

Abstract
Genetic neurodevelopmental disorders are characterized by abnormal neurophysiological and behavioral phenotypes, affecting individuals worldwide. While the subject has been heavily researched, current treatment options relate mostly to alleviating symptoms, rather than targeting the altered genome itself. In this review, we address the neurogenetic basis of neurodevelopmental disorders, genetic tools that are enabling precision research of these disorders in animal models, and postnatal gene-therapy approaches for neurodevelopmental disorders derived from preclinical studies in the laboratory.
Keywords: adeno-associated virus, CRISPR-Cas9, developmental window, genetic therapy, mouse models, neurodevelopmental disorders, non-human primates, preclinical studies
Neurodevelopmental Disorders
Most neurodevelopmental disorders (NDDs) are caused by gene mutations or deletions that result in abnormal expression or regulation of a single gene or set of genes (Deciphering Developmental Disorders Study, 2015). NDDs pose an immense challenge for scientists who are seeking to decipher the mechanisms governing the various abnormalities. Furthermore, physicians can only help alleviate the symptomatic conditions, while cognitive properties remain damaged. The prevalence of NDDs varies greatly. For example, the prevalence of autism spectrum disorder (ASD) and pervasive developmental disorders is believed to be around 60 out of 10,000 successful births (Elsabbagh et al., 2012). Rett syndrome (RTT) incidence is evaluated at 1 out of 10,000–15,000 successful births (Hagberg, 1985), while that of Williams syndrome (WS) is 1 out of 7500 successful births (Strømme et al., 2002) and Angelman syndrome (AS) prevalence is estimated at around 1 out of 20,000 successful births (Williams et al., 1995). The financial cost of supporting an individual with a NDD throughout his or her life is estimated at over $2 million, with special education, loss of parent productivity, and residential and general health care during adulthood being the main cost components (Buescher et al., 2014).
Due to the advances made in molecular biology over the last few decades, the genetic basis of some NDDs has been validated. For instance, the etiology and genetic causes of WS (Pober, 2010), AS (Knoll et al., 1989) and RTT (Amir et al., 1999) have been studied and discovered. However, not all NDDs can be linked to a genetic cause. For example, ASD represents a spectrum of disorders that vary in phenotypic characterization and severity, having a complex and heterogeneous etiology. Recent technological advances in molecular biology and genetic tools have led to an increasing number of studies focused on the genetic etiology of ASDs, but we still can only account for genetically related etiology in about 10–20% of ASD cases (Geschwind, 2011).
Because the differences in NDD etiology are broad, the affected neurobiological properties differ, leading to differential onset time of the altered neurodevelopment (Thurm et al., 2018). In some NDDs, phenotypic defects can be observed in as early as embryonic development, or immediately at birth, and some show normal development until a later age, such as fragile X syndrome (Garber et al., 2008). Similar to the time of symptom onset, the time of occurrence of deficits and damage to sensitive central nervous system (CNS)-related properties varies, from as early as the embryonic stage to early postnatal stages. NDDs may affect multiple systems other than the CNS, leading to vascular-related complications, muscular impairments, hormone and ion imbalances, and other physiological and systemic complications. Overall, these defects, occurring during critical time windows of development of various systems, may cause irreversible damage that persists through development and adulthood (Meredith, 2015). Thus, the developmental aspect is crucial, meaning that early intervention is preferred.
Current therapeutic approaches to NDDs integrate behavioral cognitive therapy with pharmaceutical agents to treat prominent symptoms. However, recent advances in genetic tools for postnatal genetic manipulations, such as restoration or in vivo editing of a gene of interest, offer a unique approach for therapeutic intervention, with the potential to rescue the genetic disorder directly, rather than only treating the symptoms. In this review, we present current knowledge on in vivo postnatal genetic treatments, focusing on studies performed with animal models of NDDs.
Search Strategy and Selection Criteria
Studies cited in this review were found on the PubMed and Google Scholar databases, between January and February 2020, using mainly the search terms: Neurodevelopmental disorders, Gene restoration, Genetic therapy, Adeno-associated virus, CRISPR-Cas9, Preclinical studies, Developmental window, Mouse models, Non-human primates, and various combinations of the above terms.
Neurogenetic Basis of Neurodevelopmental Disorders
For a few decades now, many NDDs have been thought to have a neurogenetic cause. Studies conducted on monozygotic and dizygotic twins, along with those on other affected family members have revealed a strong association between common genetic conditions and disorders such as ASD (Tick et al., 2016) and schizophrenia (Sullivan et al., 2003). Advances in molecular biology have offered a unique opportunity to examine the precise genetic mechanism of several disorders. For example, gene-association studies have pinpointed specific loci that are susceptible to genetic abnormalities, marking worthy candidates for further study and therapeutic targeting (Niemi et al., 2018). Together, the technological advances and related studies have enabled the identification of few main mechanisms responsible for genetic alterations in NDDs.
Alterations in gene dosage due to copy number variations
Copy number variation (CNV) may refer to either deletion, duplication or inversion of a genomic sequence. CNV is a genetic mechanism that allows the creation of genetic diversity in a population, but may also be an underlying cause for various NDDs (Sebat et al., 2007; Takumi and Tamada, 2018). CNVs cause genomic differences via structural variance, which can result in complex rearrangements of alleles and chromosomes (Malhotra and Sebat, 2012) through different mechanisms (Zhang et al., 2009). Although single-nucleotide polymorphisms (SNPs) are the main source of general genetic variation in the population, CNV effects have been shown to differ from those of SNPs on genetic variation in gene expression, such as altered gene dosage and gene regulation, thus emphasizing the importance of CNVs in genetic disorders (Stranger et al., 2007). CNVs are ubiquitously present in the human genome, resulting in different levels of gene expression across individuals (Freeman et al., 2006). The understanding of CNVs’ importance in genetic and phenotypic variance in humans has led to an increasing number of studies aimed at elucidating the etiology of different conditions, such as attention-deficit/hyperactivity disorder (ADHD) (Elia et al., 2010), ASD (Pinto et al., 2010) and schizophrenia (Xu et al., 2008).
WS, for example, is a NDD caused by a heterozygous microdeletion on chromosome 7q11.23 (Pober, 2010). The haploinsufficiency caused by the deletion of about 26 genes from the WS chromosome region (WSCR) is manifested by, among other things, a maladjusted, overly friendly personality, a unique intellectual profile and increased anxiety (Pober, 2010). The idea of specific loci being susceptible to genetic variability is manifested in chromosome 7q11.23, as microduplication of the WSCR was shown to be associated with ASD (Sanders et al., 2011; Morris et al., 2015). Therefore, CNVs such as deletion and duplications in the WSCR cause cognitive, biological, physiological and behavioral defects, the symptoms and phenotype of which depend on the genetic variation.
Specific gene mutations
Genetic alterations underlying NDDs may involve several genes. However, some of the NDDs have a monogenic cause (Renieri et al., 2003; Betancur and Buxbaum, 2013), such as SHANK3-related mutations in ASD (Peça et al., 2011) and methyl-CpG-binding protein 2 (MECP2) loss-of-function mutations in RTT (Amir et al., 1999). Monogenic NDDs relate to disorders mediated by mutations of a single gene. Suggested mechanisms of monogenic NDDs may include SNPs, which severely alter the genetic product (Zhou et al., 2016), and variations of the genetic sequence of a single gene, including deletion, duplication, inversion or silencing of the specific gene sequence (Kishino et al., 1997; Guy et al., 2001; Ramocki et al., 2010). Postnatal therapeutic approaches may offer better treatments for monogenic disorders, as genetic restoration or interference of a single target is more feasible than of a set of different genetic targets.
While fatal in males, RTT is a progressive NDD in females, usually caused by de-novo dominant loss-of-function mutations in the X-linked gene MECP2 (Amir et al., 1999), characterized by normal development up to 6 to 18 months, followed by a severe decline in psychiatric, motor and lingual abilities (Hagberg et al., 1983). MECP2 is a risk gene, as its duplication is associated with infantile hypotonia, mental retardation, epilepsy, speech impairment and autistic features in males, whereas females remain mostly unaffected (Ramocki et al., 2010). Fragile X syndrome is another example of a NDD associated with a dominant X-linked aberration. Most cases of fragile X syndrome are caused by transcriptional silencing of the FMR1 gene (Fu et al., 1991). Affected individuals are mostly males, and they present symptoms that vary in severity and character, including mental and emotional disabilities (Garber et al., 2008).
SHANK3 is a post-synaptic scaffolding protein that forms the platform for postsynaptic density complexes in glutamatergic synapses (Baron et al., 2006; Monteiro and Feng, 2017). SHANK3 haploinsufficiency is associated with ASD, specifically Phelan–McDermid syndrome (Wilson et al., 2003; Durand et al., 2007). Moreover, a single-nucleotide insertion mutation in one copy of the SHANK3 gene was shown to cause severe speech impairment and mental retardation in two brothers (Durand et al., 2007). The monogenic contribution of SHANK3 to ASD and ASD-like syndromes has been validated by a few studies, affirming the need for further exploration of this risk gene and its effects on NDDs (Moessner et al., 2007; Gauthier et al., 2009; Peça et al., 2011; Mei et al., 2016; Zhou et al., 2016; Amal et al., 2018).
Epigenetic alterations
Epigenetic modifications are considered as the interface plane between biology and environmental influences (Feil and Fraga, 2012). Main epigenetic mechanisms include DNA methylation (Moore et al., 2013) and histone modification (Bannister and Kouzarides, 2011). Although the original DNA sequence remains unchanged, epigenetics alterations affect gene expression and cell function (Dall’Aglio et al., 2018). Epigenetics and several NDDs have been linked, including ASD (Ladd-Acosta et al., 2014) and ADHD (Wilmot et al., 2016).
Prominent example of a NDD caused by epigenetic alterations is AS, characterized by unique cognitive, personality and physiological profiles, including mental retardation, frequent seizures and increased smiling and laughing (Kishino et al., 1997). AS is caused by a genetic mechanism called imprinting, in which the maternal and paternal alleles differ in their contribution to the phenotypic effect due to methylation of the inactive allele (Kishino et al., 1997). AS is characterized with a defect in the maternal copy of the UBE3A gene which may either be deleted (70%), mutated (10%), non-existent (uniparental disomy, 2–3%) or malfunctioning due to an imprinting defect (3–5%) (Williams et al., 1995). Similar to AS, Prader–Willi syndrome (PWS) is also caused by defects in UBE3A. However, in PWS, the paternal copy of UBE3A is inactive (due to deletion or other causes) (Cassidy, 1984). Both AS and PWS share malfunctions in 15q11.2-q13, but they differ in which allele is inactive due to methylation; the maternal copy in AS and the paternal copy in PWS (Knoll et al., 1989; Reis et al., 1994). AS and PWS support the idea that not only does the type of genetic variation (deletion, duplication, inversion) dictate the effects on biological and behavioral aspects, but also the identity of the damaged allele is of importance.
Advances in molecular biology have enabled the scientific community to gain a clear insight of the genetic basis responsible for certain NDDs. For example, individuals with WS have similar genetic deficits in 95% of the cases (Korenberg et al., 2000). However, the genetic architecture of most NDDs is highly complex (Devlin and Scherer, 2012; Lesch, 2016; Deciphering Developmental Disorders Study, 2017), thus requiring the development of multiple genetic therapeutic approaches, to maximize the arsenal of possible treatments. Furthermore, genetic deficits include not only germline transmitted mutations, but also postzygotic de-novo mutations that may contribute to somatic brain mosaicism, shown to result in abnormal brain development and NDDs (D’Gama and Walsh, 2018). Additionally, the genetic basis of NDDs may be polygenetic, affect multiple organ systems and brain regions, and cause abnormal cognitive and physiological development throughout life. Moreover, while risk genes for NDD such as ASD have been classified as having synaptic activity or developmental importance, recent studies have shown this division is not accurate, an example of how genetic mutations may result in multiple deficits (Heavner and Smith, 2020). Acknowledging these barriers is of extreme importance, as it will enable the scientific community to better prioritize the animal model research in order to dissect the underlying genetic causation of NDDs and better pivot the development of genetic therapeutic tools.
The broad heterogeneity of genetic deficits such as CNVs, SNPs and epigenetic alterations limits our efforts to link specific genetic deficits with corresponding behavioral and neurological phenotypes (Devlin and Scherer, 2012). These limitations also exist in the clinic, where ASD, for example, is diagnosed mostly based on behavioral examination of the individual and following DSM guidelines, and to a much less extent by a genetic screening or biological examination.
Animal Models for Neurodevelopmental Disorders
To better dissect the mechanisms affected by genetic alterations, animal models offer a suitable platform to perform extensive research on the etiology, characteristics, deficits and potential therapeutic approaches related to NDDs. Different animal models, such as rodents (Crawley, 2012) and non-human primates (NHPs) (Kishi et al., 2014; Miller et al., 2016), retain some well-conserved biological and behavioral characteristics that are also present in humans. Thus, modeling human genetic conditions using laboratory animals has advanced, in recent years, our understanding of the mechanisms underlying different symptoms and phenotypes in various genetic disorders.
Genetic tools to dissect neurodevelopmental disorders
Genetic tools such as the Cre-LoxP system (Schwenk et al., 1995) and CRISPR-Cas9 (clustered, regularly interspaced, short palindromic repeats and CRISPR-associated protein) (Ran et al., 2013) have enabled the creation of animal models that express a specific human, or non-human genetic condition, responsible for mediating NDDs. By using these genetic tools at different stages during development, a mechanistic understanding of the developmental trajectory of these genetic alterations and an understanding of abnormal development in a variety of NDDs can be acquired (Amir et al., 1999; Sakurai et al., 2011; Segura-Puimedon et al., 2014; Enkhmandakh et al., 2016; Zhou et al., 2016). For example, precise targeting of specific genes that are deleted in WS, using these genetic tools, helped dissect the role and downstream interactions of genes deleted from the WSCR in humans, improving our ability to analyze which deleted gene in WS is associated with the different phenotypes associated with the disorder (Sakurai et al., 2011; Segura-Puimedon et al., 2014; Barak et al., 2019). However, the same genetic manipulation in humans and animal models (rodents or NHPs) does not always result in the same biological and behavioral phenotypes (Osborne, 2010).
Overall, genetic modeling of NDDs in animal models sheds light on the underlying mechanism by which the genetic aberration affects the individual, and pivots research toward possible therapeutic targets in the associated disorders (Zoghbi and Bear, 2012).
Rodent models
Key benefits in studying rodents as animal models are the variety of developed genetic tools that specifically suit their genetic background. Rodent models with targeted mutations in genes homologous to those found altered in human patients aid us in preclinical screening for possible therapeutic agents and approaches (Crawley, 2012; Kaiser and Feng, 2015).
Different NDDs, such as ASD and schizophrenia, have been shown to share a similar genetic basis (de Lacy and King, 2013). At the molecular level, it has been suggested that defective synapse development and maintenance are key to the emergence of these disorders (Kenny et al., 2014). Shank3 is a gene of interest when observing the interaction between ASD and schizophrenia. Recently, different mutations of Shank3 in mice were shown to result in synaptic dysfunction and either ASD or schizophrenia-like motifs (Zhou et al., 2016).
The mouse model enables us to gain more mechanistic information on the etiology of NDDs. For example, using a mouse model for WS, we recently revealed novel aspects in how WS manifests in the CNS (Bey and Jiang, 2014; Barak et al., 2019). Results of that study indicated a key role for neuronal Gtf2i, a gene known to be related to the hypersocial phenotype (Dai et al., 2009; Antonell et al., 2010; Sakurai et al., 2011; Borralleras et al., 2015), in myelin formation and maintenance. Abnormalities in white matter integrity that were found in our mouse model for WS were then validated in human tissues, shedding new light on the disorder’s mode of operation in the CNS and offering a new perspective on potential therapeutic agents for WS in humans (Barak et al., 2019).
Another NDD that has been studied mechanistically in mouse models is fragile X syndrome. Excess activation of mGluR5, a metabotropic glutamate receptor, was shown to contribute to the fragile X syndrome pathology (Dölen et al., 2007). Additionally, RTT was also modeled in mouse models, where mice with a truncated allele of Mecp2 exhibited learning and memory impairments, synaptic dysfunction, and synaptic ultrastructural abnormalities during early stages of development, suggesting an early time window for the described pathogenesis in RTT (Moretti et al., 2006).
Non-human primates models
Due to the remarkable differences between rodents and humans, research conducted on the former may not be replicable in clinical trials (Hyman, 2014; Izpisua Belmonte et al., 2015). To fill the gap, genetic tools for the creation of NHP models of various medical conditions have recently been studied (Okano et al., 2012; Kishi et al., 2014; Kumita et al., 2019). NHP models are thought to more accurately represent the human condition, in cognitive and biological aspects, reducing the evolutionary distance between the animal model and the patient. However, genetic tools for NHPs are still lacking compared to those available for rodent research. As such, the study of NDDs in NHPs has yet to reach its full potential.
In 2009, germline transmission was achieved in the common marmoset (Callithrix jacchus), providing researchers with an alternative model for human conditions that offers a greater translational value than rodent models (Sasaki et al., 2009). The common marmoset is a valuable candidate for modeling NDDs as it shows unique behavioral and cognitive traits, similar to those found in humans (Dell’Mour et al., 2009), and shares biological, anatomical and developmental characteristics with humans (Abbott et al., 2003).
Following the common marmoset, other NHP models for NDDs were created (Zhou et al., 2019), using genetic tools such as viral gene expression and gene editing by CRISPR-Cas9. For example, overexpression of Mecp2 (Liu et al., 2016) or Shank3 deficiency (Zhou et al., 2017) were modeled in cynomolgus monkeys (Macaca fascicularis), in an attempt to better dissect and characterize neuronal and behavioral defects of ASD. RTT models were also developed in cynomolgus monkeys, using TALEN editing. The similarity between the model and the manifestation of RTT in humans was remarkable, including structural, behavioral and immunological aspects (Chen et al., 2017).
Nevertheless, advances in developing NHP models are essential, and can be achieved by creating a wide array of genetic tools, and by the development of behavioral tasks that are specifically suited to NHPs.
In order to understand whether the genetic basis and behavioral alterations in NDDs are shared between animal models and humans, scientists have developed techniques to examine prominent behavioral characteristics in various NDDs in animals, such as repetitive behavior (Crawley, 1999, 2008; Moy et al., 2008), OCD (Burguière et al., 2015; Monteiro and Feng, 2016), cognition properties (Brooks et al., 2005; Crawley, 2008; Wolf et al., 2016), alterations in social behavior (Crawley, 1999, 2008; Moy et al., 2008; Netser et al., 2009), motor functions and anxiety (Crawley, 1985). These tests enable scientists to evaluate new animal models for various NDDs and also examine whether a potential treatment has alleviated behavioral deficits.
Postnatal Gene-Therapy Approaches in Neurodevelopmental Disorders
Current postnatal treatments offered for NDDs include mostly symptomatic and behavioral therapy. Until about a decade ago, gene therapy was an unreachable dream, which today has become a reality. However, gene-therapy techniques in humans have not yet matured enough to produce consistent results. Some of the major questions that remain unanswered are related to the critical time window for effective gene therapy in NDDs, routes of intervention, efficiency and effective duration of genetic treatment, and the need for personalized genetic therapy for every individual.
Importance of the developmental window in genetic treatment of neurodevelopmental disorders
The human nervous system is constantly changing (Marín, 2016; Silbereis et al., 2016), with specific periods along development during which the brain’s plasticity and susceptibility to change are high, also known as critical periods (Hensch, 2004; Hübener and Bonhoeffer, 2014). One of the mechanisms suggested to play a major role in NDDs is abnormal formation and regulation of synapses and protein complexes during these critical periods (Ramocki and Zoghbi, 2008; Harlow et al., 2010; Zoghbi and Bear, 2012; Meredith, 2015; Marín, 2016). A prominent example of the importance of the timing of the therapeutic intervention is a recent study in a mouse model for AS that demonstrated the differential effectiveness of Ube3a re-expression at different developmental stages (Silva-Santos et al., 2015). That study demonstrated that although synaptic plasticity in the hippocampus was restored regardless of the intervention timing, other symptoms such as motor defects, anxiety, repetitive behavior and epilepsy were alleviated only with earlier intervention onset (Silva-Santos et al., 2015).
However, some studies debate the role of the critical period in treating NDDs. In contrast to how we understood the role of critical period in NDDs, recent studies show that some of the phenotypes present in these disorders may be alleviated or reversed as a result of postnatal treatment, well after the critical period has ended (Ehninger et al., 2008; Castrén et al., 2012; Hübener and Bonhoeffer, 2014). For example, a conditional knock-in mouse model of Shank3 showed that reinstatement of Shank3 expression in adult mice results in improvements at both the molecular and behavioral levels (Mei et al., 2016). Furthermore, neurological defects in a mouse model for RTT were alleviated following reactivation of MeCP2 expression, in both adult and immature mice (Guy et al., 2007). These studies suggest that different brain regions and functions have distinct critical periods, and that postnatal genetic treatments for NDDs may be effective even when administered in advanced postnatal stages.
Routes of administration of therapeutic agents
Delivery of therapeutic agents to targeted areas throughout the body, and specifically the brain, is another challenge in the development of gene therapy. Transfer of therapeutic agents into the CNS requires passing the blood-brain barrier (BBB) (Hawkins and Davis, 2005; Abbott et al., 2010), a selective and non-permissive barrier aimed at protecting the CNS. Unfortunately, most therapeutic agents cannot pass the BBB without an additional transfer vector (Pardridge, 2005). As a result, gene delivery into the CNS is largely dependent on invasive methods, which allow the direct application of the therapeutic vector into specific brain regions, eliminating the need to cross the BBB. Methods of direct intervention into the CNS include intracranial delivery by intraparenchymal, intracisternal, intrathecal and intracerebroventricular injections (Figure 1). These invasive methods usually require stereotaxic injection and surgical intervention, with the possibility of applying long-lasting drug-releasing depots or catheters. The main advantages of using such delivery methods are bypassing of the BBB and direct application of high concentrations of the therapeutic agent in specific brain regions. However, these methods are considered less therapeutic in their general approach as they require surgical intervention, and are thus prone to infection and other surgical complications that can lead to side effects such as elevated intracranial pressure.
Figure 1.
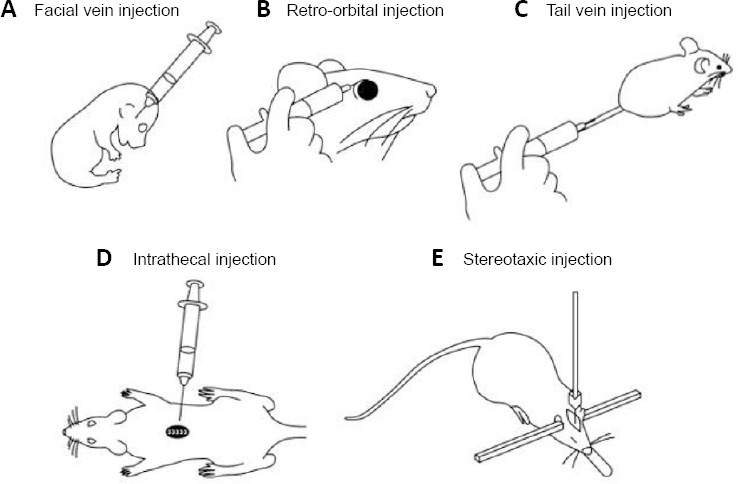
Illustrative description of routes of administration of gene-therapy agents.
(A) Facial vein injection in neonatal pup, (B) retro-orbital injection, (C) tail vein injection, (D) intrathecal injection, (E) stereotaxic injection.
The non-invasive approaches, specifically for CNS transduction, include intravenous (IV) and intranasal administration of drugs. IV administration methods (Figure 1) are as simple as taking a blood test, and has proven efficient with transfer vectors that can self-cross the BBB, such as adeno-associated virus (AAV) serotypes AAV9 (Zincarelli et al., 2008; Duque et al., 2009; Foust et al., 2009) and PHP.eB (Chan et al., 2017; Dayton et al., 2018). Systemic administration, by its very nature, is more therapeutically relevant than surgical intervention, as the latter is highly invasive, and because systemic administration can easily be performed on a continuous basis. However, systemic administration has some disadvantages, including: 1) limited biodistribution of the therapeutic agent throughout the entire body, resulting in low concentrations of therapeutic agent at the target site, and thereby requiring delivery of a higher drug dosage compared to invasive methods; 2) multisystemic immune response, depending on the transfer vector; 3) toxicity in different tissues, such as the liver, due to enhanced accumulation of the agent. Intranasal is another form of non-invasive delivery method, which allows bypassing the BBB by transporting drugs through the nasal cavity into the CNS. There is growing evidence of the efficiency of intranasal transfer of drugs to the CNS, in particular of particles that are incapable of self-passage through the BBB (Crowe et al., 2018). Overall, efficient transduction, repeatable treatments and above all, safety, are the main criteria when deciding upon a route of administration.
Main Methods and Technologies for Genetic Intervention in Neurodevelopmental Disorder Research and Treatment
In developing tools that will enable genetic intervention, one must ensure that they can efficiently and accurately manipulate a genetic sequence, and a technique needs to be harnessed to deliver the developed tool selectively to targeted tissues. While the genetic therapy research field continues to evolve, the tools generated so far are sufficient for testing their properties in preclinical and clinical trials. NDDs are a prominent family of conditions that will benefit from the emerging research in the field. Some of the fundamental techniques in gene therapy related to NDDs are discussed below, together with current gene-therapy studies in animal models for NDDs.
AAV as a gene-transfer vector
AAV is a non-pathogenic, non-immunogenic transfer agent that enables targeting specific tissues and organs through genetic engineering of the virus (Burger et al. 2005; Mandel et al., 2006; Duan, 2016; Hocquemiller et al., 2016; Srivastava, 2016; Naso et al., 2017; Hudry and Vandenberghe, 2019). The use of AAV has become widespread in many biological studies, while proving to be an efficient tool for gene transfer and manipulation (Hermonat and Muzyczka, 1984; Kay et al., 2001; Daily et al., 2011; Borralleras et al., 2015; Luoni et al., 2019), with promising results that have led to clinical success (Mingozzi and High, 2011; Ylä-Herttuala, 2012). Other viral vectors, such as lentivirus—a subtype of retrovirus, are also candidates for gene therapy (Dull et al., 1998; Galimi and Verma, 2002; Park, 2007; Cockrell and Kafri, 2011); however, this review will focus on AAV, as retroviral characteristics include adverse side effects such as malintegration in the genome (Hacein-Bey-Abina et al., 2003a, b) and genotoxicity (Montini et al., 2006). One example of the use of AAV as a possible therapeutic agent in NDD is the introduction of the exogenous Ube3a in a mouse model of AS (Daily et al., 2011). Direct injection of a Ube3a-transducing AAV9 vector to the hippocampus improved early-phase long-term potentiation (LTP) and associative learning in manipulated AS mice compared to controls (Daily et al., 2011). Another example of a gene-restoration study by administration of an AAV directly into the CNS was performed in a mouse model for WS (Borralleras et al., 2015). Intracisternal injection of a Gtf2i-transducing vector into a mouse with complete deletion of the WSCR improved behavioral attributes such as motor coordination, sociability and anxiety, and normalized the expression levels of molecular factors such as brain-derived neurotrophic factor (Bdnf) in specific brain areas (Borralleras et al., 2015). Although a prime candidate for serving as a therapeutic agent, AAV suffers from a few limitations. First, the capacity of an engineered AAV vector is limited, where the insert between the two inverted terminal repeats can be no longer than 4.7 kb (Wu et al., 2010). Second, even though AAV is mostly non-pathogenic and non-immunogenic, some studies have found evidence of hepatic genotoxicity of AAV following in vivo gene manipulation by AAV vector, resulting in hepatocellular carcinoma (Donsante et al., 2007; Chandler et al., 2015) and liver failure (Hinderer et al., 2018). The administration method of AAV is a crucial limitation for NDDs which are caused by genetic defects in the CNS, as until recently, AAV penetration through the BBB was limited. This limitation required direct administration of the vector into the CNS by surgical intervention, thus lowering the translational value of such treatment to the clinic. However, recently, a new and exciting AAV capsid, PHP.eB, was shown to have improved efficacy at penetrating the BBB via systemic administration of the virus (Chan et al., 2017). Interestingly, RTT mice with a genetically encoded Mecp2 mutation were treated with IV systemic administration of Mecp2-transducing PHP.eB virus at 4 weeks of age, resulting in delayed progression of symptoms and improved lifespan (Luoni et al., 2019).
AAV offers a great opportunity for alleviating monogenic disorders. However, NDDs pose a challenge for gene therapy by AAV, as many of them manifesting with systemic defects are not monogenic and require the therapeutic agent to reach the CNS.
Naturally occurring AAVs have limited tropism properties, and therefore a method of cell-type-specific AAV selection is required. Cre recombinase-based AAV targeted evolution (CREATE) is a method of developing AAV capsids with high transduction efficiency, in a cell-type-specific manner (Deverman et al., 2016). By harnessing CREATE, development of CNS-specific capsids has improved dramatically in recent years, including successful transduction of neurons with PHP.B and PHP.eB serotypes, and transduction of astrocytes with the PHP.S serotype (Chan et al., 2017). In addition to capsid engineering, specificity of transduction is increased via promoter-dependent transgene expression (Shevtsova et al., 2005; Gray et al., 2011). Therefore, transduction of specific CNS cell types through IV systemic administration is now feasible with the correct vector, capsid and promoter design. Because the genetic defects in various disorders may be tissue- and cell-type-specific, the need to develop tissue-specific capsids and identify cell-type-specific promoters is high.
Expanding the capacity of the AAV transgene-transduction cassette is also of great importance in facilitating genetic treatment by AAV. Transgene expression of vectors including the gene of interest, cell-specific promoter and reporter sequence are limited to the current AAV capacity of 4.7 kb between inverted terminal repeats, resulting in limited potential to reintroduce longer genes. Currently, dual- and triple-vector approaches, each expressing a fragment of a split transgene, are in use (Duan et al., 2001, 2003; Hirsch et al., 2016; Pate et al., 2019). Furthermore, gene therapy studies utilizing AAV technology in mice are very common, however, NHP studies utilizing AAV as a gene therapy approach are scarce, especially studies focused on NDDs. AAV has been shown to successfully transduce gene expression in NHP studies (Samaranch et al., 2012; Hinderer et al., 2014; György et al., 2019). Further studies of NDDs models in NHP incorporating AAV as potential gene transfer vector could dramatically increase the relevance of this technology in the lab and clinic.
CRISPR-Cas9 as a gene-editing tool
CRISPR-Cas9 is a valuable modifier of gene sequence and function, with great potential clinical implications. Genome editing by CRISPR is largely believed to be a one-time, long-lasting treatment, unlike other gene therapies. This type II bacterial system utilizes a combination of a single guide RNA (sgRNA) to target specific loci in the genome, and the Cas9 protein for precise gene editing by either nonhomologous end joining or precise homology-directed repair (HDR) using a DNA-repair template (Cong et al., 2013; Doudna and Charpentier, 2014; Sander and Joung, 2014). CRISPR has been largely utilized in generating transgenic laboratory animal models (Wang et al., 2013; Yang et al., 2013; Niu et al., 2014; Kumita et al., 2019; Offen et al., 2019; Qiu et al., 2019; Zhou et al., 2019) and ex vivo genetic manipulations, enabling the creation of genetically modified cell lines in 2–3 weeks (Ran et al., 2013). However, CRIPSR technology is faced with a few issues when implemented in in vivo systems, such as limited HDR efficiency and delivery methods.
To overcome delivery limitations, AAV was suggested as a transfer vector for Streptococcus pyogenes Cas9 (SpCas9, 4.2 kb) and the sgRNA (Senís et al., 2014). However, due to AAV’s limited packaging ability, dual-vector studies were suggested, each vector carrying either the SpCas9 or the sgRNA molecule. In 2014, in vivo genetic editing by CRISPR-Cas9 was achieved in a dual-vector study, manipulating the levels of a few genes in the CNS of mice, including Mecp2 which is associated with RTT and intellectual disability disorders (Swiech et al., 2015). Chimeric serotypes of different AAV vectors were also modified to improve CNS-transduction efficiency, allowing for the transfer of sgRNA targeting the specific schizophrenia risk gene miR137 and deleting it in a CNS-specific manner in CRISPR-Cas9 knock-in mice (Murlidharan et al., 2016). In a different study, CRISPR-mediated suppression of the expression of a mutant Htt gene (mHtt) in the striatum of an HD140Q knock-in mouse model for Huntington’s disease alleviated motor deficits (Yang et al., 2017).
Shorter orthologues of the Cas9 molecule have been recently discovered, such as the Staphylococcus aureus Cas9 (SaCas9, 3.16 kb) (Ran et al., 2015) and Campylobacter jejuni Cas9 (CjCas9, 2.95 kb) (Kim et al., 2017), increasing the potential for proper packaging in AAV and the latter’s potential to act as an expression vector for CRISPR, enabling combined packaging of the Cas9 nuclease and the sgRNA in a single AAV vector.
Genome editing of somatic cells outside the CNS in a therapeutic manner was shown to be feasible, with the correction of Duchenne muscular dystrophy in an adult mouse model for the disorder, by administering AAV-CRISPR treatment based on SaCas9 (Nelson et al., 2016). Non-viral delivery approaches are also a viable option for the transfer of CRISPR-related products into nuclei in a tissue-specific manner. For example, combined AAV and lipid nanoparticle delivery of CRISPR products to the liver gave a therapeutic outcome (Yin et al., 2016). However, tools for the systemic delivery of lipid nanoparticles and other non-viral molecules to the CNS are still under development. Moreover, more studies employing CNS-targeted genome editing are required to examine the potential of CRISPR-mediated gene therapy for the treatment of NDDs.
Antisense oligonucleotides
Antisense oligonucleotides (ASOs) are short nucleic acid sequences that are capable of hybridizing to a specific mRNA molecule and interfering with its downstream translation into a protein (De Mesmaeker et al., 1995). As such, ASOs are prime candidates for modifying protein levels and to act as therapeutic agents in NDDs caused by the misexpression of genes. For example, MECP2 duplication syndrome is a common NDD in males, estimated to cause 1% of unexplained X-linked mental retardation (Lugtenberg et al., 2009). Affected individuals present seizures, epilepsy, recurrent infections, intellectual disability and delayed psychomotor development (Ramocki et al., 2010; Van Esch, 2011). In a study from 2015, postnatal use of ASO treatment ameliorated symptoms in an adult transgenic Mecp2 duplication mouse model for RTT (Sztainberg et al., 2015). Furthermore, ASO treatment corrected MECP2 mRNA expression levels in lymphoblastoid cells from human patients with MECP2 duplication (Sztainberg et al., 2015).
ASOs were also shown to be an effective tool for harnessing the intact and silenced paternal allele of Ube3a in AS by targeting the Ube3a antisense transcript (Ube3a-ATS) (Meng et al., 2015). Treatment with a sequence-specific ASO against Ube3a-ATS resulted in reduced Ube3a-ATS and increased Ube3a levels, both in vivo and in vitro. Of clinical relevance, the ASO treatment unsilenced the paternal Ube3a allele throughout the brain and ameliorated cognitive deficits in an animal model for AS (Meng et al., 2015).
ASOs have also been shown to be an effective course of treatment in other CNS-related conditions, such as rescuing hearing in a mouse model of human deafness (Lentz et al., 2013) and ameliorating severe spinal muscular atrophy (Passini et al., 2011). Together, these findings strengthen the therapeutic potential of ASO treatment in various CNS-related conditions, including NDDs.
Conclusions and Future Directions in Gene-Therapy Approaches for Neurodevelopmental Disorders
The prospect of gene therapy for certain NDDs is tangible. The genomic revolution and constant progress in the research of NDDs and neuropsychiatric conditions are providing the scientific community with a better perspective on the underlying mechanisms of these disorders, with a large number of them having genetic etiology. These advances are enabling better design of animal models and of preclinical therapeutic studies.
While the field of gene therapy is rapidly progressing, the number of successful clinical trials is still limited, suggesting that further exploration is required to improve the bench-to-bedside success rate. A few promising aspects for future progress in the field should be further developed to maximize the chances of developing an effective tool, among them those mentioned above, as well as RNA interference (RNAi) and non-viral methods of gene-therapy induction.
RNAi is a process in which small, non-coding RNA sequences, such as short interfering RNA (siRNA) (Chang et al., 2009) and microRNA (miRNA) (Lee et al., 1993; Lee and Ambros, 2001; Shomron, 2010), regulate gene expression via mRNA degradation or inhibition of translation to proteins in a sequence-specific manner. Dysregulation of RNAi molecules has been associated with NDDs (Chang et al., 2009; Xu et al., 2010; Meza-Sosa et al., 2012; Sun and Shi, 2015), emphasizing their spatiotemporal importance for normal brain development. Directing the knowledge accumulated in extensive research on the roles and mechanisms of RNAi processes to possible innovative therapeutic approaches seems to be the next step.
In parallel to advances in gene-therapy strategies, those for administering these therapies to the CNS are lacking in therapeutic potential, as they usually require surgical intervention and/or recurring treatments. Promising vehicles for the introduction of gene therapy into the CNS include nanoparticles (Lee et al., 2018), which are able to carry several therapeutic agents, e.g., nucleic acids and proteins. Furthermore, nanoparticles offer cell-specific delivery of their cargo and reduced toxicity (Mizrahy et al., 2019). Relevant subsets of nanoparticles carriers are polymeric nanoparticles (Dikpati et al., 2012), solid lipid nanoparticles (Hou et al., 2003) and nanoliposomes (Allen and Cullis, 2013), all of which have been shown to have BBB penetrating capabilities.
Acknowledgments
We would like to thank our laboratory members: Sari S. Trangle, Meitar Grad, Inbar Fischer, Ariel Nir, Ela Bar, and May Rokach for their support during the writing of the manuscript. In addition, the authors would like to thank Idan Rooze for his help in creating the illustrations and figure. Finally, the author would like to thank the Ministry of Science and Technology, Israel for financial support during the writing of the manuscript.
Footnotes
C-Editors: Zhao M, Li JY; T-Editor: Jia Y
Conflicts of interest: Both authors declare no conflicts of interest.
Financial support: This work was supported by grants from Fritz Thyssen Stiftung, BrainBoost Innovation Center by Sagol School of Neuroscience at TAU, and SPARK Tel Aviv. GL was supported by the Eshkol Fellowship from The Ministry of Science and Technology, Israel. BB was the recipient of The Alon Fellowship for outstanding young researchers awarded by the Israeli Council for Higher Education.
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This work was supported by grants from Fritz Thyssen Stiftung, BrainBoost Innovation Center by Sagol School of Neuroscience at TAU, and SPARK Tel Aviv. GL was supported by the Eshkol Fellowship from The Ministry of Science and Technology, Israel. BB was the recipient of The Alon Fellowship for outstanding young researchers awarded by the Israeli Council for Higher Education.
References
- 1.Abbott DH, Barnett DK, Colman RJ, Yamamoto ME, Schultz-Darken NJ. Aspects of common marmoset basic biology and life history important for biomedical research. Comp Med. 2003;53:339–350. [PubMed] [Google Scholar]
- 2.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 3.Allen TM, Cullis PR. Liposomal drug delivery systems: From concept to clinical applications. Adv Drug Deliv Rev. 2013;65:36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 4.Amal H, Barak B, Bhat V, Gong G, Joughin BA, Wang X, Wishnok JS, Feng G, Tannenbaum SR. Shank3 mutation in a mouse model of autism leads to changes in the S-nitroso-proteome and affects key proteins involved in vesicle release and synaptic function. Mol Psychiatry. 2018 doi: 10.1038/s41380-018-0113-6. doi: 10.1038/s41380-018-0113-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 6.Antonell A, Del Campo M, Magano LF, Kaufmann L, de la Iglesia JM, Gallastegui F, Flores R, Schweigmann U, Fauth C, Kotzot D, Pérez-Jurado LA. Partial 7q11.23 deletions further implicate GTF2I and GTF2IRD1 as the main genes responsible for the Williams-Beuren syndrome neurocognitive profile. J Med Genet. 2010;47:312–320. doi: 10.1136/jmg.2009.071712. [DOI] [PubMed] [Google Scholar]
- 7.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barak B, Zhang Z, Liu Y, Nir A, Trangle SS, Ennis M, Levandowski KM, Wang D, Quast K, Boulting GL, Li Y, Bayarsaihan D, He Z, Feng G. Neuronal deletion of Gtf2i, associated with Williams syndrome, causes behavioral and myelin alterations rescuable by a remyelinating drug. Nat Neurosci. 2019;22:700–708. doi: 10.1038/s41593-019-0380-9. [DOI] [PubMed] [Google Scholar]
- 9.Baron MK, Boeckers TM, Vaida B, Faham S, Gingery M, Sawaya MR, Salyer D, Gundelfinger ED, Bowie JU. An architectural framework that may lie at the core of the postsynaptic density. Science. 2006;311:531–535. doi: 10.1126/science.1118995. [DOI] [PubMed] [Google Scholar]
- 10.Betancur C, Buxbaum JD. SHANK3 haploinsufficiency: a “common” but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Mol Autism. 2013;4:17. doi: 10.1186/2040-2392-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bey AL, Jiang YH. Overview of mouse models of autism spectrum disorders. Curr Protoc Pharmacol. 2014;66:5. doi: 10.1002/0471141755.ph0566s66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borralleras C, Sahun I, Pérez-Jurado LA, Campuzano V. Intracisternal Gtf2i gene therapy ameliorates deficits in cognition and synaptic plasticity of a mouse model of Williams-Beuren syndrome. Mol Ther. 2015;23:1691–1699. doi: 10.1038/mt.2015.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brooks SP, Pask T, Jones L, Dunnett SB. Behavioural profiles of inbred mouse strains used as transgenic backgrounds. II: cognitive tests Genes. Brain Behav. 4:307–317. doi: 10.1111/j.1601-183X.2004.00109.x. [DOI] [PubMed] [Google Scholar]
- 14.Buescher AV, Cidav Z, Knapp M, Mandell DS. Costs of autism spectrum disorders in the United Kingdom and the United States. JAMA Pediatr. 2014;168:721–728. doi: 10.1001/jamapediatrics.2014.210. [DOI] [PubMed] [Google Scholar]
- 15.Burger C, Nash K, Mandel RJ. Recombinant adeno-associated viral vectors in the nervous system. Hum Gene Ther. 2005;16:781–791. doi: 10.1089/hum.2005.16.781. [DOI] [PubMed] [Google Scholar]
- 16.Burguière E, Monteiro P, Mallet L, Feng G, Graybiel AM. Striatal circuits, habits, and implications for obsessive-compulsive disorder. Curr Opin Neurobiol. 2015;30:59–65. doi: 10.1016/j.conb.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassidy SB. Prader-Willi syndrome. Curr Probl Pediatr. 1984;14:1–55. doi: 10.1016/0045-9380(84)90043-4. [DOI] [PubMed] [Google Scholar]
- 18.Castrén E, Elgersma Y, Maffei L, Hagerman R. Treatment of neurodevelopmental disorders in adulthood. J Neurosci. 2012;32:14074–14079. doi: 10.1523/JNEUROSCI.3287-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, Sánchez-Guardado L, Lois C, Mazmanian SK, Deverman BE, Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chandler RJ, LaFave MC, Varshney GK, Trivedi NS, Carrillo-Carrasco N, Senac JS, Wu W, Hoffmann V, Elkahloun AG, Burgess SM, Venditti CP. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest. 2015;125:870–880. doi: 10.1172/JCI79213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang S, Wen S, Chen D, Jin P. Small regulatory RNAs in neurodevelopmental disorders. Hum Mol Genet. 2009;18:R18–26. doi: 10.1093/hmg/ddp072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Yu J, Niu Y, Qin D, Liu H, Li G, Hu Y, Wang J, Lu Y, Kang Y, Jiang Y, Wu K, Li S, Wei J, He J, Wang J, Liu X, Luo Y, Si C, Bai R, et al. Modeling Rett syndrome using TALEN-edited MECP2 mutant cynomolgus monkeys. Cell. 2017;169:945–955e10. doi: 10.1016/j.cell.2017.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cockrell AS, Kafri T. Gene delivery by lentivirus vectors. Mol Biotechnol. 2007;36:184–204. doi: 10.1007/s12033-007-0010-8. [DOI] [PubMed] [Google Scholar]
- 24.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crawley JN. Exploratory behavior models of anxiety in mice. Neurosci Biobehav Rev. 1985;9:37–44. doi: 10.1016/0149-7634(85)90030-2. [DOI] [PubMed] [Google Scholar]
- 26.Crawley JN. Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res. 1999;835:18–26. doi: 10.1016/s0006-8993(98)01258-x. [DOI] [PubMed] [Google Scholar]
- 27.Crawley JN. Behavioral phenotyping strategies for mutant mice. Neuron. 2008;57:809–818. doi: 10.1016/j.neuron.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Crawley JN. Translational animal models of autism and neurodevelopmental disorders. Dialogues Clin Neurosci. 2012;14:293–305. doi: 10.31887/DCNS.2012.14.3/jcrawley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crowe TP, Greenlee MHW, Kanthasamy AG, Hsu WH. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018;195:44–52. doi: 10.1016/j.lfs.2017.12.025. [DOI] [PubMed] [Google Scholar]
- 30.Dai L, Bellugi U, Chen XN, Pulst-Korenberg AM, Järvinen-Pasley A, Tirosh-Wagner T, Eis PS, Graham J, Mills D, Searcy Y, Korenberg JR. Is it Williams syndrome. GTF2IRD1 implicated in visual-spatial construction and GTF2I in sociability revealed by high resolution arrays? Am J Med Genet A. 2009;149A:302–314. doi: 10.1002/ajmg.a.32652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daily JL, Nash K, Jinwal U, Golde T, Rogers J, Peters MM, Burdine RD, Dickey C, Banko JL, Weeber EJ. Adeno-associated virus-mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PLoS One. 2011;6:e27221. doi: 10.1371/journal.pone.0027221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dall’Aglio L, Muka T, Cecil CAM, Bramer WM, Verbiest MMPJ, Nano J, Hidalgo AC, Franco OH, Tiemeier H. The role of epigenetic modifications in neurodevelopmental disorders: A systematic review. Neurosci Biobehav Rev. 2018;94:17–30. doi: 10.1016/j.neubiorev.2018.07.011. [DOI] [PubMed] [Google Scholar]
- 33.Dayton RD, Grames MS, Klein RL. More expansive gene transfer to the rat CNS: AAV PHP. EB vector dose–response and comparison to AAV PHPB. Gene Ther. 25:392–400. doi: 10.1038/s41434-018-0028-5. [DOI] [PubMed] [Google Scholar]
- 34.De Mesmaeker A, Altmann KH, Waldner A, Wendeborn S. Backbone modifications in oligonucleotides and peptide nucleic acid systems. Curr Opin Struct Biol. 1995;5:343–355. doi: 10.1016/0959-440x(95)80096-4. [DOI] [PubMed] [Google Scholar]
- 35.de Lacy N, King BH. Revisiting the relationship between autism and schizophrenia: toward an integrated neurobiology. Annu Rev Clin Psychol. 2013;9:555–87. doi: 10.1146/annurev-clinpsy-050212-185627. [DOI] [PubMed] [Google Scholar]
- 36.Deciphering Developmental Disorders Study (2015) Large-scale discovery of novel genetic causes of developmental disorders. Nature. 519:223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deciphering Developmental Disorders Study (2017) Prevalence and architecture of de novo mutations in developmental disorders. Nature. 542:433–438. doi: 10.1038/nature21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dell’Mour V, Range F, Huber L. Social learning and mother’s behavior in manipulative tasks in infant marmosets. Am J Primatol. 2009;71:503–509. doi: 10.1002/ajp.20682. [DOI] [PubMed] [Google Scholar]
- 39.Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, Wu WL, Yang B, Huber N, Pasca SP, Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev. 2012;22:229–237. doi: 10.1016/j.gde.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 41.D’Gama AM, Walsh CA. Somatic mosaicism and neurodevelopmental disease. Nat Neurosci. 2018;21:1504–1514. doi: 10.1038/s41593-018-0257-3. [DOI] [PubMed] [Google Scholar]
- 42.Dikpati A, Madgulkar AR, Kshirsagar SJ, Bhalekar MR, Chahal AS. Targeted drug delivery to CNS using nanoparticles. J Adv Pharm Sci. 2012;2:179–191. [Google Scholar]
- 43.Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Donsante A, Miller DG, Li Y, Vogler C, Brunt EM, Russell DW, Sands MS. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317:477. doi: 10.1126/science.1142658. [DOI] [PubMed] [Google Scholar]
- 45.Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 46.Duan D. Systemic delivery of adeno-associated viral vectors. Curr Opin Virol. 2016;21:16–25. doi: 10.1016/j.coviro.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duan D, Yue Y, Engelhardt JF. Expanding AAV packaging capacity with trans-splicing or overlapping vectors: a quantitative comparison. Mol Ther. 2001;4:383–391. doi: 10.1006/mthe.2001.0456. [DOI] [PubMed] [Google Scholar]
- 48.Duan D, Yue Y, Engelhardt JF. Dual Vector Expansion of the Recombinant AAV Packaging Capacity. In: J.M. Metzger., editor. Cardiac Cell and Gene Transfer: Principles, Protocols, and Applications. New York: Totowa, NJ: Springer; 2003. pp. 29–51. [DOI] [PubMed] [Google Scholar]
- 49.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duque S, Joussemet B, Riviere C, Marais T, Dubreil L, Douar AM, Fyfe J, Moullier P, Colle MA, Barkats M. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol Ther. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Rogé B, Héron D, Burglen L, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ehninger D, Li W, Fox K, Stryker MP, Silva AJ. Reversing neurodevelopmental disorders in adults. Neuron. 2008;60:950–960. doi: 10.1016/j.neuron.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elsabbagh M, Divan G, Koh YJ, Kim YS, Kauchali S, Marcín C, Montiel-Nava C, Patel V, Paula CS, Wang C, Yasamy MT, Fombonne E. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012;5:160–179. doi: 10.1002/aur.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elia J, Gai X, Xie HM, Perin JC, Geiger E, Glessner JT, D’arcy M, deBerardinis R, Frackelton E, Kim C, Lantieri F, Muganga BM, Wang L, Takeda T, Rappaport EF, Grant SF, Berrettini W, Devoto M, Shaikh TH, Hakonarson H, et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry. 2010;15:637–646. doi: 10.1038/mp.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Enkhmandakh B, Stoddard C, Mack K, He W, Kaback D, Yee SP, Bayarsaihan D. Generation of a mouse model for a conditional inactivation of Gtf2i allele. Genesis. 2016;54:407–412. doi: 10.1002/dvg.22948. [DOI] [PubMed] [Google Scholar]
- 56.Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13:97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- 57.Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Freeman JL, Perry GH, Feuk L, Redon R, McCarroll SA, Altshuler DM, Aburatani H, Jones KW, Tyler-Smith C, Hurles ME, Carter NP, Scherer SW, Lee C. Copy number variation: New insights in genome diversity. Genome Res. 2006;16:949–961. doi: 10.1101/gr.3677206. [DOI] [PubMed] [Google Scholar]
- 59.Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG, Jr, Warren ST. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 60.Galimi F, Verma IM. Opportunities for the Use of Lentiviral Vectors in Human Gene Therapy, in Lentiviral Vectors. In: D. Trono., editor. Vol. 2002. Berlin, Heidelberg: Springer Berlin Heidelberg; 2002. pp. 245–254. [DOI] [PubMed] [Google Scholar]
- 61.Garber KB, Visootsak J, Warren ST. Fragile X syndrome. Eur J Hum Genet. 2008;16:666–672. doi: 10.1038/ejhg.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gauthier J, Spiegelman D, Piton A, Lafrenière RG, Laurent S, St-Onge J, Lapointe L, Hamdan FF, Cossette P, Mottron L, Fombonne E, Joober R, Marineau C, Drapeau P, Rouleau GA. Novel de novo SHANK3 mutation in autistic patients. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:421–424. doi: 10.1002/ajmg.b.30822. [DOI] [PubMed] [Google Scholar]
- 63.Geschwind DH. Genetics of autism spectrum disorders. Trends Cogn Sci. 2011;15:409–416. doi: 10.1016/j.tics.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gray SJ, Foti SB, Schwartz JW, Bachaboina L, Taylor-Blake B, Coleman J, Ehlers MD, Zylka MJ, McCown TJ, Samulski RJ. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum Gene Ther. 2011;22:1143–1153. doi: 10.1089/hum.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 67.György B, Meijer EJ, Ivanchenko MV, Tenneson K, Emond F, Hanlon KS, Indzhykulian AA, Volak A, Karavitaki KD, Tamvakologos PI, Vezina M, Berezovskii VK, Born RT, O’Brien M, Lafond JF, Arsenijevic Y, Kenna MA, Maguire CA, Corey DP. Gene transfer with AAV9-PHP-B rescues hearing in a mouse model of Usher syndrome 3A and transduces hair cells in a non-human primate. Mol Ther Methods Clin Dev. 13:1–13. doi: 10.1016/j.omtm.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Ann Neurol. 1983;14:471–479. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- 69.Heavner WE, Smith SEP. Resolving the synaptic versus developmental dichotomy of autism risk genes. Trends Neurosci. 2020;43:227–241. doi: 10.1016/j.tins.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC, Cavazzana-Calvo M, Fischer A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003a;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 71.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003b;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 72.Hagberg B. Rett’s syndrome: prevalence and impact on progressive severe mental retardation in girls. Acta Paediatr Scand. 1985;74:405–408. doi: 10.1111/j.1651-2227.1985.tb10993.x. [DOI] [PubMed] [Google Scholar]
- 73.Harlow EG, Till SM, Russell TA, Wijetunge LS, Kind P, Contractor A. Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron. 2010;65:385–398. doi: 10.1016/j.neuron.2010.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 75.Hensch TK. Critical period regulation. Annu Rev Neurosci. 2004;27:549–579. doi: 10.1146/annurev.neuro.27.070203.144327. [DOI] [PubMed] [Google Scholar]
- 76.Hermonat PL, Muzyczka N. Use of adeno-associated virus as a mammalian DNA cloning vector: transduction of neomycin resistance into mammalian tissue culture cells. Proc Natl Acad Sci U S A. 1984;81:6466–6470. doi: 10.1073/pnas.81.20.6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hinderer C, Bell P, Vite CH, Louboutin JP, Grant R, Bote E, Yu H, Pukenas B, Hurst R, Wilson JM. Widespread gene transfer in the central nervous system of cynomolgus macaques following delivery of AAV9 into the cisterna magna. Mol Ther Methods Clin Dev. 2014;1:14051. doi: 10.1038/mtm.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, Richman LK, Wilson JM. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther. 2018;29:285–298. doi: 10.1089/hum.2018.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hirsch ML, Wolf SJ, Samulski RJ. Delivering Transgenic DNA Exceeding the Carrying Capacity of AAV Vectors. In: F. P. Manfredsson., editor. Gene Therapy for Neurological Disorders: Methods and Protocols. New York, NY: Springer New York; 2016. pp. 21–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hocquemiller M, Giersch L, Audrain M, Parker S, Cartier N. Adeno-associated virus-based gene therapy for CNS diseases. Hum Gene Ther. 2016;27:478–496. doi: 10.1089/hum.2016.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hou D, Xie C, Huang K, Zhu C. The production and characteristics of solid lipid nanoparticles (SLNs) Biomaterials. 2003;24:1781–1785. doi: 10.1016/s0142-9612(02)00578-1. [DOI] [PubMed] [Google Scholar]
- 82.Hübener M, Bonhoeffer T. Neuronal plasticity: beyond the critical period. Cell. 2014;159:727–737. doi: 10.1016/j.cell.2014.10.035. [DOI] [PubMed] [Google Scholar]
- 83.Hudry E, Vandenberghe LH. Therapeutic AAV gene transfer to the nervous system: a clinical reality. Neuron. 2019;101:839–862. doi: 10.1016/j.neuron.2019.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hyman SE. How far can mice carry autism research. Cell. 2014;158:13–14. doi: 10.1016/j.cell.2014.06.032. [DOI] [PubMed] [Google Scholar]
- 85.Izpisua Belmonte JC, Callaway EM, Caddick SJ, Churchland P, Feng G, Homanics GE, Lee KF, Leopold DA, Miller CT, Mitchell JF, Mitalipov S, Moutri AR, Movshon JA, Okano H, Reynolds JH, Ringach D, Sejnowski TJ, Silva AC, Strick PL, Wu J. Brains, genes, and primates. Neuron. 2015;86:617–631. doi: 10.1016/j.neuron.2015.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kaiser T, Feng G. Modeling psychiatric disorders for developing effective treatments. Nat Med. 2015;21:979–988. doi: 10.1038/nm.3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med. 2001;7:33–40. doi: 10.1038/83324. [DOI] [PubMed] [Google Scholar]
- 88.Kenny EM, Cormican P, Furlong S, Heron E, Kenny G, Fahey C, Kelleher E, Ennis S, Tropea D, Anney R, Corvin AP, Donohoe G, Gallagher L, Gill M, Morris DW. Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol Psychiatry. 2014;19:872–879. doi: 10.1038/mp.2013.127. [DOI] [PubMed] [Google Scholar]
- 89.Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY, Song DW, Lee KJ, Jung MH, Kim S, Kim JH, Kim JH, Kim JS. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun. 2017;8:14500. doi: 10.1038/ncomms14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kishi N, Sato K, Sasaki E, Okano H. Common marmoset as a new model animal for neuroscience research and genome editing technology. Dev Growth Differ. 2014;56:53–62. doi: 10.1111/dgd.12109. [DOI] [PubMed] [Google Scholar]
- 91.Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 92.Knoll JH, Nicholls RD, Magenis RE, Graham JM, Jr, Lalande M, Latt SA. Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in parental origin of the deletion. Am J Med Genet. 1989;32:285–290. doi: 10.1002/ajmg.1320320235. [DOI] [PubMed] [Google Scholar]
- 93.Korenberg JR, Chen XN, Hirota H, Lai Z, Bellugi U, Burian D, Roe B, Matsuoka R. VI. Genome structure and cognitive map of Williams syndrome. J Cogn Neurosci. 2000;12:89–107. doi: 10.1162/089892900562002. [DOI] [PubMed] [Google Scholar]
- 94.Kumita W, Sato K, Suzuki Y, Kurotaki Y, Harada T, Zhou Y, Kishi N, Sato K, Aiba A, Sakakibara Y, Feng G, Okano H, Sasaki E. Efficient generation of Knock-in/Knock-out marmoset embryo via CRISPR/Cas9 gene editing. Sci Rep. 2019;9:12719. doi: 10.1038/s41598-019-49110-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 96.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 97.Lee B, Lee K, Panda S, Gonzales-Rojas R, Chong A, Bugay V, Park HM, Brenner R, Murthy N, Lee HY. Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile X syndrome from exaggerated repetitive behaviours. Nat Biomed Eng. 2018;2:497–507. doi: 10.1038/s41551-018-0252-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lentz JJ, Jodelka FM, Hinrich AJ, McCaffrey KE, Farris HE, Spalitta MJ, Bazan NG, Duelli DM, Rigo F, Hastings ML. Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat Med. 2013;19:345–350. doi: 10.1038/nm.3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ladd-Acosta C, Hansen KD, Briem E, Fallin MD, Kaufmann WE, Feinberg AP. Common DNA methylation alterations in multiple brain regions in autism. Mol Psychiatry. 2014;19:862–871. doi: 10.1038/mp.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lesch KP. Maturing insights into the genetic architecture of neurodevelopmental disorders – from common and rare variant interplay to precision psychiatry. J Child Psychol Psychiatry. 2016;57:659–661. doi: 10.1111/jcpp.12574. [DOI] [PubMed] [Google Scholar]
- 101.Liu Z, Li X, Zhang JT, Cai YJ, Cheng TL, Cheng C, Wang Y, Zhang CC, Nie YH, Chen ZF, Bian WJ, Zhang L, Xiao J, Lu B, Zhang YF, Zhang XD, Sang X, Wu JJ, Xu X, Xiong ZQ, et al. Autism-like behaviours and germline transmission in transgenic monkeys overexpressing. MeCP2 Nature. 2016;530:98. doi: 10.1038/nature16533. [DOI] [PubMed] [Google Scholar]
- 102.Lugtenberg D, Kleefstra T, Oudakker AR, Nillesen WM, Yntema HG, Tzschach A, Raynaud M, Rating D, Journel H, Chelly J, Goizet C, Lacombe D, Pedespan JM, Echenne B, Tariverdian G, O’Rourke D, King MD, Green A, van Kogelenberg M, Van Esch H, et al. Structural variation in Xq28: MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy. Eur J Hum Genet. 2009;17:444–453. doi: 10.1038/ejhg.2008.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Luoni M, Giannelli S, Indrigo MT, Niro A, Massimino L, Iannielli A, Passeri L, Russo F, Morabito G, Calamita P, Gregori S, Deverman B, Broccoli V. Whole brain delivery of an instability-prone Mecp2 transgene improves behavioral and molecular pathological defects in mouse models of Rett syndrome. bioRxiv. 2019 doi: 10.7554/eLife.52629. doi:10.7554/eLife.52629.sa1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mandel RJ, Manfredsson FP, Foust KD, Rising A, Reimsnider S, Nash K, Burger C. Recombinant adeno-associated viral vectors as therapeutic agents to treat neurological disorders. Mol Ther. 2006;13:463–483. doi: 10.1016/j.ymthe.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 106.Marín O. Developmental timing and critical windows for the treatment of psychiatric disorders. Nat Med. 2016;22:1229–1238. doi: 10.1038/nm.4225. [DOI] [PubMed] [Google Scholar]
- 107.Mei Y, Monteiro P, Zhou Y, Kim JA, Gao X, Fu Z, Feng G. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature. 2016;530:481–484. doi: 10.1038/nature16971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518:409–412. doi: 10.1038/nature13975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Meredith RM. Sensitive and critical periods during neurotypical and aberrant neurodevelopment: A framework for neurodevelopmental disorders. Neurosci Biobehav Rev. 2015;50:180–188. doi: 10.1016/j.neubiorev.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 110.Meza-Sosa KF, Valle-García D, Pedraza-Alva G, Pérez-Martínez L. Role of microRNAs in central nervous system development and pathology. J Neurosci Res. 2012;90:1–12. doi: 10.1002/jnr.22701. [DOI] [PubMed] [Google Scholar]
- 111.Miller CT, Freiwald WA, Leopold DA, Mitchell JF, Silva AC, Wang X. Marmosets: a neuroscientific model of human social behavior. Neuron. 2016;90:219–233. doi: 10.1016/j.neuron.2016.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- 113.Mizrahy S, Gutkin A, Decuzzi P, Peer D. Targeting central nervous system pathologies with nanomedicines. J Drug Target. 2019;27:542–554. doi: 10.1080/1061186X.2018.1533556. [DOI] [PubMed] [Google Scholar]
- 114.Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, Scherer SW. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Monteiro P, Feng G. Learning from animal models of obsessive-compulsive disorder. Biol Psychiatry. 2016;79:7–16. doi: 10.1016/j.biopsych.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Monteiro P, Feng G. SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat Rev Neurosci. 2017;18:147–157. doi: 10.1038/nrn.2016.183. [DOI] [PubMed] [Google Scholar]
- 117.Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C, Sergi Sergi L, Benedicenti F, Ambrosi A, Di Serio C, Doglioni C, von Kalle C, Naldini L. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24:687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- 118.Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Morris CA, Mervis CB, Paciorkowski AP, Abdul-Rahman O, Dugan SL, Rope AF, Bader P, Hendon LG, Velleman SL, Klein-Tasman BP, Osborne LR. 7q11.23 Duplication syndrome: Physical characteristics and natural history. Am J Med Genet A. 2015;167A:2916–2935. doi: 10.1002/ajmg.a.37340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Moy SS, Nadler JJ, Young NB, Nonneman RJ, Segall SK, Andrade GM, Crawley JN, Magnuson TR. Social approach and repetitive behavior in eleven inbred mouse strains. Behav Brain Res. 2008;191:118–129. doi: 10.1016/j.bbr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Murlidharan G, Sakamoto K, Rao L, Corriher T, Wang D, Gao G, Sullivan P, Asokan A. CNS-restricted transduction and CRISPR/Cas9-mediated gene deletion with an engineered AAV vector. Mol Ther Nucleic Acids. 2016;5:e338. doi: 10.1038/mtna.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Naso MF, Tomkowicz B, Perry WL, Strohl WR. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. 2017;31:317–334. doi: 10.1007/s40259-017-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, Madhavan S, Pan X, Ran FA, Yan WX, Asokan A, Zhang F, Duan D, Gersbach CA. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Netser S, Haskal S, Magalnik H, Wagner S. A novel system for tracking social preference dynamics in mice reveals sex- and strain-specific characteristics. Mol Autism. 2017;8:53. doi: 10.1186/s13229-017-0169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Niemi MEK, Martin HC, Rice DL, Gallone G, Gordon S, Kelemen M, McAloney K, McRae J, Radford EJ, Yu S, Gecz J, Martin NG, Wright CF, Fitzpatrick DR, Firth HV, Hurles ME, Barrett JC. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature. 2018;562:268–271. doi: 10.1038/s41586-018-0566-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, Kang Y, Zhao X, Si W, Li W, Xiang AP, Zhou J, Guo X, Bi Y, Si C, Hu B, Dong G, Wang H, Zhou Z, Li T, et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell. 2014;156:836–843. doi: 10.1016/j.cell.2014.01.027. [DOI] [PubMed] [Google Scholar]
- 128.Offen D, Anidjar A, Simonovitch S, Ben-Zur T, Michaelson D. Increase in autophagy and amyloid beta uptake in apoe expressing astrocytes after calpain knock down by CRISPR-Cas9. Cytotherapy. 2019;21:e6. [Google Scholar]
- 129.Okano H, Hikishima K, Iriki A, Sasaki E. The common marmoset as a novel animal model system for biomedical and neuroscience research applications. Semin Fetal Neonatal Med. 2012;17:336–340. doi: 10.1016/j.siny.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 130.Osborne LR. Animal models of Williams syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:209–219. doi: 10.1002/ajmg.c.30257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pardridge WM. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Park F. Lentiviral vectors: are they the future of animal transgenesis. Physiol Genomics. 2007;31:159–173. doi: 10.1152/physiolgenomics.00069.2007. [DOI] [PubMed] [Google Scholar]
- 133.Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, Hua Y, Rigo F, Matson J, Hung G, Kaye EM, Shihabuddin LS, Krainer AR, Bennett CF, Cheng SH. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Patel A, et al. Design of AAV Vectors for Delivery of Large or Multiple Transgenes. In: MJ Castle., editor. Adeno-Associated Virus Vectors: Design and Delivery. New York, NY: Springer New York; 2019. pp. 19–33. [DOI] [PubMed] [Google Scholar]
- 135.Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pober BR. Williams-Beuren syndrome. N Engl J Med. 2010;362:239–252. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- 138.Qiu P, Wang L, Ni J, Zhang Y. BMAL1 knockout macaque monkeys display reduced sleep and psychiatric disorders. Nat Sci Rev. 2019;6:87–100. doi: 10.1093/nsr/nwz002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet A. 2010;152A:1079–1088. doi: 10.1002/ajmg.a.33184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Reis A, Dittrich B, Greger V, Buiting K, Lalande M, Gillessen-Kaesbach G, Anvret M, Horsthemke B. Imprinting mutations suggested by abnormal DNA methylation patterns in familial Angelman and Prader-Willi syndromes. Am J Hum Genet. 1994;54:741–747. [PMC free article] [PubMed] [Google Scholar]
- 145.Renieri A, Meloni I, Longo I, Ariani F, Mari F, Pescucci C, Cambi F. Rett syndrome: the complex nature of a monogenic disease. J Mol Med (Berl) 2003;81:346–354. doi: 10.1007/s00109-003-0444-9. [DOI] [PubMed] [Google Scholar]
- 146.Sakurai T, Dorr NP, Takahashi N, McInnes LA, Elder GA, Buxbaum JD. Haploinsufficiency of Gtf2i, a gene deleted in Williams Syndrome, leads to increases in social interactions. Autism Res. 2011;4:28–39. doi: 10.1002/aur.169. [DOI] [PubMed] [Google Scholar]
- 147.Samaranch L, Salegio EA, San Sebastian W, Kells AP, Foust KD, Bringas JR, Lamarre C, Forsayeth J, Kaspar BK, Bankiewicz KS. Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum Gene Ther. 2012;23:382–389. doi: 10.1089/hum.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PB, Choi M, Crawford EL, Davis L, Wright NR, Dhodapkar RM, DiCola M, DiLullo NM, et al. Multiple recurrent de novo CNVs, including duplications of the 7q1123 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sasaki E, Suemizu H, Shimada A, Hanazawa K, Oiwa R, Kamioka M, Tomioka I, Sotomaru Y, Hirakawa R, Eto T, Shiozawa S, Maeda T, Ito M, Ito R, Kito C, Yagihashi C, Kawai K, Miyoshi H, Tanioka Y, Tamaoki N, et al. Generation of transgenic non-human primates with germline transmission. Nature. 2009;459:523. doi: 10.1038/nature08090. [DOI] [PubMed] [Google Scholar]
- 151.Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimäki T, Ledbetter D, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Segura-Puimedon M, Sahún I, Velot E, Dubus P, Borralleras C, Rodrigues AJ, Valero MC, Valverde O, Sousa N, Herault Y, Dierssen M, Pérez-Jurado LA, Campuzano V. Heterozygous deletion of the Williams–Beuren syndrome critical interval in mice recapitulates most features of the human disorder. Hum Mol Genet. 2014;23:6481–6494. doi: 10.1093/hmg/ddu368. [DOI] [PubMed] [Google Scholar]
- 154.Senís E, Fatouros C, Große S, Wiedtke E, Niopek D, Mueller AK, Börner K, Grimm D. CRISPR/Cas9-mediated genome engineering: An adeno-associated viral (AAV) vector toolbox. Biotechnol J. 2014;9:1402–1412. doi: 10.1002/biot.201400046. [DOI] [PubMed] [Google Scholar]
- 155.Shevtsova Z, Malik JM, Michel U, Bähr M, Kügler S. Promoters and serotypes: Targeting of adeno-associated virus vectors for gene transfer in the rat central nervous system in vitro and in vivo. Exp Physiol. 2005;90:53–59. doi: 10.1113/expphysiol.2004.028159. [DOI] [PubMed] [Google Scholar]
- 156.Shomron N. MicroRNAs and pharmacogenomics. Pharmacogenomics. 2010;11:629–632. doi: 10.2217/pgs.10.26. [DOI] [PubMed] [Google Scholar]
- 157.Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N. The cellular and molecular landscapes of the developing human central nervous system. Neuron. 2016;89:248–268. doi: 10.1016/j.neuron.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Silva-Santos S, van Woerden GM, Bruinsma CF, Mientjes E, Jolfaei MA, Distel B, Kushner SA, Elgersma Y. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J Clin Invest. 2015;125:2069–2076. doi: 10.1172/JCI80554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Srivastava A. In vivo tissue-tropism of adeno-associated viral vectors. Curr Opin Virol. 2016;21:75–80. doi: 10.1016/j.coviro.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, Tyler-Smith C, Carter N, Scherer SW, Tavaré S, Deloukas P, Hurles ME, Dermitzakis ET. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Strømme P, Bjørnstad PG, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol. 2002;17:269–271. doi: 10.1177/088307380201700406. [DOI] [PubMed] [Google Scholar]
- 162.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60:1187–1192. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- 163.Sun E, Shi Y. MicroRNAs: Small molecules with big roles in neurodevelopment and diseases. Exp Neurol. 2015;268:46–53. doi: 10.1016/j.expneurol.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 164.Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y, Trombetta J, Sur M, Zhang F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol. 2015;33:102–106. doi: 10.1038/nbt.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sztainberg Y, Chen HM, Swann JW, Hao S, Tang B, Wu Z, Tang J, Wan YW, Liu Z, Rigo F, Zoghbi HY. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature. 2015;528:123–126. doi: 10.1038/nature16159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Takumi T, Tamada K. CNV biology in neurodevelopmental disorders. Curr Opin Neurobiol. 2018;48:183–192. doi: 10.1016/j.conb.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 167.Thurm A, Powell EM, Neul JL, Wagner A, Zwaigenbaum L. Loss of skills and onset patterns in neurodevelopmental disorders: Understanding the neurobiological mechanisms. Autism Res. 2018;11:212–222. doi: 10.1002/aur.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tick B, Bolton P, Happé F, Rutter M, Rijsdijk F. Heritability of autism spectrum disorders: a meta-analysis of twin studies. J Child Psychol Psychiatry. 2016;57:585–595. doi: 10.1111/jcpp.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Van Esch H. MECP2 duplication syndrome. Mol Syndromol. 2011;2:128–136. doi: 10.1159/000329580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Williams CA, Zori RT, Hendrickson J, Stalker H, Marum T, Whidden E, Driscoll DJ. Angelman syndrome. Curr Probl Pediatr. 1995;25:216–231. doi: 10.1016/s0045-9380(06)80036-8. [DOI] [PubMed] [Google Scholar]
- 172.Wilmot B, Fry R, Smeester L, Musser ED, Mill J, Nigg JT. Methylomic analysis of salivary DNA in childhood ADHD identifies altered DNA methylation in VIPR2. J Child Psychol Psychiatry. 2016;57:152–160. doi: 10.1111/jcpp.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, Hu S, Marshall J, McDermid HE. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet. 2003;40:575–584. doi: 10.1136/jmg.40.8.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wolf A, Bauer B, Abner EL, Ashkenazy-Frolinger T, Hartz AM. A comprehensive behavioral test battery to assess learning and memory in 129S6/Tg2576 mice. PLoS One. 2016;11:e0147733. doi: 10.1371/journal.pone.0147733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther. 2010;18:80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Xu B, Karayiorgou M, Gogos JA. MicroRNAs in psychiatric and neurodevelopmental disorders. Brain Res. 2010;1338:78–88. doi: 10.1016/j.brainres.2010.03.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Xu B, Roos JL, Levy S, van Rensburg EJ, Gogos JA, Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 178.Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yang S, Chang R, Yang H, Zhao T, Hong Y, Kong HE, Sun X, Qin Z, Jin P, Li S, Li XJ. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J Clin Invest. 2017;127:2719–2724. doi: 10.1172/JCI92087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yin H, Song CQ, Dorkin JR, Zhu LJ, Li Y, Wu Q, Park A, Yang J, Suresh S, Bizhanova A, Gupta A, Bolukbasi MF, Walsh S, Bogorad RL, Gao G, Weng Z, Dong Y, Koteliansky V, Wolfe SA, Langer R, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol. 2016;34:328–333. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ylä-Herttuala S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Mol Ther. 2012;20:1831–1832. doi: 10.1038/mt.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451–481. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhao H, Tu Z, Xu H, Yan S, Yan H, Zheng Y, Yang W, Zheng J, Li Z, Tian R, Lu Y, Guo X, Jiang YH, Li XJ, Zhang YQ. Altered neurogenesis and disrupted expression of synaptic proteins in prefrontal cortex of SHANK3-deficient non-human primate. Cell Res. 2017;27:1293–1297. doi: 10.1038/cr.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhou Y, Kaiser T, Monteiro P, Zhang X, Van der Goes MS, Wang D, Barak B, Zeng M, Li C, Lu C, Wells M, Amaya A, Nguyen S, Lewis M, Sanjana N, Zhou Y, Zhang M, Zhang F, Fu Z, Feng G. Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron. 2016;89:147–162. doi: 10.1016/j.neuron.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhou Y, Sharma J, Ke Q, Landman R, Yuan J, Chen H, Hayden DS, Fisher JW, Jiang M, Menegas W, Aida T, Yan T, Zou Y, Xu D, Parmar S, Hyman JB, Fanucci-Kiss A, Meisner O, Wang D, Huang Y, et al. Atypical behaviour and connectivity in SHANK3-mutant macaques. Nature. 2019;570:326–331. doi: 10.1038/s41586-019-1278-0. [DOI] [PubMed] [Google Scholar]
- 186.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 187.Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. 2012 doi: 10.1101/cshperspect.a009886. doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]