

Abstract
The adaptive immune system can diversify the antigen receptors to eliminate various pathogens through programmed DNA lesions at antigen receptor genes. In immune diversification, general DNA repair machineries are applied to transform the programmed DNA lesions into gene mutation or recombination events with common and unique features. Here we focus on antibody class switch recombination (CSR), and review the initiation of base damages, the conversion of damaged base to DNA double-strand break, and the ligation of broken ends. With an emphasis on the unique features in CSR, we discuss recent advances in the understanding of DNA repair/replication coordination, and ERCC6L2-mediated deletional recombination. We further elaborate the application of CSR in end-joining, resection and translesion synthesis assays. In the time of the COVID-19 pandemic, we hope it help to understand the generation of therapeutic antibodies.
Keywords: DNA repair, Class switch recombination, NHEJ, AID, ERCC6L2, Rev7, Shieldin, Deletional recombination
Introduction
Due to the continuous challenges by a variety of pathogens in the environment, our adaptive immune system has evolved an immune diversification process to diversify the antigen receptors in lymphocytes (Alt et al. 2013). The antigen receptors include T cell receptor (TCR) which serves as the basis for cell-mediated immune response, and B cell receptor (BCR) which can be secreted as antibody or immunoglobulin (Ig) in humoral immunity. The diversity of antigen receptors or immune repertoires, is initiated by programmed DNA lesions and channeled into either recombination or mutation events by the general DNA repair machineries with common and unique features.
Immune diversity is mainly achieved through two steps: antigen-independent and antigen-dependent steps. The first step of immune diversification involved the joining of the variable (V), diversity (D) and joining (J) gene segments to form the variable regions exons of immunoglobulin heavy (IgH) and light (IgL) chains or TCR-α/β/γ/δ chains. This antigen-independent process occurs during lymphocyte development and is called V(D)J recombination (Gellert 2002; Helmink and Sleckman 2012; Schatz et al. 1992). Upon antigen stimulation, the BCR undergoes additional diversification processes such as IgH class switch recombination (CSR), Ig variable (IgV) exon somatic hypermutation (SHM), or gene conversion (GCV) in mature B cells. CSR allows switching of the BCR isotype from IgM to other classes, which changes the effector function of a particular BCR and elicits different downstream immune functions (Chaudhuri et al. 2007; Stavnezer et al. 2008). SHM introduces point mutations or small insertions and deletions (indels) in the IgV exon, allowing antibody affinity mutation (Di Noia and Neuberger 2007; Peled et al. 2008). GCV, which exchanges the expressing rearranged V with an upstream V gene segment, is observed in birds and rabbits (Maizels 1987).
V(D)J recombination is initiated by RAG endonuclease, while CSR, SHM and GCV are initiated by the same cytidine deaminase called activation-induced deaminase (AID) (Arakawa et al. 2002; Muramatsu et al. 2000; Revy et al. 2000). Activities of RAG and AID are highly regulated in lymphocytes to ensure an ordered diversification of antigen receptors (Teng and Schatz 2015; Yeap and Meng 2019). The programmed DNA lesions of either DNA double strand breaks (DSBs) or modified base (deoxyurindine, U) caused by RAG or AID can activate the general cellular DNA damage response factors and be repaired efficiently by general DNA repair factors. In this context, the RAG-generated ends are exclusively ligated through the classic nonhomologous end-joining (c-NHEJ) pathway (Boboila, Alt, et al. 2012; Deriano and Roth 2013). In SHM, base excision repair (BER) and mismatch repair (MMR) pathways cooperatively channel the U’s into mutations or indels (Peled et al. 2008). While in CSR, AID-initiated U’s are first converted into DSBs (Stavnezer et al. 2008). CSR utilizes both c-NHEJ and alternative end-joining (Alt-EJ) pathways to ligate the ends (Boboila, Alt, et al. 2012; Deriano and Roth 2013; Yan et al. 2007), while GCV uses homologous recombination (HR)-based mechanism to diverse the antibody repertoire (Arakawa and Buerstedde 2004). Immune diversification happens in a physiological context and utilizes the general DNA repair pathways. Thus, the V(D)J recombination and CSR are frequently used as reporter assays to characterize DSB response factors or NHEJ factors, as exampled in the studies of Shieldin (Dev et al. 2018; Ghezraoui et al. 2018; Noordermeer et al. 2018).
Here we use antibody CSR as an exemplar to discuss the common and unique features in the repair of programmed DNA lesions. The readers are encouraged to read the recent reviews on V(D)J recombination (Schatz and Ji 2011), SHM and CSR (Methot and Di Noia 2017; Yeap and Meng 2019) for the general mechanisms of immune diversity, and the roles of particular DNA repair pathways in immune diversity (Saha et al. 2020; Wang et al. 2020).
Introduction of programmed DNA lesions by DNA deaminase
CSR, SHM and GCV were considered as three distinct biological processes, until the discovery of AID (Muramatsu et al. 2000; Revy et al. 2000). AID was first revealed with a cDNA subtraction approach by comparing cytokine-stimulated and unstimulated murine B cell line CH12F3 (Muramatsu et al. 1999). Later, data from both gene knockout mouse model (Muramatsu et al. 2000) and hyper IgM syndrome (a genetic disorder with defective CSR) patients (Revy et al. 2000) suggest AID is required for CSR and SHM (Honjo et al. 2002), and GCV (Arakawa et al. 2002; Fugmann and Schatz 2002). AID is a member of AID/APOBEC cytidine deaminase superfamily, which was originally considered as an RNA editing enzyme based on its homology to the RNA editing enzyme APOBEC1 (Honjo et al. 2002), until more lines of direct evidence that AID is an authentic DNA mutator (Chaudhuri et al. 2003; Petersen-Mahrt et al. 2002; Pham et al. 2003). It turns out that in the AID/APOBEC superfamily, APOBEC1 is the only RNA editing enzyme while the other catalytic-active enzymes all work on single-strand DNA (ssDNA) (Harris and Dudley 2015; Liu and Meng 2018). AID preferentially converts the cytidine in the context of WRC (W = A/T; R = G/A) motif to uridine on ssDNA, leading to an U:G mismatch on the genomic DNA (Fig. 1a).
Fig. 1.

IgH class switch recombination (CSR). Schematic illustration of IgH constant genes and AID targeting (a), processing of U’s into DSBs by BER and MMR (b), activation of DSBR (c) and end ligation by c-NHEJ and Alt-EJ (d). See text for more details
Biochemical evidence clearly suggests that AID works on ssDNA that are generated during gene transcription (Chaudhuri et al. 2003; Pham et al. 2003), correlating with the observed deamination in transcribed regions in vivo (Yoshikawa et al. 2002) (Fig. 1a). Many cis- and trans-factors are involved in the regulation of AID activity in vivo as we recently discussed (Yeap and Meng 2019). Particularly, both RNA exosome and ssDNA binding complex RPA are required for AID’s access to the ssDNA substrates (Basu et al. 2011; Chaudhuri et al. 2004). Although the AID C-terminus was proposed to interact or recruit several DNA repair factors including BER/MMR (Ranjit et al. 2011) or DSB response factors (Zahn et al. 2014), genetic evidence suggests the introduction and processing of DNA lesions are separate steps in CSR (Zarrin et al. 2007).
During CSR, AID extensively targets the long repetitive S regions (Hackney et al. 2009; Yu and Lieber 2003), which locate at the first intron of each IgH constant (IgH C) gene (Fig. 1a). During evolution, AID-preferred substrate motif AGCT is selective enriched in S region, which motif offers an ideal substrate for AID since C’s on both strands can be deaminated (Han et al. 2011). In mammalian cells, S regions harbor additional repetitive G-rich motifs (Hackney et al. 2009; Yu and Lieber 2003). Besides the Ig loci, AID is found to target many off-target proto-oncogenes and its ectopically expression or mis-regulation contributes to lymphomagenesis (Casellas et al. 2016). A few high-throughput approaches were developed to track the AID off-target sites in CSR, including high-throughput genome-wide translocation sequencing (HTGTS) (Chiarle et al. 2011; Meng et al. 2014), translocation-capture sequencing (TC-Seq) (Klein et al. 2011), resected ssDNA ChIP-seq (Qian et al. 2014), DNA capture and sequencing (Alvarez-Prado et al. 2018), etc. The chromatin features associated with AID targeting shows that AID prefers to target the divergent/convergent transcribed regions (Meng et al. 2014; Pefanis et al. 2014) in the intersection regions of super-enhancer and gene (Meng et al. 2014; Pefanis et al. 2015; Qian et al. 2014). The specific AID targeting sites contribute to its mutagenic outcomes, as elegantly demonstrated in germinal center B cells undergoing SHM and/or CSR (Liu et al. 2008). How the DNA sequence or other cis-elements contribute to the differential DNA repair is still an open question in the field. Machine learning was applied to predict AID targeting based on the mutation frequency (Alvarez-Prado et al. 2018) and recently applied to predict base editing outcomes (Arbab et al. 2020). However, whether DNA sequence or chromatin location affects DNA repair is still unclear.
Generation of DSBs through BER and MMR
In CSR, AID is specifically targeted to the IgH S regions and the deamination products were channeled into DSBs through BER and MMR pathways (Fig. 1b). UNG is found to be the major glycosylase for U excision in S regions. In this context, mouse genetic studies revealed that UNG-deficiency significantly abolished CSR (Rada et al. 2002) and SMUG1-deficiency further decreased CSR (Di Noia et al. 2006; Dingler et al. 2014), which is consistent with the fact that UNG deleterious mutants cause hyper IgM syndrome Type IV in human patients (Imai et al. 2003). However, single UNG deficiency does not completely abolish CSR (Rada et al. 2002, 2004). On the other hand, the U:G pair is also a substrate for MMR. Even before the discovery of AID, several studies have already revealed a critical role of MMR protein in SHM (Phung et al. 1998; Rada et al. 1998; Wiesendanger et al. 2000). The discovery of AID explained the role of MMR proteins in processing AID-initiated lesions, and a panel of MMR proteins were found vital to CSR (Li et al. 2004; Martin et al. 2003; Martomo et al. 2004; Schrader et al. 2002, 2003). Later, it was found that combined UNG and MMR deficiencies completely abolish CSR in different mouse models (Rada et al. 2004; Shen et al. 2006), suggesting the combined effort of BER and MMR in processing AID-lesions.
BER and MMR are important cellular pathways to maintain genome integrity, which use two distinct sets of proteins to process damaged base or mismatch into single-strand nick or gap. In BER, the damaged base is removed by glycosylase to generate an apurinic/apyrimidinic site (AP), which is further processed into a nick by apurinic/apyrimidinic endonuclease 1 (APE1) (Jacobs and Schar 2012). In MMR, the mismatch is recognized by MutS complex, which further recruits a cascade of scaffold and nuclease proteins to generate a single-strand gap (Li 2008). The nick/gap is further subjected to DNA polymerase fill-up and XRCC1-Lig1/3-mediated ligation. However, instead of working in an error-free manner, BER and MMR process the U’s into DSBs in CSR. In this context, B cells could utilize the first few steps of BER/MMR to achieve the goal. Consistent with this thought, deficiency of XRCC1 which forms complex with Lig1/3 in the ligation step enhances CSR in mouse B cells (Han et al. 2012; Saribasak et al. 2011). Although, XRCC1 could function through its role in Alt-EJ, it is tempting to speculate that XRCC1’s role in sealing the gap in single-strand break repair could inhibit the DSB generation in CSR. Furthermore, the sequence features of S regions could aid the DSB generation (Yu et al. 2004). The enriched palindrome AGCT motif offers a convenient way to generate DSB: UNG-processed APs can be cut by APE1to form two closely single-strand nicks which can turn into DSB (Han et al. 2011). Consistently, APE1 is required for CSR in B cells (Masani et al. 2013). Last but not least, the unique features of S region sequence also offer other DSB generation ways. The G-rich S region can form G4 (Dempsey et al. 1999) or R-loop structure (Yu et al. 2003), which could contribute to the genomic breaks observed in S region in the absence of AID (Chiarle et al. 2011).
Ligation of DSB ends by end-joining pathways
The AID-initiated DSBs can activate DSB response (DSBR) in activated B cells (Fig. 1c), as NBS1/gamma-H2AX foci are quickly formed at IgH locus (Petersen et al. 2001). Roles of many of the DSBR factors in CSR were dissected in mouse models, including MRN complex (Rass et al. 2009; Reina-San-Martin et al. 2005; Xie et al. 2009; Yin et al. 2009; Zha et al. 2009), ATM (Lumsden et al. 2004; Reina-San-Martin et al. 2004), H2AX (Franco et al. 2006; Reina-San-Martin et al. 2003), etc. Among the DSBR factors, 53BP1 plays a unique role in CSR, as its deficiency leads to the most dramatic CSR decrease (Manis et al. 2004; Ward et al. 2004) among deficiencies for its upstream, e.g., RNF8/RNF168 (Ramachandran et al. 2010; Santos et al. 2010) or downstream factors, e.g., Rif1 (Di Virgilio et al. 2013), PTIP (Daniel et al. 2010; Starnes et al. 2016), Rev7/Shieldin (Boersma et al. 2015; Dev et al. 2018; Ghezraoui et al. 2018; Noordermeer et al. 2018; Xu et al. 2015). Beyond its role in inhibiting end-resection, 53BP1 could affect DSB repair at chromatin level by affecting chromatin synapsis or movement (Difilippantonio et al. 2008; Dimitrova et al. 2008; Kilic et al. 2019; Lukas et al. 2011; Ochs et al. 2019), potentially through its oligomer domain (Lottersberger et al. 2013; Sundaravinayagam et al. 2019).
The end-joining pathways finally ligate the two DSBs at upstream and downstream S regions in CSR (Fig. 1d). Different from the end-joining in V(D)J recombination which DSB ends are exclusively ligated by classic-NHEJ pathway (Deriano and Roth 2013), the ends can be ligated by both c-NHEJ and Alt-EJ in CSR (Boboila, Alt, et al., 2012). NHEJ contains several evolutionarily conserved factors which are named as the “core” subunits (Lieber 2010). Contrast to the completed abolished V(D)J recombination, CSR is decreased but not abolished in the core NHEJ subunit deficiency (Yan et al. 2007), which lead to the concept of Alt-EJ pathways. Alt-EJ is defined as end-joining in the absence of certain NHEJ core subunits, which may represent many parallel pathways (Boboila, Jankovic, et al. 2010; Boboila, Yan, et al. 2010; Boboila, Oksenych, et al. 2012). Alt-EJ frequently uses micro-homology at their ligation junctions, which was also observed in yeast and termed as microhomology-mediated end-joining (MMEJ). In CSR, Alt-EJ plays an important role and a few factors are proposed in this pathway, including PARP, XRCC1/Lig1/3, FEN1, polymerase theta (POLQ), etc. (Audebert et al. 2004; Boboila, Oksenych, et al. 2012; Ceccaldi et al. 2015; Della-Maria et al. 2011; Frit et al. 2014; Han et al. 2012; Mateos-Gomez et al. 2015; Saribasak et al. 2011; Schreiber et al. 2002). It is of note that the end-joining pathway factors work in an iterative and redundant manner (Lieber 2010). The CSR end-joining of single gene deficiency could be complemented by redundant factors. For example, neither absence of POLQ nor catalytic-inactive POLQ causes defective CSR level (Li et al. 2011; Masuda et al. 2006; Zeng et al. 2004). Instead, POLQ deficiency leads to minor changes of CSR-junctions (Yousefzadeh et al. 2014).
Coordination of DNA repair and replication in CSR
Activated B cells undergo extensive cell proliferation in vivo and ex vivo, and defective cell proliferation results to decreased CSR level in various abovementioned mouse models. In this context, replicative helicase Mcm complex is found to be required for optimal CSR (Wiedemann et al. 2016). AID initiates a variety of DNA damages on genomic DNA, including U’s, AP sites, SSBs and DSBs. How B cells tolerate all these damages during DNA replication was a long-sought question in the field. Lines of evidence suggest that activated B cells have decreased p53 through a BCL6-dependent way, and tolerate the physiological DNA breaks (Phan and Dalla-Favera 2004). However, how damaged base or AP sites are tolerated was not clear. In the course of studying Rev7’s roles in CSR, we unexpectedly revealed such a role of translesion synthesis (TLS) in this process (Yang et al. 2020).
Rev7 is an adaptor protein and plays multiple roles in different cellular processes through protein interaction, including DNA translesion synthesis (Lawrence, Das, et al. 1985; Nelson et al. 1996), APC/C inhibition (Chen and Fang 2001), spindle assembly (Bhat et al. 2015), DNA resection end inhibition (Boersma et al. 2015; Xu et al. 2015) as a subunit of Shieldin (Dev et al. 2018; Ghezraoui et al. 2018; Gupta et al. 2018; Mirman et al. 2018; Noordermeer et al. 2018). We found that Rev7-deficient activated B cells undergo cell death in an AID-UNG-dependent manner (Yang et al. 2020), suggesting the Rev7 assists DNA replication in the presence of AID-UNG generated AP sites. In this context, we tested a panel of DNA repair/replication factors involved in DSBR, end resection, and TLS, and conlcude that the cell death phenotype is caused by the defective function of DNA Polymerase Zeta (PolZ). PolZ contains Rev7, Rev3l and other subunits, and is the major extender enzyme in TLS (Lange et al. 2016; Yang and Gao 2018). Combined with the previous observations in Rev3l-deficient mouse model (Schenten et al. 2009), our findings show that TLS enzyme not only diversifies DNA sequence but also ensures B cell proliferation, coordinating the processes of DNA repair and replication.
In this context, CSR serves as a valuable assay to test the functions of many DNA repair pathways. First, the end-joining efficiency can be directly visualized by flow cytometry study of surface-expressed IgG/IgM ratios, making it a robust NHEJ reporter for physiological end-joining. Second, the end resection can be showed by the location of switch junctions in IgH locus by applying HTGTS technology. In the same line, the choice of c-NHEJ and Alt-EJ can be reflected by the microhomology usage of switch junctions. Third, the mutation spectrum (e.g., transition C > T, or transversion C > A/G) can be displayed by sequencing of the 5’S regions. As an exemplar, we characterized Rev7 with CSR assays (Yang et al. 2020) (Fig. 2). Consistent with other reports (Boersma et al. 2015; Dev et al. 2018; Ghezraoui et al. 2018; Gupta et al. 2018; Leland et al. 2018; Mirman et al. 2018; Noordermeer et al. 2018; Tomida et al. 2018; Xu et al. 2015), Rev7 deficiency dramatically increases the end resection and affects optimal CSR (Yang et al. 2020). Moreover, we found that Rev7 promotes C > G transversion in S regions (Yang et al. 2020), consistent with its role in TLS (Lawrence, Das, et al., 1985; Lawrence, Nisson, et al. 1985; Torpey et al. 1994; Zhao et al. 2006).
Fig. 2.
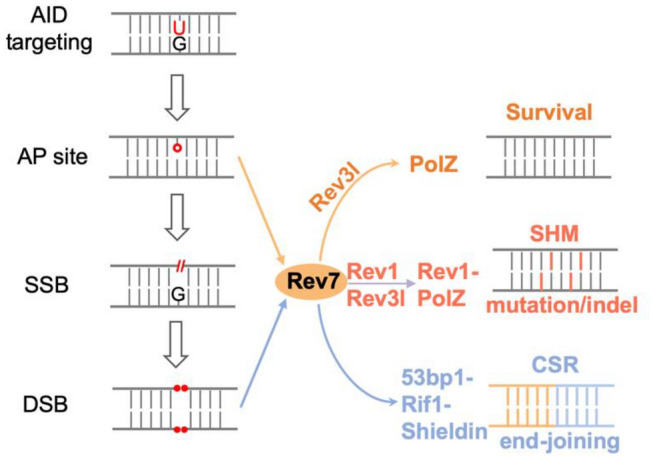
Multiple roles of Rev7 in CSR. Rev7 is involved in DSB response, transversion mutation, and toleration of AID-initiated AP sites in CSR. See text for more details
Molecular basis of deletional CSR
CSR is described as a deletional recombination event, as only the deletional end-joining yields a productive IgH locus and inversional event is non-productive. It was taken for granted that productive Ig expression could select the deletional recombination events in activated B cells, until elegant studies using high-throughput technology reveal that CSR happens intrinsically in a deletional manner (Dong et al. 2015; Panchakshari et al. 2018). The abovementioned HTGTS was adapted into CSR-HTGTS-seq using a primer annealing to the 5’Sµ region, which can capture the endogenous S region junctions in CSR (Dong et al. 2015). The junction numbers in CSR-HTGTS-seq authentically reflect the CSR levels and can serve as a molecular assay to measure CSR (Dong et al. 2015). Strikingly, CSR-HTGTS-seq reveal that deletion recombination happens nearly ten times higher than the inversion event (Dong et al. 2015).
In cis, the chromatin architecture offers a convenient way for deletional recombination. IgH locus locates in its own topological associated domain (TAD), in which context the CTCF-binding elements (CBEs) are key cis-elements in regulating either V(D)J recombination or CSR (Guo et al. 2011; Lin et al. 2015; Zhang et al. 2021). Cohesin-mediated loop extrusion facilitates chromatin contacts and the formation of TADs (Vian et al. 2018), and assists both V(D)J recombination and CSR (Zhang et al. 2019a, 2019b). In activated B cells, the activation of IgHC promoters drives stepwise cohesin loading on the pre-assembled CSR centre in naïve B cells (Zhang et al. 2019a, ), providing a directional alignment of donor and acceptor S regions for deletional CSR in cis (Zhang et al. 2019a, ). In trans, the deletion recombination replies on DSBR and end-joining factors (Dong et al. 2015; Panchakshari et al. 2018), as deletion of 53BP1 or Lig4 partially abolished the dominant deletional recombination. However, the loss of directional bias is associated with excessive resection of DSB ends in 53BP1 deficiency (Bunting et al. 2010), large amounts of unrepaired broken ends in Lig4 deficiency (Canela et al. 2016), and severe block of CSR in both genotype (Bunting et al. 2010; Han and Yu 2008). The unbias joining could reflect that the escaped broken ends from the joining complex are joined randomly in a manner resmebling that of translocation (Dong et al. 2015) and/or in a diffusional manner at low levels (Zhang et al. 2019a, b).
To search for such a determining factor in deletional CSR, we applied a combintion approach of CRISPR-screening and chemical treatment, which yield a new end-joinining factor ERCC6L2 (Liu et al. 2020). ERCC6L2 belows to the Snf2-like ERCC6 family which also include ERCC6 (CSB, important factor in transcription-coupled nucleotide excision repair) and ERCC6L (PICH, invloved in spindle assembly checkpoint). Deleterious mutations of ERCC6L2 have been identified in inherited bonee marrow failure (BMF) patients (Bluteau et al. 2018; Jarviaho et al. 2018; Shabanova et al. 2018; Tummala et al. 2014; Zhang et al. 2016) and a subtype of inherited acute myloid leukemia (Douglas et al. 2019). We found that ERCC6L2 is quickly recruited to DNA damage sites and the deficient cells are sensitive to IR or IR-mimic treatment (Liu et al. 2020). We found that ERCC6L2 is required for optimal CSR, and V(D)J recombination in XLF-defieicent background (Liu et al. 2020). Combining several lines of evidence, we concluded that ERCC6L2 is an authentic NHEJ factor (Liu et al. 2020). The conclusion is further supported by two other contemporaneous reports (Francica et al. 2020; Olivieri et al. 2020).
ERCC6L2 plays a vital role in NHEJ, and, as we showed, deteremines the orientation-specifc joining in CSR (Liu et al. 2020) (Fig. 3). Using CSR-HTGTS-seq, we found that in the absence of ERCC6L2, the deletional CSR is completely abolished and S region ends join in a 1:1 ratio in term of deletion:inversion. ERCC6L2 does not greatly affect end resection and the CSR level is decreased to half of wildtype level, making its role in deletional-CSR distinct from 53BP1 or Lig4/XRCC4. Further investigation showed that ERCC6L2’s catalytic activity is required for the orientation-specifc joining, leading to a model that ERCC6L2 could clean the DSB end for the Lig4/XRCC4 sliding, and/or affecting the long-range chromatin synapsis or DSB end tethering.
Fig. 3.

ERCC6L2-depedent deletional CSR. A working model to explain the molecular basis for deletional CSR. See text for more details
Perspective
In antigen receptor diversification, the repairs of programmed DNA lesions utilize the general cellular DNA repair factors and also show several unique features including an error-prone manner and higher efficiency. We use antibody CSR as an example to illustrate the introduction of base damage, the processing of base damage into DSBs, the ligation of DSBs, the coordination of DNA repair and replication, and the molecular basis for the unique deletional recombination. With the development of new technologies, such as CSR-HTGTS-seq, IgH deep sequencing and 3C-HTGTS-seq, we will be able to discover and elaborate new features in immune diversity. CSR is an ideal assay to examine many aspects of DNA repair including NHEJ, end resection, BER, MMR and TLS. We believe that the assay will continually aid genome instability studies in the future. Furthermore, AID/APOBEC deaminase is combined with CRISPR/Cas9 into cytidine base editing (CBE) tools (Rees and Liu 2018). However, the repair of CBE editing is less explored. Whether similar pathways in repairing AID-lesions function downstream of CBE editing are waiting to be solved.
Acknowledgements
We thank Drs. Junchao Dong and Leng-Siew Yeap for critical reading of the manuscript. Research in the Meng lab is supported by the National Key R&D Program of China (2017YFA0506700), Strategic Priority Research Program of Chinese Academy of Sciences (XDB19030000), National Natural Science Foundation of China (81622022, 32090042).
Declaration
Conflict of interest
The authors declare no competing financial interests.
References
- Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;152(3):417–429. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Prado AF, Perez-Duran P, Perez-Garcia A, Benguria A, Torroja C, de Yebenes VG, et al. A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets. Journal of Experimental Medicine. 2018;215(3):761–771. doi: 10.1084/jem.20171738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa H, Buerstedde JM. Immunoglobulin gene conversion: Insights from bursal B cells and the DT40 cell line. Developmental Dynamics. 2004;229(3):458–464. doi: 10.1002/dvdy.10495. [DOI] [PubMed] [Google Scholar]
- Arakawa H, Hauschild J, Buerstedde JM. Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science. 2002;295(5558):1301–1306. doi: 10.1126/science.1067308. [DOI] [PubMed] [Google Scholar]
- Arbab M, Shen MW, Mok B, Wilson C, Matuszek Z, Cassa CA, et al. Determinants of base editing outcomes from target library analysis and machine learning. Cell. 2020;182(2):463–480. doi: 10.1016/j.cell.2020.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. Journal of Biological Chemistry. 2004;279(53):55117–55126. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144(3):353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat A, Wu Z, Maher VM, McCormick JJ, Xiao W. Rev7/Mad2B plays a critical role in the assembly of a functional mitotic spindle. Cell Cycle. 2015;14(24):3929–3938. doi: 10.1080/15384101.2015.1120922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluteau O, Sebert M, Leblanc T, Peffault de Latour R, Quentin S, Lainey E, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood. 2018;131(7):717–732. doi: 10.1182/blood-2017-09-806489. [DOI] [PubMed] [Google Scholar]
- Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Advances in Immunology. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- Boboila C, Jankovic M, Yan CT, Wang JH, Wesemann DR, Zhang T, et al. Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(7):3034–3039. doi: 10.1073/pnas.0915067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boboila C, Oksenych V, Gostissa M, Wang JH, Zha S, Zhang Y, et al. Robust chromosomal DNA repair via alternative end-joining in the absence of X-ray repair cross-complementing protein 1 (XRCC1) Proceedings of the National Academy of Sciences of the United States of America. 2012;109(7):2473–2478. doi: 10.1073/pnas.1121470109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boboila C, Yan C, Wesemann DR, Jankovic M, Wang JH, Manis J, et al. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. Journal of Experimental Medicine. 2010;207(2):417–427. doi: 10.1084/jem.20092449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma V, Moatti N, Segura-Bayona S, Peuscher MH, van der Torre J, Wevers BA, et al. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature. 2015;521(7553):537–540. doi: 10.1038/nature14216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela A, Sridharan S, Sciascia N, Tubbs A, Meltzer P, Sleckman BP, et al. DNA breaks and end resection measured genome-wide by end sequencing. Molecular Cell. 2016;63(5):898–911. doi: 10.1016/j.molcel.2016.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF, Di Noia JM. Mutations, kataegis and translocations in B cells: Understanding AID promiscuous activity. Nature Reviews: Immunology. 2016;16(3):164–176. doi: 10.1038/nri.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature. 2015;518(7538):258–262. doi: 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Advances in Immunology. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430(7003):992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422(6933):726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- Chen J, Fang G. MAD2B is an inhibitor of the anaphase-promoting complex. Genes & Development. 2001;15(14):1765–1770. doi: 10.1101/gad.898701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147(1):107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JA, Santos MA, Wang Z, Zang C, Schwab KR, Jankovic M, et al. PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science. 2010;329(5994):917–923. doi: 10.1126/science.1187942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della-Maria J, Zhou Y, Tsai MS, Kuhnlein J, Carney JP, Paull TT, et al. Human Mre11/human Rad50/Nbs1 and DNA ligase IIIalpha/XRCC1 protein complexes act together in an alternative nonhomologous end joining pathway. Journal of Biological Chemistry. 2011;286(39):33845–33853. doi: 10.1074/jbc.M111.274159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey LA, Sun H, Hanakahi LA, Maizels N. G4 DNA binding by LR1 and its subunits, nucleolin and hnRNP D, A role for G-G pairing in immunoglobulin switch recombination. Journal of Biological Chemistry. 1999;274(2):1066–1071. doi: 10.1074/jbc.274.2.1066. [DOI] [PubMed] [Google Scholar]
- Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annual Review of Genetics. 2013;47:433–455. doi: 10.1146/annurev-genet-110711-155540. [DOI] [PubMed] [Google Scholar]
- Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nature Cell Biology. 2018;20(8):954–965. doi: 10.1038/s41556-018-0140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annual Review of Biochemistry. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- Di Noia JM, Rada C, Neuberger MS. SMUG1 is able to excise uracil from immunoglobulin genes: Insight into mutation versus repair. EMBO Journal. 2006;25(3):585–595. doi: 10.1038/sj.emboj.7600939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339(6120):711–715. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456(7221):529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456(7221):524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingler FA, Kemmerich K, Neuberger MS, Rada C. Uracil excision by endogenous SMUG1 glycosylase promotes efficient Ig class switching and impacts on A: T substitutions during somatic mutation. European Journal of Immunology. 2014;44(7):1925–1935. doi: 10.1002/eji.201444482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Panchakshari RA, Zhang T, Zhang Y, Hu J, Volpi SA, et al. Orientation-specific joining of AID-initiated DNA breaks promotes antibody class switching. Nature. 2015;525(7567):134–139. doi: 10.1038/nature14970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas SPM, Siipola P, Kovanen PE, Pyorala M, Kakko S, Savolainen ER, et al. ERCC6L2 defines a novel entity within inherited acute myeloid leukemia. Blood. 2019;133(25):2724–2728. doi: 10.1182/blood-2019-01-896233. [DOI] [PubMed] [Google Scholar]
- Francica P, Mutlu M, Blomen VA, Oliveira C, Nowicka Z, Trenner A, et al. Functional radiogenetic profiling implicates ERCC6L2 in non-homologous end joining. Cell Reports. 2020;32(8):108068. doi: 10.1016/j.celrep.2020.108068. [DOI] [PubMed] [Google Scholar]
- Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Molecular Cell. 2006;21(2):201–214. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Frit P, Barboule N, Yuan Y, Gomez D, Calsou P. Alternative end-joining pathway(s): Bricolage at DNA breaks. DNA Repair (Amst) 2014;17:81–97. doi: 10.1016/j.dnarep.2014.02.007. [DOI] [PubMed] [Google Scholar]
- Fugmann SD, Schatz DG. Immunology. One AID to unite them all. Science. 2002;295(5558):1244–1245. doi: 10.1126/science.1070023. [DOI] [PubMed] [Google Scholar]
- Gellert M. V(D)J recombination: RAG proteins, repair factors, and regulation. Annual Review of Biochemistry. 2002;71:101–132. doi: 10.1146/annurev.biochem.71.090501.150203. [DOI] [PubMed] [Google Scholar]
- Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature. 2018;560(7716):122–127. doi: 10.1038/s41586-018-0362-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Yoon HS, Franklin A, Jain S, Ebert A, Cheng HL, et al. CTCF-binding elements mediate control of V(D)J recombination. Nature. 2011;477(7365):424–430. doi: 10.1038/nature10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, et al. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell. 2018;173(4):972–988. doi: 10.1016/j.cell.2018.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney JA, Misaghi S, Senger K, Garris C, Sun Y, Lorenzo MN, et al. DNA targets of AID evolutionary link between antibody somatic hypermutation and class switch recombination. Advances in Immunology. 2009;101:163–189. doi: 10.1016/S0065-2776(08)01005-5. [DOI] [PubMed] [Google Scholar]
- Han L, Mao W, Yu K. X-ray repair cross-complementing protein 1 (XRCC1) deficiency enhances class switch recombination and is permissive for alternative end joining. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(12):4604–4608. doi: 10.1073/pnas.1120743109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Masani S, Yu K. Overlapping activation-induced cytidine deaminase hotspot motifs in Ig class-switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(28):11584–11589. doi: 10.1073/pnas.1018726108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Yu K. Altered kinetics of nonhomologous end joining and class switch recombination in ligase IV-deficient B cells. Journal of Experimental Medicine. 2008;205(12):2745–2753. doi: 10.1084/jem.20081623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Dudley JP. APOBECs and virus restriction. Virology. 2015;479–480:131–145. doi: 10.1016/j.virol.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Sleckman BP. The response to and repair of RAG-mediated DNA double-strand breaks. Annual Review of Immunology. 2012;30:175–202. doi: 10.1146/annurev-immunol-030409-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjo T, Kinoshita K, Muramatsu M. Molecular mechanism of class switch recombination: Linkage with somatic hypermutation. Annual Review of Immunology. 2002;20:165–196. doi: 10.1146/annurev.immunol.20.090501.112049. [DOI] [PubMed] [Google Scholar]
- Imai K, Catalan N, Plebani A, Marodi L, Sanal O, Kumaki S, et al. Hyper-IgM syndrome type 4 with a B lymphocyte-intrinsic selective deficiency in Ig class-switch recombination. Journal of Clinical Investigation. 2003;112(1):136–142. doi: 10.1172/JCI18161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs AL, Schar P. DNA glycosylases: in DNA repair and beyond. Chromosoma. 2012;121(1):1–20. doi: 10.1007/s00412-011-0347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarviaho T, Halt K, Hirvikoski P, Moilanen J, Mottonen M, Niinimaki R. Bone marrow failure syndrome caused by homozygous frameshift mutation in the ERCC6L2 gene. Clinical Genetics. 2018;93(2):392–395. doi: 10.1111/cge.13125. [DOI] [PubMed] [Google Scholar]
- Kilic S, Lezaja A, Gatti M, Bianco E, Michelena J, Imhof R, et al. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO Journal. 2019;38(16):e101379. doi: 10.15252/embj.2018101379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147(1):95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange SS, Tomida J, Boulware KS, Bhetawal S, Wood RD. The polymerase activity of mammalian DNA pol zeta is specifically required for cell and embryonic viability. PLoS Genetics. 2016;12(1):e1005759. doi: 10.1371/journal.pgen.1005759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence CW, Das G, Christensen RB. REV7, a new gene concerned with UV mutagenesis in yeast. Molecular and General Genetics. 1985;200(1):80–85. doi: 10.1007/BF00383316. [DOI] [PubMed] [Google Scholar]
- Lawrence CW, Nisson PE, Christensen RB. UV and chemical mutagenesis in rev7 mutants of yeast. Molecular and General Genetics. 1985;200(1):86–91. doi: 10.1007/BF00383317. [DOI] [PubMed] [Google Scholar]
- Leland BA, Chen AC, Zhao AY, Wharton RC, King MC. Rev7 and 53BP1/Crb2 prevent RecQ helicase-dependent hyper-resection of DNA double-strand breaks. Elife. 2018 doi: 10.7554/eLife.33402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GM. Mechanisms and functions of DNA mismatch repair. Cell Research. 2008;18(1):85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- Li Y, Gao X, Wang JY. Comparison of two POLQ mutants reveals that a polymerase-inactive POLQ retains significant function in tolerance to etoposide and gamma-irradiation in mouse B cells. Genes to Cells. 2011;16(9):973–983. doi: 10.1111/j.1365-2443.2011.01550.x. [DOI] [PubMed] [Google Scholar]
- Li Z, Scherer SJ, Ronai D, Iglesias-Ussel MD, Peled JU, Bardwell PD, et al. Examination of Msh6- and Msh3-deficient mice in class switching reveals overlapping and distinct roles of MutS homologues in antibody diversification. Journal of Experimental Medicine. 2004;200(1):47–59. doi: 10.1084/jem.20040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual Review of Biochemistry. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SG, Guo C, Su A, Zhang Y, Alt FW. CTCF-binding elements 1 and 2 in the Igh intergenic control region cooperatively regulate V(D)J recombination. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(6):1815–1820. doi: 10.1073/pnas.1424936112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451(7180):841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- Liu X, Liu T, Shang Y, Dai P, Zhang W, Lee BJ, et al. ERCC6L2 promotes DNA orientation-specific recombination in mammalian cells. Cell Research. 2020;30(9):732–744. doi: 10.1038/s41422-020-0328-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Meng FL. Generation of genomic alteration from cytidine deamination. Advances in Experimental Medicine and Biology. 2018;1044:49–64. doi: 10.1007/978-981-13-0593-1_5. [DOI] [PubMed] [Google Scholar]
- Lottersberger F, Bothmer A, Robbiani DF, Nussenzweig MC, de Lange T. Role of 53BP1 oligomerization in regulating double-strand break repair. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(6):2146–2151. doi: 10.1073/pnas.1222617110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nature Cell Biology. 2011;13(3):243–253. doi: 10.1038/ncb2201. [DOI] [PubMed] [Google Scholar]
- Lumsden JM, McCarty T, Petiniot LK, Shen R, Barlow C, Wynn TA, et al. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. Journal of Experimental Medicine. 2004;200(9):1111–1121. doi: 10.1084/jem.20041074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maizels N. Diversity achieved by diverse mechanisms: gene conversion in developing B cells of the chicken. Cell. 1987;48(3):359–360. doi: 10.1016/0092-8674(87)90182-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nature Immunology. 2004;5(5):481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- Martin A, Li Z, Lin DP, Bardwell PD, Iglesias-Ussel MD, Edelmann W, et al. Msh2 ATPase activity is essential for somatic hypermutation at a-T basepairs and for efficient class switch recombination. Journal of Experimental Medicine. 2003;198(8):1171–1178. doi: 10.1084/jem.20030880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martomo SA, Yang WW, Gearhart PJ. A role for Msh6 but not Msh3 in somatic hypermutation and class switch recombination. Journal of Experimental Medicine. 2004;200(1):61–68. doi: 10.1084/jem.20040691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masani S, Han L, Yu K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Molecular and Cellular Biology. 2013;33(7):1468–1473. doi: 10.1128/MCB.00026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda K, Ouchida R, Hikida M, Nakayama M, Ohara O, Kurosaki T, et al. Absence of DNA polymerase theta results in decreased somatic hypermutation frequency and altered mutation patterns in Ig genes. DNA Repair (Amst) 2006;5(11):1384–1391. doi: 10.1016/j.dnarep.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, Sfeir A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature. 2015;518(7538):254–257. doi: 10.1038/nature14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng FL, Du Z, Federation A, Hu J, Wang Q, Kieffer-Kwon KR, et al. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell. 2014;159(7):1538–1548. doi: 10.1016/j.cell.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot SP, Di Noia JM. Molecular mechanisms of somatic hypermutation and class switch recombination. Advances in Immunology. 2017;133:37–87. doi: 10.1016/bs.ai.2016.11.002. [DOI] [PubMed] [Google Scholar]
- Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, et al. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature. 2018;560(7716):112–116. doi: 10.1038/s41586-018-0324-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102(5):553–563. doi: 10.1016/S0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. Journal of Biological Chemistry. 1999;274(26):18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- Nelson JR, Lawrence CW, Hinkle DC. Thymine-thymine dimer bypass by yeast DNA polymerase zeta. Science. 1996;272(5268):1646–1649. doi: 10.1126/science.272.5268.1646. [DOI] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature. 2018;560(7716):117–121. doi: 10.1038/s41586-018-0340-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs F, Karemore G, Miron E, Brown J, Sedlackova H, Rask MB, et al. Stabilization of chromatin topology safeguards genome integrity. Nature. 2019;574(7779):571–574. doi: 10.1038/s41586-019-1659-4. [DOI] [PubMed] [Google Scholar]
- Olivieri M, Cho T, Alvarez-Quilon A, Li K, Schellenberg MJ, Zimmermann M, et al. A genetic map of the response to DNA damage in human cells. Cell. 2020;182(2):481–496. doi: 10.1016/j.cell.2020.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchakshari RA, Zhang X, Kumar V, Du Z, Wei PC, Kao J, et al. DNA double-strand break response factors influence end-joining features of IgH class switch and general translocation junctions. Proceedings of the National Academy of Sciences of the United States of America. 2018;115(4):762–767. doi: 10.1073/pnas.1719988115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pefanis E, Wang J, Rothschild G, Lim J, Chao J, Rabadan R, et al. Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature. 2014;514(7522):389–393. doi: 10.1038/nature13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pefanis E, Wang J, Rothschild G, Lim J, Kazadi D, Sun J, et al. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell. 2015;161(4):774–789. doi: 10.1016/j.cell.2015.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, et al. The biochemistry of somatic hypermutation. Annual Review of Immunology. 2008;26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236. [DOI] [PubMed] [Google Scholar]
- Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414(6864):660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418(6893):99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424(6944):103–107. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432(7017):635–639. doi: 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- Phung QH, Winter DB, Cranston A, Tarone RE, Bohr VA, Fishel R, et al. Increased hypermutation at G and C nucleotides in immunoglobulin variable genes from mice deficient in the MSH2 mismatch repair protein. Journal of Experimental Medicine. 1998;187(11):1745–1751. doi: 10.1084/jem.187.11.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon KR, Resch W, et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell. 2014;159(7):1524–1537. doi: 10.1016/j.cell.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Molecular Cell. 2004;16(2):163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Rada C, Ehrenstein MR, Neuberger MS, Milstein C. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity. 1998;9(1):135–141. doi: 10.1016/S1074-7613(00)80595-6. [DOI] [PubMed] [Google Scholar]
- Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Current Biology. 2002;12(20):1748–1755. doi: 10.1016/S0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- Ramachandran S, Chahwan R, Nepal RM, Frieder D, Panier S, Roa S, et al. The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(2):809–814. doi: 10.1073/pnas.0913790107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjit S, Khair L, Linehan EK, Ucher AJ, Chakrabarti M, Schrader CE, et al. AID recruits UNG and Msh2 to Ig switch regions dependent upon the AID C terminus [corrected] Journal of Immunology. 2011;187(5):2464–2475. doi: 10.4049/jimmunol.1101406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nature Structural & Molecular Biology. 2009;16(8):819–824. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nature Reviews: Genetics. 2018;19(12):770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC. ATM is required for efficient recombination between immunoglobulin switch regions. Journal of Experimental Medicine. 2004;200(9):1103–1110. doi: 10.1084/jem.20041162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-San-Martin B, Difilippantonio S, Hanitsch L, Masilamani RF, Nussenzweig A, Nussenzweig MC. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. Journal of Experimental Medicine. 2003;197(12):1767–1778. doi: 10.1084/jem.20030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-San-Martin B, Nussenzweig MC, Nussenzweig A, Difilippantonio S. Genomic instability, endoreduplication, and diminished Ig class-switch recombination in B cells lacking Nbs1. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(5):1590–1595. doi: 10.1073/pnas.0406289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102(5):565–575. doi: 10.1016/S0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- Saha T, Sundaravinayagam D, Di Virgilio M. Charting a DNA repair roadmap for immunoglobulin class switch recombination. Trends in Biochemical Sciences. 2020 doi: 10.1016/j.tibs.2020.10.005. [DOI] [PubMed] [Google Scholar]
- Santos MA, Huen MS, Jankovic M, Chen HT, Lopez-Contreras AJ, Klein IA, et al. Class switching and meiotic defects in mice lacking the E3 ubiquitin ligase RNF8. Journal of Experimental Medicine. 2010;207(5):973–981. doi: 10.1084/jem.20092308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribasak H, Maul RW, Cao Z, McClure RL, Yang W, McNeill DR, et al. XRCC1 suppresses somatic hypermutation and promotes alternative nonhomologous end joining in Igh genes. Journal of Experimental Medicine. 2011;208(11):2209–2216. doi: 10.1084/jem.20111135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nature Reviews: Immunology. 2011;11(4):251–263. doi: 10.1038/nri2941. [DOI] [PubMed] [Google Scholar]
- Schatz DG, Oettinger MA, Schlissel MS. V(D)J recombination: molecular biology and regulation. Annual Review of Immunology. 1992;10:359–383. doi: 10.1146/annurev.iy.10.040192.002043. [DOI] [PubMed] [Google Scholar]
- Schenten D, Kracker S, Esposito G, Franco S, Klein U, Murphy M, et al. Pol zeta ablation in B cells impairs the germinal center reaction, class switch recombination, DNA break repair, and genome stability. Journal of Experimental Medicine. 2009;206(2):477–490. doi: 10.1084/jem.20080669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader CE, Vardo J, Stavnezer J. Role for mismatch repair proteins Msh2, Mlh1, and Pms2 in immunoglobulin class switching shown by sequence analysis of recombination junctions. Journal of Experimental Medicine. 2002;195(3):367–373. doi: 10.1084/jem.20011877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader CE, Vardo J, Stavnezer J. Mlh1 can function in antibody class switch recombination independently of Msh2. Journal of Experimental Medicine. 2003;197(10):1377–1383. doi: 10.1084/jem.20022190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber V, Ame JC, Dolle P, Schultz I, Rinaldi B, Fraulob V, et al. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. Journal of Biological Chemistry. 2002;277(25):23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- Shabanova I, Cohen E, Cada M, Vincent A, Cohn RD, Dror Y. ERCC6L2-associated inherited bone marrow failure syndrome. Molecular Genetics & Genomic Medicine. 2018;6(3):463–468. doi: 10.1002/mgg3.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Tanaka A, Bozek G, Nicolae D, Storb U. Somatic hypermutation and class switch recombination in Msh6(-/-)Ung(-/-) double-knockout mice. Journal of Immunology. 2006;177(8):5386–5392. doi: 10.4049/jimmunol.177.8.5386. [DOI] [PubMed] [Google Scholar]
- Starnes LM, Su D, Pikkupeura LM, Weinert BT, Santos MA, Mund A, et al. A PTIP-PA1 subcomplex promotes transcription for IgH class switching independently from the associated MLL3/MLL4 methyltransferase complex. Genes & Development. 2016;30(2):149–163. doi: 10.1101/gad.268797.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annual Review of Immunology. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaravinayagam D, Rahjouei A, Andreani M, Tupina D, Balasubramanian S, Saha T, et al. 53BP1 supports immunoglobulin class switch recombination independently of its DNA double-strand break end protection function. Cell Reports. 2019;28(6):1389–1399. doi: 10.1016/j.celrep.2019.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng G, Schatz DG. Regulation and evolution of the RAG recombinase. Advances in Immunology. 2015;128:1–39. doi: 10.1016/bs.ai.2015.07.002. [DOI] [PubMed] [Google Scholar]
- Tomida J, Takata KI, Bhetawal S, Person MD, Chao HP, Tang DG, et al. FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1-defective cells. EMBO Journal. 2018 doi: 10.15252/embj.201899543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torpey LE, Gibbs PE, Nelson J, Lawrence CW. Cloning and sequence of REV7, a gene whose function is required for DNA damage-induced mutagenesis in Saccharomyces cerevisiae. Yeast. 1994;10(11):1503–1509. doi: 10.1002/yea.320101115. [DOI] [PubMed] [Google Scholar]
- Tummala H, Kirwan M, Walne AJ, Hossain U, Jackson N, Pondarre C, et al. ERCC6L2 mutations link a distinct bone-marrow-failure syndrome to DNA repair and mitochondrial function. American Journal of Human Genetics. 2014;94(2):246–256. doi: 10.1016/j.ajhg.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vian L, Pekowska A, Rao SSP, Kieffer-Kwon KR, Jung S, Baranello L, et al. The energetics and physiological impact of cohesin extrusion. Cell. 2018;175(1):292–294. doi: 10.1016/j.cell.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XS, Lee BJ, Zha S. The recent advances in non-homologous end-joining through the lens of lymphocyte development. DNA Repair (Amst) 2020;94:102874. doi: 10.1016/j.dnarep.2020.102874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, et al. 53BP1 is required for class switch recombination. Journal of Cell Biology. 2004;165(4):459–464. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann EM, Peycheva M, Pavri R. DNA replication origins in immunoglobulin switch regions regulate class switch recombination in an R-loop-dependent manner. Cell Reports. 2016;17(11):2927–2942. doi: 10.1016/j.celrep.2016.11.041. [DOI] [PubMed] [Google Scholar]
- Wiesendanger M, Kneitz B, Edelmann W, Scharff MD. Somatic hypermutation in MutS homologue (MSH)3-, MSH6-, and MSH3/MSH6-deficient mice reveals a role for the MSH2-MSH6 heterodimer in modulating the base substitution pattern. Journal of Experimental Medicine. 2000;191(3):579–584. doi: 10.1084/jem.191.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nature Structural & Molecular Biology. 2009;16(8):814–818. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521(7553):541–544. doi: 10.1038/nature14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449(7161):478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- Yang D, Sun Y, Chen J, Zhang Y, Fan S, Huang M, et al. REV7 is required for processing AID initiated DNA lesions in activated B cells. Nature Communications. 2020;11(1):2812. doi: 10.1038/s41467-020-16632-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Gao Y. Translesion and repair DNA polymerases: Diverse structure and mechanism. Annual Review of Biochemistry. 2018;87:239–261. doi: 10.1146/annurev-biochem-062917-012405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeap LS, Meng FL. Cis- and trans-factors affecting AID targeting and mutagenic outcomes in antibody diversification. Advances in Immunology. 2019;141:51–103. doi: 10.1016/bs.ai.2019.01.002. [DOI] [PubMed] [Google Scholar]
- Yin FF, Bailey S, Innis CA, Ciubotaru M, Kamtekar S, Steitz TA, et al. Structure of the RAG1 nonamer binding domain with DNA reveals a dimer that mediates DNA synapsis. Nature Structural & Molecular Biology. 2009;16(5):499–508. doi: 10.1038/nsmb.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa K, Okazaki IM, Eto T, Kinoshita K, Muramatsu M, Nagaoka H, et al. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. 2002;296(5575):2033–2036. doi: 10.1126/science.1071556. [DOI] [PubMed] [Google Scholar]
- Yousefzadeh MJ, Wyatt DW, Takata K, Mu Y, Hensley SC, Tomida J, et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genetics. 2014;10(10):e1004654. doi: 10.1371/journal.pgen.1004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunology. 2003;4(5):442–451. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- Yu K, Huang FT, Lieber MR. DNA substrate length and surrounding sequence affect the activation-induced deaminase activity at cytidine. Journal of Biological Chemistry. 2004;279(8):6496–6500. doi: 10.1074/jbc.M311616200. [DOI] [PubMed] [Google Scholar]
- Yu K, Lieber MR. Nucleic acid structures and enzymes in the immunoglobulin class switch recombination mechanism. DNA Repair (Amst) 2003;2(11):1163–1174. doi: 10.1016/j.dnarep.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Zahn A, Eranki AK, Patenaude AM, Methot SP, Fifield H, Cortizas EM, et al. Activation induced deaminase C-terminal domain links DNA breaks to end protection and repair during class switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(11):E988–997. doi: 10.1073/pnas.1320486111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrin AA, Del Vecchio C, Tseng E, Gleason M, Zarin P, Tian M, et al. Antibody class switching mediated by yeast endonuclease-generated DNA breaks. Science. 2007;315(5810):377–381. doi: 10.1126/science.1136386. [DOI] [PubMed] [Google Scholar]
- Zeng X, Negrete GA, Kasmer C, Yang WW, Gearhart PJ. Absence of DNA polymerase eta reveals targeting of C mutations on the nontranscribed strand in immunoglobulin switch regions. Journal of Experimental Medicine. 2004;199(7):917–924. doi: 10.1084/jem.20032022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha S, Boboila C, Alt FW. Mre11: Roles in DNA repair beyond homologous recombination. Nature Structural & Molecular Biology. 2009;16(8):798–800. doi: 10.1038/nsmb0809-798. [DOI] [PubMed] [Google Scholar]
- Zhang S, Pondarre C, Pennarun G, Labussiere-Wallet H, Vera G, France B, et al. A nonsense mutation in the DNA repair factor Hebo causes mild bone marrow failure and microcephaly. Journal of Experimental Medicine. 2016;213(6):1011–1028. doi: 10.1084/jem.20151183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yoon HS, Chapdelaine-Williams AM, Kyritsis N, Alt FW. Physiological role of the 3'IgH CBEs super-anchor in antibody class switching. Proceedings of the National Academy of Sciences of the United States of America. 2021 doi: 10.1073/pnas.2024392118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang Y, Ba Z, Kyritsis N, Casellas R, Alt FW. Fundamental roles of chromatin loop extrusion in antibody class switching. Nature. 2019;575(7782):385–389. doi: 10.1038/s41586-019-1723-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang X, Ba Z, Liang Z, Dring EW, Hu H, et al. The fundamental role of chromatin loop extrusion in physiological V(D)J recombination. Nature. 2019;573(7775):600–604. doi: 10.1038/s41586-019-1547-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wang J, Geacintov NE, Wang Z. Poleta, Polzeta and Rev1 together are required for G to T transversion mutations induced by the (+)- and (−)-trans-anti-BPDE-N2-dG DNA adducts in yeast cells. Nucleic Acids Research. 2006;34(2):417–425. doi: 10.1093/nar/gkj446. [DOI] [PMC free article] [PubMed] [Google Scholar]