

Abstract
Millions of people worldwide are affected by traumatic spinal cord injury, which usually results in permanent sensorimotor disability. Damage to the spinal cord leads to a series of detrimental events including ischaemia, haemorrhage and neuroinflammation, which over time result in further neural tissue loss. Eventually, at chronic stages of traumatic spinal cord injury, the formation of a glial scar, cystic cavitation and the presence of numerous inhibitory molecules act as physical and chemical barriers to axonal regrowth. This is further hindered by a lack of intrinsic regrowth ability of adult neurons in the central nervous system. The intracellular signalling molecule, cyclic adenosine 3′,5′-monophosphate (cAMP), is known to play many important roles in the central nervous system, and elevating its levels as shown to improve axonal regeneration outcomes following traumatic spinal cord injury in animal models. However, therapies directly targeting cAMP have not found their way into the clinic, as cAMP is ubiquitously present in all cell types and its manipulation may have additional deleterious effects. A downstream effector of cAMP, exchange protein directly activated by cAMP 2 (Epac2), is mainly expressed in the adult central nervous system, and its activation has been shown to mediate the positive effects of cAMP on axonal guidance and regeneration. Recently, using ex vivo modelling of traumatic spinal cord injury, Epac2 activation was found to profoundly modulate the post-lesion environment, such as decreasing the activation of astrocytes and microglia. Pilot data with Epac2 activation also suggested functional improvement assessed by in vivo models of traumatic spinal cord injury. Therefore, targeting Epac2 in traumatic spinal cord injury could represent a novel strategy in traumatic spinal cord injury repair, and future work is needed to fully establish its therapeutic potential.
Keywords: astrocytes, axonal regeneration, cAMP, central nervous system regeneration, Epac, glial scar, microglia, neuroinflammation, neurons, spinal cord, spinal cord injury, traumatic spinal cord injury
A Brief Overview of Central Nervous System Trauma
Central nervous system (CNS) trauma commonly occurs in the forms of traumatic spinal cord and brain injuries (TSCI and TBI). It is estimated that around 27 million people live with TSCI worldwide (GBD 2016 Traumatic Brain Injury and Spinal Cord Injury Collaborators, 2019). CNS injuries often result in permanent sensorimotor disability. The poor ability of adult mammalian CNS neurons to regrow in response to injuries is known to be associated with their very limited intrinsic growth capacity and the inhibitory environment at the lesion site post-injury (Curcio and Bradke, 2018). This review aims to discuss the importance of exchange protein directly activated by cAMP (Epac) in overcoming these barriers, in conjunction with its role in neural development, regeneration and cell death as well as modulation of the post-injury environment.
The pathophysiology of TSCI
The events following TSCI can be classified into four phases: acute (< 48 hours), subacute (48 hours to 14 days), intermediate-to-chronic (14 days to 6 months) and chronic (> 6 months) (Badhiwala et al., 2018). During the acute phase, the initial physical trauma causes damage to neural cells and the disruption of vasculature and the blood spinal cord barrier leading to haemorrhages and ischemia (Tator and Koyanagi, 1997; Mautes et al., 2000). Neurons and oligodendrocytes in the injury area undergo necrosis due to haemorrhagic/ischemic events and are also subjected to a further cascade of outcomes, which arise from changes in cell permeabilization and pro-apoptotic signaling (Choo et al., 2007). At the same time, peripheral inflammatory cells including macrophages, neutrophils and lymphocytes infiltrate the injury area and secrete mediators such as tumour necrosis factor-α and interleukin-1β (IL-1β) (Pineau and Lacroix, 2007). These cells remain beyond the subacute phase. The exposed myelin from damaged oligodendrocytes is a source of myelin-associated inhibitors, such as Nogo, oligodendrocyte myelin glycoprotein and myelin-associated glycoprotein (MAG), which are known to prevent axonal regrowth after TSCI (Berry, 1982; Caroni and Schwab, 1988). All these molecules signal through the NgR-p75NTR receptor complex via the RhoA protein, which leads to activation of Rho kinase and inhibition of axonal regrowth (Filbin, 2003; Fournier et al., 2003).
In the subacute phase, the secondary SCI cascade is characterized by prolonged inflammation and further death of neurons and oligodendrocytes (Kwon et al., 2004; Badhiwala et al., 2018). The underlying mechanisms for apoptosis include ischemia, which disrupts ionic balance of K+, Na+ and Ca2+ leading to depolarization of cell membranes, ATPase failure and increase of intracellular calcium (Agrawal and Fehlings, 1996; Badhiwala et al., 2018), as well as increased levels of the excitatory neurotransmitter, glutamate (Farooque et al., 1996; McAdoo et al., 1999). Activated microglia, together with infiltrated inflammatory cells, facilitate ongoing apoptosis and produce free radicals, which are formed as by-products of debris clearance by immune cells, caused by lipid membrane peroxidation, DNA oxidative damage and protein oxidation, leading to further apoptosis (Dizdaroglu et al., 2002; Hausmann, 2003). Preclinical studies have shown that neuronal and oligodendrocyte apoptosis can last up to 4 weeks and 1 year, respectively, post-TSCI (Beattie et al., 2002; Huang et al., 2007).
The chronic phase of the injury is characterized by reorganization of neural circuits and vascularization, demyelination and alterations in the composition of the extracellular matrix. In humans and some animal models, the continued cell death during the acute phase leads to the formation of cystic cavities containing cellular debris, extracellular fluid, and macrophages (O’Shea et al., 2017; Bradbury and Burnside, 2019). These cavities are a poor substrate for neural regeneration and therefore act as a physical barrier for axonal regrowth (O’Shea et al., 2017). The cavities are lined by scar tissue that is generally classified into two components: fibrotic and glial. The fibrotic, inner component of the scar is primarily a product of fibroblasts migrating to the lesion site from the disrupted vasculature and meningeal layers (Klapka and Muller, 2006; Fawcett et al., 2012). These cells deposit dense collagen depots forming a condensed structure, which adds up to a physical barrier for axonal regrowth (Klapka and Muller, 2006; Goritz et al., 2011; Fawcett et al., 2012). The glial, outer component of the scar is made up of reactive astrocytes, which proliferate, undergo hypertrophic changes and overlap their processes around the cavities creating a mesh-like array that forms walls of neural tissue from the harmful environment of the cavities (Yuan and He, 2013). During the chronic phase, activated glial cells (e.g., microglia, astrocytes and macrophages), together with oligodendrocyte progenitor cells (OPCs), secrete extracellular matrix proteins that are inhibitory to axonal growth, such as chondroitin sulphate proteoglycans (CSPGs) (McKeon et al., 1991, 1999; Silver and Miller, 2004; Busch and Silver, 2007). CSPGs may also affect OPCs, as they prevent the outgrowth of OPC processes and differentiation, therefore leading to weakened remyelination and an observable decrease in axonal regeneration (Siebert and Osterhout, 2011). The key players and the formation of the glial scar and cavity are demonstrated in Figure 1.
Figure 1.
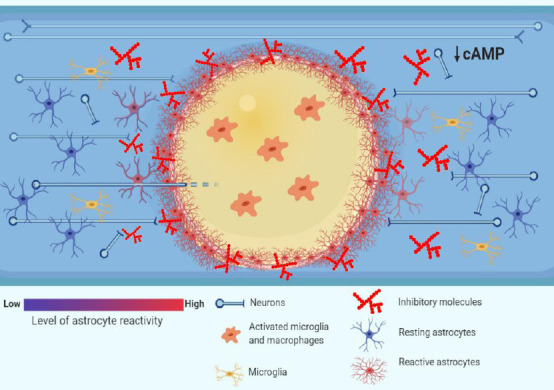
Pathophysiology of traumatic spinal cord injury (TSCI): barriers to axonal regrowth.
Following TSCI, the resulting ischemia, hemorrhage and inflammation generate zones of dying tissue that, with time, transform into cystic cavities (yellow zone in the picture). The cytotoxic environment in the cavities, which are filled with activated inflammatory cells such as microglia and macrophages, renders them a poor substrate to axonal regrowth (O’Shea et al., 2017). The cystic cavities are encircled by a glial scar that is formed by reactive astrocytes. The astrocytes undergo hypertrophy and wall off the cytotoxic environment of the cysts from the spared neural tissue, at the same time forming a physical barrier to axonal regrowth (Sofroniew, 2014). Moreover, the chemical barrier to axonal regrowth is set up by activated glial cells such as astrocytes, macrophages and oligodendrocyte progenitor cells (OPCs), which secrete inhibitory molecules such as chondroitin sulphate proteoglycans (CSPGs) (Busch and Silver, 2007). Finally, the intrinsic abilities of axons to regrow are undermined by low levels of cyclic adenosine 3′,5′-monophosphate (cAMP), which drop postnatally and decrease further after spinal cord injury (Pearse et al., 2004), and which govern a response of growth cones to extracellular guidance molecules such as myelin-associated glycoprotein (MAG) (Peace and Shewan, 2011).
Search Strategy and Selection Criteria
A first broad search was done by AGB in December 2018 using PubMed and key words such as ‘’spinal cord injury’’, ‘’CNS trauma’’, ‘’axonal regeneration’’, ‘’cAMP’’, ‘’Epac’’, ‘’neuroinflammation’’ and combinations of those words. No limits were used. A second broad search was performed by DD between September 2019 and January 2020 using PubMed with the aim to update information and add new studies. Special emphasis was given to the use of key words such as ‘’cAMP AND Microglia OR astrocytes’’ and ‘’Epac AND Microglia OR astrocytes’’. No limits were used.
Roles of Cyclic Adenosine 3′,5′-Monophosphate in the Central Nervous System
Cyclic adenosine 3’,5’-monophosphate (cAMP) is one of the most widely studied intracellular second messengers, as it exists in all types of cells. Over the decades, cAMP signaling has increasingly become a focal point for neural regeneration studies. In the nervous systems, cAMP is known to participate in the regulation of many neuronal behaviors such as axonal growth and guidance as well as neuronal survival and differentiation.
Role of cAMP signaling in CNS development
During CNS development, cAMP is vital for the growth of neurons and is expressed at high levels. However, neuronal levels of cAMP decline at postnatal age and reach a minimum in adult animals (Cai et al., 2001; Shewan et al., 2002). It is known that embryonic CNS neurons transplanted into adult CNS tissue grow axons extensively (Li and Raisman, 1993), which contrasts to adult CNS neurons transplanted on to immature CNS tissue, which fail to extend neurites in vitro (Shewan et al., 1995). These events can be attributed to differential cAMP levels in embryonic versus adult neurons, which regulate axonal guidance. Growth cones can read environmental signals which lead to either attraction or repulsion of axons. The variation of cAMP levels in growth cones can determine the cone response to external guidance cues such as Netrin-1 or MAG, which are guidance molecules important during neural development or after injury (Song et al., 1998). For example, during retinotectal development, high cAMP levels in Xenopus retinal ganglion axons at early stages lead to the attraction of axons to Netrin-1, whilst a decline of cAMP at later stages results in the repulsion of axons (Shewan et al., 2002). In general, this cAMP-mediated chemotactic behaviour was proposed to be dependent on the cAMP/cyclic guanosine monophosphate ratio across the growth cone and asymmetric calcium signaling (Nishiyama et al., 2003; Murray et al., 2009a).
Role of cAMP in CNS regenerative capability
The adult mammalian CNS is characterized by very poor intrinsic neuronal cell regenerative abilities. The so-called ‘conditioning lesion’ is a traditional approach to augment the intrinsic machinery for regeneration in CNS neurons, in which a peripheral injury enhances central regeneration of primary sensory neurons (McQuarrie and Grafstein, 1973; Richardson and Issa, 1984). This phenomenon has been associated with cAMP levels since the injection of a cAMP analogue into the dorsal root ganglion (DRG) can mimic the effects of the conditioning lesion (Neumann et al., 2002; Qiu et al., 2002). These studies led to the understanding of the involvement of cAMP levels in regeneration. Therefore, a major biochemical event linked to the poor regeneration capacity of the CNS is a significant decrease of intrinsic neuronal cAMP levels that occurs postnatally. For example, adult rat DRG neurons that are capable of growth following peripheral nerve lesion have relatively high concentrations of cAMP, while the opposite is observed in adult rat DRG neurons that do not regenerate readily following spinal cord bilateral lesion (Qiu et al., 2002). Notably, the cAMP levels are likely to further decrease after TSCI, as following thoracic contusion injury of the spinal cord of adult rats it was found that cAMP levels decreased by over 60% in the spinal cord as well as in brainstem and cortex (Pearse et al., 2004). Therefore, deteriorating neuronal regenerative abilities after TSCI could be attributed to a further drop in cAMP levels. Previous studies have then focused on the effects of artificially elevating neuronal cAMP levels to promote regrowth after injuries.
In vitro evidence of stimulating cAMP activity to promote neurite outgrowth
The elevation of cAMP using forskolin in cultured adult rat sensory neurons was reported to enhance neurite outgrowth (Neumann et al., 2002). Furthermore, the modulation of cAMP activity by a permeable analogue of cAMP was found to alter the responses of Xenopus spinal axons to several inhibitory axonal guidance cues, shifting growth cone responses from repulsion to attraction (Song et al., 1998). It was previously reported that the upregulation of intracellular cAMP increased neurite outgrowth in cultured embryonic rat motor neurons while antagonizing cAMP resulted in a profound decrease in neurite outgrowth (Aglah et al., 2008). Moreover, cAMP was also shown to positively affect neuronal survival. It was shown that cAMP elevation by forskolin sufficed to promote short term survival of embryonic rat spinal motor neurons in vitro (Hanson et al., 1998). Therefore, it has been suggested that cAMP is important not only in neurite outgrowth but also in neuronal survival.
In vivo evidence of cAMP elevation to promote axonal regrowth
A number of research groups have targeted cAMP and elements of its signaling pathways to promote axonal regrowth. Two studies showed that manipulating levels of cAMP activity, by the injection of the cAMP analogue, dibutyryl cAMP (db-cAMP; Table 1), into adult rat DRG before spinal cord lesioning, resulted in an increase in the number of axons regrowing across the lesion site (Neumann et al., 2002; Qiu et al., 2002). However, neurons in those studies were conditioned prior to lesion, which is not a clinically relevant approach.
Table 1.
Drugs manipulating cAMP and its downstream signaling molecules
| Drug type | Activity | Outcome | Reference(s) |
|---|---|---|---|
| cAMP analogues | |||
| 8-Br-cAMP | Cyclic adenosine monophosphate (cAMP) analogue | Mouse neonatal mature astrocytes upregulated antioxidant-related genes and downregulated cell death-related genes after treatment | Paco et al., 2016 |
| db-cAMP | Injection into adult rat dorsal root ganglion (DRG) before spinal cord injury (SCI) resulted in an increase in the number of axons regrowing across the injury | Neumann et al., 2002; Qiu et al., 2002 | |
| Improved functional recovery after spinal contusion in adult female rats | Pearse et al., 2004 | ||
| Increase in axonal sprouting within the glial scar | Xia et al., 2017 | ||
| Sp-cAMPs | Adult rat DRG neurons switched their repulsive response to myelin-associated glycoprotein (MAG) gradients to attraction after treatment | Murray et al., 2009b | |
| Epac agonists | |||
| 8-Me-cAMP | General exchange protein activated by cAMP (Epac) agonist | Agonist first described | Enserink et al., 2002 |
| Rat neonatal mature astrocytes increased intracellular calcium levels after treatment | Di Cesare et al., 2006 | ||
| Adult rat DRG neurons cultured on spinal cord tissue increased neurite outgrowth after treatment | Murray and Shewan, 2008 | ||
| Adult rat DRG neurons switched their repulsive response to MAG gradients to attraction after treatment | Murray et al., 2009b | ||
| BV-2 cells significantly decreased their phagocytotic behaviour after treatment | Steininger et al., 2011 | ||
| In vitro embryonic rat spinal cord culture model of SCI increased neurite outgrowth after treatment | Boomkamp et al., 2014 | ||
| Adult rat DRG neurons increased neurite outgrowth after treatment | Wei et al., 2016 | ||
| S-220 | Specific Epac2 agonist | Agonist first described | Schwede et al., 2015 |
| Neonatal rat DRG neurons showed a strong turning behaviour towards treatment | Guijarro-Belmar et al., 2019 | ||
| Neonatal rat DRG and cortical neurons increased neurite outgrowth after treatment, even in an inhibitory environment | |||
| Ex vivo model of SCI increased neurite outgrowth and reduced glial activation after treatment | |||
| -Improved locomotor recovery after treatment in an in vivo rat SCI model | |||
| Epac antagonists | |||
| ESI-05 | Specific Epac2 antagonist | Antagonist first described | Tsalkova et al., 2012 |
| Adult rat DRG neurons reduced neurite outgrowth after treatment | Wei et al., 2016 | ||
| Neonatal rat DRG and cortical neurons decreased neurite outgrowth after treatment | Guijarro-Belmar et al., 2019 | ||
| ESI-09 | General Epac antagonist | Antagonist first described | Almahariq et al., 2013 |
| Embryonic hippocampal neurons decreased neurite outgrowth after treatment | Munoz-Llancao et al., 2015 | ||
| Others | |||
| Rolipram | Phosphodiesterase type 4 inhibitor | Improved functional recovery after treatment in spinal contusion in adult female rats | Pearse et al., 2004 |
| Increased axonal plasticity and improved recovery after treatment in spinal hemisection in adult rats | Nikulina et al., 2004 | ||
| Improved locomotor abilities after treatment in spinal contusion in adult rats | Costa et al., 2013 | ||
| In vitro embryonic rat spinal cord culture model of SCI increased neurite outgrowth after treatment | Boomkamp et al., 2014 | ||
| KT-5720 | Protein kinase A antagonist | Discussion of antagonist specificity | Murray, 2008 |
| In vitro embryonic rat spinal cord culture model of SCI increased neurite outgrowth after treatment | Boomkamp et al., 2014 |
Subsequently, more clinically relevant experiments used rolipram, which inhibits phosphodiesterase 4 (PDE 4; Table 1), mainly localized in neural tissue and degrades cAMP. The Filbin group administered rolipram by intrathecal infusion combined with embryonic neural tissue grafts, showing substantial increase in axonal plasticity and improved recovery after hemisection at C3/4 level in adult rats when compared to the control group with tissue transplant only (Nikulina et al., 2004). Further work from the same group showed that in vivo rolipram treatment increased the number of spared central myelinated axons in lateral cord after spinal contusion in adult female rats (Pearse et al., 2004). Moreover, in the same study a cAMP combinatorial approach was applied in which rolipram and/or db-cAMP were administered together with Schwann cell transplants, resulting in enhancement of axonal regrowth and improved functional recovery after spinal contusion in adult female rats (Pearse et al., 2004). Furthermore, Costa et al. (2013) administered rolipram alone for 2 weeks following contusion injury in adult female rats and observed an improvement in locomotor abilities as compared to the control group that was infused with DMSO. Although no significant differences in either lesion length nor volume were reported, a significant increase in spared white matter at the lesion epicentre was noted, suggesting that increasing cAMP levels prevents excessive degeneration of myelinated fibres (Costa et al., 2013).
More recently, combinatorial approaches to promote spinal cord repair have become more common. In a hemi-transection model in adult female rats, a sustained delivery of db-cAMP and chondroitinase ABC (an enzyme degrading CSPGs) incorporated in poly-microfibres placed over the transection gap was shown to promote axonal sprouting within the glial scar (Xia et al., 2017). Importantly, the delivery of db-cAMP only to the lesion site by the microfibers increased the number of GAP-43 (neuronal regeneration marker) positive cells when compared to the control group with no treatment. Moreover, a significant increase in axonal sprouting within the glial scar demonstrated by neurofilament-200 staining, a general neuronal marker, was reported when either db-cAMP or db-cAMP in combination with chondroitinase ABC was administered by microfibers (Xia et al., 2017).
Although the attempts to manipulate cAMP levels in order to achieve a substantial recovery after TSCI showed robust results, none of the treatment options such as cAMP analogues or PDE4 inhibitors have been translated to the clinic. Factors to be considered for clinical translation using cAMP elevation strategies include cAMP’s ubiquitous expression by all types of cells and potential side effects e.g. persistent nausea caused by systemic rolipram treatment (Hebenstreit et al., 1989; Scott et al., 1991). Therefore, scientists are actively investigating more specific agents targeting downstream cAMP pathways that could limit systemic side effects of cAMP signaling manipulation. The potential key molecular mechanisms and pathways of cAMP elevation on neuronal/axonal regrowth are illustrated in Figure 2.
Figure 2.
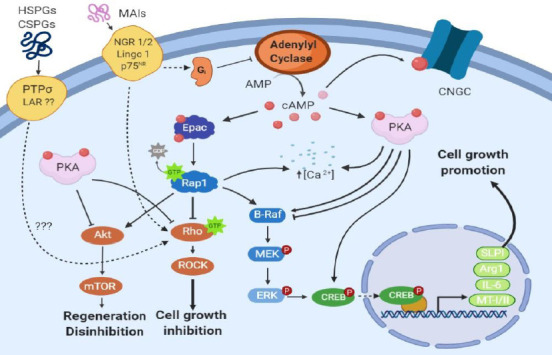
Potential mechanisms of cAMP elevation on neuronal regeneration: promoting growth and overcoming inhibition.
The effect of elevated cAMP activity on axonal regeneration was previously hypothesised to occur in two phases; a transcription-independent phase, which covers the first 7 days of regeneration post-spinal cord injury, and a transcription-dependent phase that takes over after 7 days post-injury (Batty et al., 2017). The transcription-independent phase is thought to be regulated by PKA, acting through inhibition of Rho GTPase (Bandtlow, 2003). Rho/ROCK signaling was found to mediate the inhibitory effects on axonal growth of extracellular matrix proteins such as CSPGs and heparan sulphate proteoglycans (HSPGs), and inhibition of the Rho/ROCK pathway was reported to improve axonal regeneration (McKerracher and Higuchi, 2006). Of note, Rho/ROCK signaling is also inhibited by Epac (Boomkamp et al., 2014). The transcription-dependent phase is conceived to be mediated by cAMP-response element-binding protein (CREB), activated beforehand by PKA, which enhances transcription of arginase I (ArgI), interleukin-6 (IL-6), metallothionein (MT-I/II) and secretory leukocyte protease inhibitor (SLPI) (Siddiq and Hannila, 2015). However, Epac/Rap1 activates the B-raf/ERK pathway (Vossler et al., 1997) that also results in phosphorylation of CREB (Grewal et al., 2000) and transcription of ArgI as well as other polyamines that support axonal growth. Moreover, as Epac/Rap1 also activates Akt, it can potentially trigger the Akt/mTOR pathway, which was found to induce disinhibition of axonal growth, initiation of regeneration and maintenance of a regenerative state in retinal ganglion cells after inflammatory stimulation (Leibinger et al., 2012). AMP: Adenosine monophosphate; cAMP: cyclic adenosine 3′,5′-monophosphate; CNGC: cyclic nucleotide gated channel; Epac: exchange protein directly activated by cAMP; ERK: extracellular signal-related kinase; LAR: leukocyte common antigen-related phosphatase; LINGO 1: immunoglobin-like domain-containing protein 1; MAIs: myelin associated inhibitors; MEK: MAP kinase/ERK kinase; mTOR: mammalian target of rapamycin; NgR: Nogo receptor; PKA: protein kinase A; PTPσ: transmembrane protein tyrosine phosphatase; Rap1: Ras-proximate-1; ROCK: Rho-associated kinase.
Epac: a Downstream Effector of Cyclic Adenosine 3′,5′-Monophosphate
The downstream effectors of cAMP include cyclin nucleotide receptor involved in sperm function, Popeye domain-containing proteins, cyclic nucleotide-gated ion channels, protein kinase A (PKA), and Epac (Brand and Schindler, 2017). In recent years, the latter two have been shown to play major roles in regulating the responses of the nervous system to physical trauma. Epac has been shown to play a role in a wide range of cellular functions such as cell growth, adhesion, differentiation, division, inflammation and neurotransmission (Peace and Shewan, 2011). It is known that there are two mammalian isoforms of Epac, of which Epac1 is expressed ubiquitously throughout the body, whereas Epac2 is mainly expressed in postnatal CNS tissue such as brain and spinal cord, but is also found in adrenal gland, heart, pancreas, small intestine and testis (Peace and Shewan, 2011; Ramos and Antonetti, 2017). Furthermore, recent data show that Epac2 has at least 3 different sub-isoforms: Epac2A, 2B, and 2C, which are differentially expressed in brain/heart/adrenal glands, adrenal gland/pancreas, and liver respectively (Hoivik et al., 2013). Moreover, the protein levels of Epac1 and Epac2 have been found to be developmentally regulated in the rat nervous system. For example, high-level Epac1 protein expression is observed in the embryonic and neonatal brain, spinal cord and DRG, which significantly decreases at adult stage (Murray and Shewan, 2008). In contrast, Epac2 expression is low at embryonic and neonatal stages, however it becomes dramatically upregulated at adult stage (Murray and Shewan, 2008).
Structurally, the two isoforms are different in that Epac2 has an extra nucleotide binding domain and a functional Ras association domain, which are absent in Epac1 (Bos et al., 2000). Epac1 is found to be located at the perinuclear envelope, whereas Epac2 is found diffusely throughout the cytosol but can translocate to the plasma membrane upon activation (Li et al., 2006; Liu et al., 2010). As a result of differences in tissue distribution and structures, the physiological functions of Epac1 and Epac2 might vary accordingly. Thus, Epac2 targeting may potentially result in greater tissue specificity due to postnatal CNS expression leading to greater efficacy. This gives hope for neural tissue-restricted manipulation of Epac2 and resultant localized effects.
Only very recently, specific tools have been developed to specifically discern between Epac1 and Epac2 manipulation (Schwede et al., 2015). For instance, both in vitro and in vivo evidence shows that Sp-8-BnT-cAMPS (S-220) is the most potent and selective activator of Epac2 over Epac1, whereas 8-pCPT-2′-O-Me-cAMP (8-Me-cAMP) is a general Epac agonist, and neither activate PKA (Table 1). However, a specific antagonist is currently only available for Epac2 (ESI-05; Table 1) (Tsalkova et al., 2012; Zhu et al., 2015).
Epac and axonal guidance during neural development
As Epac serves as a major effector of cAMP, its role in positive chemotactic behaviour in growing axons has been investigated. For example, using siRNA against both Epac1 and Epac2, the attraction of embryonic rat DRG neurons to a gradient of a cAMP analogue (Sp-cAMPs; Table 1) or a general Epac agonist (8-Me-cAMP; Table 1) in growth cone turning assays was switched to repulsion (Murray et al., 2009b). In addition, a recent study showed that neonatal rat DRG neurons also exhibited a strong turning behaviour (12.7 ± 4.1° at 30 minutes) towards a gradient of the Epac2 agonist, S-220 (Guijarro-Belmar et al., 2019), which is similar to previous results using similar setups for Rolipram (19.2 ± 4.9° at 30 minutes) and 8-Me-cAMP (16.7 ± 4.3° at 30 minutes) (Murray and Shewan, 2008). Therefore, the evidence indicates that unilateral Epac activation is responsible for mediating positive turning behaviors in growing axons.
The finding that Epac mediates positive turning behaviour induced by cAMP prompts the question of whether Epac also mediates attractive chemotactic behaviour to extracellular guidance cues that is brought about by increased levels of cAMP. This indeed seems to be true as the growth cones of embryonic rat DRG neurons are attracted to netrin-1 and MAG, and knocking down Epac1 and Epac2 using siRNA resulted in growth cone repulsion (Murray et al., 2009b). In contrast, adult rat DRG growth cones that were repelled by gradients of netrin-1 or MAG were not affected by knocking down Epac1 and Epac2 expression using siRNA. Instead, elevation of intracellular Epac activity by using 8-Me-cAMP switched their repulsive response to MAG gradients to attraction, which further supports the notion that Epac is responsible for mediating positive chemotactic behaviour. Furthermore, FRET imaging confirmed that when netrin-1 is added to culture media, embryonic rat DRG growth cones increased selective activation of Epac and not PKA (Murray et al., 2009b). The above evidence, together with existing evidence showing that Epac1 expression is prominent during embryonic/neonatal stages whereas Epac2 expression increases postnatally, suggest that Epac1 mediates attractive axonal guidance during neural development, which is opposite to the repulsive role exerted by PKA (Murray et al., 2009b).
In vitro evidence of Epac in promoting axonal growth
Epac activation has been found to promote neurite outgrowth in vitro, which can also be achieved by cAMP elevation. The general Epac agonist 8-Me-cAMP was able to induce significant neurite outgrowth of cultured adult rat DRG neurons, comparable to that seen when these neurons where treated with cAMP agonists in vitro (Murray and Shewan, 2008; Wei et al., 2016). Moreover, 8-Me-cAMP treatment led to an increase in neurite density and myelination similar to that of Rolipram in an in vitro model of SCI using embryonic rat myelinating spinal cord cultures (Boomkamp et al., 2014). Recently, in vitro work has revealed that Epac2 activation by the agonist S-220 significantly enhances neurite outgrowth of neonatal rat cortical and DRG neurons (Guijarro-Belmar et al., 2019) (Figure 3A–C). Moreover, the administration of a cAMP analogue, Sp-cAMPs, to adult rat DRG neurons that were transfected with Epac1/2 siRNA did not rescue neurite outgrowth (Murray and Shewan, 2008). The above evidence strongly suggests that Epac2 is a key player when it comes to mediating cAMP-induced neurite growth at postnatal and adult stages. The specific involvement of Epac2 in neurite outgrowth was further verified by recent findings in which neonatal rat DRG and cortical neurons transfected with Epac2 siRNA, or cultured with the specific Epac2 antagonist ESI-05, showed significantly reduced neurite outgrowth (Guijarro-Belmar et al., 2019) (Figure 3D–I). Furthermore, treatment of embryonic hippocampal neurons with a general Epac antagonist, ESI-09, also reduced neurite outgrowth (Munoz-Llancao et al., 2015).
Figure 3.
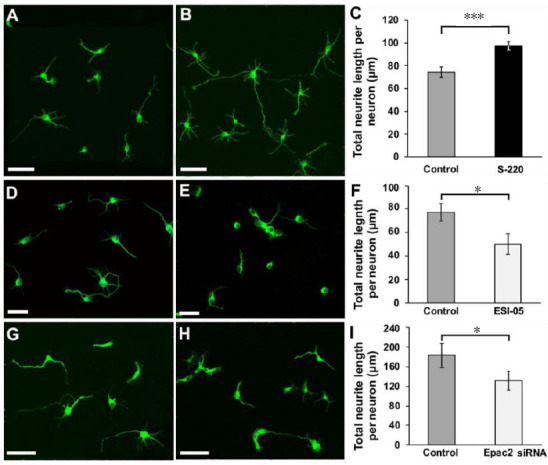
The effects of Epac2 modulation on cortical neurite outgrowth.
(A–C) Epac2 agonist S-220 promoted significant neurite outgrowth. (A) Control. (B) Treated with S-220. (C) Quantification of total neurite length shows that S-220-treated neurons had significantly longer neurites. (D–F) Epac2 antagonist ESI-05 significantly decreased cortical neurite outgrowth. (D) Control. (E) Treated with ESI-05. (F) Quantification of total neurite length shows that ESI-05-treated neurons had significantly shorter neurites. (G–I) siRNA knockdown of Epac2 significantly decreased cortical neurite outgrowth. G, Scrambled siRNA control. (H) Epac2 siRNA-treated. (I) Quantification of total neurite length shows that Epac2 siRNA-treated neurons had significantly shorter neurites. A, B, D, E, Cultures were grown for 24 hours. G, H, Cultures were grown for 48 hours. All cultures were stained for β-tubulin-III. *P < 0.05, ***P < 0.001. n = 3/group. Scale bars: A–E, 50 µm; G and H, 100 µm. Adapted from Guijarro-Belmar et al. (2019).
The effect of Epac activation on neurite outgrowth can also be revealed when neurons are cultured in an inhibitory environment. For example, when neonatal and adult rat DRG neurons were cultured on inhibitory adult rat spinal cord slices and received treatment with 8-Me-cAMP, a significant increase in the percentage of neurons with long neurites was observed, which did not occur when adult rat neurons were treated with Rolipram or Sp-cAMPs (Murray and Shewan, 2008). Moreover, after knocking down Epac1/2 expression with siRNA in embryonic DRG neurons that normally grow long neurites on adult spinal cord sections, the number of cells growing long processes was significantly reduced (Murray and Shewan, 2008). In a recent study, it has been shown that CSPGs significantly inhibit neurite outgrowth by 35% of cultured neonatal rat cortical neurons. However, treatment with S-220 induces the neurons to overcome CSPG inhibition and thereby a significantly increased neurite outgrowth was reported (Guijarro-Belmar et al., 2019; Figure 4A–D). These results are similar to what was previously found with cerebellar neurons, in which the use of MAG substrates reduced neurite outgrowth by 40% and was rescued by the addition of the cAMP agonist db-cAMP (Cai et al., 1999). Guijarro-Belmar et al. (2019) used neonatal DRG neurons co-cultured with inhibitory mature astrocytes and showed that, on contact with those astrocytes, less than 20% of DRG neurites grew over the astrocyte surface, with most neurites exhibiting contact-mediated avoidance. These results are consistent with those shown by Adcock et al. (2004) where only 15.1 ± 2.3% of postnatal rat DRG neurons cross a Schwann cell-astrocyte boundary in vitro. In their study, the addition of the phosphodiesterase inhibitor, rolipram, increased the proportion of neurites expressing crossover behaviour to 58 ± 4.1%. This four-fold increase is in agreement with the findings for S-220 treatment, (S-220 versus control: 45.7 ± 10.3% versus 11.2 ± 4.2%; Guijarro-Belmar et al., 2019; Figure 4E–G). These results strongly suggest that targeting Epac2 is just as effective in mediating neuronal behavioral changes as targeting cAMP.
Figure 4.
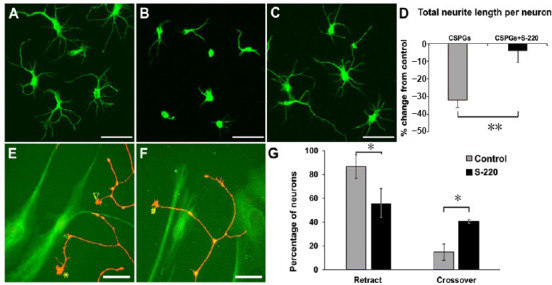
The Epac2 agonist overcomes inhibitory environments for cortical and DRG neuron growth.
(A) β-Tubulin-III positive cortical neurons grew neurites on PDL coated coverslips; (B) cortical neurons treated with CSPGs showed significantly shorter neurite lengths compared to control. (C) Epac2 agonist S-220 attenuated the inhibitory effect of CSPGs on cortical neurite outgrowth. (D) Quantification represents the percentage of change from control and shows a significant reduction in inhibition by CSPGs when S-220 was simultaneously applied. Cultures were grown for 48 hours. (E–G) S-220 also showed the effect in overcoming the astrocyte inhibition. Three neurite growth cone behaviours of DRG neurons co-cultured with mature astrocytes were observed: retract (▽ in E), reflect (* in E) and crossover (# in F) using time-lapse live cell microscopy. G, the quantification showed a significant reduction in the retract/reflect behaviors of neurites and a significant increase in the crossover behavior of neurites in cells treated with the Epac2 agonist when compared to control. (D, G) Mann-Whitney Rank Sum test, *P < 0.05, **P < 0.01, n = 3/group. Scale bars: A–C, 50 µm; E and F, 25 µm. Adapted from Guijarro-Belmar et al. (2019). CSPGs: Chondroitin sulphate proteoglycans; DRG: dorsal root ganglion; PDL: poly(D,L-lactide).
Furthermore, Boomkamp et al. (2014) showed that the positive effects of rolipram in enhancing neurite outgrowth in an in vitro embryonic rat spinal cord culture model of SCI were mediated through the activation of Epac, as demonstrated by 8-Me-cAMP treatment, and not PKA. In fact, the inhibition of PKA in this study (by using KT- 5720; Table 1) led to an enhanced outgrowth comparable to Epac activation and Rolipram treatment in this model (Boomkamp et al., 2014).
Moreover, in cultured adult rat DRG neurons, inhibition of PKA using Rp-cAMPs has been shown to increase neurite outgrowth, which can be reduced by using the specific Epac2 antagonist ESI-05 (Wei et al., 2016). Therefore, these findings strongly suggest that selective activation of Epac2, avoiding PKA signaling, mediates adult neurite outgrowth in vitro.
Ex vivo/in vivo evidence of Epac in promoting axonal growth
While there is plenty of in vivo evidence that cAMP promotes axonal regrowth after injury, such evidence with Epac has only emerged recently. The effects of the Epac2 agonist, S-220, were explored in an ex vivo model of SCI using cultured organotypic neonatal rat spinal cord slices. Addition of S-220 to the culture media or its local release from a self-assembling Fmoc-based hydrogel at the lesion site resulted in a significant enhancement of axonal outgrowth across the lesion gap as measured by the amount of β-tubulin-III+ processes (Guijarro-Belmar et al., 2019) (Figure 5). Therefore, ex vivo evidence provides further confirmation to the in vitro findings where Epac2 activation promotes neurite outgrowth. The potential of Epac2 activation was further explored in a clinically relevant adult rat contusion SCI model, in which the Fmoc hydrogel incorporated with S-220 was directly injected into the lesion cavity 3 weeks after the injury, representing a subacute stage of SCI (Guijarro-Belmar et al., 2019). Over a 4-week period post-gel injection, animals receiving treatment with S-220 delivered by the gel showed significant improvement in locomotor behaviour, reaching on average 3 BBB scales higher than contusion-only animals. Although axonal regrowth was not assessed in that study, it is likely that S-220, locally delivered and released by the gel, promoted axonal regrowth mediated by Epac2 activation. The potential key molecular mechanisms and pathways of Epac2 signaling that aid neuronal/axonal regrowth are illustrated in Figure 2.
Figure 5.
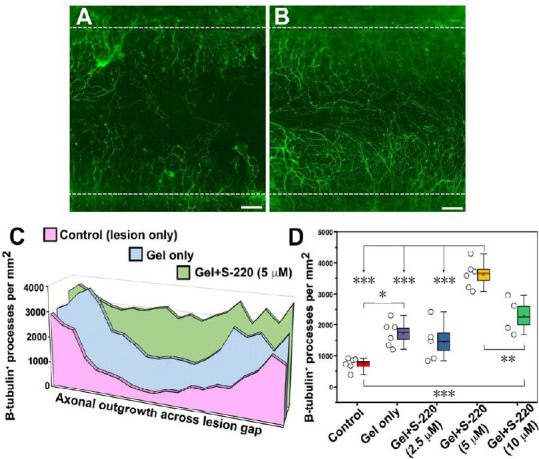
S-220 incorporated into the hydrogel promotes neurite outgrowth.
(A, B) Representative images showing significant difference in neurite outgrowth between gel-only (A) and gel + 5 µM S-220 (B). White dashed lines indicate lesion margins. (C) Neurite growth profiles across the lesion gap, showing a progressive increase with gel-only and gel + 5 µM S-220. (D) Quantification showing the numbers of β-tubulin-III+ processes per square millimetre between control and different concentrations of S-220 delivered in the gel, with 5 µM having the greatest effect. (D) Data are expressed as mean ± SEM (box limits). Bars above and below each box represent 5% and 95% confidence limits. Circles represent individual biological replicates (n = 4–6). **P < 0.01, ***P < 0.001. Scale bars: 100 µm in A and B. Adapted from Guijarro-Belmar et al. (2019).
Epac and Neuronal Death
In vitro evidence of Epac-mediated neuronal death
Although convincing evidence shows that Epac activation promotes axonal growth in vitro, ex vivo and in vivo, recent evidence suggests that an increased level of Epac2 activity in cortical neurons after neural trauma might lead to neuronal death (Zhang et al., 2018; Zhuang et al., 2019). Application of OxyHb, a chemical used to induce intracranial cerebral haemorrhage in animal models, has been shown to elevate Epac2 protein levels in cultured embryonic rat cortical neurons, which coincides with increased neuronal apoptosis (Zhuang et al., 2019). In vitro treatment with the Epac2 inhibitor, ESI-05, in the presence of OxyHb resulted in significantly reduced apoptotic death in those cells. However, this study was carried out on embryonic neurons, which were previously shown to express significantly lower levels of Epac2 protein than their adult counterparts (Murray and Shewan, 2008); therefore, the underlying mechanism of this increased Epac2 in OxyHb-exposed embryonic neurons is unclear.
In vivo evidence of Epac-mediated neuronal death
In vivo evidence has also suggested that increased Epac2 protein levels in the brain following neural trauma could lead to neuronal apoptosis. Following intracranial cerebral haemorrhage or traumatic brain injury in adult rats, Epac2 levels have been shown to increase significantly in cortical neurons, leading to neuronal apoptosis (Zhang et al., 2018; Zhuang et al., 2019). However, treatment with ESI-05 was shown to attenuate Epac2-mediated neuronal apoptosis in these studies and result in improved neurological functions. The underlying mechanism could be via a p38-mediated cell death pathway, as treatment with ESI-05 also led to reduced levels of p-p38 (Zhang et al., 2018; Zhuang et al., 2019). Thus, activation of Epac2 might lead to enhanced activity of Rap1, which could potentially activate p38/MAPK, thereby contributing to apoptosis. However, it is important to note that other evidence suggests that Epac2 could be important for cell survival. For example, the protective effects of urocortin-1, whose receptor is positively linked to adenylyl cyclase and increases cAMP production, in an ex vivo rat model of heart ischaemia using adult rat cardiac myocytes were lost when ESI-05 was applied to culture media, suggesting a protective role of Epac2 in this model (Calderon-Sanchez et al., 2016). Moreover, our preliminary evidence showed that immediate treatment with S-220 delivered by the Fmoc hydrogel in a hemi-transection model of SCI in adult rats improved locomotor function recovery acutely, suggesting a potential neuroprotective role of Epac2 activation (unpublisehd data). Therefore, these observations indicate that differential regulation of cell death and survival via Epac2 may depend on different tissues/cells and their age, as well as different neural injury pathogenesis.
Cyclic Adenosine 3′,5′-Monophosphate/Epac and Glial Cells
Glial cells, such as astrocytes and microglia, play important roles in the inflammatory and reparatory responses to neural trauma in the CNS. Since cAMP signaling is common in all types of cells, it is likely that manipulation of cAMP signaling might also affect these cells’ functions, including their activation after neural trauma.
CAMP and astrocytes
Astrocytes play vital roles during CNS development and injuries (Markiewicz and Lukomska, 2006; Silver et al., 2014). In particular, it is now known that following SCI, there are heterogeneous astrocyte populations in and around the lesion site (Sofroniew, 2014). Therefore, previous literature using cAMP elevation strategies in in vivo SCI models as discussed in Section 3 might have also impacted on astrocytes. However, direct evidence of this is lacking.
An elevated cAMP level is crucial for the differentiation of astrocytes from neural precursor cells (NPCs). In cultured embryonic rat cortical NPCs, treatment with pituitary adenylate cyclase-activating peptide increases cAMP levels, which leads to a profound increase in the percentage of cells (~80%) expressing GFAP and S-100β and having a typical astrocyte-like stellate morphology, with no upregulation of neuronal or oligodendrocyte markers (Vallejo and Vallejo, 2002; (Figure 6B). Moreover, cAMP has been shown to promote differentiation of astrocytes from C6 glioma cells in the presence of interleukin-6 (Takanaga et al., 2004).
Figure 6.
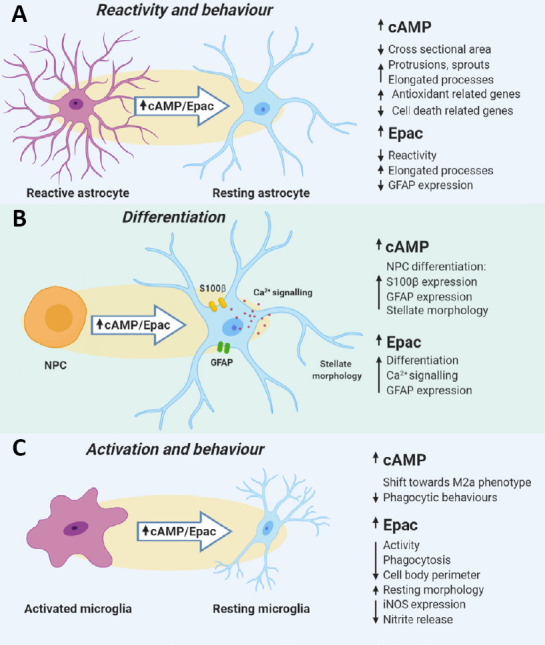
Effects of elevated cAMP and increased Epac activity on glial cells.
(A) In resting astrocytes, elevation of cAMP results in morphological changes such as a decrease in cross sectional area, increased elongation of processes and an increase in the number of sprouts and protrusions from the cell. What is more, cAMP elevation can shift a genetic profile of astrocytes towards a pro-survival state by inducing expression of antioxidant-related genes and downregulation of cell death-related genes. In reactive astrocytes, activation of Epac2 led to a decrease in GFAP expression as well as induced morphology that resembled resting astrocytes. (B) The elevation of cAMP or Epac was found to induce differentiation of NPCs to astrocytes. The upregulation of S100β and GFAP expression, as well as a shift to a stellate morphology, was observed in NPCs treated with a cAMP elevating agent or Epac2 agonist. (C) In microglia, an increase in cAMP levels was found to be essential to shift M1 to M2 microglia in the presence of IL-4, which suggests that cAMP is crucial to controlling microglial behaviour. What is more, either elevation of cAMP or activation of Epac decreased the phagocytic behaviours of the BV-2 cells. In the ex vivo model of SCI, treatment of the tissue with Epac2 agonist resulted in a decrease in microglial iNOS expression and nitrite release, as well as an induction of resting-like morphology, which indicates that Epac controls not only the reactivity of astrocytes, but also the activation of microglia. cAMP: Cyclic adenosine 3′,5′-monophosphate; GFAP: glial fibrillary acidic protein; iNOS: inducible nitric oxide synthase; IL: interleukin; NPCs: neural precursor cells; SCI: spinal cord injury.
Increased intracellular cAMP levels by treating cultured neonatal rat astrocytes with adrenaline, forskolin or db-cAMP for 30–60 minutes are associated with reduced cell cross-sectional areas and increased cell perimeters (Vardjan et al., 2014). Moreover, Increased intracellular cAMP levels in these cultured cells are also associated with increased sprouts, protrusions, and elongated processes on the membrane surface (Vardjan et al., 2014, 2016). Activation of β-adrenoreceptors with adrenaline in cultured neonatal rat astrocytes can also reduce cell swelling induced by hypotonic conditions, an effect that is also observable with an increase in cAMP level (Vardjan et al., 2016; Figure 6A).
Furthermore, elevation of cAMP levels in astrocytes is known to induce a pro-survival state. After treatment with 8-Br-cAMP, a cAMP analogue (Table 1), cultured mature astrocytes from neonatal mouse cortex showed an upregulation of antioxidant-related genes and a downregulation of cell death-related genes (Paco et al., 2016; Figure 6A).
CAMP and microglia
Microglia are CNS innate immune cells. They participate in neurogenesis, programmed cell death and synapse elimination, as well as the establishment and remodeling of neuronal circuits during development (Li and Barres, 2018). At adult stages, microglia are known to be responsible for phagocytosis, secretion of growth factors and propagation of immune responses. Microglia, upon activation, can resume similar phenotypes as macrophages, i.e., M1, which is a pro-inflammatory and neurotoxic state, and M2, which is a pro-regenerative phenotype (Colonna and Butovsky, 2017).
Increasing intracellular cAMP levels in cultured microglial cell line BV-2 cells by forskolin, IBMX, or β-adrenergic agonist isoproterenol decreases phagocytic behaviors of the cells (Steininger et al., 2011; Figure 6C). Treatment of BV-2 cells with lipopolysaccharide (LPS) resulted in induction of the M1 phenotype characterized by upregulation of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 proteins and downregulation of Arg1, transglutaminase-2 and RELM-α (Ghosh et al., 2016). When they were exposed to LPS and treated with IL-4 and db-cAMP simultaneously, cells showed a shift towards a M2 phenotype, which is characterized by iNOS/cyclooxygenase-2 downregulation and Arg1 upregulation (Figure 6C). However, neither IL-4 nor db-cAMP on its own was sufficient to induce the M2 phenotype in LPS-exposed BV2 cells. Moreover, systemic co-treatment with IL-4 and db-cAMP at 15 minutes following a contusion SCI in adult rats resulted in significantly increased Arg1 in ED1-labelled microglia and macrophages (Ghosh et al., 2016). These findings suggest that cAMP might play an important role in regulating microglia phenotype and its effects on BV-2 cell and microglial activation might require the presence of IL-4.
Epac and astrocytes
Both Epac1 and Epac2 have been implicated in astrocyte biology. In mature astrocytes cultured from neonatal rat cortex, treatment with the Epac general agonist 8-Me-cAMP causes increased intracellular calcium levels (Di Cesare et al., 2006). In mice with global knockout of Epac2, cerebral GFAP expression was decreased at birth (Seo and Lee, 2016), suggesting that Epac2 might be important for astrocytic differentiation. Moreover, when NPCs cultured from Epac2-KO mice were treated with pituitary adenylate cyclase-activating peptide to increase cAMP, these cells failed to increase GFAP expression. Therefore, it is likely that cAMP elevation might act through Epac2 to modulate astrocytic differentiation.
Recent evidence shows that activation of Epac2 reverses LPS-induced activation of astrocytes in vitro (Guijarro-Belmar et al., 2019; Figure 7A–D). Upon treatment with LPS, mature astrocytes cultured from neonatal rat cortex became activated with hypertrophic cell bodies, shortened/thickened processes and increased GFAP expression (Figure 7B). However, when the cells were simultaneously exposed to LPS and the Epac2 agonist S-220, they showed non-reactive morphology and reduced GFAP expression, similar to the control samples (Figure 7C). Furthermore, when S-220 was delivered by a Fmoc hydrogel at the lesion site in the ex vivo model of SCI, astrocytes adopted a morphology with elongated processes and reduced expression of GFAP and Nestin (an activation marker for astrocytes) (Guijarro-Belmar et al., 2019; Figure 8A–K). Notably, regrowing axons accompanied the astrocyte processes, suggesting that S220-treated astrocytes might provide guidance to regrowing axons (Figure 8G and H).
Figure 7.
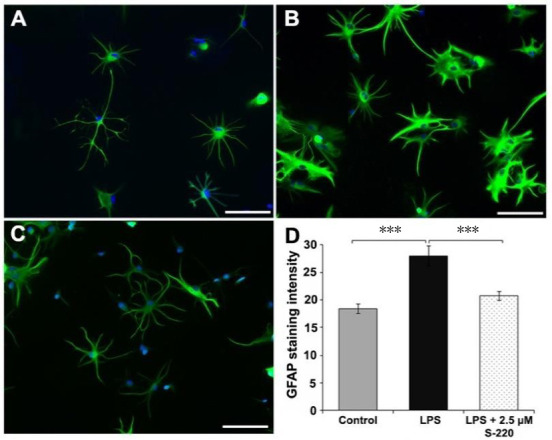
Epac2 agonist S-220 attenuates LPS induced astrocyte activation in vitro.
Representative fluorescent images of control (A), LPS-treated (4 hours) (B) and LPS + S-220-treated (4 hours) (C) astrocytes. Scale bars: A–C 100 µm. (D) Quantification of the mean fluorescence intensity of GFAP showed significant difference between control and LPS-treated astrocytes, and between LPS and LPS + S-220-treated astrocytes. n = 3/group. ***P < 0.001. GFAP: Glial fibrillang acidic protein; LPS: lipopolysaccharide. Adapted from Guijarro-Belmar et al. (2019).
Figure 8.
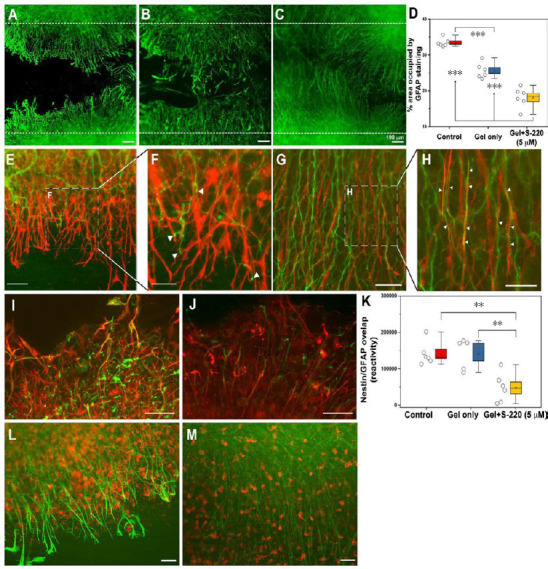
S-220 incorporated into the hydrogel promotes neurite outgrowth and suppresses astrocyte activation.
(A–D) Representative images exhibit astrocyte activation in the lesion using GFAP staining as an astrocyte marker. (A) Control. (B) Gel only. (C) Gel + 5 µM S-220. (D) Quantification of the GFAP immunoreactivity intensity showed a significant reduction of mean grey value (OD) in gel + 5 µM S-220 compared to the control and the gel only treatment. E, Representative image of the relationship between GFAP, (red) and β-tubulin-III (green) immunoreactive processes in a control injury condition. (F) Higher magnification image showing the collapse of growth cones (white arrowheads) when they meet activated astrocytes. (G) Representative image of the relationship between GFAP and β-tubulin-III immunoreactive processes in a lesion with combined treatment with gel + 5 µM S-220. (H) Higher magnification image showing the alignment of the astrocytes and β-tubulin-III immunoreactive processes (white arrowheads). (I–K) Levels of astrocyte reactivity were estimated by the overlapping of GFAP (red) and nestin (green). (I) Representative image of GFAP/nestin reactivity in lesion sites of non-treated slices. (J) Representative image of GFAP/nestin overlapping in slices treated with S-220 delivered by the hydrogel. (K) Quantification of GFAP/nestin pixel overlapping. (L) Representative image of the relationship between GFAP and Iba-1 immunoreactive cells in an injured control. (M) Representative image of the relationship between GFAP and Iba-1 immunoreactive cells in injured slices treated with a combination of hydrogel + 5 µM S-220. Circles represent individual biological replicates (n= 4–6). Scale bars: A–C, I and J, 100 µm; E and G, 50 µm; F and H, 25 µm; L and M, 50 µm. GFAP: Glial fibrillang acidic protein. Adapted from Guijarro-Belmar et al. (2019).
Epac and microglia
Epac1 and Epac2 have also been shown to influence microglial activation. When treated with the Epac general agonist-8-Me-cAMP, BV-2 cells significantly decreased their phagocytotic behaviour (Steininger et al., 2011). In vitro activation of Epac2 in LPS-exposed microglia cultured from neonatal rat cortex significantly attenuated their activation by inducing resting morphology and reducing iNOS expression and nitrite release (Guijarro-Belmar et al., 2019; Figures 6C and 9). Treatment with S-220 in the ex vivo model also led to a reduced activation of microglia at the lesion site, with decreased cell body perimeter and a morphology resembling microglia in non-injured areas (Guijarro-Belmar et al., 2019; (Figures 6C and 8L, H). Taken together, these findings indicate that activation of Epac2 is able to impact on the activation of both astrocytes and microglia at the lesion site in the ex vivo model, thereby providing a more permissive environment to allow axonal regrowth (Figure 8–M).
Figure 9.
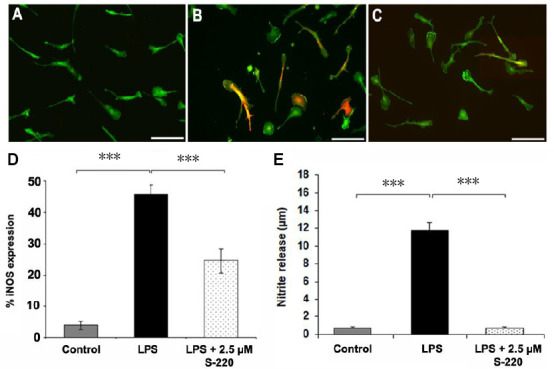
Epac2 agonist S-220 attenuates LPS induced microglial activation in vitro.
Representative fluorescent images of control (A), LPS-treated (4 hours) (B) and LPS + S-220-treated (4 hours) (C) primary microglia cultured from postnatal rat cortex and immunostained for Iba-1 (green) and iNOS (red). Scale bars: 100 µm. (D) Quantitative image analysis showing significant differences in iNOS-immunoreactive cell numbers among control, LPS-treated, and LPS + S-220-treated microglia. (E) The Griess assay demonstrated a significant increase of nitrate concentration in the supernatant collected from LPS-treated microglial cultures compared with that of the control. n = 3/group. iNOS: Inducible nitric oxide synthase; LPS: lipopolysaccharide. Adapted from Guijarro-Belmar et al. (2019).
Conclusions
Although cAMP plays an important role in regulating the post-lesion environment after TSCI, as well as neuronal chemotactic behaviour, growth and survival, the direct modulation of cAMP signalling is unlikely to find a use in the clinic due to the ubiquity of the cAMP pathways in humans. Instead, a downstream effector of cAMP, Epac2, represents a promising target to manipulate the outcome of TSCI. Epac2 is mainly expressed postnatally in the CNS and mediates positive effects of cAMP on neuronal growth and guidance. Epac2 can also modulate microglial/astrocyte activation and astrocyte morphology after SCI to be more supportive to axonal regrowth. Therefore, Epac2 activation is likely to positively affect cells that play crucial roles following TSCI and make the post-lesion environment more supportive for the regrowing axons. As Epac2 expression is largely limited to adult CNS and new sub-isoforms are being discovered, it brings hope for CNS tissue-specific effects and reduction of possible side effects, which occur with treatment options that manipulate cAMP levels. Moreover, further combinations with other strategies such as locomotor training (which also enhances cAMP signalling) could maximize and further promote functional recovery. However, further investigation of the molecular events downstream of Epac/Rap1 is essential to reveal the mechanisms that lead to the modulation of the inhibitory environment and axonal regeneration.
Additional file: Open peer review report 1 (81.8KB, pdf) .
Footnotes
P-Reviewer: Doncel-Pérez E; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: This work was supported by Scottish Rugby Union funding to WH and DS, the NRB PhD scholarship from the International Spinal Rsesarch Trust to AGB, and a Hot-Start Scholarship from the University of Aberdeen to DD.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Ernesto Doncel-Pérez, Grupo de Plasticidad Neural, Spain.
Funding: This work was supported by Scottish Rugby Union funding to WH and DS, the NRB PhD scholarship from the International Spinal Rsesarch Trust to agb , and a Hot-Start Scholarship from the University of Aberdeen to DD.
References
- 1.Adcock KH, Brown DJ, Shearer MC, Shewan D, Schachner M, Smith GM, Geller HM, Fawcett JW. Axon behaviour at Schwann cell - astrocyte boundaries: manipulation of axon signalling pathways and the neural adhesion molecule L1 can enable axons to cross. Eur J Neurosci. 2004;20:1425–1435. doi: 10.1111/j.1460-9568.2004.03573.x. [DOI] [PubMed] [Google Scholar]
- 2.Aglah C, Gordon T, Posse de Chaves EI. cAMP promotes neurite outgrowth and extension through protein kinase A but independently of Erk activation in cultured rat motoneurons. Neuropharmacology. 2008;55:8–17. doi: 10.1016/j.neuropharm.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Agrawal SK, Fehlings MG. Mechanisms of secondary injury to spinal cord axons in vitro: role of Na+, Na(+)-K(+)-ATPase, the Na(+)-H+ exchanger, and the Na(+)-Ca2+ exchanger. J Neurosci. 1996;16:545–552. doi: 10.1523/JNEUROSCI.16-02-00545.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almahariq M, Tsalkova T, Mei FC, Chen H, Zhou J, Sastry SK, Schwede F, Cheng X. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol Pharmacol. 2013;83:122–128. doi: 10.1124/mol.112.080689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Badhiwala JH, Ahuja CS, Fehlings MG. Time is spine: a review of translational advances in spinal cord injury. J Neurosurg Spine. 2018;30:1–18. doi: 10.3171/2018.9.SPINE18682. [DOI] [PubMed] [Google Scholar]
- 6.Bandtlow CE. Regeneration in the central nervous system. Exp Gerontol. 2003;38:79–86. doi: 10.1016/s0531-5565(02)00165-1. [DOI] [PubMed] [Google Scholar]
- 7.Batty NJ, Fenrich KK, Fouad K. The role of cAMP and its downstream targets in neurite growth in the adult nervous system. Neurosci Lett. 2017;652:56–63. doi: 10.1016/j.neulet.2016.12.033. [DOI] [PubMed] [Google Scholar]
- 8.Beattie MS, Hermann GE, Rogers RC, Bresnahan JC. Cell death in models of spinal cord injury. Prog Brain Res. 2002;137:37–47. doi: 10.1016/s0079-6123(02)37006-7. [DOI] [PubMed] [Google Scholar]
- 9.Berry M. Post-injury myelin-breakdown products inhibit axonal growth: an hypothesis to explain the failure of axonal regeneration in the mammalian central nervous system. Bibl Anat. 1982;23:1–11. [PubMed] [Google Scholar]
- 10.Boomkamp SD, McGrath MA, Houslay MD, Barnett SC. Epac and the high affinity rolipram binding conformer of PDE4 modulate neurite outgrowth and myelination using an in vitro spinal cord injury model. Br J Pharmacol. 2014;171:2385–2398. doi: 10.1111/bph.12588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bos JdR, Holger R, Miranda van T, Robert HC, Alfred W, Johannes L. Mechanism of Regulation of the Epac Family of cAMP-dependent RapGEFs. J Biol Chem. 2000;275:20829–20836. doi: 10.1074/jbc.M001113200. [DOI] [PubMed] [Google Scholar]
- 12.Bradbury EJ, Burnside ER. Moving beyond the glial scar for spinal cord repair. Nat Commun. 2019;10:3879. doi: 10.1038/s41467-019-11707-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brand T, Schindler R. New kids on the block: The Popeye domain containing (POPDC) protein family acting as a novel class of cAMP effector proteins in striated muscle. Cell Signal. 2017;40:156–165. doi: 10.1016/j.cellsig.2017.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Busch SA, Silver J. The role of extracellular matrix in CNS regeneration. Curr Opin Neurobiol. 2007;17:120–127. doi: 10.1016/j.conb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT. Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci. 2001;21:4731–4739. doi: 10.1523/JNEUROSCI.21-13-04731.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai D, Shen Y, De Bellard M, Tang S, Filbin MT. Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron. 1999;22:89–101. doi: 10.1016/s0896-6273(00)80681-9. [DOI] [PubMed] [Google Scholar]
- 17.Calderon-Sanchez E, Diaz I, Ordonez A, Smani T. Urocortin-1 mediated cardioprotection involves XIAP and CD40-ligand recovery: role of EPAC2 and ERK1/2. PLoS One. 2016;11:e0147375. doi: 10.1371/journal.pone.0147375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caroni P, Schwab ME. Two membrane protein fractions from rat central myelin with inhibitory properties for neurite growth and fibroblast spreading. J Cell Biol. 1988;106:1281–1288. doi: 10.1083/jcb.106.4.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choo AM, Liu J, Lam CK, Dvorak M, Tetzlaff W, Oxland TR. Contusion, dislocation, and distraction: primary hemorrhage and membrane permeability in distinct mechanisms of spinal cord injury. J Neurosurg Spine. 2007;6:255–266. doi: 10.3171/spi.2007.6.3.255. [DOI] [PubMed] [Google Scholar]
- 20.Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441–468. doi: 10.1146/annurev-immunol-051116-052358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costa LM, Pereira JE, Filipe VM, Magalhaes LG, Couto PA, Gonzalo-Orden JM, Raimondo S, Geuna S, Mauricio AC, Nikulina E, Filbin MT, Varejao AS. Rolipram promotes functional recovery after contusive thoracic spinal cord injury in rats. Behav Brain Res. 2013;243:66–73. doi: 10.1016/j.bbr.2012.12.056. [DOI] [PubMed] [Google Scholar]
- 22.Curcio M, Bradke F. Axon regeneration in the central nervous system: facing the challenges from the inside. Annu Rev Cell Dev Biol. 2018;34:495–521. doi: 10.1146/annurev-cellbio-100617-062508. [DOI] [PubMed] [Google Scholar]
- 23.Di Cesare A, Del Piccolo P, Zacchetti D, Grohovaz F. EP2 receptor stimulation promotes calcium responses in astrocytes via activation of the adenylyl cyclase pathway. Cell Mol Life Sci. 2006;63:2546–2553. doi: 10.1007/s00018-006-6262-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H. Free radical-induced damage to DNA: mechanisms and measurement. Free Radic Biol Med. 2002;32:1102–1115. doi: 10.1016/s0891-5849(02)00826-2. [DOI] [PubMed] [Google Scholar]
- 25.Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Doskeland SO, Blank JL, Bos JL. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- 26.Farooque M, Hillered L, Holtz A, Olsson Y. Changes of extracellular levels of amino acids after graded compression trauma to the spinal cord: an experimental study in the rat using microdialysis. J Neurotrauma. 1996;13:537–548. doi: 10.1089/neu.1996.13.537. [DOI] [PubMed] [Google Scholar]
- 27.Fawcett JW, Schwab ME, Montani L, Brazda N, Muller HW. Defeating inhibition of regeneration by scar and myelin components. Handb Clin Neurol. 2012;109:503–522. doi: 10.1016/B978-0-444-52137-8.00031-0. [DOI] [PubMed] [Google Scholar]
- 28.Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- 29.Fournier AE, Takizawa BT, Strittmatter SM. Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci. 2003;23:1416–1423. doi: 10.1523/JNEUROSCI.23-04-01416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.GBD. Traumatic Brain Injury and Spinal Cord Injury Collaborators (2019) Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2016;18:56–87. doi: 10.1016/S1474-4422(18)30415-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghosh M, Xu Y, Pearse DD. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J Neuroinflammation. 2016;13:9. doi: 10.1186/s12974-015-0463-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisen J. A pericyte origin of spinal cord scar tissue. Science. 2011;333:238–242. doi: 10.1126/science.1203165. [DOI] [PubMed] [Google Scholar]
- 33.Grewal SS, Fass DM, Yao H, Ellig CL, Goodman RH, Stork PJ. Calcium and cAMP signals differentially regulate cAMP-responsive element-binding protein function via a Rap1-extracellular signal-regulated kinase pathway. J Biol Chem. 2000;275:34433–34441. doi: 10.1074/jbc.M004728200. [DOI] [PubMed] [Google Scholar]
- 34.Guijarro-Belmar A, Viskontas M, Wei Y, Bo X, Shewan D, Huang W. Epac2 elevation reverses inhibition by chondroitin sulfate proteoglycans in vitro and transforms post-lesion inhibitory environment to promote axonal outgrowth in an ex vivo model of spinal cord injury. J Neurosci. 2019;39:8330–8346. doi: 10.1523/JNEUROSCI.0374-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanson MG, Jr, Shen S, Wiemelt AP, McMorris FA, Barres BA. Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro. J Neurosci. 1998;18:7361–7371. doi: 10.1523/JNEUROSCI.18-18-07361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hausmann ON. Post-traumatic inflammation following spinal cord injury. Spinal Cord. 2003;41:369–378. doi: 10.1038/sj.sc.3101483. [DOI] [PubMed] [Google Scholar]
- 37.Hebenstreit GF, Fellerer K, Fichte K, Fischer G, Geyer N, Meya U, Sastre-y-Hernandez M, Schony W, Schratzer M, Soukop W. Rolipram in major depressive disorder: results of a double-blind comparative study with imipramine. Pharmacopsychiatry. 1989;22:156–160. doi: 10.1055/s-2007-1014599. [DOI] [PubMed] [Google Scholar]
- 38.Hoivik EA, Witsoe SL, Bergheim IR, Xu Y, Jakobsson I, Tengholm A, Doskeland SO, Bakke M. DNA methylation of alternative promoters directs tissue specific expression of Epac2 isoforms. PLoS One. 2013;8:e67925. doi: 10.1371/journal.pone.0067925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang WL, George KJ, Ibba V, Liu MC, Averill S, Quartu M, Hamlyn PJ, Priestley JV. The characteristics of neuronal injury in a static compression model of spinal cord injury in adult rats. Eur J Neurosci. 2007;25:362–372. doi: 10.1111/j.1460-9568.2006.05284.x. [DOI] [PubMed] [Google Scholar]
- 40.Klapka N, Muller HW. Collagen matrix in spinal cord injury. J Neurotrauma. 2006;23:422–435. doi: 10.1089/neu.2006.23.422. [DOI] [PubMed] [Google Scholar]
- 41.Kwon BK, Tetzlaff W, Grauer JN, Beiner J, Vaccaro AR. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J. 2004;4:451–464. doi: 10.1016/j.spinee.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 42.Leibinger M, Andreadaki A, Fischer D. Role of mTOR in neuroprotection and axon regeneration after inflammatory stimulation. Neurobiol Dis. 2012;46:314–324. doi: 10.1016/j.nbd.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18:225–242. doi: 10.1038/nri.2017.125. [DOI] [PubMed] [Google Scholar]
- 44.Li Y, Asuri S, Rebhun JF, Castro AF, Paranavitana NC, Quilliam LA. The RAP1 guanine nucleotide exchange factor Epac2 couples cyclic AMP and Ras signals at the plasma membrane. J Biol Chem. 2006;281:2506–2514. doi: 10.1074/jbc.M508165200. [DOI] [PubMed] [Google Scholar]
- 45.Li Y, Raisman G. Long axon growth from embryonic neurons transplanted into myelinated tracts of the adult rat spinal cord. Brain Res. 1993;629:115–127. doi: 10.1016/0006-8993(93)90489-a. [DOI] [PubMed] [Google Scholar]
- 46.Liu C, Takahashi M, Li Y, Dillon TJ, Kaech S, Stork PJ. The interaction of Epac1 and Ran promotes Rap1 activation at the nuclear envelope. Mol Cell Biol. 2010;30:3956–3969. doi: 10.1128/MCB.00242-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Markiewicz I, Lukomska B. The role of astrocytes in the physiology and pathology of the central nervous system. Acta Neurobiol Exp (Wars) 2006;66:343–358. doi: 10.55782/ane-2006-1623. [DOI] [PubMed] [Google Scholar]
- 48.Mautes AE, Weinzierl MR, Donovan F, Noble LJ. Vascular events after spinal cord injury: contribution to secondary pathogenesis. Phys Ther. 2000;80:673–687. [PubMed] [Google Scholar]
- 49.McAdoo DJ, Xu GY, Robak G, Hughes MG. Changes in amino acid concentrations over time and space around an impact injury and their diffusion through the rat spinal cord. Exp Neurol. 1999;159:538–544. doi: 10.1006/exnr.1999.7166. [DOI] [PubMed] [Google Scholar]
- 50.McKeon RJ, Jurynec MJ, Buck CR. The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J Neurosci. 1999;19:10778–10788. doi: 10.1523/JNEUROSCI.19-24-10778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McKeon RJ, Schreiber RC, Rudge JS, Silver J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. 1991;11:3398–3411. doi: 10.1523/JNEUROSCI.11-11-03398.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McKerracher L, Higuchi H. Targeting Rho to stimulate repair after spinal cord injury. J Neurotrauma. 2006;23:309–317. doi: 10.1089/neu.2006.23.309. [DOI] [PubMed] [Google Scholar]
- 53.McQuarrie IG, Grafstein B. Axon outgrowth enhanced by a previous nerve injury. Arch Neurol. 1973;29:53–55. doi: 10.1001/archneur.1973.00490250071008. [DOI] [PubMed] [Google Scholar]
- 54.Munoz-Llancao P, Henriquez DR, Wilson C, Bodaleo F, Boddeke EW, Lezoualc’h F, Schmidt M, Gonzalez-Billault C. Exchange protein directly activated by cAMP (EPAC) regulates neuronal polarization through Rap1B. J Neurosci. 2015;35:11315–11329. doi: 10.1523/JNEUROSCI.3645-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murray AJ. Pharmacological PKA inhibition: all may not be what it seems. Sci Signal. 2008;1:re4. doi: 10.1126/scisignal.122re4. [DOI] [PubMed] [Google Scholar]
- 56.Murray AJ, Peace AG, Shewan DA. cGMP promotes neurite outgrowth and growth cone turning and improves axon regeneration on spinal cord tissue in combination with cAMP. Brain Res. 2009a;1294:12–21. doi: 10.1016/j.brainres.2009.07.071. [DOI] [PubMed] [Google Scholar]
- 57.Murray AJ, Shewan DA. Epac mediates cyclic AMP-dependent axon growth, guidance and regeneration. Mol Cell Neurosci. 2008;38:578–588. doi: 10.1016/j.mcn.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 58.Murray AJ, Tucker SJ, Shewan DA. cAMP-dependent axon guidance is distinctly regulated by Epac and protein kinase A. J Neurosci. 2009b;29:15434–15444. doi: 10.1523/JNEUROSCI.3071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI. Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron. 2002;34:885–893. doi: 10.1016/s0896-6273(02)00702-x. [DOI] [PubMed] [Google Scholar]
- 60.Nikulina E, Tidwell JL, Dai HN, Bregman BS, Filbin MT. The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci U S A. 2004;101:8786–8790. doi: 10.1073/pnas.0402595101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nishiyama M, Hoshino A, Tsai L, Henley JR, Goshima Y, Tessier-Lavigne M, Poo MM, Hong K. Cyclic AMP/GMP-dependent modulation of Ca2+ channels sets the polarity of nerve growth-cone turning. Nature. 2003;423:990–995. doi: 10.1038/nature01751. [DOI] [PubMed] [Google Scholar]
- 62.O’Shea TM, Burda JE, Sofroniew MV. Cell biology of spinal cord injury and repair. J Clin Invest. 2017;127:3259–3270. doi: 10.1172/JCI90608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paco S, Hummel M, Pla V, Sumoy L, Aguado F. Cyclic AMP signaling restricts activation and promotes maturation and antioxidant defenses in astrocytes. BMC Genomics. 2016;17:304. doi: 10.1186/s12864-016-2623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peace AG, Shewan DA. New perspectives in cyclic AMP-mediated axon growth and guidance: The emerging epoch of Epac. Brain Res Bull. 2011;84:280–288. doi: 10.1016/j.brainresbull.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 65.Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, Bunge MB. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10:610–616. doi: 10.1038/nm1056. [DOI] [PubMed] [Google Scholar]
- 66.Pineau I, Lacroix S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comp Neurol. 2007;500:267–285. doi: 10.1002/cne.21149. [DOI] [PubMed] [Google Scholar]
- 67.Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 2002;34:895–903. doi: 10.1016/s0896-6273(02)00730-4. [DOI] [PubMed] [Google Scholar]
- 68.Ramos CJ, Antonetti DA. The role of small GTPases and EPAC-Rap signaling in the regulation of the blood-brain and blood-retinal barriers. Tissue Barriers. 2017;5:e1339768. doi: 10.1080/21688370.2017.1339768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Richardson PM, Issa VM. Peripheral injury enhances central regeneration of primary sensory neurones. Nature. 1984;309:791–793. doi: 10.1038/309791a0. [DOI] [PubMed] [Google Scholar]
- 70.Schwede F, Bertinetti D, Langerijs CN, Hadders MA, Wienk H, Ellenbroek JH, de Koning EJP, Bos JL, Herberg FW, Genieser H-G, Janssen RAJ, Rehmann H. Structure-guided design of selective Epac1 and Epac2 agonists. PLoS Biol. 2015;13:e1002038. doi: 10.1371/journal.pbio.1002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scott AI, Perini AF, Shering PA, Whalley LJ. In-patient major depression: is rolipram as effective as amitriptyline. Eur J Clin Pharmacol. 1991;40:127–129. doi: 10.1007/BF00280065. [DOI] [PubMed] [Google Scholar]
- 72.Seo H, Lee K. Epac2 contributes to PACAP-induced astrocytic differentiation through calcium ion influx in neural precursor cells. BMB Rep. 2016;49:128–133. doi: 10.5483/BMBRep.2016.49.2.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shewan D, Berry M, Cohen J. Extensive regeneration in vitro by early embryonic neurons on immature and adult CNS tissue. J Neurosci. 1995;15:2057–2062. doi: 10.1523/JNEUROSCI.15-03-02057.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shewan D, Dwivedy A, Anderson R, Holt CE. Age-related changes underlie switch in netrin-1 responsiveness as growth cones advance along visual pathway. Nat Neurosci. 2002;5:955–962. doi: 10.1038/nn919. [DOI] [PubMed] [Google Scholar]
- 75.Siddiq MM, Hannila SS. Looking downstream: the role of cyclic AMP-regulated genes in axonal regeneration. Front Mol Neurosci. 2015;8:26. doi: 10.3389/fnmol.2015.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Siebert JR, Osterhout DJ. The inhibitory effects of chondroitin sulfate proteoglycans on oligodendrocytes. J Neurochem. 2011;119:176–188. doi: 10.1111/j.1471-4159.2011.07370.x. [DOI] [PubMed] [Google Scholar]
- 77.Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 78.Silver J, Schwab ME, Popovich PG. Central nervous system regenerative failure: role of oligodendrocytes, astrocytes, and microglia. Cold Spring Harb Perspect Biol. 2014;7:a020602. doi: 10.1101/cshperspect.a020602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sofroniew MV. Astrogliosis. Cold Spring Harb Perspect Biol. 2014;7:a020420. doi: 10.1101/cshperspect.a020420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Song H, Ming G, He Z, Lehmann M, McKerracher L, Tessier-Lavigne M, Poo M. Conversion of neuronal growth cone responses from repulsion to attraction by cyclic nucleotides. Science. 1998;281:1515–1518. doi: 10.1126/science.281.5382.1515. [DOI] [PubMed] [Google Scholar]
- 81.Steininger TS, Stutz H, Kerschbaum HH. Beta-adrenergic stimulation suppresses phagocytosis via Epac activation in murine microglial cells. Brain Res. 2011;1407:1–12. doi: 10.1016/j.brainres.2011.06.050. [DOI] [PubMed] [Google Scholar]
- 82.Takanaga H, Yoshitake T, Hara S, Yamasaki C, Kunimoto M. cAMP-induced astrocytic differentiation of C6 glioma cells is mediated by autocrine interleukin-6. J Biol Chem. 2004;279:15441–15447. doi: 10.1074/jbc.M311844200. [DOI] [PubMed] [Google Scholar]
- 83.Tator CH, Koyanagi I. Vascular mechanisms in the pathophysiology of human spinal cord injury. J Neurosurg. 1997;86:483–492. doi: 10.3171/jns.1997.86.3.0483. [DOI] [PubMed] [Google Scholar]
- 84.Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, Holz GG, Woods VL, Jr, Cheng X. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc Natl Acad Sci U S A. 2012;109:18613–18618. doi: 10.1073/pnas.1210209109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vallejo I, Vallejo M. Pituitary adenylate cyclase-activating polypeptide induces astrocyte differentiation of precursor cells from developing cerebral cortex. Mol Cell Neurosci. 2002;21:671–683. doi: 10.1006/mcne.2002.1189. [DOI] [PubMed] [Google Scholar]
- 86.Vardjan N, Horvat A, Anderson JE, Yu D, Croom D, Zeng X, Luznik Z, Kreft M, Teng YD, Kirov SA, Zorec R. Adrenergic activation attenuates astrocyte swelling induced by hypotonicity and neurotrauma. Glia. 2016;64:1034–1049. doi: 10.1002/glia.22981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vardjan N, Kreft M, Zorec R. Dynamics of beta-adrenergic/cAMP signaling and morphological changes in cultured astrocytes. Glia. 2014;62:566–579. doi: 10.1002/glia.22626. [DOI] [PubMed] [Google Scholar]
- 88.Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- 89.Wei D, Hurd C, Galleguillos D, Singh J, Fenrich KK, Webber CA, Sipione S, Fouad K. Inhibiting cortical protein kinase A in spinal cord injured rats enhances efficacy of rehabilitative training. Exp Neurol. 2016;283:365–374. doi: 10.1016/j.expneurol.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 90.Xia T, Huang B, Ni S, Gao L, Wang J, Wang J, Chen A, Zhu S, Wang B, Li G, Zhu S, Li X. The combination of db-cAMP and ChABC with poly(propylene carbonate) microfibers promote axonal regenerative sprouting and functional recovery after spinal cord hemisection injury. Biomed Pharmacother. 2017;86:354–362. doi: 10.1016/j.biopha.2016.12.045. [DOI] [PubMed] [Google Scholar]
- 91.Yuan YM, He C. The glial scar in spinal cord injury and repair. Neurosci Bull. 2013;29:421–435. doi: 10.1007/s12264-013-1358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang L, Zhang L, Liu H, Jiang F, Wang H, Li D, Gao R. Inhibition of Epac2 attenuates neural cell apoptosis and improves neurological deficits in a rat model of traumatic brain injury. Front Neurosci. 2018;12:263. doi: 10.3389/fnins.2018.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhu Y, Chen H, Boulton S, Mei F, Ye N, Melacini G, Zhou J, Cheng X. Biochemical and pharmacological characterizations of ESI-09 based EPAC inhibitors: defining the ESI-09 “therapeutic window”. Sci Rep. 2015;5:9344. doi: 10.1038/srep09344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhuang Y, Xu H, Richard SA, Cao J, Li H, Shen H, Yu Z, Zhang J, Wang Z, Li X, Chen G. Inhibition of EPAC2 attenuates intracerebral hemorrhage-induced secondary brain injury via the p38/BIM/Caspase-3 pathway. J Mol Neurosci. 2019;67:353–363. doi: 10.1007/s12031-018-1215-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.