a minimum rather than a maximum error. Shown in Figure S1B is a plot of the ratio error as a function of different ratio values, R, for example, theoretical and observed peak relative intensities. Indicated in red is the log2 ratio 0.21 corresponding to the Ropt value obtained by the above formula, corresponding to the minimum value of the error curve.
For each cross-link MS2 spectrum, it is often possible to quantify several ratios from peptides of different charge states and from stump-containing fragment ions. In addition, because the same cross-link can be identified in multiple scans in a run, and in separate replicate runs, peptide and fragment ratios are collected in a log2 form from all scans for each cross-link and combined together at a final step. If normalization is specified, all contributing cross-link ratios are divided by the median value of combined peptide and fragment ratios of all sample cross-links. Outlier removal is then applied for each cross-link whereby contributing ratios with values greater than 1.5 times the difference between values of the second and third quartile from the third quartile, or less than 1.5 times the difference between values of the second and third quartile from the first quartile, are removed in up to 2 iterations. Finally, the cross-link log2 ratio mean, standard deviation, T-statistic, and p-value are computed.
RESULTS AND DISCUSSION
Design and Synthesis of iqPIR Cross-linkers.
The peptide composition and modular design features of PIR cross-linkers are advantageous for exploring many new concepts in cross-linker technologies, such as photocleavage,23,24 electron-base cleavage,25 and multidimensional cross-linkers.26 These PIR attributes also enabled the synthesis of first generation isobaric quantitative PIR molecules. Initial iqPIR molecules were based on a modified form of the BDP-NHP cross-linker which has been successfully applied to a number of complex biological systems.2,4,6,9 iqPIR cross-linkers were produced with solid-phase peptide synthesis by coupling the following amino acids in sequence Gly, Gly, biotin-Lys, Lys, Pro, Asp, and succinate. The chemical structure of the iqPIR is illustrated in Figure 1A. A total of four 13C atoms were incorporated into the reporter region of the cross-linker via 13C2-Gly to generate the RH version of the iqPIR reagent. For the corresponding isobaric partner, the SH iqPIR, heavy isotopes were incorporated with 13C2-succinic anhydride. Both forms of the iqPIR reagent have the same monoisotopic mass of 1531.574678 Da. Direct infusion ESI-MS analysis of separate samples of the RH iqPIR and SH iqPIR resulted in the measurement of identical precursor ion m/z values within the expected mass accuracy of the instrument, 7T Velos-FTICR at 5E4 resolving power at m/z 400 (Figure 1B). Upon isolation and CID fragmentation of the precursor ion at 1532.584, differences in the fragmentation patterns were observed in the MS2 for the RH and SH iqPIR reagents (Figure 1C). Notably, the measured m/z values of the reporter ions differed by 4.014 Da, while the long arm fragments resulting from the cleavage of a single Asp–Pro bond differed by 2.007 Da.
Figure 1.
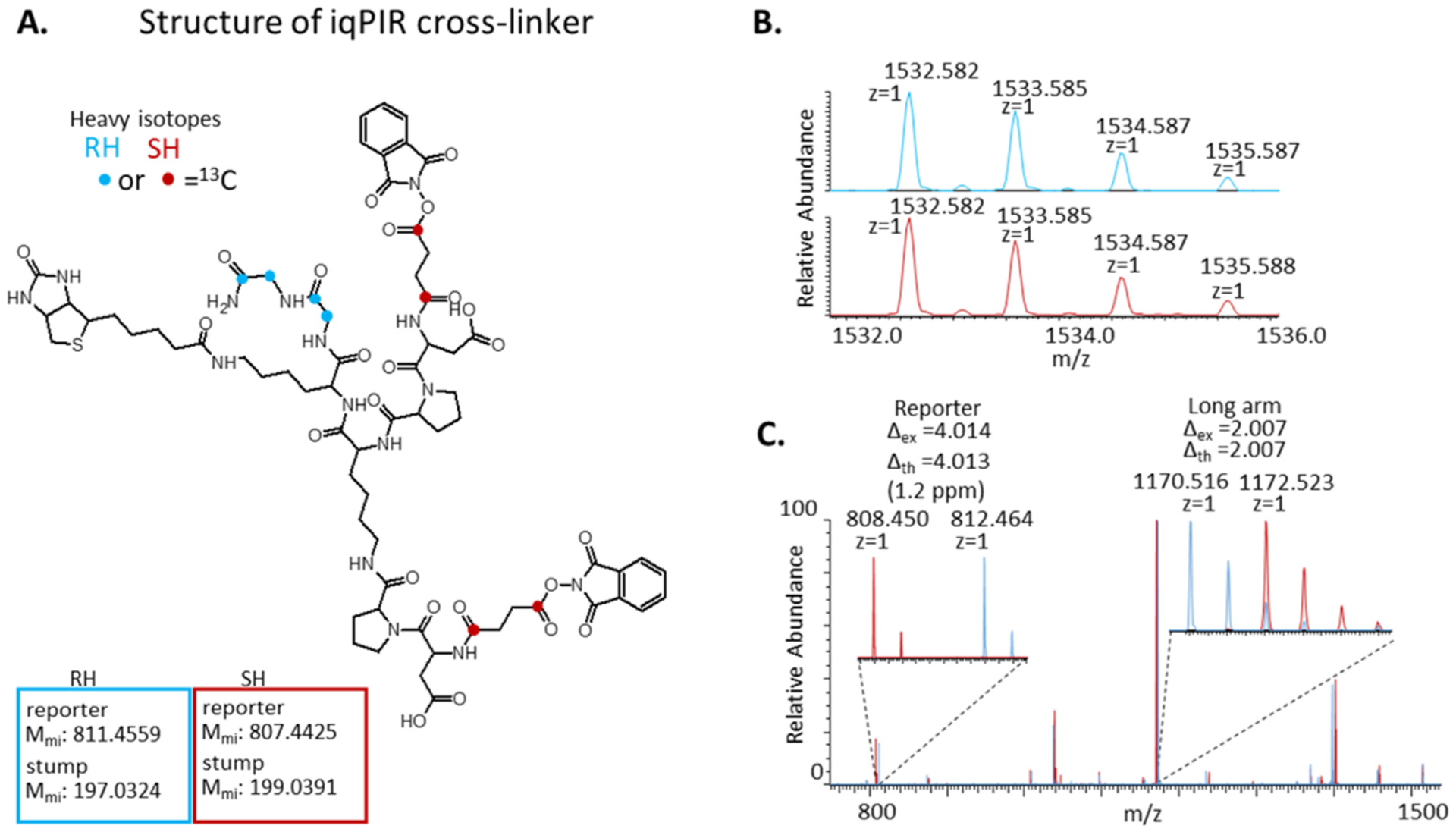
Structure and MS characterization of iqPIR cross-linker. (A) Chemical structure of iqPIR cross-linker with the positions of 13C atoms indicated with blue circles for the RH version and with red circles for the SH version. (B) MS1 spectra for the [M + H]+ ions for the RH (top, blue) and SH (bottom, red) iqPIR cross-linkers demonstrating they are isobaric. (C) Overlaid MS2 spectra for the RH (blue) and SH (red) iqPIR cross-linkers with zoomed insets indicating the mass differences observed for the reporter ion and the long arm fragment ion (resulting from cleavage of a single Asp–Pro bond).
Evaluation of iqPIR with Purified Proteins.
Initially, we evaluated the quantitative performance of iqPIR using samples of three purified proteins, including bovine serum albumin (ALBU_BOVIN), alcohol dehydrogenase from Saccharomyces cerevisiae (ADH1_YEAST), and myoglobin from horse (MYG_HORSE). Each protein was cross-linked independently with the RH and the SH iqPIR cross-linkers as described in the methods. After the cross-linking reaction, each of the three proteins were mixed at 1:1 (RH/SH) ratios prior to tryptic digestion. Upon collision-induced dissociation, each iqPIR cross-linked peptide pair precursor ion produces a number of fragment ions containing quantitative information as demonstrated by the MS2 spectrum of a 1:1 (RH/SH) mixture of the cross-linked peptide pair linking K207 with K226 of ADH1_YEAST (EK2 2 6DIVGAVLK-VLGIDGGEG-K207EELFR), as shown in Figure 2.
Figure 2.
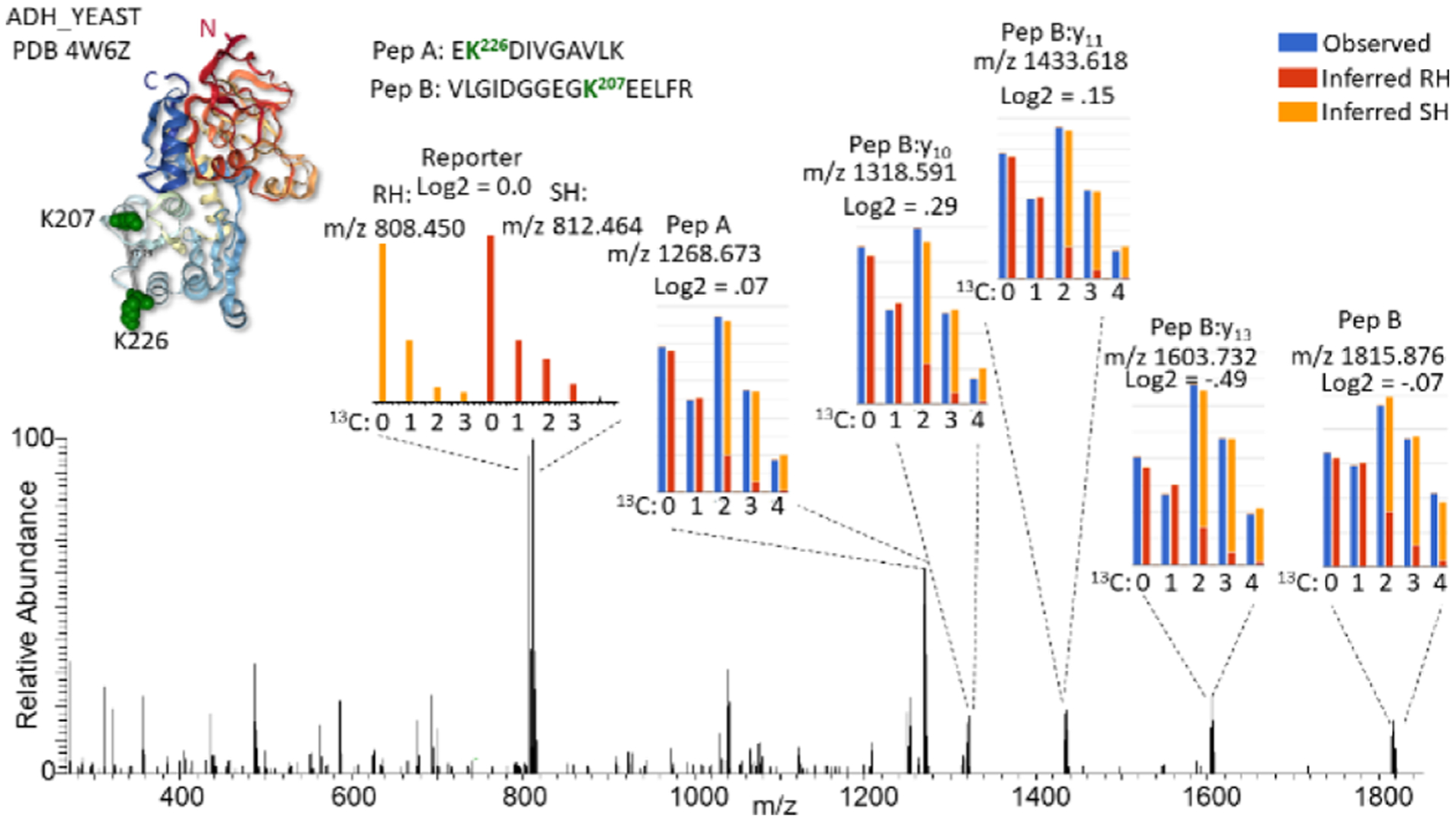
Example fragmentation spectrum of an iqPIR cross-linked peptide pair. MS2 spectrum of the iqPIR cross-linked peptide pair linking residues 207–226 of ADH1_YEAST a 1:1 mixture of RH/SH cross-linked. The PDB structure 4w6z is shown as a ribbon structure colored red from the N-terminus to blue at the C-terminus with the cross-linked Lys shown as green space filled residues. Insets show expanded views of selected fragment ions, illustrating the isotopic differences which are used for quantification. For fragment ions differing by two 13C, the observed signal is shown in blue while the deconvoluted signal from the RH and SH are shown in red and orange, respectively. The reporter ion signal differs by four 13C, requiring no deconvolution, and follows the red/orange color scheme.
Notably, these include the intact released peptide fragment ions at m/z 1268.6732 and 1815.8759, as well as peptide backbone fragment ions which contain the residual stump of the iqPIR cross-linker at the cross-linked residue. Each of these ions exist as a light and heavy isotopic pair differing by the mass of two 13C. Additionally, the iqPIR reporter ion is observed as an isotopic pair at m/z 808.450 and 812.464 differing by four 13C atoms. Because of the placement of 13C atoms within the iqPIR molecule, upon fragmentation, the RH cross-linked peptide pair generates a heavy reporter ion (m/z 812.464) and corresponding light released peptide fragment ions, while the SH cross-linked peptide pair produces the light reporter ion (m/z 808.464) and the heavy released peptide fragment ions. It is important to note that while the light and heavy reporter ion isotope envelopes do not overlap with one another allowing for a straight forward ratio calculation from the signal, the light and heavy released peptide ions differing by the mass of two 13C atoms require deconvolution of the natural occurrence of second and third 13C isotope contributions of the light peptide from the monoisotopic and first 13C peak of the heavy peptide prior to quantification. Quantification of the signals from the MS2, as shown in Figure 2, accurately represents the 1:1 mixture with an average log2 (RH/SH) value of −0.05.
In addition to the 1:1 mixtures, three samples were generated consisting of varying RH/SH mixtures of the three iqPIR cross-linked proteins, as illustrated in Figure 3. In total, 229 nonredundant cross-linked peptide pair sequences were identified from these samples at an estimated FDR of less than 1%. To allow for automated quantification of a large number of iqPIR fragmentation spectra, we developed an informatics strategy as described in detail in the methods. Briefly, the peak intensities for each m/z containing quantitative information including reporter ion signal, released peptide ions, and any fragment ions containing the iqPIR stump mass are extracted from the MS2 spectra. The signal from the released peptide ions and peptide backbone fragments are deconvolved utilizing the atomic composition of each fragment ion and the natural abundances of heavy isotopes.22 Ratios (RH/SH) are then calculated for each quantifiable peak and used to compute an average ratio for each nonredundant cross-linked peptide pair. To investigate any potential ratio compression because of co-isolation in the MS2 spectra, the ratios derived from the reporter ion signals were evaluated separately from those calculated from the released peptide and backbone fragment ions. Ratios originating from the reporter ion signal displayed compression becoming more severe with increasing expected ratios, as shown in Figure S2A−G, Table S1. In contrast, ratios derived from the released peptide and backbone fragment ions remained accurate relative to the expected ratios despite any co-isolation effects. This is similar to the use of complement ions to overcome the ratio compression observed in traditional isobaric mass tag quantitative proteomics.27 Therefore, the reporter ion signal was not included in the calculation of average ratios for cross-linked peptide pairs. Utilizing this strategy with the three mixture samples, as illustrated in Figure 3, resulted in 93% (212/229) of the cross-links being quantified to produce relative log2 (SH/RH) ratios. To access the accuracy of the iqPIR quantification approach, the measured log2 ratios vs the expected mixed ratios for these 212 cross-linked peptide pairs are displayed in the violin plot in Figure 4. Normalizing all of the measured log2 ratios to their expected values results in a mean log2 value of 0.026 with a standard deviation of 0.49 and a 95% confidence interval of 0.052, as shown in Figure S2H.
Figure 3.
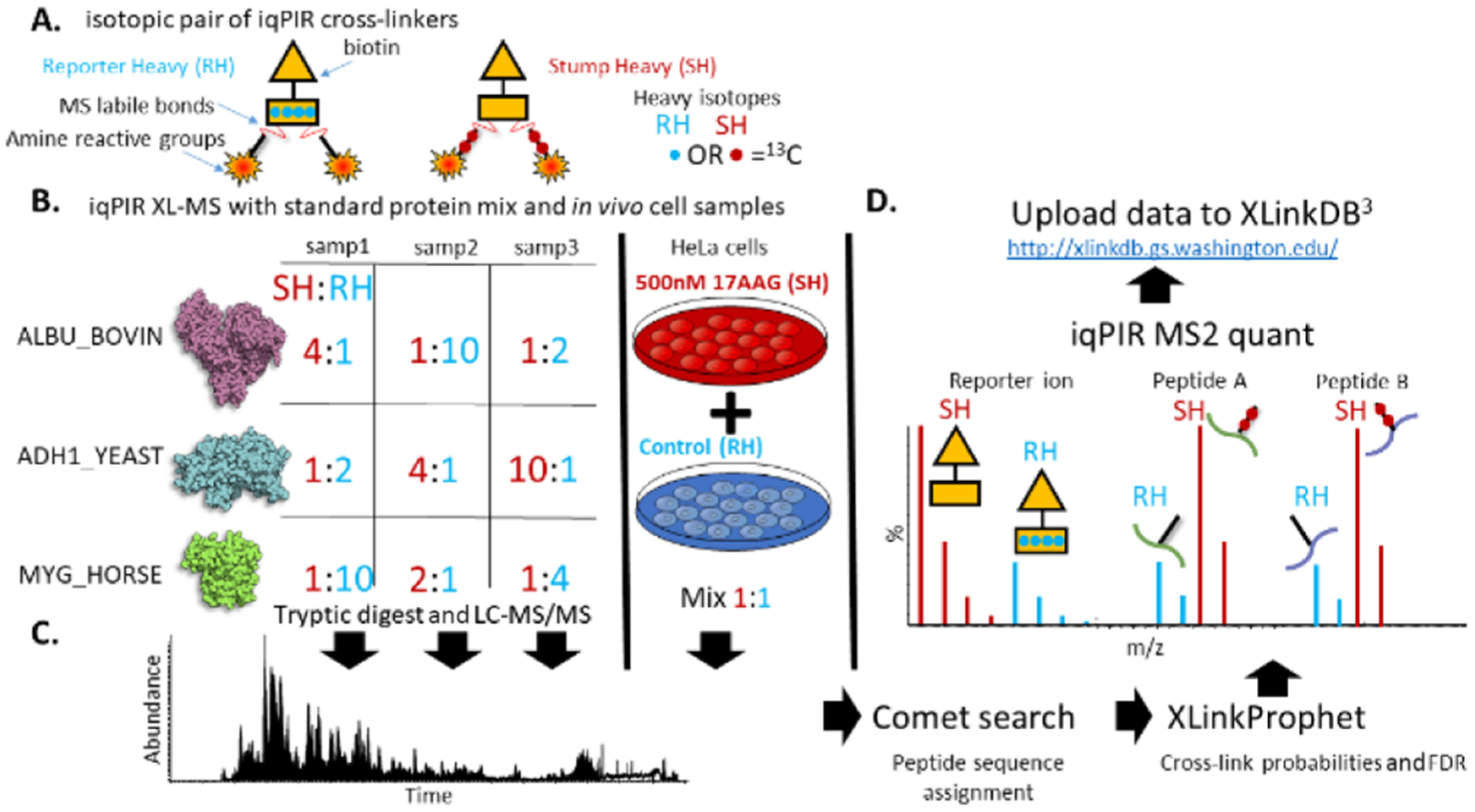
Experimental overview of iqPIR application to cross-linked protein standards and 17-AAG treated HeLa cells. (A) Cartoon depiction of the RH and SH iqPIR cross-linkers illustrating the primary molecular features including MS-labile bonds, amine reactive groups, location of 13C isotopes (red & blue circles) and biotin affinity tag. (B) Preparation of iqPIR cross-linked standard protein samples consisting of ADH1_YEAST, ALBU_BOVIN, and MYG_HORSE mixed at various RH/SH ratios as indicated, well as an in vivo cross-linked 17-AAG treated vs vehicle (0.1% DMSO) HeLa cell sample. (C) Example total ion chromatogram representing LC–MS/MS analysis of cross-linked peptide pair samples. (D) Data analysis consisted of a Comet database search, XLinkProphet FDR estimation and filtering, quantification of relative abundance of fragment ions in the MS2 spectra and upload of data to XLinkDB.
Figure 4.
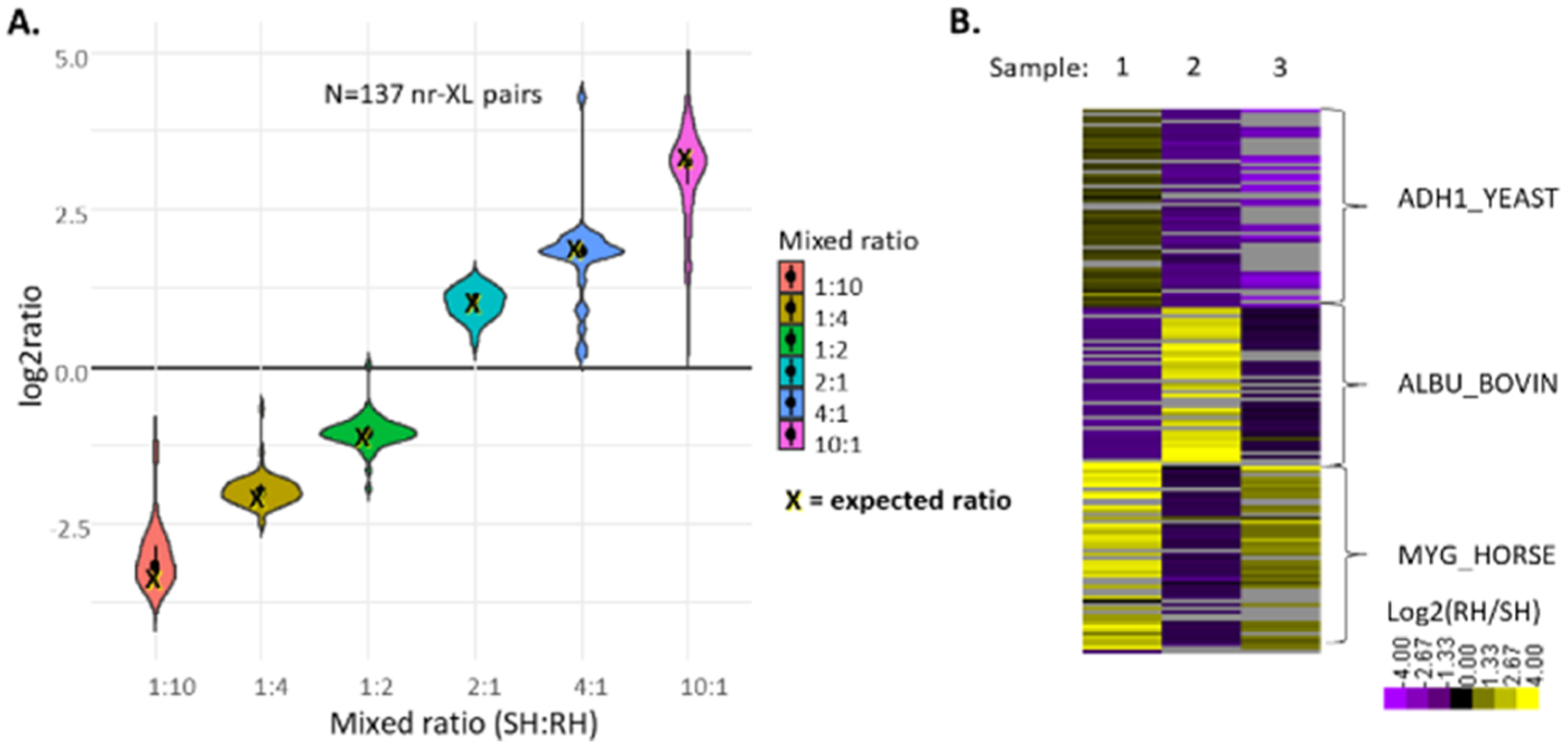
Evaluation of iqPIR with mixture of cross-linked protein standards. (A) Violin plot illustrating the distribution of the computed log2 ratios for cross-linked peptide pairs vs their expected values. (B) Heatmap illustrating the quantified cross-linked peptide pairs according to sample and protein.
In addition to the quantitative information contained in the cross-linked peptide pairs, we set out to investigate whether quantitative information from dead-end monolinks could be utilized. Dead-end monolinks originate when one reactive group of the cross-linker reacts with an amino acid residue while the other reacts with water. These hydrolysis products are generally presumed to form at much higher levels than cross-links. Although they are generally not utilized in XL-MS studies, monolinks can potentially provide information on solvent accessibility, residue reactivity, and protein abundance. A recent study demonstrated the effective use of monolinks in modeling protein structures.28 Being that they are present in every sample prepared for XL-MS, the use of monolink information can come as an added bonus to complement the cross-link information. Searching the data collected on the iqPIR cross-linked samples analyzed above for dead-end monolinks using the mass offset feature in Comet4 and subsequent FDR estimation with PeptideProphet29 resulted in the identification of 83 nonredundant monolink sequences at less than 1% FDR (Table S2). Nearly 60% (49/83) of the monolink peptides were also identified as a cross-linked peptide pair (Figure S3A). Those peptides that were identified in a cross-linked peptide pair but not as a monolink tended to be shorter in length than those identified as both cross-linked and monolinked or only monolinked (Figure S3B). It is likely they were not identified because of the charge state exclusion setting for four plus and higher charge state precursor ions used during LC–MS acquisition. Quantification based on the fragment ions present in the monolink MS2 spectra exhibited excellent agreement with the cross-link levels, indicating their quantitative values reflect the relative protein abundances in the samples (Figure S3C). The data from the iqPIR standard protein samples are available in XLinkDB with table name iqPIR_std_proteins_Chavez2020_Bruce.
Application of iqPIR to 17-AAG-Treated HeLa Cells.
To evaluate the quantitative capabilities of iqPIR with large-scale in vivo XL-MS, we cross-linked HeLa cells treated with the heat shock protein 90 (Hsp90) inhibitor 17-AAG. A previous qXL-MS study utilizing SILAC, quantified changes to cross-linked peptide pairs with varying concentrations of 17-AAG.12 It was found that the 17-AAG treatment induced a compact conformation of Hsp90 in which the N-terminal domain (NTD) folds down to make contact with the middle domain. It has been postulated that this compact conformation represents a transition state during the hydrolysis of ATP to ADP during the catalytic Hsp90 cycle.30 17-AAG mimics the binding of ADP in the Hsp90 NTD31 and similarly induces the formation of a compact conformation.
Application of iqPIR to HeLa cells treated with 500 nM 17-AAG for 18 h resulted in a total of 3445 cross-linked peptide pairs at less than 1% FDR. These links were 19% inter-protein, corresponding to 3173 Lys residue pairs and 980 protein pairs (Figure 5, Table S3). Data including a cross-link-derived interaction network and iqPIR quantitative information are available in XLinkDB (xlinkdb.gs.washington.edu) with table name iqPIR_HeLa_17AAG_Chavez2020_Bruce. Eighty-six percent (2977/3445) of the cross-linked peptide pairs were able to be quantified utilizing relative intensities of the released peptide and backbone fragment ions. Analysis of dead-end monolinks resulted in the identification of 2910 nonredundant peptide sequences, 2859 of which a relative log2 ratio was obtained and used to estimate relative protein levels for 610 proteins. On average, proteins had a median of 3 dead-ends each, with a maximum of 21 observed for the DNA-dependent protein kinase catalytic subunit (PRKDC_HUMAN), with a minimum threshold of two quantified dead-ends for protein level estimation. Focusing on the cross-links involving the primary targets of 17-AAG, the cytoplasmic isoforms of Hsp90 (HS90A, HS90B), reveals a subnetwork consisting of 14 proteins, 294 nonredundant cross-linked peptide pairs linking 220 lysine residues (Figure 5). Multiple intraprotein and interprotein cross-links involving HS90A, HS90B, HS71A, STIP1, and CHRD1 were quantified with altered levels with 17-AAG drug treatment. These include links between K513 of STIP1 and K443 of HS90A and the homologous site, K435 of HS90B, which displayed increased levels (log2 values of 1.41 and 1.36 respectively) with 17-AAG treatment. Additionally, the link between K486 of STIP1 and K204 of HS90B was quantified with a log2 value of 2.7 despite the minimal effect of 17-AAG on the protein expression levels of STIP1 and HS90B (Figure 5).
Figure 5.
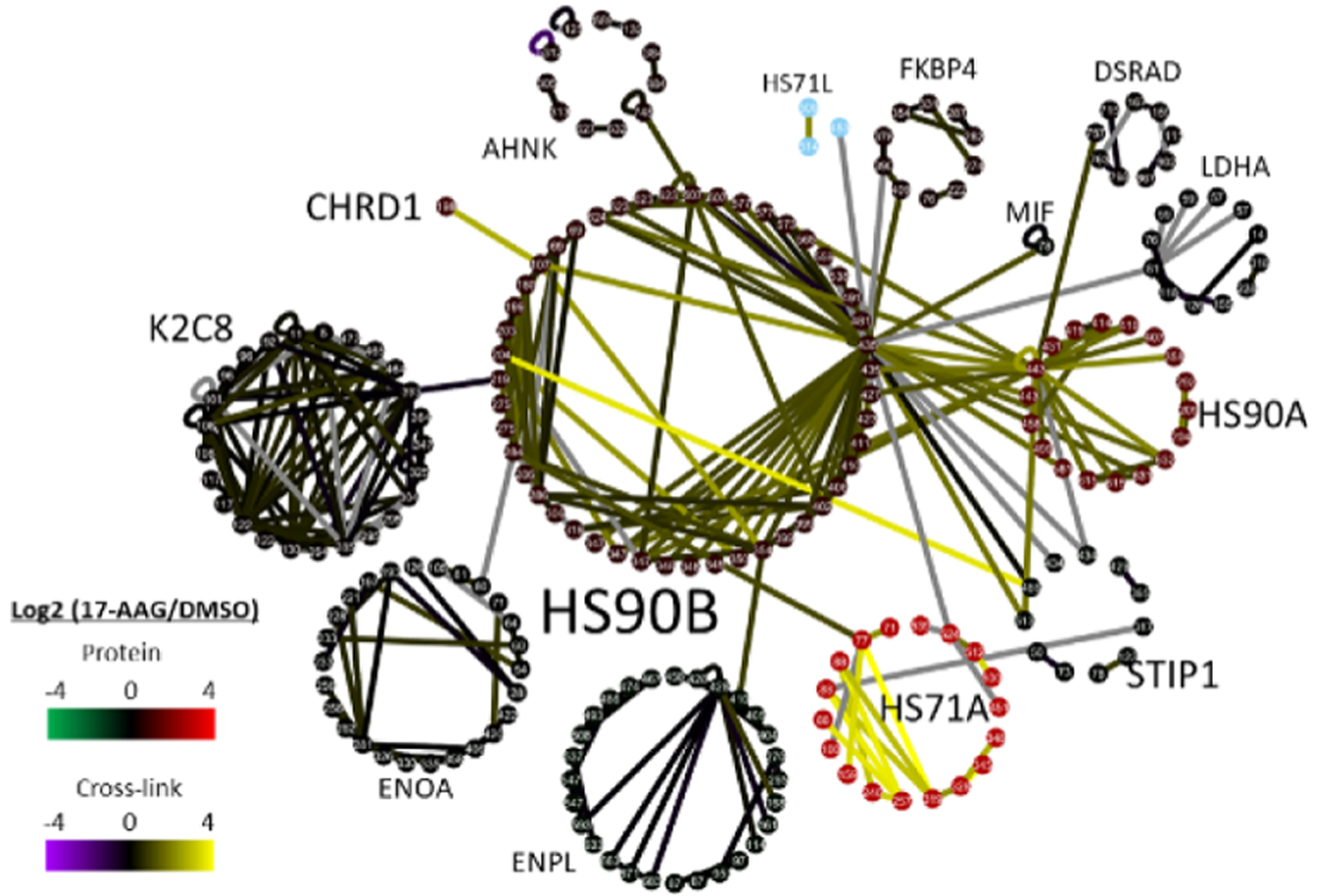
Hsp90 inhibitor 17-AAG quantitative in vivo XL-MS subnetwork. (A) Subnetwork of cross-linked peptide pairs involving thirteen cross-linked proteins including HS90A & HS90B and proteins they were identified as cross-linked to. The network consists of 220 nodes representing cross-linked Lys residues and 294 edges representing cross-links between Lys residues. Edges are colored on a purple/yellow scale according to the cross-linked peptide pair log2(17AAG/DMSO) values. Nodes are clustered into circles according to their assigned proteins and colored according to the estimated protein ratios based on the quantified dead-end monolinks for each protein.
Comparison of the iqPIR data on 17-AAG-treated HeLa with previously generated qXL-MS SILAC data indicated multiple cross-linked peptide pairs displaying similar trends because of 17-AAG treatment. The overall 1353 cross-linked peptide pairs were common between the two studies (Figure S4A). Comparing the log2 ratios between the two studies for the 500 nM 17AAG treatment conditions resulted in 1005 common quantified cross-links with approximately half (455) having an absolute difference of a log2 of 0.5 or less (Figure S4B). The majority of cross-links related to a 17AAG-induced heat shock response displayed excellent agreement between the SILAC and iqPIR experiments (Figure S4B). These include links indicative of the compact conformation of Hsp90 including a link between K107–K435 of HS90B (Figure 5). Additionally, cross-links indicating increased levels of the HS90A homodimer (K443–K443) as well as the HS90A-B heterodimer (K443–K435) were also observed with excellent agreement between SILAC and iqPIR data sets (Figure 5). Induction of a heat shock response because of NTD inhibition of Hsp90 was evident in the increased relative expression levels of proteins including HS71A, HS90A, and HS105 (Table S3).
CONCLUDING REMARKS
Here, we have described the synthesis and application of iqPIR cross-linking reagents and developed informatics to enable quantification of isotope-encoded released peptide and fragment ions from tandem mass spectra of iqPIR cross-linked species. The isobaric nature of the iqPIR strategy affords multiple benefits to qXL-MS experiments akin to those afforded by quantitative isobaric mass tags including TMT and iTRAQ. As demonstrated by Yu et al., the use of TMT labeling with chemical cross-linking in a strategy termed QMIX offered benefits to qXL-MS experiments, including the ability for multiplexed quantification of cross-linked peptides using widely available, commercially produced reagents.14 However, the QMIX strategy requires additional chemical labeling steps, requires mixing of samples after digestion which introduces additional experimental variance, and suffers from ratio compression of the reporter ion signal at the MS2 level and low sensitivity at the MS3 level. In contrast, the iqPIR strategy described here allows for sensitive and accurate quantification at the MS2 level and requires no additional labeling steps beyond the cross-linking reaction. Furthermore, iqPIR-labeled samples can be mixed immediately post-cross-linking, thereby minimizing variances introduced in subsequent experimental steps. Automated solid phase peptide synthesis of iqPIR reagents can be performed in individual laboratories or these reagents can be purchased from the many custom peptide synthesis companies.32 As a proof of concept, the results presented here were generated using a binary pair of iqPIR reagents, but due to the flexibility of the peptide structure of iqPIR and the availability of stable isotope-labeled amino acids, the concept is extensible to allow a greater level of multiplexing. These features make iqPIR well suited to improve future qXL-MS studies, evaluating the dynamic nature of protein structures and interactions. Beyond the results presented here, iqPIR has been successfully applied in a recent study evaluating the role of the natural inhibitor of ATP synthase (ATIF1), in a murine model of heart failure (unpublished work, University of Washington 2020). Therein, iqPIR-quantified cross-linked peptide pairs supported the increased formation of ATIF1 complexed with ATP synthase in an inactivated tetramer conformation, as identified by a recent cryo-EM study.33 In summary, iqPIR represents a new strategy for qXL-MS studies and will serve as a primary tool for dynamic interactome studies ongoing in our laboratory.
Supplementary Material
ACKNOWLEDGMENTS
We would also like to thank the members of the Bruce Lab for helpful input during these experiments. This work was supported by the following grants from the National Institutes of Health 5R01GM086688, 5R01HL144778, and 1R35GM136255.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c03128.
Theoretical relative intensities, histogram distributions, Venn diagram, distributions of peptide length, and scatter plot (PDF)
iqPIR cross-links from standard proteins (XLSX)
Standard protein dead-ends (XLSX)
Cross-links from 17-AAG treated HeLa cells (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c03128
The authors declare no competing financial interest.
REFERENCES
- (1).Tang X; et al. Anal. Chem 2005, 77, 311–318. [DOI] [PubMed] [Google Scholar]
- (2).Weisbrod CR; et al. J. Proteome Res 2013, 12, 1569–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Navare AT; et al. Structure 2015, 23, 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chavez JD; et al. Mol. Cell. Proteomics 2013, 12, 1451–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Schweppe DK; et al. Chem. Biol 2015, 22, 1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Schweppe DK; et al. Proc Natl Acad Sci U S A 2017, 114, 1732–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fasci D; et al. Mol. Cell. Proteomics 2018, 17, 2018–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Makepeace KAT; et al. Mol. Cell. Proteomics 2020, 19, 624–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chavez JD; et al. Cell Syst. 2018, 6, 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhong X; et al. J. Proteome Res 2017, 16, 720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chavez JD; et al. Nat. Commun 2015, 6, 7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chavez JD; et al. Cell Chem. Biol 2016, 23, 716–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chavez JD; et al. Cell Rep. 2019, 29, 2371–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yu C; et al. Anal. Chem 2016, 88, 10301–10308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chavez JD; et al. PLoS One 2016, 11, No. e0167547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Xu W; et al. Nat. Commun 2019, 10, 2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ross PL; et al. Mol. Cell. Proteomics 2004, 3, 1154–1169. [DOI] [PubMed] [Google Scholar]
- (18).Thompson A; et al. Anal. Chem 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- (19).Savitski MM; et al. J. Proteome Res 2013, 12, 3586–3598. [DOI] [PubMed] [Google Scholar]
- (20).Mohr JP; et al. Anal. Chem 2018, 90, 6028–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Keller A; Chavez JD; Bruce JE Bioinformatics 2019, 35, 895–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yergey JA Int. J. Mass Spectr. Ion Phys 1983, 52, 337–349. [Google Scholar]
- (23).Yang L; et al. J. Proteome Res 2012, 11, 1027–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yang L; et al. Anal. Chem 2010, 82, 3556–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chowdhury SM; et al. Anal. Chem 2006, 78, 8183–8193. [DOI] [PubMed] [Google Scholar]
- (26).Mohr JP; Chavez JD; Bruce JE Multidimensional Cross-Linking with a Tetra-Reactive Cross-Linker. 67th ASMS Proceedings, 2019. [Google Scholar]
- (27).Wühr M; et al. Anal. Chem 2012, 84, 9214–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Sinnott M; et al. Structure 2020, 28, 1061–1070. [DOI] [PubMed] [Google Scholar]
- (29).Keller A; et al. Anal. Chem 2002, 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- (30).Mayer MP; Le Breton L Mol. Cell 2015, 58, 8–20. [DOI] [PubMed] [Google Scholar]
- (31).Roe SM; et al. J. Med. Chem 1999, 42, 260–266. [DOI] [PubMed] [Google Scholar]
- (32).Chavez JD; et al. Nat. Protoc 2019, 14, 2318–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gu J; et al. Science 2019, 364, 1068–1075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.