

Abstract
BglX is a heretofore uncharacterized periplasmic glycoside hydrolase (GH) of the human pathogen Pseudomonas aeruginosa. X-ray analysis identifies it as a protein homodimer. The two active sites of the homodimer comprise catalytic residues provided by each monomer. This arrangement is seen in <2% of the hydrolases of known structure. In vitro substrate profiling shows BglX is a catalyst for β-(1→2) and β-(1→3) saccharide hydrolysis. Saccharides with β-(1→4) or β-(1→6) bonds, and the β-(1→4) muropeptides from the cell-wall peptidoglycan, are not substrates. Additional structural insights from X-ray analysis (including structures of a mutant enzyme-derived Michaelis complex, two transition-state mimetics, and two enzyme–product complexes) enabled the comprehensive description of BglX catalysis. The half-chair (4H3) conformation of the transition-state oxocarbenium species, the approach of the hydrolytic water molecule to the oxocarbenium species, and the stepwise release of the two reaction products were also visualized. The substrate pattern for BglX aligns with the [β-(1→2)-Glc]x and [β-(1→3)-Glc]x periplasmic osmoregulated periplasmic glucans, and possibly with the Psl exopolysaccharides, of P. aeruginosa. Both polysaccharides are implicated in biofilm formation. Accordingly, we show that inactivation of the bglX gene of P. aeruginosa PAO1 attenuates biofilm formation.
Graphical Abstract
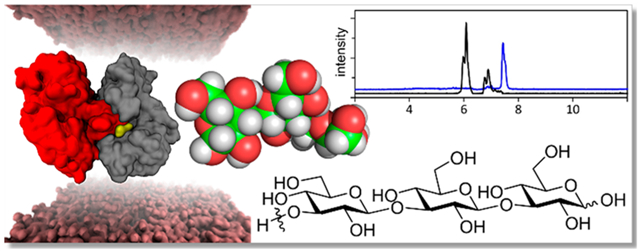
The periplasm is the structurally complex matrix between the inner cytoplasmic membrane and the outer membrane of Gram-negative bacteria. The physiological importance of the periplasm in regulating solute ingress and egress, and toward the creation of the cell wall that is found between these membranes, belies its small size (perhaps only 8–16% by volume, compared to the cytoplasm).1,2 The periplasm is the locus for four different glycan pathways. In addition to the cell-wall peptidoglycan polymer, the glycan components of the secreted extracellular polysaccharides and the lipopolysaccharides of the outer leaflet of the outer membrane transit through the periplasm. Fourth, the periplasm contains glucose-derived polymers that are associated with the essential function of osmoregulation.3 These osmoregulation saccharides play a role in biofilm formation and contribute to the inherent antibiotic resistance of the Pseudomonas aeruginosa biofilm.4 Accordingly, the presence in the periplasm of enzymes with glycoside hydrolase (GH) activity is hardly surprising. We previously investigated the remarkable conformational dynamics of the cytoplasmic GH family 3 (GH3) enzyme, NagZ.5 NagZ recycles the critical glycan component of the periplasmic peptidoglycan (Figure 1). However, the loss of NagZ function by gene ablation in Pseudomonas fluorescens significantly impairs extracellular polysaccharide secretion.6 A possible explanation, as observed in Neisseria gonorrheae, is a moonlighting activity of NagZ as a secreted enzyme for the purpose of degradation of the extracellular polysaccharide of its biofilm.7 NagZ recognition of its peptidoglycan-derived substrate and its catalytic mechanism involve the dramatic conformational mobility of the loops surrounding the active site.5 Our attention was drawn to a second GH3 enzyme, BglX, encoded by the P. aeruginosa genome. Although BglX is broadly similar in structure and sequence to NagZ, the encoding of a leader sequence in its gene indicates that its cellular location is the periplasm. Here, we provide the first structural, mechanistic, and functional understanding of BglX using mass spectrometry, substrate screening, X-ray crystallography, site-directed mutagenesis, computational analyses, and a microbiological assay.
Figure 1.

Periplasm of Gram-negative bacteria contains abundant carbohydrate polymers. BglX acts catalytically on β-glucose saccharides linked by β-(1→2)- and β-(1→3)-glycosidic bonds. Cell-wall peptidoglycan is recycled by lytic transglycosylases (LTs), and their product GlcNAc-anhMurNAc is transported to the cytoplasm, where it is processed by NagZ to release GlcNAc and anhMurNAc.5
RESULTS AND DISCUSSION
The bglX gene (PA1726) from P. aeruginosa PAO1 (amino acid residues 21—774; lacking the N-terminal periplasmic signal peptide) was cloned (Table S1). The expressed BglX protein was purified to homogeneity (Supporting Information). Our first objective was characterization of the carbohydrate structure recognized by BglX (Figure 2 and Figures S1-S3 and Table S2). We examined first the substrates for NagZ, the distinctive cell-wall-derived muropeptides. Seven synthetic peptidoglycans containing the GlcNAc-β-(1→4)-MurNAc or GlcNAc-β-(1→4)-anhMurNAc (with or without peptide) glycosidic links, as well as chitin [a polymer of GlcNAc with a β-(1→4) linkage], were tested. None was turned over by BglX. These results argue that BglX was not a periplasmic equivalent of the cytoplasmic glucosaminidase NagZ.8 We examined seven p-nitrophenolate monosaccharides having varied pyranoside [Glc, GlcNAc, GlcA (glucuronic acid), Gal, and Man] structure and anomeric stereochemistry (α- or β-glycosides). Five—Glc-α-PNP, GlcNAc-β-PNP, 4-nitrophenyl β-D-glucuronide (GlcA-β-PNP), Man-α-PNP, and Man-β-PNP—were not substrates. Two, Glc-β-PNP and Gal-β-PNP, were substrates (Figure S1). We evaluated diverse disaccharides. α,β-Trehalose [α,β-(1→1)-glycosidic bond], cellobiose [β-(1→4)-], and gentiobiose [β-(1→6)-] were not substrates. Sophorose and laminaribiose, with β-(1→2)- and β-(1→3)-glycosidic bonds, respectively, were substrates. Laminaritriose [LMT, a trisaccharide with β-(1→3)-glycosidic-linked glucose] was a substrate. Laminarin, a soluble polymer containing [β-(1→3)-Glc]x with β-(1→6) branches, was used to test the ability of BglX to process a more complex structure. The possible products of BglX turnover of laminarin would be Glc from its [β-(1→3)-Glc] region and gentiobiose from its β-(1→6)-branched region. Indeed, glucose and Glc-Glc were observed (Figure 2). Alginate, a polymer comprised of β-(1→4)-D-mannuronate and α-(1→4)-L-guluronate, was evaluated, as it is a biofilm component of P. aeruginosa. Neither alginate, nor its disaccharide and trisaccharide components [β-(1→4)-D-mannuronate and α-(1→4)-L-guluronate], was a substrate. We asked whether BglX had phosphorylase activity, as is seen in other GH3 family enzymes.9 BglX was incubated with sophorose either with or without sodium phosphate with LC/MS evaluation of the reaction products (Supporting Information). The presence of phosphate neither enhanced BglX activity nor gave glucose 1-phosphate as a product. BglX would appear to lack phosphorylase activity under these conditions, similar to the cases of other reported GH3 enzyme.10
Figure 2.

(A) Structures of a selected set of saccharides (boxed) that are substrates of BglX. Other saccharide structures (unboxed) and a transition-state mimetic (1-deoxynojirimycin) structure are also shown (see also the Supporting Information). (B) LC/MS extracted-ion chromatogram traces of the BglX reaction with laminaritriose (blue), laminaribiose (red), and (C) the ladder of turned over saccharides from polymeric laminarin. The number (1–26) given on top of each peak in panel C is the number of glucoses of the product polysaccharide. The inset shows the increased level of formation of Glc and Glc-Glc at higher BglX concentrations expanded for the first 5 min of elution.
The β-(1→2)- and β-(1→3)-glycosidic pattern for substrate recognition presumptively matches BglX with the [β-(1→2)-Glc]x and [β-(1→3)-Glc]x structures of the P. aeruginosa osmoregulated periplasmic glucans (OPG) as possible substrates.11,12 OPG influence biofilm formation in P. aeruginosa PAO1.4,11,12 Intriguingly, the bglX gene (PA1726) is adjacent to the mucR gene (PA1727) in the P. aeruginosa PAO1 genome. MucR is involved in c-di-GMP-dependent P. aeruginosa biofilm formation.13,14 This proximity suggested a potential role for BglX in biofilm formation. This premise was tested. Comparison of the bglX::Tn (transposon-insertion mutant of bglX, strain PW4042 from the Seattle P. aeruginosa PAO1 transposon mutant library) strain to wild-type (wt) PAO1 using the crystal violet biofilm assay15 showed attenuation of biofilm formation (A595 average of 20 replicates each, 0.8 ± 0.1 for bglX::Tn and 1.7 ± 0.2 for wt) after incubation for 24 h (Figure S4). As a complementation assay, the bglX::Tn strain harboring the pBglX plasmid with the full-length bglX gene restored the level of biofilm formation to the wild-type level (A595 average of 20 replicates of bglX::Tn/pBglX, 1.8 ± 0.3) (Figure S4).
We determined the wild-type apo X-ray structure of BglX [1.8 Å resolution (Table S3)]. BglX crystallized as a homodimer (Figure 3A). Each monomer has three domains: an (α/β)8TIM barrel domain, a domain with an α/β sandwich, and a fibronectin type III-like domain. The two active sites are formed by the convergence of nine loops of varying length, distributed throughout the sequence of each of the monomers [L1–L9 (Figure S5 and Table S4)]. An extended L9 loop (F585–E649) of the second monomer (M-II) inserts and contributes to the active site of the other monomer [M-I (Figure 3)]. The dimer interface, accounting for 19% of the entire molecular surface of each monomer, is formed predominantly by the folding of seven loops (L2–L5 and L7–L9) that also contribute to the active site (Figure S5).
Figure 3.

(A) Crystal structure of the wild-type apo homodimer structure of BglX (M-I monomer colored gray and M-II monomer colored red). The dimer duplicates a second active site on the opposite face. (B) Close-up stereoview of the active site with the saccharide-binding subsites labeled from position −1 to +2.
We evaluated the dimer using molecular dynamics (MD) simulation. The dimer was immersed in a box of water molecules and energy-minimized, and its dynamics examined for 500 ns with AMBER1616 by a methodology reported previously (Supporting Information).5,17 The embrace of the L9 loop completes the active site, while the L7 loop acts as a lid atop the active site (Movie S1). The MD trajectories showed stabilization of the dimer by five salt bridges and several hydrogen bonds between the monomers (Figure S6). These contacts suggested dimer stability also in solution. Nondenaturing mass spectrometry of wild-type BglX (as described previously)18 verified the dimer structure (Figure S7). The Protein Data Bank was probed for homodimeric structural homologues of BglX (Supporting Information). The phylogenetic tree generated from the sequence alignment clustered a similar homodimeric active site to a single group with merely four enzymes in addition to BglX (Figure S8 and Table S5). These four homodimeric enzymes are Lin1840 from Listeria innocua (PDB entry 4ZO6),19 BT_3567 from Bacteroides thetaiotaomicron (PDB entry 5XXL),20 a homologue enzyme from Bacteroides intestinalis (PDB entry 5TF0), and JMB19063 (PDB entry 3U48) isolated from compost using metagenomic analysis.21 Similar to BglX, the Lin1840 and BT_3567 enzymes from B. thetaiotaomicron hydrolyze glucose polymers with β-(1→2)- and β-(1→3)-glycosidic bond specificity.20 Notwithstanding the overall structural similarity with BglX, JMB19063 is different. It hydrolyzes glycosides with β-(1→4) linkages.
We sought to understand the catalytic cycle of BglX. As a GH3 family enzyme, BglX was anticipated to proceed via a two-step, double-displacement mechanism with participation of a covalent intermediate derived from an Asp as the catalytic nucleophile.5,22,23 Structure–sequence comparison to NagZ (Figure S5) indicated D286 as the candidate. We cloned the D286N variant. This variant was inactive, which strengthened the assertion (Table S6). The X-ray structure of the D286N variant in complex with the substrate LMT (2.15 Å resolution) showed three substrate recognition subsites [−1, +1, and +2 (Figure 4A)]. Substrate recognition at the −1 subsite, the buried end of the active site, demonstrated a chair (4C1) conformation for glucose involving six hydrogen bonds with four amino acids (D110, R168, K207, and H208). This network anchors the −1 saccharide throughout the conformational itinerary for catalysis (discussed below). The L9 loop of M-II gives roles in substrate recognition for residues Y626 and R616 at the +1/+2 subsites. R616 (M-II) hydrogen bonds with the 2-hydroxyl of the +1 saccharide, and residue Y626 interacts with the saccharide at the +1 subsite and partially with the +2 subsite. In our BglX-D286N:LMT complex, E517 (the complementary general acid/base) is positioned for donation of a proton to the glycosidic oxygen (O3), a requisite for glycosidic bond cleavage (Figure 4A). The native wild-type enzyme–substrate complex (Michaelis complex) was modeled computationally by changing the crystallographic asparagine at position 286 to the aspartate of the wild type. This energy-minimized enzyme was evaluated by a 500 ns MD simulation. A carboxylate oxygen of the catalytic nucleophile D286 was poised suitably for nucleophilic addition to the C1 atom of the glucose at the −1 subsite. During the MD simulation, a D286…C1 distance of ~3.5 Å was maintained, while the E517…O3 distance showed flexibility (Movie S2). The saccharide at the +1 subsite remained in contact with aromatic residues Y626 and H287 throughout the simulation with energetically favorable CH–π interactions.
Figure 4.

Sequential steps in the catalysis of BglX delineated by X-ray crystallography, coupled with computational modeling based on the X-ray structures of wild-type and active-site variants. Each box depicts a discrete step in the catalytic cycle, with the bottom panel showing the threedimensional structure. The wild-type apo X-ray structure is shown in Figure 3. (A) X-ray structure of the D286N variant in complex with the substrate laminaritriose (BglX-D286N:LMT) that represents the Michaelis complex. (B) Proposed first high-energy species and the corresponding X-ray complex of BglX with the transition-state mimic 1-deoxynojirimycin (green carbon-capped sticks) (BglX:NOJ). Crystallographic waters are represented as red spheres. (C) Computational model of the covalent glycosyl species. (D) Snapshot from MD simulation of the model in panel C showing a water molecule (sphere representation) between E517 and C1 of the glycosyl species. (E) X-ray structure of two products bound to the active site (BglX-D286N:BGC:BGC). (F) X-ray structure of β-D-glucose at the −1 subsite (BglX-D286N:BGC) that represents the state after the departure of the first product. The completion of catalysis by departure of the β-D-glucose from the −1 subsite leads to the apo state (Figure 3).
We performed reaction profile energetics using quantum mechanics/molecular mechanics (QM/MM) with semiempirical PM6-D in AMBER16 (Supporting Information), based on the X-ray structure, to investigate the transition species (TS) encountered in the catalytic mechanism. The side chains of E517, D286, and the substrate/product atoms were included in the QM region. The rest of the solvated system was treated with MM. In the energy-minimized Michaelis complex, the substrate saccharides at all three subsites (−1, +1, and +2) are in 4C1 conformations as per Cremer–Pople parameters.24 The glycosidic oxygen atom (O3) is 1.85 Å from the proton of E517. The angle of the proton with the oxygen atom of E517 and glycosidic O3 is 156.9° and appropriately placed for proton transfer. The distance between the oxygen atom of nucleophile D286 and the anomeric C1 atom is 3.09 Å. The oxygen atoms of D286 and E517 are 5.9 Å apart, which is characteristic of “retaining” (double-inversion) GH mechanism.25,26 Previous studies of the catalytic mechanism of the GH3 family show an oxocarbenium-ion transition species (Figure 4B).22,23,27 Dynamic trajectories were sampled as the proton from E517 was gradually steered toward the glycosidic oxygen atom (O3) and the simultaneous breaking of the glycosidic bond. The formation of a covalent bond between C1 of glucose at the −1 site and the oxygen atom of D286 results in the covalent–enzyme species and the first product (laminaribiose) at the +1/+2 site (Movie S3 and Figure S9). The Cremer–Pople ring conformation24 of the −1 subsite saccharide undergoes a transition from 4C1 in the Michaelis complex to 4H3 in the first TS and to 4E in the covalent–enzyme intermediate. The parameters for this TS (Figure S9C) demonstrate a nearly perfect angle of approach of the nucleophile with the departing group, which is a 178.4° O3–C1–Oδ2(D286) angle. The Bürgi–Dunitz trajectory αBD angle of the nucleophile–C1–O5 structure of 99.6° is in the range that allows optimal overlap between molecular orbitals.28 To evaluate experimentally the structural interactions of the transition state, wild-type apo BglX crystals were soaked with the inhibitor 1-deoxynojirimycin (Ki = 4.4 ± 0.5 mM), a mimetic of the oxocarbenium species (Figure 2). The X-ray complex of BglX with 1-deoxynojirimycin (2.0 Å resolution) demonstrated occupancy of a single molecule at the −1 subsite (Figure 4B). Its electron density fits a half-chair conformation (Figures S10C and S11C) as expected for a mimetic of the oxocarbenium species.29
The first step of the reaction (glycosylation of the enzyme) culminates in the concurrent formation of the covalent glycosyl species at the −1 subsite and formation of the first product (in the present example, the disaccharide laminaribiose at +1 and +2 subsites) (Figure 4C). We modeled this stage of the reaction on the basis of our previous study of NagZ5 (Supporting Information) and a reported X-ray structure of a GH3 homologue.30 Molecular dynamics simulation showed a shift of the laminaribiose product (in the +1 and +2 subsites) to allow entry of a water molecule (Movie S4). The water molecule closely approached (3 Å) the C1 atom of the glycosyl–enzyme species (Figure 4D and Movie S4). Interestingly, clear density for a crystallographic water molecule in the complex with 1-deoxynojirimicin is observed at this location (Figure 4B and Figures S10C and S11C). This water molecule is positioned between the general base E517 and the C1 atom of the glycosyl species and thus coincides with the hydrolytic water for deglycosylation. E517 abstracts a proton from the water to promote formation of the second oxocarbenium-like species (Figure 4D). Here, a bond forms between the oxygen of the water molecule and the C1 atom as the bond between D287 and C1 breaks (Figure 4E). This deglycosylation step was evaluated by a second set of QM/MM calculations (Movie S5). Like the glycosylation step, there is a single TS. Near the deglycosylation TS, the covalent bond between anomeric C1 and the oxygen of D286 [C1–Oδ2(D286)] is broken (distance of 2.10 Å) while the oxygen atom of the incoming water molecule (OW) is at a distance of 2.02 Å (Figure S9B). The OW–C1–H1 and Oδ2(D286)–C1–H1 angles are 91.1° and 80.7°, respectively. The Bürgi–Dunitz angle (αBD) for nucleophilic attack trajectory is 88.9°, while the angle formed by the nucleophile and the departing group [OW–C1–Oδ2(D286)] is 161.0°. The 4H3 ring conformation of the second TS is the same as that seen in the first TS. Moreover, the second TS superimposes on the 1-deoxynojirimiycin crystal structure and validates the locus of a water molecule in the TS model. The OW…OE2(E517) and OW…C1 distances in the crystal structure are 2.4 and 2.8 Å, respectively, while the counterpart distances in the TS model are 2.70 and 2.02 Å, respectively. The OW–C1–Oδ2(D286) angle of 149.7° in the 1-deoxynojirimycin complex is also comparable (161.0°). The reaction from the TS proceeds with formation of β-D-glucose in the 4C1 conformation as the second product at the −1 subsite, while the first product (the disaccharide laminaribiose from the first step of the BglX reaction) occupies the +1/+2 sites. Double soaking of both reaction products, β-D-glucose and laminaribiose, with the BglX crystals was unsuccessful toward giving this complex. The X-ray structure showed occupancy of two β-D-glucose molecules at the −1 and +1 sites (Figure 4E). Additional soaking of BglX crystals with β-D-glucose gave an X-ray structure with a single occupancy of β-D-glucose at the −1 subsite (Figure 4F), indicating the release of the first product from the +1/+2 subsites. Subsequent release of the second product (β-D-glucose) from the −1 subsite regenerates the apo state (Figure 3) to complete the catalytic cycle.
We investigated the structural basis for the selective recognition of saccharides by BglX. Although BglX demonstrates a hydrogen bonding pattern at the −1 subsite similar to that in NagZ, it excludes the occupancy of the N-acetamido group of cell-wall GlcNAc by an elegantly repositioned salt bridge (Figure S12). The inability of GlcNAc-β-PNP to act as a BglX substrate in our assay, while Glc-β-PNP is found to be substrate, confirms the structural incompatibility. On the other hand, the basis for the exclusion of other β-(1→4)-glycosides as substrates is less obvious. We determined the X-ray structures of three nonsubstrate β-(1→4)-glycosides—cellobiose, lactose, and xylotriose (Figure 2 and Figures S1 and S2)—in complex with BglX [resolutions of 1.65, 1.60, and 1.80 Å, respectively (Figure 5)]. In each structure, contact between E517 (as the source of the critical proton) and the glycosidic oxygen atom of the ligands is lost. Cellobiose and lactose bind nonproductively in a flipped directionality with the glucose toward the reducing end, seated at the −1 site (Figure 5A,B), as opposed to the +1 site in the productive binding in the BglX-D286N:LMT complex (Figure 5D). Importantly, the lone pairs of electrons on the glycosidic oxygens in the bound nonsubstrate ligands point away from the source of proton, in contrast to the case of the substrate LMT. The requisite protonation by E517 is possible only for the substrate saccharide.
Figure 5.

X-ray structures of BglX-D286N in complex with nonsubstrate saccharides. (A) Cellobiose [Glc-β-(1→4)-Glc], (B) lactose [Gal-β-(1→4)-Glc], and (C) xylotriose [Xyl-β-(1→4)2-Xyl; two saccharide rings were visible], compared to (D) the substrate laminaritriose (LMT). Relevant residues are labeled. Distances between acid/base residue E517 and the glycosidic oxygen atom of the disaccharide are labeled and depicted as yellow dashed lines. Cellobiose, lactose, and xylotriose are weak inhibitors of BglX (Ki > 50 mM).
The intact BglX dimer, not the individual monomer, emerges as the catalytic unit. Protein dimerization, and especially heterodimerization, is a prevalent structural motif. Homodimerization creation of a functional active site is less common. In contrast to the notable example of symmetric homodimerization of HIV protease assembling a single active site,31 each monomer of BglX contributes unequally to the two active sites. Apart from the four GH3 enzymes mentioned earlier, the homodimeric active site has been reported in other GH families.32,33 Analysis of the determined hydrolase structures in the PDB (close to 29000 in July 2019) identified close to 2000 homodimeric structures (Table S7). The Protein Interfaces, Surfaces and Assemblies (PISA) program34 showed that <2% of these hydrolases have ligand occupancy at the interface of the dimer (Supporting Information), suggesting a relatively uncommon structural feature seen for BglX. Additionally, a possible regulation mechanism of the function of BglX may be deduced. Because the periplasmic space of P. aeruginosa has an abundance of a variety of carbohydrates, the enzymes that act on carbohydrates must have both high specificity for their substrates and regulation of their catalytic activities. Homodimerization could be a mechanism of regulation in periplasmic BglX, while in contrast, the cytoplasmic GH3 enzyme NagZ is functional as a monomer and its regulation involves a dramatic conformational change following substrate binding.5
As reported herein, catalysis by BglX plays a role in biofilm formation. Given the correlation of biofilm formation to virulence by P. aeruginosa, these results support further inquiry for BglX in the assembly (or disassembly) of either (or both) the mixed (alginate, Pel, Psl) exopolysaccharide matrix of the P. aeruginosa biofilm and the OMP oligosaccharides of the periplasm. The distribution of BglX homologues over multiple genera [such as the Gram-positive Clostridium and the Gram-negative Vibrio (Figure S13)] suggests a fundamental function for BglX enzymes in the bacterial kingdom.
METHODS
Substrate Screening.
We analyzed the ability of BglX to process a variety of carbohydrate molecules (structures are shown in Figure 2A and Figure S1). Eleven peptidoglycans were synthesized in our laboratories.8,35-37 The remaining saccharides are commercial (Sigma-Aldrich, Carbosynth). Reaction of each compound with recombinant wild-type BglX was carried out in 20 mM HEPES, 0.1 M NaCl, pH 7.0 buffer at 37 °C. After 18 h, the reaction mixtures were flash-frozen. The reaction mixtures were analyzed by LC/MS (Figure 2 and Figures S2 and S3). The LC/MS instrument conditions are listed in Table S2.
Crystallization, Data Collection, and Structure Determination.
A solution of native BglX at 12 mg mL−1 in 50 mM Tris, 300 mM NaCl, 200 mM imidazole, pH 8 buffer was prepared. Crystals grew using the sitting drop vapor-diffusion method at 291 K with 50 mM Bis-Tris, 30% pentaerythritol ethoxylate, 50 mM ammonium sulfate, pH 6.5 buffer as the precipitant solution. Equal volumes of the protein and precipitant solutions (1 μL) were combined and equilibrated against 150 μL of the reservoir solution. The crystals were cryoprotected by a mixture of 70% paratone and 30% parafine before being flash-cooled at 100 K. Crystals of BglX with ligands were obtained by soaking as described in the Supporting Information. All diffraction data sets were collected using synchrotron radiation on beamline XALOC of the ALBA synchrotron (Barcelona, Spain) using a Pilatus 6M detector. The images were processed and scaled using XDS,38 iMOSFLM,39 and AIMLESS.40 The wild-type BglX structure was determined by the molecular-replacement method using the structure of the homologue glycosyl hydrolase of family 3 from B. intestinalis (PDB entry 5TF0) as a search model followed by manual modeling using COOT41 and refinement with PHENIX.42 Structures for the different complexes were determined by the molecular-replacement method using the coordinates of the BglX native structure. Rotational and translational searches were performed using PHASER43 followed by manually modeling using COOT41 and refined with PHENIX.42 The statistics for data processing and refinement are listed in Table S3.
Computational Methods.
Protein structures for the simulation [X-ray structures or computational models (see details in the Supporting Information)] were prepared using Maestro (version 2018). Molecular dynamics simulations were performed with the Amber16 package. The Amber ff14SB force field provided parameters for amino acid residues, while GLYCAM0644 provided the parameters for the carbohydrates. For the glycosyl-modified D286 residue, charges were derived quantum-mechanically at the HF/6-31G(d) level of theory with Gaussian09 (Gaussian, Inc.) and fit using RESP methodology.45 Sequential energy minimization and equilibration were performed as previously reported.5,17 Production simulations in the NPT ensemble were carried out with the GPU-accelerated PMEMD module for 500 ns durations following the published protocol.5,17 For QM/MM analysis, snapshots were extracted from MD trajectories and subjected to steered MD (sMD) based on the predefined reaction coordinate (RC). The Amber16 package with QM/MM supports performed MD with the semiempirical method PM6-D for treating the QM region and Amber ff14SB force field for treating the MM region.46
Complete descriptions of the cloning, protein purification, site-directed mutagenesis, assays, X-ray crystallography methods, and the computational analyses are given in the Supporting Information.
Supplementary Material
ACKNOWLEDGMENTS
The work at the University of Notre Dame was supported by grants from the National Institutes of Health (GM61629 and GM131685), and that in Spain by a grant from MICIU Ministry (BFU2017-90030-P). The authors thank the staff from the ALBA (Barcelona, Spain) synchrotron facility for help in X-ray data collection and CRC of the University of Notre Dame for the computing resources. The authors acknowledge Grant P30 DK089507 from the National Institutes of Health for the BglX transposon mutant of P. aeruginosa.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.9b00754.
Methods of cloning, protein purification, site-directed mutagenesis, substrate screening, activity assays, X-ray crystallography, computational analysis, and additional figures (PDF)
The authors declare no competing financial interest.
Contributor Information
Kiran V. Mahasenan, University of Notre Dame, Notre Dame, Indiana.
María T. Batuecas, Institute of Physical Chemistry “Rocasolano”, CSIC, Madrid, Spain.
Stefania De Benedetti, University of Notre Dame, Notre Dame, Indiana.
Choon Kim, University of Notre Dame, Notre Dame, Indiana.
Neha Rana, University of Notre Dame, Notre Dame, Indiana.
Mijoon Lee, University of Notre Dame, Notre Dame, Indian.
Dusan Hesek, University of Notre Dame, Notre Dame, Indiana.
Jed F. Fisher, University of Notre Dame, Notre Dame, Indiana
Julia Sanz-Aparicio, Institute of Physical Chemistry “Rocasolano”, CSIC, Madrid, Spain.
Juan A. Hermoso, Institute of Physical Chemistry “Rocasolano”, CSIC, Madrid, Spain.
Shahriar Mobashery, University of Notre Dame, Notre Dame, Indiana.
REFERENCES
- (1).Graham LL, Beveridge TJ, and Nanninga N (1991) Periplasmic space and the concept of the periplasm. Trends Biochem. Sci 16, 328–329. [DOI] [PubMed] [Google Scholar]
- (2).Asmar AT, Ferreira JL, Cohen EJ, Cho SH, Beeby M, Hughes KT, and Collet JF (2017) Communication across the bacterial cell envelope depends on the size of the periplasm. PLoS Biol. 15, No. e2004303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bontemps-Gallo S, Bohin JP, and Lacroix JM (2017) Osmoregulated periplasmic glucans. EcoSal Plus 7, 7. [DOI] [PubMed] [Google Scholar]
- (4).Mah TF, Pitts B, Pellock B, Walker GC, Stewart PS, and O’Toole GA (2003) A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 426, 306–310. [DOI] [PubMed] [Google Scholar]
- (5).Acebron I, Mahasenan KV, De Benedetti S, Lee M, Artola-Recolons C, Hesek D, Wang H, Hermoso JA, and Mobashery S (2017) Catalytic cycle of the N-acetylglucosaminidase NagZ from Pseudomonas aeruginosa. J. Am. Chem. Soc 139, 6795–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ertesvag H, Sletta H, Senneset M, Sun YQ, Klinkenberg G, Konradsen TA, Ellingsen TE, and Valla S (2017) Identification of genes affecting alginate biosynthesis in Pseudomonas fluorescens by screening a transposon insertion library. BMC Genomics 18, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bhoopalan SV, Piekarowicz A, Lenz JD, Dillard JP, and Stein DC (2016) NagZ triggers Gonococcal biofilm disassembly. Sci. Rep 6, 22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lee M, Hesek D, Dik DA, Fishovitz J, Lastochkin E, Boggess B, Fisher JF, and Mobashery S (2017) From genome to proteome to eucidation of reactions for all eleven known lytic transglycosylases from Pseudomonas aeruginosa. Angew. Chem., Int. Ed 56, 2735–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Macdonald SS, Blaukopf M, and Withers SG (2015) N-acetylglucosaminidases from CAZy family GH3 are really glycoside phosphorylases, thereby explaining their use of histidine as an acid/base catalyst in place of glutamic acid. J. Biol. Chem 290, 4887–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ducatti DR, Carroll MA, and Jakeman DL (2016) On the phosphorylase activity of GH3 enzymes: A (-N-acetylglucosaminidase from Herbaspirillum seropedicae SmR1 and a glucosidase from Saccharopolyspora erythraea. Carbohydr. Res 435, 106–112. [DOI] [PubMed] [Google Scholar]
- (11).Bontemps-Gallo S, and Lacroix JM (2015) New insights into the biological role of the osmoregulated periplasmic glucans in pathogenic and symbiotic bacteria. Environ. Microbiol. Rep 7, 690–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lequette Y, Rollet E, Delangle A, Greenberg EP, and Bohin JP (2007) Linear osmoregulated periplasmic glucans are encoded by the opgGH locus of Pseudomonas aeruginosa. Microbiology 153, 3255–3263. [DOI] [PubMed] [Google Scholar]
- (13).Hay ID, Remminghorst U, and Rehm BH (2009) MucR, a novel membrane-associated regulator of alginate biosynthesis in Pseudomonas aeruginosa. Appl. Environ. Microbiol 75, 1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Li Y, Heine S, Entian M, Sauer K, and Frankenberg-Dinkel N (2013) NO-induced biofilm dispersion in Pseudomonas aeruginosa is mediated by an MHYT domain-coupled phosphodiesterase. J. Bacteriol 195, 3531–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).O’Toole GA (2011) Microtiter dish biofilm formation assay. J. Visualized Exp, No. e2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Case DA, Betz R, Cerutti DS, Cheatham T, Darden T, Duke R, Giese TJ, Gohlke H, Götz A, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee T-S, LeGrand S, Li P, Lin C, Luchko T, and Kollman PA (2016) AMBER 16, University of California, San Francisco, San Francisco. [Google Scholar]
- (17).Mahasenan KV, Molina R, Bouley R, Batuecas MT, Fisher JF, Hermoso JA, Chang M, and Mobashery S (2017) Conformational dynamics in penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus, allosteric communication network and enablement of catalysis. J. Am. Chem. Soc 139, 2102–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dik DA, Dominguez-Gil T, Lee M, Hesek D, Byun B, Fishovitz J, Boggess B, Hellman LM, Fisher JF, Hermoso JA, and Mobashery S (2017) Muropeptide binding and the X-ray structure of the effector domain of the transcriptional regulator AmpR of Pseudomonas aeruginosa. J. Am. Chem. Soc 139, 1448–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nakajima M, Yoshida R, Miyanaga A, Abe K, Takahashi Y, Sugimoto N, Toyoizumi H, Nakai H, Kitaoka M, and Taguchi H (2016) Functional and structural analysis of a b-glucosidase involved in b-1,2-glucan metabolism in Listeria innocua. PLoS One 11, No. e0148870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ishiguro R, Tanaka N, Abe K, Nakajima M, Maeda T, Miyanaga A, Takahashi Y, Sugimoto N, Nakai H, and Taguchi H (2017) Function and structure relationships of a h-1,2-glucooligosaccharide-degrading b-glucosidase. FEBS Lett. 591, 3926–3936. [DOI] [PubMed] [Google Scholar]
- (21).McAndrew RP, Park JI, Heins RA, Reindl W, Friedland GD, D’Haeseleer P, Northen T, Sale KL, Simmons BA, and Adams PD (2013) From soil to structure, a novel dimeric S-glucosidase belonging to glycoside hydrolase family 3 isolated from compost using metagenomic analysis. J. Biol. Chem 288, 14985–14992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chiba S (2012) A historical perspective for the catalytic reaction mechanism of glycosidase; so as to bring about breakthrough in confusing situation. Biosci., Biotechnol., Biochem 76, 215–231. [DOI] [PubMed] [Google Scholar]
- (23).Zechel DL, and Withers SG (2000) Glycosidase mechanisms: anatomy of a finely tuned catalyst. Acc. Chem. Res 33, 11–18. [DOI] [PubMed] [Google Scholar]
- (24).Cremer D, and Pople JA (1975) General definition of ring puckering coordinates. J. Am. Chem. Soc 97, 1354–1358. [Google Scholar]
- (25).Davies G, and Henrissat B (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3, 853–859. [DOI] [PubMed] [Google Scholar]
- (26).McCarter JD, and Withers SG (1994) Mechanisms of enzymatic glycoside hydrolysis. Curr. Opin. Struct. Biol 4, 885–892. [DOI] [PubMed] [Google Scholar]
- (27).Geronimo I, Payne CM, and Sandgren M (2018) Hydrolysis and transglycosylation transition states of glycoside hydrolase family 3 -Glucosidases differ in charge and puckering conformation. J. Phys. Chem. B 122, 9452–9459. [DOI] [PubMed] [Google Scholar]
- (28).Bürgi HB, Dunitz JD, Lehn JM, and Wipff G (1974) Stereochemistry of reaction paths at carbonyl centres. Tetrahedron 30, 1563–1572. [Google Scholar]
- (29).Gloster TM, and Davies GJ (2010) Glycosidase inhibition: assessing mimicry of the transition state. Org. Biomol. Chem 8, 305–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bacik JP, Whitworth GE, Stubbs KA, Vocadlo DJ, and Mark BL (2012) Active site plasticity within the glycoside hydrolase NagZ underlies a dynamic mechanism of substrate distortion. Chem. Biol 19, 1471–1482. [DOI] [PubMed] [Google Scholar]
- (31).Navia MA, Fitzgerald PM, McKeever BM, Leu CT, Heimbach JC, Herber WK, Sigal IS, Darke PL, and Springer JP (1989) Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature 337, 615–620. [DOI] [PubMed] [Google Scholar]
- (32).Nurizzo D, Nagy T, Gilbert HJ, and Davies GJ (2002) The structural basis for catalysis and specificity of the Pseudomonas cellulosaa-glucuronidase, GlcA67A. Structure 10, 547–556. [DOI] [PubMed] [Google Scholar]
- (33).O’Neill EC, Stevenson CE, Paterson MJ, Rejzek M, Chauvin AL, Lawson DM, and Field RA (2015) Crystal structure of a novel two domain GH78 family (-rhamnosidase from Klebsiella oxytoca with rhamnose bound. Proteins: Struct., Funct., Genet 83, 1742–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Krissinel E, and Henrick K (2007) Inference of macro-molecular assemblies from crystalline state. J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- (35).Hesek D, Lee M, Zhang W, Noll BC, and Mobashery S (2009) Total synthesis of N-acetylglucosamine-1,6-anhydro-N-acetylmuramylpentapeptide and evaluation of its turnover by AmpD from Escherichia coli. J. Am. Chem. Soc 131, 5187–5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lee M, Hesek D, Lastochkin E, Dik DA, Boggess B, and Mobashery S (2017) Deciphering the Nature of Enzymatic Modifications of Bacterial Cell Walls. ChemBioChem 18, 1696–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lee M, Hesek D, Shah IM, Oliver AG, Dworkin J, and Mobashery S (2010) Synthetic peptidoglycan motifs for germination of bacterial spores. ChemBioChem 11, 2525–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kabsch W (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Battye TG, Kontogiannis L, Johnson O, Powell HR, and Leslie AG (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr., Sect. D: Biol. Crystallogr 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Evans PR, and Murshudov GN (2013) How good are my data and what is the resolution? Acta Crystallogr., Sect. D: Biol. Crystallogr 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr., Sect. D: Biol. Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Read RJ, and McCoy AJ (2016) A log-likelihood-gain intensity target for crystallographic phasing that accounts for experimental error. Acta Crystallogr. D Struct. Biol 72, 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeirino J, Daniels CR, Foley BL, and Woods RJ (2008) GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J. Comput. Chem 29, 622–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Cornell WD, Cieplak P, Bayly CI, and Kollman PA (1993) Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J. Am. Chem. Soc 115, 9620–9631. [Google Scholar]
- (46).de M. Seabra G, Walker RC, Elstner M, Case DA, and Roitberg AE (2007) Implementation of the SCC-DFTB method for hybrid QM/MM simulations within the amber molecular dynamics package. J. Phys. Chem. A 111, 5655–5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.