

Abstract
Cysteine-directed covalent ligands have emerged as a versatile category of chemical probes and drugs that leverage thiol nucleophilicity to form permanent adducts with proteins of interest. Understanding the scope of cysteines that can be targeted by covalent ligands, as well as the types of electrophiles that engage these residues, represent important challenges for fully realizing the potential of cysteine-directed chemical probe discovery. Although chemical proteomic strategies have begun to address these important questions, only a limited number of electrophilic chemotypes have been explored to date. Here, we describe a diverse set of candidate electrophiles appended to a common core 6-methoxy-1,2,3,4-tetrahydroquinoline fragment and evaluate their global cysteine reactivity profiles in human cancer cell proteomes. This work uncovered atypical reactivity patterns for a discrete set of cysteines, including residues involved in enzymatic catalysis and located in proximity to protein–protein interactions. These findings thus point to potentially preferred electrophilic groups for site-selectively targeting functional cysteines in the human proteome.
Keywords: Cysteine, Chemical proteomics, Electrophile, Covalent, Activity-based protein profiling
Cysteine residues play important roles in diverse biological processes including enzyme catalysis and as sites of post-translational regulation by redox signaling pathways [1–5]. Nucleophilic, but non-catalytic cysteines can also reside in functional pockets of proteins, offering a way to target these proteins with cysteine-directed covalent ligands. This has been well-demonstrated for protein kinases, including BTK and EGFR, for which cysteine-directed covalent inhibitors have been developed as targeted cancer therapeutics [6–9]. Covalent ligands can offer advantages as chemical probes and drugs over reversibly acting small molecules, including increased pharmacological action due to longer residency time and improved affinity for shallow binding pockets expanding the number of ‘druggable’ proteins [10–13]. Nonetheless, understanding the scope of cysteine residues in the proteome that can be covalently engaged by electrophilic small molecules and, conversely, the types of reactive groups that preferentially modify these cysteines, remains poorly understood.
Chemical proteomic methods, such as activity-based protein profiling (ABPP), have been introduced to characterize the global reactivity of electrophilic small molecules in native biological systems. In this strategy, the reactivity of electrophilic compounds is assessed across thousands of nucleophilic residues in the proteome using a broad-spectrum, chemoselective (e.g., cysteine-, serine-, or lysine-directed) probe and quantitative mass spectrometry (MS) analysis [14–17]. Previous ABPP studies have illuminated many hundreds of ligandable cysteines in diverse human cell types, including cancer cell lines and primary immune cells, and revealed how covalent ligands targeting these cysteines can inhibit the activity of proteins [15,18–20], perturb protein–protein interactions [13,21] and promote the degradation of proteins [22–24]. To date, however, only a limited number of cysteine-directed reactive groups have been explored by chemical proteomics, mostly constituting acrylamides and alpha-chloroacetamides (αCAs), with some exceptional cases noted, such as heteroaromatic sulfones [25,26], bicyclobutane carboxamides [27], chloromethyl triazoles [20], α-cyanoacrylamides (reversible) [28], and acyoxymethyl ketones [29].
The potency and selectivity of covalent ligands are influenced by at least two major properties of the compounds: 1) the recognition group, which participates in reversible binding interactions to the protein target; and 2) the reactive group, which forms a covalent bond with a cysteine residue within or proximal to the binding pocket on the protein target. As noted above, a number of advanced covalent probes and drugs have been developed for diverse proteins, and most of these medicinal chemistry efforts have focused on optimization of the recognition group while preserving a mainstay reactive group (e.g., acrylamide) [18,30–32]. A recent report examined the electrophilicity of an array of candidate reactive groups combined with a common recognition group and uncovered differences in the reactivity of these compounds with cysteines in purified proteins [33]. Here, we sought to build on and extend these investigations to explore candidate electrophiles for cysteine reactivity on a proteome-wide scale using gel- and MS-ABPP.
We appended candidate electrophiles to a constant recognition group with the goal of better understanding the reactivity preferences of cysteines in the proteome. A 6-methoxy-1,2,3,4-tetrahydroquinoline core was selected as the recognition group, as the corresponding αCA containing this group (KB02, Fig. 1) has been found to serve as a versatile ‘scout’ fragment capable of engaging a large fraction of the total cysteines liganded by larger collections of fragment and/or elaborated electrophilic compounds [15,21,24]. The αCA was replaced by a diverse set of candidate reactive groups (Fig. 1 and Supplementary Fig. S1A), including established electrophiles, such as a carbamate (1), epoxide (5), and a benzoyloxymethyl amide (6, analogous to an acyloxymethyl ketone), as well as less characterized chemical groups that were anticipated to show more tempered electrophilicity (2, 3, 4, 7). Their synthesis and characterization are provided in the ESI.
Fig. 1.
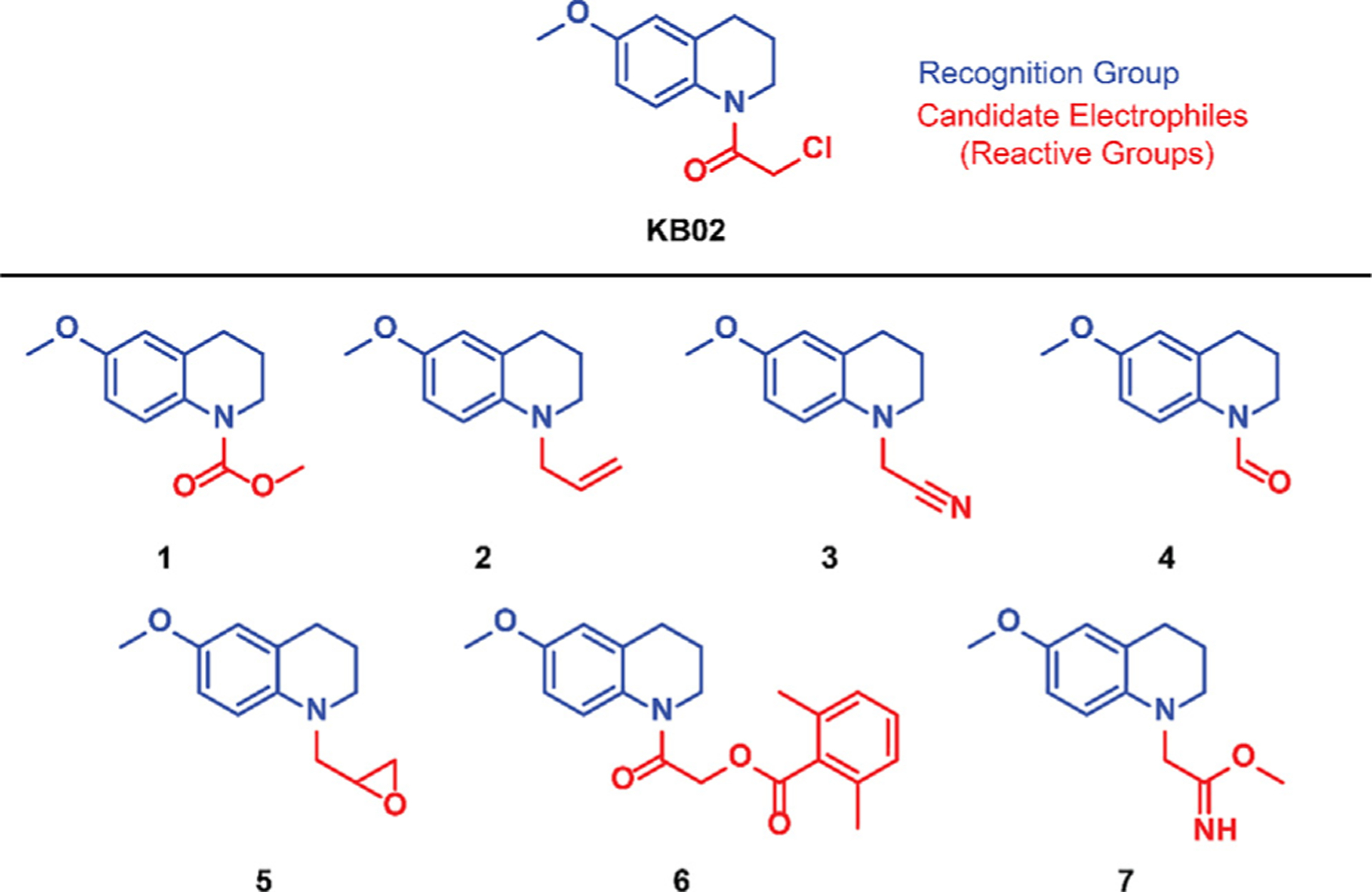
Structures of the α-chloroacetamide (αCA) scout fragment (KB02) and candidate electrophilic compounds 1–7. The recognition group (blue) remains constant throughout and the reactive group (red) was varied.
The compound collection was initially screened in a gel-ABPP assay in which proteomic lysate of Ramos cells (a human B cell lymphoma line) was treated with DMSO or compounds (500 μM, 1 h) followed by exposure to the broad cysteine-reactive probe iodoacetamide-alkyne (IA-alkyne, 1 μM, 1 h). IA-alkyne-labeled proteins were then conjugated to a rhodamine-azide tag using copper-catalyzed azide-alkyne cycloaddition (CuAAC) [34] and analyzed via SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and in-gel fluorescence scanning [35]. Compared to KB02, a subset of compounds (e.g., 1–7; Fig. 2) exhibited more selective and restricted blockade of IA-alkyne-protein interactions, whereas other compounds (e.g., SI-1, SI-4, SI-6) did not show evidence of disrupting IA-alkyne reactivity with proteins visible by gel-ABPP (Supplementary Fig. S1B). Based on these initial gel-ABPP results, combined with our interest in exploring less extensively characterized candidate electrophilic groups, we selected compounds 1–7 for more in-depth analysis of cysteine interactions in the proteome.
Fig. 2.
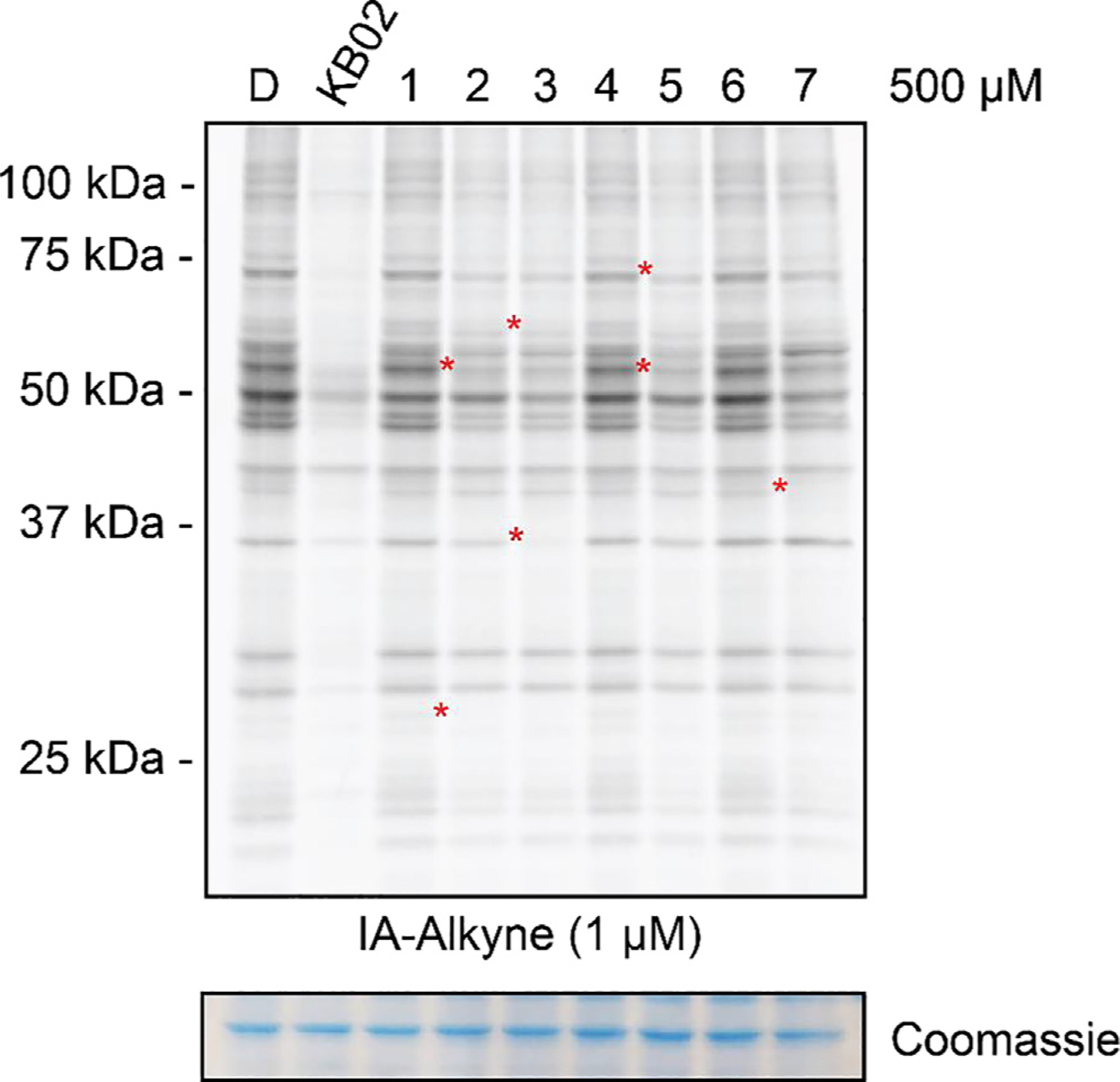
In-gel fluorescence image depicting competitive blockade of IA-alkyne (1 μM, 1 h) labeling of proteins in Ramos cell lysate by DMSO, KB02, or compounds 1–7 (500 μM, 1 h). Red asterisks highlight proteins that showed impairments in IA-alkyne reactivity in 1–7-treated lysates. Coomassie stained gel is shown as a loading control.
We proceeded to identify cysteine residues targeted by compounds 1–7 using MS-ABPP, using previously described methods [24]. In brief, Ramos proteome (2 mg protein/mL) was treated with DMSO, 1–7, or KB02, (500 μM, 1 h) followed by an IA-desthiobiotin probe (IA-DTB, 100 μM, 1 h). Proteins were then digested with trypsin and IA-DTB-labeled peptides enriched with streptavidin beads, eluted, and modified with isobaric tandem mass tagging (TMT-10plex™) to allow for multiplexed liquid chromatography (LC)-MS identification and quantification. Cysteines showing reductions in TMT signals, or R values, greater than 4 in a given compound-treated sample compared to DMSO control (reflecting > 75% inhibition of IA-DTB labeling) were considered to be liganded. From greater than 15,000 quantified cysteines, 1,005 cysteines were liganded by KB02, while compounds 1–7 showed much more restricted profiles that ranged from 0 to 60 liganded cysteines (Fig. 3A and Supplementary Dataset 1). Examples are provided of cysteines liganded by KB02 and compounds 1–7 (catalytic cysteine C319 in ALDH2; Fig. 3B) [36,37] versus those liganded by only KB02 (catalytic cysteine C113 in PIN1; Fig. 3C) [38]. In both cases, the liganding events were site-selective, as other cysteines quantified for ALDH2 and PIN1 were unaffected by compound treatment (Fig. 3B, C).
Fig. 3.
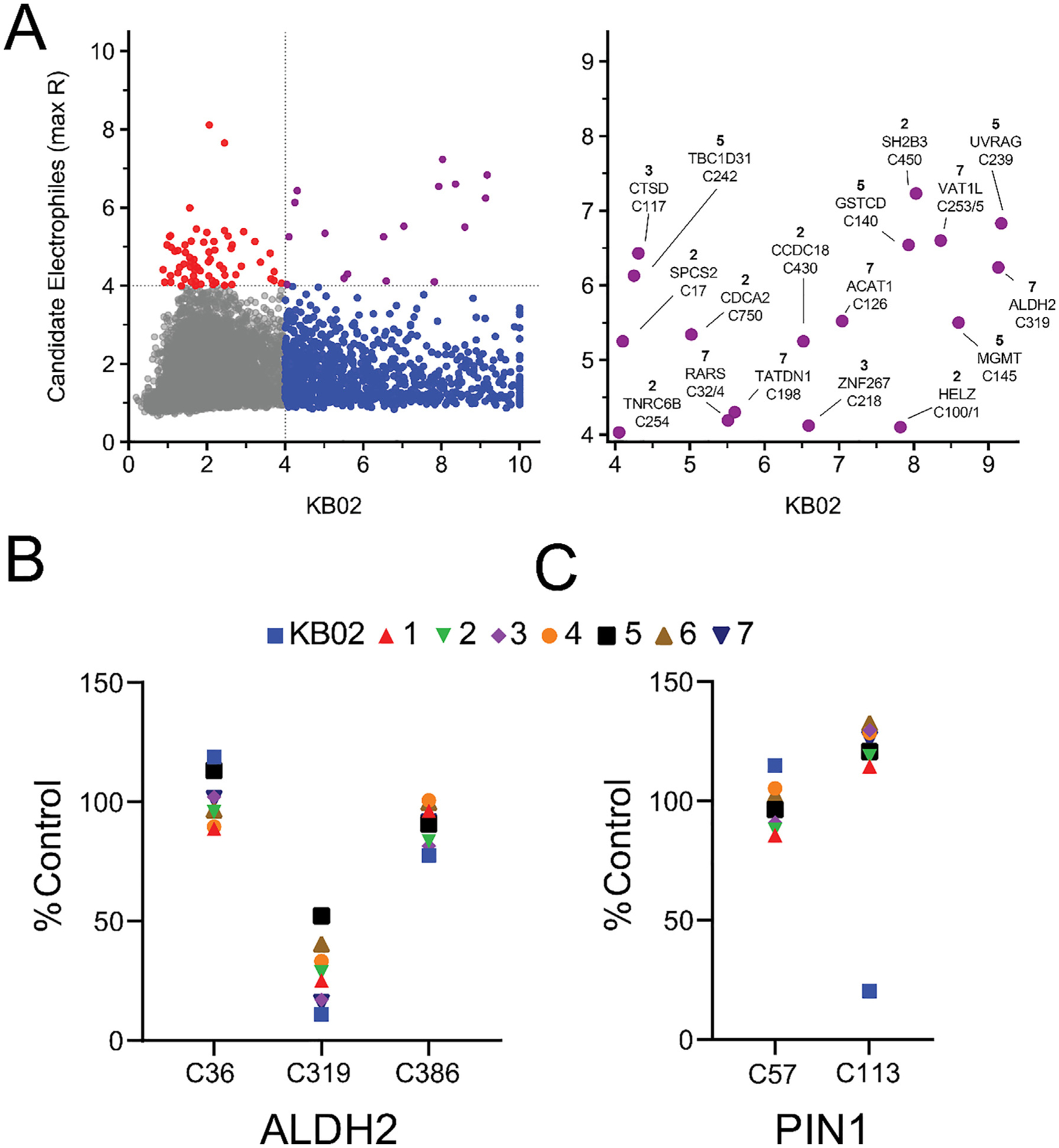
Chemical proteomic (MS-ABPP) analysis of cysteine ligandability in Ramos cell lysate treated with candidate electrophilic compounds. (A) Left, plot of cysteine reactivity values (or R values) in DMSO vs compound-treated samples. Ramos proteome was treated with DMSO or compounds (500 μM, 1 h), and cysteines with R values ≥ 4 (DMSO/compound) were considered liganded. Red and blue dots represent cysteines that showed R values ≥ 4 for one or more of compounds 1–7 or KB02, respectively. Purple dots represent cysteines that showed R values ≥ 4 for both KB02 and one or more of compounds 1–7 (compound with highest R value is shown for each cysteine). Right, enlarged upper right quadrant of left panel highlighting cysteine residues that are liganded by both KB02 and 1–7. (B) ALDH2_C319 as an example of a cysteine that was liganded by all tested compounds. (C) PIN1_C113 as an example of a cysteine that was only liganded by KB02.
As highlighted in purple in Fig. 3A, a handful of cysteines were liganded by both KB02 and varying subsets of compounds 1–7. Interestingly, these liganded cysteines included not only active site residues in enzymes (C150 in MGMT (Fig. 4A) and C126 in ACAT1 (Fig. 4B)) [39,40], but also cysteines located at protein–protein interfaces (e.g., C239 of UVRAG; Fig. 4C). For both MGMT and ACAT1, it is noteworthy that, while KB02 liganded multiple active-site cysteines in each protein, compounds 5 and 7 selectively engaged one active-site cysteine – C150 in MGMT and C126 in ACAT1 – respectively (Fig. 4A, B). These data suggest that the more tempered electrophiles examined herein have the potential to provide greater selectivity over the αCA group for site-specifically targeting cysteines in the active sites of proteins. Indeed, compound 7 showed very limited overall reactivity across the cysteine proteome, pointing to the O-methyl imidate electrophile as a potentially privileged reactive group for the design of future chemical probes that selectively target the catalytic C126 of ACAT1. UVRAG is a key regulatory component of autophagy complex II, and C239, which was strongly liganded by both KB02 and compound 5, is located at a protein–protein interface involving UVRAG and Beclin 1 (Fig. 4C) [41,42]. Considering this liganded event was uniquely observed with KB02 and 5, the interaction may show a reactivity preference for nucleophilic attack by C239 on a sp3 hybridized carbon compared to a sp or sp2 hybridized carbon.
Fig. 4.
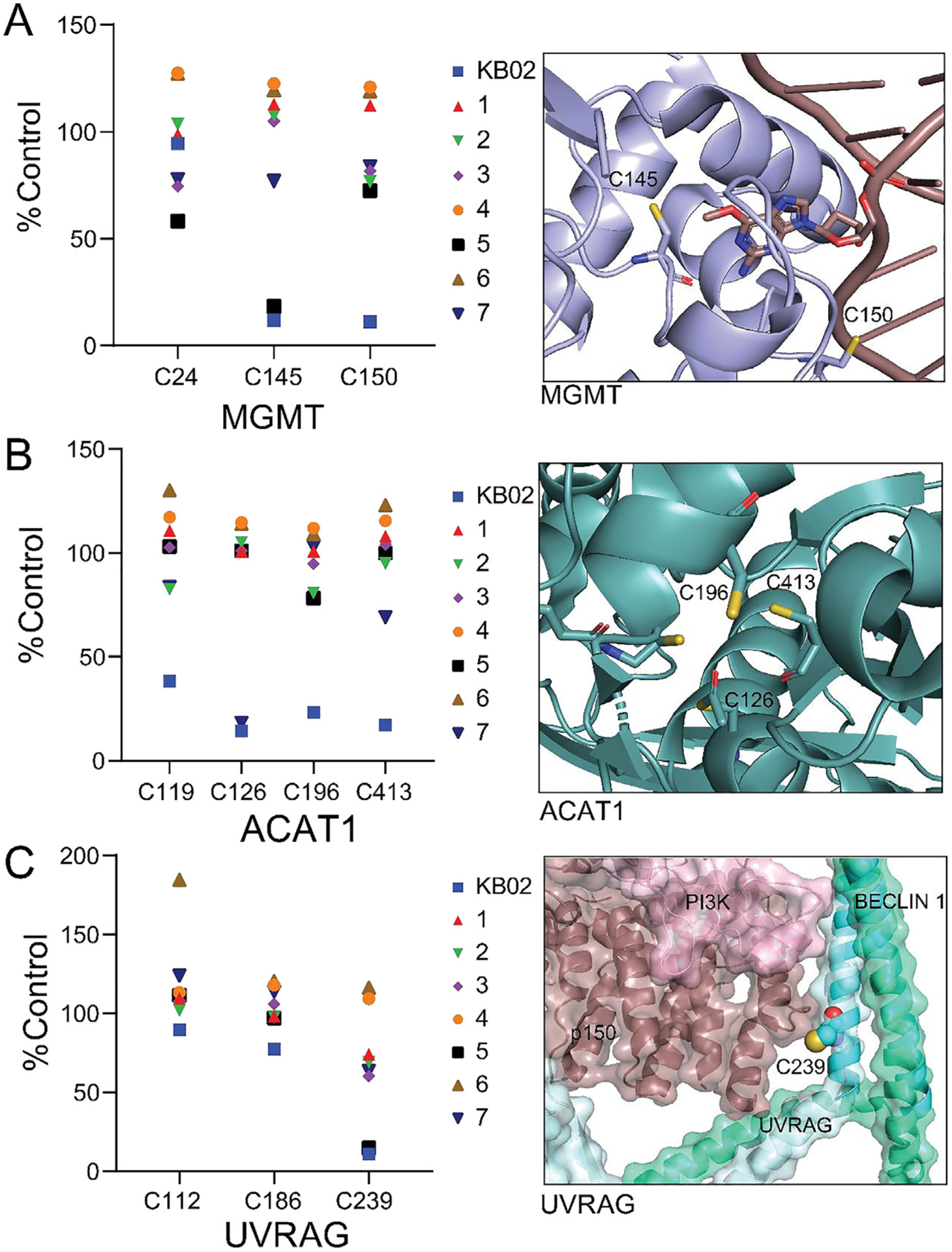
Examples of cysteines that were selectively liganded by individual candidate electrophilic compounds. (A) Left graph, the cysteine nucleophile (C145) of MGMT (methylated DNA-protein-cysteine methyltransferase) was site-specifically liganded by compound 5. In contrast, KB02 liganded both C150 and another active-site cysteine C145. Right image, crystal structure highlighting C145 interaction with a strand of DNA (PDB ID: 1T38). C150 is located near the active site at the DNA-protein interface. (B) Left graph, the cysteine nucleophile (C126) of ACAT1 (acetyl-CoA acetyltransferase) was site-specifically liganded by compound 7. In contrast, KB02 liganded both C126 and additional actives-site cysteines (C196 and C413). Right image, crystal structure of ACAT1 highlighting the active-site cysteine residues (PDB ID: 2F2S). (C) Left graph, C239 in UVRAG was site-specifically liganded by compound 5 and KB02. Right image, UVRAG_C239 is found at the protein–protein interface of UVRAG (light blue, yeast: VPS38), Beclin 1 (green, yeast: VPS30), p150 (brown, yeast: Vps15), and PI3K (pink, yeast: Vps34). Shown is the mouse UVRAG/Beclin 1 structure (PDB ID: 5YR0) aligned with the yeast Beclin 1 ortholog (VPS30) of yeast complex structure (PDB ID: 5DFZ) to show localization of UVRAG_C239 at the PPI interface of this complex.
A final group of cysteine reactivity changes was observed that reflected shared apparent engagement by amine containing (2, 3, 5, 7) compounds compared to (form)amide/carbamate containing (1, 4, 6) compounds, and many of these cysteines were also not engaged by KB02 (red dots, Fig. 3A). This subset of differentially reactive cysteines was unanticipated considering the presumed reduced electrophilicity of several amine compounds (e.g., 2, 3, 7), as reflected in their lower thiol (2-nitro-5-thiobenzoic acid) reactivity rates compared to more electrophilic compounds (KB02, 5) (Supplementary Fig. S2) [43]. We therefore hypothesized that the amine compounds may be blocking cysteine reactivity through reversible binding mechanisms, for instance, if the charged, protonated amine provided enhanced affinity for certain protein pockets that also happen to harbor a reactive cysteine. As potential support for this premise, we highlight bleomycin hydrolase (BLMH) as an example of a protein with an active site cysteine (C73) that was site-selectively blocked in IA-DTB reactivity by 2, 3, and 5, but not other compounds (Supplementary Fig. S3A) [44]. The BLMH active site has multiple negatively charged residues, including D143 and the carboxy terminus (A454, Supplementary Fig. S3B) [45] which may promote binding to amine compounds and impairment of IA-DTB reactivity with C73. Other cases of amine compound sensitivity could reflect non-specific induction of protein aggregation or denaturation by the compounds, as reflected in proteins for which several quantified cysteines were impaired in IA-DTB reactivity (e.g., KDM3A; JCHAIN, BACH2; Supplementary Dataset 1).
In summary, by assessing the proteomic cysteine reactivity profiles of a set of fragments bearing diverse candidate reactive groups appended to a common recognition group, we identified rare cysteines that show preferential engagement by otherwise tempered electrophiles. These cysteines included catalytic residues in the actives sites of enzymes like ACAT1 (C126) and MGMT (C145), which contain multiple reactive cysteines that are indiscriminately engaged by more electrophilic compounds such as the αCA KB02, but were site-specifically sensitive to O-methyl imidate 7 and epoxide 5, respectively. We are also encouraged that other cysteines engaged by epoxide 5 included residues at protein-protein interfaces (e.g., C239 of UVRAG), pointing to the potential for attenuated electrophiles to target more challenging sites of druggability in the proteome. Our findings, including other recent and complementary studies on purified proteins [33], as well as past success with identifying tunable electrophiles for other nucleophilic amino acids (e.g., serine [46]), should encourage the continued exploration of candidate electrophilic groups for cysteine reactivity, which we imagine will continue to expand the proportion of proteins in the human proteome that can be more widely or more selectively targeted by covalent chemistry for chemical probe and drug development.
Supplementary Material
Acknowledgements
This work was supported by the NIH: CA2319991 (B.F.C.), AI14278 (B.F.C.), CA212467 (V.M.C.), and DA15648 (D.L.B.).
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.tetlet.2021.152861.
References
- [1].Pace NJ, Weerapana E, Diverse functional roles of reactive cysteines, ACS Chem Biol 8 (2) (2013) 283–296. [DOI] [PubMed] [Google Scholar]
- [2].Chapman HA, Riese RJ, Shi GP, Emerging roles for cysteine proteases in human biology, Annu. Rev. Physiol 59 (1997) 63–88. [DOI] [PubMed] [Google Scholar]
- [3].Scheffner M, Nuber U, Huibregtse JM, Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade, Nature 373 (6509) (1995) 81–83. [DOI] [PubMed] [Google Scholar]
- [4].Zhang DD, Hannink M, Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress, Mol. Cell. Biol 23 (22) (2003) 8137–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sirover MA, Role of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in normal cell function and in cell pathology, J. Cell. Biochem 66 (2) (1997) 133–140. [PubMed] [Google Scholar]
- [6].Brooks ND,R; Boulet S; Lu Z; Kays L; Cavitt R; Gomez S; Strelow J; Milligan P; Roth K; Bauer R; Antonysamy S; Hahn P; Rankovic Z; McCann D; Mo G; Tiu R; Burkholder T; Geeganage S; Gilmour R In Identification and characterization of LY3410738, a novel covalent inhibitor of cancer-associated mutant Isocitrate Dehydrogenase 1 (IDH1), Proceedings of the American Association for Cancer Research Annual Meeting 2019, Atlanta, GA. Philadelphia (PA), AACR: Atlanta, GA. Philadelphia (PA), 2019. [Google Scholar]
- [7].Davids MS, Brown JR, Ibrutinib: a first in class covalent inhibitor of Bruton’s tyrosine kinase, Future Oncol. 10 (6) (2014) 957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Niessen S, Dix MM, Barbas S, Potter ZE, Lu S, Brodsky O, Planken S, Behenna D, Almaden C, Gajiwala KS, Ryan K, Ferre R, Lazear MR, Hayward MM, Kath JC, Cravatt BF, Proteome-wide map of targets of T790M-EGFR-directed covalent inhibitors, Cell Chem. Biol 24 (11) (2017) 1388–1400 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu R, Yue Z, Tsai CC, Shen J, Assessing lysine and cysteine reactivities for designing targeted covalent kinase inhibitors, J. Am. Chem. Soc 141 (16) (2019) 6553–6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Singh J, Petter RC, Baillie TA, Whitty A, The resurgence of covalent drugs, Nat Rev Drug Discov 10 (4) (2011) 307–317. [DOI] [PubMed] [Google Scholar]
- [11].Bradshaw JM, McFarland JM, Paavilainen VO, Bisconte A, Tam D, Phan VT, Romanov S, Finkle D, Shu J, Patel V, Ton T, Li X, Loughhead DG, Nunn PA, Karr DE, Gerritsen ME, Funk JO, Owens TD, Verner E, Brameld KA, Hill RJ, Goldstein DM, Taunton J, Prolonged and tunable residence time using reversible covalent kinase inhibitors, Nat. Chem. Biol 11 (7) (2015) 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang X, Chemical proteomics for expanding the druggability of human disease, ChemBioChem 21 (2020) 3319–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cheng SS, Yang GJ, Wang W, Leung CH, Ma DL, The design and development of covalent protein-protein interaction inhibitors for cancer treatment, J Hematol Oncol 13 (1) (2020) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF, Quantitative reactivity profiling predicts functional cysteines in proteomes, Nature 468 (7325) (2010) 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF, Proteome-wide covalent ligand discovery in native biological systems, Nature 534 (7608) (2016) 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hacker SM, Backus KM, Lazear MR, Forli S, Correia BE, Cravatt BF, Global profiling of lysine reactivity and ligandability in the human proteome, Nat. Chem 9 (12) (2017) 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu Y, Patricelli MP, Cravatt BF, Activity-based protein profiling: the serine hydrolases, Proc Natl Acad Sci U S A 96 (26) (1999) 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee HY, Suciu RM, Horning BD, Vinogradova EV, Ulanovskaya OA, Cravatt BF, Covalent inhibitors of nicotinamide N-methyltransferase (NNMT) provide evidence for target engagement challenges in situ, Bioorg. Med. Chem. Lett 28 (16) (2018) 2682–2687. [DOI] [PubMed] [Google Scholar]
- [19].Chung CY, Shin HR, Berdan CA, Ford B, Ward CC, Olzmann JA, Zoncu R, Nomura DK, Covalent targeting of the vacuolar H(+)-ATPase activates autophagy via mTORC1 inhibition, Nat. Chem. Biol 15 (8) (2019) 776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang C, Abegg D, Hoch DG, Adibekian A, Chemoproteomics-enabled discovery of a potent and selective inhibitor of the DNA repair protein MGMT, Angew. Chem. Int. Ed. Engl 55 (8) (2016) 2911–2915. [DOI] [PubMed] [Google Scholar]
- [21].Bar-Peled L, Kemper EK, Suciu RM, Vinogradova EV, Backus KM, Horning BD, Paul TA, Ichu TA, Svensson RU, Olucha J, Chang MW, Kok BP, Zhu Z, Ihle NT, Dix MM, Jiang P, Hayward MM, Saez E, Shaw RJ, Cravatt BF, Chemical proteomics identifies druggable vulnerabilities in a genetically defined cancer, Cell 171 (3) (2017) 696–709 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang X, Crowley VM, Wucherpfennig TG, Dix MM, Cravatt BF, Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16, Nat. Chem. Biol 15 (7) (2019) 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Spradlin JN, Hu X, Ward CC, Brittain SM, Jones MD, Ou L, To M, Proudfoot A, Ornelas E, Woldegiorgis M, Olzmann JA, Bussiere DE, Thomas JR, Tallarico JA, McKenna JM, Schirle M, Maimone TJ, Nomura DK, Harnessing the anti-cancer natural product nimbolide for targeted protein degradation, Nat. Chem. Biol 15 (7) (2019) 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vinogradova EV, Zhang X, Remillard D, Lazar DC, Suciu RM, Wang Y, Bianco G, Yamashita Y, Crowley VM, Schafroth MA, Yokoyama M, Konrad DB, Lum KM, Simon GM, Kemper EK, Lazear MR, Yin S, Blewett MM, Dix MM, Nguyen N, Shokhirev MN, Chin EN, Lairson LL, Melillo B, Schreiber SL, Forli S, Teijaro JR, Cravatt BF, An activity-guided map of electrophile-cysteine interactions in primary human T cells, Cell 182 (4) (2020) 1009–1026 e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zambaldo C, Vinogradova EV, Qi X, Iaconelli J, Suciu RM, Koh M, Senkane K, Chadwick SR, Sanchez BB, Chen JS, Chatterjee AK, Liu P, Schultz PG, Cravatt BF, Bollong MJ, 2-Sulfonylpyridines as tunable, cysteine-reactive electrophiles, J. Am. Chem. Soc 142 (19) (2020) 8972–8979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Motiwala HF, Kuo YH, Stinger BL, Palfey BA, Martin BR, Tunable heteroaromatic sulfones enhance in-cell cysteine profiling, J. Am. Chem. Soc 142 (4) (2020) 1801–1810. [DOI] [PubMed] [Google Scholar]
- [27].Tokunaga K, Sato M, Kuwata K, Miura C, Fuchida H, Matsunaga N, Koyanagi S, Ohdo S, Shindo N, Ojida A, Bicyclobutane carboxylic amide as a cysteine-directed strained electrophile for selective targeting of proteins, J. Am. Chem. Soc 142 (43) (2020) 18522–18531. [DOI] [PubMed] [Google Scholar]
- [28].Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, Frodin M, Taunton J, Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles, Nat. Chem. Biol 8 (5) (2012) 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fonovic M, Bogyo M, Activity-based probes as a tool for functional proteomic analysis of proteases, Expert Rev Proteomics 5 (5) (2008) 721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Thorarensen A, Dowty ME, Banker ME, Juba B, Jussif J, Lin T, Vincent F, Czerwinski RM, Casimiro-Garcia A, Unwalla R, Trujillo JI, Liang S, Balbo P, Che Y, Gilbert AM, Brown MF, Hayward M, Montgomery J, Leung L, Yang X, Soucy S, Hegen M, Coe J, Langille J, Vajdos F, Chrencik J, Telliez JB, Design of a Janus Kinase 3 (JAK3) specific inhibitor 1-((2S,5R)-5-((7H-pyrrolo[2,3-d] pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop −2-en-1-one (PF-06651600) allowing for the interrogation of JAK3 signaling in humans, J. Med. Chem 60 (5) (2017) 1971–1993. [DOI] [PubMed] [Google Scholar]
- [31].Jiang J, Jiang B, He Z, Ficarro SB, Che J, Marto JA, Gao Y, Zhang T, Gray NS, Discovery of covalent MKK4/7 dual inhibitor, Cell Chem. Biol (2020), 10.1016/j.chembiol.2020.08.014. [DOI] [PubMed] [Google Scholar]
- [32].Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Janne PA, Novel mutant-selective EGFR kinase inhibitors against EGFR T790M, Nature 462 (7276) (2009) 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Petri L, Abranyi-Balogh P, Imre T, Palfy G, Perczel A, Knez D, Hrast M, Gobec M, Sosic I, Nyiri K, Vertessy BG, Jansch N, Desczyk C, Meyer-Almes FJ, Ogris I, Grdadolnik SG, Iacovino LG, Binda C, Gobec S, Keseru GM, Assessment of tractable cysteines by covalent fragments screening, ChemBioChem (2020), 10.1002/cbic.202000700, in press. [DOI] [PubMed] [Google Scholar]
- [34].Rostovtsev VV, Green LG, Fokin VV, Sharpless KB, A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes, Angew. Chem. Int. Ed. Engl 41 (14) (2002) 2596–2599. [DOI] [PubMed] [Google Scholar]
- [35].Speers AE, Adam GC, Cravatt BF, Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition, J. Am. Chem. Soc 125 (16) (2003) 4686–4687. [DOI] [PubMed] [Google Scholar]
- [36].Buchman CD, Hurley TD, Inhibition of the aldehyde dehydrogenase 1/2 family by psoralen and coumarin derivatives, J. Med. Chem 60 (6) (2017) 2439–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Farres J, Wang TT, Cunningham SJ, Weiner H, Investigation of the active site cysteine residue of rat liver mitochondrial aldehyde dehydrogenase by site-directed mutagenesis, Biochemistry 34 (8) (1995) 2592–2598. [DOI] [PubMed] [Google Scholar]
- [38].Chen CH, Li W, Sultana R, You MH, Kondo A, Shahpasand K, Kim BM, Luo ML, Nechama M, Lin YM, Yao Y, Lee TH, Zhou XZ, Swomley AM, Butterfield DA, Zhang Y, Lu KP, Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer’s disease, Neurobiol. Dis 76 (2015) 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Daniels DS, Woo TT, Luu KX, Noll DM, Clarke ND, Pegg AE, Tainer JA, DNA binding and nucleotide flipping by the human DNA repair protein AGT, Nat. Struct. Mol. Biol 11 (8) (2004) 714–720. [DOI] [PubMed] [Google Scholar]
- [40].Min JR, Dombrovski L, Antoshenko T, Wu H, Loppnau P, Weigelt J, Sundstrom M, Arrowsmith CH, Edwards AM, Bochkarev A, Plotnikov AN, Structural Genomics Consortium (SGC), Human mitochondrial acetoacetyl-CoA thiolase (2005), 10.2210/pdb2F2S/pdb. [DOI] [Google Scholar]
- [41].Rostislavleva K, Soler N, Ohashi Y, Zhang L, Pardon E, Burke JE, Masson GR, Johnson C, Steyaert J, Ktistakis NT, Williams RL, Structure and flexibility of the endosomal Vps34 complex reveals the basis of its function on membranes, Science 350 (6257) (2015) aac7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wu S, He Y, Qiu X, Yang W, Liu W, Li X, Li Y, Shen HM, Wang R, Yue Z, Zhao Y, Targeting the potent Beclin 1-UVRAG coiled-coil interaction with designed peptides enhances autophagy and endolysosomal trafficking, Proc. Natl. Acad. Sci. U.S.A 115 (25) (2018) E5669–E5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Resnick E, Bradley A, Gan J, Douangamath A, Krojer T, Sethi R, Geurink PP, Aimon A, Amitai G, Bellini D, Bennett J, Fairhead M, Fedorov O, Gabizon R, Gan J, Guo J, Plotnikov A, Reznik N, Ruda GF, Diaz-Saez L, Straub VM, Szommer T, Velupillai S, Zaidman D, Zhang Y, Coker AR, Dowson CG, Barr HM, Wang C, Huber KVM, Brennan PE, Ovaa H, von Delft F, London N, Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening, J. Am. Chem. Soc 141 (22) (2019) 8951–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].O’Farrell PA, Gonzalez F, Zheng W, Johnston SA, Joshua-Tor L, Crystal structure of human bleomycin hydrolase, a self-compartmentalizing cysteine protease, Structure 7 (6) (1999) 619–627. [DOI] [PubMed] [Google Scholar]
- [45].Crnovcic I, Gan F, Yang D, Dong LB, Schultz PG, Shen B, Activities of recombinant human bleomycin hydrolase on bleomycins and engineered analogues revealing new opportunities to overcome bleomycin-induced pulmonary toxicity, Bioorg. Med. Chem. Lett 28 (16) (2018) 2670–2674. [DOI] [PubMed] [Google Scholar]
- [46].Otrubova K, Chatterjee S, Ghimire S, Cravatt BF, Boger DL, N-Acyl pyrazoles: Effective and tunable inhibitors of serine hydrolases, Bioorg. Med. Chem 27 (8) (2019) 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.