

Abstract
Objectives:
This research explores the use of polymer brushes for surface treatment of fillers used in polymer-based dental restoratives with focus on shrinkage stress reduction. The influence of interfacial reactive groups on shrinkage stress is explored.
Methods:
Oligomers of varying lengths and with varying number of reactive groups along the length were synthesized by modifying commercial oligomers. Surface of silica fillers (OX50) was treated with methylaminopropyltrimethoxysilane and this was further reacted with the synthesized oligomers to obtain a series of polymer brushes on the surface. Fillers modified with γ-methacryloxypropyltrimethoxysilane were used as a control. Filler surface treatment was confirmed using diffuse reflectance spectroscopy and thermogravimetric analysis. Fillers were added at 30 wt % to a resin made of BisGMA/TEGDMA and polymerization kinetics, shrinkage stress, volumetric shrinkage, flexural strength and modulus, viscosity were measured.
Results:
Composites with polymer brush functionalized fillers showed up to a 30 % reduction in shrinkage stress as compared to the control, with no reduction in flexural strength and modulus. Shrinkage stress reduced with increasing length of the polymer brush and increased with increase in number of reactive groups along the length of the polymer brush.
Significance:
The interface between inorganic fillers and an organic polymer matrix has been utilized to reduce shrinkage stress in a composite with no compromise in mechanical properties. This study gives insights into the stress development mechanism at the interface.
Keywords: Shrinkage stress, shrinkage, conversion, Photopolymerization, filler, surface modification, polymer brush, Mechanical properties, dental restorative, silanes, interface, interphase
Graphical Abstract
1. Introduction
Polymer-based dental composite materials have been in use for more than 50 years [1][2] and now account for the majority of more than 100 million restorations that are placed each year in the US. These esthetic amalgam alternatives were introduced for anterior restorations as an advancement over the existing silicate cements that do not bond to dentin and erode quite rapidly [3]. Despite their widespread use and steady improvements [4] there remain major issues with the use of polymeric dental restoratives regarding their contraction during polymerization, which in turn promotes considerable shrinkage stress [5][6]. The placement of a shrinking composite in a confined, adhesively bonded setting, as is the case when a restoration is photocured in-situ, exacerbates the stress levels within the composite and can cause failure of the restoration or even damage the adjacent tooth structure. There has been a tremendous amount of research directed toward addressing the shrinkage, stress and mechanical properties of composite materials based on modification to the resin phase [7], but new studies focused on addressing these concerns based on alteration of the filler component of the composites are lagging behind.
Most efforts in literature for shrinkage and stress reduction in dental composite materials have focused on modification of the resin matrix. Lower shrinkage and consequently, lower shrinkage stress can be achieved by using high molecular weight monomers or oligomers, thus reducing the reactive group concentration [8]. Monomers with ring-opening functional groups that have lower shrinkage upon polymerization have also been used [9]. Radical-mediated thiol-ene and thiol-Michael click reactions have been used due to their step-growth nature to delay the gelation and slow down the stress build up during polymerization leading to a lower final shrinkage stress [10–13]. More recently, polymeric nanogels with highly internally crosslinked structures have been used as fillers in composite formulations to reduce shrinkage and shrinkage stress [14,15]. Covalent adaptable networks that are able to undergo reversible exchange or reversible addition of bonds during or even after polymerization upon use of an appropriate stimulus have been used to relieve stresses built up during polymerization [16]. Addition fragmentation chain transfer agents as monomers have been used to alter reaction kinetics by the generation of stable radicals that delay network gelation, thus lowering the final stress value [17,18].
Dental composite materials rely on the interface or interphase that covalently couples the dispersed, high-modulus inorganic filler phase to the continuous resin matrix. Effective stress transfer across this interface promotes composite mechanical integrity critical for clinical function [19]. Good interfacial bonding between the filler and resin phases is necessary to achieve the bulk mechanical reinforcing effect of the filler required for the challenging application of in-situ polymerized restorative materials [20,21]. The absence of filler-resin interfacial adhesion can negatively affect composite properties like wear resistance [22], fracture toughness [23] and bulk strength [24]. In the dental composite literature in particular, surprisingly little attention has been paid to the optimization of the interphase created by the presence of a coupling agent that binds the two phases together [25]. γ-Methacryloxypropyltrimethoxysilane (γ-MPS) has been the silane coupling agent of choice in the dental industry since the development of the modern composites due to its hybrid functionality capable of binding to both the filler surface and the organic resin via the respective presence of a surface condensable and methacrylate groups.
The conditions of silane application to substrates vary widely, which results in huge variations in the order and orientation of the silanes in the interphase region. For poorly controlled surface treatment of filler surfaces with γ-MPS, it has been reported that much of the methacrylate functionality remains unreacted and buried in a disordered multi-layered interphase [26].Aqueous conditions typically lead to the formation of multiple layers of the silane on the surface of the particles due to condensation reactions that occur prior to surface addition [27]. The 3-carbon spacer of γ-MPS is relatively rigid and when combined with the multiple siloxane-interconnected layers of the silane with restricted mobility near the surface of the filler. There have been a limited number of attempts to modify the interfacial region to improve composite properties for dental materials and no detailed study on the efficiency of polymerization reaction at the matrix-filler interface. A mixture of functional and non-functional silanes on the filler surface was used in order to improve the hydrolytic stability of the composites [28][29]. Longer spacers than γ-MPS (up to C10) have also been tried with the effect of creating more hydrophobic interfaces and enhanced potential for filler loading [30]. Dendrimers have been used to functionalize the filler surface to successfully reduce polymerization shrinkage stress [31]. More recently, fillers modified with thiourethane oligomers have been used to reduce polymerization stress [32]. Attaching polymeric nanogels to the surface of fillers has also been found to alleviate shrinkage stress [33]. Dynamic covalent chemistry has been utilized at the interface to actively reduce shrinkage stress at the filler-polymer interface during curing [34]. The focus of most of the studies related to alternative approaches to coupling agents, however, is to promote matrix-filler integration, composite stability and mechanical properties, such as fracture strength and modulus, rather than to control shrinkage stress and study the interfacial reaction characteristics.
With this background in mind, it is evident that exploring the design of the interface in dental restorative materials can help in advancing the field and determining the parameters needed for improving material properties. It has been reported that the interface between a solid surface and a cross-linked polymer can be strengthened considerably by the addition of polymer chains that are tethered at one end to the solid surface and extend into the polymer matrix [35]. The level of adhesion depends on the grafting density of these chains, which can be called “polymer brushes” that can be referred to as an assembly of polymer chains that are tethered to one end of a surface or an interface [36]. The use of a chemically grafted polymer brush with controlled spatial geometry and chemical functionality produces a significant increase in the strength and fracture energy of the interphase [37]. It has also been reported that a polymer brush layer exhibits greater wear resistance as compared to a conventional surface coating [38].
The reported benefits of polymer brushes encouraged our exploration of this generic approach in dental composites to design a novel surface treatment that involved tethering oligomeric chains on the surface of filler particles in order to enhance various properties in comparison to the traditional γ-MPS treatment. The primary targeted property studied here is polymerization shrinkage stress in the dental composites with the acknowledgement that properties such as conversion and mechanical strength must not be compromised. Another goal of the research was to study the effect of the interphase on material property development in dental composites and apply the knowledge gained to the design of improved materials.
2. Materials and Methods -
2.1. Materials
Untreated silica nanoparticles (Aerosil OX50, average diameter = 40 nm, surface area 50 m2/g) were donated by Evonik (Germany). OX50 silanized with γ-MPS (γ-methacryloxypropyl-trimethoxysilane) was donated by Septodont/Confi-Dental (Louisville, Colorado, USA). N-Methylaminopropyltrimethoxysilane (MAPTS) was purchased from Gelest. 2,2-Bis[4-(2-hydroxy-3-methacryloyloxypropyl-oxy)phenyl]propane (BisGMA) and triethylene glycol dimethacrylate (TEGDMA) monomers were donated by Esstech. Isocyanatoethyl methacrylate (IEM), poly(bisphenol A-co-epichlorohydrin), glycidyl end-capped, (referred to here as epoxy oligomers, with number average molecular weights of 1075, 4000, and 6100 g/mol), methylene chloride, anhydrous N,N-dimethyl formamide (DMF), anhydrous toluene, and dibutyltin dilaurate were obtained from Sigma-Aldrich (USA).
2.2. Modification of epoxy oligomers
The structure of the epoxy oligomers used is shown in Figure 1 where R’ = OH for the unmodified oligomer. The epoxy oligomers were varied in chain length, with n in Figure 1 ranging from an average of 2 to 20. Three oligomers were used with molecular weights 1075 (n~2), 4000 (n~12) and 6100 (n~20) g/mol. The hydroxyl groups on the oligomers were reacted with IEM in order to introduce pendant urethane methacrylate functionality along their length. Two series of modified oligomers were created using this synthetic approach; one series utilized 10 % of all possible hydroxyl groups in the urethane-forming reaction with IEM, while the other had all of the hydroxyls converted to the urethane methacrylate functionality. The representation of the final oligomer structure is shown in Figure 1.
Figure 1.

Structure of epoxy oligomer series used to modify the OX50 surface where n varies from 2 to 20. Modification of oligomers – Case 1 – Low methacrylate group concentration on oligomer backbone – 10 % of OH groups are reacted with IEM, leaving 90 % OH groups unmodified. Case 2 – High methacrylate group concentration on oligomer backbone – all OH groups are reacted with IEM.
2.3. Modification of the filler surface
Bare silica particles were first treated with MAPTS and then the epoxy-terminated, pre-formed oligomers were attached to the amine end groups of the silane (Figure 2). Untreated OX50 particles were first dried in a rotary dryer at about 165 °C under vacuum for 3 h to remove surface bound moisture that can potentially react with the silanes and promote the formation of multiple layers of the silane. Anhydrous toluene was used as a solvent for the silanization process. It was refluxed for 2 h and then the particles were added and further refluxed for an hour to ensure good dispersion and removal of any remaining moisture. The MAPTS was added to the solution so that the final concentration in the mixture was 1.5 % by weight. This reaction mixture was allowed to reflux for 6 h after which the particles were separated from the solvent by centrifugation. The particles were washed three times with toluene and two times with methylene chloride to ensure that unreacted silane was removed. The silanized particles were then dried overnight under vacuum at 90 °C to remove all traces of solvent. The dried and silanized particles were then added to anhydrous DMF, which contained a five-fold excess of the modified epoxy oligomers to minimize potential head and tail surface coupling. The reaction mixture was heated to 45 °C and the reaction was allowed to continue for 24 h. The particles were separated and isolated by following a similar centrifugation and washing procedure as described above.
Figure 2.

Attachment of oligomers to silica particle surface – OX50 particles were silanized in dry conditions with MAPTS. MAPTS was further reacted with modified epoxy oligomers to yield particles with oligomers on the surface.
2.4. Composite Formulation
The resin matrix for the composites was a mixture of BisGMA and TEGDMA in a 70:30 mass ratio. The resin included a free radical visible light initiating system comprising 0.3 wt% camphorquinone and 0.8 wt% ethyl 4-N,N-dimethylaminobenzoate. All fillers were added to the resin and homogeneously dispersed with a centrifugal speedmixer (DAC 150 FVZ, Flakteck Inc.). All composite pastes had a final filler loading level of 30 wt%, based on the inorganic mass of the fillers. Composites with γ-MPS-treated OX50 particles were used as the control. The freshly prepared composites were allowed to sit under vacuum to ensure elimination of air bubbles prior to use.
2.5. Characterization of fillers
Thermogravimetric analysis (pyris 7 TGA, Perkin Elmer) was used to determine the amount of surface functionalization on the OX50 particles after each stage of modification. Samples were placed in platinum pans in a nitrogen atmosphere and heated from 50 °C to 850 °C at 10 °C/min while the mass loss as a function of temperature was recorded.
2.6. Shrinkage stress measurements
Shrinkage stress was measured using a cantilever beam-based instrument referred to as a tensometer (American Dental Association, Volpe Research Center, MD, USA). This instrument measures the force generated by an actively polymerizing material that is partially constrained from a free shrinkage state. The material is bonded to the opposing ends of two diametrically placed quartz rods. The polished ends of the glass rods are freshly treated with γ-MPS, to promote bonding with the polymerizing material. The upper quartz rod is attached to the cantilever beam and can move along with the shrinking composite. The lower quartz rod is fixed to the tensometer base and provides the shrinkage constraint. A dental curing light (IQ2, Caulk, Dentsply), with an output wavelength maximum of 465 nm, was adapted to the bottom of the lower quartz rod, which serves as a light guide to transmit the curing light to the material. A detailed description of the stress measurement involving the tensometer has been published previously [39]. The testing protocol uses specimen discs of 1.5 mm thickness and 6 mm diameter. For the initial shrinkage stress measurements, the light intensity reaching the sample was measured at 350 mW/cm2. The samples were irradiated for 40 s with the dynamic stress development profile continuously monitored for 20 min. Real-time double bond conversion measurements based on simultaneous near-IR spectroscopy were made during the stress measurements. The samples were irradiated for 40 s and measurements were taken continuously for 20 min.
2.7. Shrinkage measurements
Polymerization shrinkage was measured using a linometer (ACTA Foundation, Netherlands). In this method, composite paste specimens were placed between an aluminum disc and a glass slide that were greased to allow the free shrinkage of the sample along the radial direction. Specimens were 1.5 mm in thickness and 6 mm in diameter. The sample was irradiated from the top of the glass slide (350 mW/cm2 for 40 s). The displacement of the freely moving aluminum disc was measured by linear variable differential transformer (LVDT) and converted to a dynamic volumetric shrinkage plot that extended to 10 min to capture the initial post-cure shrinkage behavior.
2.8. Flexural modulus and strength measurements
Flexural modulus and ultimate strength were measured in a 3-point bending mode (858 Mini Bionix II, MTS Systems, Eden Prairie, MA). The composite pastes were injected into a cavity measuring 2×2×10 mm. The mold cavity was formed by Teflon spacers on the sides and clamped glass slides on the top and bottom. The composite samples were cured using the dental curing light at 550 mW/cm2 for 3 min on both the sides to promote uniform conversion throughout the sample. The crosshead speed used was 1 mm/min.
2.9. Infrared spectroscopic characterization
Infrared spectroscopy was used to monitor the conversion of the resin and the surface modification of the fillers using an FTIR (Nexus 670, Nicolet Instruments, Madison, WI). Double bond conversion was monitored in transmission mode using near-infrared (NIR) spectroscopy. The area of the peak at 6165 cm−1 representing the methacrylate =CH2 absorption was used to track conversion. For the simultaneous measurement of conversion with shrinkage stress and also during rheometry measurements, the NIR signal from the source was transmitted through the sample and back to the detector using 1 mm diameter optical fibers. Diffuse reflectance mid-infrared Fourier transform spectroscopy (DRIFTS) was used to study the filler surfaces. For the DRIFTS experiments OX50 filler particles representing the various surface treatments, including the unmodified filler, were mixed with KBr powder and spectra were taken at a resolution of 4 cm−1, using KBr as a background.
3. Results
The attachment of the silane on the surface of the OX50 particles and the subsequent attachment of oligomers to the silane molecules on the surface was confirmed using DRIFTS (Figure 3). The deposition of the high molecular weight oligomer series (6100–10% and 6100–100%) is shown as an example. Unmodified, dried OX50 particles showed a sharp peak at about 3747 cm−1 representing free silanol groups on the silica surface. Upon silanization with MAPTS, this peak disappeared, indicating reaction of the surface silanols. A broad peak representing the N-H stretching appears around 3346 cm−1. The multiple peaks at 2871–2972 cm−1 correspond to the stretching mode of C-H in -CH, –CH2 and –CH3. When the epoxy oligomers were reacted with the MAPTS layer, a carbonyl peak appeared at about 1720 cm−1 and the C=C peak appeared at 1635 cm−1. These two peaks are more intense in the filler with the 6100–100% oligomer, confirming the presence of a greater number of methacrylate groups than the 6100–10% oligomer. The amount of surface coverage due to the epoxy oligomer attachment was determined by thermogravimetric analysis (TGA). Figure 4 represents the TGA curves for the various systems. The nomenclature of the brushes used here is as follows: OX50 Mw-10% or OX50 Mw-100%, where Mw represents the molecular weight of the brush and 10% or 100% represents the percentage of hydroxyl groups on the oligomer that were pre-reacted with IEM. It can be seen that there is an increase in the weight loss from unmodified OX50 particles to silanized particles and then a further increase for those modified with the epoxy brushes. There is an approximately 1 % mass loss difference between the unmodified and silanized particles. Technical data (Evonik) reveals that the OX50 particles have a surface silanol group density of 2.2 per nm2. Assuming that each silanol group reacts with a MAPTS molecule and using the molecular weight of the silane, the estimated weight loss should be about 3 %, which indicates that about a third of the silanol sites have reacted with the MAPTS. Using this data, the relative coverage of the different epoxy brushes on the surface were estimated. Table 1 gives the net weight loss of each of the epoxy brushes and fraction of the silane sites that are estimated to have reacted.
Figure 3 –

Diffuse reflectance spectra of OX50 particles – 1) Unmodified OX50 2) OX50 treated with MAPTS 3) OX50 with MAPTS treatment followed by addition of epoxy oligomer 6100–10% 4) OX50 with MAPTS treatment followed by addition of epoxy oligomer 6100–100%.
Figure 4 –

Thermogravimetric analysis of untreated OX50 and modified OX50 particles with heating at 10 °C/min to 800 °C. The weight loss is representative of the amount of organic material on the filler surface. The fraction of mass lost increases with increase in length of oligomer.
Table 1 –
Mass loss contribution on the filler and fraction of the surface silanol sites covered by the modified epoxy oligomer series.
| Sample | % Mass Loss | Fraction of surface silanol sites covered |
|---|---|---|
| OX50_1075–10% | 3.4 | 0.17 |
| OX50_1075_100% | 3.4 | 0.16 |
| OX50_4000–10% | 4.5 | 0.06 |
| OX50_4000–100% | 4.6 | 0.05 |
| OX50_6100_ 10% | 4.6 | 0.04 |
| OX50_6100–100% | 5.4 | 0.03 |
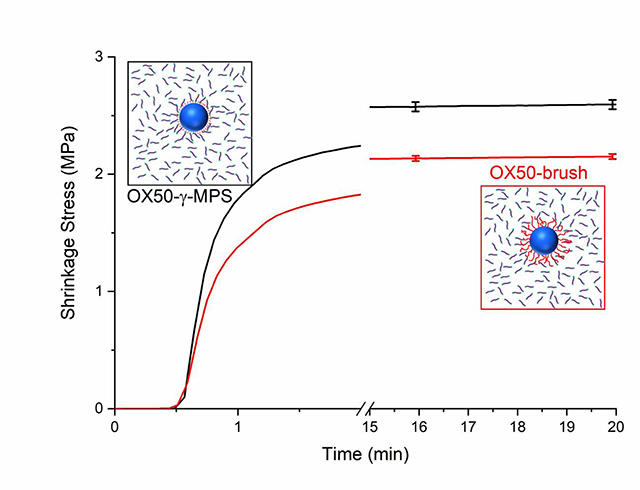
Overall, the experimental composite exhibited lower shrinkage stress upon polymerization as compared to the γ-MPS control, which had the highest shrinkage stress at 2.59 ± 0.05 MPa. The modified composites show a reduction in the shrinkage stress that follows the increase in the molecular weight of the epoxy oligomers with the OX50_1075–10% at 2.20 ± 0.02 MPa, OX50_4000–10% at 2.08 ± 0.04 MPa and OX50_6100–10% at 1.81 ± 0.11 MPa (Figure 5(a)). The same clear linear progression of the shrinkage stress with respect to the molecular weight of the oligomers is not observed for the series with all hydroxyl groups reacted with IEM. The OX50_4000–100% shows the least reduction in stress with the final stress at 2.36 ± 0.04 MPa, followed by OX50_1075–100% at 2.19 ± 0.03 MPa and OX50–6000-100% at 2.15 ± 0.02 MPa. The matrix double bond conversion of all the samples was around 65% without significant differences between different materials (Figure 5 (b)). Shrinkage measurements revealed no significant difference between any of the composite materials (Figure 5 (c)). The composites contracted by the same amount, about 5 vol%, under the standardized irradiation conditions. The rate of shrinkage development in the initial stages was found to be similar for all the composites. The conversion of all the composites at the end of the shrinkage run was also at similar levels.
Figure 5.


(a) Real time shrinkage stress measurements (b) Double bond conversion and (c) Volumetric shrinkage measurements of composites with 30 wt% OX50 particles with different surface treatments, as indicated.
Flexural modulus and strength of the experimental composites were similar to that of the control material (Figure 6).
Figure 6 -.

(a) Flexural strength and (b) flexural modulus of composites with OX50 modified with different surface treatments.
4. Discussion
The surface of OX50 silica nanofillers was treated with oligomers of varying lengths and concentration of functional groups to create an alternate surface treatment methodology for use in polymeric dental restorative materials. The oligomeric series was chosen such that their structures were similar to BisGMA, which is the major component of the model resin system. The structural similarity was expected to enhance the compatibility of the oligomers with the resin system, allowing for good wetting of the particle surface with the resin monomers. The range of molecular weights chosen for the oligomers allowed study of the effect of length of the polymer brush on the properties of the composite. The epoxy oligomers had pendant hydroxyl groups along their length that afforded a controllable and convenient way to introduce reactive methacrylate groups along the length of the oligomers. The reactive groups are necessary in order for covalent bonding to occur between the interface and the resin, thus completing the coupling between the filler and the matrix[19,30,40]. A covalent attachment usually results in enhanced bulk mechanical properties in a composite, primarily in flexure or tension rather than compression. Hence it is viewed as necessary to maintain at least some degree of reactivity at the surface of the fillers. For this reason, the hydroxyl groups on the oligomer backbone were reacted with IEM to give a low level (10% of total hydroxyl groups) and 100% (all hydroxyl groups) of methacrylate functionality along the length of the oligomer. It should be mentioned that for the low molecular weight oligomers, since there are fewer hydroxyl groups on the shorter brush backbones, there will be oligomer chains without any methacrylate functionality while some may have all the hydroxyl groups reacted with IEM. Shrinkage stress development in polymerizing composites is a complicated process, with dependence on factors such as resin matrix conversion, shrinkage, filler loading level, viscoelastic properties, modulus of the composite and kinetics[41,42]. Unlike the comparisons drawn in the dental materials literature involving polymerization stress of different commercial composite formulations, here most of these factors like filler loading, resin formulation and photoinitiator type and amount were held constant with the focus on the filler/resin interface. Factors within each measurement technique such as sample geometry, irradiation conditions and filler loading level were also kept constant for all the samples. The only difference between the samples was the surface treatment of the fillers. By maintaining the other factors constant, we could better elucidate the effect of the surface treatment on the properties of the dental composites. It should be noted that the polymer brush treatment resulted in a surface with low grafting density, due to the grafting-to nature of the functionalization process. This is viewed as critical to provide a more random coil orientation rather than the extended brush structure that would accompany a dense surface attachment configuration. As the oligomers synthesized are of a relatively low molecular weight and at low concentration, the brush layer, which is also infiltrated with the resin matrix, does not contribute a significant difference to the organic volume within the composite as compared with conventionally silane-treated filler.
Overall, significant decrease in the shrinkage stress of the experimental composites was observed in comparison with the γ-MPS-based control, while the degree of conversion and shrinkage as well as flexural modulus and strength values were maintained. In general, shrinkage stress reduction of the composites was influenced by both the length of the oligomer and the amount of reactive functional groups along the length of the oligomer. Shrinkage stress was found to be inversely proportional to the length of the oligomer attached to the surface and, for a particular length of oligomer, the shrinkage stress was proportional to the number of reactive groups along the oligomer. For the control particles, there is a very high concentration of short, surface-bound methacrylate groups due to the γ-MPS silane (Table 1). During polymerization, the methacrylate groups in the resin phase copolymerize with these surface methacrylate groups. As the dense, surface-bound groups are relatively immobile [26,43] and there is a shrinkage-related force that tends to pull away from the surface towards the bulk, stress is built up at the interface between the particles and the resin matrix [44–46]. The amount of stress can be high as the surface area of the nanofillers used here is very large, leading to development of high shrinkage stress for the bulk material. The hypothesis behind the current work was that oligomeric brushes on the particle surface, instead of the more compact and rigid gamma-MPS layer, can create a compliant interface that increases mobility and reduces shear between the high modulus fillers and the lower modulus polymer phase and hence reduces the accumulation of interfacial stress. Increasing the length of the oligomer should have the effect of increasing the interfacial compliance, potentially leading to a lower shrinkage stress.
The number and concentration of functional groups on the surface also had a significant effect on shrinkage stress. The greater the number of methacrylate groups on brush oligomers of a given length, the greater the shrinkage stress. As the functional group concentration increases, the connection points via covalent bonds between the resin matrix and the oligomers increase, thus increasing the restriction of compliance within the developing polymeric resin matrix and likely leading to less stress reduction. As demonstrated, the final stress value is dependent on the balance between the brush chain length and the reactive group density. Comparing Figures 6(a) and 6(b), the 6100 and 4000 molecular weight oligomers show stress increases as the methacrylate functionality on the chains increases. In the case of the 1075 molecular weight oligomer with an average of two hydroxyl groups per chain, the stress for both the high and low reactive group composites are similar most likely due to the relatively limited compliance that can be accommodated; although, these stress values are still significantly less than that available with the more rigid γ-MPS control material. A point to note is that filler particles with no surface treatment are known to result in significant stress reduction when added to a resin matrix, as compared to functionalized particles [47]. However, as there is loss in mechanical properties due to interfacial defects in these composites, untreated fillers have not been considered here as an alternate control material.
Stress reduction can result from a reduction in either shrinkage or modulus due to reduction in conversion or other factors such as effective crosslink density of the polymer network [41]. It was verified that similar levels of conversion (Figure 5(b)), final shrinkage (Figure 5(c)) and modulus (Figure 6(b)) were achieved with all the composite materials. A plot of real-time shrinkage stress versus conversion for the composites is shown in Figure 7. Only composites with the 10 % reactive group modification on the polymer brush structures are compared with the γ-MPS control here. In all cases, the shrinkage stress begins to develop gradually beyond the gel point but there are subtle differences between the control and the experimental composites evident over the intermediate conversion range with stress development in the γ-MPS control increasing at a higher rate. As vitrification is approached, a precipitous increase in the stress values with respect to small changes in the conversion is observed in all the composites. Vitrification is reached at a significantly earlier stage of conversion for the control, which also reaches a higher value of final stress. A delay in the vitrification could be due to the enhanced mobility afforded by the flexible oligomer layer in the brush-modified composites.
Figure 7.

Shrinkage stress plotted as a function of double bond conversion. The γ-MPS control exhibits an earlier exponential rise in stress resulting in a higher ultimate stress as compared to the polymer brush-based composites.
Viscosity of all the composite pastes was measured to study the effect the particles have on the rheological properties. The lowest viscosity is shown by the γ-MPS control (Figure 8). All the experimental composites exhibit higher and length-dependent viscosity levels, which indicates a wider sphere of influence around the particles due to the nature of the surface-bound oligomers that would also apply in the final polymeric state. Within each oligomeric molecular weight, the 10 % reactive group versions display higher viscosity than their 100 % functionalized counterparts. This is most likely due to the presence of the residual hydroxyl groups along the pendant chains in the 10 % functional brushes that interact with the BisGMA component of the resin. Higher viscosity composites would be expected to have an earlier gelation and consequently an earlier stress development with higher final stress. However, the opposite trend is observed here where higher viscosity composites have a lower shrinkage stress but equivalent modulus compared with the lower viscosity composites. This is likely due to a delay in effective transfer of stress between the matrix and filler as conversion progresses since potentially multiple reactions of the brush into the resin matrix are required to fix its position in the polymeric network. The aspect of a physically entangled as well as covalently reacted brush structure within the polymer matrix could also provide a degree of reinforcement and toughness along with its stress relief potential.
Figure 8.

Viscosity of composite pastes with 30 wt% OX50 filler loading. Viscosity increases with increase in length of oligomer on the filler surface.
SEM images of cryogenically fractured photopolymerized composite bars were obtained to get information on the morphological effects due to the filler surface treatments. The fractured surface of the γ-MPS based control and the 4000–100% based composite sample (Figure 9) demonstrate that the experimental composite has a much rougher topography. This implies that in the experimental composites, the propagating crack follows a more tortuous path and dissipates more energy while doing so. This would potentially lead to an increase in the fracture toughness and possibly wear resistance of the composites but those factors were not tested here.
Figure 9.

SEM micrograph of cryogenically fractured composite surface (a) OX50-γMPS control and (b) OX50–4000-100% sample. (Scale bar – 1 μm).
5. Conclusions
A novel surface modification technique was employed in order to improve polymerization shrinkage stress in dental composites. Oligomers with different surface-tethered brush chain lengths and different levels of reactivity were used to modify nanofillers in a dimethacrylate resin matrix. The results show that the experimental composites have reduced shrinkage stress by as much as 30 % as compared with the traditionally used γ-MPS-based control while otherwise maintaining similar final conversion, shrinkage and modulus behavior. The surface modification procedure also has the potential to affect other properties such as dynamic modulus development and fracture toughness. It can be concluded that the surface modification technique used here has the potential to improve dental composites in terms of the critical property of polymerization stress development and it highlights potential benefits associated with an intentionally designed interphase rather than a sharp interface in composite materials.
Highlights.
Polymer brushes can be used for surface modification of filler particles to reduce polymerization shrinkage stress compared to a silane used for conventional surface modification
Level of shrinkage stress reduction is proportional to length of oligomers and inversely proportional to number of reactive groups along the oligomers
Reduce shrinkage stress is due to delayed development of stress with respect to double bond conversion development
Acknowledgements
Funding for this project was provided by NIH/NIDCR R01DE023197 and R21DE028444. The authors declare no potential conflicts of interest with respect to authorship and/or publication of this article.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Bowen RL, Marjenhoff WA. Dental Composites/Glass Ionomers: the Materials. Adv Dent Res 1992;6:44–9. 10.1177/08959374920060011601. [DOI] [PubMed] [Google Scholar]
- [2].Lovell LG, Berchtold K a., Elliott JE, Lu H, Bowman CN. Understanding the kinetics and network formation of dimethacrylate dental resins. Polym Adv Technol 2001;12:335–45. 10.1002/pat.115. [DOI] [Google Scholar]
- [3].Anusavice KJ. Phillips’ Science of Dental Materials. 11th ed. Elsevier; 2003. [Google Scholar]
- [4].Sadowsky SJ. An overview of treatment considerations for esthetic restorations: a review of the literature. J Prosthet Dent 2006;96:433–42. 10.1016/j.prosdent.2006.09.018. [DOI] [PubMed] [Google Scholar]
- [5].Davidson CL, Feilzer AJ. Polymerization shrinkage and shrinkage stress in polymer-based restoratives. J Dent 1997;25:435–40. [DOI] [PubMed] [Google Scholar]
- [6].Rosin M, Urban AD, Gärtner C, Bernhardt O, Splieth C, Meyer G. Polymerization shrinkage-strain and microleakage in dentin-bordered cavities of chemically and light-cured restorative materials. Dent Mater 2002;18:521–8. [DOI] [PubMed] [Google Scholar]
- [7].Bowman CN, Cramer NB, Sansbury JW. Recent Advances and Developments in Composite Dental Restorative Materials. J Dent Res 2010;90:402–16. 10.1177/0022034510381263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ge J, Trujillo M, Stansbury J. Synthesis and photopolymerization of low shrinkage methacrylate monomers containing bulky substituent groups. Dent Mater 2005;21:1163–9. 10.1016/j.dental.2005.02.002. [DOI] [PubMed] [Google Scholar]
- [9].Weinmann W, Thalacker C, Guggenberger R. Siloranes in dental composites. Dent Mater 2005;21:68–74. 10.1016/j.dental.2004.10.007. [DOI] [PubMed] [Google Scholar]
- [10].Boulden JE, Cramer NB, Schreck KM, Couch CL, Bracho-Troconis C, Stansbury JW, et al. Thiol-ene-methacrylate composites as dental restorative materials. Dent Mater 2011;27:267–72. 10.1016/j.dental.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cramer NB, Couch CL, Schreck KM, Carioscia JA, Boulden JE, Stansbury JW, et al. Investigation of thiol-ene and thiol-ene-methacrylate based resins as dental restorative materials. Dent Mater 2010;26:21–8. 10.1016/j.dental.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Song HB, Sowan N, Shah PK, Baranek A, Flores A, Stansbury JW, et al. Reduced shrinkage stress via photo-initiated copper(I)-catalyzed cycloaddition polymerizations of azide-alkyne resins. Dent Mater 2016;32. 10.1016/j.dental.2016.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Podgorski M, Becka E, Claudino M, Flores A, Shah PK, Stansbury JW, et al. Ester-free thiol-ene dental restoratives - Part A: Resin development. Dent Mater 2015;31. 10.1016/j.dental.2015.08.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu J, Cao D, Zhang L. Static and dynamic properties of model elastomer with various cross-linking densities: a molecular dynamics study. J Chem Phys 2009;131:034903. 10.1063/1.3179691. [DOI] [PubMed] [Google Scholar]
- [15].Moraes RR, Garcia JW, Barros MD, Lewis SH, Pfeifer CS, Liu J, et al. Control of polymerization shrinkage and stress in nanogel-modified monomer and composite materials. Dent Mater 2012;27:509–19. 10.1016/j.dental.2011.01.006.Control. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Park HY, Kloxin CJ, Scott TF, Bowman CN. Covalent adaptable networks as dental restorative resins: Stress relaxation by addition-fragmentation chain transfer in allyl sulfide-containing resins. Dent Mater 2010;26:1010–6. 10.1016/j.dental.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schoerpf S, Catel Y, Moszner N, Gorsche C, Liska R. Enhanced reduction of polymerization-induced shrinkage stress: Via combination of radical ring opening and addition fragmentation chain transfer. Polym Chem 2019;10:1357–66. 10.1039/c8py01540f. [DOI] [Google Scholar]
- [18].Shah PK, Stansbury JW, Bowman CN. Application of an addition-fragmentation-chain transfer monomer in di(meth)acrylate network formation to reduce polymerization shrinkage stress. Polym Chem 2017;8:4339–51. 10.1039/c7py00702g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Antonucci JM, Dickens SH, Fowler BO. Chemistry of Silanes : Interfaces in Dental Polymers and Composites. J Res Natl Inst Stand Technol 2005;110:541–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bréchet Y, Cavaillé J-Y, Chabert E, Chazeau L, Dendievel R, Flandin L, et al. Polymer Based Nanocomposites: Effect of Filler-Filler and Filler-Matrix Interactions. Adv Eng Mater 2001;3:571. . [DOI] [Google Scholar]
- [21].Harada M, Morimoto M, Ochi M. Influence of network chain orientation on the mechanical property of epoxy resin filled with silica particles. J Appl Polym Sci 2003;87:787–94. 10.1002/app.11454. [DOI] [Google Scholar]
- [22].Lim B Effect of filler fraction and filler surface treatment on wear of microfilled composites. Dent Mater 2002;18:1–11. 10.1016/S0109-5641(00)00103-2. [DOI] [PubMed] [Google Scholar]
- [23].Nakamura S, Pavlovic E, Kramer E. Fracture Energy of Epoxy Interfaces with Layers of Different Silane Coupling Agents. J Adhes 2007;83:351–65. 10.1080/00218460701282372. [DOI] [Google Scholar]
- [24].Vallittu PK. Curing of a silane coupling agent and its effect on the transverse strength of autopolymerizing polymethylmethacrylate-glass fibre composite. J Oral Rehabil 1997;24:124–30. [DOI] [PubMed] [Google Scholar]
- [25].Lung CYK, Matinlinna JP. Aspects of silane coupling agents and surface conditioning in dentistry: an overview. Dent Mater 2012;28:467–77. 10.1016/j.dental.2012.02.009. [DOI] [PubMed] [Google Scholar]
- [26].Halvorson RH, Erickson RL, Davidson CL. The effect of filler and silane content on conversion of resin-based composite. Dent Mater 2003;19:327–33. [DOI] [PubMed] [Google Scholar]
- [27].Qiao B, Wang TJ, Gao H, Jin Y. High density silanization of nano-silica particles using γ-aminopropyltriethoxysilane (APTES). Appl Surf Sci 2015;351:646–54. 10.1016/j.apsusc.2015.05.174. [DOI] [Google Scholar]
- [28].Craig RG, Dootz ER. Effect of mixed silanes on the hydrolytic stability of composites. J Oral Rehabil 1996;23:751–6. [DOI] [PubMed] [Google Scholar]
- [29].Nihei T, Kurata S, Kondo Y, Umemoto K, Yoshino N, Teranaka T. Enhanced Hydrolytic Stability of Dental Composites by Use of Fluoroalkyltrimethoxysilanes. J Dent Res 2002;81:482–6. 10.1177/154405910208100710. [DOI] [PubMed] [Google Scholar]
- [30].Wilson KS, Zhang K, Antonucci JM. Systematic variation of interfacial phase reactivity in dental nanocomposites. Biomaterials 2005;26:5095–103. 10.1016/j.biomaterials.2005.01.008. [DOI] [PubMed] [Google Scholar]
- [31].Ye S, Azarnoush S, Smith IR, Cramer NB, Stansbury JW, Bowman CN. Using hyperbranched oligomer functionalized glass fillers to reduce shrinkage stress. Dent Mater 2012;28:1004–11. 10.1016/j.dental.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Faria-e-Silva AL, dos Santos A, Tang A, Girotto EM, Pfeifer CS. Effect of thiourethane filler surface functionalization on stress, conversion and mechanical properties of restorative dental composites. Dent Mater 2018;34:1351–8. 10.1016/j.dental.2018.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fronza BM, Rad IY, Shah PK, Barros MD, Giannini M, Stansbury JW. Nanogel-Based Filler-Matrix Interphase for Polymerization Stress Reduction. J Dent Res 2019;98:779–85. 10.1177/0022034519845843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sowan N, Cox LM, Shah PK, Song HB, Stansbury JW, Bowman CN. Dynamic Covalent Chemistry at Interfaces : Development of Tougher, Healable Composites through Stress Relaxation at the Resin – Silica Nanoparticles Interface 2018;1800511:1–10. 10.1002/admi.201800511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Brochard-Wyart F, De Gennes PG , Leger L, Marciano Y, Raphael E. Adhesion promoters. J Phys Chem 1994;98:9405–10. 10.1021/j100089a009. [DOI] [Google Scholar]
- [36].Zhao B Polymer brushes: surface-immobilized macromolecules. Prog Polym Sci 2000;25:677–710. 10.1016/S0079-6700(00)00012-5. [DOI] [Google Scholar]
- [37].Gutowski WS, Li S, Filippou C, Hoobin P, Petinakis S . Interface/interphase engineering of polymers for adhesion enhancement: Part II. Theoretical and technological aspects of surface-engineered interphase-interface systems for adhesion enhancement. J Adhes 2003;79:483–519. 10.1080/00218460309562. [DOI] [Google Scholar]
- [38].Julthongpiput D, LeMieux M, Tsukruk V. Micromechanical properties of glassy and rubbery polymer brush layers as probed by atomic force microscopy. Polymer (Guildf) 2003;44:4557–62. 10.1016/S0032-3861(03)00404-X. [DOI] [Google Scholar]
- [39].Lu H, Stansbury JW, Dickens SH, Eichmiller FC, Bowman CN. Probing the origins and control of shrinkage stress in dental resin-composites: I. Shrinkage stress characterization technique. J Mater Sci Mater Med 2004;15:1097–103. 10.1023/B:JMSM.0000046391.07274.e6. [DOI] [PubMed] [Google Scholar]
- [40].Wilson KS, Antonucci JM. Interphase structure-property relationships in thermoset dimethacrylate nanocomposites. Dent Mater 2006;22:995–1001. 10.1016/j.dental.2005.11.022. [DOI] [PubMed] [Google Scholar]
- [41].Shah PK, Stansbury JW. Role of filler and functional group conversion in the evolution of properties in polymeric dental restoratives. Dent Mater 2014;30:586–93. 10.1016/j.dental.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Boaro LCC, Gonçalves F, Guimarães TC, Ferracane JL, Versluis A, Braga RR. Polymerization stress, shrinkage and elastic modulus of current low-shrinkage restorative composites. Dent Mater 2010;26:1144–50. 10.1016/j.dental.2010.08.003. [DOI] [PubMed] [Google Scholar]
- [43].Wilson KS, Allen AJ, Washburn NR, Antonucci JM. Interphase effects in dental nanocomposites investigated by small-angle neutron scattering. J Biomed Mater Res Part A 2006. 10.1002/jbm.a. [DOI] [PubMed] [Google Scholar]
- [44].Fu S, Feng X, Lauke B, Mai Y. Effects of particle size, particle/matrix interface adhesion and particle loading on mechanical properties of particulate–polymer composites. Compos Part B Eng 2008;39:933–61. 10.1016/j.compositesb.2008.01.002. [DOI] [Google Scholar]
- [45].Feng L, Suh BI, Shortall AC. Formation of gaps at the filler-resin interface induced by polymerization contraction stress: Gaps at the interface. Dent Mater 2010;26:719–29. 10.1016/j.dental.2010.03.004. [DOI] [PubMed] [Google Scholar]
- [46].Obaid N, Kortschot MT, Sain M. Understanding the stress relaxation behavior of polymers reinforced with short elastic fibers. Materials (Basel) 2017;10:1–15. 10.3390/ma10050472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Condon JR, Ferracane JL. Reduced polymerization stress through non-bonded nanofiller particles. Biomaterials 2002;23:3807–15. [DOI] [PubMed] [Google Scholar]