

Abstract
With Remdesivir being approved by FDA as a drug for the treatment of Corona Virus Disease 2019 (COVID-19), nucleoside drugs have once again received widespread attention in the medical community. Herein, we summarized modification of traditional nucleoside framework (sugar + base), traizole nucleosides, nucleoside analogues assembled by other drugs, macromolecule-modified nucleosides, and their bioactivity rules. 2′-“Ara”-substituted by –F or –CN group, and 3′-“ara” substituted by acetylenyl group can greatly influence their anti-tumor activities. Dideoxy dehydrogenation of 2′,3′-sites can enhance antiviral efficiencies. Acyclic nucleosides and L-type nucleosides mainly represented antiviral capabilities. 5-F Substituted uracil analogues exihibit anti-tumor effects, and the substrates substituted by –I, –CF3, bromovinyl group usually show antiviral activities. The sugar coupled with 1-N of triazolid usually displays anti-tumor efficiencies, while the sugar coupled with 2-N of triazolid mainly represents antiviral activities. The nucleoside analogues assembled by cholesterol, polyethylene glycol, fatty acid and phospholipid would improve their bioavailabilities and bioactivities, or reduce their toxicities.
Keywords: Nucleosides scaffold, Antiviral, Anti-tumor, Triazoles, Prodrugs
Graphical abstract
This review mainly summarize the effects of structure modification of traditional nucleoside framework (sugar + base), traizole nucleosides, nucleoside analogues assembled by other drugs, macromolecule-modified nucleosides on the antiviral/anti-tumor activity.
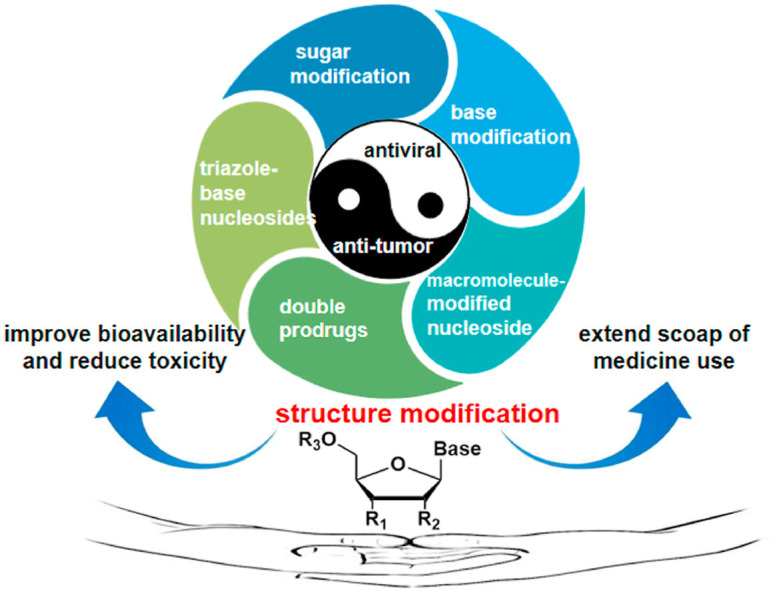
1. Introduction
Nucleosides play a key role in biology processes, involving in the retention, replication and transcription of gene information. Nucleoside analogues can be used as a nucleic acid metabolism inhibitor to interfere with virus reproduction and tumor cell growth, showing good antiviral and anti-tumor effects.
Nucleoside drugs are mainly divided into enzyme inhibitors and DNA chain terminators. They are recognized by specific proteins on the cell membrane and then transferred to the cells, which can be monophosphorylated, diphosphorylated and then triphosphorylated by the kinases, including thymidine kinases (TKs), adenosine kinase (AK), deoxycytidine kinase (dCK), and deoxyguanosine kinase (dGK). A triphosphorylated nucleoside drug can act as a cellular polymerase inhibitor or a reverse transcriptase inhibitor, or participate in the replication or transcription of a nucleic acid. Monophosphorylation is the rate-limiting step in its metabolism [1]. Anti-tumor nucleoside drugs can not only serve as a competitive substrate for cell polymerase, but also tightly bind to the DNA-enzyme complex, thereby activating genotoxic stress response, leading to apoptosis [2].
Table 1 lists currently available antiviral and anti-tumor nucleoside drugs. It is worth noting that Remdesivir (RDV, GS-5734) shows good inhibition effect on COVID-19 [[3], [4], [5], [6]], and has been approved by FDA as a drug for the treatment of COVID-19. The nucleoside drugs in clinical trial include Valtorcitabine (anti-HBV), Elvucitabin (Fd4C) (anti-HBV, anti-HIV). Besides, some nucleoside drugs show inhibitory activities on SARS virus, Ebola virus and Zika virus [[7], [8], [9], [10]].
Table 1.
Antiviral and anti-tumor nucleoside drugs currently on the market.
| Bioactivities | Drug names | |
|---|---|---|
| Antiviral | Broad-spectrum antiviral drug | Ribavirin |
| Anti-HIV | Zidovudine, Didanosine, Zalcitabine, Stavudine, Lamivudine, Abacavir, Tenofovir, Emtricitabine | |
| Anti-HBV | Lamivudine, Telbivudine, Adefovir dipivoxil, Emtricitabine, Entecavir, Ttenofovir disoproxil, Clevudine | |
| Anti-HCV | Sofosbuvir | |
| Anti-herpes virus | Idoxuridine, Trifluorothymidine, Brivudine, Acyclovir, Valacyclovir, Penciclovir, Famciclovir, Ganciclovir, Formimivir, Cidofovir | |
| Anti-COVID-19 | Remdesivir | |
| Anti-tumor | 6-Mercaptopurine, 6-Mercaptoguanine, Fludarabine, Clofarabine, Cladribine, Adenine arabinoside, Nelarabine, Cytarabine, Decitabine, Enocitabine, Troxacitabine, 5-FU, Gemcitabine, Zalcitabine, Capecitabine, CNDAC | |
Different from the progress of nucleoside derivatives [[11], [12], [13]], herein we mainly focus on different structural characteristics of nucleoside compounds with antiviral or anti-tumor effects, and sum up triazole nucleosides, drug assembly nucleosides, and macromolecule-modified nucleosides, to provide useful information for development of nucleoside analogues. It should be pointed out that, from structure of nucleosides, there is no absolutely strict distinction between the antiviral and anti-tumor effect.
2. Modification of sugar
2.1. Ribose/deoxyribose nucleoside analogues
2.1.1. 2′-Site modification
2.1.1.1. Antiviral compounds
Modification at 2′-site mainly involves in: (I) Introduction of “ara”-substituent (“ara” means “up” on the ring plane). (II) Dehydroxylation at 2′-site or substitution of 2′-OH with a fluoro-methylene group. The 2′-site methyl substituted nucleosides 2′-methylcytidine (active form of Valopicitabine, 1), 2′-methyluridine (2), 2′-methyladenosine (3) and 2′-methylguanosine (4) show anti-HCV activities (3 > 1 > 4 > 2), but these 2′-methyl substituted derivatives have poor bioavailabilities [13,14]. Introducing valine ester at 3′-O-site of 1 could generate Valopicitabine (NM283, 5) with better bioavailability [15]. The presence of F at 2′-site (compound 6) could reduce cytotoxicity [16]. 2′-Hydroxymethyl adenosine (7) obtained by substituting methyl with hydroxymethyl also shows anti-HCV activity [17]. Besides, compound 1 exhibits efficacy against foot-and-mouth disease virus, yellow fever virus, norovirus, dengue virus, hepatitis E virus and parechoviruses A (HPeV) [[18], [19], [20], [21], [22], [23]].
Fluoriodine Cytarabine (FIAC, 8) and Fluoroiodine Uridine (FIAU, 9) are 2′-F and 5-I substituted pyrimidine derivatives. Compound 9 displays good anti-HBV activity in vitro [24] and 8 has broad-spectrum antiviral efficiency, which not only has effect against HBV, but also against herpes simplex virus (HSV), varicella zoster virus (VZV), human cytomegalovirus (HCMV), and Epstein-Barr virus (EBV) [25]. Adenosine arabinoside (ara-A, 10) is used clinically to treat simplex virus encephalitis, herpes zoster and varicella infections in immunosuppressed patients and its monophosphate has inhibitory effect on HBV.
2′-Deoxynucleoside analogues such as Iodouridine (IDU, 11), Trifluridine (TFT, 12), Brivudin (13) present anti-HSV activities, and (E)-2′-deoxy-2′-fluoromethylene cytidine (MDL101731, 14) is a ribonucleotide reductase inhibitor, which shows anti-HSV potential [26].
When O atom in sugar is replaced by S, and H atom of 5-site in pyrimidine is substituted by I, ethyl or chloroethyl, the resulting 2′-deoxy-2′-F-4′-thio-nucleoside analogues 15, 16, 17, 18 exhibit anti-HSV activities. Besides, purine nucleoside analogues 19 and 20 also demonstrate outstanding anti-HSV efficacies, but they are cytotoxic [27] (Fig. 1 ).
Fig. 1.

2′-Modified nucleoside analogues with antiviral activities.
2.1.1.2. Anti-tumor compounds
The 2′-site substitution “X” (-OH, –CN, –F) of anti-tumor ones is always “up” on the ring plane. For example, Cytarabine (21) is mainly used for treatment of acute leukemia, and has a certain effect on malignant lymphoma, and cancer of lung, digestive tract, head and neck. Cyclocytidine (Ancitabine, 22) is prodrug of 1. Its 2′-OH on sugar dehydrates and condenses with the O atom at pyrimidine to form a ring and this special structure makes it stable to cytosine nucleoside deaminase, being effective orally. It has similar anti-leukemia activity as 21 in clinic, but lower toxic side effect [25]. The –CN group in CNDAC (23) possesses a small volume, special electronic effect, and can act as a receptor of hydrogen bond, which makes CNDAC have ability to inhibit growth of tumor cells by cutting off DNA chain and stopping cell replication in the G2 phase. It is effective for hematologic malignancies and solid tumors (the anti-tumor range is wider than 21) [28,29]. Gemcitabine (24) with two F atoms at 2′-site, has a different anti-tumor spectrum from 21 and is effective for a variety of solid tumors. Clearly, –F, –CN substituents at 2′-C are closely related to solid tumors and blood systemic tumors. When –OH is maintained, it is effective for acute leukemia.
Cladribine (25) is a deoxydehydro nucleoside at 2′-site, which has a good therapeutic effect on lymphoproliferative diseases, but is not ideal for solid tumors. On this basis, 2′-site is substituted by –F group to give Clofarabine (26), which not only inherits advantages of 25, but also displays superior safety and broader anti-tumor range (involving solid tumors) than 25. Compound 26 is the first drug approved by FDA for specific treatment of childhood leukemia. Fludarabine (27) is F derivative at 2-site on the base moiety, which is similar to 25 and 26. It is the first adenosine analogue for the treatment of chronic B-lymphocytic leukemia (B-CLL) and clinically used in combination with 21 to achieve a synergistic effect. Compound 21 is mainly used to treat leukemia, while it is effective for a few solid tumors. 4′-Thio-arabinofuranosyl cytosine (T-araC, 28) is an analogue of 21 where the 4′-O atom was replaced by S atom, showing active against many different solid tumors as well as leukemia and lymphoma (wider anti-tumor spectrum) [30]. This compound has a certain effect on DNA damage bypass polymerase and is active on a xenograft human solid tumor mouse model [31]. As analogues of 24, 2′-F-4′-thio-arabinofuranosyl cytosine (29) and 2′-F-4′-thio-5-F-arabinofuranosyl cytosine (30) have shown effective inhibitory acitivities against leukemia and solid tumor cells, but introduction of 5-F slightly decreases anti-tumor activity [27] (Fig. 2 ).
Fig. 2.

2′-Modified nucleoside analogues with anti-tumor activities.
2.1.2. 3′-Site modification
Modification at 3′-site usually presents anti-tumor activity. For example, Ethynylcytidine (ECyd, 31) and EUrd (32) display very potent strengths in adenocarcinoma breast cancer cells (MCF-7) and prostate cancer cells (PC-3) in vitro [32]. Compound 31 can significantly restrain growth of various human solid tumor cells in vivo and in vitro and has now entered clinical trials [33]. Adenosine derivative 3′-methyladenosine (33) has a good inhibitory effect on leukemia and solid tumors (human colon carcinoma HT-29 and human breast carcinoma MCF-7 cell lines) [34] (Fig. 3 ).
Fig. 3.

3′-Modified nucleoside analogues with anti-tumor activities.
2.1.3. Simultaneous modification on 2′and 3′-site
2.1.3.1. Antiviral compounds
Thymidine analogue Zidovudine (AZT, 34) is the first nucleoside drug approved by FDA for treatment of AIDS. Its 3′-azide group, which is compatible with N-containing natural nucleosides, has a linear structure, resulting in small hindrance and electronegativity. This phenomenon can improve the stability of nucleoside and make the drug not easy to be recognized by viruses. FLT (35) is obtained by replacing 3′-OH in natural thymidine with a F atom, which is an effective HIV nucleoside reverse transcriptase inhibitor (NRTI), but is also highly toxic [35]. Stavudine (d4T, 36), where its sugar ring is dehydrogenated and deoxygenated simultaneously to form a C C bond at 2′, 3′-site, is an effective HIV-1 NRTI with high oral availability, and has been approved for treatment of HIV infection [36]. Uridine analogue 3′-F-2′,3′-dideoxy-5-chlorouridine (3′-F-ddClU, 37) with Cl substitution at 5-site, displays good anti-HIV activity. Its “selectivity index (SI)” is close to 34, whereas cytotoxicity is lower than 34 [37]. FLG (38), which is similar to 35, shows weaker anti-HIV activity, but has a good resistance potential to HBV [35]. The anti-HIV activity follows 35 > 34 > 36 > 37 > 38 order, and the cytotoxicity follows 35 > 34 > 36 > 38 > 37 trend [[35], [36], [37]].
Dedanoxin (ddI, 39) is a NRTI against HIV. 2′,3′-Dideoxyadenosine (ddA, 40) can be metabolized to 39 in the body so has anti-HIV potential. 2′-F-2′,3′-Dideoxy-arabinoside (2′-F-ara-ddA, 41) has anti-HIV activity close to 39, where the introduction of 2′-“ara” F improves metabolic and chemical stability and is currently in pre-clinical trials [38]. The 3′-hydroxymethylpurine derivative (42) of 39 presents comparable capability to 39 and has no significant toxicity for H9 cells [39].
Zalcitabine (ddC, 43) is also a NRTI that is used for the treatment of AIDS patients ineffectively treated by 41 and 34, but long-term use will inhibit the synthesis of mitochondrial DNA and cause peripheral neuropathy. Its 5-F substituted analogue 44 has stronger pharmaceutical activity but greater cytotoxicity. When configuration is changed to L-type, antiviral spectrum expands. For instance, 45 and 46 present anti-HBV effects. The order of anti-HIV activity is 46 > 43 > 44 > 45, and cytotoxicity: 44 > 43 > 46 > 45 [40].
Many L-type nucleosides demonstrate good momentum in antiviral therapy. Lamivudine (3TC, 47) is a classic anti-HBV and anti-HIV drug. Emtricitabine (FTC, 48) is obtained by replacing 5-H with F atom, which can improve its anti-virus bioactivity and decrease the toxicity. L-Fd4C (49) is a deoxydehydrogenation derivative at 2′,3′-site, its anti-HBV and HIV activities are stronger than 48, and anti-HBV potential is 5–10 times than that of 47 [41]. The phase I/II clinical trials showed that 49 had good tolerance, low side effect and superior safety, and clinical combination therapy could be considered. Interestingly, F atom introduced in the base or sugar can change the antiviral efficiency. Compared with 47, the fluorinated substances 48, 49, and L-2′-F-d4C (50) show more intensive effects [42]. Anti-HBV potencies of 50 and L-2′-F-5-Fd4C (51) are at the same level as 14, and higher than 13 [43]. L-d4C (52) and L-5-FddC (46) also exhibit anti-HBV and anti-HIV potential [44]. The order of anti-HBV activity follows 49 ≈ 50 ≈ 51 > 52 ≈ 48 > 47 ≈ 46, and the anti-HIV capability follows 49 > 50 > 46 > 52 ≈ 48 > 47 >51 [[41], [42], [43], [44]]. Seen from Fig. 4 : (I) Formation of a C C bond at 2′,3′-site of sugar ring has the greatest effect on anti-HBV activity, the IC50 value of 49 against HBV is only about one-tenth that of 46. (II) Presence of F at 5-site of base exhibits good potency, the IC50 value of 48 against HBV is only about one-third of that of 47, and its anti-HIV value is about one-quarter. (III) Introduction of F at 2′-site slightly reduces the antiviral efficacy, the IC50 value of 49 against HIV is only about one-eighteenth of 51, and its anti-HBV value is about one-quarter. In addition, β-L-3′-F-d4C (53) is inhibitory against 3 TC-resistant strains in vitro, and can be used as a candidate for the treatment of HIV and HBV infection, especially patients resistant to 3 TC (47) [45]. Anti-HBV effect of Hyd4C (54) is comparable to 47 [46]. Valtorcitabine (55) is modified as an amino acid ester that has been improved in oral bioavailability [47].
Fig. 4.

2′,3′-Modified nucleoside analogues with antiviral activities.
Inspired by 3 TC (47) and FTC (48), 2′-deoxy-3′-oxa-4′-thiocytidine (dOTC, BCH-10652, 56) and its 5-F analogue 57 (dOTFC) display anti-HIV-1 potential. Though anti-HIV activity of 56 is not as good as 47. This substance is effective against strains resistant to 47, and introduction of 5-F has slightly effect on the antiviral activity since there is no order of magnitude difference in the anti-HIV IC50 values of compound 56 and 57 for Cord Blood Mononuclear cells, MT-4 cells, and H9 cells [48,49]. As D-configuration compound of Troxacitabine (67) (L-type, anti-tumor, Fig. 5 ), (+)-(2′R, 4′R)-dioxolane cytidine (58) presents anti-HIV potency, but its cytotoxicity is high. Its 5-F analogue (β-D-FDOC, 59) has anti-HIV activity, and lower cytotoxicity [50,51]. 2,6-Diamino-9-(2-dioxolane) purine (DAPD, 60) is prodrug of (−)-β-d-dioxolane guanine (DXG, 61), which displays anti-HIV potential (weaker than AZT, 34). Compound 60 is less active than 61, but less cytotoxic and more water soluble [52,53].
Fig. 5.
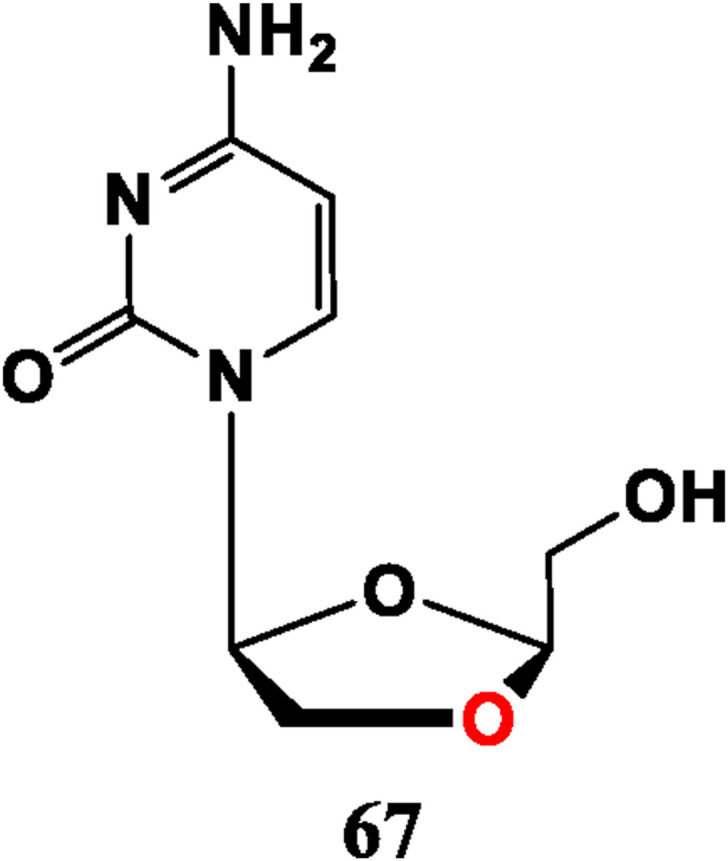
L-type Nucleoside analogue with anti-tumor activity.
Telbivudine (62) is a thymine nucleoside, and its mechanism of action is similar to that of 3 TC (47). It enters the cell to form a 5′-hydroxy-triphosphorylated active molecule and then competes with the natural nucleoside of viral DNA to participate in and terminate DNA strand replication for the treatment of chronic HBV, but carries a risk of myopathy. An “ara”-F atom is introduced at 2′-site of 62 to give Clavudine (L-FMAU, 63). It has anti-EB virus capabilities in vitro, and has strong and long-lasting anti-HBV ability in clinical trials, but long-term therapy may result in the development of considerable resistance and myopathy. Introduction of “ara”-F enhances bioactivity and toxicity [54,55]. In D-configuration, such as Gemcitabine (24), the introduction of “ara”-F at 2′-bit has a great impact on the anti-tumor efficiency, but when the drug configuration becomes L-type, such functional group no longer shows the reinforce to anti-tumor functioning. It hints “ara”-F cannot change the configuration’s antiviral effect, thus verifying the important role of L-configuration on antiviral activity. Meanwhile, anti-HBV capability of 63 is less than 46, and it has no activity against HIV [41]. It can be preliminarily concluded that cytosine bases are more effective than thymine bases, which may be related to the gene sequence structure of the open reading frame of HBV.
Among L-d4N and L-d2N (N = A/I/G), L-d4A (64) presents the strongest antiviral potential, but is far less than cytosine nucleoside 52, while L-d4A’s vigour is much greater than L-ddA (65) [56]. Inspired by Gemcitabine (24) and L-FMAU (23), two F atoms are introduced at 2′-site to form β-L-2′,2′-2F-ddA (66), which shows anti-HIV-1 potential [31]. The anti-HIV activity follows 64 > 66 > 65 trend (EC50 = 0.38, 3.4, 8.2 μM, respectively) and anti-HBV activity follows 64 > 65 > 66 trend (EC50 = 1.2, 6, 20 μM, respectively) [56,57], revealing such patterns: (I) Deoxydehydrogenation at 2′,3′-site to form a C C bond is a significant factor to promote the antiviral activity, the anti-HIV EC50 value of compound 64 is only about one-twentieth of 65, and the anti-HBV value is about one-fifth. (II) Introduction of F at 2′-site has a positive or negative influence on the antiviral effect, the anti-HIV EC50 value of compound 66 is about half of that of 65, while the anti-HBV value is about 3 times. (III) L-Configuration is an important factor influencing the antiviral activity, and substitution of “ara”-F at 2′-site cannot change this effect, since compound 66 possesses antiviral potential but its D-configuration (compound 24) is an anti-tumor substance (Fig. 4).
2.1.3.2. Anti-tumor compounds
Troxacitabine (L-OddC, BCH-4556, 67) is an anti-tumor drug under clinical trials, which has effects on leukemia, lung cancer, liver cancer, and prostate cancer [[58], [59], [60]]. The 3′-C of its sugar ring is replaced by an O atom to become a dioxolane, which may be an important factor affecting its anti-tumor activity, while 3 TC (47) and FTC (48) in which 3′-C atom is replaced by an S atom, show antiviral effects (Fig. 5).
2.1.4. 4′-Site modification
4′-Site modification of nucleosides mainly involved 4′-site substitution with ethynyl, cyano, azidosubstituents or 2′-dehydroxylation, usually exhibiting antiviral efficacy.
2′-Deoxy-4′-ethynyl-5-F cytidine (68) and 2′-deoxy-4′-ethynyl adenosine (72) have anti-HIV-1 activity similar to AZT (34). 2′-Deoxy-4′’-ethynyl arabinofuranosyl cytidine (69) shows higher activity and cytotoxicity than AZT. It indicates that introduction of 2′-“ara”-OH enhances anti-HIV capability and the cytotoxicity. 2′-Deoxy-4′-ethynylthymidine (70) also has anti-HIV potential, but its ability is weaker than AZT [61]. 2′,3′-Dideoxydehydroge -4′-ethynylthymidine (4′-Ed4T, 71) shows more persistent anti-HIV efficiency than d4T (36) or AZT in cell culture. The presence of 4′-ethynyl greatly improves potency and reduced side effects of d4T in inhibiting mitochondrial DNA synthesis and cell growth [62,63]. It may be due to the additional binding of the 4′-ethynyl group at a presumed hydrophobic pocket in the reverse transcriptase active site. 4′-Ethylnyl-2-F-2′-deoxyadenosine (EFdA, 73) demonstrates highly potent anti-HIV activity. The introduce of 4′-ethynyl and 2-F also cause significant efficacy against NRTI-resistant strains [64]. AZT was used as a control substance to record the sequence of anti-HIV activity in MTT experiment: 73 > 69 > 72 > 68 ≈ AZT > 70 [61], or 73 > 68 ≈ AZT > 69 > 72 >70 [64]. Although there is a difference in sorting, it can be determined that 73 is the most specific and potent, while 70 is the least potent. But their therapeutic windows are small (much smaller than AZT) except 71 and 73 [[61], [62], [63], [64]]. Therefor 73 is the most potential anti-HIV-1 compound, which is stable to intracellular enzymatic catabolism and acidic degradation. It has a very long intracellular T1/2, does not greatly inhibit DNA polymerase, and does not have acute mouse toxicity [64]. Furthermore, 4′-ethynyl-2′-deoxy-4′-thioguanosine (74) has inhibitory effect on reverse transcriptase with low cytotoxicity [65].
Thymine derivative 4′-cyano-2′-deoxythymidine (75) presents anti-HIV efficacy, but great toxicity in a mouse model [66]. Cytosine analogue 4′-cyano-2′-deoxycytidine (76) has ability to resist HIV and HSV, and also has an inhibitory effect on L1210 tumor cells, without obvious cytotoxicity. The cell experiments show that its anti-HIV activity is slightly stronger than AZT, but the treatment window is much smaller [67]. The purine derivatives CAdA (77) and CdG (79) have anti-HIV effect, but the toxicity is also obvious. It is later found that they both have anti-HBV potential. CdA (78) and CdI (80) without –NH2 substituent at 2-site have no obvious toxicity, but their anti-HIV ability is weakened. The order of anti-HIV ability follows 78 > 77 > AZT > 79 ≈ 80, and cytotoxicity: 77 ≈ 78 > 79 > 80 > AZT [68]. Clearly, the –NH2 substituent at 2-site reduces toxicity and bioactivity.
Guanosine analogue 4′-azido-2′-deoxyguanosine (81) and cytidine analogue 4′-azido-2′-deoxycytidine (82) have stronger anti-HIV potency than AZT, but are highly toxic. Uracil derivatives 4′-azido-2′-deoxyuridine (83) and 4′-azido-2′-deoxy-5-chlorouridine (84) have anti-HIV effect. Introduction of -Cl at the 5-site can enhance anti-HIV potential and reduce toxicity. Thymine derivative 4′-azido-2′-deoxythymidine (85) shows anti-HIV activity equivalent to AZT (34) for A3.01 cell line and is effective against HIV-1 strains resistant to AZT, but its cytotoxicity is also stronger than AZT [69]. The order of anti-HIV activity is 81 > 82 > 95 ≈ AZT > 84 > 83, but therapeutic window of these compounds are much smaller than AZT. Interestingly, cytosine derivatives R1479 (86), RO-9187 (87) and RO-0622 (Azvudine, FNC, 88), where the 2′-substituent are α-OH, “ara” –OH and “ara”-F, respectively, have anti-HCV efficacy. Compounds 87 and 88 with 2′-“ara” substituents are stronger than 86 [70,71]. Compound 87 with 2′-“ara” OH has resistance to tick-borne encephalitis virus (TEBV) [72]. More interestingly, the 2′-“ara” F substituted compound 88 also has strong anti-HIV activity and is currently in clinical phase III trial [73,74]. It is worth noting that 87, 88, and Gemcitabine (24), Cytarabine (21) all belong to cytarabine derivatives. Compounds 87 and 88 possess antiviral potential, which can be guessed that the –N3 at 4′-site is an important influencing group. Benefits of introduction of 4′-N3 can be summarized as follows: the 3′ and 5′-hydroxy groups of the nucleoside will form a five-membered ring during enzymatic and acidolysis, but when –N3 is introduced at the 4′-site, it will hinder the formation of the five-membered ring, thereby increasing stability in organisms [75] (Fig. 6 ).
Fig. 6.

4′-Site modified nucleoside analogues with antiviral activities.
2.1.5. 5′-Site modification
Nucleosides usually need to convert into active substrate of 5′-triphosphates in the body, and the formation of monophosphates is the speed-determining step. Therefore, most of the modifications at 5′-site are focused on the phosphorylation of 5′-site which does not affect its anti-tumor or antiviral activity, but rather improve the oral bioavailability and efficacy.
2.1.5.1. Antiviral compounds
Sofosbuvir (89) approved by FDA for the treatment of HCV has a broad spectrum and good therapeutic effect [76]. In addition, fluorine-containing nucleic acid 4′-fluorouracil-5′-O-triphosphate (90) also shows potency for HCV [77] (Fig. 7 ).
Fig. 7.

5′-Site modified nucleoside analogues with antiviral activities.
2.1.5.2. Anti-tumor compounds
5′-Diphosphonates nucleoside APCP (91) and “Clofarabine” PCP (92) are emerging tumor target CD73 inhibitors, and the latter is more potent than 91 [78]. The 3′,5′-site modified compound 93 shows significantly greater efficacy in the Capan2 cell line compared with Gemcitabine [79]. Fosteabine (YNK 01, 94) is a fatty phosphate esterified derivative of Cytarabine (21), which can be taken orally, and is also effective for leukemia [80]. In addition, 5′-ureido modified Cl-IB-MECA (95) is an adenosine A3 receptor agonist, and is effective against leukemia and various solid tumors in vitro with excellent safety. It is currently undergoing a Ⅱ-phase clinical trial in patients with liver cancer [81]. 5-Fluorouracil (5-FU, 146) is a rare nucleoside drug without sugar but has a broad anti-tumor spectrum. When it is glycosylated by furanose, and the 5′-OH is replaced by a methyl group, the prodrug Furtulon (96) is generated. It is an oral anti-tumor drug on the market. The introduction of furanose achieves the purpose of improving oral absorption. The structural advantage of compound 96 is that without 5′-OH, it could not be converted into nucleotides and incorporated by polymerase, but could be treated by pyrimidine nucleoside phosphorylase in tumor tissue to release 5-FU, and then exerts anti-tumor effect. The presence of 4′-F in compound 97 makes the glycosidic bond abnormally unstable and increases bond cleavage of uridine phosphorylase, so more 5-FU can be delivered to the tumor site to enhance anti-tumor effect. Its IC50 value in L1210 cell line is one order of magnitude lower than that of 96 [82] (Fig. 8 ).
Fig. 8.

5′-Site modified nucleoside analogues with anti-tumor activities.
2.2. Carbocyclic sugar nucleoside analogues
Natural carbocyclic nucleosides (−)-Aristeromycin (98) and (−)-Neplanocin A (99) display antibacterial activity. Five-membered carbocyclic nucleoside Carbovir (100) and Abacavir (ABC, 101) have anti-HIV potential, and Entecavir (102) is effective against HBV. Besides, Neplanocin F (103) can be regarded as the allyl rearranged isomer of Neplanocin A, which is much less cytotoxic [83]. Four-membered carbocyclic nucleoside Lobucavir (Cyclobut-G, 104) and Cyclobut-A (105) have broad-spectrum antiviral activities, involving in HIV, HCMV, HSV and other viruses to varying degrees. In 2′-F-Cyclobut-A (106), displacement of a hydroxyl group at 2′-site to F diminishes anti-HIV efficacy [84]. Three-membered carbocyclic nucleoside A-5021 (107) can effectively inhibit the replication of HSV and VZV [85]. Synadenol (108) and synguanol (109) exhibit potential anti-HCMV capabilities [86] (Fig. 9 ).
Fig. 9.

Carbocyclic nucleoside analogues.
2.3. Acyclic nucleoside analogues
Acyclic nucleosides mainly present antiviral effects.
2.3.1. Adenine nucleoside analogues (PMEA)
Tenofovir (TFV, 110) is a NRTI used to treat HIV infection. However, it is almost not absorbed through the gastrointestinal tract, so it is made into different prodrugs. Tenofovir disoproxil fumarate (TDF, 111) and Tenofovir alafenamide (TAF, 112) have been made into oral preparations. Adefovir dipivoxil (ADV, 113) is mainly used to treat HBV infection and has some resistance to HIV. Long-term use can easily cause side effects such as renal dysfunction and lactic acidosis. To decrease its renal toxicity, researchers modified the sugar chain and obtained the following drugs. Pradefovir (PDV, 114) is a liver-targeting phosphonate prodrug of Adefovir including a 4-aryl cyclic substituent in the structure. Compared with 113, 114 shows higher antiviral efficiency, stronger resistance and lower nephrotoxicity [87]. Alamifovir (MCC478, Ly582563, 115) has anti-HBV ability that is much higher than 3 TC (47), and the clinical trial is underway [88]. Adefovir dip-l-isoleucine ethyl ester (116) is a representative compound of Adefovir amino acid ester prodrug. Its in vitro anti-HBV activity is 5 times that of 113. The most feature of its structure is that the phosphono group is connected with the amino acid by a two-carbon chain, and the side chain is an amino acid ester, so it will not produce formaldehyde and pivalic acid as in 113 metabolism. So kidney toxicity can be reduced [89,90] (Fig. 10 ).
Fig. 10.

“PMEA” type acyclic nucleoside analogues.
2.3.2. Guanine nucleoside analogues (PMEG)
With Acyclovir (ACV, 117) as matrix, continuous modification of the ring-opening sugar chain can give lots of compounds. Ganciclovir (GCV, 118), Penciclovir (PCV, 119), Famciclovir (FCV, 120), Valacyclovir (VGV, 121) and Valganciclovir (122) are used to treat symptoms such as herpes virus and cytomegalo virus infection, with increased bioavailability and reduced toxicity. Compounds 120, 121, and 122 are orally effective. Cycloganciclovir (123) is a ring-shaped prodrug of 118, which can mimic the second messenger cGMP in structure and exhibit broad-spectrum antiviral efficacy. Besifovir converted from LB80380 (124) is a new type of guanine nucleoside phosphonate prodrug. It is equivalent to Entecavir (102) in the treatment of HBV infection and has no nephrotoxicity. It was approved by Korean Drug Administration in June 2018 (Fig. 11 ).
Fig. 11.

“PMEG” type acyclic nucleoside analogues.
2.3.3. Pyrimidine nucleoside analogues
Cidofovir (HPMPC, 125) was approved by FDA in June 1996 for treatment of cytomegalovirus (CMV) infections and treatment of diffuse HSV in AIDS patients. But it has a low oral bioavailability and is nephrotoxic. Cyclo-cidofovir (126) is a lactone-type prodrug formed by cyclization. It is chemically stable and can be converted into 125 by phosphatase in the cell. Its oral bioavailability is improved and the renal toxicity is weakened (Fig. 12 ).
Fig. 12.
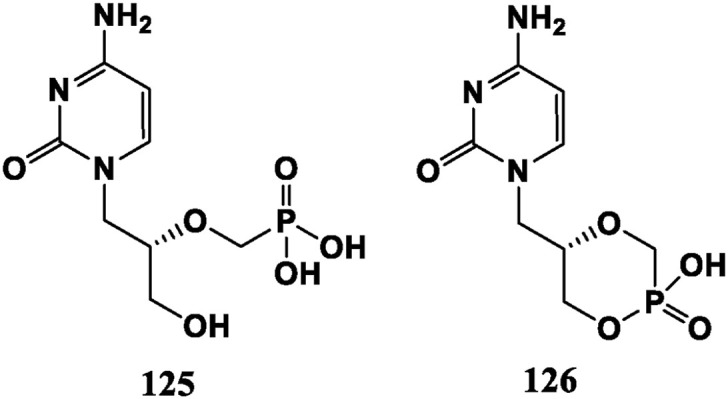
Pyrimidine acyclic nucleoside analogues.
3. Modification of base
3.1. Purine nucleoside analogues
3.1.1. Antiviral compounds
To increase the lipophilicity of anti-HIV drugs and better penetrate the blood-brain barrier, 6-site of the purine can be usually modified. For example, adenosine deaminase active prodrugs 6-methylmercaptopurine riboside (6MMPr, 127) exhibits anti-Zika virus (ZIKV) potency [91]. 2′-F-6-Ethylaminoadenosine (128) and 2′-F-6-dimethylaminoadenosine (129) are much more lipophilic than their unmodified compound ddI (39) or its 2′-F analogue, although the anti-HIV activity is slightly reduced [74]. 2′-F-6-Azido adenosine (130) has brain targeting property as a prodrug of anti-HIV nucleoside 2′-F-ara-ddA (41) [92]. 2′-F-2′,3′-Dideoxy-6-ethoxyinosine (131) has good lipophilicity and acid stability, but the anti-HIV activity is lower than its final active form ddI in vivo [93].
Adenine analogue BCX-4430 (132) shows broad-spectrum antiviral capability against a wide range of RNA viruses but does not include HIV [94]. It is worth noting that its glycosidic bonds are not traditional C–N bonds, but “C–C” bonds (C-nucleoside). Remdesivir (GS-5734, 133), which is in the limelight in the 2020 new coronavirus pandemic, belongs to this type of nucleoside. It is a viral RNA-dependent RNA polymerase (RdRp) inhibitor that works by hindering the synthesis of viral RNA during the replication stage [8]. GS-441524 (134) could completely inhibit infectious peritonitis (FIP) virus replication in CRFK cells and naturally infected feline peritoneal macrophages in vitro without obvious toxicity [95]. Remdesivir (133) is phosphorylated product of 134 and its phosphorylation design is similar to that of Sofosbuvir (89). This feature can make 133 have stronger activity and better cell permeability, and be more effectively metabolized into active nucleoside triphosphate (NTP). When nucleoside analogues are integrated into the viral RNA, they will be cut out by COVID-19 exonuclease. However, Remdesivir (133) is resistant to exonuclease. This should be an important reason why this drug is more effective against COVID-19 than other nucleoside analogues [96]. NITD008 (135) can inhibit proliferation of Enterovirous (EV71) [97]. 3′-Ethynyl-β-D-ribofuranose purine nucleoside (136) shows potential against HCMV [98]. 6-Methyl-7-substituted-7-deaza purine nucleoside (137) exhibits anti-influenza virus effect in MDCK cells [99] (Fig. 13 ).
Fig. 13.

Purine modified nucleoside analogues with antiviral activities.
3.1.2. Anti-tumor compounds
Mercaptopurine (6-MP, 138) is suitable for the treatment of chorionic epithelial cancer, malignant hydatidiform mole and leukemia. Mercaptoguanine (6-MG, 139) is suitable for leukemia treatment. While 127 (Fig. 13) linked to furanose, exhibits anti-Zika virus (ZIKV) efficiency [91].
2-Site halogenated nucleosides Cladribine (25), Clarabine (26) and Fludarabine (27) are suitable for leukemia or lymphoma. Nairabine (140) is a water-soluble prodrug of 9-β-D-arabinofuran guanine, which is effective against T-cell malignant tumor (T-LBL) and T-cell leukemia (T-ALL) and has excellent therapeutic effect, which was approved by FDA in 2006 [100]. 3′-Ethynyl-isonucleoside (141) has an inhibitory activity on metastatic breast tumor [98]. Guanine analogue Forodesine (Immucillin-H, 142) displays the potency of inhibiting leukemia T cells and has been approved by FDA for the treatment of T-cell non-Hodgkin’s lymphoma including T-cell skin cancer, leukemia, and acute lymphocytic leukemia (B-ALL) [101]. Tricyclic nucleoside analogue Triciribine phosphate (VQD-002, 143) is an anti-tumor drug developed in the 1990s, but was abandoned for various reasons. It has recently been discovered that it can selectively inhibit the Akt signaling pathway by restraining the phosphorylation of protein kinase Akt, thereby inducing the apoptosis of tumor cells with abnormal expression of Akt. It is currently in the phase II clinical stage of various tumor treatments [102] (Fig. 14 ).
Fig. 14.

Purine modified nucleoside analogues with anti-tumor activities.
3.2. Pyrimidine nucleoside analogues
3.2.1. Antiviral compounds
5-Substituted derivatives IDU (11), TFT (12), Brivudin (13) are anti-herpes virus drugs. FIAC (8) is a broad-spectrum antiviral medicine and FIAU (9) has an inhibitory effect on HBV [24]. 1-(2-Hydroxyethoxy methyl)-6-phenylthio thymidine (HEPT, 144) is an acyclic NRTI, showing strong selective inhibitory activity against HIV [103]. 4-Amino acid derivative (145) desmonstrates promising efficacy against coxsackie virus B (CVB) [104] (Fig. 15 ).
Fig. 15.

Pyrimidine modified nucleoside analogues with antiviral activities.
3.2.2. Anti-tumor compounds
5-FU (146) is the first pyrimidine anti-tumor metabolism drug, which is easy to cause bone marrow suppression and serious digestive tract reactions such as emesis, diarrhea. To reduce the adverse reactions, and improve the selectivity, heterocycles, amino acids, sugar rings and other means are introduced to derive a series of nucleoside anti-tumor drugs. For example, Carmofur (147) is obtained by connecting alkyl chain with an amide bond on the basis of 5-FU. Furtulon (96), Tegafur (148) and Difuradin (149) are modified by linking sugar with 5-FU derivatives, and their oral bioavailability has been improved. They have all been made into oral dosage forms in clinic, which can gradually release 5-FU in the body and play an anti-cancer effect. In 149, the presence of tetrahydrofuran ring can reduce toxicity. Capecitabine (150) is an oral fluoropyrimidine nucleoside drug, which is catalyzed by enzyme to metabolize 5-FU in the body. Its efficacy is comparable to other intravenously administered pyrimidine drugs, and its toxicity and side effects, especially bone marrow suppression, are less than 5-FU. These may be related to the tissue distribution of the catalytic enzyme in the bone marrow [105]. Compared with Cytarabine (21), 4-amide cytosine nucleoside, such as Enocitabin (151), is modified into a compound with amide group. Its half-life is significantly prolonged, and anti-tumor activity has been significantly improved. It is currently in the stage of clinical trials for treating leukemia [106]. Decitabine (152) is a hypomethylating agent, mainly acts on DNA methyltransferase (DNMT) and was approved in 2006 by FDA for treatment of myelodysplastic syndromes (MDS) and chronic myelomonocytic leukemia (CMML) [107] (Fig. 16 ).
Fig. 16.

Pyrimidine modified nucleoside analogues with anti-tumor activities.
3.3. Triazole nucleoside analogues
Triazole is an important class of five-membered heterocyclic compounds with two common isomers, 1,2,3-triazole and 1,2,4-triazole. The unique structure makes it interact with various bio-macromolecules in the form of coordination bonds, hydrogen bonds, ion-dipole, cation-π, π-π stacking, hydrophobicity and van der Waals forces [108].
3.3.1. 1,2,4-Triazole nucleoside analogues
3.3.1.1. Ribavirin and its analogues
1,2,4-Triazole nucleoside is a novel analogue with various biological activities and it is not easy to be metabolized after entering the organism [108]. Ribavirin (153) with broad-spectrum antiviral activity is an important milestone in the development of nucleoside drugs and belongs to hypoxanthine nucleoside dehydrogenase (IMPDH) inhibitors. It has an antiviral effect by reducing guanylate levels and is effective against a variety of RNA and DNA viruses. However, its side effects are large, and long-term use can cause anemia and teratogenicity. Besides, Ribavirin has an inhibitory efficacy on a variety of solid tumor cells [109].
Most of Ribavirin derivatives are antiviral compounds. For example, Levovirin (154), the l-enantiomer of Riavirin, retains the immunomodulatory effects and has less adverse reaction. Viramidine (155) is a prodrug of Ribavirin in which the phthalimide group of Ribavirin is replaced by an amidino group. It can be taken orally and has the characteristics of liver targeting. It will be converted to Ribavirin under the action of alanine aminotransferase in the liver, rather than in other tissues and organs, and its side effects are significantly reduced [110]. The ribose 5′-OH substituted triazole riboside compounds 156, 157, 158 and the above three compounds have different degrees of inhibition against influenza A virus [[111], [112], [113]]. ETAR (159) is an analogue of Ribavirin obtained by replacing the carboxamide group with an ethynyl group. The in vitro and in vivo experiments show that it is superior to Ribavirin for Hantavirus [114], and also demonstrates significant inhibition against flaviviruses in vitro [115]. This phonoment indicates that ethynyl substitution could change antiviral spectrum.
Ribavirin’s iminoester compounds 160 and 161 do not show virus inhibitory activity at the cellular level and anti-tumor activity in vitro, but exhibit potential in mice with leukemia [116]. Furthermore, Ribavirin can be used as an inhibitor of eukaryotic translation initiation factor eIF4E, and has a certain effect on leukemia [117,118] (Fig. 17 ).
Fig. 17.

Ribavirin and its analogues with antiviral activities (black ones)and anti-tumor activities (blue ones).
3.3.1.2. Aryl-modified triazole nucleoside analogues
Alkynyl triazole ribonucleosides 162, 163 and the aryl alkynyl open-loop triazole ribonucleoside 164 have better anti-HCV potential than Ribavirin [119,120]. Furthermore, bis-triazole-containing derivatives have similar activity to Ribavirin against tobacco mosaic virus (TMV) [[121], [122], [123]] (Fig. 18 ).
Fig. 18.

Aryl-modified triazole nucleoside analogues with antiviral activities (black ones) and anti-tumor activities (blue ones).
The other compounds not only have good inhibitory effect on the proliferation of tumor cells, but also show different mechanisms of action from those of conventional nucleoside drugs, such as restriction of heat shock-related proteins, androgen receptors, transcription, and immune regulation [[124], [125], [126], [127], [128], [129]]. The alkenyl compounds 165, 166, 168, 169 and the N aryl compounds 167, 170, 171, 172 exhibit potential viabilities without significant side effects on the drug-resistant pancreatic cancer MiaPaCa-2 cell line, of which 166, 167, 170, 171 and 172 are with a better potency than Gemcitabine (24). Compound 167 does not inhibit DNA synthesis, but exerts an anti-tumor effect by down-regulating expression of heat shock protein heat shock protein 27 (HSP27). Compound 168 could down-regulate heat shock transcription factor 1 (HSF1), thereby down-regulating HSP27, HSP79, HSP90, reduce presence of client oncoproteins such as STAT3 and AKt, thereby inhibiting the proliferation of mouse pancreatic cancer tumors. In addition to its inhibitory effect on pancreatic cancer, compound 173 can play an anti-prostate cancer role by down-regulating HSF1 and HSPs and restricting expression and transcription of androgen receptor (AR). Compounds 174 and 175 have certain activities on BxPC-3, Pancake-1, HepG2, PC-3 and SKOV3 tumor cell lines, and the effects is superior to Ribavirin [130].
The anti-tumor characteristics are summarized as follows: (I) N-aryl triazoles. (II) C–N glycosidic bonds formed by 1′-C of sugar ring and 1-N of triazolyl. (III) Ribose sugar moiety protection. (IV) Para-substituted alkynyl groups, the substituents on aryl may be long alkyl chain, halogen, trifluoromethyl, etc. Different substituents have effects on the mechanism of action, such as long alkyl chains can reduce the expression of HSPs.
Interestingly, whether the sugar is ring-opened or not has no significant effect on anti-tumor or antiviral properties of these compounds, which is different from the antiviral capabilities of traditional nucleoside derivatives (purines and pyrimidines) mentioned above (Fig. 18).
3.3.2. 1, 2, 3-Triazole nucleoside analogues
SRO-91 (176) is very similar to Ribavirin and presents a promising anti-tumor activity of the same order as Ribavirin with a lower cytotoxicity on the MeT-5A mesothelial cells in vitro and no lethal effect on mice in vivo [109]. 4′-Azido-2′-deoxy-2′-F- [[1], [2], [3]]-triazole-4-amide nucleoside analogue 177 can effectively restrain HBV replication and is effective against Lamivudine resistant strains [131]. Compounds 178, 179 with benzoyl (Tol-) protection exhibit anti-HCV potential against single-stranded RNA viruses and less toxicity [132].
N Atom on triazole base can be substituted with C, S, Se, etc., giving some anti-tumor compounds. Mizoribine (180), Tiazofurin (181), Selenazofurin (182) belong to hypoxanthine IMPDH inhibitor, as does Ribavirin, and act as antiviral drugs by reducing guanylate levels. Compound 180 is clinically used as an anti-rejection drug for treatment of organ transplantation. Compounds 181 and 182 have inhibitory effects on lymphoma, toga viruses, bunya viruses, and arena viruse. In particular, 181 is the only one of IMPDH inhibitors developed for clinical use as an FDA-approved anti-cancer agent and is an orphan drug for chronic myeloid leukemia (CML) primitive cell crisis (Fig. 19 ).
Fig. 19.

1,2,3-Triazole nucleoside analogues.
In some cases, triazole can be regarded as a modification group of the sugar. Compound 183 shows strong suppressive activity against human liver cancer cells (HepG2), lung cancer cells (A549 and LAC) and cervical cancer cells (Hela) [133]. Compound 184, which with 3-triazolium alkyl substitution chain length of 7–17 carbon atoms exhibits good anti-tumor potency against human gastric cancer cell line (MGC0803) [134]. 1,2,3-Triazole thymidine derivative (185) presents inhibiting effect against HIV or influenza virus A549 cells [135]. 2′-Deoxygen-2′-F-4′-(1,2,3-triazole) cytosine nucleoside derivative (186), displays similar inhibitory efficiency against HIV-1 as the control drug Zidovudine, and it also demonstrates inhibitory action against HBV [136]. 1,2,3-Triazole acyclic nucleoside phosphate compounds 187, 188 possess significant anti-HCV potential without obvious toxicity [137] (Fig. 20 ).
Fig. 20.

1,2,3-Triazole nucleoside derivatives with antiviral activities (black ones) and anti-tumor activities (blue ones).
4. Drug assembled nucleoside analogues
The same or different active fragments can be assembled as a new hybrid molecule, i.e. “double prodrugs”, which not only have characteristics of individual active fragments, but also synergistically enhance biological activity and reduce toxic side effects [138].
4.1. Antiviral “double prodrugs”
Compound 189 and deoxydibase nucleoside 190 can inhibit NS5B polymerase in hepatitis C, because the phosphate two-base nucleoside binds stably to the lysine and arginine residues of polymerase through magnesium ions [139,140]. “d4T-MKC442” Dual-prodrugs 191, 192 and 193 are active against Y181C mutant strain of wild type HIV-1 and against HIV-2 [141]. “AZT-HEPT” Complexes 194, 195 and 196 have anti-HIV-1 potency, but no effect against HIV-2 or Y181C mutant of HIV-1. The ddC-HEPT conjugate 197 is active against HIV-1 and HIV-2 [142]. The dual prodrug 198 assembled from foscarnet (PFA, anti-reverse transcriptase pyrophosphate analogue) and AZT (34) exhibits good antiviral efficacy in vitro, and has resistance to HIV, HSV-1, human megasalivirus (HCMV) and human herpesvirus-6B (HHV-6) [143] (Fig. 21 ).
Fig. 21.

Antiviral “double prodrugs”.
4.2. Anti-tumor “double prodrugs”
The quinoxaline group (QX) is a known DNA intercalator that prevents DNA replication and repair. 3′-Deoxythymidine phenylquinoxaline conjugate (dT-QX) (199) can selectively kill liver cancer cells and has low lethality to normal hepatocytes [144]. But its high EC50 value limits clinical application [145]. Halogen atoms are further introduced into the QX part, and the synthesized halogen dT-QX 2Cl/2Br (200) shows stronger anti-tumor activity than dT-QX under low power UVA irradiation [146]. Sunitinib, a multi-target tyrosine kinase inhibitor, with multiple effect against tumor cell growth and tumor angiogenesis, has been approved by FDA as a first-line treatment for advanced renal cell carcinoma. However, the “dT-Sunitinib” conjugate does not show significant anti-tumor property [147].
Compounds 201 (obtained by the combination of triazole and aminodithioformate), 202 (obtained by combination of thiosemicarbazide, alkynyl and pyrimidine), 203, 204 and 205 (obtained by combination of fluorenyl heterocyclic and pyrimidotriazole skeleton), could effectively inhibit tumor growth and metastasis by targeting silencing histone lysine specific demethylase 1 (LSD1) expression or inhibiting the enzyme activity of LSD1 [[148], [149], [150]]. Compound 206 assembled from triazole and pyrimidine has good inhibitory activity against gastric cancer cell MGC-803 and is little toxic to normal cell line GES-1 [151]. Similar derivative compound 207 has significant inhibitory activity on lung cancer cells H1650 and A549, and little toxicity on normal cell lines GES-1 [152]. Thiazolo-[5,4-d]-pyrimidine derivative 208 presents good anti-proliferative potential against gastric cancer cell HGC-27, and low toxicity to GES-1 cells [153]. Similar compound 209 demonstrates high potency to gastric cancer cells and selective inhibition to MGC-803 cells [154].
Moreover, 5-FdU-ECyd (5′–5′) (210) can fight against gastric adenocarcinoma cell lines. 5-FdU-ECyd (3′–5′) (211) can resist against hepatoblastoma cell lines, and ECyd-lipid-5-FdU (212) can confront against cervical cancer cells [[155], [156], [157]] (Fig. 22 ).
Fig. 22.

Anti-tumor “double prodrugs”.
5. Macromolecular carrier conjugated nucleosides
Most nucleoside analogues, as hydrophilic compounds, cannot pass freely through the cell membrane, especially through the blood-brain barrier to inhibit viral replication in the brain tissue. The nucleoside modified with a lipophilic macromolecular carrier to form a prodrug is a common strategy. Under the action of free diffusion or directed active transport system of cell membrane, these prodrugs can pass through the corresponding biological barrier and release active molecules thencausing antiviral activity [158,159]. The lipophilic macromolecular carriers include cholesterol, polyethylene glycol, fatty acids, and phospholipids.
5.1. Cholesterol conjugated nucleosides
Some cholesterol-polyamine amide conjugates display antibacterial effect in addition to increasing its solubility. The steroid-polyamine conjugate can be used as an ionophore and has a strong interaction with the negatively charged lipid membrane. The lipophilic cholesterol conjugated with nucleosides ACV (118), ddI (39), and AZT (34) can form a battery of novel self-assembling drug delivery systems. Among them, the cholesterol-AZT conjugate (213) shows good anti-HIV activity in vitro, and EC50 value reaches 1 nM in the MT-4 cell model, which was 1/5 of AZT. The cholesterol-ddI conjugate 214 exhibits good sustained release in vivo, but it is too stable to be degraded to release the active drug ddI, so it does not exhibit significant potential [160,161] (Fig. 23 ).
Fig. 23.

Cholesterol conjugated nucleosides.
5.2. Polyethylene glycol conjugated nucleosides
Polyethylene glycol and its derivatives have become significant chemical modifiers due to their water-fat amphiphilicity, biocompatibility, non-immunogenicity, and no toxic side effects. The Gemcitabine-folate-PEG conjugate 215 shows a sustained release effect relative to Gemcitabine (24), and significantly increases oral bioavailability. It also presents higher anti-proliferative potency in early cervical cancer cells (HeLa cells) and nasopharyngeal epithelial cancer cells (KB-3-1) and exhibits lower toxicity than Gemcitabine [162]. mPEG750-AZT (216) has good stability in vitro, meanwhile its in vitro activity of original AZT against wild strains of HIV-1 and HIV-2 is well retained, and the toxicity is significantly reduced. Pharmacokinetic evaluation shows that the half-life of compound 216 is extended to 2.3 times of AZT, and the oral bioavailability increased to 224% of AZT [163] (Fig. 24 ).
Fig. 24.

Polyethylene glycol conjugated nucleosides.
5.3. Fatty acid modified nucleosides
Many nucleoside analogue drugs have low oral bioavailability, poor cell penetration and high nephrotoxicity. To reduce these therapeutic defects, alkoxyalkyl groups can be used to modify nucleotides. A macromolecular nucleotide prodrug conjugated with alkoxyalkyl can be readily absorbed by gastrointestinal tract as an analogue of lysophospholipid and its half-life in blood can be prolonged. This macromolecular prodrug can reduce its accumulation in renal tubules since it cannot be recognized by the transport system, thus reducing the renal toxicity of the drug. Since the macromolecular modified prodrug is more permeable to the cell membrane, the alkoxyalkyl nucleotide modifier also exhibits stronger activity in vitro than the original drug [164,165]. Among nucleotide conjugates, compounds CMX001 (217) (Cidofovir (125) modified by cetyl propoxy) and CMX157 (218) (TFV (110) modified by cetyl propoxy) have now entered clinical studies [166,167]. The bioavailability of TFV diester prodrug TDF (111) has been improved to some extent, but it is only 25%–40%, depending on the fat content in the diet [168]. Octadecyloxyethyl-Tenofovir (ODE-TFV, 219) is obtained by introducing ether-soluble long chains onto one of the hydroxyl groups of the phosphonates in the TFV molecule. It shows higher bioavail ability and chemical stability than TDF, and exhibits higher inhibitory activity than TDF and 3 TC in the human hepatoma cell line 2.2.15 cells transfected with HBV gene. DHBV-infected duckling model is used to evaluate the antiviral potency in vivo and results indicate that medium and high-dose ODE-TFV (100, 200 mg/kg) could significantly inhibit replication of DHBV-DNA [169].
To solve the defects of low oral bioavailability of cytarabine and short half-life, Cytarabine (21) is modified with long-chain alkyl groups to form cytarabine ocfosfate (Fosteabine, YNK01, 94) which can be effectively taken orally. This modification can improve survival rate, reduce the recurrence rate in clinical phase II, but its side effects make the patient less tolerable [170]. CP-4055 (Elacytarabine, 220) is a prodrug connected between oleic acid and 5-OH of cytarosine by ester bond, which can gradually increase fat solubility and drug accumulation in the nucleus, and has an inhibitory effect on both leukemia and solid tumor [[171], [172], [173]]. CP-4126 (221) has entered clinical stage III, and patients with solid tumors have a good tolerance to it without obvious side effects [173].
The Cytarabine (21) connected with six carbon hexanoic acid (HA), twelve carbon lauric acid (LA), eighteen carbon oleic acid (OA), can construct the multifunctional prodrugs HA-Ara (222), LA-Ara (223), OA-Ara (224), DTA-Ara (225) [[174], [175], [176], [177], [178]]. These four conjugates show significant suppressive activities against acute myelogenous leukemia cell line HL60 and human chronic myelogenous leukemia cell line K562. They could self-assemble into nanomolymers and significantly increase drug loading, and their inhibitory efficiency and oral bioavailability on cancer cells are higher than that of raw material drug Cytarabine. LA-Ara (223) nanofibers have inhibitory effect on solid tumor-derived breast cancer cells (4T1) and can avoid the clinical toxic side effects such as hand-foot syndrome and mucositis [178]. The amphiphilic molecular conjugates Ara-AOT (226) and Cytarabine prodrug with double hydrophilic head groups Ara-R-Ara (227) have stronger cell sensitivity and increased toxicity among leukemic HL60 cells and K562 cells than the original drug Cytarabine, and have a rapid onset of action [179,180] (Fig. 25 ).
Fig. 25.

Fatty acid modified nucleosides.
5.4. Phospholipid conjugated nucleosides
Phospholipids have a wide range of physiological activities, and have targeted affinity to tumor tissues. To this end, a large number of nucleoside glycerophospholipid conjugates have been developed to promote the distribution of nucleoside drugs in tumor tissues so to improve drug selectivity and reduce the minimum concentration required for treatment, and at the same time produce synergistic effects with nucleosides to decrease its toxic and side effects in normal tissues. Cytarabine phospholipid conjugates (228, 229) are better for mouse lymphocytic leukemia L1210 than the parent drug Cytarabine [181]. Thioether phospholipid conjugates of Cytarabine (230, 231) show good effect on transplanted mouse leukemia L1210 [182]. Anti-HIV activity of AZT phospholipid conjugate (232) is only 1/10 of AZT, but its anticancer activity is 10 times that of AZT [183].
The anticancer drug Tegafur (148) is modified by phospholipid and active atomic selenium. The resulting compound 233 has a good inhibitory effect on the malignant proliferation of bladder cancer cell PGA1 [184]. Compound 234 displays good growth inhibition efficacy on both gastric cancer cells BGC-823 and bladder cancer cells T-24, but shows highly toxic on normal liver epithelial cells [185]. Tegafur-selenophospholipid conjugate (235) has stronger suppressive effect on bladder cancer cell PGA1 than Tegafur [186] (Fig. 26 ).
Fig. 26.

Phospholipid conjugated nucleosides.
6. Conclusion
With Remdesivir being approved by FDA as a drug for the treatment of COVID-19, nucleoside drugs have once again received widespread attention in the medical community. Herein, we summarized structural modifications of nucleosides scaffold and the bioactivity rules. 2′-“Ara”-substitution by –F, or –CN group, and 3′-“ara” substitution by acetylenyl group in sugar have important influence on the anti-tumor activities. The presence of “ara”-OH or the absence of 2′-OH has good effect on non-solid tumors such as leukemia. When the substituent is the other groups, it can not only treat non-solid tumors, but also enhance the sensitivity to solid tumors. Fluoroiodine cytarabine (FIAC) is an exception, possibly because the substitution of 5-I on cytosine base has a greater impact on biological activity. Dideoxy dehydrogenation of 2′,3′-sites of sugar ring can promote the bioactivity. For example, the antiviral capabilities of L-Fd4C (49) and L-d4A (64) are 10–20 times stronger than the 2′,3′-saturated analogues L-FddC (46) and L-ddA (65). Acyclic nucleosides and L-type nucleosides mainly represent antiviral potential. Nearly all of acyclic nucleosides can fight with DNA viruses, but only adenosine-acyclic nucleosides are effective for RNA viruses (HIV).
The 5-F substitution of uracil bases is a key factor for bioactivity, such as 5-FU (146) and its derivatives with anti-tumor properties. While 5-site is substituted by –I, –CF3, bromovinyl group, it usually shows antiviral activity (such as FIAC (8), FIAU (9), IDU (11), TFT (12), Brivudin (13)).
C–N Glycosidic bond is an important factor affecting potentials of triazolid nucleoside analogues. When 1′-C of sugar coupled with 1-N of triazolid the analogues usually show anti-tumor activities. Whereas 1′-C of sugar ring coupled with 2-N of triazolid the analogues mainly represent antiviral efficiencies. Triazole nucleosides modified by long alkyl chain, halogen, or trifluoromethyl group would usually affect their action mechanism. For example, long alkyl chain can reduce the expression of HSPs. Whether the sugar is cycled or not has no significant effect on the anti-tumor or antiviral properties of triazoles, which is different from the traditional nucleoside derivatives of antiviral drugs.
The nucleoside analogues assembled by cholesterol, polyethylene glycol, fatty acid and phospholipid would improve their bioavailabilities and bioactivities. For example, alkoxyalkyl conjugated Cytarabine achieves oral efficacy. The anti-HIV activity of the cholesterol conjugated AZT is 5 times that of the original drug, while the anti-tumor activity of ether phospholipid conjugated AZT is 10 times that of AZT.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
This work was supported by the National Natural Science Foundation of China (No. 21967003), 2017 basic ability improvement project for young and middle-aged teachers in colleges and universities in Guangxi (No. 2017KY1226).
References
- 1.Jazayeri A., Falck J., Lukas C., Bartek J., Smith G.C.M., Lukas J., Jackson S.P. ATM-and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 2.Ewald B., Sampath D., Plunkett W. Nucleoside analogs: molecular mechanisms signaling cell death. Oncogene. 2008;27:6522–6537. doi: 10.1038/onc.2008.316. [DOI] [PubMed] [Google Scholar]
- 3.Lo1 M.K., Feldmann F., Gary J.M., Jordan R., Bannister R., Cronin J., Patel N.R., Klena1 J.D., Nichol S.T., Cihlar T., Zaki S.R., Feldmann H., Spiropoulou C.F., de Wit E., Remdesivir G.S.- 5734) protects African green monkeys from Nipah virus challenge. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aau9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tchesnokov E.P., Feng J.Y., Porter D.P., Götte M. Mechanism of inhibition of Ebola virus RNA-dependent RNA polymerase by Remdesivir. Viruses. 2019;11:e326. doi: 10.3390/v11040326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheahan T.P., Sims A.C., Leist S.R., Schäfer A., Won J., Brown A.J., Montgomery S.A., Hogg A., Babusis D., Clarke M.O., Spahn J.E., Bauer L., Sellers S., Porter D., Feng J.Y., Cihlar T., Jordan R., Denison M.R., Baric R.S. Comparative therapeutic efficacy of Remdesivir and combination Lopinavir, Ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020;11:e222. doi: 10.1038/s41467-019-13940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang M., Cao R., Zhang L., Yang X.L., Liu J., Xu M.Y., Shi Z.L., Hu Z.H., Zhong W., Xiao G.F. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ikejiri M., Saijo M., Morikawa S., Fukushi S., Mizutani T., Kurane I., Maruyama T. Synthesis and biological evaluation of nucleoside analogueshaving 6-Chloropurine as anti-SARS-CoV agents. Bioorg. Med. Chem. Lett. 2007;17:2470–2473. doi: 10.1016/j.bmcl.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor R., Kotiana P., Warrenb T., Panchal Rekha, Bavari S., Julander J., Doboa S., Rosea A., El-Kattana Y., Taubenheimd B., Babua Y., Sheridan W.P. BCX4430-A broad-spectrum antiviral adenosine nucleoside analog under development for the treatment of Ebola virus disease. J. Infect. Public. Heal. 2016;9:220–226. doi: 10.1016/j.jiph.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y.L., Yokokawa F., Shi P.Y. The search for nucleoside/nucleotide analog inhibitors of Dengue virus. Antivir. Res. 2015;122:12–19. doi: 10.1016/j.antiviral.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 10.Bernatchez J.A., Coste M., Beck S., Wells G.A., Luna L.A., Clark A.E., Zhu Z., Hecht D., Rich J.N., Sohl C.D., Purse B.W., Siqueira-Neto J.L. Viruses11; 2019. Activity of Selected Nucleoside Analogue Protides against Zika Virus in Human Neural Stem Cells; p. e365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jordheim L.P., Durantel D., Zoulim F., Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. 2013;12:447–464. doi: 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- 12.Seley-Radtke K.L., Yates M.K. The evolution of nucleoside analogue antivirals: a review for chemists and non-chemists. Part 1: early structural modifications to the nucleoside scaffold. Antivir. Res. 2018;154:66–86. doi: 10.1016/j.antiviral.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yates M.K., Seley-Radtke K.L. The evolution of antiviral nucleoside analogues: a review for chemists and non-chemists. Part II: complex modifications to the nucleoside scaffold. Antivir. Res. 2019;162:5–21. doi: 10.1016/j.antiviral.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pierra C., Amador A., Badaroux E., Storer R., Gosselin G. Synthesis of 2′-C-methylcytidine and 2′-C-methyluridine derivatives modified in the 3′-position as potential antiviral agents. Collect. Czech Chem. Commun. 2006;71:991–1010. [Google Scholar]
- 15.Pierra C., Amador A., Benzaria S., Cretton-Scott E., D’Amours M., Mao J., Mathieu S., Moussa A., Bridges E.G., Standring D.N., Sommadossi J.P., Storer R., Gosselin G. Synthesis and pharmacokinetics of Valopicitabine (NM283), an efficient prodrug of the potent anti-HCV agent 2′-C-methylcytidine. J. Med. Chem. 2006;49:6614–6620. doi: 10.1021/jm0603623. [DOI] [PubMed] [Google Scholar]
- 16.Clark J.L., Hollecker L., Mason J.C., Stuyver L.J., Tharnish P.M., Lostia S., McBrayer T.R., Schinazi R.F., Watanabe K.A., Otto M.J., Furman P.A., Stec W.J., Patterson S.E. Design, synthesis, and antiviral activity of 2′-deoxy-2′-fluoro-2′-C-methylcytidine, a potent inhibitor of hepatitis C virus replication. J. Med. Chem. 2005;48:5504–5508. doi: 10.1021/jm0502788. [DOI] [PubMed] [Google Scholar]
- 17.Yoo B.N., Kin H.O., Moon H.R., Seol S.K., Jang S.K., Leea K.M., Jeong L.S. Synthesis of 2-C-hydroxy-methylribofuranosyl purines as potent anti-hepatitis C virus (HCV) agent. Bioorg. Med. Chem. Lett. 2006;16:4190–4194. doi: 10.1016/j.bmcl.2006.05.089. [DOI] [PubMed] [Google Scholar]
- 18.Nesya G., Armando D.P., Jean-Francois T., Ina M., Johan N., Kris D.C. 2′-C-Methylcytidine as a potent and selective inhibitor of the replication offoot-and-mouth disease virus. Antivir. Res. 2007;73:161–168. doi: 10.1016/j.antiviral.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 19.Julander J.G., Jha A.K., Choi J.A., Junga K.H., Smeea D.F., Morreya J.D., Chu C.K. Efficacy of 2′-C-methylcytidine against yellow fever virus in cell culture and in a hamster model. Antivir. Res. 2010;86:261–267. doi: 10.1016/j.antiviral.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rocha-Pereira J., Jochmans D., Neyts J. Prophylactic treatment with the nucleoside analogue 2′-C-methylcytidine completely prevents transmission of norovirus. J. Antimicrob. Chemother. 2015;70:190–197. doi: 10.1093/jac/dku363. [DOI] [PubMed] [Google Scholar]
- 21.Lee J.C., Tseng C.K., Wu Y.H., Kaushik-Basu N., Lin C.K., Chen W.C., Wu H.N. Characterization of the activity of 2′-C-methylcytidine against Dengue virus replication. Antivir. Res. 2015;116:1–9. doi: 10.1016/j.antiviral.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Qu C., Xu L., Yin Y.B., Peppelenbosch1 M.P., Pan Q.W., Wang W.S. Nucleoside analogue 2′-C-methylcytidine inhibits hepatitis E virus replication but antagonizes Ribavirin. Arch. Virol. 2017;162:2989–2996. doi: 10.1007/s00705-017-3444-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamko K., Ma Y., Delang L., Mirabelli G., Neyts J. Antiviral effects of selected nucleoside analogues against human parechoviruses A1 and A3. Antivir. Res. 2019;162:51–53. doi: 10.1016/j.antiviral.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 24.Staschke K.A., Colacino J.M., Mabry T.E., Jones C.D. The in vitro anti-hepatitis B virus activity of FIAU[1-(2′-deoxy-2′-fluoro-1-β-D-arabinofuranosyl-5-iodo) uracil] is selective, reversible, and determined, at least in part, by the host cell. Antivir. Res. 1994;23:45–61. doi: 10.1016/0166-3542(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 25.Ho D.H.W. Biochemical studies of a new antitumor agent, O2, 2′-cyclocytidine. Biochem. Pharmacol. 1974;23:1235–1244. doi: 10.1016/0006-2952(74)90327-x. [DOI] [PubMed] [Google Scholar]
- 26.Bridges C.G., Ahmed S.P., Sunkara P.S., McCarthy J.R., Tyms A.S. The ribonucleotide reductase inhibitor (E)-2′-fluoromethylene-2′-deoxycytidine (MDL 101,731): a potential topical therapy for herpes simplex virus infection. Antivir. Res. 1995;27:325–334. doi: 10.1016/0166-3542(95)00015-e. [DOI] [PubMed] [Google Scholar]
- 27.Yoshimura Y., Kitano K., Yamada K., Sakata S., Ashida N., Machida H. Synthesis and biological activities of 2′-deoxy-2′-fluoro-4′-thioarabinofuranosylpyrimidine and purine nucleosides. Bioorg. Med. Chem. 2000;8:1545–1558. doi: 10.1016/s0968-0896(00)00065-1. [DOI] [PubMed] [Google Scholar]
- 28.Fleming F.F., Yao L.H., Ravikumar P.C., Funk L., Shook B.C. Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore. J. Med. Chem. 2010;53:7902–7917. doi: 10.1021/jm100762r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azuma A., Huang P., Matsuda A., Plunkett W. 2′-C-Cyano-2′-deoxy-1-β-D-arabino-pentofuranosylcytosine: a novel anticancer nucleoside analog that causes both DNA strand breaks and G2 arrest. Mol. Pharmacol. 2001;59:725–731. doi: 10.1124/mol.59.4.725. [DOI] [PubMed] [Google Scholar]
- 30.Parker W.B., Shaddix S.C., Gilbert K.S., Shepherd R.V., Waud W.R. Enhancement of the in vivo antitumor activity of clofarabine by 1-beta-D-[4-thio-arabinofuranosyl]-cytosine, Cancer Chemother. Pharmacology. 2009;64:253–261. doi: 10.1007/s00280-008-0862-z. [DOI] [PubMed] [Google Scholar]
- 31.Chen Y.W., Chou K.M. DNA lesion bypass polymerases and 4′-thio-beta-darabinofuranosylcytosine (T-araC) Int. J. Biochem. Mol. Biol. 2011;2:340–346. [PMC free article] [PubMed] [Google Scholar]
- 32.Hrdlicka P.J., Andersen N.K., Jepsen J.S., Hansen F.G., Haselmann K.F., Nielsend C., Wengel J. Synthesis and biological evaluation of branched and conformationally restricted analogs of the anticancer compounds 3′-C-ethynyluridine (EUrd) and 3′-C-ethynylcytidine (ECyd) Bioorg. Med. Chem. 2005;13:2597–2621. doi: 10.1016/j.bmc.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 33.Lui V.W.Y., Lau C.P.Y., Cheung C.S.F., Ho K., Ng M.H.L., Cheng S.H., Hong B., Tsao S.W., Tsang C.M., Lei K.I.K., Yamasaki Y., Mita A., Chan A.T.C. An RNA-directed nucleoside anti-metabolite, 1-(3-C-ethynyl-beta-D-ribopentofuranosyl)cytosine (ECyd), elicits antitumor effect via TP53-induced glycolysis and apoptosis regulator (TIGAR) downregulation. Biochem. Pharmacol. 2010;79:1772–1780. doi: 10.1016/j.bcp.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 34.Franchetti P., Cappellacci L., Pasqualini M., Petrelli R., Vita P., Jayaram H.N., Horvath Z., Szekeres T., Grifantini M. Antitumor activity of C-methyl-β-d-ribofuranosyl adenine nucleoside ribonucleotide reductase inhibitors. J. Med. Chem. 2005;48:4983–4989. doi: 10.1021/jm048944c. [DOI] [PubMed] [Google Scholar]
- 35.Hartmann H., Vogt M.W., Durno A.G., Hirsch M.S., Hunsmann G., Eckstein F. Enhanced in vitro inhibition of HIV-1 replication by 3′-fluoro-3′-deoxythymidine compared to several other nucleoside analogs. AIDS Res. Hum. Retrovir. 1988;4:457–466. doi: 10.1089/aid.1988.4.457. [DOI] [PubMed] [Google Scholar]
- 36.Pokrovsky A.G., Pronayeva T.R., Fedyuk N.V., Shirokova E.A., Khandazhinskaya A.L., Tarusova N.B., Karpenko I.L., Krayevsky A.A. Anti-HIV activity of novel phosphonate derivatives of AZT, d4T, and ddA. Nucleos. Nucleot.Nucl. 2001;20:767–769. doi: 10.1081/NCN-100002426. [DOI] [PubMed] [Google Scholar]
- 37.Aerschot A.V., Herdewijn P., Balzarini J., Pauwels R., Clercq E.D. 3′-Fluoro-2′, 3′-dideoxy-5-chlorouridine: most selective anti-HIV-1 agent among a series of new 2′- and 3′-fluorinated 2′, 3′-dideoxynucleoside analogues. J. Med. Chem. 1989;32:1743–1749. doi: 10.1021/jm00128a013. [DOI] [PubMed] [Google Scholar]
- 38.Roth J.S., F H., Jr., Tanaka M., Mitsuya H., Kelley J.A. Determination of 2′-β-fluoro-2′, 3′-dideoxyadenosine, an experimental anti-AIDS drug, in human plasma by highperformance liquid chromatography. J. Chromatogr. B. 1998;712:199–210. doi: 10.1016/s0378-4347(98)00144-3. [DOI] [PubMed] [Google Scholar]
- 39.Lee-Ruff E., Ostrowski M., Ladha A., Stynes D.V., Vernik I., Jiang J.L., Wan W.Q., Ding S.F., Joshi S. Synthesis and HIV inhibition activity of 2′, 3′-dideoxy-3′-C-hydroxymethyl nucleosides. J. Med. Chem. 1996;39:5276–5280. doi: 10.1021/jm950822k. [DOI] [PubMed] [Google Scholar]
- 40.Lin T.S., Luo M.Z., Liu M.C., Pai S.B., Dutschman G.E., Cheng Y.C. Antiviral activity of 2′, 3′-dideoxy-β-L-5-fluorocytidine (β-L-FddC) and 2′, 3′-dideoxy-cytidine (β-L-ddC) against hepatitis B virus and human immunodeficiency virus 1 in vitro. Biochem. Pharmacol. 1994;47:171–174. doi: 10.1016/0006-2952(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 41.Chen S.H. Comparative evaluation of L-Fd4C and related nucleoside analogs as promising antiviral agents. Curr. Med. Chem. 2002;9:899–912. doi: 10.2174/0929867024606696. [DOI] [PubMed] [Google Scholar]
- 42.Chen H.C., Pai S.B., Hurwitz S.J., Chu C.K., Glazkova Y., McClure H.M., Feitelson M., Schinazi R.F. Antiviral activity and pharmacokinetics of 1-(2,3-dideoxy-2-fluoro-L-glyceropent-2-enofuranosyl) cytosine. Antimicrob. Agents Chemother. 2003;47:1922–1928. doi: 10.1128/AAC.47.6.1922-1928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pai S.B., Pai R.B., Xie M.Y., Beker T., Shi J., Tharnish P.M., Chu C.K., Schinazi R.F. Characterization of hepatitis B virus inhibition by novel 2′-fluoro-2′,3′-unsaturated beta-D and L-nucleosides. Antivir. Chem. Chemother. 2005;16:183–192. doi: 10.1177/095632020501600304. [DOI] [PubMed] [Google Scholar]
- 44.Lin T.S., Luo M.Z., Liu M.C., Zhu Y.L., Gullen E., Dutschman G.E., Cheng Y.C. Design and synthesis of 2′,3′-dideoxy-2′,3′-didehydro-β-L-cytidine (β-L-d4C) and 2′,3′-dideoxy-2′,3′-didehydro-β-L-5-fluorocytidine (β-L-Fd4C), two exceptionally potent inhibitors of human hepatitis B virus (HBV) and potent inhibitors of human immunodeficiency virus (HIV) in vitro. J. Med. Chem. 1996;39:1757–1759. doi: 10.1021/jm950836q. [DOI] [PubMed] [Google Scholar]
- 45.Asif G., Hurwitz S.J., Gumina G., Chu C.K., McClure H.M., Schinazi R.F. Pharmacokinetics of the antiviral agent β-L-3′-fluoro-2′,3′-didehydro-2,3-dideoxycytidine in rhesus monkeys. Antimicrob. Agents Chemother. 2005;49:560–564. doi: 10.1128/AAC.49.2.560-564.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matthes E., Funk A., Krahn I., Gaertner K., von Janta-Lipinski M., Lin L., Will H., Sirma H. Strong and selective inhibitors of hepatitis B virus replication among novel N4-hydroxy-and 5-methyl-β-L-deoxycytidine analogues. Antimicrob. Agents Chemother. 2007;51:2523–2530. doi: 10.1128/AAC.00001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hewlett G., Hallenberger S., Rubsamen-Waigmann H. Antivirals against DNA viruses (hepatitis B and the herpes viruses) Curr. Opin. Pharmacol. 2004;4:453–464. doi: 10.1016/j.coph.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 48.Mansour T.S., Jin H., Wang W., Hooker E.U., Ashman C., Cammack N., Salomon H., Belmonte A.R., Wainberge M.A. Anti-human immunodeficiency virus and anti-hepatitis-B virus activities and toxicities of the enantiomers of 2′-deoxy-3′-oxa-4′-thiocytidine and their 5-fluoro analogs in vitro. J. Med. Chem. 1995;38:1–4. doi: 10.1021/jm00001a001. [DOI] [PubMed] [Google Scholar]
- 49.Stoddar C.A., Moreno M.E., Linquist-stepps V.D., Bare C., Bogan M.R., Gobbi A., Buckheit R.W., JR., Bedard J., Rando R.F., Mccune J.M. Antiviral activity of 2′-deoxy-3′-oxa-4′-thiocytidine (BCH-10652) against Lamivudine-resistant human immunodeficiency virus type 1 in SCID-hu Thy/Liv mice. Antimicrob. Agents Chemother. 2000;44:783–786. doi: 10.1128/aac.44.3.783-786.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mao S.L., Bouygues M., Welch C., Biba M., Chilenski J., Schinazib R.F., Liotta D.C. Synthesis of enantiomerically pure D-FDOC, an anti-HIV agent. Bioorg. Med. Chem. Lett. 2004;14:4991–4994. doi: 10.1016/j.bmcl.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 51.Wilson L.J., Choi W.B., Spurting T., Uotta D.C., Bchinazi R.F., Cannon D., Painter G.R., Clair M.St, Furman P.A. The synthesis and anti-HIV activity of pyrimidine dioxolanyl nucleosides. Bioorg. Med. Chem. Lett. 1993;3:169–174. [Google Scholar]
- 52.Furman P.A., Jeffrey J., Kiefer L.L., Feng J.Y., Anderson K.S., Borroto-esoda K., Hill E., Copeland W.C., Chu C.K., Sommadossi J.P., Liberman I., Schinazi R.F., Painter G.R. Mechanism of action of 1-β-D-2,6-Diaminopurine Dioxolane, a Prodrug of the human immunodeficiency virus type 1 inhibitor 1-β-D-dioxolane guanosine, Antimicrob. Age. 2001;45:158–165. doi: 10.1128/AAC.45.1.158-165.2001. Ch. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim H.O., Schinazi R.F., Nampalli S., Shanmuganathan K., Cannon D.L., Alves A.J., Jeong L.S., Beach J.W., Chu C.K. 1,3-Dioxolanylpurine nucleosides (2R,4R) and (2R,4S) with selective anti-HIV- 1 activity in human lymphocytes. J. Med. Chem. 1993;36:30–37. doi: 10.1021/jm00053a004. [DOI] [PubMed] [Google Scholar]
- 54.Jang J.H., Kim J.W., Jeong S.H., Myung H.J., Kim H.S., Park Y.S., Lee S.H., Hwang J.H., Kim N., Lee D.H. Clevudine for chronic hepatitis B: antiviral response, predictors of response, and development of myopathy. J. Viral Hepat. 2011;18:84–90. doi: 10.1111/j.1365-2893.2010.01281.x. [DOI] [PubMed] [Google Scholar]
- 55.Yoo B.C., Kim J.H., Chung Y.H., et al. Twenty-four-week Clevudine therapy showed potent and sustained antiviral activity in HBeAg-positive chronic hepatitis B. Hepatology. 2007;45:1172–1178. doi: 10.1002/hep.21629. [DOI] [PubMed] [Google Scholar]
- 56.Bolon J.P., Wang P.Y., Chu C.K. Anti-human immunodeficiency and anti-hepatitis B virus activities of β-l-2′,3′-dideoxy purine nucleosides. Bioorg. Med. Chem. Lett. 1996;6:1657–1662. [Google Scholar]
- 57.Kotra L.P., Xiang Y.J., Newton M.G., Schinazi R.F., Cheng Y.C., Chu C.K. Structure-activity relationships of 2′-deoxy-2′,2′-difluoro-lerythro-pentofuranosyl nucleosides. J. Med. Chem. 1997;40:3635–3644. doi: 10.1021/jm970275y. [DOI] [PubMed] [Google Scholar]
- 58.Jimeno A., Messersmith W.A., Lee C.K., Ma W.W., Laheru D., Donehower R.C., Baker S.D., Hidalgo M. Phase I study of Troxacitabine administered by continuous infusion in subjects with advanced solid malignancies. Ann. Oncol. 2008;19:374–379. doi: 10.1093/annonc/mdm572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Townsley C.A., Chi K., Ernst D.S., Belanger K., Tannock I., Bjarnason G.A., Stewart D., Goel R., Ruether J.D., Siu L.L., Jolivet J., McIntosh L., Seymour L., Moore M.J. Phase II study of Troxacitabine (BCH-4556) in patients with advanced and/or metastatic renal cell carcinoma: a trial of the National Cancer Institute of Canada-Clinical Trials Group. J. Clin. Oncol. 2003;21:1524–1529. doi: 10.1200/JCO.2003.03.057. [DOI] [PubMed] [Google Scholar]
- 60.Giles F.J., Garcia-Manero G., Cortes J.E., Baker S.D., Miller C.B., O’Brien S.M., Thomas D.A., Andreeff M., Bivins C., Jolivet J., Kantarjian H.M. Phase II study of Troxacitabine, a novel dioxolane nucleoside analog, in patients with refractory Leukemia. J. Clin. Oncol. 2002;20:6564–6664. doi: 10.1200/JCO.2002.20.3.656. [DOI] [PubMed] [Google Scholar]
- 61.Ohrui H., Kohgo S., Kitano K., Sakata S., Kodama E., Yoshimura K., Matsuoka M., Shigeta S., Mitsuya H. Syntheses of 4′-C-ethynyl-β-D-arabino- and 4′-C-ethynyl-2′-deoxy-β-D-ribo-pento-furanosyl pyrimidines and -purines and evaluation of their anti-HIV activity. J. Med. Chem. 2000;43:4516–4525. doi: 10.1021/jm000209n. [DOI] [PubMed] [Google Scholar]
- 62.Yang G.W., Elijah P., Dutschman G.E., Grill S.P., Wang C.J., Wang J., Tanaka H., Hamasaki T., Baba M., Cheng Y.C. Impact of novel human immunodeficiency virus type 1 reverse transcriptase mutations P119S and T165A on 4-ethynylthymidine analog resistance profile. Antimicrob. Agents Chemother. 2009;53:4640–4646. doi: 10.1128/AAC.00686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang X., Hiromichi T., Baba M., Cheng Y.C. Retention of metabolites of 2,3-didehydro-3-deoxy-4-ethynylthymidine, a novel anti-human immunodeficiency virus type 1 thymidine analog, in cells, Antimicrob. Age. 2009;53:3317–3324. doi: 10.1128/AAC.00302-09. Ch. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ohrui H. 2′-Deoxy-4′-C-ethynyl-2-fluoroadenosine, a nucleoside reverse transcriptase inhibitor, is highly potent against all human immunodeficiency viruses type 1 and has low toxicity. Chem. Rec. 2006;6:133–143. doi: 10.1002/tcr.20078. [DOI] [PubMed] [Google Scholar]
- 65.K. Haraguchi, H. Shimada, K. Kimura, G. Akutsu, H. Tanaka, H. Abe, T. Hamasaki, M. Baba, E.A. Gullen, G.E. Dutschman, Y.C. Cheng, J. Balzarini,2′-Deoxy-4′-thioribonucleosides and discovery of a highly potent and less toxic NRTI, ACS Med. Chem. Lett. 2 (20119) 692-697. [DOI] [PMC free article] [PubMed]
- 66.O-Yang C., Wu H.Y., Fraser-Smith E.B., Walker K.A.M. Syntbeuia of 4′-cyanothymidine and analogs as potent inhibitors of HIV. Tetrahedron Lett. 1992;33:37–40. [Google Scholar]
- 67.Nomura M., Shuto S., Tanaka M., Sasaki T., Mori S., Shigeta S., Matsuda A. Nucleosides and nucleotides. 185. synthesis and biological activities of4′α-C-branched-chain sugar pyrimidine nucleosides. J. Med. Chem. 1999;42:2901–2908. doi: 10.1021/jm990050i. [DOI] [PubMed] [Google Scholar]
- 68.Kohgo S., Yamada K., Kitano K., Iwai Y., Sakata S., Ashida N., Hayakawa H., Nameki D., Kodama E., Matsuoka M., Mitsuya H., Ohrui H. Design, efficient synthesis, and anti-HIV activity of 4′-C-cyano- and 4′-C-ethynyl-2′-deoxy purine nucleosides. Nucleos. Nucleot.Nucl. 2004;23:671–690. doi: 10.1081/NCN-120037508. [DOI] [PubMed] [Google Scholar]
- 69.Maag H., Rydzewski R.M., McRoberta M.J., Crawford-Ruth D., Verheyden J.P.H., Prisbe E.J. Synthesis and anti-HIV activity of 4′-azido- and 4′-methoxynucleoside. J. Med. Chem. 1992;35:1440–1451. doi: 10.1021/jm00086a013. [DOI] [PubMed] [Google Scholar]
- 70.Smith D.B., Martin J.A., Klumpp K., et al. Design, synthesis, and antiviral properties of 4′-substituted ribonucleosides as inhibitors of hepatitis C virus replication: the discovery of R1479. Bioorg. Med. Chem. Lett. 2007;17:2570–2576. doi: 10.1016/j.bmcl.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 71.Klumpp K., Kalayanov G., Ma H., et al. 2′-Deoxy-4′-azido nucleoside analogs are highly potent inhibitors of hepatitis C virus replication despite the lack of 2′-α-hydroxyl groups. J. Biol. Chem. 2008;283:2167–2175. doi: 10.1074/jbc.M708929200. [DOI] [PubMed] [Google Scholar]
- 72.Eyer L., Smídkova M., Nencka R., Neca J., Kastl T., Palus M., Clercq E.D., Ruzek D. Structure-activity relationships of nucleoside analogues for inhibitionof Tick-borne Encephalitis virus. Antivir. Res. 2016;133:119–129. doi: 10.1016/j.antiviral.2016.07.018. [DOI] [PubMed] [Google Scholar]
- 73.Wang R.R., Yang Q.H., Luo R.H., Peng Y.M., Dai S.X., Zhang X.J., Chen H., Cui X.Q., Liu Y.J., Huang J.F., Chang J.B., Zheng Y.T. Azvudine, a novel nucleoside reverse transcriptase inhibitor showed good drug combination features and better inhibition on drug-resistant strains than Lamivudine in vitro. PloS One. 2014;9 doi: 10.1371/journal.pone.0105617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Driscoll J.S., Siddiqui M.A., Ford H., Jr., Kelley J.A., Roth J.S., Mitsuya H., Tanaka M., Marquez V.E. lipophilic, acid-stable,adenosine deaminase-activated anti-HIV prodrugs for central nervous system delivery. 3.6-aminoprodrugs of 2′-β-fluoro-2′,3′-dideoxyinosine. J. Med. Chem. 1996;39:1619–1625. doi: 10.1021/jm9509197. [DOI] [PubMed] [Google Scholar]
- 75.Wang T. Liaoning Normal University; Dalian: 2007. Synthesis of 5′-Deoxy-Adenosine Derivatives and Reaseach on Their Biological Activities, Master Thesis. [Google Scholar]
- 76.Herbst D.A., Reddy K.R. Sofosbuvir, a nucleotide polymerase inhibitor, for the treatment of chronic hepatitis C virus infection. Expet Opin. Invest. Drugs. 2013;22:527–536. doi: 10.1517/13543784.2013.775246. [DOI] [PubMed] [Google Scholar]
- 77.Ivanov M.A., Ludva G.S., Mukovnya A.V., Kochetkov S.N., Tunitskaya V.L., Alexandroval L.A. Synthesis and biological properties of pyrimidine 4′-fluoronucleosides and 4′-fluorouridine 5′-O-triphosphate. Russ. J. Bioorg. Chem. 2010;36:488–496. [Google Scholar]
- 78.Dumontet C., Peyrottes S., Rabeson C., Cros-Perrial E., Geant P.Y., Chaloin L., Jordheim L.P. CD73 Inhibition by purine cytotoxic nucleoside analogue-based diphosphonates. Eur. J. Med. Chem. 2018;157:1051–1055. doi: 10.1016/j.ejmech.2018.08.035. [DOI] [PubMed] [Google Scholar]
- 79.Labbéa M.O., Collinsc L., Lefebvrea C.A., Maharsyc W., Beauregardc J., Dostiea S., Prévosta M., Nemerc M., Guindon Y. Identification of a C3′-nitrile nucleoside analogue inhibitor of pancreatic cancer cell line growth, Bioorg. Med. Chem. Lett. 2020;30:126983. doi: 10.1016/j.bmcl.2020.126983. [DOI] [PubMed] [Google Scholar]
- 80.Ohno R., Tatsumi N., Hirano M., et al. Treatment of myelodysplastic syndromes with orally administered 1-β-D-arabinofuranosylcytosine-5′-stearylphosphate. Oncology. 1991;48:451–455. doi: 10.1159/000226979. [DOI] [PubMed] [Google Scholar]
- 81.Borea P.A., Varani K., Vincenzi F., Baraldi P.G., Tabrizi M.A., Merighi S., Gessi S. The A3 adenosine receptor: history and perspectives. Pharmacol. Rev. 2015;67:74–102. doi: 10.1124/pr.113.008540. [DOI] [PubMed] [Google Scholar]
- 82.Ajmera S., Bapat A.R., Stephanian E., Danenberg P.V. Synthesis and interaction with uridine phosphorylase of 5′-deoxy-4′,5-difluorouridine, a new prodrug of 5-fluorouracil. J. Med. Chem. 1998;31:1094–1098. doi: 10.1021/jm00401a008. [DOI] [PubMed] [Google Scholar]
- 83.Zhang H.W., Schinazi R.F., Chu C.K. Synthesis of neplanocin F analogues as potential antiviral agents. Bioorg. Med. Chem. 2006;14:8314–8322. doi: 10.1016/j.bmc.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 84.Sato Y., Ueyama K., Mauryama T., Richman D.D. Synthesis of the fluoromethyl derivatives of carbocyclic oxetanocin A. Nucleos. Nucleot.Nucl. 1996;15:109–119. [Google Scholar]
- 85.Neyts J., Naesens L., Ying C., Bolle L.D., Clercq E.D. Anti-herpes virus activity of (1′S, 2′R)-9-[[1′, 2′-bis(hydroxymethyl)-cycloprop-1′-yl]methyl] guanine (A-5021) in vitro and in vivo. Antivir. Res. 2001;49:115–120. doi: 10.1016/s0166-3542(00)00144-3. [DOI] [PubMed] [Google Scholar]
- 86.Baldanti F., Sarasini A., Drach J.C., Zemlicka J., Gerna G. Z-isomers of 2-hydroxymethylcyclopropylidenemethyl adenine (Synadenol) and guanine (Synguanol) are active against Ganciclovir- and Foscarnet-resistant human Cytomegalovirus UL97 mutants. Antivir. Res. 2002;56:273–278. doi: 10.1016/s0166-3542(02)00129-8. [DOI] [PubMed] [Google Scholar]
- 87.Reddy K.R., Matelich M.C., Ugarkar B.G., Gómez-Galeno J.E., DaRe J., Ollis K., Sun Z.L., Craigo W., Colby T.J., Fujitaki James M., Boyer S.H., van Poelje P.D., Erion M.D. Pradefovir: a prodrug that targets Adefovir to the liver for the treatment of hepatitis B. J. Med. Chem. 2008;51:666–676. doi: 10.1021/jm7012216. [DOI] [PubMed] [Google Scholar]
- 88.Kamiya N., Kubota A., Iwase Y., Sekiya K., Ubasawa M., Yuasa S. Antiviral activities of MCC-478, a novel and specific inhibitor of hepatitis B virus. Antimicrob. Agents Chemother. 2002;46:2872–2877. doi: 10.1128/AAC.46.9.2872-2877.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fu X.Z., Jiang S., Li C., Xin J., Yanga Y., Ji R. Design and synthesis of novel bis (l-amino acid) ester prodrugs of 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA) with improved anti-HBV activity. Bioorg. Med. Chem. Lett. 2002;17:465–470. doi: 10.1016/j.bmcl.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 90.Pertusati F., Serpi M., McGuigan C. Medicinal Chemistry of nucleoside phosphonate prodrugs for antiviral therapy. Antivir. Chem. Chemother. 2011;22:181–203. doi: 10.3851/IMP2012. [DOI] [PubMed] [Google Scholar]
- 91.de Carvalho O.V., Félix D.M., de Mendonça L.R., de Araújo C.M.C.S., de Oliveira Franca R.F., Cordeiro M.T., Júnior A.S., Pena L.J. The thiopurine nucleoside analogue 6-Methylmercaptopurine Riboside (6MMPr) effectively blocks Zika Virus replication. Int. J. Antimicrob. Agents. 2017;50:718–725. doi: 10.1016/j.ijantimicag.2017.08.016. [DOI] [PubMed] [Google Scholar]
- 92.Koudriakova T., Manouilov K.K., Shanmuganathan K., Kotra L.P., Boudinot F.D., Cretton-Scott E., Sommadossi J.P., Schinazi R.F., Chu C.K. In Vitro and in vivoevaluation of 6-azido-2′,3′-dideoxy-2′-fluoro-β-d-arabinofuranosylpurine and N6-methyl-2′,3′-dideoxy-2′-fluoro-β-d-arabinofuranosyladenine as prodrugs of the anti-HIV nucleosides 2′-F-ara-ddA and 2′-F-ara-ddI. J. Med. Chem. 1996;39:4676–4681. doi: 10.1021/jm960140c. [DOI] [PubMed] [Google Scholar]
- 93.Ford H., Jr., Siddiqui M.A., Driscoll J.S., Marquez V.E., Kelley J.A., Mitsuya H., Shirasakat T. Lipophilic, acid-stable, adenosine deaminase-activated anti-HIV prodrugs for central nervous system delivery. 2.6-halo and 6-alkoxy prodrugs of 2′-β-fluoro-2′,3′-dideoxyinosine. J. Med. Chem. 1995;38:1189–1195. doi: 10.1021/jm00007a015. [DOI] [PubMed] [Google Scholar]
- 94.Warren T.K., Wells J., Panchal1 R.G., Stuthman1 K.S., Garza1 N.L., Tongeren S.A.V., et al. Protection against Filovirus Diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature. 2014;508:402–405. doi: 10.1038/nature13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Murphya B.G., Perronc M., Murakamic E., Bauera K., Parkc Y., Eckstranda C., Liepnieksa M., Pedersenb N.C. The nucleoside analog GS-441524 strongly inhibits feline infectious peritonitis (FIP) virus in tissue culture and experimental cat infection studies. Vet. Microbiol. 2018;219:226–233. doi: 10.1016/j.vetmic.2018.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.de Wilde A.H., Snijder E.J., Kikkert M., van Hemert M.J. Host factors in coronavirus replication. Curr. Top. Microbiol. Immunol. 2018;419:1–42. doi: 10.1007/82_2017_25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shang L.Q., Wang Y.X., Qing J., Shu B., Cao L., Luo Z.Y., Gong P., Sun Y., Yin Z. An adenosine nucleoside analogue NITD008 inhibits EV71 proliferation. Antivir. Res. 2014;112:47–58. doi: 10.1016/j.antiviral.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 98.Hulpia F., Noppen S., Schols D., Andrei G., Snoeck R., Liekens S., Vervaeke P., Calenbergh S.V. Synthesis of a 3′-C-ethynyl-β-D-ribofuranose purine nucleoside library: discovery of C7-deazapurine analogs as potent antiproliferative nucleosides. Eur. J. Med. Chem. 2018;157:248–267. doi: 10.1016/j.ejmech.2018.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lin C., Sun C., Liu X., Zhou Y., Hussain M., Wan J., Li M., Li X., Jin R., Tu Z., Zhang J. Design, synthesis, and in vitro biological evaluation of novel 6-methyl-7-substituted-7-deaza purine nucleoside analogs as anti-influenza A agents. Antivir. Res. 2016;129:13–20. doi: 10.1016/j.antiviral.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 100.Winter S.S., Dunsmore K.P., Devidas M., Eisenberg N., Asselin B.L., Wood B.L., Leonard Rn M.S., Murphy J., Gastier-Foster J.M., Carroll A.J., Heerema N.A., Loh M.L., Raetz E.A., Winick N.J., Carroll W.L., Hunger S.P. Safe integration of Nelarabine into intensive chemotherapy in newly diagnosed T-cell acute lymphoblastic leukemia: children’s Oncology Group Study AALL0434. Pediatr. Blood Canc. 2015;62:1176–1183. doi: 10.1002/pbc.25470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Balakrishnan K., Ravandi F., Bantia S., Franklin A., Gandhi V. Preclinical and clinical evaluation of Forodesine in pediatric and adult B-cell acute lymphoblastic leukemia. Cl. Lymph. Myelom.Leuk. 2013;13:458–466. doi: 10.1016/j.clml.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.A Ibrahim M., Johnson H.W.B., Jeong J.W., Lewis G.L., Shi X., Noguchi R.T., Williams M., Leahy J.W., Nuss J.M., Woolfrey J., Banica M., Bentzien F., Chou Y.C., Gibson A., Heald N., Lamb P., Mattheakis L., Matthews D., Shipway A., Wu X., Zhang W.T., Zhou S., Shankar G. Discovery of a novel class of potent and orally bioavailable sphingosine l-phosphate receptor 1 antagonists. J. Med. Chem. 2012;55:1368–1381. doi: 10.1021/jm201533b. [DOI] [PubMed] [Google Scholar]
- 103.Baba M., Tanaka H., De Clercy E., Pauwels R., Balzarini J., Schols D., Nakashima H., Perno C.F., Walker R.T., Miyasaka T. Highly specific inhibition of human immunodeficiency virus type-1 by a novel 6-subustituted acyclouridine derivative. Biochem.Bioph. R. 1989;165:1375–1381. doi: 10.1016/0006-291x(89)92756-3. [DOI] [PubMed] [Google Scholar]
- 104.Tao L., Li L.J., Guo X.H., Dong L.H., Liu L.P., Wang Q., Yu X.J., Song C.J., Chang J.B. Synthesis and anti-CBV3 activity of 4′-amino acid derivative substituted pyrimidine nucleoside analogues. Bioorg. Med. Chem. Lett. 2020;30:126770. doi: 10.1016/j.bmcl.2019.126770. [DOI] [PubMed] [Google Scholar]
- 105.Ishitsuka H., Shimma N. Capecitabine preclinical studies: from discovery to translational research,Piet Herdewijn,Modified nucleosides: in biochemistry. Biotechnology and Medicine. 2008:585–600. [Google Scholar]
- 106.Ito Y., Wakita A., Takada S., Mihara M., Gotoh M., Ohyashiki K., Ohtake S., Miyawaki S., Ohnishi K., Naoe T. Phase I trial of Gemtuzumab Ozogamicin in combination with Enocitabine and Daunorubicin for elderly patients with relapsed or refractory acute myeloid leukemia: Japan adult leukemia study group (JALSG)-GML208 study. Int. J. Hematol. 2012;96:485–491. doi: 10.1007/s12185-012-1165-z. [DOI] [PubMed] [Google Scholar]
- 107.Jabbour E., Issa J.P., Garcia-Manero G., Kantarjian H. Evolution of decitabine development. Cancer. 2008;112:2341–2351. doi: 10.1002/cncr.23463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhou C.H., Wang Y. Recent researches in Triazole compounds as medicinal drugs. Curr. Med. Chem. 2012;19:239–280. doi: 10.2174/092986712803414213. [DOI] [PubMed] [Google Scholar]
- 109.Sabat N., Migianu-Griffoni E., Tudela T., Lecouvey M., Kellouche S., Carreiras F., Gallier F., Uziel J., Lubin-Germain N. Synthesis and antitumor activities investigation of a C-nucleoside analogue of Ribavirin. Eur. J. Med. Chem. 2020;188:112009. doi: 10.1016/j.ejmech.2019.112009. [DOI] [PubMed] [Google Scholar]
- 110.Fang C., Srivastava P., Lin C.C. Effect of Ribavirin, Levovirin and Viramidine on liver toxicological gene expression in rats. J. Appl. Toxicol. 2003;23:453–459. doi: 10.1002/jat.938. [DOI] [PubMed] [Google Scholar]
- 111.Li Z.C., Chen S.H., Jiang N., Cui G. Synthesis of Triazole nucleoside derivatives. Nucleos. Nucleot.Nucl. 2003;22:419–435. doi: 10.1081/NCN-120022032. [DOI] [PubMed] [Google Scholar]
- 112.Li Q.H., Li Z.C., Chen S.H., Jiang N. Studies on synthesis and antiviral activity of Ribavirin and its derivatives. Chin. J. Org. Chem. 2004;24:1432–1435. [Google Scholar]
- 113.Sidwell R.W., Bailey K.W., Wong M.-H., Barnard D.L., Smee D.F. In vitro and in vivoinfluenza virus-inhibitory effects of Viramidine. Antivir. Res. 2005;68:10–17. doi: 10.1016/j.antiviral.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 114.Chung D.H., Kumarapperuma S.C., Sun Y., Li Q., Chua Y.K., Arterburnb J.B., Parker W.B., Smithc J., Spikc K., Ramanathand H.N., Schmaljohnc C.S., Jonsson C.B. Synthesis of 1-beta-D-ribofuranosyl-3-ethynyl-[1,2,4] Triazole and its in vitro and in vivo efficacy against Hantavirus. Antivir. Res. 2008;79:19–27. doi: 10.1016/j.antiviral.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mcdowell M., Gonzales S.R., Kumarapperuma S.C., Jeselnik M., Arterburnb J.B., Hanley K.A. A novel nucleoside analog, 1-beta-D-ribofuranosyl-3-ethynyl-[1,2,4] Triazole (ETAR), exhibits efficacy against a broad range of flaviviruses in vitro. Antivir. Res. 2010;87:78–80. doi: 10.1016/j.antiviral.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kini G.D., Robins R.K., Avery T.L. Synthesis and antitumor activity of Ribavirin imidates. A new facile synthesis of Ribavirin amidine (1-beta-D-ribofuranosyl-1,2,4-triazole-3-carboxamidine hydrochloride) J. Med. Chem. 1989;32:1447–1449. doi: 10.1021/jm00127a008. [DOI] [PubMed] [Google Scholar]
- 117.Borden K.L.B., Culjkovic-Kraljacic B. Ribavirin as an anti-cancer therapy: acute myeloid leukemia and beyond? Leuk. Lymphoma. 2010;51:1805–1815. doi: 10.3109/10428194.2010.496506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kraljacic B.C., Arguello M., Amri A., Cormack G., Borden K. Inhibition of eIF4E with Ribavirin cooperates with common chemotherapies in primary acute myeloid leukemia specimens. Leukemia. 2011;25:1197–1200. doi: 10.1038/leu.2011.57. [DOI] [PubMed] [Google Scholar]
- 119.Wan J.Q., Xia Y., Liu Y., Wang M.H., Rocchi P., Yao J.H., Qu F.A., Neyts J., Iovanna J.L., Peng L. Discovery of novel arylethynyltriazole ribonucleosides with selective and effective antiviral and antiproliferative activity. J. Med. Chem. 2009;52:1144–1155. doi: 10.1021/jm800927r. [DOI] [PubMed] [Google Scholar]
- 120.Zhu R.Z., Wang M.H., Xia Y., Qu F.Q., Neytsc J., Peng L. Arylethynyltriazole acyclonucleosides inhibit hepatitis C virus replication. Bioorg. Med. Chem. Lett. 2008;18:3321–3327. doi: 10.1016/j.bmcl.2008.04.026. [DOI] [PubMed] [Google Scholar]
- 121.Xia Y., Li W., Qu F.Q., Fan Z.J., Liu X.F., Berro C., Rauzy E., Peng L. Synthesis of bitriazolyl nucleosides and unexpectedly different reactivity of azidotriazole nucleoside isomers in the huisgen reaction. Org. Biomol. Chem. 2007;5:1695–1701. doi: 10.1039/b703420b. [DOI] [PubMed] [Google Scholar]
- 122.Li W., Xia Y., Fan Z.J., Qu F.Q., Wu Q.Y., Peng L. Bitriazolyl acyclonucleosides with antiviral activity against tobacco mosaic virus. Tetrahedron Lett. 2008;49:2804–2809. [Google Scholar]
- 123.Wang M.H., Zhu R.Z., Fan Z.J., Fu Y.F., Feng L., Yao J.H., Maggiani A., Xia Y., Qu F.Q., Peng L. Bitriazolyl acyclonucleosides synthesized via huisgen reaction using internal alkynes show antiviral activity against tobacco mosaic virus. Bioorg. Med. Chem. Lett. 2011;21:354–357. doi: 10.1016/j.bmcl.2010.10.141. [DOI] [PubMed] [Google Scholar]
- 124.Wang M.H., Xia Y., Fan Y.T., Rocchi P., Qu F.Q., Iovanna J.L., Peng L. A novel arylethynyltriazole acyclonucleoside inhibits proliferation of drug-resistant pancreatic cancer cells, Bioorg. Med. Chem. Lett. 2010;20:5979–5983. doi: 10.1016/j.bmcl.2010.08.093. [DOI] [PubMed] [Google Scholar]
- 125.Xia Y., Liu Y., Wan J.Q., Wang M.H., Rocchi P., Qu F.Q., Iovanna J.L., Peng L. Novel triazole ribonucleoside down-regulates heat shock protein 27 and induces potent anticancer activity on drug-resistant pancreatic cancer. J. Med. Chem. 2009;52:6083–6096. doi: 10.1021/jm900960v. [DOI] [PubMed] [Google Scholar]
- 126.Xia Y., Liu Y., Rocchi P., Wang M.H., Fan Y.T., Qu F.Q., Iovanna J.L., Peng L. Targeting heat shock factor 1 with a triazole nucleoside analog to elicit potent anticancer activity on drug-resistant pancreatic cancer. Canc. Lett. 2012;318:145–153. doi: 10.1016/j.canlet.2011.09.043. [DOI] [PubMed] [Google Scholar]
- 127.Xia Y., Wang M.H., Beraldi E., Cong M., Zoubeidi A., Gleave M., Peng L. A novel triazole nucleoside suppresses prostate cancer cell growth by inhibiting heat shock factor 1 and androgen receptor. Anticancer Agents Med. Chem. 2015;15:1333–1340. doi: 10.2174/1871520615666150617110943. [DOI] [PubMed] [Google Scholar]
- 128.Liu Y., Xia Y., Fan Y.T., Maggiani A., Rocchi P., Qu F.A., Iovanna J.L., Peng L. N-Aryltriazole ribonucleosides with potent antiproliferative activity against drug-resistant pancreatic cancer. Bioorg. Med. Chem. Lett. 2010;20:2503–2507. doi: 10.1016/j.bmcl.2010.02.104. [DOI] [PubMed] [Google Scholar]
- 129.Fan Y.T., Xia Y., Tang J.J., Rocchi P., Qu F.Q., Iovanna J., Peng L. Ligand-mediated highly effective and selective C-N coupling for synthesizing bioactive N-aryltriazole acyclonucleosides. Org. Lett. 2010;12:5712–5715. doi: 10.1021/ol102537p. [DOI] [PubMed] [Google Scholar]
- 130.Chen M.M., Zhou Z.W., Sou Y.X., Li M.Y., Yao J.H., Peng L., Xia Y. Acyclonucleosides bearing coplanar arylethynyltriazole nucleobases: synthesis, structural analysis, and biological evaluation. New J. Chem. 2017;41:8509–8519. [Google Scholar]
- 131.Liu Y., Peng Y.M., Lu J.J., Wang J.W., Ma H.R., Song C.J., Liu B.J., Qiao Y., Yu W.Q., Wu J., Chang J.B. Design, synthesis and biological evaluation of new 1,2,3-triazolo-2′-deoxy-2′-fluoro-4′-azido nucleoside derivatives ans potent anti-HBV agents. Eur. J. Med. Chem. 2018;143:137–149. doi: 10.1016/j.ejmech.2017.11.028. [DOI] [PubMed] [Google Scholar]
- 132.Chittepu P., Sirivolu V.R., Seela F. Nucleosides and oligonucleotides containing 1,2,3-triazole residues with nucleobase tethers: synthesis via the azide-alkyne ‘Click’ reaction. Bioorg. Med. Chem. 2008;16:8427–8439. doi: 10.1016/j.bmc.2008.08.026. [DOI] [PubMed] [Google Scholar]
- 133.Yu J.L., Wu Q.P., Zhang Q.S., Xi X.D., Liu N.N., Li Y.Z., Liu Y.H., Yin H.Q. Synthesis and antitumor activity of novel 2′,3′-diethanethio-2′,3′,5′-trideoxy-5′-triazolonucleoside analogues. Eur. J. Med. Chem. 2010;45:3219–3222. doi: 10.1016/j.ejmech.2010.03.038. [DOI] [PubMed] [Google Scholar]
- 134.Guo M.L. Zhengzhou University; Zhengzhou: 2016. Design, Synthesis and Antitumor Activity Research of Novel Tricyclic Higher Carbon Sugar Nucleoside Analogues, Master Thesis. [Google Scholar]
- 135.Vernekar S.K.V., Qiu L., Zacharias J., Geraghty R.J., Wang Z.Q. Synthesis and antiviral evaluation of 4′-(1,2,3-triazol-1-yl) thymidines. Med. Chem. Comm. 2014;5:603–608. [Google Scholar]
- 136.Wu J., Yu W.Q., Fu L L.X., He W., Wang Y., Chai B.S., Song C.J., Chang J.B. Design, synthesis, and biological evaluation of new 2′-deoxy-2′-fluoro-4′-triazole cytidine nucleosides as potent antiviral agents. Eur. J. Med. Chem. 2013;63:739–745. doi: 10.1016/j.ejmech.2013.02.042. [DOI] [PubMed] [Google Scholar]
- 137.Elayadi H., Smietana M., Pannecouque C., Leyssen P., Neyts J., Vasseur J.J., Lazrek H.B. Straightforward synthesis of triazoloacyclonucleotide phosphonates as potential HCV inhibitors. Bioorg. Med. Chem. Lett. 2010;20:7365–7368. doi: 10.1016/j.bmcl.2010.10.046. [DOI] [PubMed] [Google Scholar]
- 138.Gediya L.K., Njar V.C. Promise and challenges in drug discovery and development of hybrid anticancer drugs, Exper.Opin. Drug Discov. 2009;4:1099–1111. doi: 10.1517/17460440903341705. [DOI] [PubMed] [Google Scholar]
- 139.Zlatev I., Dutartre H., Barvik I., Neyts J., Canard B., Vasseur J.J., Alvarez K., Morvan F. Phosphoramidate dinucleosides as hepatitis C virus polymerase inhibitors. J. Med. Chem. 2008;51:5745–5757. doi: 10.1021/jm800617c. [DOI] [PubMed] [Google Scholar]
- 140.Priet S., Zlatev I., Barvik I., Jr., Geerts K., Leyssen P., Neyts J., Dutartre H., Canard B., Vasseur J.J., Morvan F., Alvarez K. 3′-Deoxy phosphoramidate dinucleosides as improved inhibitors of hepatitis C virus subgenomic replicon and NS5B polymerase activity. J. Med. Chem. 2010;53:6608–6617. doi: 10.1021/jm100102v. [DOI] [PubMed] [Google Scholar]
- 141.Petersen L., Jorgensen P.T., Nielsen C., Hansen T.H., Nielsen J., Pedersen E.B. Synthesis and evaluation of double-prodrugs against HIV conjugation of d4T with 6-benzyl-l-(ethoxymethyl)-5-isopropyluracil (mkc-442, emivirine)-type reverse transcriptase inhibitors via the SATE prodrug approach. J. Med. Chem. 2005;48:1211–1220. doi: 10.1021/jm040845b. [DOI] [PubMed] [Google Scholar]
- 142.Pontikis R., Dolle V., Guillaumel J., Dechaux E., Note R., Nguyen C.H., Legraverend M., Bisagni E., Aubertin A.M., Grierson D.S., Monneret C. Synthesis and evaluation of “AZT-HEPT”, “AZT-Pyridinone”, and “ddC-HEPT” conjugates as inhibitors of HIV reverse transcriptase. J. Med. Chem. 2000;43:1927–1939. doi: 10.1021/jm991125l. [DOI] [PubMed] [Google Scholar]
- 143.Yao K., Hoset C., Rashti F., Schott T.C., Jacobson S. Effect of (r)-9-[4-hydroxy-2-(hydroxymethyl)butyl]guanine (H2G) and AZT–lipid–PFA on human herpesvirus-6B infected cells. J. Clin. Virol. 2009;46:10–14. doi: 10.1016/j.jcv.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wei Q., Zhang D.J., Yao A., Mai L.Y., Zhang Z.W., Zhou Q.B. Design, Synthesis, and in vitro and in vivobiological studies of a 3′-deoxythymidine conjugate that potentially kills cancer cells selectively. PloS One. 2012;7 doi: 10.1371/journal.pone.0052199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wei Q., Liu H.J., B Zhou Q., Zhang D.J., Zhang Z.W., Zhou Q.B. Anticancer activity of a thymidine auinoxaline conjugate is modulated by cytosolic thymidine pathways. BMC Canc. 2015;15:159–169. doi: 10.1186/s12885-015-1149-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhang D.J., Liu H.M., Wei Q., Zhou Q.B. Structure-activity relationship study of anticancer thymidine-auinoxaline conjugates under the low radiance of long wavelength ultraviolet light for photodynamic therapy. Eur. J. Med. Chem. 2016;107:180–191. doi: 10.1016/j.ejmech.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 147.Liu H.M. Huazhong University of Science and Technology; Wuhan: 2016. Investigation of the Liver Cancer Chemotherapeatic Agent dT-QX Analogs and Their Activity under UVA Irradiation, Master Thesis. [Google Scholar]
- 148.Zheng Y.C., Duan Y.C., Ma J.L., Xu R.M., Zi X.L., Lv W.L., Wang M.M., Ye X.W., Z S., Mobley D., Zhu Y.Y., Wang J.W., Li J.F., Wang Z.R., Zhao W., Liu H.M. Triazole-dithiocarbamate based selective lysine specific demethylase 1 (LSD1) inactivators inhibit gastric cancer cell growth, invasion, and migration. J. Med. Chem. 2013;56:8543–8560. doi: 10.1021/jm401002r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ma L.Y., Zheng Y.C., Wang S.Q., Wang B., Wang Z.R., Pang L.P., Zhang M., Wang J.W., Ding L., Li J., Wang C., Hu B., Liu Y., Zhang X.D., Wang J.J., Wang Z.J., Zhao W., Liu H.M. Design, synthesis and structure-activity relationship of novel LSD1 inhibitors based on pyrimidine-thiorea hybrids as potent, orally active antitumor agents. J. Med. Chem. 2015;58:1705–1716. doi: 10.1021/acs.jmedchem.5b00037. [DOI] [PubMed] [Google Scholar]
- 150.Wang S., Zhao L.J., Zheng Y.C., Shen D.D., Miao E.F., Qiao X.P., Zhao L.J., Liu Y., Huang R.L., Yu B., Liu H.M. Design, synthesis and biological evaluation of [1,2,4] triazolo [1,5-a]pyrimidines as potent lysine specific demethylase 1 (LSD1/KDM1A) inhibitors. Eur. J. Med. Chem. 2017;125:940–951. doi: 10.1016/j.ejmech.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 151.Li Z.H., Yang D.X., Geng P.F., Zhang J., Wei H.M., Hu B., Guo Q., Zhang X.H., Guo W.G., Zhao B., Yu B., Ma L.Y., Liu H.M. Design, synthesis and biological evaluation of [1,2,3] triazolo [4,5-d] pyrimidine derivatives possessing a hydrazone moiety as antiproliferative agents. Eur. J. Med. Chem. 2016;124:967–980. doi: 10.1016/j.ejmech.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 152.Li Z.H., Liu X.Q., Zhao T.Q., Geng P.F., Guo W.G., Yu B., Liu H.M. Design, synthesis and preliminary biological evaluation of new [1,2,3] triazolo [4,5-d] pyrimidine/thiourea hybrids as antiproliferative agents. Eur. J. Med. Chem. 2017;139:741–749. doi: 10.1016/j.ejmech.2017.08.042. [DOI] [PubMed] [Google Scholar]
- 153.Li Z.H., Liu X.Q., Geng P.F., Zhang J., Ma J.L., Wang B., Zhao T.Q., Zhao B., Zhang X.H., Yu B., Liu H.M. Design, synthesis and antiproliferative activity of thiazolo [5,4-d] pyrimidine derivatives through the atom replacement strategy. Eur. J. Med. Chem. 2017;138:1034–1041. doi: 10.1016/j.ejmech.2017.07.039. [DOI] [PubMed] [Google Scholar]
- 154.Li Z.H., Zhang J., Liu X.Q., Geng P.F., Ma J.L., Wang B., Zhao T.Q., Zhao B., Wei H.M., Wang C., Fu D.J., Yu B., Liu H.M. Identification of thiazolo [5,4-d] pyrimidine derivatives as potent antiproliferative agents through the drug repurposing strategy. Eur. J. Med. Chem. 2017;135:204–212. doi: 10.1016/j.ejmech.2017.04.056. [DOI] [PubMed] [Google Scholar]
- 155.Jurgen W., Sarah S., Königsrainer I., Zieker D., Königsrainer A., Schott H. Cytostatic activity of the duplex drug linking 2′-deoxy-5-fluorouridine (5FdU) with 3′-C-ethynylcytidine (ECyd) against gastric adenocarcinoma cell lines. Invest. N. Drugs. 2011;29:1294–1302. doi: 10.1007/s10637-010-9483-6. [DOI] [PubMed] [Google Scholar]
- 156.Carmen E., Alexander D., Ellerkamp V., Fuchs J., Schott S., Armeanu-Ebinger S. Effect of duplex drugs linking 2′-deoxy-5-fluorouridine (5-FdU) with 3′-C-ethynylcytidine (ECyd) on hepatoblastoma cell lines. Pediatr. Surg. Int. 2013;29:121–127. doi: 10.1007/s00383-012-3192-5. [DOI] [PubMed] [Google Scholar]
- 157.Saran S., Ansgar B. Induction of apoptosis in cervical cancer cells by the duplex drug 5-FdU–ECyd, coupling 2′-deoxy-5-fluorouridine and 3′-C-ethinylcytidine. Gynecol. Oncol. 2014;135:342–348. doi: 10.1016/j.ygyno.2014.08.034. [DOI] [PubMed] [Google Scholar]
- 158.Bae S., Lakshman M.K. A novel polymer supported approach to nucleoside modification. J. Org. Chem. 2008;73:3707–3713. doi: 10.1021/jo702558n. [DOI] [PubMed] [Google Scholar]
- 159.Mezzina E., Mariani P., Itri R., Masiero S., Pieraccini S., Spada G.P., Spinozzi F., Davis J.T., Gottarelli G. The self-assembly of a lipophilic guanosine nucleoside into polymeric columnar aggregates: the nucleoside strucutre contains sufficient information to drive the process towards a strikingly regular polymer. Chem. Eur J. 2001;7:388–395. doi: 10.1002/1521-3765(20010119)7:2<388::aid-chem388>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 160.Jin Y.G., Xin R., Ai P., Chen D.W. Self-assembled drug delivery systems: 2. Cholesteryl derivatives of antiviral nucleoside analogues: synthesis, properties and the vesicle formation. Chem. Int. J. Pharm. 2008;350:330–337. doi: 10.1016/j.ijpharm.2007.08.037. [DOI] [PubMed] [Google Scholar]
- 161.Jin Y.G., Xing L., Tian Y., Li M., Gao C.S., Du L.N., Dong J.X., Chen H.X. Self-assembled drug delivery systems. Part 4.In vitro/in vivo studies of the self-assemblies of cholesteryl-phosphonyl Zidovudine. Int. J. Pharm. 2009;381:40–48. doi: 10.1016/j.ijpharm.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 162.Pasut G., Canal F., Via L.D., Arpicco S., Veronese F.M., Schiavon O. Antitumoral activity of PEG–Gemcitabine prodrugs targeted by folic acid. J. Contr. Release. 2008;127:239–248. doi: 10.1016/j.jconrel.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 163.Li W.J., Wu J.D., Zhan P., Chang Y., Pannecouque C., Clercqb E.D., Liu X.Y. Synthesis, drug release and anti-HIV activity of a series of PEGylated Zidovudine conjugates. Int. J. Biol. Macromol. 2012;50:974–980. doi: 10.1016/j.ijbiomac.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 164.Valiaeva N., Prichard M.N., Buller R.M., Beadle J.R., Hartline C.B., Keith K.A., Schriewer J., Trahana J., Hostetler K.Y. Antiviral evaluation of octadecyloxyethyl esters of (S)-3-hydroxy-2-(phosphonomethoxy)propyl nucleosides against herpesviruses and orthopoxviruses. Antivir. Res. 2009;84:254–259. doi: 10.1016/j.antiviral.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wyles D.L., Kaihara K.A., Korba B.E., Schooley R.T., Beadle J.R., Hostetler K.Y. The octadecyloxyethyl ester of (S)-9-[3-hydroxy-2-(phosphonomethoxy) propyl]adenine is a potent and selective inhibitor of hepatitis C virus replication in genotype 1A, 1B, and 2A replicons. Antimicrob. Agents Chemother. 2009;53:2660–2662. doi: 10.1128/AAC.01546-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lanie E.R., Painter W., Tippin T.K., Anderson M., Lampert B.M., Mommeja-Marin H., Trost L.C. CMX001 (Hexadecyloxypropyl Cidofovir) antiviral activity against adenovirus in patients correlates with drug levels and viral sensitivity. Antivir. Res. 2011;90:A27–A28. [Google Scholar]
- 167.Painter G.R., Almond M.R., Trost L.C., Lampert B.M., Neyts J., Clercq E.D., Korba B.E., Aldern K.A., Beadle J.R., Hostetler K.Y. Evaluation of hexadecyloxypropyl-9-R-[2-(Phosphonomethoxy)propyl]- adenine, CMX157, as a potential treatment for human immunodeficiency virus type 1 and hepatitis B virus infections. Antimicrob. Agents Chemother. 2007;51:3505–3509. doi: 10.1128/AAC.00460-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jenh A.M., Pham P. Tenofovir disoproxil fumarate in the treatment of chronic hepatitis B. Expert Rev. Anti Infect.Ther. 2010;8:1079–1092. doi: 10.1586/eri.10.91. [DOI] [PubMed] [Google Scholar]
- 169.Li S.Y., Li Z., Chen S.Z., Zhang T., Wang S.Q., Li Y., Tao P.Z., Li Z.R. Anti-hepatitis B virus in vivo and in vitro of a novel nucleoside derivative Octadecyloxyethyl Tenofovir Disoproxil. Chin. Med. Biotechnol. 2013;8:173–177. [Google Scholar]
- 170.Maloisel F., Guerci A., Guyotat D., Ifrah N., Michallet M., Reiffers J., Tertain G., Blanc M., Bauduer F., Briere J., et al. Results of a phase II trial of a combination of oral Cytarabine Ocfosfate (YNK01) and interferon α-2b for the treatment of chronic myelogenous leukemia patients in chronic phase. Leukemia. 2002;16:573–580. doi: 10.1038/sj.leu.2402433. [DOI] [PubMed] [Google Scholar]
- 171.DiNardo C.D., O′Brien S., Gandhi V.V., Ravandi F. Elacytarabine (CP-4055) in the treatment of acute myeloid leukemia. Future Oncol. 2013;9:1073–1082. doi: 10.2217/fon.13.130. [DOI] [PubMed] [Google Scholar]
- 172.Dueland S., Aamdal S., Lind M.J., Thomas H., Sandvold M.L., Gaullier J.M., Rasch W. Intravenous administration of CP-4055 (ELACYTTM) in patients with solid tumours. A phase I study. Acta Oncol. 2009;48:137–145. doi: 10.1080/02841860802183620. [DOI] [PubMed] [Google Scholar]
- 173.Adema A.D., Smid K., Losekoot N., Honeywell R.J., Verheul H.M., Myhren F., Sandvold M.L., Peters G.J. Metabolism and accumulation of the lipophilic deoxynucleoside analogs Elacytarabine and CP-4126. Invest. N. Drugs. 2012;30:1908–1916. doi: 10.1007/s10637-011-9756-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Liu J., Zhao D.J., He W.X., Zhang H.Y., Li Z.H., Luan Y.X. Nanoassemblies from amphiphilic Cytarabine prodrug for leukemia targeted therapy. J. Colloid Interface Sci. 2017;487:239–249. doi: 10.1016/j.jcis.2016.10.041. [DOI] [PubMed] [Google Scholar]
- 175.Liu J., Liu J., Zhao D.J., Ma N.X., Luan Y.X. Highly enhanced leukemia therapy and oral bioavailability from a novel amphiphilic prodrug of Cytarabine. RSC Adv. 2016;6:35991–35999. [Google Scholar]
- 176.Liu J., Ma N.X., Zhao D.J., Li Z.H., Luan Y.X. Spiral assembly of amphiphilic cytarabine prodrug assisted by probe sonication: enhanced therapy index for leukemia. Colloids Surf., B. 2015;136:918–927. doi: 10.1016/j.colsurfb.2015.10.034. [DOI] [PubMed] [Google Scholar]
- 177.Liu R.L., Zhang J., Zhang D., Wang K.M., Luan Y.X. Self-assembling nanoparticles based on Cytarabine prodrug for enhanced leukemia treatment. J. Mol. Liq. 2018;251:178–184. [Google Scholar]
- 178.Liu R.L., Jiang Y., Hu X., Wu J.L., Jiang W., Jin G.X., Luan Y.X. A preclinical evaluation of Cytarabine prodrug nanofibers assembled from Cytarabine-Lauric Acid conjugate toward solid tumors. Int. J. Pharm. 2018;552:111–118. doi: 10.1016/j.ijpharm.2018.09.043. [DOI] [PubMed] [Google Scholar]
- 179.Liu J., Jiang Y., et al. Cui Y.T., Xu C.S., Ji X.Q., Luan Y.X. Cytarabine-AOT catanionic vesicle-loaded biodegradable thermosensitive hydrogel as an efficient Cytarabine delivery system. Int. J. Pharm. 2014;473:560–571. doi: 10.1016/j.ijpharm.2014.07.032. [DOI] [PubMed] [Google Scholar]
- 180.Li F.F., Liu J., Shi J.X., Luan Y.X. Synthesis and biological evaluation of a fatty acyl di-Cytarabine prodrug. RSC Adv. 2015;5:25382–25388. [Google Scholar]
- 181.Ryu E.K., Ross R.J., Matsushita T., MacCoss M., Hong C.I., West C.R. Phosphopipid-nucleoside conjugates.3. Syntheses and preliminary biological evaluation of 1-β-D-arabinofuranosyl-cytosine 5′-monophosphate-L-1, 2-dipalmitin and selected 1-β-D-arabino-furanosylcytosine 5′-diphosphate-L-1, 2-di-acylglycerols. J. Med. Chem. 1982;25:1322–1329. doi: 10.1021/jm00353a010. [DOI] [PubMed] [Google Scholar]
- 182.Hong C.I., Kirisits A.J., Nechaev A., Buchheit D.J., West C.R. Nucleoside conjugats. 11. Synthesis and antitumor activity of 1-β-D-arabinofuranosylcytosine and Cytidine conjugates of thioether lipids. J. Med. Chem. 1990;33:1380–1386. doi: 10.1021/jm00167a016. [DOI] [PubMed] [Google Scholar]
- 183.Mavromoustakos T., Calogeropoulou T., Koufaki M., Kolocouris A., Daliani I., Demetzos C., Meng Z.X., Makriyannis A., Balzarini J., Clercq E.D. Ether phospholipid-AZT conjugates possessing anti-HIV and antitumor cell activity. Synthesis, conformational analysis, and study of their thermal effects on membrane bilayers. J. Med. Chem. 2001;44:1702–1709. doi: 10.1021/jm001121c. [DOI] [PubMed] [Google Scholar]
- 184.Xu X.H., Chen R.Y. Design, synthesis and anti-cancer activity of lecithin analogs containing Tegafur. Acta Pharm. Sin. 2002;37:858–862. [Google Scholar]
- 185.Zhang Q.L., Zeng J.C., Xu X.H., Zhou B., Lu R.L., Chen X., Li Y.J. Synthesis and biological activity of Tegafur thiophospholipid conjugates containing aromatic selenium group on glycerin skeleton. Chem. J. Chin. Univ. 2005;26:2046–2051. [Google Scholar]
- 186.Zang Z.L., Liu S.Q., Chen X., Li Y.J., Zhou B., Xu X.H. Synthesis and anti-cancer activity of selenium lecithins of Tegafur. Acta Pharm. Sin. 2006;41:1184–1187. [PubMed] [Google Scholar]