

Abstract
Ischemic stroke is the most common type of cerebrovascular disease and is caused by an interruption of blood flow in the brain. In this disease, two different damage areas are identifying: the lesion core, in which cells quickly die; and the penumbra (surrounding the lesion core), in which cells are functionally weakened but may recover and restore their functions. The currently approved treatments for ischemic stroke are the recombinant tissue plasminogen activator and endovascular thrombectomy, but they have a short therapeutic window (4.5 and 6 hours after stroke onset, respectively) and a low percentage of stroke patients actually receive these treatments. Memantine is an approved drug for the treatment of Alzheimer’s disease. Memantine is a noncompetitive, low affinity and use-dependent antagonist of N-methyl-D-aspartate glutamate receptor. Memantine has several advantages over developing a new drug to treat focal ischemic stroke, but the most important is that it has sufficient safe probes in preclinical models and humans, and if the preclinical studies provide more evidence about pharmacological actions in tissue protection and repair, this could help to increase the number of clinical trials. The present review summarizes the physiopathology of isquemic stroke and the pharmacological actions in neuroprotection and neuroplasticity of memantine in the post stroke stage of preclinical stroke models, to illustrate their potential to improve functional recovery in human patients.
Keywords: focal ischemic stroke, memantine, neuroplasticity, neuroprotection, stroke therapy
Introduction
Among cerebrovascular diseases, stroke is the most frequent. Worldwide, stroke is the second most common cause of death and the third most common cause of disability (Feigin et al., 2017). There are two main types of stroke, hemorrhagic and ischemic. The former represents about 15% while the latter accounts for the remaining 85%. Focal ischemic stroke is the most common and is caused by the occlusion of a cerebral artery and can be local or distal. Locally, the main cause is the formation of a thrombus or an atherosclerotic plaque. Distally, an embolus (clot) forms in another blood vessel and travels to the brain and occludes a cerebral artery (Brouns and De Deyn, 2009; Moskowitz et al., 2010).
Currently approved treatments for ischemic stroke are the recombinant tissue plasminogen activator and endovascular thrombectomy, which focus on saving the penumbra cells and limit the infarct core enlargement by removing the occlusion in the artery, but they are effective only if started within a few hours of the onset of the stroke. Treatment with recombinant tissue plasminogen activator, a fibrinolytic agent, is effective if applied within the first four and a half hours of stroke onset, however, there is a potential risk of hemorrhage (del Zoppo, 2013). Mechanical thrombectomy, on the other hand, shows benefits when applied within the first six hours of ischemic stroke onset, but the patients should be selected by appropriate imaging and treated by experienced operators (Asadi et al., 2015). Both treatments are effective but have a very limited therapeutic window and few patients have access to them or are suitable candidates. Moreover, in some cases, these treatments may cause reperfusion injury, leading to brain blood barrier disruption and hemorrhagic transformation or massive brain edema (Molina and Alvarez-Sabín, 2009). Therefore, finding new treatments with wider therapeutic windows is necessary to help more patients. Hundreds of drugs have been studied in animal stroke models, many with positive results but there is not yet an approved medication for helping recovery rehabilitation after a stroke. Many aspects are involved in this failure, including the high cost of developing a new drug, insufficient basic research, including no beneficial effect or serious side effects in clinical trials and lack of safety testing. Drug repurposing (or reprofiling) is a strategy to find a new therapeutic use for a drug approved for a different condition. This approach has many advantages over developing a new drug. For example, approved drugs are sufficiently safe in preclinical models and humans, there is a lot information regarding cellular and molecular effects generally available, the drug is probably available in the market and could be used in a clinical trial (phase II and III). Taking these advantages into consideration, we will analyze the effects of memantine and its potential as a repurposed drug to help human stroke patients. In the followings sections, we will briefly describe the physiopathology of ischemic stroke and the evidence of the benefits of memantine used after stroke in experimental animal models.
Electronic databases were searched for articles to collect all data since 2000, related to preclinical studies, involving memantne and focal ischemic stroke, using the following terms: Ischemic stroke OR Cerebral ischemia OR Oxygen-glucose deprivation AND memantine, using Pubmed. For ongoing clinical trials, the ClinicalTrials.gov electronic database was searched with the terms Stroke and Memantine. If the abstract meets the inclusion criteria, the full-text article was obtained and reviewed.
Stroke
The brain depends on a continuous and regulated blood supply to support its functional integrity. It receives nearly 15% of the cardiac output and consumes more than 20% of the metabolic substrates (oxygen and glucose), though neurons have very limited energy reserves. This suggests that neurons extract oxygen and glucose from the blood on an as-needed basis to support ongoing neural function to meet the physiological demands imposed by neural activation (Lourenço et al., 2017). The reduction or cessation of blood flow in the brain may produce different levels of damage depending on the time elapsed, cell resistance and magnitude of the ischemia, and activates a very complex cascade of interrelated cellular and molecular events with a temporal overlapping profile that evolves over minutes, hours or days, inducing injury in all cell types and damage that can range from transient to irreversible (e.g., cell death) (Brouns and De Deyn, 2009; Deb et al., 2010). This event produces two different areas of damage: the ischemic core and the penumbra. In the ischemic core, blood flow is abruptly reduced and the cells are permanently injured and die rapidly by necrosis. The size of this area will depend primarily on the duration and magnitude of the ischemia and the location of the stroke. The penumbra is located around the ischemic core. This zone is perfused by collateral blood vessels which allow cells that are structurally intact but functionally weakened (Brouns and De Deyn, 2009; Deb et al., 2010). These functionally impaired cells can be slowly incorporated into the ischemic stroke or saved. Some studies have shown a certain degree of spontaneous recovery in the weeks or months after a stroke in both humans and mice (Clarkson et al., 2013; Bergsma et al., 2017; Delavaran et al., 2017). This recovery involves adaptive changes in neurons and glial cells to partially reverse the alterations caused by an ischemic stroke. Many factors can delay or impair recovery, for example, tissue susceptibility, age, and comorbidities.
Ischemic Cascade
Ischemic cascade refers to several interrelated and overlapped pathological mechanisms that are activated after a few minutes of blood vessel occlusion and whose progress occurred on a different time scale. The first event of the ischemic cascade is the reduction of oxygen and glucose which leads to a failure to produce high-energy molecules to maintain the cellular homeostasis. This sets off several mechanisms that include ionic imbalance, excitotoxicity, calcium overload, cytotoxic and vasogenic edema, peri-infarct depolarization, oxidative and nitrosative stress, blood-brain barrier (BBB) disruption, inflammation, and apoptosis. These mechanisms are briefly described below.
Energy Failure and Ionic Imbalance
Severe decrease of blood perfusion produces a depletion of oxygen and glucose and a consequent decline of ATP levels in neurons and glial cells, which leads to the failure of ion transport pumps (e.g. sodium/potassium pump) and depolarization. Subsequently, this causes an influx of sodium, chloride and calcium ions along with water and the output of potassium ions. Moreover, there is an increase of neurotransmitter release (e.g., excitatory amino acids) and inhibition of neurotransmitter reuptake systems that enhance the accumulation of neurotransmitters in the extracellular space (Nicholls and Attwell, 1990; Katsura et al., 1994; Martin et al., 1994). Additionally, the low availability of oxygen induces anaerobic glycolysis and increases the levels of lactate, which can cause acidosis cell damage (Brouns et al., 2008).
Excitotoxicity and Calcium Overload
Excitotoxicity refers to damage caused by pathological depolarization due to the abnormal release of excitatory neurotransmitter glutamate and calcium overload (Lai et al., 2014). The extracellular glutamate interacts with ionotropic receptor subtypes N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-propionate or kainate, which depolarize the cell membrane and this event, open voltage-gated ion channels, and dramatically contributes to increase the influx of sodium, chloride and calcium ions (Meldrum et al., 1985; Rothman and Olney, 1986; Choi, 1988; Chen et al., 2008). Height calcium influx activates several calcium-dependent enzymes such as proteases, lipases, and nucleases that cause damage to macromolecules resulting in extensive cell damage that lead to cell death from liquefactive necrosis (Choi, 1988, 1995).
Cytotoxic and Vasogenic Edema
Cytotoxic edema is described as cell swelling and is caused by passive water influx that follows the abnormal accumulation of sodium ion. This type of edema is observed a few hours after ischemic stroke onset and declines within 1 day. The dramatic increase the influx of sodium, chloride and calcium ions is passively followed by water resulting in cell swelling that can cause cell death. Vasogenic edema is due to extravasation and extracellular accumulation of fluid into the cerebral parenchyma and is caused by the disruption of the BBB. Vasogenic edema develops within two or three days and last several days. Abnormal accumulation of fluids produces increased brain volume and intracranial pressure, which can induce hypoxia and blood flow reduction (Michinaga and Koyama, 2015).
Peri-Infarct Depolarizations
Peri-infarct depolarizations are spontaneous depolarization waves that propagate at slow velocity (3–5 mm/min) and occur in the penumbra due to ionic imbalance. Moreover, these waves are accompanied by prolonged recovery of cortical function, or no recovery at all, and are a causal factor in the expansion of experimental infarcts (Lauritzen et al., 2011). The number of peri-infarct depolarizations and the cumulative duration have a positive correlation with the expansion of infarct size (Mies et al., 1993; Dijkhuizen et al., 1999). The extracellular accumulation of potassium ions, glutamate, and increase of intracellular calcium in astrocytes are important conditions to trigger peri-infarct depolarizations (Fabricius et al., 1993; Duffy and MacVicar, 1996; Murphy et al., 2008).
Oxidative and nitrosative stress
Stroke activates several pathways that produce reactive oxygen and nitrogen that surpass the cellular mechanism to free radical scavenging. Calcium overload produces several effects in relation to oxidative stress, such as mitochondria depolarization and a large amount of superoxide. It also activates NADPH oxidase which produces superoxide and induces the conversion of xanthine dehydrogenase to xanthine oxidase, which generates different oxygen species, for instance, superoxide and hydrogen peroxide (Abramov et al., 2007; Brennan et al., 2009; Girouard et al., 2009). Oxidative stress can produce lipid peroxidation, protein dysfunction and DNA damage (Allen and Bayraktutan, 2009). Nitrosative stress is due mainly to the generation of the strong oxidant peroxynitrite from the combination of superoxide and nitric oxide, produced by nitric oxide synthase activated during the stroke (Beckman et al., 1990).
Brain-blood barrier disruption
BBB disruption starts with an inflammatory process caused by stagnant blood flow, due to the release of proinflammatory mediators from the endothelium activation of intravascular leukocytes, and brain parenchyma. The intravascular inflammation establishes the conditions for BBB breakdown and leukocyte invasion of the ischemic tissue (Anrather and Iadecola, 2016). Other factors released by glia and neuron cells also contribute to BBB disruption such as oxidative stress and inflammatory responses that change the endothelial tight junctions (Michinaga and Koyama, 2015).
Inflammation
After parenchyma injury, damaged cells release damage-associated molecular patterns and these molecules activate receptors such as toll-like receptors in microglia and astrocytes to initiate immune responses. A few minutes after damage onset, microglia become reactive and accumulate at the lesion core and in the penumbra. The gradual alteration in the BBB allows the infiltration of leucocytes, which occurs hours after ischemic stroke onset (Gelderblom et al., 2009; Grønberg et al., 2013). On the first day, reactive microglia show an anti-inflammatory profile or M2 phenotype, but after this period, they switch to M1 phenotype with a pro-inflammatory profile (Hu et al., 2012). Microglial cells release several pro-inflammatory mediators, such as reactive oxygen species and tumor necrosis factor-α, which promote the infiltration of circulating leukocytes (Ritzel et al., 2015). Cytokines released by M1 microglia can induce the reactivity of astrocytes (astrogliosis) within 4 to 24 hours after stimulation (Zhu et al., 2000; Nowicka et al., 2008). Moreover, leukocytes release several pro-inflammatory cytokines, such as interleukin 1β and interferon γ to damage neural structure directly or indirectly and contribute to the enlargement of the lesion core (Lambertsen et al., 2004; Allan et al., 2005). The peak inflammatory response is within 7 days after stroke onset and then slowly declines (Dirnagl et al., 1999).
Apoptosis
In the penumbra, cells die generally in a regulated manner known as apoptosis which presents a defined morphological pattern that is characterized by cell shrinkage, membrane-bound vesicles (apoptotic bodies) and pyknosis (chromatin condensation) (Puig et al., 2018). This process can be triggered by several events such as DNA damage, excessive activation of the NMDA receptor, acid-sensing ion channels, and transient receptor potential channels, among other factors (Budd et al., 2000; Broughton et al., 2009). Apoptosis can be initiated by internal (intrinsic pathway) or external events (extrinsic pathway). The intrinsic pathway involves calcium overload, disruption of mitochondria and the release of the cytochrome C, which leads to the downstream activation of caspases that damage the DNA. The extrinsic pathway is activated by specific extracellular ligands (e.g. Fas ligand) that bind to transmembrane death receptors such as Fas, TNFR-1 and p75NTR. The extracellular Fas ligand binds to Fas death receptors which trigger a series of events, including caspase 3 to damage DNA (Broughton et al., 2009). Pro and anti-apoptotic proteins have been observed in the penumbra of experimental stroke after 0.5–1 hour of model cerebral artery occlusion (Ferrer et al., 2003) and markers of apoptosis peaked at 1–2 days and persisted for 4 weeks after stroke onset (Li et al., 1995). Figures 1 and 2 show the interaction between different pathological processes in the ischemic core and penumbra, respectively.
Figure 1.
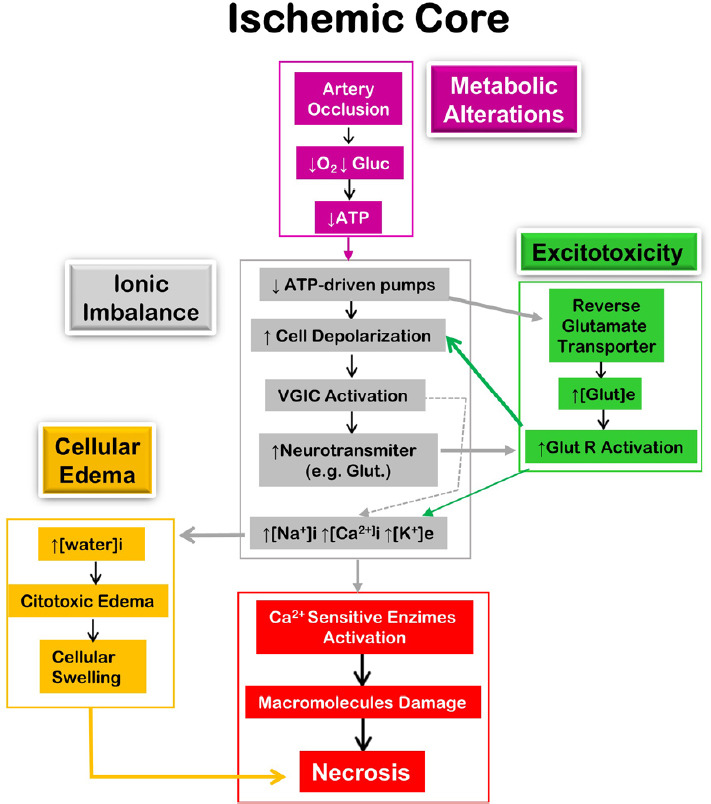
Diagrammatic representation of the main pathological mechanism in the ischemic core leading to cell death by necrosis.
[ ]: Concentration; e: extracellular; Glu: glucose; Glut: glutamate; Glut R: glutamate receptor;i: intracellular; VGIC: voltage-gated ion channels. Adapted from López-Valdés et al. (2020).
Figure 2.
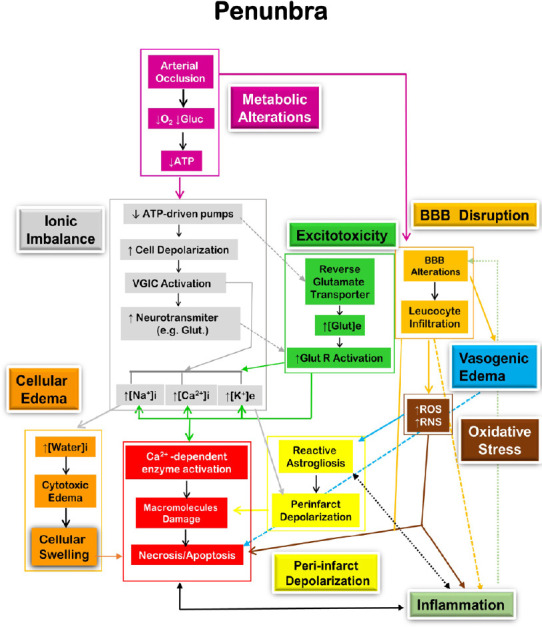
Diagrammatic representation of the main pathological mechanism in the penumbra leading to cell death by apoptosis.
[ ]: Concentration; e: extracellular; Glu: glucose; Glut: glutamate; Glut R: glutamate receptor; i: intracellular; RNS: reactive nitrogen species; ROS, reactive oxygen species; VGIC: voltage-gated ion channels. Adapted from López-Valdés et al. (2020).
Mechanism of neuroprotection and neuroplasticity
Although after a stroke neuroprotective and neuroplastic mechanisms overlap, they can be differentiated as follows: after damage, all the cells activate a series of mechanisms against the harmful factors to avoid damage (neuroprotection). In the infarct core, these mechanisms exceed their capacity to protect and the cells die. In the penumbra this mechanism contributes to limit the expansion of the infarct core and include: 1) homeostatic regulation of ions and fluids, 2) free radical scavenging, 3) neurotransmitter re-uptake, 4) production and release of anti-inflammatory cytokines, 5) BBB and neurovascular unit repair, 6) isolation of the injury site through scar formation, and 7) phagocytosis and debris clearance (Li et al., 2008; Sofroniew and Vinters, 2010; Burda and Sofroniew, 2014; Pekny and Pekna, 2014). Neuroplasticity involves the activation of mechanisms to restore the homeostatic function of the tissue previously damaged through reorganization and compensation of structural and functional deficiencies and includes: 1) production and release of growth factors, 2) angiogenesis, gliogenesis, and neurogenesis, 3) reorganization of microvascular and neuronal networks, and 4) synaptic contact growth (Ko and Yoon, 2013; Dąbrowski et al., 2019).
Stages of ischemic stroke
The classification of stroke stages used here is commonly utilized in imaging studies (Birenbaum et al., 2011). The main characteristics of these stages are summarized following.
Acute stage
The acute stage starts immediately after injury and covers the first 24 hours. In the ischemic core, the events progress rapidly, and within minutes or hours there is depletion of cellular energy stores, ionic imbalance, release of excitatory neurotransmitters and inhibition of their reuptake, peri-infarct depolarization, BBB disruption, cellular edema, and excitotoxicity, causing irreversible injury and cell death, mainly caused by excitotoxicity, ionic imbalance and oxidative and nitrosative stress (Brouns and De Deyn, 2009; Deb et al., 2010). In the penumbra, the damage is less severe since the collateral blood flow provides sufficient oxygenated blood for cells to partially preserve their energy metabolism and survive, and all cells are exposed to several deleterious mechanisms, such as excitotoxicity, oxidative and nitrosative stress, cytotoxic edema, peri-infarct depolarization, inflammation and BBB dysfunction, and apoptosis which can eventually cause cell death (Brouns and De Deyn, 2009; Deb et al., 2010).
Subacute stage
This stage includes the time period from 24 hours to 5 days after stroke onset. All the events occur in the penumbra. Several pathological mechanisms present in the acute phase are still active in this area as vasogenic edema, BBB disruption, oxidative and nitrosative stress, inflammation and apoptosis (Brouns and De Deyn, 2009; Deb et al., 2010).
Chronic stage
This stage includes the period of time after 5 days to months following stroke onset and all the events occur in the penumbra. Several processes are still active, such as vasogenic edema, apoptosis, astrogliosis, and inflammation (Brouns and De Deyn, 2009; Deb et al., 2010). On day 6 after injury, a subset of reactive astrocyte, NG2 cells, and reactive microglia start the glial scar formation (Ding, 2014; Adams and Gallo, 2018), which is completed between 2 and 4 weeks after stroke onset, beginning a chronic state that lasts many months. The perimeter of astrocyte scars has a variety of cells formed by reactive microglia, hypertrophic reactive astrocytes, viable neurons and other cell types with a long-term potential for remodeling and forming new circuits (Clarkson et al., 2013; Li et al., 2010; Overman et al., 2012), and in fact, considerable functional recovery can occur in the first weeks after stroke, mostly due to spontaneous active mechanisms (Cramer, 2008; Grefkes and Fink, 2014).
Memantine
Memantine or memantine hydrochloride, are the generic names for 3,5-dimethyladamantan-1-amine hydrochloride (PubChem CID: 181458). The drug was officially launched in Germany in 1989 to treat dementia and in 2003, the FDA approved it to treat patients with moderate to severe Alzheimer’s disease (Rogawski and Wenk, 2003; FDA, 2004). Memantine is absorbed by the gastrointestinal tract with an absolute bioavailability of 100% and follows a linear dose–concentration pattern. The peak plasma levels are reached within 3–7 hours and the elimination half-life in man is between 60 and 80 (Schmitt et al., 2007). Memantine crosses the BBB to distribute to many areas within the brain (Kornhuber and Quack, 1995; Parsons et al., 1999). Three major metabolites of memantine have been characterized and include an N-glucuronide conjugate, 6-hydroxymemantine, and 1-nitroso memantine, but between 57–82% of the memantine dose is eliminated unchanged in the urine, which suggests that the drug does not undergo extensive metabolism, besides, all major metabolites have minimal NMDA receptor activity (Schmitt et al., 2007). Due to memantine being an uncompetitive, voltage dependent NMDA receptor antagonist with moderate affinity and fast on−off kinetics, one of its main therapeutic actions is it selectively blocks the pathological over-activation of the NMDA receptor in excitotoxicity (Parsons et al., 1999; Lipton, 2006). However, evidence from in vivo and in vitro experiments, showed that memantine has pleiotropic effects that include brain-derived neurotrophic factor (BDNF) (Marvanová et al., 2001; Meisner et al., 2008; Jantas et al., 2009; Réus et al., 2010) and glial cell-derived neurotrophic factor upregulation (Wu et al., 2009), anti-apoptotic effects (Jantas et al., 2009) and reduction of neuroinflammation (Rosi et al., 2009; Wu et al., 2009).
Memantine in experimental stroke
In this review, we will analyze separately studies that applied memantine after focal permanent vessel occlusion in adults, because this is more relevant to translate into clinical stroke trials, and then, we will look at those that applied the drug in the model of occlusion/reperfusion in adults, which is similar to interventional thrombectomy (Hossmann, 2012; Sommer, 2017).
Memantine in permanent vessel occlusion models
Neuroprotective and neuroplasticity effects have been demonstrated when memantine is given acutely and chronically. Culmsee et al. (2004) showed that memantine (20 mg/kg) intraperitoneally (IP) decreased the infarct area by 10% when the drug is applied 5 minutes after stroke in mice, but failed to protect when administered at 30 or 90 minutes after stroke. Another study conducted in rabbits (Lapchak, 2006), showed improved behavior when the memantine (25 mg/kg) was applied intravenously (IV) acutely (≤ 60 minutes after stroke). López-Valdés et al., (2014) treated mice with memantine (30 mg/kg) in drinking water for 28 days after stroke and the treatment significantly improved their functional locomotor performance, increased the forepaw sensory map area, vascular density, BDNF, and phosphorylated-tropomyosin–related kinase-B receptor in the penumbra and a decrease in reactive astrogliosis was observed in the same area. Lastly, no differences were identified in infarct and glial scar size between memantine and control animals.
Memantine in occlusion/reperfusion models
Seif el Nasr et. al. (1990) used a rat model of transient forebrain ischemia induced by clamping both carotid arteries for 10 minutes to study the effects when the drug (10 and 20 mg/kg) is applied I.P. immediately after ischemia. They found that only 10 mg/kg significantly decreased the neuronal damage. Another study also in rats showed that memantine (30 mg/kg) administered via nasogastric intubations after reperfusion, showed significant reductions in the infarct volume and significant improvement in the neurological score at 24 and 72 hours after treatment (Aluclu et al., 2008). Killic et al. (2013) analyzed the effects of memantine (20 mg/kg) alone or in combination with melatonin on pathways related to vascular leakage after stroke, when applied immediately after reperfusion. They evaluated the effects 24 hours after treatment and found that memantine significantly decreases the stroke volume, the BBB permeability, and the fragmented DNA. Another research group used memantine (20 mg/kg) immediately after artery occlusion, followed by a maintenance dose of 1 mg/kg at 12 hours intervals. After 24 hours of artery occlusion, they found that memantine improved neurological deficits, decreased infarct volume, decreased apoptosis and calpain (a calcium dependent protease) in the penumbra (Chen et al., 2017). Wang et al. (2017) studied the effect of memantine (20 mg/kg per day) applied in the subacute stage (72 hours after stroke) and delivered it subcutaneously over 28 days. They found that memantine significantly decreased astrogliosis and increased concentrations of BDNF, glial cell-derived neurotrophic factor and the vascular endothelial growth factor in the cortex and striatum (Wang et al., 2017). Some of the effects mentioned above, can be supported by the results obtained in vitro with brain slices and cell culture exposed to oxygen-glucose deprivation, a common model of stroke in vitro (Holloway and Gavins, 2016). Rat hippocampal slices exposed to oxygen-glucose deprivation and treated with memantine (1–30 μM) produced a concentration-dependent neuroprotection (Öz and Saybaşılı, 2016; Landucci et al., 2018). In the same preparation, memantine (10 μM) reduced lactate dehydrogenase (LDH) by 40%, a marker of tissue damage (Sobrado et al., 2004). In primary cultured hippocampal neurons, ATP-depleted by sodium azide (5 mM NaN3) treated with memantine (10 or 50 μM) significantly increased survival neurons and decreased the lactate dehydrogenase release (Chen et al., 2017). In another study in primary cultured cortical neurons, memantine (10 μM) significantly decreased the apoptosis and increased cell viability (Chen et al., 2016). Another source that can also help, are the results obtained in experiments that applied memantine before the artery occlusion and the results show that memantine decreased oxidative stress (Tanaka et al., 2018), the number of reactive microglia and astrocytes, endothelial cells damaged, matrix metallopeptidase 9 secretion, an enzyme involved in the degradation of extracellular matrix, and improved the BBB integrity (Chen et al., 2016). Furthermore, indirect evidence from the effects of memantine in another disease can also help. For example, in preclinical models of Alzheimer’s disease, chronic treatment with memantine decreases neuroinflammation, oxidative stress, improves memory, and increases synapse density and BDNF concentration (Folch et al., 2018).
Conclusions
Memantine, an approved drug to treat patients with moderate to severe Alzheimer’s disease, showed pleiotropic effects in models of experimental stroke (Figure 3), suggesting that it participates in different regulatory pathways of neuroprotection and neuroplasticity. The protective role of memantine is expressed as the reduction of infarct volume and neurological deficits if the drug is applied (IP) immediately or a few minutes after artery occlusion, which suggests its utility in the acute phase as a complement to thrombolytic therapy and endovascular thrombectomy. Moreover, some results suggest that memantine decreases apoptosis, proteases, and BBB permeability, which can contribute to the protective effect. Long-term use (28 days, oral administration) showed neuroplastic effects such as reconfiguration of optical maps, significant increase of BDNF, angiogenesis, and functional recovery, decrease of astrogliosis, but no difference in the infarct size, which suggests that memantine could be useful in the chronic poststroke stage, which is a more realistic possibility. Although the results of the experiments included in this analysis showed that memantine has neuroprotection and neuroplasticity effects, more experiments are needed to confirm and expand the knowledge of the molecular mechanism involved in the therapeutic actions of memantine, particularly at the subacute and chronic stages. It is clear that all the evidence presented above contains some caveats because all the experiments were performed using young male animals without comorbidities and it is known that stroke is more severe in older animals and humans, and human patients generally show comorbidities (Sohrabji et al., 2013; Barnes, 2015), consequently animals with these conditions and females should be included in the experiments. Finally, at this time, only two ongoing clinical trials are testing the efficacy of memantine on patients with ischemic stroke, one is recruiting patients (ClinicalTrials.gov identifiers: NCT02144584) and the status of the other (NCT02535611) is unknown. Therefore, more clinical trials are needed to evaluate the effects of this drug in improving the outcomes of stroke patients .
Figure 3.
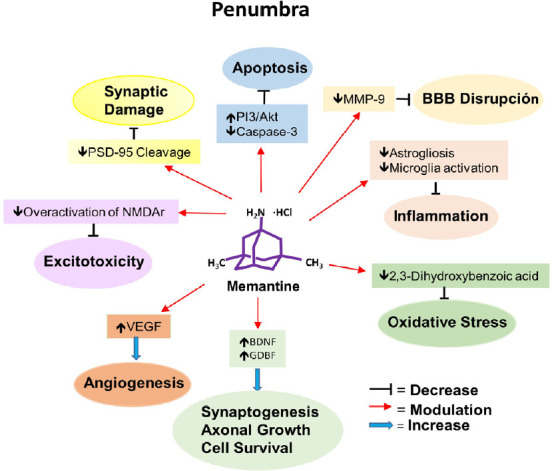
Diagrammatic representation of the main therapeutic effects of memantine on penumbra.
Akt: Protein kinase B; BBB: brain-blood barrier; BDNF: brain-derived neurotrophic factor; GDNF: glial cell-derived neurotrophic factor; MMP-9: matrix metallopeptidase 9; NMDAr: N-methyl-D-aspartate receptor; PI3-K: phosphatidylinositol-3 kinase; PSD-95: postsynaptic density protein 95; VEGF: vascular endothelial growth factor.
Footnotes
C-Editors: Zhao M, Li JY; T-Editor: Jia Y
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: None.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
References
- 1.Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams KL, Gallo V. The diversity and disparity of the glial scar. Nat Neurosci. 2018;21:9–15. doi: 10.1038/s41593-017-0033-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- 4.Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4:461–470. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- 5.Anrather J, Iadecola C. Inflammation and stroke: an overview. Neurother J Am Soc Exp Neurother. 2016;13:661–670. doi: 10.1007/s13311-016-0483-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aluclu MU, Arslan S, Acar A, Guzel A, Bahceci S, Yaldiz M. Evaluation of effects of memantine on cerebral ischemia in rats. Neurosciences (Riyadh) 2008;13:113–116. [PubMed] [Google Scholar]
- 7.Asadi H, Dowling R, Yan B, Wong S, Mitchell P. Advances in endovascular treatment of acute ischaemic stroke. Intern Med J. 2015;45:798–805. doi: 10.1111/imj.12652. [DOI] [PubMed] [Google Scholar]
- 8.Barnes PJ. Mechanisms of development of multimorbidity in the elderly. Eur Respir J. 2015;45:790–806. doi: 10.1183/09031936.00229714. [DOI] [PubMed] [Google Scholar]
- 9.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergsma DP, Elshout JA, van den Berg AV. Segregation of spontaneous and training induced recovery from visual field defects in subacute stroke patients. Front Neurol. 2017;8:681. doi: 10.3389/fneur.2017.00681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Birenbaum D, Bancroft LW, Felsberg GJ. Imaging in acute stroke. West J Emerg Med. 2011;12:67–76. [PMC free article] [PubMed] [Google Scholar]
- 12.Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- 14.Brouns R, De Deyn PP. The complexity of neurobiological processes in acute ischemic stroke. Clin Neurol Neurosurg. 2009;111:483–495. doi: 10.1016/j.clineuro.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Brouns R, Sheorajpanday R, Wauters A, De Surgeloose D, Mariën P, De Deyn PP. Evaluation of lactate as a marker of metabolic stress and cause of secondary damage in acute ischemic stroke or TIA. Clin Chim Acta. 2008;397:27–31. doi: 10.1016/j.cca.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 16.Budd SL, Tenneti L, Lishnak T, Lipton SA. Mitochondrial and extramitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc Natl Acad Sci U S A. 2000;97:6161–6166. doi: 10.1073/pnas.100121097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen B, Wang G, Li W, Liu W, Lin R, Tao J, Jiang M, Chen L, Wang Y. Memantine attenuates cell apoptosis by suppressing the calpain-caspase-3 pathway in an experimental model of ischemic stroke. Exp Cell Res. 2017;351:163–172. doi: 10.1016/j.yexcr.2016.12.028. [DOI] [PubMed] [Google Scholar]
- 19.Chen M, Lu TJ, Chen XJ, Zhou Y, Chen Q, Feng XY, Xu L, Duan WH, Xiong ZQ. Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke. 2008;39:3042–3048. doi: 10.1161/STROKEAHA.108.521898. [DOI] [PubMed] [Google Scholar]
- 20.Chen ZZ, Yang DD, Zhao Z, Yan H, Ji J, Sun XL. Memantine mediates neuroprotection via regulating neurovascular unit in a mouse model of focal cerebral ischemia. Life Sci. 2016;150:8–14. doi: 10.1016/j.lfs.2016.02.081. [DOI] [PubMed] [Google Scholar]
- 21.Choi DW. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988;11:465–469. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- 22.Choi DW. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- 23.Clarkson AN, López-Valdés HE, Overman JJ, Charles AC, Brennan KC, Thomas Carmichael S. Multimodal examination of structural and functional remapping in the mouse photothrombotic stroke model. J Cereb Blood Flow Metab. 2013;33:716–723. doi: 10.1038/jcbfm.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cramer SC. Repairing the human brain after stroke. II Restorative therapies. Ann Neurol. 2008;63:549–560. doi: 10.1002/ana.21412. [DOI] [PubMed] [Google Scholar]
- 25.Culmsee C, Junker V, Kremers W, Thal S, Plesnila N, Krieglstein J. Combination therapy in ischemic stroke: synergistic neuroprotective effects of memantine and clenbuterol. Stroke. 2004;35:1197–1202. doi: 10.1161/01.STR.0000125855.17686.6d. [DOI] [PubMed] [Google Scholar]
- 26.Dąbrowski J, Czajka A, Zielińska-Turek J, Jaroszyński J, Furtak-Niczyporuk M, Mela A, Poniatowski ŁA, Drop B, Dorobek M, Barcikowska-Kotowicz M, Ziemba A. Brain functional reserve in the context of neuroplasticity after stroke. Neural Plast. 2019;2019:9708905. doi: 10.1155/2019/9708905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deb P, Sharma S, Hassan KM. Pathophysiologic mechanisms of acute ischemic stroke: An overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology. 2010;17:197–218. doi: 10.1016/j.pathophys.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 28.del Zoppo GJ. Plasminogen activators and ischemic stroke: conditions for acute delivery. Semin Thromb Hemost. 2013;39:406–425. doi: 10.1055/s-0033-1338126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delavaran H, Aked J, Sjunnesson H, Lindvall O, Norrving B, Kokaia Z, Lindgren A. Spontaneous recovery of upper extremity motor impairment after ischemic stroke: implications for stem cell-based therapeutic approaches. Transl Stroke Res. 2017;8:351–361. doi: 10.1007/s12975-017-0523-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dijkhuizen RM, Beekwilder JP, van der Worp HB, Berkelbach van der Sprenkel JW, Tulleken KA, Nicolay K. Correlation between tissue depolarizations and damage in focal ischemic rat brain. Brain Res. 1999;840:194–205. doi: 10.1016/s0006-8993(99)01769-2. [DOI] [PubMed] [Google Scholar]
- 31.Ding S. Dynamic reactive astrocytes after focal ischemia. Neural Regen Res. 2014;9:2048–2052. doi: 10.4103/1673-5374.147929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 33.Duffy S, MacVicar BA. In vitro ischemia promotes calcium influx and intracellular calcium release in hippocampal astrocytes. J Neurosci. 1996;16:71–81. doi: 10.1523/JNEUROSCI.16-01-00071.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fabricius M, Jensen LH, Lauritzen M. Microdialysis of interstitial amino acids during spreading depression and anoxic depolarization in rat neocortex. Brain Res. 1993;612:61–69. doi: 10.1016/0006-8993(93)91644-8. [DOI] [PubMed] [Google Scholar]
- 35.Feigin VL, Norrving B, Mensah GA. Global burden of stroke. Circ Res. 2017;120:439–448. doi: 10.1161/CIRCRESAHA.116.308413. [DOI] [PubMed] [Google Scholar]
- 36.Ferrer I, Friguls B, Dalfó E, Justicia C, Planas AM. Caspase-dependent and caspase-independent signalling of apoptosis in the penumbra following middle cerebral artery occlusion in the adult rat. Neuropathol Appl Neurobiol. 2003;29:472–481. doi: 10.1046/j.1365-2990.2003.00485.x. [DOI] [PubMed] [Google Scholar]
- 37.FDA. First drug for moderate-to-severe Alzheimer’s. FDA Consum. 2004;38:3. [PubMed] [Google Scholar]
- 38.Folch J, Busquets O, Ettcheto M, Sánchez-López E, Castro-Torres RD, Verdaguer E, Garcia ML, Olloquequi J, Casadesús G, Beas-Zarate C, Pelegri C, Vilaplana J, Auladell C, Camins A. (n. d.) Memantine for the Treatment of Dementia: A Review on its Current and Future Applications. J Alzheimers Dis. 62:1223–1240. doi: 10.3233/JAD-170672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe C-U, Siler DA, Arumugam TV, Orthey E, Gerloff C, Tolosa E, Magnus T. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–1857. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- 40.Girouard H, Wang G, Gallo EF, Anrather J, Zhou P, Pickel VM, Iadecola C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J Neurosci. 2009;29:2545–2552. doi: 10.1523/JNEUROSCI.0133-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grefkes C, Fink GR. Connectivity-based approaches in stroke and recovery of function. Lancet Neurol. 2014;13:206–216. doi: 10.1016/S1474-4422(13)70264-3. [DOI] [PubMed] [Google Scholar]
- 42.Grønberg NV, Johansen FF, Kristiansen U, Hasseldam H. Leukocyte infiltration in experimental stroke. J Neuroinflammation. 2013;10:115. doi: 10.1186/1742-2094-10-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holloway PM, Gavins FN. Modeling ischemic stroke in vitro: status Quo and future perspectives. Stroke. 2016;47:561–569. doi: 10.1161/STROKEAHA.115.011932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hossmann KA. The two pathophysiologies of focal brain ischemia: implications for translational stroke research. J Cereb Blood Flow Metab. 2012;32:1310–1316. doi: 10.1038/jcbfm.2011.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke J Cereb Circ. 2012;43:3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- 46.Jantas D, Szymanska M, Budziszewska B, Lason W. An involvement of BDNF and PI3-K/Akt in the anti-apoptotic effect of memantine on staurosporine-evoked cell death in primary cortical neurons. Apoptosis Int J Program Cell Death. 2009;14:900–912. doi: 10.1007/s10495-009-0370-6. [DOI] [PubMed] [Google Scholar]
- 47.Katsura K, Kristián T, Siesjö BK. Energy metabolism, ion homeostasis, and cell damage in the brain. Biochem Soc Trans. 1994;22:991–996. doi: 10.1042/bst0220991. [DOI] [PubMed] [Google Scholar]
- 48.Kilic U, Yilmaz B, Reiter RJ, Yüksel A, Kilic E. Effects of memantine and melatonin on signal transduction pathways vascular leakage and brain injury after focal cerebral ischemia in mice. Neuroscience. 2013;237:268–276. doi: 10.1016/j.neuroscience.2013.01.059. [DOI] [PubMed] [Google Scholar]
- 49.Ko SB, Yoon BW. Mechanisms of functional recovery after stroke. Front Neurol Neurosci. 2013;32:1–8. doi: 10.1159/000346405. [DOI] [PubMed] [Google Scholar]
- 50.Kornhuber J, Quack G. Cerebrospinal fluid and serum concentrations of the N-methyl-D-aspartate (NMDA) receptor antagonist memantine in man. Neurosci Lett. 1995;195:137–139. doi: 10.1016/0304-3940(95)11785-u. [DOI] [PubMed] [Google Scholar]
- 51.Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157–188. doi: 10.1016/j.pneurobio.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 52.Lambertsen KL, Gregersen R, Meldgaard M, Clausen BH, Heibøl EK, Ladeby R, Knudsen J, Frandsen A, Owens T, Finsen B. A role for interferon-gamma in focal cerebral ischemia in mice. J Neuropathol Exp Neurol. 2004;63:942–955. doi: 10.1093/jnen/63.9.942. [DOI] [PubMed] [Google Scholar]
- 53.Landucci E, Filippi L, Gerace E, Catarzi S, Guerrini R, Pellegrini-Giampietro DE. Neuroprotective effects of topiramate and memantine in combination with hypothermia in hypoxic-ischemic brain injury in vitro and in vivo. Neurosci Lett. 2018;668:103–107. doi: 10.1016/j.neulet.2018.01.023. [DOI] [PubMed] [Google Scholar]
- 54.Lapchak PA. Memantine, an uncompetitive low affinity NMDA open-channel antagonist improves clinical rating scores in a multiple infarct embolic stroke model in rabbits. Brain Res. 2006;1088:141–147. doi: 10.1016/j.brainres.2006.02.093. [DOI] [PubMed] [Google Scholar]
- 55.Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31:17–35. doi: 10.1038/jcbfm.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Ståhlberg A, Aprico K, Larsson K, Yabe T, Moons L, Fotheringham A, Davies I, Carmeliet P, Schwartz JP, Pekna M, Kubista M, Blomstrand F, Maragakis N, et al. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab. 2008;28:468–481. doi: 10.1038/sj.jcbfm.9600546. [DOI] [PubMed] [Google Scholar]
- 57.Li Y, Chopp M, Jiang N, Yao F, Zaloga C. Temporal profile of in situ DNA fragmentation after transient middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1995;15:389–397. doi: 10.1038/jcbfm.1995.49. [DOI] [PubMed] [Google Scholar]
- 58.Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- 59.López-Valdés HE, Clarkson AN, Ao Y, Charles AC, Carmichael ST, Sofroniew MV, Brennan KC. Memantine enhances recovery from stroke. Stroke. 2014;45:2093–2100. doi: 10.1161/STROKEAHA.113.004476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.López-Valdés HE, Mendoza-Rojas MX, Martínez-Coria H. Physiology and pathology of neuroglia. 2020. Astrogliosis in stroke; pp. 144–176. Daniel Reyes Haro, INB-UNAM. [Google Scholar]
- 61.Lourenço CF, Ledo A, Barbosa RM, Laranjinha J. Neurovascularneuroenergetic coupling axis in the brain: master regulation by nitric oxide and consequences in aging and neurodegeneration. Free Radic Biol Med. 2017;108:668–682. doi: 10.1016/j.freeradbiomed.2017.04.026. [DOI] [PubMed] [Google Scholar]
- 62.Martin RL, Lloyd HG, Cowan AI. The early events of oxygen and glucose deprivation: setting the scene for neuronal death. Trends Neurosci. 1994;17:251–257. doi: 10.1016/0166-2236(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 63.Marvanová M, Lakso M, Pirhonen J, Nawa H, Wong G, Castrén E. The neuroprotective agent memantine induces brain-derived neurotrophic factor and trkB receptor expression in rat brain. Mol Cell Neurosci. 2001;18:247–258. doi: 10.1006/mcne.2001.1027. [DOI] [PubMed] [Google Scholar]
- 64.Meisner F, Scheller C, Kneitz S, Sopper S, Neuen-Jacob E, Riederer P, ter Meulen V, Koutsilieri E German Competence Network HIV/AIDS. Memantine upregulates BDNF and prevents dopamine deficits in SIV-infected macaques: a novel pharmacological action of memantine. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2008;33:2228–2236. doi: 10.1038/sj.npp.1301615. [DOI] [PubMed] [Google Scholar]
- 65.Meldrum B, Evans M, Griffiths T, Simon R. Ischaemic brain damage: the role of excitatory activity and of calcium entry. Br J Anaesth. 1985;57:44–46. doi: 10.1093/bja/57.1.44. [DOI] [PubMed] [Google Scholar]
- 66.Michinaga S, Koyama Y. Pathogenesis of brain edema and investigation into anti-edema drugs. Int J Mol Sci. 2015;16:9949–9975. doi: 10.3390/ijms16059949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mies G, Iijima T, Hossmann KA. Correlation between peri-infarct DC shifts and ischaemic neuronal damage in rat. Neuroreport. 1993;4:709–711. doi: 10.1097/00001756-199306000-00027. [DOI] [PubMed] [Google Scholar]
- 68.Molina CA, Alvarez-Sabín J. Recanalization and reperfusion therapies for acute ischemic stroke. Cerebrovasc Dis Basel Switz 27 Suppl. 2009;1:162–167. doi: 10.1159/000200455. [DOI] [PubMed] [Google Scholar]
- 69.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murphy TH, Li P, Betts K, Liu R. Two-photon imaging of stroke onset in vivo reveals that NMDA-receptor independent ischemic depolarization is the major cause of rapid reversible damage to dendrites and spines. J Neurosci. 2008;28:1756–1772. doi: 10.1523/JNEUROSCI.5128-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nicholls D, Attwell D. The release and uptake of excitatory amino acids. Trends Pharmacol Sci. 1990;11:462–468. doi: 10.1016/0165-6147(90)90129-v. [DOI] [PubMed] [Google Scholar]
- 72.Nowicka D, Rogozinska K, Aleksy M, Witte OW, Skangiel-Kramska J. Spatiotemporal dynamics of astroglial and microglial responses after photothrombotic stroke in the rat brain. Acta Neurobiol Exp (Warsz) 2008;68:155–168. doi: 10.55782/ane-2008-1685. [DOI] [PubMed] [Google Scholar]
- 73.Overman JJ, Clarkson AN, Wanner IB, Overman WT, Eckstein I, Maguire JL, Dinov ID, Toga AW, Carmichael ST. A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc Natl Acad Sci U S A. 2012;109:E2230–2239. doi: 10.1073/pnas.1204386109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Öz P, Saybaşılı H. Data on pharmacological applications and hypothermia protection against in vitro oxygen-glucose-deprivation-related neurodegeneration of adult rat CA1 region. Data Brief. 2016;10:373–376. doi: 10.1016/j.dib.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist--a review of preclinical data. Neuropharmacology. 1999;38:735–767. doi: 10.1016/s0028-3908(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 76.Pekny M, Pekna M. Astrocyte Reactivity and Reactive Astrogliosis: Costs and Benefits. Physiological Reviews. 2014;94:1077–1098. doi: 10.1152/physrev.00041.2013. [DOI] [PubMed] [Google Scholar]
- 77.Puig B, Brenna S, Magnus T. Molecular communication of a dying neuron in stroke. Int J Mol Sci. 2018;19:2834. doi: 10.3390/ijms19092834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Réus GZ, Stringari RB, Kirsch TR, Fries GR, Kapczinski F, Roesler R, Quevedo J. Neurochemical and behavioural effects of acute and chronic memantine administration in rats: Further support for NMDA as a new pharmacological target for the treatment of depression. Brain Res Bull. 2010;81:585–589. doi: 10.1016/j.brainresbull.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 79.Ritzel RM, Patel AR, Grenier JM, Crapser J, Verma R, Jellison ER, McCullough LD. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation. 2015;12:106. doi: 10.1186/s12974-015-0329-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rogawski MA, Wenk GL. The neuropharmacological basis for the use of memantine in the treatment of Alzheimer’s disease. CNS Drug Rev. 2003;9:275–308. doi: 10.1111/j.1527-3458.2003.tb00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rosi S, Ramirez-Amaya V, Vazdarjanova A, Esparza EE, Larkin PB, Fike JR, Wenk GL, Barnes CA. Accuracy of hippocampal network activity is disrupted by neuroinflammation: rescue by memantine. Brain J Neurol. 2009;132:2464–2477. doi: 10.1093/brain/awp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic--ischemic brain damage. Ann Neurol. 1986;19:105–111. doi: 10.1002/ana.410190202. [DOI] [PubMed] [Google Scholar]
- 83.Schmitt F, Ryan M, Cooper G. A brief review of the pharmacologic and therapeutic aspects of memantine in Alzheimer’s disease. Expert Opin Drug Metab Toxicol. 2007;3:135–141. doi: 10.1517/17425255.3.1.135. [DOI] [PubMed] [Google Scholar]
- 84.Seif el Nasr M, Peruche B, Rossberg C, Mennel HD, Krieglstein J. Neuroprotective effect of memantine demonstrated in vivo and in vitro. Eur J Pharmacol. 1990;185:19–24. doi: 10.1016/0014-2999(90)90206-l. [DOI] [PubMed] [Google Scholar]
- 85.Sobrado M, Roda JM, López MG, Egea J, García AG. Galantamine and memantine produce different degrees of neuroprotection in rat hippocampal slices subjected to oxygen-glucose deprivation. Neurosci Lett. 2004;365:132–136. doi: 10.1016/j.neulet.2004.04.067. [DOI] [PubMed] [Google Scholar]
- 86.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sohrabji F, Bake S, Lewis DK. Age-related changes in brain support cells: Implications for stroke severity. Neurochem Int. 2013;63:291–301. doi: 10.1016/j.neuint.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sommer CJ. Ischemic stroke: experimental models and reality. Acta Neuropathol. 2017;133:245–261. doi: 10.1007/s00401-017-1667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tanaka A, Ito Y, Kawasaki H, Kitabayashi C, Nishioka R, Yamazato M, Ishizawa K, Nagai T, Hirayama M, Takahashi K, Yamamoto T, Araki N. Effects of memantine on nitric oxide production and hydroxyl radical metabolism during cerebral ischemia and reperfusion in mice. J Stroke Cerebrovasc Dis. 2018;27:1609–1615. doi: 10.1016/j.jstrokecerebrovasdis.2018.01.014. [DOI] [PubMed] [Google Scholar]
- 90.Wang Y, Sanchez-Mendoza EH, Doeppner TR, Hermann DM. Post-acute delivery of memantine promotes post-ischemic neurological recovery, peri-infarct tissue remodeling, and contralesional brain plasticity. J Cereb Blood Flow Metab. 2017;37:980–993. doi: 10.1177/0271678X16648971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu HM, Tzeng NS, Qian L, Wei SJ, Hu X, Chen SH, Rawls SM, Flood P, Hong JS, Lu RB. Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology. 2009;34:2344–2357. doi: 10.1038/npp.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhu Y, Roth-Eichhorn S, Braun N, Culmsee C, Rami A, Krieglstein J. The expression of transforming growth factor-beta1 (TGF-beta1) in hippocampal neurons: a temporary upregulated protein level after transient forebrain ischemia in the rat. Brain Res. 2000;866:286–298. doi: 10.1016/s0006-8993(00)02240-x. [DOI] [PubMed] [Google Scholar]