

Abstract
Deregulation of the cell cycle is a common strategy employed by many DNA and RNA viruses to trap and exploit the host cell machinery toward their own benefit. In many coronaviruses, the nucleocapsid protein (N protein) has been shown to inhibit cell cycle progression although the mechanism behind this is poorly understood. The N protein of severe acute respiratory syndrome-coronavirus (SARS-CoV) bears signature motifs for binding to cyclin and phosphorylation by cyclin-dependent kinase (CDK) and has recently been reported by us to get phosphorylated by the cyclin-CDK complex (Surjit, M., Kumar, R., Mishra, R. N., Reddy, M. K., Chow, V. T., and Lal, S. K. (2005) J. Virol. 79, 11476–11486). In the present study, we prove that the N protein of SARS-CoV can inhibit S phase progression in mammalian cell lines. N protein expression was found to directly inhibit the activity of the cyclin-CDK complex, resulting in hypophosphorylation of retinoblastoma protein with a concomitant down-regulation in E2F1-mediated transactivation. Coexpression of E2F1 under such conditions could restore the expression of S phase genes. Analysis of RXL and CDK phosphorylation mutant N protein identified the mechanism of inhibition of CDK4 and CDK2 activity to be different. Whereas N protein could directly bind to cyclin D and inhibit the activity of CDK4-cyclin D complex; inhibition of CDK2 activity appeared to be achieved in two different ways: indirectly by down-regulation of protein levels of CDK2, cyclin E, and cyclin A and by direct binding of N protein to CDK2-cyclin complex. Down-regulation of E2F1 targets was also observed in SARS-CoV-infected VeroE6 cells. These data suggest that the S phase inhibitory activity of the N protein may have major significance during viral pathogenesis.
Mitotic cells undergo repeated cycles of division in order to produce daughter cells, a process essential for the maintenance of tissue homeostasis. Different steps of a cell division event typically include the G1 phase (preparation for DNA synthesis), followed by the S phase (genome replication), the G2 phase (preparation for cell division), and the M phase (mitosis). One of the most critical phases during cell cycle progression is the S phase, since it involves precise duplication of the whole genome, which carries all genetic messages for the next generation. Any abnormality during the replication step or thereafter would be disastrous for the organism. Hence, cells employ multiple strategies to ensure accurate and error-free genome replication. First, cells synthesize adequate amounts of raw materials that would be utilized during the S phase; second, cells strategically employ multiple check points (in the form of inhibitory factors) to block the S phase progression should there be a hostile environment arising due to any intracellular or extracellular factors.
The majority of the events during cell cycle progression are driven by enzymes called cyclin-dependent kinases (CDKs),2 which are dependent on a series of cyclins to remain catalytically active. Regulating the activity of these cyclin-CDK complexes constitutes the crux of cell cycle regulation, which remains under the scrutiny of inhibitors of CDK. On the other hand, this regulatory network has been an attractive target for pathogens to exploit the cellular machinery toward their benefit. For example, herpes simplex virus gene products ICP0 (1, 2), and ICP27 induce G1 cell cycle arrest and shut off host gene expression during infection (3). Similarly, IE2 (4) and UL69 (5) of cytomegalovirus, Zta of Epstein-Barr virus (6), p28 of mouse hepatitis virus (7), and K-bZIP of Kaposi sarcoma-associated herpes virus (8) have been shown to arrest cell cycle progression at the G1 phase. It has been postulated that imposing a G1 block may help these pathogens in utilizing the cellular raw materials to replicate their own genome or provide them shelter for a longer duration to complete their life cycle and bud off.
The mechanisms of virus-induced cell cycle arrest differ from one virus to the other. Both Epstein-Barr virus Zta and IE2 of cytomegalovirus bind to and stabilize p53 (3, 9, 10), which in turn up-regulates the expression of p21, leading to the inhibition of CDK2 and CDK4 activity, whereas the K-bZIP gene product of Kaposi sarcoma-associated herpes virus directly associates with and inhibits the activity of the cyclin-CDK2 complex (8). Among the coronaviruses, the p28 protein of mouse hepatitis virus has been shown to accumulate hypophosphorylated retinoblastoma (Rb), stabilize p53, and up-regulate the levels of p21, thus inducing cell cycle arrest at the G1 phase (7).
The virus responsible for severe acute respiratory syndrome (SARS) is a recently discovered coronavirus, which shares significant homology with the mouse hepatitis virus. However, no information exists regarding the involvement of this virus in host cell cycle modulation. We have recently shown that the nucleocapsid protein (N protein) of the SARS-CoV localizes to the nucleus as well as cytoplasm and bears signature motifs for binding to the cyclin box and phosphorylation by the CDK, and we have experimentally demonstrated it to be a substrate of the cyclin-CDK complex (11). However, its involvement in modulation of the host cell cycle remains unknown.
In this report, we provide substantial evidence that the N protein of the SARS-CoV binds to and inhibits the activity of the cyclin-CDK complex, resulting in down-regulation of the S phase gene products and subsequent inhibition of S phase progression. Mutational analysis identified the mechanism of inhibition to be different for G1 and late G1/S phase cyclins. In addition, expression of the S phase gene products were found to be down-regulated in SARS-CoV-infected cells, further supporting the above data. The possible significance of this phenomenon during the natural course of SARS-CoV infection is discussed.
EXPERIMENTAL PROCEDURES
Plasmids and Reagents—pCDNA3.1N has been described earlier (12). RXL and RGNSPAR mutants (denoted as C and K, respectively) were constructed on pCR-XL-TOPO-N backbone by site-directed mutagenesis at Bangalore Genei Corp. (Bangalore, India). Mutants were confirmed by sequencing the entire gene. C and K mutants were cloned into pCDNA3.1 Myc vector at BamHI and ApaI restriction sites. The CK dual mutant was created by cloning the HindIII fragment from pCR-XL-TOPO-K into the HindIII restriction site of the pCR-XL-TOPO-C vector construct. The orientation of this insert was checked by restriction enzyme mapping. The pCDNA3.1-CK dual mutant was created by cloning a BamHI-ApaI fragment from pCR-XL-CK into BamHI and ApaI-digested pCDNA3.1 vector backbone. Final clone was again verified by sequencing. pRC/cytomegalovirus cyclin D1-HA plasmid that expresses HA-tagged cyclin D1 protein was a gift from Dr. Mark Ewen (13). pHisTrx-cyclin A plasmid and the pMM vector bearing coding sequences for CDK2 and CIV1 was a gift from Dr. Anindya Dutta (14). Wild-type and mutant cyclin E reporter constructs were obtained from Dr. J. R. Nevins (15). pSGI-E2F1 plasmid was a gift from Dr. Vijay Kumar (International Centre for Genetic Engineering and Biotechnology, New Delhi, India).
Histone H1 and acetyl-CoA were purchased from Calbiochem. All antibodies and glutathione S-transferase Rb protein were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). A rabbit polyclonal anti-N antibody was used to detect N protein expression. [14C]chloramphenicol, [γ-32P]ATP, [35S]cysteine/methionine promix was obtained from PerkinElmer Life Sciences. The rabbit reticulocyte coupled transcription-translation (TNT) kit was obtained from Promega Corp. (Madison, WI). Bromodeoxyuridine and other biochemicals were obtained from Sigma.
Cell Culture and Transfection—COS-7 and Huh7 cells were maintained in Dulbecco's modified Eagle's medium supplemented with penicillin, streptomycin, and 10% fetal bovine serum. Cells were transfected with Lipofectamine or Fugene 6 reagent (Invitrogen or Roche Applied Science, respectively) as per the manufacturer's instructions. Mock-transfected cells were transfected with the empty vector. For synchronization experiments, 24 h postseeding, cells were starved for 34 h in serum-free medium followed by stimulation with 10% serum-containing medium for the indicated time periods.
Fluorescence-activated Cell Sorting (FACS)—FACS analysis of cell cycle progression was done as described by Krishan (16) using the propidium iodide staining method.
Metabolic Labeling and Immunoprecipitation—Forty hours post-transfection, cells were starved for 1 h in cysteine/methionine-deficient medium and then labeled with 100 μCi of [35S]Cys/Met promix for 4 h. After labeling, cells were washed once in phosphate-buffered saline and lysed in immunoprecipitation buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton, 2.5 mm sodium pyrophosphate, 1 mm β-glycerolphosphate, 1 mm Na3VO4) with protease inhibitor mixture. For immunoprecipitation, equal amounts of protein were incubated overnight with 1 μg of the corresponding antibody. This was followed by a 1-h incubation with 100 μl of 10% protein A-Sepharose suspension. The beads were washed four times with lysis buffer, and the protein was eluted by boiling the samples in 2× SDS dye. Proteins were then resolved by SDS-PAGE. Data obtained are representative of three independent sets of experiments conducted. Results were quantified, and normalized values were calculated using the NIH Image version 1.32 program. The graphs represent ± S.E. of three independent sets of experiments. Nuclear extracts were prepared as described earlier (11).
Protein Expression by Coupled in Vitro Transcription-Translation—DNA was isolated using a miniscale DNA isolation kit (Qiagen Corp.). Transcription of the plasmid DNA was initiated from the T7 promoter, and the resultant transcript was translated by the translation machinery present in the rabbit reticulocyte lysate. The reaction was conducted using a commercially available TNT kit (Promega Corp.), following the manufacturer's protocol. The resultant protein was stored at –20 °C. An aliquot of the lysate was mixed with equivalent amounts of 2× SDS-loading dye and boiled for 5 min, and protein bands were visualized by SDS-PAGE followed by staining in Coomassie Brilliant Blue or by autoradiography.
In Vitro Phosphorylation Assay—Immunoprecipitated cell lysates were washed twice with kinase buffer (25 mm Tris, pH 7.5, 5 mm β-glycerolphosphate, 2 mm dithiothreitol, 0.1 mm Na3VO4, 10 mm MgCl2) and then incubated with the indicated substrate along with 100 μm ATP and 10 μCi of [γ-32P]ATP for 45 min at 30 °C. Samples were subsequently boiled for 5 min in 10 μl of 4× SDS dye. Protein bands were resolved on 12% SDS-PAGE and detected by autoradiography.
Chloramphenicol Acetyltransferase (CAT) Assay—Cells cultured in 60-mm dishes were transfected with respective plasmids. The total amount of transfected DNA was kept equal for each sample by adjusting the amount with respective empty vectors. 34 h poststarvation, cells were stimulated with 10% fetal bovine serum (FBS) for the indicated time period and harvested in phosphate-buffered saline. The CAT assay was done as described by Kalra et al. (16).
Cell Lysate Preparation and Immunoblotting—Cells were washed once in phosphate-buffered saline and harvested in 1× SDS dye followed by vigorous vortexing and incubation in a boiling water bath for 5 min. Protein amount was equalized using the Bio-Rad protein assay kit. Samples were resolved by SDS-PAGE, followed by electrotransfer into nitrocellulose membrane (Amersham Biosciences) and incubated with respective antibodies. Protein bands were developed by the enhanced chemiluminiscence method using a commercially available kit (Cell Signaling Technology).
Virus Infection and Lysate Preparation—Vero E6 cells (ATCC number CRL-1586) were cultured in M199 medium (Invitrogen) supplemented with 10% fetal calf serum, 2.2 g/liter sodium bicarbonate, 10 mm HEPES, 0.1 mm nonessential amino acids, 1 mm sodium pyruvate at 37 °C under 5% CO2 in a humidified incubator. When cells reached ∼90% confluence, one batch of Vero E6 cells that served as the control was mock-infected with sterile medium (18, 19). Another batch was infected with SARS-CoV (strain 2003VA2774 isolated from a SARS patient in Singapore) at a multiplicity of infection of 1 (20), with a virus inoculum volume of 0.75 ml diluted with 1.25 ml of maintenance medium with only 3% fetal calf serum. After adsorption for 1 h, the inoculum was removed, and 20 ml of maintenance medium was added. At 12 h postinfection, cytopathic effects were observed in infected cells. Following incubation at 37 °C for 12 h, both uninfected and infected Vero E6 cells were treated with 0.2 ml of 5% formalin (Merck) and then incubated at 4 °C for 24 h. The formalin-treated cells were further subjected to heat at 60 °C for 30 min and UV for 5 min to ensure complete inactivation. Finally, 2 ml of 10% Triton X-100 was added to both mock-infected and SARS-CoV infected Vero E6 cells and stored at –80 °C until further use. Lysate was prepared from these samples as described by Ikeda et al. (21) with minor modifications. Briefly 200 μl of sample was mixed with 50 μl of 5× sample buffer (1× sample buffer: 1 m sodium dihydrogen phosphate, 10 mm disodium hydrogen phosphate, 154 mm sodium chloride, 1% Triton X-100, 12 mm sodium deoxycholate, 0.2% sodium azide, 0.95 mm fluoride, 2 mm phenylmethylsulfonyl fluoride, 50 mg/ml aprotinin, 50 mm leupeptin, 2% SDS, pH 7.6), and the contents were incubated under different conditions as follows: at 0 °C for 2 h, at 37 °C for 2 h, at 60 °C for 2 h, and at 100 °C for 20 min, followed by incubation at 60 °C for 2 h. After incubation, the tissue lysates were centrifuged at 15,000 × g for 20 min at 4 °C. The supernatants were equalized for protein content, boiled for 5 min in 6× SDS dye, and loaded onto SDS-polyacrylamide gel, followed by Western immunoblotting.
Bacterial Expression and Purification of Cyclin-CDK Complex—Bacterial expressed cyclin-CDK complex was reconstituted as described earlier by Wohlschlegel et al. (14).
Bromodeoxyuridine (BrdUrd) Incorporation Assay—Cells seeded on the coverslip were transfected with respective pCDNA3.1N plasmid. 34 h post-transfection, cells were stimulated with 10% FBS for 14 h and further incubated for 1 h in the presence of 10 mm BrdUrd. The medium was then aspirated, and cells were fixed with ice-cold fix (70 ml of ethanol plus 30 ml of glycine, pH 2.0) for 30 min at –20 °C. The cells were then washed in phosphate-buffered saline, and an immunofluorescence assay was conducted as described earlier (12). BrdUrd staining was observed using anti-BrdUrd antibody (1:100 dilution), and N protein expression was checked using anti-Myc antibody (1:100 dilution). Nucleus was stained with 4′,6-diamidino-2-phenylindole. BrdUrd-positive cells were counted by using a Nikon TE 2000U immunofluorescence microscope.
RESULTS
Exogenous Expression of the N Protein Inhibits the Activity of Cyclin-CDK Complex—Earlier experiments done in our laboratory have demonstrated that the N protein bears the structural motif necessary for cyclin binding and phosphorylation by cyclin-dependent kinases. Accordingly, the N protein was found to be a substrate of the cyclin-CDK complex (11). We thus asked whether the possession of signature motifs and phosphorylation by the CDK is a strategy employed by the N protein to manipulate the host cell cycle machinery toward its benefit by acting as a competitive inhibitor to in vivo substrates of the cyclin-CDK complex. Hence, experiments were designed to check the effect of N protein expression on the kinase activity of different cyclin-CDK complex.
Initially, control experiments were done to check the temporal profile of different phases of cell cycle in COS7 (African green monkey kidney) and Huh7 (human hepatoma) cells. For this, cells were maintained in the absence of growth factors for different time periods to arrest majority of the cell population at the G0 phase and then stimulated with 10% FBS to allow reentry into the cell cycle. Poststimulation, cells were harvested at every 3-h interval up to 30 h and the total cell lysate was immunoblotted to check the expression profile of different factors like cyclin D, cyclin E, cyclin A, cyclin B, p27, P-p27, CDK2, and CDK1. Also, aliquots of the lysate were used for FACS analysis to check percentage distribution of different phases of cell cycle and for in vitro phosphorylation assay to check the activity profile of cyclin D, cyclin E, cyclin A, and cyclin B, whose activities start at early G1,G1/S, S, and M phase, respectively. Approximately 30–48-h starvation (without FBS) was sufficient for arresting the majority of the Huh7 cells at the G0 phase, since no cyclin E, cyclin A, or cyclin B activity was observed. However, some level of cyclin D activity was observed during starvation too. In COS7 cells, all cyclins demonstrated basal activity at the same time period, which may be attributed to the presence of large T antigen in those cells. After the addition of 10% FBS, activity of G1 and S phase cyclins peaked at 6–9 and 12–15 h, respectively (data not shown). These experiments gave us a rough idea regarding the cell cycle profile of COS7 and Huh7 cells, and the same experimental condition was followed in all of the subsequent experiments.
Based on the above observations, Huh7 cells were transfected with empty vector only (mock) or with pCDNA3.1N (N). Post-transfection, cells were starved for 34 h and then stimulated for different time periods in order to harvest different cyclin-CDK complexes during the peak of their activity. An in vitro phosphorylation assay using histone H1 as a substrate was used to check the activity of the holoenzyme. First we tested the activity of CDK4-cyclin D complex. Its activity was found to be significantly inhibited in N protein-expressing cells (Fig. 1A , first and second panels) at all three time points chosen. To ensure that an equal amount of protein was used in each reaction, aliquots of the lysate were immunoblotted with CDK4 antibody (third panel). The graph represents a quantitative estimation of the normalized band intensities with reference to loading control. As a control, a parallel experiment was conducted to check the CDK4 activity in enhanced green fluorescent protein-expressing cells. There was no difference in CDK4 activity of enhanced green fluorescent protein-transfected cells in comparison with mock-transfected cells (data not shown), thus ruling out the possibility that the observed CDK4 inhibitory activity of the N protein is an artifact of the transient transfection method. Similarly, we checked the activity of CDK6. The inhibitory effect of the N protein on CDK6 kinase activity appeared to be less intense as compared with that of CDK4 (fourth panel). The fifth panel shows the protein level of CDK6 as a loading control. Aliquots of the lysate were immunoblotted with anti-Myc (9E10) antibody to confirm the expression of N protein (sixth panel). The bottom panel shows schematics of cell cycle distribution at the respective time points as judged by subjecting control cells to fluorescence-assisted cell sorting analysis. A similar set of experiment was performed using COS7 cells, which showed similar results (data not shown).
FIGURE 1.

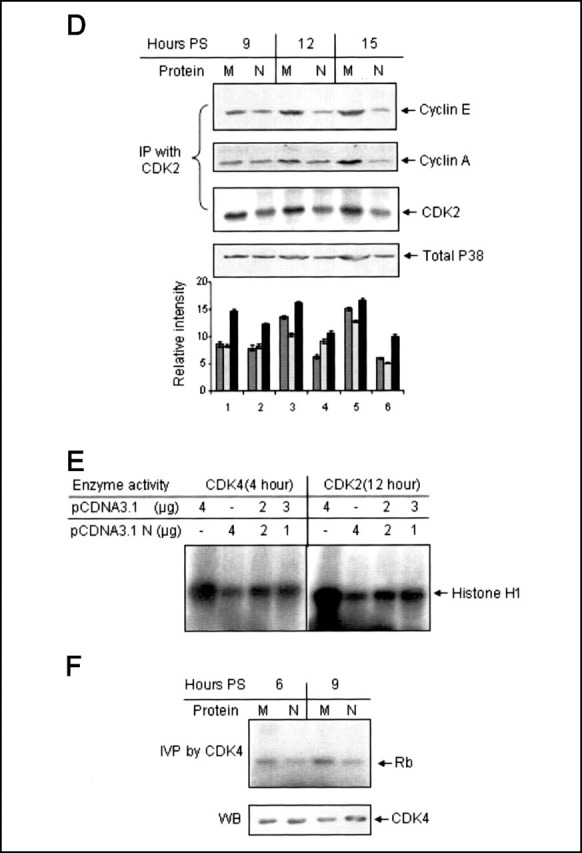
N protein expression down-regulates cyclin-CDK activity.A, Huh7 cells transfected with pCDN3.1 (M) or pCDNA3.1N (N) plasmid were starved for 34 h followed by stimulation with 10% bovine serum for the indicated time periods. Aliquots of the lysate were immunoprecipitated with CDK4 (first panel), cyclin D (second panel), and CDK6 (fourth panel) antibody and used for in vitro phosphorylation assay (IVP). PS, poststimulation. A fraction of the total cell lysate was immunoblotted (WB) with CDK4 (third panel), CDK6 (fifth panel), or anti-Myc (sixth panel) antibody. The graph represents mean ± S.D. relative band intensity from three independent experiments. In the graph, each set of bars represents the corresponding lane in the gel above. Numbers 1–6 represent mock- and pCDNA3.1N-transfected sample at the 3, 6, and 9 h time point, respectively. Dark gray, light gray, and black bars represent CDK4, cyclin D, and CDK6 band intensity, respectively. The seventh panel represents FACS analysis of cell cycle status at 3, 6, and 9 h after the stimulation period. Numbers represent the percentage of cells in that particular phase. B, cells maintained and harvested as described in A were immunoprecipitated with cyclin D antibody, and aliquots of the lysate were immunoblotted with CDK4 (second panel) or p27 (third panel) antibody. The p27 blot was stripped and reprobed with cyclin D antibody (first panel). A fraction of the total cell lysate was immunoblotted with total ERK antibody to check equal loading (fourth panel). C, cells maintained as described above were harvested at the indicated time periods, and in vitro phosphorylation was done using CDK2 (first panel), cyclin E (second panel), and cyclin A (third panel) antibody. Aliquots of the total cell lysate were immunoblotted with CDK4 (fourth panel) or anti-Myc (sixth panel) antibody. In the graph, dark gray, light gray, and black bars represent CDK2, cyclin E, and cyclin A activity, respectively. Numbers 1–6 represent mock- and pCDNA3.1N-transfected sample at the 9, 12, and 15 h time point, respectively. The bottom panel represents FACS analysis of cell cycle status at 9, 12, and 15 h after the stimulation period. Numbers represent percentage of cells in that particular phase. D, cells maintained and harvested as described in C were immunoprecipitated with CDK2 antibody, and aliquots of the lysate were immunoblotted with cyclin E (first panel) and cyclin A (second panel) antibody. Cyclin A and cyclin E blot were stripped and reprobed with CDK2 (third panel) and total p38 (fourth panel) antibody, respectively. In the graph, the dark gray, light gray, and black bars represent cyclin E, cyclin A, and CDK2 protein level, respectively. Numbers 1–6 represent mock- and pCDNA3.1N-transfected sample at the 9, 12, and 15 h time point, respectively. E, Huh7 cells were transfected with the indicated amounts of respective plasmids and harvested 4 h (lanes 1–3) or 12 h (lanes 4–6) poststimulation with 10% bovine serum. An in vitro phosphorylation assay was done from an equal amount of lysate using CDK4 (lanes 1–3) or CDK2 (lanes 4–6) antibody. F, CDK4 in vitro phosphorylation as described in A using Rb as a substrate (first panel). Aliquots of the total cell lysate were immunoblotted with CDK4 antibody.
Since down-regulation of the CDK4-cyclin D activity may be a result of decreased association of CDK4 with cyclin D or p27 with the CDK4-cyclin D complex, we checked these associations in N protein-expressing cells. As shown in Fig. 1B, immunoprecipitation of mock- or N protein-expressing cell lysate with cyclin D antibody and immunoblotting with CDK4 or p27 antibody revealed that there was no interference in the assembly of cyclin D-CDK4-p27 complex in N protein-expressing cells as compared with mock-transfected cells. Aliquots of total cell lysate were immunoblotted with total ERK antibody to ensure that equal amounts of lysate were used for each immunoprecipitation reaction (Fig. 1B, bottom panel).
Having observed that N protein expression could down-regulate CDK4 activity without destabilizing the formation of the CDK4-cyclin D complex, we next investigated whether N protein expression inhibited CDK2 activity as well. An in vitro phosphorylation assay using CDK2, cyclin E, and cyclin A antibodies clearly indicated that the N protein could effectively inhibit CDK2 activity as well (Fig. 1C, first, second, and third panels and accompanying graph). The fourth panel shows an immunoblot of the total CDK4 protein, used as a loading control for this set of experiments. Aliquots of the lysate were also immunoblotted with anti-Myc antibody to check the expression of N protein (fifth panel). The bottom panel shows schematics of cell cycle distribution at the respective time points as judged by subjecting control cells to FACS analysis.
Further, we checked the effect of N protein expression on the association of CDK2 and cyclin E-cyclin A complex. In contrast to cyclin D-CDK4 association, assembly of the cyclin E-CDK2 as well as cyclin A-CDK2 complex was found to be inhibited in pCDNA3.1N-transfected cells (Fig. 1D, first and second panel). Also, the level of total CDK2 protein used for immunoprecipitation was found to be decreased in N protein-expressing cells (third panel). Hence, it may be possible that expression of N protein actually inhibited the expression of cyclin E, cyclin A, and CDK2, resulting in overall decreased assembly of the complex. An aliquot of the total cell lysate was immunoblotted with total p38 antibody to verify equal protein amounts in the above immunoprecipitation reactions (fourth panel).
In order to further ensure that the CDK-inhibitory activity was a property specific to the N protein, a titration experiment was done using increasing amounts of pCDNA3.1N plasmid. Cells transfected with 1, 2, or 4 μgof the N protein-expressing plasmid construct were starved for 34 h, followed by stimulation with 10% FBS for 4 or 12 h. CDK4 or CDK2 activity was assayed by their ability to in vitro phosphorylate histone H1. As seen in Fig. 1E, CDK4 and CDK2 activity was found to gradually decrease with increasing amounts of the N protein expression plasmid. Further, purified recombinant retinoblastoma (Rb) protein was used as a substrate instead of histone H1 in the in vitro phosphorylation assay to rule out bias for a specific substrate. Rb phosphorylation too was effectively inhibited by lysate immunoprecipitated with CDK2 antibody from N protein-expressing cells (Fig. 1F). These experiments confirmed that the CDK-inhibitory property exhibited by the N protein of SARS-CoV was specific to this protein.
Exogenous Expression of the N Protein Inhibits S Phase Progression—Since expression of the N protein could effectively block the activity of both G1 and S phase cyclins, we next checked its effect on the progression of the S phase. Mock- or N protein-expressing cells were starved for 34 h, followed by stimulation with 10% FBS for 14 h. Cellular incorporation of BrdUrd was checked by an immunofluorescence assay. BrdUrd is a uracil analog that gets incorporated into newly synthesized DNA and thus serves as a sensitive indicator of S phase progression. As shown in Table 1 , ∼65% of mock-transfected cells showed BrdUrd incorporation. The absence of BrdUrd staining in ∼35% of cells may be attributed to a nonsynchronized population of cells. Among N protein-expressing cells, only ∼7.3% cells showed BrdUrd incorporation, thus indicating that the majority of N protein-expressing cells were unable to enter the S phase. The data were derived by counting the respective number of fluorescent cells from different samples. An average from three independent experiments is shown in Table 1.
TABLE 1.
Statistical analysis of N protein-expressing cells in S phase as judged by BrdUrd incorporation assay
A total of 200 cells were counted in each category. Data represent the average of three experiments. Only cells expressing N protein and showing BrdUrd staining were counted as positive in the pCDNA3.1N category. pCDNA3.1N expression was checked by staining with anti-Myc antibody, followed by labeling with anti-rabbit fluorescein isothiocyanate. BrdUrd incorporation was checked by staining with anti-BrdUrd antibody followed by labeling with anti-mouse Texas Red.
| Plasmid | Cells counted | N protein-expressing cells | BrdUrd +ve cells | Percentage of BrdUrd +ve cells |
|---|---|---|---|---|
| % | ||||
| pCDNA3.1 | 200 | 0 | 130 ± 9 | ∼65 |
| pCDNA3.1N | 200 | 150 ± 12 | 11 ± 3 | ∼7.3 |
The N Protein Inhibits Rb Phosphorylation and Down-regulates the Expression of E2F1 Targets—One of the major target of CDK4 and CDK2 holoenzyme complex during G1 and late G1 phase of the cell cycle is Rb. Phosphorylation of Rb by the activity of CDK4 and CDK2 has been shown to release E2F1 from the inhibitory activity of former, thus enabling it to drive the transcription of S phase genes. Many viral proteins have been shown to inhibit E2F1 activity, leading to cell cycle arrest (21). Therefore, our subsequent experiments aimed at testing whether exogenous expression of the N protein could inhibit Rb phosphorylation. Immunoblot analysis using an antibody specific for the phosphoserine 795 residue of Rb protein (which is one of the amino acid residues commonly phosphorylated by the CDKs) revealed decreased Ser795 phosphorylation in N protein-expressing cells (Fig. 2A , upper panel). The same blot was stripped and immunoblotted with anti-Akt antibody to reveal total cellular Akt as a gel loading control (lower panel).
FIGURE 2.

N protein expression leads to hypophosphorylation of Rb and down-regulation of E2F1 targets.A, Huh7 cells transfected with pCDN3.1 (M) or pCDNA3.1N (N) plasmid were starved for 34 h, followed by simulation with 10% bovine serum for the indicated time periods. Total cell lysate was immunoblotted with phospho-Rb (Ser795)(upper panel) or total Akt antibody (lower panel). P-Rb band intensities were quantified using the NIH Image program, normalized with reference to that of total Akt, and the graph was plotted. PS, poststimulation. B, cells maintained as in A for the indicated time periods were used to prepare nuclear extracts. Equal amounts of nuclear lysate from each sample were immunoprecipitated with E2F1 antibody and immunoblotted with Rb (first panel) and HDAC1 (third panel) antibody. Rb blot was stripped and immunoblotted with E2F1 antibody (second panel). Aliquots of the total nuclear lysate were immunoblotted with phospho-p38 antibody (fourth panel). C, cells maintained as in A for the indicated time periods were harvested in SDS lysis buffer, and aliquots of the lysate were immunoblotted with cyclin E (first panel), CDK2 (second panel), and CDK4 (third panel) antibody. The CDK4 blot was stripped and immunoblotted with total p38 antibody (fourth panel). D, cells transfected with wild type (lanes 1–4) or E2F element mutants (lanes 5 and 6) cyclin E CAT reporter along with pCDNA3.1N (lanes 2, 3, 4, and 6) and pSGI E2F1 (lanes 5 and 6) were harvested 15 h poststimulation in phosphate-buffered saline and processed for the CAT reporter assay. The total amount of DNA transfected was kept constant for each sample, using pCDNA3.1 DNA. Samples were resolved by thin layer chromatography, and spot intensities were quantified and graph-plotted. The graph represents mean ± S.D. of three experiments. E, cells transfected with pCDNA3.1 (lane 1) or pCDNA3.1N (lane 2) or pCDNA3.1N along with the indicated amount of pSGI E2F1 plasmid (lanes 3 and 4) were starved for 34 h and harvested 12 h poststimulation with 10% bovine serum. Aliquots of the lysate were immunoprecipitated with CDK2 antibody and processed for the in vitro phosphorylation assay (IVP; upper panel). A fraction of the lysate was immunoblotted with CDK2 (second panel) or total p38 (third panel) antibody. The total amount of transfected DNA was equalized for each sample by using pCDNA3.1 DNA. In the graph, dark gray and light gray bars represent CDK2 activity and CDK2 protein level, respectively.
Next, we checked the association of Rb with E2F1 and HDAC1 in nuclear extracts prepared from mock or N protein-expressing cells at different time points. As shown in Fig. 2B, Rb and HDAC1 were stably associated with E2F1 in N protein-expressing cells at 6 h poststimulation, whereas coprecipitation of Rb and HDAC1 with E2F1 was significantly reduced in mock-transfected cells under similar conditions. Phospho-p38 mitogen-activated protein level was used as a control to ensure that equal amounts of protein were used for immunoprecipitation. Prolonged association of Rb with E2F1 in N protein-expressing cells indicated inhibition of E2F1 activity, which would result in decreased expression of E2F1-responsive proteins. Hence, we checked the protein levels of some of the E2F1 targets, such as cyclin E and CDK2, by immunoblot analysis. Levels of cyclin E and CDK2 were found to be significantly reduced in N protein-expressing cells (Fig. 2C, first and second panels). Similar was the effect on CDK1 level (data not shown). The fact that the observed effect was specific for CDK2 and CDK1 was confirmed by immunoblotting aliquots of the lysate with CDK4 antibody, which remains unaltered despite the presence or absence of the N protein (Fig. 2C, third panel). Aliquots of the lysate were immunoblotted with total p38 antibody to check for equal loading in the above experiment (fourth panel).
Inhibition of E2F1 activity was further confirmed by assaying the activity of an E2F1-responsive promoter in N protein-expressing cells. A cyclin E promoter-driven CAT reporter plasmid bearing a wild type or mutated E2F1 response element was cotransfected along with vector only or with the pCDNA3.1N plasmid. Cells were starved for 34 h, followed by stimulation with 10% FBS for 15 h, and CAT reporter activity was measured by assaying acetylation of [14C]chloramphenicol by thin layer chromatography. CAT activity of triplicate samples were quantitated, and a graph was plotted (Fig. 2D). N protein expression resulted in inhibition of wild-type cyclin E promoter activity, whereas no effect was observed in samples containing the E2F mutant promoter. Further, exogenous expression of E2F1 was able to restore cyclin E reporter expression in N protein-expressing cells, thus confirming the role of E2F1 in inhibiting cyclin E promoter activity (Fig. 2D). Expression of E2F1 and N protein was confirmed by immunoblotting aliquots of the sample with respective antibodies (data not shown).
We subsequently assayed CDK2 kinase activity in both E2F1- and N protein-overexpressing cells. Cells were transfected with different plasmids, as shown in the Fig. 2E, starved for 34 h, and stimulated for 12 h, followed by in vitro phosphorylation assay. As expected, overexpression of E2F1 could alleviate N-mediated down-regulation of CDK2 activity in a dose-dependent manner (Fig. 2E, first panel). Aliquots of the lysate were immunoblotted with CDK2 antibody. Protein levels of CDK2 were restored to that of control cells in E2F1-overexpressing cells (Fig. 2E, second panel). Aliquots of the lysate were immunoblotted with total p38 antibody to check equal loading (Fig. 2E, third panel). A quantitative estimation of the normalized band intensity is shown in the graph. These experiments suggest that the N protein down-regulates E2F1 activity by inhibiting Rb phosphorylation, resulting in decreased expression of the S phase genes, or overexpression of E2F1 is sufficient to override the CDK-inhibitory activity of the N protein.
The N Protein-mediated Inhibition of CDK Activity Is Independent of p16, p27, and p21—CDK inhibitors like p16, p27, and p21 are known to be the major factors that regulate endogenous cyclin-CDK activity under different circumstances by binding to the CDK only (p16) or to the cyclin box of different cyclins (p27 and p21), leading to inhibition of their kinase activity. Hence, expression level of these molecules is enhanced or protein level is stabilized by a wide variety of chemical or genotoxic agents in order for them to induce cell cycle arrest. Also, many viral factors that modulate host cell cycle are known to up-regulate the expression of these molecules in order to block CDK activity (8). We thus wanted to check whether N protein expression followed a similar path. Mock- or pCDNA3.1N-transfected cells were starved for 34 h, followed by stimulation with 10% FBS for different time periods. Cell lysates were immunoblotted with p16, p27, and p21 antibody (Fig. 3, A (first panel) and B (first and second panels)). Protein levels of neither p16 nor p27 and p21 were altered in N protein-expressing cells with reference to control. As a control to confirm that CDK2 activity was inhibited in N protein-expressing cell lysate used for immunoblotting of p27 and p21 protein, aliquots of the lysate were immunoblotted with P-p27 (Thr187) specific antibody, which showed decreased phosphorylation of p27 at the threonine 187 residue at all time points (Fig. 3B, third panel). The P-p27 blot was stripped and reprobed with total ERK antibody to ensure equal loading (Fig. 3B, fourth panel). Aliquots of the lysate were immunoblotted with anti-Myc (9E10) antibody to check expression of the N protein in these cells (data not shown). Thus, it was clear that the CDK-inhibitory activity of the N protein is not mediated through the up-regulation of p16, p27, or p21.
FIGURE 3.
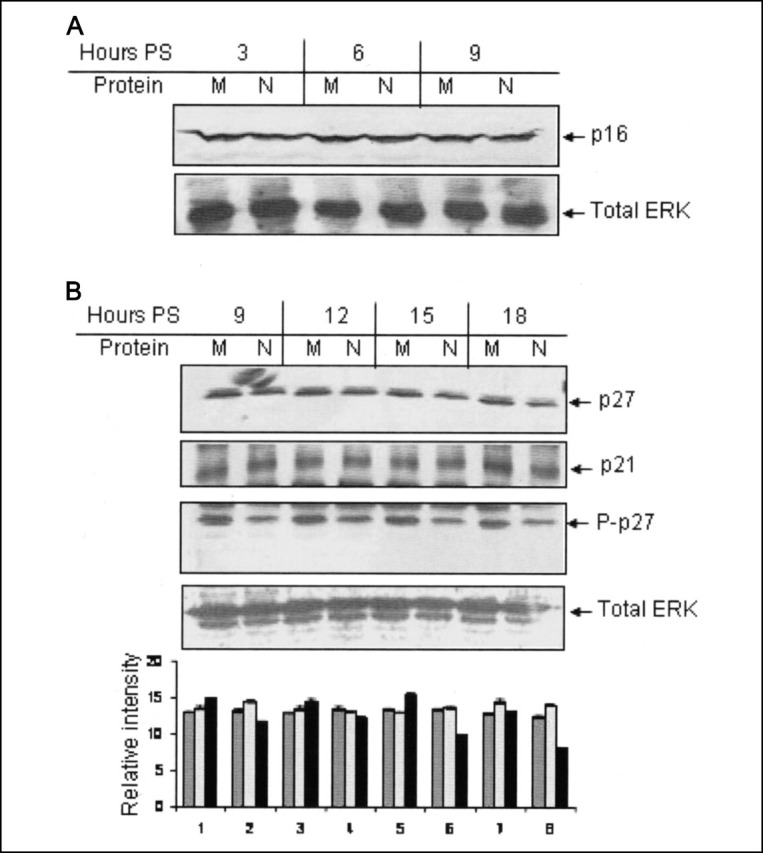
Down-regulation of CDK2 activity is independent of p16, p27, and p21.A, Huh7 cells transfected with pCDN3.1 (M) or pCDNA3.1N (N) plasmid were starved for 34 h, followed by simulation with 10% bovine serum for the indicated time periods. Total cell lysate was immunoblotted with total p16 antibody (first panel). PS, poststimulation. The same blot was stripped and reprobed with total ERK antibody (second panel). B, Huh7 cells transfected with pCDNA3.1 (M) or pCDNA3.1N (N) plasmid were starved for 34 h, followed by simulation with 10% bovine serum for the indicated time periods. Total cell lysate was immunoblotted with total p27 (first panel), total p21 (second panel), and phospho-p27 (Thr187) (third panel) antibody. P-p27 blot was stripped and reprobed with total ERK (fourth panel) antibody. Band intensities were normalized with reference to that of total ERK and graph-plotted. The image is representative of three experiments. In the graph, each set of bars represents the corresponding lane in the gel above; dark gray, light gray, and black bars represent p27, p21, and P-p27 band intensity, respectively.
Transaddition of N Protein Can Block CDK Activity—The N protein has been shown to be a substrate of cyclin-CDK complex and it bears the signature sequence for binding to the cyclin box (RXL motif), which is generally seen in high affinity substrates or inhibitors of the cyclin-CDK complex, such as E2F1, CDC6, SSeCKs, p27, and p21 (14). Since the above data revealed that N protein-mediated inhibition of CDK activity did not involve CDK inhibitors (CKIs), we reasoned that N protein itself might be mimicking the role of CKIs by directly binding to the cyclin box, or it might be acting as a competitive inhibitor to natural substrates of cyclins. Thus, we tested whether exogenously added N protein can inhibit CDK activity in an in vitro phosphorylation assay. One set of cells in a 60-mm dish were transfected with vector only or with pCDNA3.1N plasmid and harvested at 48 h post-transfection in coimmunoprecipitation buffer. In a parallel setup, cells were seeded at 50% confluence in a 60-mm dish, starved for 34 h, stimulated with 10% FBS for 4 or 12 h (to check CDK4 or CDK2 activity, respectively), and harvested in the same buffer as above. Cell lysate was equally divided into two tubes and mixed with equivalent amounts of mock- or pCDNA3.1N-transfected cell lysate. 1 μg of CDK4 or CDK2 antibody was added to respective tubes and incubated overnight with rocking at 4 °C, and an in vitro phosphorylation assay was done using histone H1 as a substrate. As shown in Fig. 4A , in the N protein-added sample, CDK4 activity was significantly inhibited (lane 2) as compared with the control. Similarly, CDK2 activity was decreased in the N protein added sample (compare lane 4 with lane 3), although to a lesser extent with reference to that of CDK4. N protein expression was verified by immunoblotting one set of the sample with anti-Myc (9E10) antibody (Fig. 4B). The above observation was further confirmed by including TNT (in vitro transcribed and translated) expressed N protein in the in vitro phosphorylation assay. Control cell lysate was prepared as described above and mixed with 5 μl(lanes 2 and 5) or 10 μl(lanes 3 and 6) of N protein-expressing TNT lysate or 10 μl of mock-translated TNT lysate (lanes 1 and 4). 1 μg of CDK4 or CDK2 antibody was added to each sample and incubated overnight at 4 °C with rocking, and in vitro phosphorylation was done. The addition of N protein-expressing lysate resulted in down-regulation of the activity of both CDK4 and CDK2 (Fig. 4C). As a control to check whether the observed phenomenon is a specific property of the N protein or is commonly observed among all substrates of cyclin-CDK complex, we included different concentrations of purified recombinant Rb protein with cell lysate instead of N protein in a parallel set of experiments, and after overnight rocking at 4 °C, the in vitro phosphorylation assay was conducted using histone H1 as a substrate. However, no inhibition of histone H1 phosphorylation was observed in the presence of Rb protein (Fig. 4D). This experiment indicated that the CDK-inhibitory activity was specific to the N protein of SARS-CoV. Further, since inhibition of CDK activity was observed despite washing the lysate three times in immunoprecipitation and kinase buffer, we reasoned that it may be possible that the N protein binds to the cyclin-CDK complex and inhibits its activity in a manner analogous to that of CKIs. In order to further study this possibility, a site-directed mutagenesis approach was undertaken to alter the RXL and RGNSPAR motifs of the N protein.
FIGURE 4.
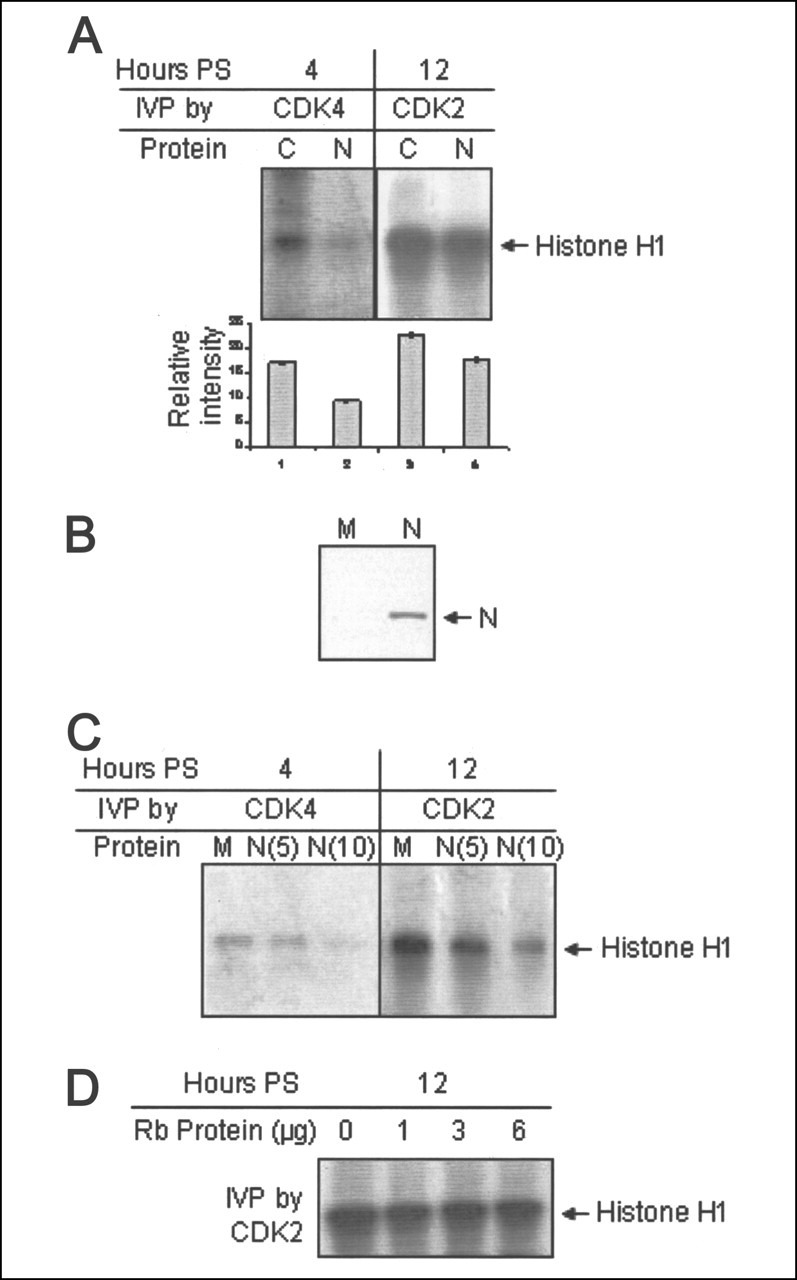
Exogenously added N protein can inhibit CDK activity.A, in vitro phosphorylation (IVP) assay using CDK4 (lanes 1 and 2) or CDK2 (lanes 3 and 4) and trans expressed N protein added sample (lanes 3 and 4). Band intensities were quantified and plotted in the graph. PS, poststimulation. B, pCDNA3.1 (M, lane 1) or pCDNA3.1N (N, lane 2) transfected cells were harvested 48 h post-transfection, and aliquots of the lysate were immunoblotted with anti-Myc antibody. C, COS7 cells seeded at 50% confluence were starved for 34 h, followed by stimulation with 10% bovine serum for 4 h (lanes 1–3) or 12 h (lanes 4–6), and total cell lysate was prepared in immunoprecipitation buffer. The lysate was equally divided into three tubes and mixed with 10 μl of mock TNT lysate (lanes 2 and 5 and lanes 3 and 6, respectively). In vitro phosphorylation was done using indicated antibodies. D, cells maintained as in C were harvested 12 h poststimulation, and total cell lysate prepared in immunoprecipitation buffer was mixed with the indicated amount of purified Rb protein. In vitro phosphorylation was done using CDK2 antibody.
The N Protein Utilizes Different Mechanisms to Inhibit CDK4 and CDK2 Activity—In an attempt to further prove whether the N protein behaves as a CDK inhibitor, we mutated the KELSP (RXL motif) and RGNSPAR (CDK phosphorylation motif) amino acid sequence to AEVGP and RGNAALG, respectively, by site-directed mutagenesis. Fig. 5A depicts the schematics of the altered amino acid residues and their corresponding nucleotide sequences. Expression of these mutants was confirmed by labeling the protein with [35S]cysteine/methionine promix and immunoprecipitation using anti-Myc (9E10) antibody (Fig. 5B). The RXL mutant is denoted as C, RGNSPAR mutant is shown as K, and the construct bearing both the mutations is denoted as CK, for the remainder of this work. All mutant clones were tested positive for efficient expression in COS7 and Huh7 cells.
FIGURE 5.
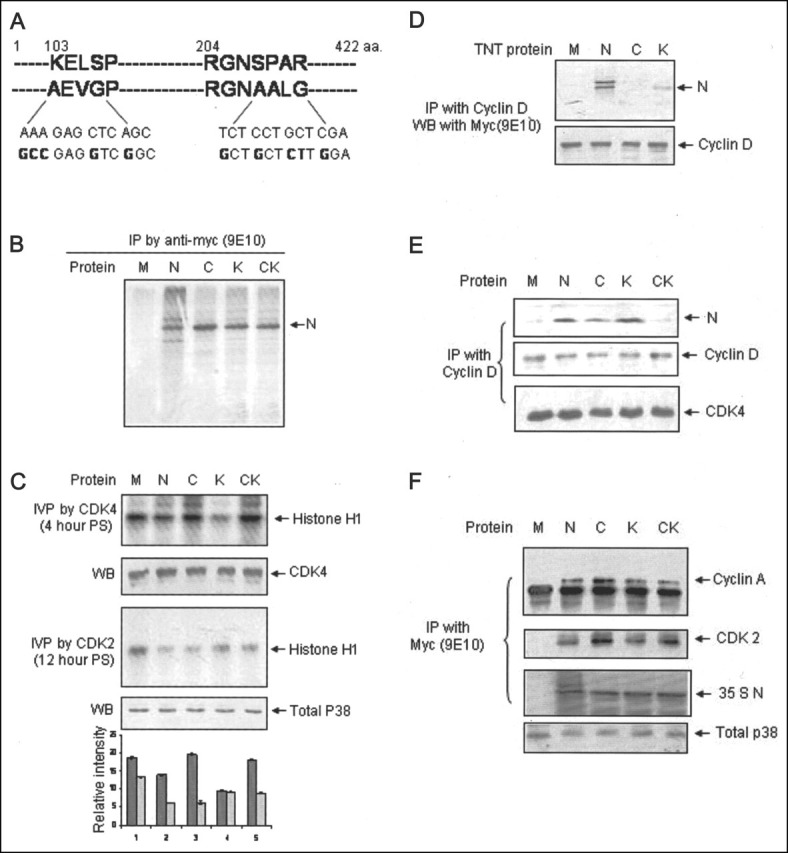
Mutants of N protein demonstrate different mechanisms for inhibition of CDK4 and CDK2 activity.A, schematic representation of RXL and RGNSPAR motifs of N protein and respective mutated amino acid residues (aa). B, expression of wild type and mutant N proteins as judged by labeling with [35S]cysteine/methionine promix and immunoprecipitation (IP) with anti-Myc (9E10) antibody. 4 μg each of the DNA was transfected into Huh7 cells; 48 h post-transfection, cells were harvested and immunoprecipitated, samples were resolved by 12% SDS-PAGE, and bands were detected by fluorography. M, pCDNA3.1-transfected sample; N, wild type nucleocapsid; C,RXL mutant nucleocapsid; K, CDK motif mutant nucleocapsid; CK,RXL and CDK motif dual mutant nucleocapsid, respectively. C, in vitro phosphorylation (IVP) of histone H1 in mutant N protein expressing cell lysate by CDK4 (first panel) or CDK2 (third panel). Aliquots of the lysate were immunoblotted (WB) with CDK4 and total p38 (second and fourth panel, respectively) antibody. The graph represents ± S.E. of three independent experiments. In the graph, dark gray and light gray bars represent CDK4 and CDK2 activity, respectively. D, association of the N protein with cyclin D. 10 μl each of the TNT lysate separately expressing cyclin D and different N proteins were mixed in immunoprecipitation buffer. Samples were immunoprecipitated with cyclin D antibody and immunoblotted with anti-Myc (9E10) antibody (first panel). The same blot was stripped and reprobed with cyclin D antibody (second panel). E, association of the N protein with endogenous cyclin D-CDK4 complex. Cells expressing different N protein mutants were immunoprecipitated with cyclin D antibody 4 h post-stimulation, and aliquots of the sample were immunoblotted with anti-Myc (9E10) (first panel) and CDK4 (third panel) antibody. Nucleocapsid blot was stripped and reprobed with cyclin D (second panel) antibody. F, association of N protein with endogenous cyclin A-CDK2 complex. Cells transfected with the indicated plasmids were starved for 34 h followed by stimulation with 10% FBS for 12 h and labeling with [35S]cysteine/methionine promix for 2 h. Equal amount of sample was immunoprecipitated with anti-Myc (9E10) antibody and immunoblotted with cyclin A antibody (first panel). The same blot was stripped and reprobed with CDK2 antibody (second panel). CDK2 blot was air-dried and exposed to x-ray film (third panel). Aliquots of the total cell lysate were immunoblotted with total p38 antibody (fourth panel).
Next, we conducted an in vitro phosphorylation assay using CDK4 and CDK2 antibody from cell lysate expressing wild-type and different mutant N proteins. Interestingly, the C and K mutants demonstrated opposite properties on their ability to inhibit CDK4 and CDK2 activity, respectively. The C mutant lost CDK4-inhibitory activity, whereas the K mutant was more efficient in inhibiting CDK4 activity as compared with the wild-type N protein (Fig. 5C, first panel). This indicated that the CDK4-inhibitory activity of the N protein was dependent on its ability to bind to the cyclin box. The higher inhibition efficiency of the K mutant implies that phosphorylation of the N protein by the cyclin-CDK complex might be relieving its inhibitory property; hence, the phosphorylation mutant was able to bind more stably to cyclin D and inhibit its activity. As expected, the CK dual mutant was unable to inhibit CDK4 activity. An aliquot of the lysate was immunoblotted with CDK4 antibody to check equal loading (Fig. 5C, second panel). Similarly, we tested the effect of N protein mutants on CDK2 activity. Cells transfected with different N protein mutants were starved for 34 h, followed by stimulation with 10% FBS for 12 h. Cell lysate was used for the in vitro phosphorylation assay. As seen in Fig. 5C (third panel), the C mutant could inhibit CDK2 activity like the wild-type N protein, whereas the K and CK mutants lost the CDK-inhibitory activity significantly. Aliquots of the lysate were immunoblotted with total p38 antibody to check equal loading (fourth panel). This experiment indicated that the mechanism of CDK2 inhibition mediated by the N protein is different from that of CDK4. CDK2 inhibition appeared to be independent of the involvement of the RXL motif present in N protein. Since the K mutant lost the ability to inhibit the CDK2 activity, we postulated that it may be possible that the N protein acts as a competitive inhibitor to the CDK2 substrates.
In order to further clarify the mechanism, we next tested whether the N protein could directly bind to cyclins through the RXL motif. Coimmunoprecipitation assays were designed using in vitro expressed cyclin D or cyclin A and the N protein to test the association between these two. Cyclin D, cyclin A, and the three N protein mutants were expressed by an in vitro transcription translation system (TNT kit), and 10 μl of each lysate was mixed in 500 μl of immunoprecipitation buffer followed by the addition of 1 μg of the respective antibody and overnight rocking at 4 °C. As a control, mock-translated lysates were incubated along with cyclin D or cyclin A. These samples were washed four times in IP buffer and immunoblotted with anti-Myc (9E10) antibody to check the associated N protein. As expected, wild type and the K mutant of the N protein could bind to cyclin D whereas the C mutant lost its binding property (Fig. 5D). The same blot was stripped and reprobed with cyclin D antibody to check equal loading. Expression of all of the N protein mutants was verified by running aliquots of the lysate in SDS-PAGE (described in the legend to Fig. 6A ). However, the N protein was unable to coprecipitate with TNT-expressed cyclin A protein (data not shown). Next, we checked the association of N protein mutants with endogenous cyclin D-CDK4 and cyclin A-CDK2 complex by coimmunoprecipitation assay. Cells transfected with different mutant plasmids were starved for 34 h followed by a 4-h stimulation with 10% FBS and lysis in immunoprecipitation buffer. Samples were immunoprecipitated with cyclin D antibody and immunoblotted with anti-Myc (first panel) or CDK4 (third panel) antibody. The Myc blot was stripped and reprobed with cyclin D antibody (Fig. 5E, second panel). Both C and K mutants were capable of associating with cyclin D-CDK4 complex, whereas the CK mutant was unable to do so (Fig. 5E). Association of the C mutant with the cyclin D-CDK4 complex appeared weaker than the wild-type and K mutant of N protein. Similarly, we checked the association of different N protein mutants with the cyclin A-CDK2 complex. Cells transfected with different plasmids were starved for 34 h followed by a 12-h stimulation with 10% FBS and 2-h labeling with [35S]cysteine/methionine promix (due to some cross-reactivity of cyclin A antibody with the Myc antibody, it was difficult to check N bands by immunoblotting, hence radiolabeling was opted for). Cells were processed for coimmunoprecipitation using anti-Myc (9E10) antibody. As expected, the wild-type and C mutant N protein co-precipitated with the cyclin A-CDK2 complex. Interestingly, the K and CK mutant proteins were also able to associate with the complex (Fig. 5F). The same blot was stripped and reprobed with CDK2 antibody (Fig. 5F, second panel). An aliquot of the lysate was immunoblotted with total p38 antibody to check equal loading (Fig. 5F, fourth panel). The CDK2 blot was air-dried and exposed to x-ray film to check the expression of N protein mutants (Fig. 5F, third panel). These experiments confirmed that the association of the N protein with cyclin D enables it to directly inhibit CDK4 activity, and since the C mutant was unable to bind cyclin D, it lost the ability to inhibit cyclin D-CDK4 kinase activity. Further, the K mutant's ability to bind the cyclin D or cyclin D-CDK4 complex and its efficient inhibition of CDK4 activity ruled out the possibility of competitive inhibition of natural CDK4 substrates by the N protein. However, since the C mutant was efficient in inhibiting the CDK2 kinase activity and the K mutant lost that ability, the possibility exists that the CDK2-inhibitory activity may be a result of competition of the N protein with natural CDK2 substrates. This hypothesis was further investigated below.
FIGURE 6.
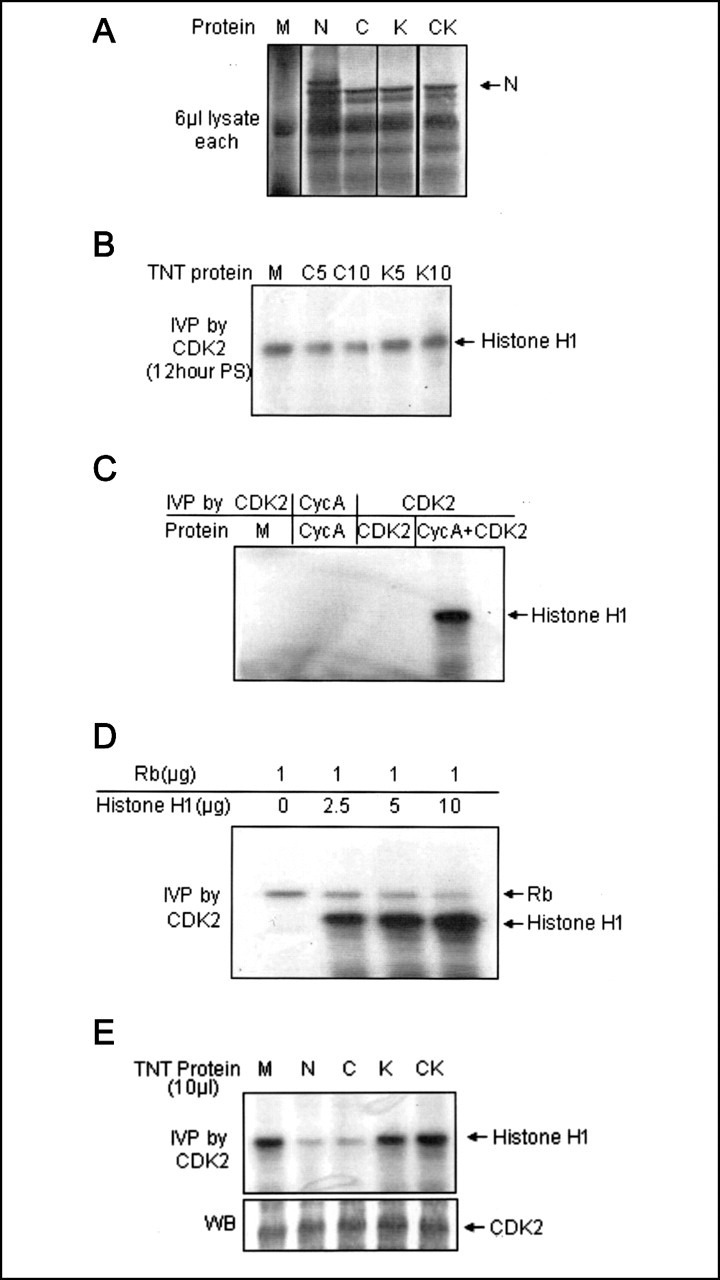
Exogenously added mutant N protein can inhibit CDK2 activity.A, expression analysis of [35S]cysteine/methionine promix-labeled mock-translated TNT lysate (lane 1) or lysate expressing different N protein mutants (lanes 2–5). 5 μl of each sample was resolved on 12% SDS-PAGE, and bands were detected by autoradiography. B, exogenously added RXL mutant N protein can inhibit CDK2 activity. Cells seeded at 50% confluence were starved for 34 h followed by 12-h stimulation with 10% bovine serum. Cell lysate was equally divided into five tubes and mixed with 5 or 10 μl of TNT-expressed C and K motif mutant N protein (mutants C5 and C10 and mutants K5 and K10, respectively) or 10 μl of mock lysate (lane 1). In vitro phosphorylation (IVP) was done with CDK2. C, bacterial lysate expressing the indicated protein was immunoprecipitated with respective antibody, and in vitro phosphorylation was done using histone H1 as a substrate. M, mock-expressed lysate. D, cyclin A-CDK2 complex reconstituted from bacterial lysate was immobilized on protein A-Sepharose beads bound to CDK2 antibody. In vitro phosphorylation was done in the presence of 1 μg of purified recombinant Rb protein or along with the indicated amount of purified histone H1 protein. E, effect of the presence of different mutant N proteins on the in vitro reconstituted CDK2 activity (upperpanel). M, mock-translated lysate. An in vitro phosphorylation assay was done as described in E. Aliquots of the lysate were immunoblotted with CDK2 antibody (lower panel). WB, Western blot.
Exogenously Added N Protein Binds to and Inhibits CDK2 Activity—N protein-mediated inhibition of CDK2 activity appeared to be regulated in two ways: first, inhibition of E2F1 activity results in decreased expression of its targets like cyclin E, cyclin A, and CDK2; second, the N protein might be acting as a competitive inhibitor to CDK2 substrates. The second possibility was predicted due to the ability of exogenously added wild-type N protein to inhibit the CDK2 activity (described in the legend to Fig. 4). This observation was further confirmed by testing the ability of exogenously added mutant N protein to inhibit the activity of COS7 cell-extracted or bacterially expressed CDK2-cyclin A complex.
The N protein mutants were expressed using the TNT kit, and expression was checked (Fig. 6A). Cells seeded at 50% confluence were starved for 34 h and stimulated with 10% FBS for 12 h, followed by lysis in immunoprecipitation buffer. Cells were equally divided into five tubes, mixed with 5 or 10 μl of C or K mutant N protein or 10 μl of mock-translated lysate, followed by the addition of 1μg of CDK2 antibody and overnight rocking at 4 °C. In vitro phosphorylation was done using histone H1 as a substrate. As expected, the C mutant could inhibit CDK2 activity, whereas the K mutant was unable to do so (Fig. 6B). Although this implied that the N protein inhibits CDK2 activity by directly binding to the cyclin-CDK2 complex, involvement of some other host factors (like recruitment of CKIs) in the above process could not be ruled out. In order to further clarify the mechanism, we tested the ability of N protein to inhibit CDK2 activity reconstituted from bacterially expressed cyclin A and CDK2.
Lysate of different BL21DE3 cells expressing cyclin A and CDK2 + Civ1 (CDK-activating kinase in vivo) were mixed in equivalent amounts, and the resulting cyclin A-CDK2 complex was immobilized on cyclin A antibody-bound protein A-Sepharose beads. Unbound proteins were removed by multiple washes in wash buffer. Expression of cyclin A and CDK2 was confirmed by immunoblotting with respective antibodies (data not shown). This complex was first assayed for CDK2 activity. Beads bearing the cyclin A-CDK2 complex were found to be enzymatically active as judged by their ability to phosphorylate histone H1 (Fig. 6C, lane 4), whereas mock-expressed or only cyclin A- or CDK2-bound beads were catalytically inactive (Fig. 6C, lanes 1–3). In order to further confirm that CDK2 activity reconstituted in vitro behaves like the native enzyme, the cyclin A-CDK2 complex immobilized on protein A-Sepharose beads was assayed for CDK2 activity in the presence of 1 μg of recombinant Rb protein and an increasing concentration of histone H1 protein. As expected, with increasing concentrations of histone H1, Rb phosphorylation was gradually decreased (Fig. 6D).
Next, CDK2-cyclin A complex bound to protein A-Sepharose was resuspended in immunoprecipitation buffer and incubated with 10 μl of TNT expressed wild-type or mutant N protein, and an in vitro phosphorylation assay was done using histone H1 as a substrate. Wild-type as well as the C mutant was capable of inhibiting the CDK2 activity, whereas the K or CK dual mutant lost this property (Fig. 6E, first panel). Aliquots of the lysate were immunoblotted with CDK2 antibody to check equal loading (Fig. 6E, second panel). This proved that the N protein could directly inhibit CDK2 activity independent of its ability to modulate CDK4 activity or the involvement of other host factors.
Down-regulation of E2F1 Target Expression and Inhibition of CDK2 Activity in SARS-CoV-infected Cells—All of the above experiments proved that exogenously expressed N protein has the property of inhibiting CDK activity and blocking S phase progression in cultured mammalian cells. However, whether the above property was functionally relevant during a natural infection was unclear. Thus, we tested the protein level of some E2F1 targets in SARS-CoV-infected cells. Vero E6 cells infected with SARS-CoV or mock-infected cells were heat- and UV-inactivated and formalin-fixed. Total cell lysate was processed as described under “Experimental Procedures” and immunoblotted with cyclin A, CDK2, P-p27 (Thr-187), nucleocapsid, and calnexin antibody. Protein levels of cyclin A and CDK2 and P-p27 were significantly down-regulated in virus-infected cells, supporting the in vitro data. Aliquots of the lysate were immunoblotted with anti-nucleocapsid antibody to check its expression in the infected sample (Fig. 7 ). Calnexin was used as a loading control. Together, these data suggest that the ability of N protein to inhibit cell cycle progression is a physiologically relevant step during the natural course of SARS CoV infection.
FIGURE 7.

Down-regulation of the expression of E2F1 targets and inhibition of CDK2 activity in SARS-CoV-infected Vero E6 cells. Mock-infected (M) or SARS-CoV-infected (S) Vero E6 cells were formalin-fixed and heat-inactivated. Aliquots of total cell lysate was immunoblotted with cyclin E (first panel), CDK2 (second panel), P-p27 (third panel), anti-nucleocapsid (N)(fourth panel), and calnexin (fifth panel) antibody, respectively. Data are representative of two independent experiments.
DISCUSSION
In this report, we provide evidence to prove that the N protein of SARS-CoV inhibits S phase progression in mammalian cells. Down-regulation of the S phase gene expression and decreased phosphorylation of a CDK2 substrate in SARS-CoV-infected cells further suggest the above property of N protein to be physiologically relevant. Thus, the study uncovers another interesting example of a process by which a viral protein manipulates the cell cycle machinery.
Many viral proteins have earlier been shown to positively or negatively modulate the cell cycle in order to create a more favorable milieu for their survival. For example, many viral gene products interact with cell cycle-regulatory proteins like cyclins and CDKs or their regulators such as p53 and Rb protein (1, 3, 9, 10, 24, 25, 26, 27, 28). Some of these interactions expedite cell cycle progression and cell proliferation, in some cases leading to the transformation of target cells. On the other hand, many other viral gene products, such as ICP0, ICP27 of herpes simplex virus, IE2 and UL69 of cytomegalovirus, and K-bZIP of KSAHV, delay cell cycle progression and transiently arrest the infected cells at the G1 stage.
Among the viral gene products that result in inhibition of CDK activity, K-bZIP protein of KSAHV has been shown to directly associate with CDK2-cyclin complex and down-regulate its kinase activity. Further, this was found to be accompanied by an increase in the protein level of p27 and p21, leading to G1 arrest (8). Similarly, in the present study, the N protein of SARS-CoV was found to associate with the cyclin-CDK complex and inhibit their activity. However, the observed phenomenon was independent of p27 and p21, since their protein level did not increase in N protein-expressing cells. This may be attributed to the fact that alternate pathways exist for the degradation of p27 at the G1 phase (29), and we did not find up-regulation of p53 protein level, which drives the expression of p21 in the event of adverse conditions.
Further, the observed phenomenon was not an experimental artifact of N protein overexpression and was ruled out in multiple experiments; for example, different N protein mutants exhibited different effects on CDK2 and CDK4, overexpression of E2F1 could alleviate the inhibitory effect of N protein in a dose-dependent manner, and exogenously added N protein could block the kinase activity of in vitro reconstituted cyclin-CDK complex.
The mechanism of the CDK inhibitory property demonstrated by N protein appeared to be unique among the viral CDK inhibitors, because nucleocapsid employs multiple mechanisms to inhibit CDK activity. This might be helpful to ensure that in the event of a failure of the ability to block CDK4 activity, CDK2 activity will still be kept under vigil so that S phase progression is prohibited. Apart from its dependence on the RXL motif to inhibit CDK4 activity and the RGNSPAR motif to inhibit CDK2 activity, N protein appeared to possess an additional property to inhibit CDK2 kinase activity, because although CK mutant was completely unable to inhibit CDK4 activity in N protein-expressing cells, some degree of inhibitory activity was elicited by the CK mutant toward CDK2 kinase activity. However, this inhibitory activity was not observed in the CDK2 kinase assay where exogenous CK mutant N protein was added, indicating that this property is independent of RXL and RGNSPAR motifs of the N protein and is dependent on other host cellular factors. It may very well be possible that the N protein also modulates host signaling pathways to inhibit CDK2 activity, thereby securing a third way to inhibit CDK2 activity. By combining all of these abilities in a single protein, the virus may be able to withstand evolutionary pressure more efficiently.
CDK4 and CDK2 play critical roles in cell cycle progression. Kinase activities of both of these proteins phosphorylate Rb or its related molecules, the p130 and p107 pocket proteins. This alleviates E2F1 sequestering by Rb, thus enabling E2F1-mediated transactivation of several S phase genes like cyclin E, cyclin A, DNA polymerase, E2F1, CDK2, histone H1, etc. (30, 31, 32, 33, 34). This is one of the crucial steps essential for S phase progression. Further, during late G1 and S phase, CDK2 phosphorylates and targets its inhibitor p27 for proteasomal degradation, which is also essential for G1 to S phase transition (33, 35). Thus, CDK4 and CDK2 activities regulate multiple pathways leading to S phase progression. Hence, by modulating the activities of both CDK4 and CDK2, the N protein doubly ensures the blockage of S phase progression. Although the physiological significance of this phenomenon during SARS-CoV infection remains to be understood, presumably it provides the virus ample time and sufficient raw materials for replication of its genome as well as for assembly and budding of progeny particles. However, further experiments using model infection systems need to be done in order to understand the exact functional relevance of the above process. Nevertheless, the present study provides possible functional relevance of nuclear localization of the N protein as well as addresses mechanistic issues by which the N protein might be inducing cell cycle arrest during SARS-CoV infection.
Acknowledgments
We thank Dr. Mark Ewen for pRC/cytomegalovirus cyclin D1-HA plasmid, Dr. Anindya Dutta for the pHis Trx-cyclin A plasmid, Dr. J. R. Nevins for the cyclin E reporter constructs, and Dr. Vijay Kumar for the pSGI-E2F1 plasmid. We are also thankful to Dr. Chetan Chitins for the immunofluorescence microscope facility and Suchi Goel and M. C. Phoon for technical help.
Footnotes
The abbreviations used are: CDK, cyclin-dependent kinase; N protein, nucleocapsid protein; SARS, severe acute respiratory syndrome; CoV, coronavirus; BrdUrd, bromodeoxyuridine, Rb, retinoblastoma, FBS, fetal bovine serum; HA, hemagglutinin; FACS, fluorescence-activated cell sorting; CAT, chloramphenicol acetyltransferase; CKI, CDK inhibitor; ERK, extracellular signal-regulated kinase.
References
- 1.Hobbs W.E., II, DeLuca N.A. J. Virol. 1999;73:8245–8255. doi: 10.1128/jvi.73.10.8245-8255.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lomonte P., Everett R.D. J. Virol. 1999;73:9456–9467. doi: 10.1128/jvi.73.11.9456-9467.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song B., Yeh K.C., Liu J., Knipe D.M. Virology. 2001;290:320–328. doi: 10.1006/viro.2001.1175. [DOI] [PubMed] [Google Scholar]
- 4.Wiebusch L., Hagemeier C. J. Virol. 1999;73:9274–9283. doi: 10.1128/jvi.73.11.9274-9283.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu M., Shenk T. J. Virol. 1999;73:676–683. doi: 10.1128/jvi.73.1.676-683.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cayrol C., Flemington E.K. EMBO J. 1996;15:2748–2759. [PMC free article] [PubMed] [Google Scholar]
- 7.Chen C.J., Sugiyama K., Kubo H., Huang C., Makino S. J. Virol. 2004;78:10410–10419. doi: 10.1128/JVI.78.19.10410-10419.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Izumiya Y., Lin S.F., Ellison T.J., Levy A.M., Mayeur G.L., Izumiya C., Kung H.J. J. Virol. 2003;77:9652–9661. doi: 10.1128/JVI.77.17.9652-9661.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flemington E.K. J. Virol. 2001;75:4475–4481. doi: 10.1128/JVI.75.10.4475-4481.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Q., Gutsch D., Kenney S. Mol. Cell. Biol. 1994;14:1929–1938. doi: 10.1128/mcb.14.3.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Surjit M., Kumar R., Mishra R.N., Reddy M.K., Chow V.T., Lal S.K. J. Virol. 2005;79:11476–11486. doi: 10.1128/JVI.79.17.11476-11486.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Surjit M., Liu B., Jameel S., Chow V.T., Lal S.K. Biochem. J. 2004;383:13–28. doi: 10.1042/BJ20040984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamb J., Ramaswamy S., Ford H.L., Contreras B., Martinez R.V., Kittrell F.S., Zahnow C.A., Patterson N., Golub T.R., Ewen M.E. Cell. 2003;114:323–334. doi: 10.1016/s0092-8674(03)00570-1. [DOI] [PubMed] [Google Scholar]
- 14.Wohlschlegel J.A., Dwyer B.T., Takeda D.Y., Dutta A. Mol. Cell. Biol. 2001;21:4868–4874. doi: 10.1128/MCB.21.15.4868-4874.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohtani K., DeGregori J., Nevins J.R. Proc. Natl. Acad. Sci. U. S. A. 1995;92:12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krishan A. J. Cell Biol. 1975;66:188–193. doi: 10.1083/jcb.66.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kramer M.F., Cook W.J., Roth F.P., Zhu J., Holman H., Knipe D.M., Coen D.M. J. Virol. 2003;77:9533–9541. doi: 10.1128/JVI.77.17.9533-9541.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munir S., Kapur V. J. Virol. 2003;77:4899–4910. doi: 10.1128/JVI.77.8.4899-4910.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ng M.L., Tan S.H., See E.E., Ooi E.E., Ling A.E. J. Gen. Virol. 2003;84:3291–3303. doi: 10.1099/vir.0.19505-0. [DOI] [PubMed] [Google Scholar]
- 21.Ikeda K., Monden T., Kanoh T., Tsujie M., Izawa H., Haba A., Ohnishi T., Sekimoto M., Tomita N., Shiozaki H., Monden M. J. Histochem. Cytochem. 1998;46:397–403. doi: 10.1177/002215549804600314. [DOI] [PubMed] [Google Scholar]
- 24.Friborg J., Jr., Kong W., Hottiger M.O., Nabel G.J. Nature. 1999;402:889–894. doi: 10.1038/47266. [DOI] [PubMed] [Google Scholar]
- 25.Godden-Kent D., Talbot S.J., Boshoff C., Chang Y., Moore P., Weiss R.A., Mittnacht S. J. Virol. 1997;71:4193–4198. doi: 10.1128/jvi.71.6.4193-4198.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagemeier C., Caswell R., Hayhurst G., Sinclair J., Kouzarides T. EMBO J. 1994;13:2897–2903. doi: 10.1002/j.1460-2075.1994.tb06584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park J., Seo T., Hwang S., Lee D., Gwack Y., Choe J. J. Virol. 2000;74:11977–11982. doi: 10.1128/jvi.74.24.11977-11982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polson A.G., Huang L., Lukac D.M., Blethrow J.D., Morgan D.O., Burlingame A.L., Ganem D. J. Virol. 2001;75:3175–3184. doi: 10.1128/JVI.75.7.3175-3184.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamura T., Hara T., Matsumoto M., Ishida N., Okumura F, Hatakeyama S., Yoshida M., Nakayama K., Nakayama K.I. Nat. Cell Biol. 2004;12:1229–1235. doi: 10.1038/ncb1194. [DOI] [PubMed] [Google Scholar]
- 30.Bruce J.L., Hurford R, Jr K., Classon M., Koh J., Dyson N. Mol. Cell. 2000;6:737–742. doi: 10.1016/s1097-2765(00)00072-1. [DOI] [PubMed] [Google Scholar]
- 31.Harbour J.W., Dean D.C. Genes Dev. 2000;14:2393–2409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- 32.Sherr C.J. Cancer Res. 2000;60:3489–3495. [PubMed] [Google Scholar]
- 33.Montagnoli A., Fiore F., Eytan E., Carrano A.C., Draetta G.F., Hershko A., Pagano M. Genes Dev. 1999;13:1181–1189. doi: 10.1101/gad.13.9.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinberg R.A. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 35.Sherr C.J., Roberts J.M. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
References
- 17.Kalra N., Kumar V. J. Biol. Chem. 2004;279:25313–25319. doi: 10.1074/jbc.M400932200. [DOI] [PubMed] [Google Scholar]
- 22.Kudoh A., Fujita M., Kiyono T., Kuzushima K., Sugaya Y., Izuta S., Nishiyama Y., Tsurumi T. J. Virol. 2003;77:851–861. doi: 10.1128/JVI.77.2.851-861.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korgaonkar C., Zhao L., Modestou M., Quelle D.E. Mol. Cell. Biol. 2002;22:196–206. doi: 10.1128/MCB.22.1.196-206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]