

Abstract
Engineered tissue models comprise a variety of multiplexed ensembles in which combinations of epithelial, stromal, and immune cells give rise to physiologic function. Engineering spatiotemporal control of cell-cell and cell-matrix interactions within these 3D multicellular tissues would represent a significant advance for tissue engineering. In this work, a new method, entitled CAMEO (Controlled Apoptosis in Multicellular tissues for Engineered Organogenesis) enables the non-invasive triggering of controlled apoptosis to eliminate genetically-engineered cells from a pre-established culture. Using this approach, the contribution of stromal cells to the phenotypic stability of primary human hepatocytes is examined. 3D hepatic microtissues, in which fibroblasts can enhance phenotypic stability and accelerate aggregation into spheroids, were found to rely only transiently on fibroblast interaction to support multiple axes of liver function, such as protein secretion and drug detoxification. Due to its modularity, CAMEO has the promise to be readily extendable to other applications that are tied to the complexity of 3D tissue biology, from understanding in vitro organoid models to building artificial tissue grafts.
Keywords: tissue engineering, biomedical applications, hydrogels
Table of Contents Entry
Like their native counterparts, bioengineered tissues often consist of multiple cell lineages to recapitulate complex physiology. Current tools are limited in resolving the dynamic contributions of multicellular interactions. Here, CAMEO (Controlled Apoptosis in Multicellular Tissues for Engineered Organogenesis) is presented as a new strategy to precisely manipulate tissue composition, which enables new insight into the complexity of 3D tissue biology.
Graphical Abstract
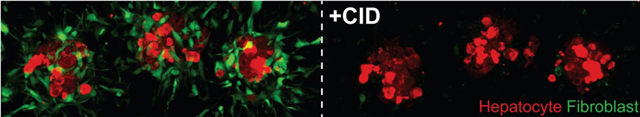
1. Introduction
Three-dimensional (3D) tissue engineered models have evolved to encompass a range of applications spanning therapeutic cell-based therapies to in vitro organoid models. In all use cases, recapitulation of physiologic functions and native tissue behavior is key to studying and harnessing complex, tissue-specific phenomena in normal and pathophysiological states.[1–7] Across these systems, it is experimentally difficult to decouple the dynamic contributions of homotypic and heterotypic cell-cell interactions, cell-matrix interactions, soluble bioactive factors, chemical cues, and mechanical stimuli. Many existing approaches were designed to decipher these interactions, including tunable hydrogel systems;[1,8] controlled manipulation via magnetic, fluidic, optical, electrical, mechanical, and intermolecular forces;[9,10] and time-lapse microscopy for tracking cell fate and cellular rearrangement.[11–13] However, the contribution of cell-cell interactions remains difficult to disentangle, especially in engineered tissues with high cellular diversity.
For the liver, the inclusion of hepatocytes, which comprise the parenchymal compartment, has been shown to be required to address numerous liver-specific synthetic and metabolic functions.[14] However, it is well known that primary hepatocytes rapidly lose viability and function upon isolation from their native microenvironment, thereby presenting an inherent challenge for creating bioengineered liver tissue. A body of work has established that the provision of heterotypic and homotypic cell-cell interactions are of particular importance in engineered livers, including 3D stem cell-derived and primary tissue-derived ensembles. Specifically, we and others have shown that co-culture with stromal cells derived from the liver or elsewhere can enhance phenotypic stability of primary hepatocytes in a biomaterial context, though whether this enhancement of hepatocyte function and longevity derives from ongoing, direct cell-cell interaction, or a more transient, even indirect mechanism is not comprehensively understood.[15–17] Multiple groups have demonstrated that the interactions between various species of hepatocytes (including human, rat, monkey, and dog) and 3T3-J2 murine fibroblasts significantly enhance the long-term phenotypic stability of hepatocytes in vitro and in vivo.[18–24] Of note, Hui et al. demonstrated that the maintenance of liver-specific phenotype and viability was dependent on transient contact-mediated signals, followed by sustained paracrine interactions with fibroblasts in a 2-dimensional (2D) format; these findings highlight that there are spatiotemporal nuances in the role that fibroblasts play in maintaining hepatocytes.[25,26] Furthermore, some evidence points toward contributions from cell-cell contacts and cell-secreted matrix molecules (e.g. T-cadherin, decorin, E-cadherin, liver-regulating protein).[19,27–29] Relatedly, we previously executed a high-throughput screen which identified stromal gene products that impact hepatocyte function in vitro.[30,31] However, these phenomena are poorly studied in 3D tissue structures since dynamic and precise manipulation of established complex, multicellular cultures is experimentally challenging.
In order to study the temporal role that fibroblasts play in maintaining liver-specific phenotype and function, we engineered a method to non-invasively trigger the removal of fibroblasts in a 3D multicellular context. In this study, we introduce a technique termed CAMEO (Controlled Apoptosis in Multicellular tissues for Engineered Organogenesis), in which a genetically-modified cell population can be induced to undergo apoptosis-driven elimination from a multicellular tissue construct. We implemented CAMEO and demonstrated non-invasive, rapid manipulation of established co-cultures in order to explore the phenotypic stability of both 2D and 3D liver models. Our efforts yielded 3D microlivers, from which fibroblasts were eliminated, that still phenocopy multiple liver functions in vitro. Besides their utility in building artificial grafts, we envision that access to the CAMEO method will also impact the field of organoid science, in which stromal feeder layers are conventionally used to promote stem cell renewal and maintenance.
2. Results and Discussion
2.1. Activation of suicide gene-expressing fibroblasts led to uniform elimination by apoptosis
We sought to design a cell line that could undergo quick, complete removal using a non-invasive trigger. We employed inducible caspase-9 (iCasp9), which is activated by treatment with a small molecule chemical inducer of dimerization (CID; also known as rapalog, an analog of rapamycin) and leads to subsequent cell death through the intrinsic apoptosis pathway (Figure 1a).[32–34] The safety and efficacy of the iCasp9 transgene and CID have previously been shown in vitro and in vivo in animals and humans.[32–34] We used a lentivirus to transduce 3T3-J2 murine fibroblasts (‘J2’), shown previously to support primary human hepatocyte function in both 2D and 3D platforms[15,35,36], with a bicistronic expression cassette encoding iCasp9 and GFP genes, and used FACS to enrich the infected population for the 15% highest-expressing GFP+ cells. By flow cytometry analysis, we confirmed that the GFP+ cell population appeared homogeneous for at least 7 passages, and the population remained >97% GFP+ even at passage 20 (Figure S1a,b of Supporting Information). After GFP+ J2 fibroblasts were exposed to CID, iCasp9 dimers were detected by staining with a caspase-9 antibody, confirming the expression of the bicistronic iCasp9-IRES-GFP vector (Figure 1b). Compared to wild-type J2s, iCasp9-GFP J2s underwent significantly increased caspase-9 cleavage at 15 (16-fold) and 30 (18-fold) minutes after CID dosing (Figure 1c). To confirm that CID-triggered caspase-9 activation led to apoptosis, unfixed cells were stained with Annexin V, which binds to an early indicator of apoptosis, and SYTOX, a general marker of cell death, and analyzed by flow cytometry. The proportion of iCasp9-GFP J2s undergoing apoptosis increased in a time-dependent manner within the first hour after CID treatment (Figure 1d). Lastly, we observed that CID-treated iCasp9-GFP J2s were efficiently removed from culture (Figure 1e) by 1 hour after exposure (Figure 1f, <1% by cell viability for ATP). Prior studies using similar inducible iCasp9-based switches have observed similar timeframes for rapid onset of apoptotic activity and subsequent cell death.[32,34,37] Taken together, these results demonstrate that an iCasp9-bearing population of J2s could be treated with CID to quickly and efficiently eliminate them from culture by activating the apoptotic pathway.
Figure 1. iCasp9-GFP J2s are activated by CID and uniformly eliminated by apoptosis.

J2 fibroblasts bearing an iCasp9 suicide gene were treated with CID to induce iCasp9 dimerization, leading to apoptosis and elimination of the cells from culture (a). CID-induced dimerization of iCasp9 unimers and was detected by immunofluorescence imaging (b, scale bar = 50 μm). CID induced activation of caspase-9 cleavage activity (c; ****p<0.0001 vs. time-matched, dose-matched J2s, n=3). CID-treated iCasp9-GFP J2s were stained with Annexin V and SYTOX and analyzed by flow cytometry to quantify the extent of apoptotic activity (d; 100,000 events). CID-treated iCasp9-GFP J2s were removed from culture by apoptosis (e, scale bar = 100 μm) in a quick and efficient manner at low concentrations of CID (f, n=3).
2.2. 2D micropatterned co-cultures depend on the sustained presence of stromal cells
Recapitulation of cues from the native hepatic microenvironment, including from cells, ECM, and soluble factors, has been found to lead to phenotypic rescue of primary hepatocytes as well as prolongation of longevity and function.[16,18,38] In our system, the incorporation of J2 fibroblasts enhanced phenotypic stability of hepatocytes.[25,35,36] To study this phenomenon, we previously engineered an actuatable 2D platform to enable the manipulation of established co-cultures. Using this platform, the dependency of hepatocytes on fibroblasts was interrogated in 2D; it was found that despite an initial priming phase of direct cell-cell contact with fibroblasts, primary human hepatocytes did not maintain phenotypic stability if fibroblast juxtacrine and paracrine support were both removed.[25] We hypothesized that CAMEO-driven removal of fibroblast support would also be disruptive to hepatocyte culture in 2D. As a first step to test this hypothesis, we cultured primary human hepatocytes and J2 fibroblasts in a micropatterned co-culture (MPCC), in which we corroborated our past findings[35] that hepatocyte phenotypic stability is enhanced by J2 co-culture (Figure 2a). iCasp9-GFP J2 and J2 fibroblasts both provided support of multiple axes of liver function, including synthesis of albumin protein (Figure 2b), production of urea as a byproduct of nitrogen metabolism (Figure 2c), and expression of drug metabolism-related enzymes (Figure S2a of Supporting Information), suggesting that genetic modification of J2s did not abrogate their capability to support hepatocytes. Throughout this work, we make use of the aforementioned readouts as a representative and stringent panel of readouts for the assessment of hepatocyte function.
Figure 2. 2D MPCC cultures depend on the sustained presence of stromal cells.

MPCCs or pure hepatocytes were assayed for albumin secretion rate (a, n=3). MPCCs containing wild-type and modified J2s were assayed for albumin secretion rate (b, n=6) and urea secretion rate (c, n=6). MPCCs comprised of primary human hepatocytes (brown) and iCasp9-GFP J2 or wild-type J2 fibroblasts (blue) were treated with CID to remove iCasp9-GFP J2s by apoptosis (d). Vehicle- or CID-treated MPCCs were stained and visualized by immunofluorescence imaging (e, scale bar = 250 μm). MPCCs were treated with CID at day 1, 3, or 7 after initiating co-culture and assayed for albumin secretion rate (f, n = 5, normalized to day 13, arrows indicate CID dose day).
We next sought to eliminate inducible apoptosis gene-bearing cells in a multicellular culture, by treating iCasp9-GFP J2-bearing MPCCs with CID (Figure 2d). CID-dosed MPCCs displayed selective removal of the iCasp9-GFP J2 population (Figure 2e, S2b of Supporting Information), whereas unmodified J2s plated in MPCCs were unaffected by CID exposure (data not shown). Furthermore, hepatocyte albumin production was not abrogated if MPCCs were cultured with conditioned ‘apoptotic’ medium, suggesting that at least one axis of liver-specific function was not affected by exposure to neighboring apoptotic cells (Figure S2c of Supporting Information). Altogether, these data suggest that CID-driven removal of stromal cells by apoptosis is a compatible system for probing phenotypic stability of hepatocytes in MPCCs.
To query the dependence of hepatocytes on fibroblasts with CAMEO, we deleted fibroblasts from MPCCs by CID treatment at various time points and assessed the albumin production rate as a surrogate marker of phenotypic stability. We observed that the deletion of stromal cells resulted in loss of hepatocyte phenotypic stability at early (day 1), intermediate (day 3), and late (day 7) time points (Figure 2f, Figure S2d of Supporting Information). Taken together, these data suggest that the function of primary human hepatocytes is heavily reliant on stromal support in this 2D MPCC configuration, which is consistent with our past 2D studies.[25,35]
2.3. iCasp9-expressing fibroblasts can be eliminated from 3D multicellular spheroid-laden hydrogels
In our prior work, we found that phenotypic stability and longevity of primary hepatocytes cultured as 3D microtissues were transiently supported by pre-aggregation to increase homotypic cell-cell interactions, and were further enhanced upon inclusion of J2 fibroblasts.[36] Thus, here we incorporate iCasp9-GFP J2 fibroblasts into these 3D hepatic ensembles, which were fabricated by plating primary human hepatocytes and fibroblasts in microwells in order to facilitate physical cell-cell contacts, as previously described.[20,21] Optimal overnight aggregation into stable spheroids was achieved by increasing the amount of fibroblasts co-seeded in the microwells (Figure S3a,b of Supporting Information). Resulting spheroids were encapsulated in a 10mg/mL fibrin hydrogel (crosslinked with 1.25 U/ml thrombin). Fibroblast co-culture, which provided supportive cell-cell interactions and increased aggregation stability, significantly improved the rate of primary human hepatocyte albumin secretion from the ensembles (Figure 3a). Spheroid-laden hydrogels containing either J2s or iCasp9-GFP J2s both exhibited enhanced synthetic (albumin production; Figure 3b), metabolic (nitrogen metabolism; Figure 3c), and detoxification (CYP3A4 activity; Figure 3d) functions of hepatocytes.
Figure 3. Fibroblasts are not required to maintain hepatocyte function in 3D spheroid-laden cultures.

3D cultures consisting of pure hepatocytes or hepatocytes and fibroblasts were assayed for albumin secretion rate (a, n=9). J2s and iCasp9-GFP J2s were co-cultured with hepatocytes in spheroid-laden hydrogels and assayed for albumin secretion rate (b, n=6), nitrogen metabolism (c, n=6), and CYP3A4 expression (d, n=6, **p<0.001). Hepatocytes were aggregated with fibroblasts in microwell molds and treated with CID to remove fibroblasts via apoptosis (e). Spheroid-laden hydrogels were treated with vehicle or CID and imaged to assess the robustness of fibroblast elimination (f, scale bar = 100 μm). Spheroid-laden hydrogels were dosed with CID on day 1 after co-culture initiation and albumin secretion rate was assayed for 3 weeks of culture (g, n=9). Fibroblast-depleted (CID) and fibroblast-intact (vehicle) cultures were treated with rifampin for 72 hours and assayed for induction of CYP3A4 activity (h, n=8–10).
We next sought to confirm the effectiveness of CID treatment for elimination of iCasp9-GFP fibroblasts embedded in a hydrogel. Encapsulated iCasp9-GFP fibroblasts were treated with CID, and fibroblast viability was undetectable after 6 hours (Figure S3c of Supporting Information). Furthermore, fibroblasts did not regrow over the course of 21 days (Figure S3d of Supporting Information). Next, to assess the specificity of CAMEO in 3D, we cultured hydrogel-encapsulated, multicellular spheroids (in which iCasp9-bearing fibroblasts are placed in close proximity to hepatocytes) and treated the ensembles with CID in an attempt to specifically eliminate iCasp9-GFP fibroblasts (Figure 3e). In these spheroid-laden hydrogel cultures, CID was able to access iCasp9-GFP J2s, leading to their robust and specific deletion throughout the hydrogel, without any apparent toxicity to co-cultured hepatocytes (Movie S1). These results suggest that CAMEO can be employed by dosing embedded co-cultures with CID to trigger the removal of inducible apoptosis gene-bearing cells in 3D multicellular ensembles.
2.4. Fibroblasts are not required to maintain hepatocyte function in 3D spheroid-laden cultures
To probe the dependence of hepatocyte phenotypic stability on fibroblast co-culture in 3D, we deleted fibroblasts from spheroid-laden hydrogel cultures using CAMEO after 1 day of hepatocyte-fibroblast co-culture. Fibroblasts were robustly removed from 3D co-cultures, as detected by immunofluorescence imaging (Figure 3f). While 2D MPCC cultures were found to be dependent on fibroblast interactions for the extent of our experiment (1.5 weeks, Figure 2f), fibroblast-depleted 3D cultures exhibited stable phenotype, as detected by albumin secretion rate, for up to 3 weeks (Figure 3g). Furthermore, fibroblast-depleted and fibroblast-intact cultures underwent similar induction of CYP3A4 activity in response to rifampin treatment (Figure 3h). Notably, when CID-triggered iCasp9-GFP J2 deletion was delayed until later time points (after 3 or 7 days of hepatocyte-fibroblast co-culture), hepatocyte function was negatively impacted, suggesting that primary hepatocytes cultured with J2s and embedded in fibrin are sensitive to deletion kinetics (Figure S3e of Supporting information). Taken together, these findings, enabled by CAMEO, demonstrate that there is a window of opportunity for fibroblast deletion in this particular tissue engineered context.
2.4. Discussion
In this work, we leverage an inducible apoptosis switch to study the temporal role of fibroblasts in the maintenance of phenotypic stability of hepatic tissue engineered models. Specifically, we engineered a mouse fibroblast line bearing a caspase-9-driven apoptotic switch to query the temporal dependence of primary human hepatocytes on stromal support. Previously, inducible caspase-9 has been deployed as a safety measure for engineered cell therapies (e.g. adoptive T cell therapy, engineered cell therapy, mesenchymal cell therapy) across a range of applications (e.g. regeneration, anti-cancer).[32,32,34] Recently, inducible apoptosis gene-engineered stromal cells were used to probe the contribution of cancer-associated fibroblasts to metastatic potential in vivo.[39] In this work, we demonstrate a new use case for inducible apoptosis genes as tools for tissue engineering. Here, we show that iCasp9-bearing fibroblasts are amenable to quick, efficient, and robust removal (Figure 1). We show that iCasp9-bearing cells can be specifically and efficiently removed from 2D co-cultures and 3D multicellular ensembles embedded in a porous hydrogel. Using CAMEO, we demonstrate that fibroblasts enable significant enhancement of the phenotypic stability of hepatocytes in 2D MPCCs (Figure 2), which is consistent with our previous findings in a related 2D model. Lastly, we used CAMEO to query the impact of fibroblast co-culture on hepatocytes in 3D spheroid-laden hydrogels, and found that supporting fibroblasts could be removed at 24 hours after initial stabilization without abrogating maintenance of liver-specific function (Figure 3).
In our prior work, in which an actuatable micromechanical comb system enabled precise manipulation of the proximity of two cell populations, hepatocytes were found to lose phenotypic stability after initial cell-cell contact priming if fibroblasts were subsequently removed from the culture.[25] Similarly, in this work, we employ CAMEO to show that the removal of fibroblasts from 2D MPCCs through CID-triggered apoptosis leads to the loss of hepatocyte maintenance (Figure 2). Together, these results suggest that proximity to fibroblasts is required in 2D hepatocyte-fibroblast co-cultures. We also employed CAMEO to study 3D hepatic cultures, and demonstrated that fibroblasts were dispensable for the maintenance of hepatocytes after 24 hours of cell-cell contact (Figure 3). In pursuit of an explanation for these differences and their potential contribution to hepatocyte function, we focused on specific changes in cell-matrix and cell-cell adhesions between 2D and 3D contexts. In particular, we perturbed candidate interactions within the initial 24-hour window of co-culture to evaluate the contribution to hepatocyte phenotypic stability in 3D.
After initial rescue with stromal support, it has been shown that presentation of certain supportive cues can enhance hepatocyte phenotypic stability.[25,29] In our 3D spheroid-laden fibrin hydrogel cultures, apoptotic debris and fibroblast-derived paracrine factors and matrix are trapped in the hydrogel after CID-triggered deletion of fibroblasts, which may constitute a persisting supportive milieu for the hepatocytes. To probe the dependence on trapped factors from the fibroblast population, we instead dosed co-cultures with CID prior to encapsulation in fibrin hydrogels and removed apoptotic debris and conditioned supernatant by differential centrifugation (Figure S4a of Supporting Information). We did not observe any significant differences in albumin secretion between the CID pre-encapsulation condition (i.e. hepatocyte-fibroblast spheroids dosed with CID prior to encapsulation), and the CID post-encapsulation conditions (i.e. hepatocyte-fibroblast spheroids dosed after encapsulation), suggesting that apoptotic debris and other retained fibroblast-derived factors were not the main drivers of hepatocyte phenotypic stability (Figure S4b of Supporting Information).
Additionally, we reduced the retention of fibroblast-secreted proteins by encapsulating hepatic spheroids (containing primary human hepatocytes and iCasp9-GFP fibroblasts) in alginate, which, compared to fibrin, is non-adhesive to cells and has a greatly reduced intrinsic binding capacity for paracrine factors, due to the lack of a promiscuous protein-binding domain (Figure S4a of Supporting Information).[40,41] Spheroid-laden alginate beads were dosed with CID and cultured for a 2 week period. We assessed the secretion of human albumin as a proxy for hepatic function over two weeks, and observed no significant differences between untreated and CID-treated cultures (Figure S4c of Supporting Information). Taken together, these results suggest that the maintenance of functional hepatocytes in 3D after elimination of fibroblasts is unlikely to be driven by the retention of either apoptotic bodies or of matrix-bound factors.
Given that the retention of sequestered paracrine factors and apoptotic bodies were not found to be critical cues for driving hepatocyte phenotypic stability in 3D, we then focused on specific cell-cell and cell-matrix adhesive factors that could be at play. Previous studies in monocultures formats suggest that E-cadherin-mediated cell-cell interactions and integrin β1-mediated cell-matrix interactions significantly contribute to hepatocyte phenotypic stability.[42,43] In our 3D co-culture, we hypothesized that homotypic cell-cell interactions (such as E-cadherin engagement) and cell-matrix integrations (which require β1 integrin) were promoted by the inclusion of fibroblasts via compaction and deposition of matrix components (Figure S5a of Supporting Information). To perturb these interactions, we incubated function-blocking monoclonal antibodies against human β1 integrin or human E-cadherin either with primary human hepatocytes before compaction (i.e. before co-culturing with fibroblasts), or after compaction and before encapsulation (Figure S5b of Supporting Information). We found that transient, pre-compaction functional blockade of β1 integrin or E-cadherin significantly reduced albumin secretion at later time points, which we measured as a proxy for hepatocyte function (Figure S5c of Supporting Information). When we blocked β1 integrin or E-cadherin post-compaction, hepatocyte function was slightly reduced but largely preserved, as compared to pre-compaction inhibition (Figure S5c of Supporting Information). Taken together, these results suggest that adhesion via E-cadherin and β1 integrin were crucial for promoting longer-term hepatocyte phenotypic stability in a 3D hydrogel-laden co-culture, and that even fleeting fibroblast co-culture is sufficient to establish these stabilizing effects early in culture. Importantly, it has been shown that cadherin and integrin-based interactions dramatically influence cellular behavior and interactions in a 3D niche,[44] which may explain why stromal cell removal from 3D hepatic cultures (Figure 3) was more readily compensated for than in a similar 2D platform (Figure 2).
Interestingly, we observed a catastrophic effect on primary hepatocyte phenotypic stability at later time points of fibroblast removal from 3D cultures (Figure S3e of Supporting Information). It is possible that irregular apoptotic activity was linked to a pathophysiological process, which has been previously described in fetal and adult liver to be implicated in phenotypic dysfunction, tumorigenesis, and fibrosis/cirrhosis.[45] Poor hepatocyte function at later stages may also be due to a bystander injury effect caused by apoptosis of target cells. In the oncology field, herpes simplex virus thymidine kinase (HSV-TK) inducible suicide gene therapy leads to a pronounced killing of neighboring cells via the “bystander effect,” which refers to the gap junction channel-related transfer of toxic ganciclovir metabolites after TK-mediated conversion.[46–48] While the bystander effect has been noted to be less relevant to suicide gene therapies driven by iCasp9,[49] it has not yet been studied whether activated intermediates from apoptotic cells can induce apoptosis following transfer via gap junction channels or uptake by neighboring cells. Additionally, it has been shown in silico and in vitro that heterotypic hepatocyte-fibroblast gap junctions are unable to form and support intercellular communication.[50] Taken together, we do not expect to observe an iCasp9-specific bystander effect due to gap junction communication and transfer of apoptotic intermediates.
In future work, we will leverage CAMEO to more closely examine the contribution of stromal cells to the phenotypic stability of hepatocytes in a variety of multicellular, tissue engineered platforms for the study of human liver biology and development of transplantable implants. From a clinical perspective, the inclusion of stroma in a co-culture can be limiting, due to the additional complexity, as well as the potential to serve as a significant nutrient sink and transport barrier. Aside from enabling removal for practical and translational purposes, we envision that inducible apoptosis genes can also be deployed in other compartments of multicellular tissue ensembles in order to study interactions and their impact on phenotype and function. Importantly, eventual clinical application of multicellular engineered livers may require incorporation of non-parenchymal cell types such as Kupffer cells, stellate cells, liver sinusoidal endothelial cells, and cholangiocytes to include an immune component, build a perfusable vascular network, and grow a biliary tree, respectively. CAMEO offers a pre-clinical tool that will enable the precise, systematic removal of individual cell types from such complex multicellular ensembles with temporal control. Furthermore, we envision that orthogonal inducible apoptosis genes could be designed to enable a multiplexed evolution of the CAMEO platform. Lastly, because the administration of CID has been previously demonstrated to be safe and efficacious in both humans and animals and because there are established clinical frameworks in development that may enable cell therapies that include xenogeneic components,[32–34,51] we posit that CAMEO may be useful for dissecting the role of various cell populations in implant integration and persistence in the host.
In addition to the primary hepatocytes discussed in our work, hiPSC, hESC, and epithelial cells are also used to build organoid models to study human biology. The growth and maintenance of these cells in vitro has traditionally relied on stromal cell-based feeder layer culture.[52] Feeder layers commonly consist of xenogeneic stromal cells (e.g. murine embryonic fibroblasts, murine 3T3 fibroblasts), which pose a significant translational challenge, thereby motivating the development of feeder-free and animal product-free strategies. Concerns with feeder layer cultures include overgrowth (i.e. limiting scale-up of tissue engineering by depleting nutrients and space), contamination of the target cell culture, and zoonosis.[1,52] Transfer of zoonotic pathogens and immunogenic components can also happen through the use of conditioned medium; Martin et al. demonstrated that mammalian sialic acid Neu5Gc from conditioned medium elicited an antibody response in humans, thereby limiting clinical use.[53] In the case of hESCs and hPSCs, acellular support strategies for cell culture consisting of defined growth factor cocktails and functionalized culture surfaces have been elucidated.[1,54] For most epithelial cells, the exact chemical and physical factors to create a perfect synthetic feeder substitute remain to be defined. Feeder cell support is thought to act through (a) growth factors, (b) detoxification of culture medium (e.g. removal of pro-apoptotic signals), (c) synthesis and provision of ECM proteins and/or (d) physical contact (e.g. mechanotransductive interactions, engagement of juxtacrine pathways). CAMEO may be useful as an additional degree of engineered control for dissecting these complex intercellular phenomena, which have been historically difficult to deconvolute.
3. Conclusions
In this work, we present a new tool, termed CAMEO, that has the potential to shed light upon biological phenomena driving cellular phenotype and function. Here we share a proof-of-concept use of CAMEO by leveraging inducible apoptosis switches to probe the temporal dependence of primary human hepatocytes on fibroblast co-culture in both 2D and 3D multicellular tissue engineered platforms. Unlike existing technologies from our lab and others that can manipulate cell-cell interactions only in 2D cultures, an inducible apoptosis switch-bearing cell population can be triggered in both 2D and 3D in vitro formats. We expect this new methodology to be readily incorporated into a variety of cell types, and envision vast utility in the investigation of multicellular microenvironments and dissection of requirements for phenotypic stability, network formation, in vivo host integration, and more.
4. Experimental Section
Cell Culture:
Primary cryopreserved human hepatocytes (Lot ZGF, 33-year-old, Caucasian, male; BioreclamationIVT) were maintained in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 g/L glucose (CellGro) containing 10% (v/v) fetal bovine serum (FBS) (Gibco), 1% (v/v) ITS supplement (insulin, transferrin, sodium selenite; BD Biosciences), glucagon (70 ng/mL), dexamethasone (0.04 μg/mL), 0.015 M HEPES, and 1% (v/v) penicillin-streptomycin (Invitrogen). 3T3-J2 murine fibroblasts were a kind gift provided by Howard Green (Harvard Medical School) and were cultured in DMEM with 4.5 g/L glucose, 10% bovine serum, and 1% (v/v) penicillin-streptomycin.
Micropatterned co-cultures (MPCC) were fabricated as described previously.[35,55] Briefly, collagen was adsorbed in each well of a 96 well plate (glass bottom), and then patterned using an elastomeric polydimethysiloxane mold and oxygen plasma gas ablation. Human hepatocytes were thawed and seeded (70k/well) on the collagen islands (500 μm with 1,200 μm center-to-center spacing). Adhered hepatocytes (~10k/well) were allowed to spread overnight before fibroblasts were seeded for co-culture (7k/well).
Hepatic spheroids were cultured as described previously.[20,21] In brief, cryopreserved human hepatocytes were thawed and immediately plated with fibroblasts in AggreWells (400 μm pyramidal microwells) and incubated overnight. Hepatic spheroids (about 150 hepatocytes per spheroid, ~100 μm diameter) were imaged and analyzed to quantify the extent of spheroid compaction. Individual spheroids were isolated manually using Fiji,[56] and greyscale erosion was applied to threshold for hepatocytes (~7 μm). Resulting morphologies were traced and measured for circularity. Resulting spheroids were embedded in fibrin (10 mg/mL bovine fibrinogen, 1.25 U/mL human thrombin; Sigma-Aldrich) using 96 microwell plates as molds. Spheroid-laden hydrogels were cultured in hepatocyte media supplemented with 10 μg/ml aprotinin, a serine protease inhibitor, to prevent hydrogel degradation. Alternatively, spheroids were cultured in alginate beads and hepatocyte media. Alginate beads were formed using sterilized 2% w/v alginate (Sigma-Aldrich) and 2% w/v CaCl2 (Sigma Aldrich), both dissolved in HEPES-buffered saline (20 mM HEPES, 150 mM NaCl in ddH2O). Spheroids were resuspended in alginate and added dropwise into a pre-warmed, stirred CaCl2 bath, then washed and collected using a 40 μm cell strainer before culturing in hepatocyte media.
Cell line generation and validation:
J2s were lentivirally transduced using the 3rd generation lentiviral system with an iCasp9-IRES-GFP plasmid (Addgene; #15567 pMSCV-F-del Casp9.IRES.GFP; cloned in-house to a lentivirus plasmid backbone with an SFFV promoter).[34] Briefly, plasmids were co-transfected into HEK-293T cells with pVSVG, pRSV-REV, and pMDLg/pRRE using the calcium phosphate transfection method. Assembled viruses were collected in the culture supernatant after 48 hours and precipitated using PEG-IT (SBI), resuspended in PBS, and stored at −80 °C. To transfect J2s, virus was added to growth media and cultured overnight. iCasp9-GFP J2s were purified (top 15% of cell population; GFP) by FACS (FACSAria II, BD Biosciences). iCasp9-GFP J2 fibroblasts at passage 22 were grown at confluence for 2 weeks to mimic experimental culture conditions, without a decrease in the percentage of the cell population with positive GFP expression (Figure S1a of Supporting Information). iCasp9-GFP J2s were plated in monolayer and dosed with ethanol vehicle or CID (B/B homodimerizer, AP20187; rapalog; Takara/ClonTech) at a concentration of 50 nM (1:10,000 dilution) unless otherwise noted in the text. Cultures were then assayed for cell viability using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega), for caspase-9 activation using the CaspGLOW™ Fluorescein Active Caspase-9 Staining Kit (Thermo Fisher Scientific) and Caspase-Glo®9 Assay Systems (Promega), or stained with the Pacific Blue™ Annexin V/SYTOX™ AADvanced™ Apoptosis Kit (Thermo Fisher) to identify apoptotic cells by flow cytometry (>100,000 cells analyzed per condition).
CID Treatment:
MPCCs and spheroid-laden hydrogels were dosed with 50 nM CID (1:10,000 dilution) for all co-culture experiments. Spheroids were dosed with CID after encapsulation in fibrin hydrogels, except where noted in the text. In the case of pre-encapsulation CID treatment, spheroids were treated with 50 nM CID prior to harvest from microwell molds. The contents of the microwell molds (including hepatocytes, fibroblast-derived apoptotic debris, and conditioned media) were collected, diluted at least 5-fold, and centrifuged at 60×g for 6 minutes for 3 washes total in order to isolate hepatocytes via differential centrifugation. Pelleted hepatocytes were then encapsulated in fibrin hydrogels and cultured in hepatocyte media.
Functional Antibody Blockade:
Primary human hepatocytes or compacted hepatocyte-fibroblast spheroids (immediately after harvest from microwell molds) were incubated with 10 μg/mL functional blocking monoclonal antibody (mouse anti-human β1 integrin, clone P5D2; mouse anti-human E-cadherin, clone 67A4; EMD Millipore) or an isotype control (Santa Cruz Biotechnology) for 20 minutes at 37 °C in hepatocyte media. Excess antibody was removed by centrifugation at 60×g for 6 minutes for 3 washes total. Pelleted hepatocytes were then encapsulated in fibrin hydrogels and cultured in hepatocyte media.
Biochemical assays:
Supernatant was collected from cultures every other day and stored at −20 °C. Human albumin was quantified using an enzyme-linked immunosorbent assay using a sheep anti-rat albumin antibody (ELISA) (Bethyl Laboratories) and 3,3’,5,5’-tetramethylbenzidine (TMB, Thermo Fisher). Urea concentration was measured using a colorimetric (diacetylmonoxime) assay with acid and heat (Stanbio Labs). CYP3A4 activity was assessed with the luminogenic P450-Glo™ CYP450 assay kit (Promega) for nonlytic assays using cultured cells. Cultures were pre-treated with 25 μM rifampin or 1:1000 DMSO vehicle control prepared in hepatocyte maintenance media for 72 hours (daily replenishment) where indicated.
Immunocytochemistry:
For immunostaining of cellular constructs, tissues were fixed in 4% paraformaldehyde. For identification of primary human hepatocytes, tissues were incubated with primary antibody against human arginase-1 (rabbit, 1:400; Sigma-Aldrich) followed by Alexa Fluor® 546-conjugated rabbit anti-human secondary antibody (1:1000; Life Technologies). Alternatively, hepatocytes were visualized by pre-labeling with 1 μM CellTracker Deep Red (Thermo Fisher) for 20 minutes at 37°C. Nuclei were stained with Hoechst (1:2000).
Imaging:
Fiji was used to uniformly adjust brightness/ contrast, pseudocolor, and merge images. Spheroid-laden hydrogels were imaged on a Zeiss 710 confocal microscope using a water immersion 40X objective or the Leica SP8 spectral confocal microscope using the 10X air or 25X water immersion objective. Live imaging was captured using a Nikon Spinning-disk Confocal Microscope with TIRF module.
Quantitation:
iCasp9-GFP J2s were encapsulated as single cells in a fibrin hydrogel. Cultures were dosed with vehicle or CID and cultured for 3 weeks. Cultures were fixed with 4% paraformaldehyde, stained with Hoechst (1:2000) and imaged with a Nikon Ti-E inverted epifluorescent microscope. Number of cells (by counting nuclei) was quantified for 3 representative 20x field of views per sample.
Statistical Analysis:
All data are expressed as mean ± standard deviation and/or visualized as dot plots (n = 3–10 as indicated). Statistical significance (ɑ = 0.05) was determined using the appropriate statistical test (unpaired 2-tailed t-test, 1-way ANOVA, 2-way ANOVA), and followed by multiple comparisons testing (Tukey’s post hoc test) (GraphPad).
Supplementary Material
Acknowledgements
The authors thank Dr. T.N. Vo, Dr. Q.B. Smith, T.R. Nash, K.N. Vyas, Dr. J.L. Bays, and Dr. L. Li for insightful discussions; the Flow Cytometry and Imaging cores of the Koch Institute Swanson Biotechnology Center; Howard Green (Harvard Medical School) for providing the 3T3-J2 fibroblast cell line; and Wilson Wong (Boston University) for his helpful guidance and discussion regarding iCasp9 constructs. A.X.C. was supported by the National Science Foundation Graduate Research Fellowship (1122374). A.C. was supported by the Paul and Daisy Soros Fellowships for New Americans. This work was supported in part by the NIH (R01 EB008396, R01 EB000262, UG3 EB017103), Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute, the National Science Foundation Cellular Metamaterials Engineering Research Center, and the Boston University Biological Design Center. S.N.B. is a Howard Hughes Medical Institute Investigator.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Amanda X. Chen, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Arnav Chhabra, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; Harvard–MIT Program in Health Sciences and Technology, Institute for Medical Engineering and Science, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
H.-H. Greco Song, Harvard–MIT Program in Health Sciences and Technology, Institute for Medical Engineering and Science, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; Biological Design Center, Department of Biomedical Engineering, Boston University, Boston, MA 02215, USA; Wyss Institute for Biologically Inspired Engineering, Boston, MA 02115, USA.
Heather E. Fleming, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139, USA Harvard–MIT Program in Health Sciences and Technology, Institute for Medical Engineering and Science, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Christopher S. Chen, Biological Design Center, Department of Biomedical Engineering, Boston University, Boston, MA 02215, USA Wyss Institute for Biologically Inspired Engineering, Boston, MA 02115, USA.
Sangeeta N. Bhatia, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139, USA Harvard–MIT Program in Health Sciences and Technology, Institute for Medical Engineering and Science, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; Wyss Institute for Biologically Inspired Engineering, Boston, MA 02115, USA; Department of Electrical Engineering and Computer Science, Massachusetts Institute of Technology, Cambridge, MA 02142, USA; Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA 02115, USA; Broad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, MA 02139, USA; Howard Hughes Medical Institute, Chevy Chase, MD 20815, USA.
References
- [1].Lutolf MP, Gilbert PM, Blau HM, Nature 2009, 462, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Takebe T, Wells JM, Science 2019, 364, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Park SE, Georgescu A, Huh D, Science 2019, 364, 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bissell MJ, Radisky D, Nat. Rev. Cancer 2001, 1, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tuveson D, Clevers H, Science 2019, 364, 952. [DOI] [PubMed] [Google Scholar]
- [6].Bonnans C, Chou J, Werb Z, Nat. Rev. Mol. Cell Biol 2014, 15, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nelson CM, Bissell MJ, Semin. Cancer Biol 2005, 15, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tibbitt MW, Anseth KS, Biotechnol. Bioeng 2009, 103, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Todhunter ME, Jee NY, Hughes AJ, Coyle MC, Cerchiari A, Farlow J, Garbe JC, LaBarge MA, Desai TA, Gartner ZJ, Nat. Methods 2015, 12, 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yi C, Li C-W, Ji S, Yang M, Anal. Chim. Acta 2006, 560, 1. [Google Scholar]
- [11].Cerchiari AE, Garbe JC, Jee NY, Todhunter ME, Broaders KE, Peehl DM, Desai TA, LaBarge MA, Thomson M, Gartner ZJ, Proc. Natl. Acad. Sci. U. S. A 2015, 112, 2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hu H, Gehart H, Artegiani B, LÖpez-Iglesias C, Dekkers F, Basak O, van Es J, Chuva SM de Sousa Lopes, Begthel H, Korving J, van den Born M, Zou C, Quirk C, Chiriboga L, Rice CM, Ma S, Rios A, Peters PJ, de Jong YP, Clevers H, Cell 2018, 175, 1591. [DOI] [PubMed] [Google Scholar]
- [13].Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, Clevers H, Science 2012, 337, 730. [DOI] [PubMed] [Google Scholar]
- [14].Bhatia SN, Underhill GH, Zaret KS, Fox IJ, Sci. Transl. Med 2014, 6, 245sr2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bhatia SN, Balis UJ, Yarmush ML, Toner M, FASEB J. 1999, 13, 1883. [DOI] [PubMed] [Google Scholar]
- [16].Godoy P, Hewitt NJ, Albrecht U, Andersen ME, Ansari N, Bhattacharya S, Bode JG, Bolleyn J, Borner C, Böttger J, Braeuning A, Budinsky RA, Burkhardt B, Cameron NR, Camussi G, Cho C-S, Choi Y-J, Craig Rowlands J, Dahmen U, Damm G, Dirsch O, Donato MT, Dong J, Dooley S, Drasdo D, Eakins R, Ferreira KS, Fonsato V, Fraczek J, Gebhardt R, Gibson A, Glanemann M, Goldring CEP, Gómez-Lechón MJ, Groothuis GMM, Gustavsson L, Guyot C, Hallifax D, Hammad S, Hayward A, Häussinger D, Hellerbrand C, Hewitt P, Hoehme S, Holzhütter H-G, Houston JB, Hrach J, Ito K, Jaeschke H, Keitel V, Kelm JM, Kevin Park B, Kordes C, Kullak-Ublick GA, LeCluyse EL, Lu P, Luebke-Wheeler J, Lutz A, Maltman DJ, Matz-Soja M, McMullen P, Merfort I, Messner S, Meyer C, Mwinyi J, Naisbitt DJ, Nussler AK, Olinga P, Pampaloni F, Pi J, Pluta L, Przyborski SA, Ramachandran A, Rogiers V, Rowe C, Schelcher C, Schmich K, Schwarz M, Singh B, Stelzer EHK, Stieger B, Stöber R, Sugiyama Y, Tetta C, Thasler WE, Vanhaecke T, Vinken M, Weiss TS, Widera A, Woods CG, Xu JJ, Yarborough KM, Hengstler JG, Arch. Toxicol 2013, 87, 1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Guguen-Guillouzo C, Guillouzo A, In Enzyme Induction and Modulation; Najjar VA., Ed.; Developments in molecular and cellular biochemistry; Springer US: Boston, MA, 1983; pp. 35–56. [DOI] [PubMed] [Google Scholar]
- [18].Chen AA, Thomas DK, Ong LL, Schwartz RE, Golub TR, Bhatia SN, Proc. Natl. Acad. Sci 2011, 108, 11842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Khetani SR, Szulgit G, Del Rio JA, Barlow C, Bhatia SN, Hepatology 2004, 40, 545. [DOI] [PubMed] [Google Scholar]
- [20].Stevens K, Ungrin M, Schwartz R, Ng S, Carvalho B, Christine K, Chaturvedi R, Li C, Zandstra P, Chen C, Bhatia S, Nat. Commun 2013, 4, 1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Stevens KR, Scull MA, Ramanan V, Fortin CL, Chaturvedi RR, Knouse KA, Xiao JW, Fung C, Mirabella T, Chen AX, McCue MG, Yang MT, Fleming HE, Chung K, de Jong YP, Chen CS, Rice CM, Bhatia SN, Sci. Transl. Med 2017, 9, eaah5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ukairo O, Kanchagar C, Moore A, Shi J, Gaffney J, Aoyama S, Rose K, Krzyzewski S, McGeehan J, Andersen ME, Khetani SR, Lecluyse EL, J. Biochem. Mol. Toxicol 2013, 27, 204. [DOI] [PubMed] [Google Scholar]
- [23].Ballard TE, Wang S, Cox LM, Moen MA, Krzyzewski S, Ukairo O, Obach RS, Drug Metab. Dispos. Biol. Fate Chem 2016, 44, 172. [DOI] [PubMed] [Google Scholar]
- [24].Lin C, Shi J, Moore A, Khetani SR, Drug Metab. Dispos. Biol. Fate Chem 2016, 44, 127. [DOI] [PubMed] [Google Scholar]
- [25].Hui EE, Bhatia SN, Proc. Natl. Acad. Sci 2007, 104, 5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hui EE, Bhatia SN, JoVE J. Vis. Exp 2007, e268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Goulet F, Normand C, Morin O, Hepatology 1988, 8, 1010. [DOI] [PubMed] [Google Scholar]
- [28].Hamilton GA, Jolley SL, Gilbert D, Coon DJ, Barros S, LeCluyse EL, Cell Tissue Res. 2001, 306, 85. [DOI] [PubMed] [Google Scholar]
- [29].Khetani SR, Chen AA, Ranscht B, Bhatia SN, FASEB J. 2008, 22, 3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shan J, Logan DJ, Root DE, Carpenter AE, Bhatia SN, J. Biomol. Screen 2016, 21, 897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shan J, Schwartz RE, Ross NT, Logan DJ, Thomas D, Duncan SA, North TE, Goessling W, Carpenter AE, Bhatia SN, Nat. Chem. Biol 2013, 9, 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK, 10.1056/NEJMoa11061522011. [DOI]
- [33].Gargett T, Brown MP, Front. Pharmacol 2014, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Straathof KC, Pulè MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, Heslop HE, Spencer DM, Rooney CM, Blood 2005, 105, 4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Khetani SR, Bhatia SN, Nat. Biotechnol 2008, 26, 120. [DOI] [PubMed] [Google Scholar]
- [36].Li CY, Stevens KR, Schwartz RE, Alejandro BS, Huang JH, Bhatia SN, Tissue Eng. Part A 2014, 20, 2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Marin V, Cribioli E, Philip B, Tettamanti S, Pizzitola I, Biondi A, Biagi E, Pule M, Comparison of Different Suicide-Gene Strategies for the Safety Improvement of Genetically Manipulated T Cells. https://home.liebertpub.com/hgtb 2012. [DOI] [PMC free article] [PubMed]
- [38].Underhill GH, Chen AA, Albrecht DR, Bhatia SN, Biomaterials 2007, 28, 256. [DOI] [PubMed] [Google Scholar]
- [39].Shen K, Luk S, Elman J, Murray R, Mukundan S, Parekkadan B, Sci. Rep 2016, 6, 21239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fischbach C, Kong HJ, Hsiong SX, Evangelista MB, Yuen W, Mooney DJ, Proc. Natl. Acad. Sci 2009, 106, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Martino MM, Briquez PS, Ranga A, Lutolf MP, Hubbell JA, Proc. Natl. Acad. Sci 2013, 110, 4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Luebke-Wheeler JL, Nedredal G, Yee L, Amiot BP, Nyberg SL, Cell Transplant. 2009, 18, 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lin R-Z, Chou L-F, Chien C-CM, Chang H-Y, Cell Tissue Res 2006, 324, 411. [DOI] [PubMed] [Google Scholar]
- [44].Baker BM, Chen CS, J. Cell Sci 2012, 125, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang K, Lin B, Int. Sch. Res. Not 2013. [Google Scholar]
- [46].Mesnil M, Yamasaki H, Cancer Res. 2000, 60, 3989. [PubMed] [Google Scholar]
- [47].Udawatte C, Ripps H, Apoptosis 2005, 10, 1019. [DOI] [PubMed] [Google Scholar]
- [48].Marconi P, Tamura M, Moriuchi S, Krisky DM, Niranjan A, Goins WF, Cohen JB, Glorioso JC, Mol. Ther. J. Am. Soc. Gene Ther 2000, 1, 71. [DOI] [PubMed] [Google Scholar]
- [49].Yagyu S, Hoyos V, Del Bufalo F, Brenner MK, Mol. Ther 2015, 23, 1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kim D, Seo Y, Kwon S, Biotechnol. Bioprocess Eng 2015, 20, 358. [Google Scholar]
- [51].Schuurman H-J, Int J Surg 2015, 23, 312. [DOI] [PubMed] [Google Scholar]
- [52].Costa‐Almeida R, Soares R, Granja PL, J. Tissue Eng. Regen. Med 2018, 12, 240. [DOI] [PubMed] [Google Scholar]
- [53].Martin MJ, Muotri A, Gage F, Varki A, Nat. Med 2005, 11, 228. [DOI] [PubMed] [Google Scholar]
- [54].Discher DE, Mooney DJ, Zandstra PW, Science 2009, 324, 1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].March S, Ramanan V, Trehan K, Ng S, Galstian A, Gural N, Scull MA, Shlomai A, Mota M, Fleming HE, Khetani SR, Rice CM, Bhatia SN, Nat. Protoc 2015, 10, 2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Nat. Methods 2012, 9, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.