

Abstract
MicroRNAs (miRNAs) are small non-coding RNA molecules that regulate post-transcriptional gene expression and contribute to all aspects of cellular function. We previously reported that the activities of several mitochondria-enriched miRNAs regulating inflammation (i.e., miR-142-3p, miR-142-5p, and miR-146a) are altered in the hippocampus at 3–12 hours following a severe traumatic brain injury. In the present study, we investigated the temporal expression profile of these inflammatory miRNAs in mitochondria and cytosol fractions at more chronic post-injury times following severe controlled cortical impact injury in rats. In addition, several inflammatory genes were analyzed in the cytosol fractions. The analysis showed that while elevated levels were observed in cytoplasm, the mitochondria-enriched miRNAs, miR-142-3p and miR-142-5p continued to be significantly reduced in mitochondria from injured hippocampi for at least 3 days and returned to near normal levels at 7 days post-injury. Although not statistically significant, miR-146a also remained at reduced levels for up to 3 days following controlled cortical impact injury, and recovered by 7 days. In contrast, miRNAs that are not enriched in mitochondria, including miR-124a, miR-150, miR-19b, miR-155, and miR-223 were either increased or demonstrated no change in their levels in mitochondrial fractions for 7 days. The one exception was that miR-223 levels were reduced in mitochondria at 1 day following injury. No major alterations were observed in sham operated animals. This temporal pattern was unique to mitochondria-enriched miRNAs and correlated with injury-induced changes in mitochondrial bioenergetics as well as expression levels of several inflammatory markers. These observations suggested a potential compartmental re-distribution of the mitochondria-enriched inflammatory miRNAs and may reflect an intracellular mechanism by which specific miRNAs regulate injury-induced inflammatory signaling. To test this, we utilized a novel peptide-based nanoparticle strategy for in vitro and in vivo delivery of a miR-146a mimic as a potential therapeutic strategy for targeting nuclear factor-kappaB inflammatory modulators in the injured brain. Nanoparticle delivery of miR-146a to BV-2 or SH-SY5Y cells significantly reduced expression of TNF receptor-associated factor 6 (TRAF6) and interleukin-1 receptor-associated kinase 1 (IRAK1), two important modulators of the nuclear factor-kappaB (NF-κB) pro-inflammatory pathway. Moreover, injections of miR-146a containing nanoparticles into the brain immediately following controlled cortical impact injury significantly reduced hippocampal TNF receptor-associated factor 6 and interleukin-1 receptor-associated kinase 1 levels. Taken together, our studies demonstrate the subcellular alteration of inflammatory miRNAs after traumatic brain injury and establish proof of principle that nanoparticle delivery of miR-146a has therapeutic potential for modulating pro-inflammatory effectors in the injured brain. All of the studies performed were approved by the University of Kentucky Institutional Animal Care and Usage Committee (IACUC protocol # 2014-1300) on August 17, 2017.
Keywords: cell permeable peptide-delivery, controlled cortical impact, inflammatory pathway, mitochondria-associated microRNA, nanoparticle, nuclear factor-kappaB, traumatic brain injury
Chinese Library Classification No. R453; R364; R741
Introduction
Traumatic brain injury (TBI) elicits a rapid and pronounced neuroinflammatory response propagated, in part, by resident microglia as well as circulating monocytes that differentiate into macrophages along a continuum ranging from cytotoxic pro-inflammatory to reparative anti-inflammatory phenotypes. These cells have been consistently linked with pro-inflammatory signaling processes underlying chronic degenerative changes after TBI (Ziebell and Morganti-Kossmann, 2010; Kumar and Loane, 2012; Woodcock and Morganti-Kossmann, 2013). The expression of reparative myeloid cells peaks at approximately one week following injury and then declines, while the pro-inflammatory phenotype may persist for months to years (Ziebell and Morganti-Kossmann, 2010; Kumar and Loane, 2012; Johnson et al., 2013; Woodcock and Morganti-Kossmann, 2013; Kumar et al., 2015). This differential expression pattern favoring prolonged pro-inflammation limits tissue repair and is associated with poor functional outcomes. Therefore, strategies that effectively limit detrimental pro-inflammatory responses, while maintaining the reparative potential of macrophages, have clear therapeutic potential (Ziebell and Morganti-Kossmann, 2010; Kumar and Loane, 2012; Woodcock and Morganti-Kossmann, 2013). In support of this, experimental approaches that mitigate pro-inflammatory signaling after severe TBI can significantly reduce tissue pathology and promote functional recovery (Lloyd et al., 2008; Hsieh et al., 2014; Bachstetter et al., 2015; Morganti et al., 2015). However, identifying tools that shift these myeloid effectors towards a reparative bias with a high degree of specificity and efficacy has been very limited. One novel strategy with high translational potential is the use of nanoparticle technologies to deliver microRNAs (miRNAs) targeting genes that regulate inflammatory signaling (O’Neill et al., 2011; Contreras and Rao, 2012; O’Connell et al., 2012).
MiRNAs are evolutionally conserved non-coding short RNA molecules that bind recognition elements in messenger RNA (mRNA) and fine tune post-transcriptional gene expression (Bartel, 2004; Huntzinger and Izaurralde, 2011). MiRNAs regulate all aspects of cellular function, and disruption of miRNA activity contributes to many disease states including neurodegeneration (Calin and Croce, 2006; Nelson et al., 2008; Liu and Xu, 2011; Hata, 2013). MiRNA-based therapeutic approaches have been tested and several are currently in clinical trials targeting hepatitis C, type 2 diabetes, and several cancer types (Rupaimoole and Slack, 2017). Therefore, the ability of miRNAs to regulate expression of genes contributing to TBI-related neuroinflammatory signaling makes them attractive therapeutic candidates. Using mouse and rat models, we and others have reported that a number of miRNAs are altered in cortex and hippocampus following TBI (Redell et al., 2009; Hu et al., 2012; Liu et al., 2014; Sabirzhanov et al., 2014; Wang et al., 2015, 2017; Harrison et al., 2017; Kumar et al., 2017). The studies from our lab are the first to demonstrate that a subset of miRNAs regulating cellular inflammatory activities (e.g., miR-146a) is enriched in brain mitochondria fractions (Wang et al., 2015, 2017). We also reported that levels of these miRNAs display a compartmental shift from the mitochondria to the cytosol as early as 3–12 hours following TBI (Wang et al., 2015, 2017), which corresponds to the temporal loss in mitochondrial function after acute TBI (Sullivan et al., 1998, 2007; Singh et al., 2006). These observations suggest an interaction between mitochondrial function and miRNA-mediated inflammatory signaling at early time points following TBI.
MiR-146a plays a significant role in regulating levels of inflammatory cytokines and related signaling events in a manner that favors expression of the reparative macrophage phenotype (Cui and Liu, 2015). However, the relationship between miRNA changes and inflammatory signaling in the hippocampus beyond 12 hours following TBI is not known. Moreover, strategies that modify miRNA signaling to effectively promote expression of the reparative macrophage phenotype are lacking. Therefore, the aim of this study was to quantify changes of select miRNAs and inflammatory genes isolated from the hippocampus at different time points following severe TBI. In addition, we tested the ability of miR-146a nanoparticle delivery to target the nuclear factor-kappaB (NF-κB) modulators TNF receptor-associated factor 6 (TRAF6) and interleukin-1 receptor-associated kinase 1 (IRAK1) in vitro as well as in vivo after severe TBI.
Materials and Methods
Animals and TBI procedures
All of the studies performed were approved by the University of Kentucky Institutional Animal Care and Usage Committee (IACUC protocol # 2014-1300) on August 17, 2017. Additionally, the Division of Laboratory Animal Resources at the University is accredited by the Association for the Assessment and Accreditation for Laboratory Animal Care, International (AAALAC, International) and all experiments were performed with its guidelines. All animal experiments are in compliance with ARRIVE guidelines and experiments are carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). Thirty young adult male (7–10 weeks old) Sprague-Dawley rats weighing ~250 g (Charles River Laboratories, Wilmington, MA, USA) were acclimated for 7 days prior to experimentation. Rats were allowed free access to food/water and housed in a temperature-controlled room on a 12-hour light/dark cycle. The surgical and brain injury procedures performed in this study followed our previously published protocols (Sullivan et al., 1999; Wang et al., 2015). The controlled cortical impact (CCI) injury is a well-established brain injury model utilized by numerous researchers and, as such, allows for comparison of outcomes across studies (Dixon et al., 1991). Briefly, experimental rats were anesthetized with a dose of 4% isoflurane (Covetrus, Portland, ME, USA) via a nose cone and were positioned in a stereotaxic frame (David Kopf, Tujunga, CA, USA). The surgical and CCI procedure was conducted under sterile conditions and continued supplied dose of 2.5% isoflurane throughout the procedure. The core body temperature of the experimental rats was kept at 37°C during the procedures and the post-surgical/CCI recovery period. An incision was made along the midline in a rostra-caudal orientation to expose the skull. A 6-mm craniotomy was then created lateral to center between bregma and lambda. The brain injury was performed by driving a 5 mm diameter rod tip at 3.5 m/s velocity to compress the cortex to a depth of 2.0 mm using a pneumatically controlled impacting device (Precision Systems and Instrumentation, Fairfax Station, VA, USA). This procedure resulted in a severe injury consistent with previous studies (Baldwin et al., 1997; Davis et al., 2008; Gilmer et al., 2009) and characterized by physical damage to the cortex and the presence of neuronal damage to the underlying hippocampus. Following injury, surgical absorbable hemostat was placed in the craniotomy and a plastic disc adhered to the skull to cover and stabilize the craniotomy site. Sham animals received identical exposure to the surgical procedures, but without brain injury. The surgeries were conducted at the same time of day, 2 days apart with three CCI and two sham operations performed at each time. Animals (total n = 6 per group for CCI, n = 4 per group for sham surgery) were euthanatized with CO2 at 1, 3, or 7 days following injury and the ipsilateral and contralateral hippocampi were rapidly removed for isolation of the mitochondria and cytosolic fractions.
Cytosolic and mitochondrial fractionation
For mitochondrial bioenergetics and the miRNA and mRNA expression studies, the mitochondria and cytosol fractions were obtained from the hippocampus following a previously described Ficoll density gradient procedure (Pandya et al., 2009). Briefly, harvested hippocampal tissue was homogenized in pre-cooled mitochondrial isolation buffer (215 mM mannitol, 75 mM sucrose, 0.1% bovine serum albumin (BSA), 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA); pH adjusted to 7.2 with KOH). The homogenate was then centrifuged at 1300 × g for 3 minutes to remove tissue debris and nuclei. The resulting supernatant was then subjected to a centrifugation at 13,000 × g for 10 minutes to obtain crude mitochondrial pellets and the supernatant was saved as cytosol (CT) fraction. The re-suspended crude mitochondria sample was then placed into a nitrogen cell disruption chamber for 10 minutes (1200 psi) to rupture and release synaptosomal mitochondria (Brown et al., 2004). The crude mitochondria fraction was then laid over a discontinuous Ficoll gradient (7.5% + 10%) and further separated by centrifugation using a SW 55Ti rotors at 100,000 × g for 30 minutes. The mitochondria pellet (MT) was rinsed in mitochondrial isolation buffer in the absence of EGTA and centrifuged at 10,000 × g at 4°C for 10 minutes and resuspended in the same buffer to achieve a concentration of ~10 mg/mL. The protein content of the various fractions was analyzed using BCA protein assay kit (ThermoFisher, Waltham, MA USA).
Mitochondria bioenergetics
Mitochondrial bioenergetics were assessed using the Seahorse Biosciences XFe24 Flux Analyzer (Agilent Technologies [Santa Clara, CA]; RRID:SCR_013575) using a procedure developed in our lab (Sauerbeck et al., 2011; Pandya et al., 2016; Hubbard et al., 2018). In these experiments, rotenone, oligomycin, and trifluoromethoxy carbonylcyanide phenylhydrazone, 2-[2-[4-(trifluoromethoxy)phenyl]hydrazinylidene]-propanedinitrile (FCCP) were purchased from Biomol (Plymouth Meeting, PA, USA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Briefly, 3.5 µg protein equivalent of isolated mitochondria were suspended in respiration buffer (RB) (125 mM KCl, 2 mM MgCl2, 0.1% BSA, 20 mM HEPES and 2.5 mM KH2 PO4, adjusted pH 7.2). The combination of substrates/inhibitors/uncoupler were prepared in RB and 75 µL of each injected as pyruvate + malate + adenosine diphosphate (ADP), oligomycin, FCCP, and rotenone + succinate sequentially through ports A–D at the final concentrations of 5 mM pyruvate, 2.5 mM malate, 1 mM ADP, 1 µg/mL oligomycin, 4 µM FCCP, 0.1 µM rotenone, and 10 mM succinate. ADP driven complex I mediated coupled respiration (State III), ATPase locked leaky respiration (State IV), complex I driven maximum uncoupled respiration (State VCI), and complex II-driven maximum respiration (State VCII) were determined with each subsequent injection. The results are presented as oxygen consumption rate (OCR) and expressed as pmol of oxygen consumed per minute.
TaqMan single-tube miRNA RT-qPCR
Isolation of total hippocampal tissue RNA from the cytosolic and mitochondrial fractions and TaqMan single-tube RT-qPCR (ThermoFisher) of select miRNA were performed as previously reported (Wang et al., 2008, 2015). Selected miRNAs for analysis included miR-146a (Assay ID: 000468), miR-142-3p (Assay ID: 000464), miR-142-5p (Assay ID: 002248), miR-155 (Assay ID: 002571), miR-150 (Assay ID: 000473), miR-223 (Assay ID: 002295), miR-124a (Assay ID: 001182), and miR-19b (Assay ID: 000396). Each miRNA analysis was performed in technical triplicates of either contralateral or ipsilateral hippocampi for a total of 6 TBI and 4 sham biological replicates from each time point (n = 6 TBI, n = 4 sham). The raw cycle threshold (CT) values were determined using QuantStudio™ Real-Time PCR Software with automatic baseline and threshold (ThermoFisher). Overall miRNA levels following TBI or sham surgery in the cytoplasmic and mitochondrial fractions at each time point were calculated using formula RQ = 2–(Ct(Ipsi)–Ct(Contra)) (Table 1) comparing the ipsilateral injured side or sham surgery side relative to the uninjured contralateral side or no surgery side (RQ: relative quantity). To evaluate the net change of miRNA enrichment in mitochondria, the following formula was used: RQ = 2–(Ct(Mito)–Ct(Cyto)) comparing the mitochondria fraction to cytoplasmic fraction of the same brain side. The calculation method followed the instructions detailed in the “Guide to Performing Relative Quantitation of Gene Expression Using Real-Time Quantitative PCR” (Applied Biosystems).
Table 1.
Changes in miRNA levels in cytoplasmic fractions of hippocampal tissue following TBI and in sham-operated control animals
| miRNA | 1 d | 3 d | 7 d | ||||
|---|---|---|---|---|---|---|---|
| Fold (range) | P-value | Fold (range) | P-value | Fold (range) | P-value | ||
| TBI | miR-124 | 0.93 (0.67–1.27) | 0.625 | 0.99 (0.87–1.13) | 0.919 | 1.03 (0.73–1.45) | 0.885 |
| miR-142-3p | 2.24 (1.61–3.11) | 0.001 | 3.49 (1.97–6.17) | 0.001 | 0.93 (0.70–1.24) | 0.886 | |
| miR-142-5p | 2.62 (2.05–3.35) | 0.0001 | 3.41 (2.47–4.70) | 0.0002 | 0.94 (0.72–1.22) | 0.878 | |
| miR-146a | 1.55 (0.98–2.47) | 0.148 | 1.93 (1.09–3.42) | 0.078 | 0.93 (0.54–1.58) | 0.852 | |
| miR-150 | 0.87 (0.59–1.29) | 0.541 | 0.88 (0.66–1.19) | 0.678 | 0.96 (0.87–1.06) | 0.548 | |
| miR-155 | 11.54 (6.3–21.3) | 0.00017 | 6.18 (3.92–9.72) | 0.020 | 1.15 (0.73–1.81) | 0.828 | |
| miR-223 | 15.0 (9.9–22.8) | 0.000002 | 3.92 (2.07–7.42) | 0.003 | 0.66 (0.32–1.35) | 0.538 | |
| miR-19b | 1.31 (1.06–1.63) | 0.072 | 1.33 (1.04–1.72) | 0.028 | 0.83 (0.70–0.97) | 0.318 | |
| Sham | miR-124 | 0.90 (0.70–1.17) | 0.711 | 0.86 (0.7–1.05) | 0.213 | 0.88 (0.27–1.77) | 0.482 |
| miR-142-3p | 0.89 (0.82–1.29) | 0.897 | 1.23 (0.86–1.76) | 0.329 | 1.50 (0.83–2.71) | 0.573 | |
| miR-142-5p | 0.87 (0.70–1.07) | 0.485 | 1.28 (0.68–2.42) | 0.520 | 1.36 (0.67–2.76) | 0.701 | |
| miR-146a | 1.05 (0.74–1.48) | 0.865 | 0.96 (0.47–1.98) | 0.941 | 1.76 (1.25–2.47) | 0.495 | |
| miR-150 | 0.87 (0.73–1.04) | 0.581 | 0.91 (0.60–1.37) | 0.719 | 1.07 (0.96–1.19) | 0.740 | |
| miR-155 | 1.89 (1.27–2.81) | 0.202 | 1.22 (0.44–3.40) | 0.735 | 3.04 (1.58–5.84) | 0.355 | |
| miR-223 | 1.67 (1.15–2.43) | 0.095 | 1.61 (0.71–1.82) | 0.494 | 2.55 (1.82–3.57) | 0.357 | |
| miR-19b | 1.00 (0.87–1.15) | 0.999 | 0.85 (0.61–1.17) | 0.379 | 1.39 (1.00–1.94) | 0.372 | |
Levels of miRNAs were calculated using the formula: fold change = 2–(Ct(Ipsi)–Ct(Contra)) comparing the ipsilateral injured/sham-operated side of the brain relative to that of the uninjured/no operated contralateral side of the same animal. Data represent the mean value of the six biological replicates for traumatic brain injury (TBI) and four biological replicates for sham, and the P-values were generated using two-tailed Student's t-test (GraphPad 8.0.2). The fold change range is shown in parentheses. Significantly different expressed miRNAs are highlighted in red (P < 0.05). 1, 3, or 7 days indicates the tissue harvesting time following controlled cortical impact or sham surgery.
Gene expression analysis using TaqMan low density array panel
A customized TaqMan low density array (TLDA) qRT-PCR panel in an 8 × 48 gene array format (ThermoFisher) was employed to analyze gene expression in cytosolic fractions from the injured ipsilateral hippocampi relative to the uninjured contralateral side. Forty-seven genes including a group of inflammatory-related marker genes (Gensel et al., 2017) and an internal control ‘house-keeping’ gene (18S) were selected for the custom array. The list of genes and their assay IDs are provided in Additional Table 1 (2.9MB, pdf) . Total cytosolic RNA (500 ng) was subjected to first-strand cDNA synthesis using SuperScript™ III First-Strand Synthesis System (ThermoFisher). The resulting cDNA from each sample was then combined with TaqMan Universal PCR Master Mix (No AmpErase UNG, ThermoFisher) and loaded into each port of the TLDA cards following manufacturer’s instructions. Quantitative real-time PCR was performed with a QuantStudio™ 7 Real-Time PCR System (ThermoFisher) using the manufacturer’s standard program (hold 2 minutes at 50°C, followed by 10 minutes at 95°C, then 40 cycles of 15 seconds at 95°C and 1 minute at 60°C).
TLDA data processing
Raw qPCR CT (cycle threshold) values from the TLDA card were first determined using QuantStudio™ Real-Time PCR Software (ThermoFisher) with automatic baseline and threshold. PCR data that failed software-defined QC and those CT values equal to or more than 35 were considered as undetectable and were given a value of 35. The CT value of 18S ribosomal RNA was used to obtain ΔCT values of a given sample and the whole dataset was then further normalized using the Global Mean Normalization method (Mestdagh et al., 2009) to minimize the technical variability among array sets. The comparative ΔCT method was used to calculate relative gene expression levels. Mean ΔΔCT values and standard deviations were calculated following the “Guide to Performing Relative Quantitation of Gene Expression Using Real-Time Quantitative PCR” (Applied Biosystems (2008)). The effect of TBI on each gene in the TLDA card was expressed as fold change in cytosolic fractions from the ipsilateral hippocampus relative to uninjured contralateral side.
Cell permeable peptide-delivery of miRNA mimic in cell cultures
Cell culture experiments were conducted to document the ability of a mellitin-derived cell permeable peptide (p5RHH) to introduce Cy5-labeled miR-146a or scrambled control (Integrated DNA Technologies, Skokie, IL, USA) to BV-2 (kind gift from Dr. Linda van Eldik of University of Kentucky) and SH-SY5H (Cat# CRL-2266, ATCC, Manassas, VA, USA, RRID:CVCL_0019) cells. The p5RHH peptide (VLTTGLPALISWIRRRHRRHC) was synthesized by NeoScientific (Neobiolabs, Cambridge, MA, USA) and a stock of 10 mM was prepared using protease and nuclease free water, aliquoted and stored at –80°C. A total of 3 × 105 cells were seeded into 12-well plates and maintained for 24 hours in standard cell culture conditions prior to transfection. Nanoparticle complexes were prepared at a ratio of 200:1 (molar ratio of p5RHH:miR-146a mimic) with 20 nM miR-146a using procedures described previously (Hou, 2014). The p5RHH+miR-146a nanoparticles were then incubated with BV-2 and SH-SY5Y cells for 24 hours. Additional plated cells were incubated with Cy5-labeled miR-146a only or transfected using Lipofectamine RNAiMAX (ThermoFisher) to assess p5RHH + miR-146a transfection efficiency. Cells were visualized and photographed using a Nikon C2+ confocal microscope (Nikon Inc, Melville, NY, USA) and then washed with PBS and harvested for RNA analysis or Western blot analysis with either TRAF6 or Actin antibodies.
Peptide-delivery of miRNA mimic to rat brain following CCI
Animals were subjected to a CCI injury and randomly assigned to receive an intrahippocampal injection of p5RHH + miR-146a mimic or scrambled RNA control nanoparticles to test in vivo effect of the miR-146a mimic on TRAF6 and IRAK1. Immediately following injury, a Hamilton syringe with a 33-gauge Nanofil (WPI, Sarasota, FL, USA) blunt needle was slowly lowered in the dorsal hippocampus ipsilateral to the injury side using stereotaxic coordinates (Paxinos and Watson, 2013) –3.3 mm from bregma, 2.4 mm from midline, and 2.8 mm ventral. The needle was left in place for 1 minute before injecting a total of 0.5 µL of p5RHH:miR-146a nanoparticles (ratio of 200:1, 40 nM miR-146a) at a rate of 10 nL/s using a WPI Ultramicropump III connected to a WPI MICRO2T SMARTouch controller (WPI, Sarasota, FL, USA). The needle was left in place for an additional 2 minutes, slowly withdrawn, and the craniotomy site and overlying skin treated and closed with sutures. The animals were then euthanatized with CO2 at 48 hours after injection and the hippocampus removed rapidly and the dorsal hippocampal tissue used to analyze TRAF6 and IRAK1 gene expression using single-tube TaqMan assays (Wang et al., 2015) and western blotting for TRAF6.
Protein isolation and western blot analysis
Protein isolation and Western blot analysis were performed using standard techniques routinely performed in our lab (Wang et al., 2015). Briefly, RIPA buffer (Cat# R0278; Sigma) was used to lyse proteins of rat hippocampal tissue, as well as total cellular protein in transfected cells. Protein concentrations were determined using BCA protein assay (Cat# 23227, Pierce Biotechnology, Rockford, IL, USA). For Western blotting, equal amounts of protein (10–20 µg) were loaded on 4–16% Mini-PROTEAN® TGXTM gels (Cat# 456-1106, Bio-Rad, Hercules, CA, USA) and run at 0.03 A. The gels were then transblotted onto polyvinylidene fluoride membranes (Cat# 1620177, Bio-Rad) and non-specific binding sites blocked by incubating the membrane with 5% nonfat milk for 1–2 hours. The blots were then incubated overnight with primary antibody at 4°C. Primary antibodies used in this study included anti-TRAF6 (1:1000 dilution; rabbit, Cat# ab33915; Abcam, Cambridge, MA, USA) and anti-actin (1:2000, rabbit, Cat# A5060; Sigma). After 3–4 rinses following primary antibody incubation, blots were incubated with goat anti-rabbit peroxidase-conjugated secondary antibody (Jackson ImmunoResearch laboratories, Inc. West Grove, Pennsylvania, USA) or IRDye® 800 goat anti-rabbit IgG (1:20,000, Cat# 925-68070, LI-COR, Lincoln, Nebraska, USA) for 1 hour at room temperature. The immunoreactive signals were visualized using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Pierce Biotechnology) or using Odyssey CLx (LI-COR, Lincoln).
Statistical analysis
Statistical analysis was carried out using GraphPad Prism 8.0.2 (GraphPad Software Inc., San Diego, CA, USA) Animal numbers were powered according to published experimental data and anticipated effect sizes. One-way analysis of variance and Student’s t-test (two-tailed) were applied to evaluate mean differences (bioenergetics and miRNA, mRNA single-tube assay data) when appropriate. A Student’s t-test was used to evaluate the difference of the mean fold change of inflammatory responsive genes between the ipsilateral and contralateral cytoplasm of qPCR data and western blot densitometry data. Data are expressed as the mean ± SD and a P value less than 0.05 was considered as statistically significant.
Results
Mitochondria bioenergetics following TBI
The post-injury bioenergetic profile of total mitochondria isolated from the hippocampus revealed significant changes in state-dependent oxygen consumption rates (OCR) over time (Figure 1). The OCR significantly decreased in all four tested respiration states (P < 0.01, for State III, State VCI, and State VCII, respectively; P < 0.05 for State IV) in mitochondria isolated from the ipsilateral hippocampus at 1 day following CCI relative to intact contralateral side (Figure 1A). At 3 days post CCI, OCR in mitochondria from the ipsilateral hippocampus continued to be significantly decreased in State IV and State VCII (P < 0.05), with near significant decreased levels in State III (P = 0.058) compared to the contralateral side. By 1 week post-injury, there were no significant differences in any respiration state in mitochondria from the injured hippocampus relative to contralateral side. Sham-surgery did not result in any significant changes in bioenergetics across all three time points (Figure 1B).
Figure 1.
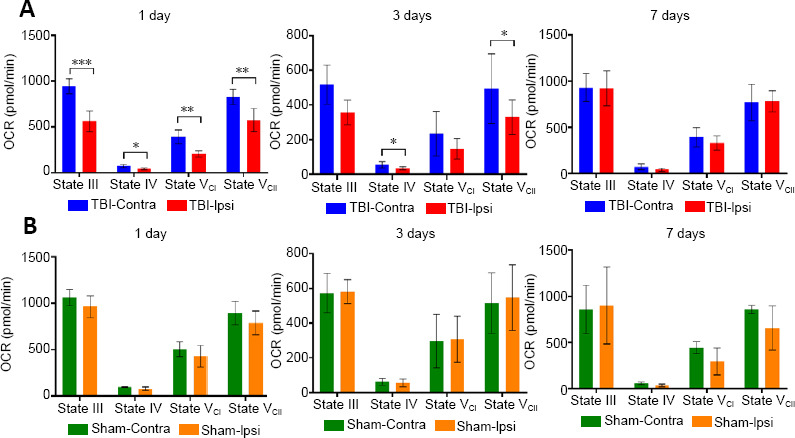
Temporal assessment of State III, IV, and VCI and VCII mitochondrial respiration rates in hippocampal mitochondria after severe TBI (A) or sham-surgery (B).
Animals (total n = 6 per group for CCI, n = 4 per group for sham surgery) were euthanatized at 1, 3, or 7 days following injury and the ipsilateral (Ipsi) and contralateral (Contra) hippocampi were rapidly removed for isolation of the mitochondria. Mitochondrial bioenergetics was assessed using the Seahorse Biosciences XFe24 Flux Analyzer. Data are expressed as the mean ± SD. The difference between the ipsilateral and the contralateral hippocampi was evaluated using one-way analysis of variance and Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001. OCR: Oxygen consumption rate; TBI: traumatic brain injury.
Alteration of the association of mitochondria-enriched miRNAs following TBI
We reported previously that levels of miR-146a, miR-142-3p, and miR-142-5p are highly enriched in the mitochondria compared to cytosol (Wang et al., 2015). We also found that the levels of these mitochondria-enriched miRNAs are decreased in the mitochondrial fraction at early times following TBI (3 and 12 hours), and that all three miRNAs increased in cytoplasmic fractions at the same time points (Wang et al., 2015, 2017). In the present study, we further investigated levels of these miRNAs at extended times up to 7 days following TBI (Figure 2). When miRNA levels in the mitochondrial fraction are normalized to that of cytoplasmic fraction, it provides a relative measure of mitochondrial enrichment. The enrichment of miR-142-3p and miR-142-5p significantly decreased (P = 0.050 and 0.002, respectively) at 1 day post TBI in mitochondria from the injured ipsilateral hippocampus compared to the uninjured contralateral hippocampus (Figure 2A). The mitochondrial enrichment of these two miRNAs remained significantly lower for up to 3 days (P < 0.01) and returned to near normal levels at 7 days post-injury. The levels of another mitochondria-enriched miRNA, miR-146a trended lower at 1 day in the mitochondrial fraction following TBI with a similar pattern being observed at 3 days. However, by 7 days post-injury, miR-146a levels in the mitochondrial fraction were found to be significantly elevated (P = 0.033; Figure 2A).
Figure 2.
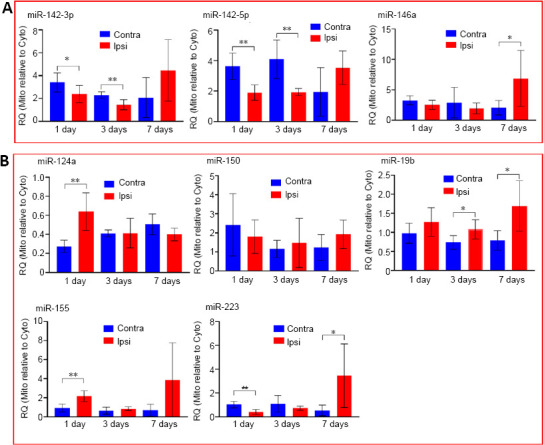
Profile of mitochondria-enrichment of miRNAs following traumatic brain injury.
Animals (total n = 6 per group) were euthanatized at 1, 3, or 7 days following injury and the ipsilateral (Ipsi) and contralateral (Contra) hippocampi were rapidly removed for isolation of the mitochondria (Mito) and cytosolic (Cyto) fractions. The fractions were then subjected to RNA isolation and miRNA analysis. The level of mitochondria-enrichment of a miRNA expressed as relative quantity (RQ) was calculated using the formula RQ = 2–(Ct(Mito)–Ct(Cyto)) where mitochondrial and cytoplasmic fractions were isolated from the same tissue. (A) Mitochondria-enriched miRNAs. (B) Other miRNAs. Data are expressed as the mean ± SD. The difference between the ipsilateral and the contralateral hippocampi was evaluated using one-way analysis of variance and Student’s t-test. *P < 0.05, **P < 0.01.
The levels of several non-mitochondria-enriched miRNAs also exhibited changes over time following TBI with different trends (Figure 2B). MiR-124a, miR-150, miR19b and a known TBI-responsive miRNA miR-155 were either increased (such as miR-124a, miR-155, miR-19b) or not significantly altered (miR-150) in mitochondria at any time examined following TBI. One exception was miR-223 (Figure 2B), which is also a TBI-responsive miRNA that showed significantly decreased levels in mitochondria at 1 day after TBI (P < 0.01). However, by 7 days post-injury, the levels of miR-223 were significantly elevated (P < 0.05) in mitochondria isolated from the injured hippocampus. No major changes in any of the above mentioned miRNAs were observed between the ipsilateral and contralateral hippocampal mitochondria isolated from the group of sham operated animals (Additional Figure 1 (1.7MB, tif) ).
Interestingly, while levels of the mitochondria-enriched miRNAs miR-142-3p and miR-142-5p were reduced in mitochondria at 1 and 3 days following TBI, their levels in cytoplasmic fractions significantly increased (P < 0.01; Table 1). By 7 days following TBI, the levels of both miR-142-3p and miR-142-5p in the cytoplasmic fraction had returned to normal. A similar trending pattern was observed for miR-146a. Finally, levels of two TBI-responsive miRNAs, miRNA miR-155 and miR-223 were significantly elevated in the cytoplasm at 1 and 3 days post-injury and returned to normal levels by 7 days (see levels and P values in Table 1).
Inflammatory gene expression at different time points following TBI
A customized TLDA was used to analyze the expression of selected inflammatory-related genes in hippocampal cytoplasm. In general, alterations in gene expression were found to be most active at 1 day following TBI (Table 2). By 3 days following TBI, the number and levels of genes significantly altered were less than that at 1 day, especially with respect to anti-inflammatory genes (Table 2). Among the tested genes, there was only one anti-inflammatory gene, Tgfb1, which remained elevated at 3 days post-injury. Among the pro-inflammatory genes examined, the chemokine (C-C motif) ligand 2 (CCL2, also called monocyte chemoattractant protein 1, MCP1) appeared to have the greatest change and was significantly elevated in the injured ipsilateral hippocampus compared to uninjured side by 160- and 15-fold respectively at 1 and 3 days post TBI (P < 0.05; Table 2). By 7 days following TBI, CCL2 levels returned to control values and we did not observe any significant differences in any of the other genes in the panel at 7 days between the ipsilateral injured and contralateral sides.
Table 2.
Response of inflammatory-related genes in hippocampus following TBI
| Gene symbol | 1 d | 3 d | 7 d | ||||
|---|---|---|---|---|---|---|---|
| Fold (range) | P-value | Fold (range) | P-value | Fold (range) | P-value | ||
| Anti-inflammatory | Arg1 | 0.37 (0.26–0.53) | 0.005 | 0.54 (0.39–0.75) | 0.025 | 1.29 (0.81–2.04) | 0.340 |
| Ccl22 | 0.69 (0.3–1.62) | 0.550 | 0.35 (0.30–0.42) | 0.151 | 1.08 (0.64–1.82) | 0.799 | |
| Epas1 | 0.61 (0.47–0.78) | 0.001 | 0.85 (0.63–1.14) | 0.301 | 1.21(0.88–1.66) | 0.271 | |
| Il4 | 0.28 (0.12–0.61) | 0.072 | 0.57 (0.27–1.23) | 0.285 | 1.71 (0.73–4.01) | 0.351 | |
| Pparg | 0.61 (0.44–0.83) | 0.032 | 0.52 (0.24–1.11) | 0.178 | 1.01 (0.75–1.37) | 0.952 | |
| Sphk1 | 6.10 (3.4–10.9) | 0.004 | 1.49 (1.03–2.16) | 0.390 | 0.83 (0.69–1.01) | 0.275 | |
| Stat6 | 0.66 (0.51–0.86) | 0.013 | 0.92 (0.78–1.09) | 0.514 | 1.22 (0.95–1.56) | 0.155 | |
| Tgfb1 | 1.53 (1.2–1.94) | 0.006 | 2.28 (1.85–2.82) | 0 | 0.60 (0.34–1.07) | 0.282 | |
| Tgm2 | 2.40 (1.78–3.24) | 0.011 | 1.17 (0.99–1.39) | 0.220 | 1.31 (0.81–2.11) | 0.442 | |
| Pro-inflammatory | Ccr2 | 10.67 (6.3–18.0) | 0.004 | 4.93 (3.54–6.88) | 0.007 | 0.65 (0.21–2.00) | 0.505 |
| Ccl2 | 160 (85.5–299.8) | 0.002 | 15.43 (8.16–29.2) | 0.001 | 0.47 (0.17–1.27) | 0.367 | |
| Ccr7 | 0.47 (0.3–0.74) | 0.079 | 0.74 (0.45–1.21) | 0.361 | 1.54 (1.14–2.07) | 0.242 | |
| Cd86 | 0.87 (0.47–1.59) | 0.646 | 2.51 (1.91–3.31) | 0.007 | 0.50 (0.28–0.92) | 0.290 | |
| Il12a | 0.30 (0.21–0.42) | 0.001 | 0.55 (0.45–0.67) | 0 | 1.26 (0.94–1.67) | 0.396 | |
| Il12b | 0.36 (0.24–0.54) | 0.010 | 0.44 (0.30–0.64) | 0.057 | 0.76 (0.51–1.14) | 0.235 | |
| Il1b | 9.46 (6.55–13.7) | 0.001 | 1.58 (1.23–2.04) | 0.053 | 0.50 (0.27–0.94) | 0.458 | |
| Il6 | 0.78 (0.51–1.18) | 0.392 | 0.76 (0.48–1.22) | 0.512 | 1.13 (0.77–1.68) | 0.737 | |
| Nos2 | 2.12 (0.47–9.6) | 0.395 | 0.51 (0.43–0.60) | 0.098 | 0.47 (0.12–1.80) | 0.702 | |
| Stat1 | 0.89 (0.7–1.14) | 0.377 | 0.98 (0.81–1.18) | 0.720 | 1.15 (0.87–1.52) | 0.314 | |
| Aif1 | 1.48 (0.9–1.45) | 0.510 | 3.13 (2.11–4.63) | 0.004 | 0.54 (0.24–1.19) | 0.313 | |
| Nlrp3 | 1.04 (0.77–1.4) | 0.801 | 2.17 (1.82–2.59) | 0.001 | 0.85 (0.54–1.32) | 0.590 | |
| Itgam | 1.56 (1.23–1.98) | 0.018 | 3.07 (2.52–3.74) | 0.001 | 0.71 (0.41–1.21) | 0.419 | |
A TLDA card consisting of selected inflammatory responsive genes was used to assess mRNA expression levels in hippocampal cytoplasm obtained from each time points following traumatic brain injury (TBI). Gene expression levels were calculated using formula: fold change = 2–(Ct(Ipsi)–Ct(Contra)) comparing the ipsilateral injured side of the brain relative to the uninjured contralateral side. Data represent the mean value of 4 biological replicates, and the P values were generated using two-tailed Student’s t-tests. The fold change range is shown in parentheses. Significantly different (P < 0.05) expression genes are highlighted in red for increased expression and in green for decreased expression. The 1, 3, and 7 days indicate the tissue harvesting time following controlled cortical impact.
Effect of miR-146a nanoparticle delivery to BV-2 and SH-SY5Y cells
MiR-146a targets TRAF6 and IRAK1, two effectors that are important regulators on the NF-κB pathway (Taganov et al., 2006). Given our previous observations demonstrating a differential expression of miR-146a at early hours following TBI (Wang et al., 2015, 2017), we selected this anti-inflammatory miRNA for the peptide-based nanoparticle delivery studies. We first tested the effects of the peptide/miR-146a nanoparticle delivery system using the mouse microglial BV-2 and human neuroblastoma SH-SY5Y cell line. These two cell lines were incubated with p5RHH + miR-146a-Cy5 mimic or scrambled control nanoparticles for 48 hours. Additional cell culture wells were incubated with miR-146a-Cy5 mimic (or scrambled control) without any carrier or in the presence of Lipofectamine RNAiMAX, the latter of which served as a positive transfection control. Confocal microscopy demonstrated effective transfection of miR-146a-Cy5 in both the BV-2 (Figure 3A) and SH-SY5Y cells (Additional Figure 2 (2.2MB, tif) ) using p5RHH peptide or Lipofectamine, while no fluorescent signal was observed in the cells where transfection carrier agent was not used. These observations document the necessity of using cell permeable carrier molecules to ensure cellular delivery of Cy5-labeled miR-146a. Western blot analysis confirmed the biological activity of miR-146a in p5RHH peptide and Lipofectamine transfected cells as reflected by a significant decrease in TRAF6 protein levels (P = 0.0005 and 0.013, respectively; Figure 3B and C). These experiments suggest that the p5RHH + miR-146a nanoparticle platform effectively delivered miR-146a mimic and down-regulated its validated target TRAF6.
Figure 3.
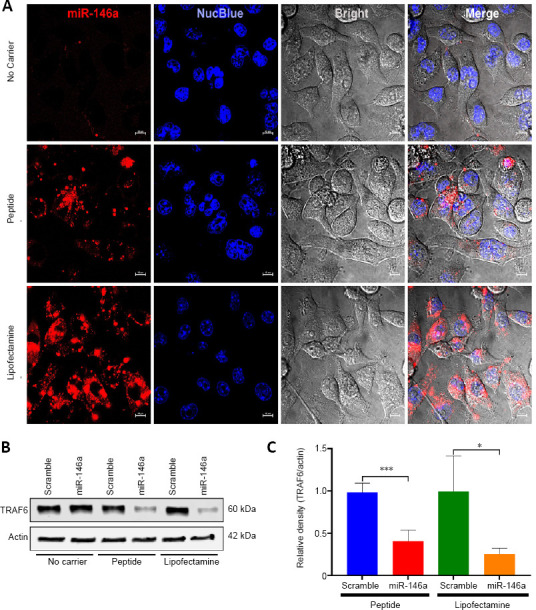
In vitro delivery of mellitin-derived cell permeable peptide (p5RHH)-based miR-146a nanoparticles to cultured BV-2 cells.
Confocal microscopy graph of miR-146a and scramble control transfections (scale bars: 10 µm) (A) and the western blot analysis of TNF receptor-associated factor 6 (TRAF6) in the transfected cells (B). Densitometry quantification of the western blot analysis from 4 independent transfection experiments (C, n = 4). Pre-seeded BV-2 cells were incubated either with miR-146a or scramble mimic/p5RHH-nanoparticle complexes or miR-146a or scramble mimic/Lipofectamine, or without carrier for 24 hours and the cells were visualized and photographed using a Nikon C2+ confocal microscope and then washed with PBS and harvested for RNA analysis or western blot analysis. The densitometry readout of TRAF6 protein signal was normalized on that of actin signal in the same protein sample. Data are expressed as the mean ± SD. The difference between miR-146a and scramble mimics was evaluated using Student’s t-test. *P < 0.05, ***P < 0.001.
Effect of miR-146a delivery to the injured rat hippocampus
The p5RHH peptide was then used to deliver miR-146a mimic or scrambled control to the rat hippocampus following severe TBI. The miR-146a mimic-nanoparticle complex was injected into the dorsal hippocampus immediately following a severe CCI and animals sacrificed 48 hours later. Hippocampi were then harvested for analysis of TRAF6 and IRAK1 gene and protein (TRAF6) expression. RT-qPCR revealed a significant reduction in IRAK1 (P = 0.048) and TRAF6 (P = 0.036) mRNA levels following miR-146a delivery in comparison to scrambled control demonstrating the efficacy of the nanoparticle delivery system (Figure 4A). Importantly, Western blot analysis demonstrated that TRAF6 protein levels were also decreased in the hippocampus of animals receiving p5RHH + miR-146a nanoparticles (Figure 4B).
Figure 4.
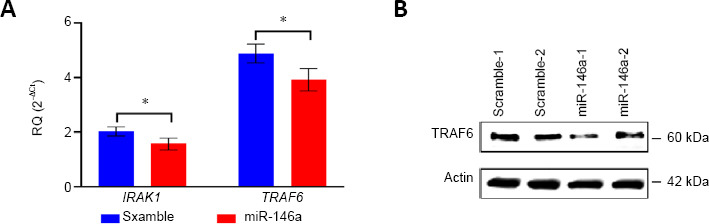
In vivo delivery of miR-146a mimic using mellitin-derived cell permeable peptide (p5RHH) effectively reduced levels of TRAF6 and IRAK1 mRNAs and protein levels.
Animals were subjected to controlled cortical impact injury (n = 6) and randomly assigned to receive an intrahippocampal injection of p5RHH + miR-146a mimic (n = 3) or scrambled RNA control (n = 3). The animals were then euthanatized at 48 hours after injection and the hippocampi removed for protein and RNA isolation and analysis. (A) TRAF6 and IRAK1 mRNA levels determined by RT-qPCR following TBI and nanoparticle injections. Data are expressed as the mean ± SD. The difference between miR-146a and scramble mimics was evaluated using Student’s t-test. *P < 0.05. (B) Western blot analysis for TRAF6 using hippocampal tissues that were injected with either miR-146a or scrambled control mimic. IRAK1: Interleukin-1 receptor-associated kinase 1; TRAF6: TNF receptor-associated factor 6.
Discussion
Our previous studies reported that several mitochondria-enriched miRNAs regulating inflammation including miR-142-3p, miR-142-5p, and miR-146a are altered in the hippocampus following a severe traumatic brain injury (Wang et al., 2015, 2017). Specifically, the levels of these miRNAs were reduced significantly in mitochondria at 3–12 hours following TBI (Wang et al., 2015, 2017). We now demonstrate that the levels of these same miRNAs continue to be reduced in the hippocampal mitochondria for at least 3 days following TBI, with a concomitant increase in expression levels in the cytosol. Interestingly, the reduction in mitochondria-enriched miRNAs coincided with decreased mitochondrial bioenergetics at the same time points following injury. The results of these experiments suggest a potential time-dependent compartmental re-distribution of mitochondria-enriched miRNAs that is related to TBI-induced loss of mitochondrial bioenergetics. We further demonstrate the utilization of a novel cell-penetrating peptide nanoparticle system for delivery of miR-146a for targeting modulators of NF-κB pro-inflammatory signaling pathway in vitro and in vivo.
We suggested previously that mitochondria and miRNA interactions may play a role in miRNA cellular function related to inflammation in the injured brain (Wang et al., 2015, 2017). This was based on our observations that levels of miR-146a, miR-142-3p and miR-142-5p were significantly decreased in the mitochondrial fraction and increased in the cytoplasmic fraction at early time points post TBI (Wang et al., 2015). We hypothesized that this compartmental shift may represent a translocation of mitochondria-enriched miRNAs in response to cellular stressors (e.g., TBI) that impact mitochondrial function. We now show that this compartmental shift is still evident up to 3 days following TBI for miR-142-3p and miR-142-5p in hippocampal tissue, and returns to normal levels by 7 days. Although not statistically significant, alterations in miR-146a showed a trend similar to miR-142-3p and miR-142-5p.
It has been well described that TBI elicits a rapid and significant loss of mitochondrial function in the hippocampus and is a key factor in determining cell survival and recovery (Sullivan et al., 1998; Singh et al., 2006). In our current study, mitochondrial bioenergetics in the hippocampus were significantly compromised for at least 3 days following TBI and recovered by 7 days post injury. Interestingly, we observed that changes in the expression patterns of the mitochondria-enriched, inflammatory miRNAs (miR-142-3p, miR-142-5p, and miR-146a) were highly correlated with the loss in mitochondria bioenergetics. Mitochondria can be considered as ‘ first-responders’ to cellular demands and stressors and can trigger various signaling pathways involving gene regulation, in which miRNAs play a critical role. Therefore, we postulate that mitochondria function may determine the subcellular localization and function of these mitochondria-enriched miRNAs. Future studies are needed to understand the correlation of translocation of these mitochondria-enriched, inflammatory miRNAs with mitochondrial bioenergetics and its functional impact on the inflammatory response following TBI.
Our miRNA and gene expression studies indicate that alterations of selected inflammatory related miRNAs and mRNAs peaked at 1 day and their activities remained elevated at 3 days. The activities were then reduced to control levels at 1 week following TBI. The inflammatory gene expression activity observed in this study is consistent with previous reports (Morganti-Kossmann et al., 2002; Semple et al., 2010; Ziebell and Morganti-Kossmann, 2010; Morganti et al., 2016) and may provide the basis for designing miRNA-based interventional strategies targeting acute times following TBI. For example, the neuroinflammatory response following TBI is characterized by a prolonged period of pro-inflammation persisting for months to years after the injury (Johnson et al., 2013). However, there also exists a reparative anti-inflammatory phase that is only present for acute time points following TBI (Jin et al., 2012; Hsieh et al., 2013; Kumar et al., 2016). Therefore, sustaining a balance of inflammatory responses that maintains expression of the reparative phase will be essential to promoting tissue survival and recovery.
We found that miR-146a levels increased in cytoplasm initially, but then returned to control levels by 7 days post-injury. These observations are intriguing for at least two reasons. First, miR-146a plays a significant role in regulating the production of certain pro-inflammatory cytokines by targeting two adaptor proteins TRAF6 and IRAK1 that function upstream of the NF-κB signaling pathway (Liu et al., 2017). Second, miR-146a expression is actually regulated by activation of the NF-κB pathway (Taganov 2006). Thus, miR-146a is activated by and also acts as a negative feedback regulator of NF-κB signaling (Figure 5), which may enable a fine-tune control of the inflammatory response. Taken together, it is postulated that the transient expression of anti-inflammatory reparative signaling may be related to the downregulation of miR-146a through NF-κB. While the mechanisms by which miR-146a modulates inflammatory signaling in the injured brain are currently not known, these observations prompted us to test whether nanoparticle delivery of miR-146a can serve as a therapeutic strategy for modulating the neuroinflammatory response following TBI. Specifically, we employed a cell-permeable peptide p5RHH to deliver miR-146a mimic to cultured cell lines and also following TBI. The p5RHH cell-permeable peptide forms a non-immunogenic nanoparticle complex with small inhibitory RNA molecules and has been shown to be an effective delivery platform (Hou et al., 2013a; Hou et al., 2013b). Our data demonstrated that p5RHH effectively delivered miR-146a mimic and reduced the expression levels of TRAF6 and IRAK1 in vitro and in vivo. The results of these studies are encouraging as they establish a proof of principle that this novel nanoparticle delivery approach has therapeutic potential for modulating pro-inflammatory effectors in the injured brain .
Figure 5.
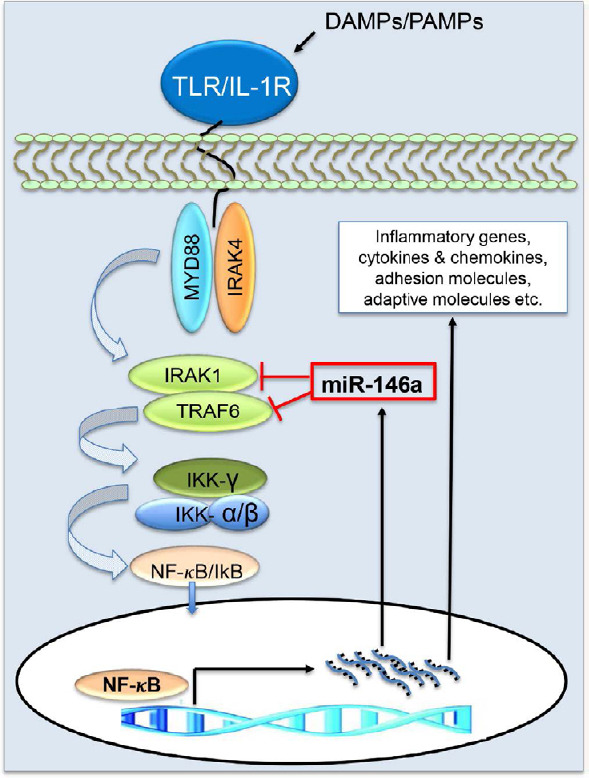
MiR-146a is a negative regulator of the NF-κB signaling pathway.
MiR-146a targets TNF receptor-associated factor 6 (TRAF6) and interleukin-1 receptor-associated kinase 1 (IRAK1), two upstream adaptor proteins on the nuclear factor-kappaB (NF-κB) pathway thus inhibiting synthesis of pro-inflammatory cytokines. Interestingly, NF-κB induces miR-146a transcription to control the miR-146a levels in the cells. This feedback mechanism may contribute to maintaining a delicate equilibrium of pro-inflammatory and anti-inflammatory states in normal physiological conditions, which is often disrupted in pathological conditions and under cellular stresses such as following TBI. Thus, manipulation of miR-146a levels may have therapeutic benefits in intervening undesirable inflammatory responses related to cellular stressors. DAMPs: Damage-associated molecular pattern molecules; IKK: IkappaB kinase; IκB: IkappaB; IL-1R: interleukin-1 receptor; MYD88: myeloid differentiation primary response 88; PAMPs: pathogen-associated molecular pattern molecules; TLR: Toll-like receptor.
Additional files:
Additional Figure 1 (1.7MB, tif) : Profile of mitochondria-enrichment of miRNAs in sham operated animals.
Profile of mitochondria-enrichment of miRNAs in sham operated animals.
Sham operated animals (total n = 4 per group) were euthanatized at 1, 3, or 7 days following surgery and the ipsilateral and contralateral hippocampi were rapidly removed for isolation of the mitochondria and cytosolic fractions. The fractions were then subjected to RNA isolation and miRNA analysis. The level of mitochondria-enrichment of a miRNA expressed as relative quantity (RQ) was calculated using the formula RQ=2-(Ct(Mito)-Ct(Cyto)) where mitochondrial and cytoplasmic fractions were isolated from the same tissue. Green and orange bars correspond to the contralateral and ipsilateral hippocampi, respectively, of the sham-surgery animals. Data are expressed as the mean ± SD.
Additional Figure 2 (2.2MB, tif) : In vitro delivery of p5RHH peptide-based miR-146a nanoparticles to cultured SH-SY5Y cells.
In vitro delivery of p5RHH peptide-based miR-146a nanoparticles to cultured SH-SY5Y cells.
Confocal microscopy graph of miR-146a and scramble control transfections (Scale bars:10 µm). Pre-seeded SH-SY5Y cells were incubated either with miR-146a or scramble mimic/p5RHH-nanoparticle complexes or miR-146a or scramble mimic/Lipofectamine, or without carrier for 24 hours and the cells were visualized and photographed using a Nikon C2+ confocal microscope.
Additional Table 1 (2.9MB, pdf) : Customized TaqMan low density array (TLDA) Genes and the assay ID.
Footnotes
C-Editors: Zhao M, Li CH; T-Editor: Jia Y
Conflicts of interest: All authors report no conflicts of interests.
Financial support: his work is supported by a grant (15-12A) from the Kentucky Spinal Cord and Head Injury Research Trust to JES and WXW.
Institutional review board statement: All of the studies performed were approved by the University of Kentucky Institutional Animal Care and Usage Committee (IACUC protocol # 2014-1300) on August 17, 2017.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This work is supported by a grant (15-12A) from the Kentucky Spinal Cord and Head Injury Research Trust to JES and WXW.
References
- 1.Bachstetter AD, Webster SJ, Goulding DS, Morton JE, Watterson DM, Van Eldik LJ. Attenuation of traumatic brain injury-induced cognitive impairment in mice by targeting increased cytokine levels with a small molecule experimental therapeutic. J Neuroinflammation. 2015;12:69. doi: 10.1186/s12974-015-0289-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baldwin SA, Gibson T, Callihan CT, Sullivan PG, Palmer E, Scheff SW. Neuronal cell loss in the CA3 subfield of the hippocampus following cortical contusion utilizing the optical disector method for cell counting. J Neurotrauma. 1997;14:385–398. doi: 10.1089/neu.1997.14.385. [DOI] [PubMed] [Google Scholar]
- 3.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 4.Brown MR, Sullivan PG, Dorenbos KA, Modafferi EA, Geddes JW, Steward O. Nitrogen disruption of synaptoneurosomes: an alternative method to isolate brain mitochondria. J Neurosci Methods. 2004;137:299–303. doi: 10.1016/j.jneumeth.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 5.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 6.Contreras J, Rao DS. MicroRNAs in inflammation and immune responses. Leukemia. 2012;26:404–413. doi: 10.1038/leu.2011.356. [DOI] [PubMed] [Google Scholar]
- 7.Cui H, Liu G. How noncoding RNAs contribute to macrophage polarization. In: Greene CM, editor. MicroRNAs and Other Non-Coding RNAs in Inflammation. Cham, Switzerland: Springer; pp. 59–84. [Google Scholar]
- 8.Davis LM, Pauly JR, Readnower RD, Rho JM, Sullivan PG. Fasting is neuroprotective following traumatic brain injury. J Neurosci Res. 2008;86:1812–1822. doi: 10.1002/jnr.21628. [DOI] [PubMed] [Google Scholar]
- 9.Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- 10.Gensel JC, Kopper TJ, Zhang B, Orr MB, Bailey WM. Predictive screening of M1 and M2 macrophages reveals the immunomodulatory effectiveness of post spinal cord injury azithromycin treatment. Sci Rep. 2017;7:40144. doi: 10.1038/srep40144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilmer LK, Roberts KN, Joy K, Sullivan PG, Scheff SW. Early mitochondrial dysfunction after cortical contusion injury. J Neurotrauma. 2009;26:1271–1280. doi: 10.1089/neu.2008.0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison EB, Emanuel K, Lamberty BG, Morsey BM, Li M, Kelso ML, Yelamanchili SV, Fox HS. Induction of miR-155 after Brain Injury Promotes Type 1 Interferon and has a Neuroprotective Effect. Front Mol Neurosci. 2017;10:228. doi: 10.3389/fnmol.2017.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hata A. Functions of microRNAs in cardiovascular biology and disease. Annu Rev Physiol. 2013;75:69–93. doi: 10.1146/annurev-physiol-030212-183737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hou KK, Pan H, Lanza GM, Wickline SA. Melittin derived peptides for nanoparticle based siRNA transfection. Biomaterials. 2013a;34:3110–3119. doi: 10.1016/j.biomaterials.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou KK, Pan H, Ratner L, Schlesinger PH, Wickline SA. Mechanisms of nanoparticle-mediated siRNA transfection by melittin-derived peptides. ACS Nano. 2013b;7:8605–8615. doi: 10.1021/nn403311c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsieh CL, Kim CC, Ryba BE, Niemi EC, Bando JK, Locksley RM, Liu J, Nakamura MC, Seaman WE. Traumatic brain injury induces macrophage subsets in the brain. Eur J Immunol. 2013;43:2010–2022. doi: 10.1002/eji.201243084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsieh CL, Niemi EC, Wang SH, Lee CC, Bingham D, Zhang J, Cozen ML, Charo I, Huang EJ, Liu J, Nakamura MC. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J Neurotrauma. 2014;31:1677–1688. doi: 10.1089/neu.2013.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu Z, Yu D, Almeida-Suhett C, Tu K, Marini AM, Eiden L, Braga MF, Zhu J, Li Z. Expression of miRNAs and their cooperative regulation of the pathophysiology in traumatic brain injury. PLoS One. 2012;7:e39357. doi: 10.1371/journal.pone.0039357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubbard WB, Harwood CL, Geisler JG, Vekaria HJ, Sullivan PG. Mitochondrial uncoupling prodrug improves tissue sparing, cognitive outcome, and mitochondrial bioenergetics after traumatic brain injury in male mice. J Neurosci Res. 2018;96:1677–1688. doi: 10.1002/jnr.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 21.Jin X, Ishii H, Bai Z, Itokazu T, Yamashita T. Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS One. 2012;7:e41892. doi: 10.1371/journal.pone.0041892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136:28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 24.Kumar A, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. Microglial/macrophage polarization dynamics following traumatic brain injury. J Neurotrauma. 2016;33:1732–1750. doi: 10.1089/neu.2015.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar A, Stoica BA, Loane DJ, Yang M, Abulwerdi G, Khan N, Kumar A, Thom SR, Faden AI. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J Neuroinflammation. 2017;14:47. doi: 10.1186/s12974-017-0819-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar RG, Boles JA, Wagner AK. Chronic inflammation after severe traumatic brain injury: characterization and associations with outcome at 6 and 12 months postinjury. J Head Trauma Rehabil. 2015;30:369–381. doi: 10.1097/HTR.0000000000000067. [DOI] [PubMed] [Google Scholar]
- 27.Liu L, Sun T, Liu Z, Chen X, Zhao L, Qu G, Li Q. Traumatic brain injury dysregulates microRNAs to modulate cell signaling in rat hippocampus. PLoS One. 2014;9:e103948. doi: 10.1371/journal.pone.0103948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu NK, Xu XM. MicroRNA in central nervous system trauma and degenerative disorders. Physiol Genomics. 2011;43:571–580. doi: 10.1152/physiolgenomics.00168.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017;2 doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lloyd E, Somera-Molina K, Van Eldik LJ, Watterson DM, Wainwright MS. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J Neuroinflammation. 2008;5:28. doi: 10.1186/1742-2094-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mestdagh P, Van Vlierberghe P, De Weer A, Muth D, Westermann F, Speleman F, Vandesompele J. A novel and universal method for microRNA RT-qPCR data normalization. Genome Biol. 2009;10:R64. doi: 10.1186/gb-2009-10-6-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care. 2002;8:101–105. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Morganti JM, Riparip LK, Rosi S. Call off the dog(ma): M1/M2 polarization is concurrent following traumatic brain injury. PLoS One. 2016;11:e0148001. doi: 10.1371/journal.pone.0148001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morganti JM, Jopson TD, Liu S, Riparip LK, Guandique CK, Gupta N, Ferguson AR, Rosi S. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci. 2015;35:748–760. doi: 10.1523/JNEUROSCI.2405-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson PT, Wang WX, Rajeev BW. MicroRNAs (miRNAs) in neurodegenerative diseases. Brain Pathol. 2008;18:130–138. doi: 10.1111/j.1750-3639.2007.00120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295–312. doi: 10.1146/annurev-immunol-020711-075013. [DOI] [PubMed] [Google Scholar]
- 37.O’Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol. 2011;11:163–175. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- 38.Pandya JD, Pauly JR, Sullivan PG. The optimal dosage and window of opportunity to maintain mitochondrial homeostasis following traumatic brain injury using the uncoupler FCCP. Exp Neurol. 2009;218:381–389. doi: 10.1016/j.expneurol.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 39.Pandya JD, Sullivan PG, Leung LY, Tortella FC, Shear DA, Deng-Bryant Y. Advanced and high-throughput method for mitochondrial bioenergetics evaluation in neurotrauma. Methods Mol Biol. 2016;1462:597–610. doi: 10.1007/978-1-4939-3816-2_32. [DOI] [PubMed] [Google Scholar]
- 40.Paxinos G, Watson C. Cambridge, MA, USA: Academic Press; 2013. The Rat Brain in Stereotaxic Coordinates. [Google Scholar]
- 41.Redell JB, Liu Y, Dash PK. Traumatic brain injury alters expression of hippocampal microRNAs: potential regulators of multiple pathophysiological processes. J Neurosci Res. 2009;87:1435–1448. doi: 10.1002/jnr.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sabirzhanov B, Zhao Z, Stoica BA, Loane DJ, Wu J, Borroto C, Dorsey SG, Faden AI. Downregulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. J Neurosci. 2014;34:10055–10071. doi: 10.1523/JNEUROSCI.1260-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sauerbeck A, Pandya J, Singh I, Bittman K, Readnower R, Bing G, Sullivan P. Analysis of regional brain mitochondrial bioenergetics and susceptibility to mitochondrial inhibition utilizing a microplate based system. J Neurosci Methods. 2011;198:36–43. doi: 10.1016/j.jneumeth.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2-/- mice. J Cereb Blood Flow Metab. 2010;30:769–782. doi: 10.1038/jcbfm.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab. 2006;26:1407–1418. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- 46.Sullivan PG, Keller JN, Mattson MP, Scheff SW. Traumatic brain injury alters synaptic homeostasis: implications for impaired mitochondrial and transport function. J Neurotrauma. 1998;15:789–798. doi: 10.1089/neu.1998.15.789. [DOI] [PubMed] [Google Scholar]
- 47.Sullivan PG, Krishnamurthy S, Patel SP, Pandya JD, Rabchevsky AG. Temporal characterization of mitochondrial bioenergetics after spinal cord injury. J Neurotrauma. 2007;24:991–999. doi: 10.1089/neu.2006.0242. [DOI] [PubMed] [Google Scholar]
- 48.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang WX, Sullivan PG, Springer JE. Mitochondria and microRNA crosstalk in traumatic brain injury. Prog Neuropsychopharmacol Biol Psychiatry. 2017;73:104–108. doi: 10.1016/j.pnpbp.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 50.Wang WX, Visavadiya NP, Pandya JD, Nelson PT, Sullivan PG, Springer JE. Mitochondria-associated microRNAs in rat hippocampus following traumatic brain injury. Exp Neurol. 2015;265:84–93. doi: 10.1016/j.expneurol.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang WX, Wilfred BR, Baldwin DA, Isett RB, Ren N, Stromberg A, Nelson PT. Focus on RNA isolation: obtaining RNA for microRNA (miRNA) expression profiling analyses of neural tissue. Biochim Biophys Acta. 2008;1779:749–757. doi: 10.1016/j.bbagrm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol. 2013;4:18. doi: 10.3389/fneur.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Profile of mitochondria-enrichment of miRNAs in sham operated animals.
Sham operated animals (total n = 4 per group) were euthanatized at 1, 3, or 7 days following surgery and the ipsilateral and contralateral hippocampi were rapidly removed for isolation of the mitochondria and cytosolic fractions. The fractions were then subjected to RNA isolation and miRNA analysis. The level of mitochondria-enrichment of a miRNA expressed as relative quantity (RQ) was calculated using the formula RQ=2-(Ct(Mito)-Ct(Cyto)) where mitochondrial and cytoplasmic fractions were isolated from the same tissue. Green and orange bars correspond to the contralateral and ipsilateral hippocampi, respectively, of the sham-surgery animals. Data are expressed as the mean ± SD.
In vitro delivery of p5RHH peptide-based miR-146a nanoparticles to cultured SH-SY5Y cells.
Confocal microscopy graph of miR-146a and scramble control transfections (Scale bars:10 µm). Pre-seeded SH-SY5Y cells were incubated either with miR-146a or scramble mimic/p5RHH-nanoparticle complexes or miR-146a or scramble mimic/Lipofectamine, or without carrier for 24 hours and the cells were visualized and photographed using a Nikon C2+ confocal microscope.