

Abstract
Gene-directed enzyme–prodrug therapy (GDEPT) is a promising approach for cancer therapy, but it suffers from poor targeted delivery in vivo. Polyethylenimine (PEI) is a cationic polymer efficient in delivering negatively charged nucleic acids across cell membranes; however, it is highly toxic in vivo. Hence, we efficiently reduced PEI toxicity without compromising its transfection efficiency by conjugating it with poly(d,l-lactic-co-glycolic acid) (PLGA) and poly(ethylene glycol) (PEG) as triblock copolymers through a multistep synthetic process. The synthesized nanoparticles showed efficient delivery of loaded nucleic acids to tumor cells in vitro and in vivo in mice. We used this nanoparticle to deliver a rationally engineered thymidine kinase (TK)–p53–nitroreductase (NTR) triple therapeutic gene against hepatocellular carcinoma (HCC), where p53 tumor suppressor gene is mutated in more than 85% of cancers. TK–p53–NTR triple gene therapy restores p53 function and potentiates cancer cell response to delivered prodrugs (ganciclovir (GCV) and CB1954). We used SP94 peptide-functionalized PLGA–PEG–PEI nanoparticles for the optimal delivery of TK–p53–NTR therapeutic gene in vivo. The nanoparticles prepared from the conjugated polymer showed high loading efficiency for the DNA and markedly enhanced TK–NTR-mediated gene therapy upon the simultaneous coexpression of p53 by the concurrent rescue of the endogenous apoptotic pathway in HCC cells of both p53-mutant and wild-type phenotypes in vitro. In vivo delivery of TK–p53–NTR genes by SP94-targeted PLGA–PEG–PEI NP in mice resulted in a strong expression of suicide genes selectively in tumors, and subsequent administration of GCV and CB1954 led to a decline in tumor growth, and established a superior therapeutic outcome against HCC. We demonstrate a highly efficient approach that exogenously supplements p53 to enable synergy with the outcome of TK–NTR suicide gene therapy against HCC.
Keywords: gene-directed enzyme–prodrug therapy (GDEPT), thymidine kinase, nitroreductase, p53, poly(lactic-co-glycolic acid), polyethylenimine, gene delivery
Graphical Abstract
INTRODUCTION
Hepatocellular carcinoma is the third leading cause of cancer-related mortality.1 The p53 cell cycle pathway is altered in the majority of hepatocellular carcinoma (HCC) patients.2 Disruption of this pathway occurs by mutations within the p53 gene itself, or by alterations such as p14ARF inactivation, or as a result of amplification/overexpression of p53 inhibitors, such as MDM2 and MDM4.3,4 Although p53-augmented chemotherapy and radiation therapy have been extensively investigated in the past, the implications of suicide gene therapy combined with the restoration of p53 function have remained unexplored.
Suicide gene therapy accounts for nearly 7.7% (157 out of 2076) of all gene therapy clinical trials conducted worldwide, as of early 2015.5 Gene-directed enzyme–prodrug therapy (GDEPT) introduces exogenous genes expressing metabolic enzymes into target cancer cells, which convert nontoxic prodrugs into activated cytotoxic agents to kill cancer cells,6 either by inducing DNA damage or by interfering with the cell cycle. Since the conception of GDEPT, many combinations of suicide genes and prodrugs have emerged as promising anticancer therapies. GDEPT, in particular, yields better therapeutic outcomes because of the bystander effect to the neighboring cancer cells,7 thus eliminating the need to deliver therapeutic genes to all cancer cells, especially in the inaccessible hypovascularized and hypoxic cores of solid tumors that are often responsible for drug resistance and tumor recurrence.8 However, clinical translation of GDEPT is challenging because, in solid human cancers, nondividing cells are much more abundant than proliferating cells, rendering most of the tumor cells insensitive to the prodrug.9 Thus, alternative enzyme/prodrug combinations that do not discriminate between dividing and nondividing cells would be highly desirable for the treatment of HCC. We previously established that a mutant version of herpes simplex virus 1-thymidine kinase (HSV1sr39TK) and the Escherichia coli nitroreductase (NTR) enzyme–prodrug combination therapy are effective against cancer in two ways: the TK-catalyzed product of the prodrug ganciclovir (GCV) causes cell death by inhibiting DNA synthesis, while the NTR-catalyzed CB1954 product kills quiescent cancer cells through DNA alkylation. They therefore produce a synergistic anticancer response.10
Over the past decades, gene therapy has become a subject of extensive research for potential applications against a broad range of diseases. Among other gene therapy approaches, gene-directed enzyme–prodrug therapy (GDEPT) has emerged as one of the most promising anticancer therapies. Although GDEPT has enormous potential for clinical applications, its use is severely limited by the lack of efficient and safe systems for the delivery of the therapeutic DNA into target cells in vivo.6 Moreover, achieving efficient gene therapy requires sufficient levels of therapeutic gene delivery into cancer cells to obtain adequate expression of therapeutic enzymes.11 Most clinical studies of GDEPT adopt viral-based vector systems that are highly efficient, but limited by immunogenicity, carcinogenicity, and toxicity. The nonviral vectors, including liposomes, dendrimers, polycationic lipids, and polymeric nanoparticles (NPs) that possess significantly lower safety risks, have been suggested as important alternative strategies.12 Among the nonviral vectors, NP vectors based on biodegradable and biocompatible poly(D,L-lactic-co-glycolic acid) (PLGA) polymers, especially the ones modified with poly(ethylene glycol) chains (PEGylated), appear attractive for DNA delivery. PLGA is an FDA-approved polymer for human medical use. For the advantages of biodegradability, stability, and low toxicity, PEGylated PLGA-based NPs (PEG–PLGA NPs) have been broadly tested as delivery systems for drug molecules and small nucleic acids in a range of preclinical studies and clinical trials.13 We also previously established that biodegradable poly(ethylene glycol)–poly(lactic-co-glycolic acid) (PEG–PLGA) nanoparticles are efficient vehicles for the delivery of microRNAs in vivo, with no significant toxicity.14 However, apart from the broad application as a drug formulation molecule and its use in small RNA delivery systems, PLGA-based NPs are far from ideal gene delivery systems owing to their low loading efficiency for large nucleic acids and poor transfection efficiency.15 To address these issues, in the present work, the conjugation of three polymers was designed based on the rationale that polymer poly(d,l-lactic-co-glycolic acid) (PLGA) is a versatile FDA-approved biodegradable and biocompatible polymer for the synthesis of nanoparticles. The PEG chains on the surface of these nanoparticles improve systemic circulation time and decrease immunogenicity. Furthermore, importantly, the cationic PEI polymer component enables the efficient loading of therapeutic genes and improves transfection efficiency into the cells. Unlike the usual nanoparticle synthesis procedure, the triblock polymer precursor cannot be used as such for nanoparticle synthesis, specifically because the conjugation of PEI to PLGA–PEG as a PLGA–PEG–PEI triblock polymer disrupts its ability to form nanoparticles in the w/o/w emulsion procedure. Thus, in this study we sought PEI conjugation post-synthesis of PLGA–PEG nanoparticles by introducing a predefined fraction of activated carboxyl groups on the nanoparticle surface to control the PEI conjugation with a limited proportion for efficient nucleic acid loading while reducing the toxicity.
In this study, we synthesized PLGA–PEG–PEI as a triblock copolymer to prepare optimal nanoparticles that have reduced toxicity while maintaining high DNA loading efficiency. To boost the therapeutic efficacy of our TK–NTR dual gene therapy and to be of particular benefit to hepatocellular carcinoma (HCC) cancers with an impaired p53 mechanism, as a first step, we here establish the therapeutic utility of TK–NTR dual gene therapy in the most prevalent p53-mutant variants of HCC.11 We then explore the merits of p53-augmented TK and NTR suicide gene therapies by constructing a triple GDEPT vector directed at HCC having either wild-type or mutant p53 backgrounds (HepG2 (p53+/+) and Hep3B (p53−/−)) to rescue the p53 function in cells that lack active p53 protein. In addition, to achieve targeted delivery of therapeutic DNA to tumor cells in vivo, we surface-modified these nanoparticles by functionalizing with SP94 (SFSIIHT-PILPL), the HCC-specific targeting peptide.16 Overall, we demonstrate a clear correlation of therapeutic outcomes of this novel treatment with TK, NTR, and p53 protein expression and establish the superior therapeutic efficacy of TK–p53–NTR triple gene therapy when compared to dual TK–NTR, in both in vitro and in vivo conditions (Scheme 1). A brief summary of the design of experiments in the present study is presented as a flow chart in Scheme S1.
Scheme 1.
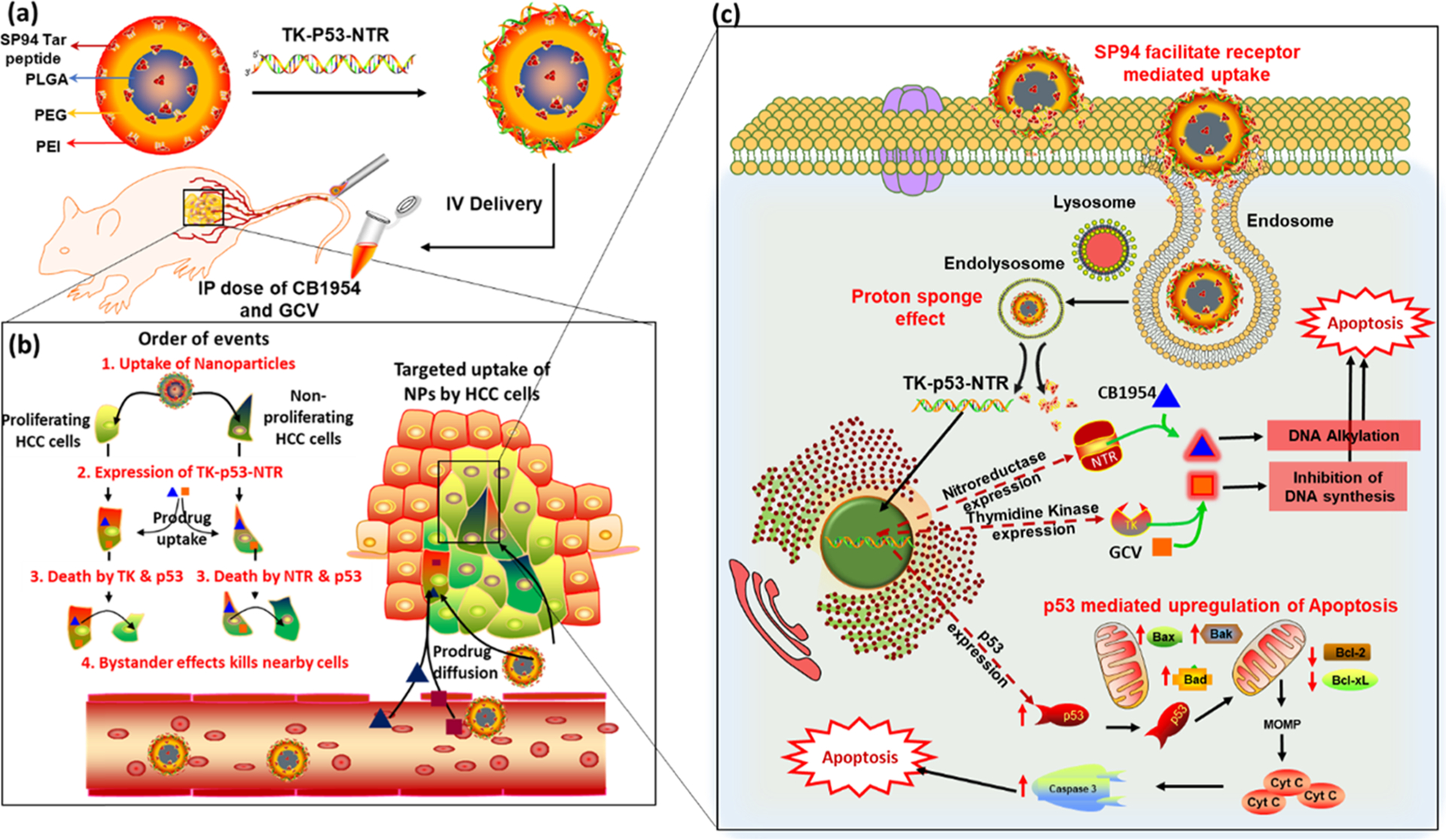
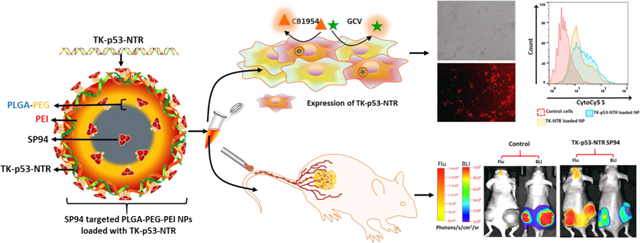
Schematic Illustration of (a) TK–p53–NTR Triple-Gene-Loaded PLGA–PEG–PEI NP Surface Functionalized with SP94; (b) Targeted Delivery and Order of Events Occur upon the Cellular Uptake of Nanoparticle and Prodrugs in HCC cells, and (c) Subsequent Intracellular Gene Expression and Signaling Cascades Activate Apoptosis in HCC Cells
MATERIALS AND METHODS
PLGA–PEG–PEI Nanoparticle Preparation and Characterization.
PLGA–PEG–PEI nanoparticles and PLGA–PEG–PEI/DNA complexes were prepared and characterized as detailed in the Supporting Methods section.
DNA Loading Capacity of PLGA–PEG–PEI Nanoparticles.
To assess the loading capacity of PLGA–PEG–PEI NPs, increasing amount of YOYO-3-labeled DNA (3 μL of 0.2 μM YOYO-3 for 1 μg of DNA) in 10% sucrose was complexed with a stipulated quantity of PLGA–PEG–PEI NPs (4 μg) for 10 min at 37 °C. The resultant complexes were analyzed by flow cytometry for DNA loading efficiency in a Green-Blue filter for 8000 events from each sample, and the results were represented as overlaid histograms in FlowJo analysis software.
Evaluation of Transfection Efficiency of PEG–PLGA–PEI Nanoparticles.
After evaluating the plasmid encapsulation efficiency of PLGA–PEG–PEI nanoparticles, its transfection efficiency at different N/P ratios was assessed in HepG2 and Hep3B HCC cells. A dual reporter plasmid construct expressing FLuc–EGFP (pcDNA–FLuc–EGFP) under a ubiquitin promoter was used in this experiment for the estimation of transfection efficiency in terms of protein expression (EGFP), as well as enzyme-catalyzed substrate conversion kinetics (FLuc assay). HepG2 and Hep3B cells were seeded in 12-well plates (60 000 cells/well) and were treated with different N/P ratio NP/DNA complexes (i.e., 4, 8, 12, and 16 μg of NPs for 2 μg of FLuc–EGFP plasmid) on the following day. After 48 h of treatment, the cells were imaged using a fluorescence microscope (Olympus IX81) under a GFP filter for the expression of EGFP. The treated cells were then assayed for bioluminescence by adding d-luciferin (150 μg/mL) as substrate and imaged for bioluminescence in a Lumina III in vivo imaging system (IVIS, PerkinElmer) for 2 min acquisition. After assay completion, the cells were subsequently rinsed with phosphate-buffered saline (PBS), trypsinized, and harvested for quantification of EGFP expression in a flow cytometer (GUAVA, Thermo Fisher). The cells treated with pcDNA-loaded PLGA–PEG–PEI nanoparticles served as control for all of the assays.
Estimation of TK–NTR and TK–p53–NTR Delivery by CytoCy5S Imaging and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR).
HepG2 cells were plated in 12-well plates (60 000 cell/well) and incubated at 37 °C. The next day, the cells were transfected with pcDNA (referred to as control pDNA), pcDNA–HSV1sr39TK–NTR (referred to as TK–NTR), and pcDNA–HSV1sr39TK–p53–NTR (referred to as TK–p53–NTR) fusion plasmid DNA using PEGylated PLGA–PEI nanoparticles at specified experimental conditions. After different time points of incubation with nanoparticle–DNA complexes, the cells were treated with CytoCy5S (0.25 μg/mL) and further incubated for 2 h. The cells were imaged for Cy5 signal using fluorescence microscopy (Olympus IX81) and analyzed by flow cytometry for the extent of NTR-catalyzed conversion of nonfluorescent substrate CytoCy5S into fluorescent Cy5. Around 5000 events were acquired for each sample, and the results were processed by FlowJo software (Tree Star, Ashland, OR) and presented as a histogram.
Quantitative Estimation of Delivered Plasmid Copy Numbers Using PCR.
The delivery of therapeutic genes in HepG2 cells was estimated quantitatively by PCR analysis of plasmid DNA isolated from HepG2 cells treated with TK–NTR- and TK–p53–NTR-loaded PEG–PLGA–PEI nanoparticles for 48 h. After the incubation period, the cells were rinsed with PBS twice, trypsinized, and the collected cells were lysed and processed further by a modified protocol using the QIAGEN plasmid DNA extraction kit. The isolated DNA was probed for TK–NTR with gene-specific amplification of TK by EVA green real-time polymerase chain reaction (RT-PCR). The forward and reverse primers for targeted amplification of the HSV1-TK segment were TACCCGAGCCGATGACTTAC and CCGATTAGAGGAGC-CAGAAC, respectively. A standard graph for both TK–NTR and TK–p53–NTR plasmids was generated by PCR amplification of serially diluted plasmids of known copy numbers, and the resultant Ct values were plotted to the corresponding log(N0 × 0.1), and the linear fit of this plot was adapted as the standard curve for calculating the copy number of TK–NTR and TK–p53–NTR plasmids delivered into cells for the respective treatments in the cellular uptake studies and in vivo in living animals.
In Vivo Study of TK–p53–NTR and TK–NTR Delivery and Therapeutic Effects.
All animal experiments were performed under the guidance and ethical approval of the Administrative Panel on Laboratory Animal Care (APLAC), Stanford University. All female nude mice (nu/nu) were obtained from Charles River laboratories (Wilmington, MA). The animals were anesthetized through nose cone with 2% isoflurane in oxygen and imaged using a Lago spectral instrument imaging system (Tucson, AZ) at the Stanford Center for Innovation in In Vivo Imaging (SCi3) core facility. The xenograft tumor model was developed in mice by injecting 0.1 mL of HepG2–FLuc cell suspension (5 × 106) containing 50 μL of medium growth factor Matrigel (Corning Discovery Labware Inc., Bedford, MA) into the left and right lower flanks. After 1 week, when the tumors grew to ~50 mm3, the animals were randomly divided into five groups ((1) PLGA–PEG–PEI nanoparticles loaded with pcDNA, (2) PLGA–PEG–PEI nanoparticles loaded with TK–NTR, (3) SP94-tagged PLGA–PEG–PEI nanoparticles loaded with TK–NTR, (4) PLGA–PEG–PEI nanoparticles loaded with TK–p53–NTR, and (5) SP94-tagged PLGA–PEG–PEI nanoparticles loaded with TK–p53–NTR). The respective PLGA–PEG/PEI nanoparticle–DNA complexes were administered via tail vein injection. Nanoparticle/DNA (200 μg of nanoparticle/50 μg of DNA) complex was prepared by incubating at room temperature for 30 min. The nanoparticle/DNA complex was intravenously injected at once, and the prodrugs were administered intraperitonially. The tumor volume was also measured using a Vernier caliper. We measured the greatest longitudinal diameter (length) and the greatest transverse diameter (width) and determined the volume by using the modified ellipsoidal formula17,18
The prodrugs (GCV (40 mg/kg)/CB1954 (40 mg/kg)) were intraperitoneally injected on days 3 and 5 of each treatment cycle continuously for 30 days. The second cycle of prodrug and nanoparticle/DNA complex was administered on day 8 and then followed up with the same protocol for four cycles of treatment. On day 28, CytoCy5S dye (10 μg) was intravenously injected and the mice were imaged for CytoCy5S fluorescent signal (640 nm excitation, 690 nm emission) followed by imaging after 24 h using a Lago spectral instrument Imaging system (Tucson, Arizona). All animals were sacrificed on day 29, and the organs and tumors were collected for ex vivo quantification of gene delivery and histological analysis.
Statistical Analyses.
Student’s t test was performed for all statistical analyses. Differences were considered to be statistically significant when the P value was less than 0.05.
RESULTS
Synthesis and Characterization of PLGA–PEG–PEI Triblock Conjugate Polymer and Optimization of PEGylated PLGA–PEI Nanoparticles for Gene Delivery.
We present the detailed methods for the preparation and characterization of PLGA–PEG–PEI nanoparticles in the Supporting Results and Discussion section. In brief, the L-PEI polymer laden with a large number of amine groups was conjugated to the terminal carboxyl group of PLGA–PEG polymer by in situ reaction during the synthesis process to obtain optimal PLGA–PEG–PEI nanoparticles for gene delivery (Figure 1a,b). The characterization by Fourier transform infrared (FTIR), dynamic light scattering (DLS), NMR, and field emission scanning electron microscopy (FE-SEM) analysis resulted in optimal nanoparticles for efficient DNA loading and delivery (Figure 1c–h; Tables S1 and S2; Figures S1 and S2).19–21 A gel retardation assay was used to measure the DNA-to-nanoparticle ratio necessary for efficient transfection. The polyplexes of different N/P ratios (0.4–4 μg) evaluated by the gel retardation assay identified the nanoparticle-to-DNA ratio of 4:1 μg as the optimal formulation for efficient DNA loading (Figure 1f). The polyplexes were assessed for DNA transfection efficiency in HepG2 and Hep3B cells using a FLuc–EGFP reporter plasmid construct (Figures S3 and S4). As outlined earlier, the inclusion of SP94 peptide on the surface of nanoparticles resulted in variation in the zeta potential, whereas the gel retardation assay did not indicate any significant change in DNA encapsulation efficiency (Table S2 and Figure S2). We performed flow cytometry and optical bioluminescence imaging for evaluating the level of transfection by measuring gene expression while evaluating the absolute copy numbers of delivered plasmid DNA to HCC (HepG2 and Hep3B) cells using real-time PCR. The results showed very high transfection efficiency with the functional expression of delivered genes with SP94 targeting specific improvement in the delivery (Figures S3–S9).
Figure 1.
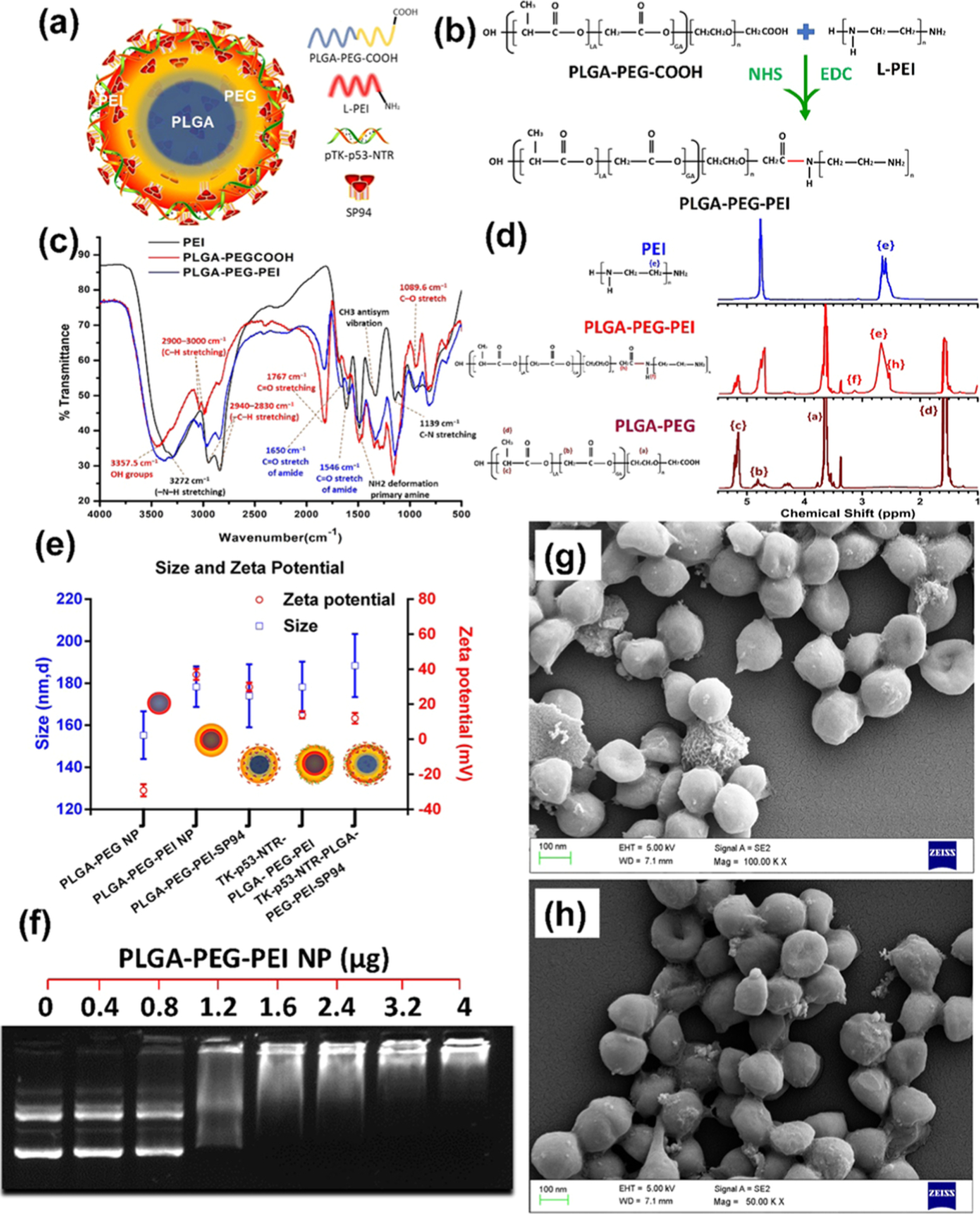
Synthesis and characterization of PLGA–PEG–PEI nanoparticles. (a) Schematic illustration of SP94-targeted PLGA–PEG–PEI (SP94–PLGA–PEG–PEI) nanoparticles loaded with TK–p53–NTR plasmid; (b) brief reaction scheme for PEI conjugation to PLGA–PEG nanoparticles; (c, d) FTIR and 1H NMR spectra of precursor and PLGA–PEG–PEI conjugate polymers; (e) size and zeta potential of PLGA–PEG–PEI nanoparticles after each modification; (f) DNA encapsulation efficiency of PLGA–PEG–PEI NPs at different N/P ratios by the gel retardation assay; (g, h) FE-SEM image of PLGA–PEG–PEI nanoparticles without (g) and with SP94 peptide (h).
Therapeutic Evaluation of TK–NTR and TK–p53–NTR Delivered Using PLGA–PEG–PEI Nanoparticles to HCC Cells of Different Phenotypes (p53+/+ and p53−/−).
The therapeutic potential of TK–NTR and TK–p53–NTR suicide genes was investigated against HepG2 (p53-wild-type) and Hep3B (p53-mutant) cells in the presence and absence of prodrugs CB1954 and/or GCV over time using flow-cytometry-based apoptosis analysis (Figures 2, S10, and S11).
Figure 2.
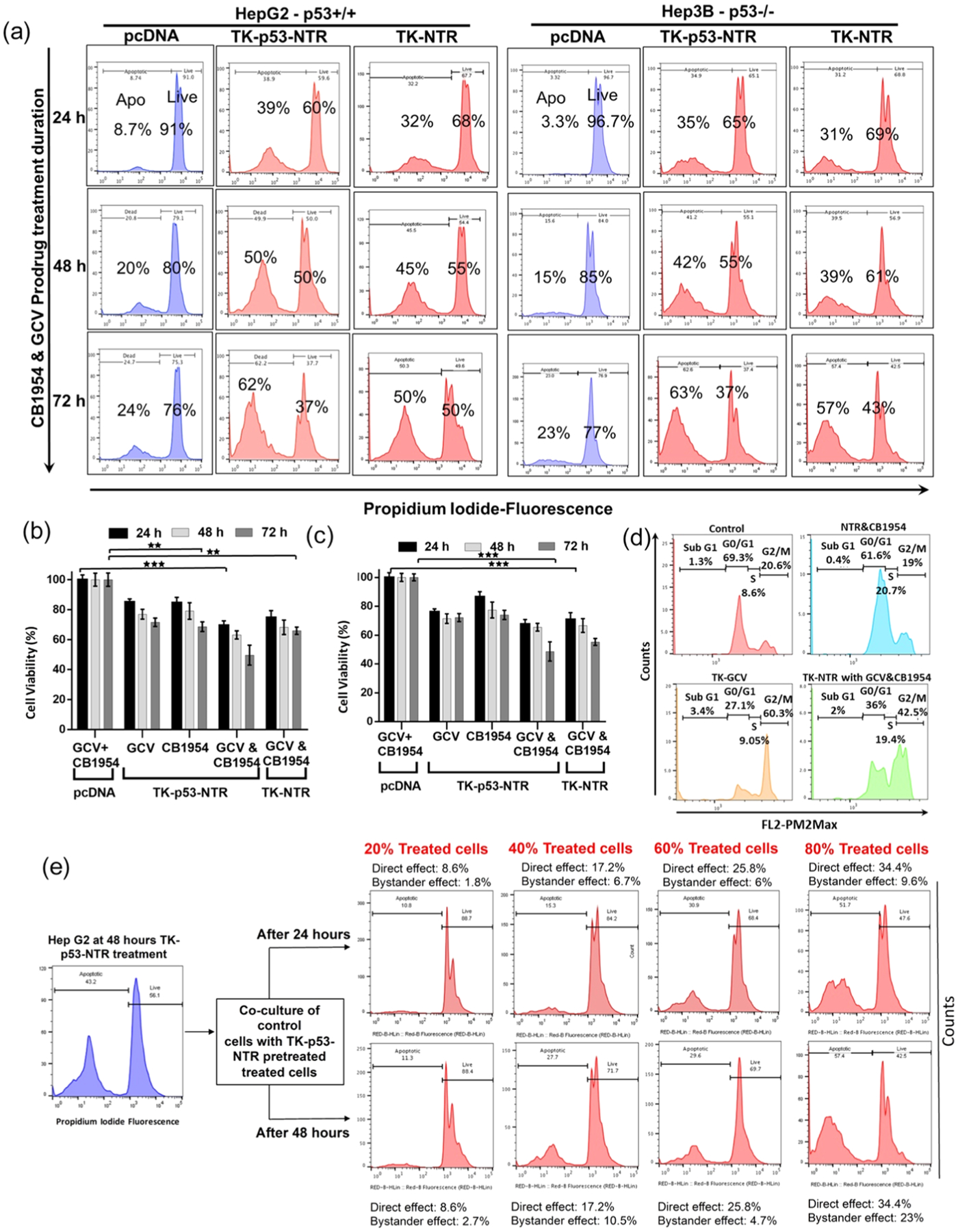
In vitro evaluation of the therapeutic efficacy of TK–p53–NTR gene delivered by SP94–PLGA–PEG–PEI nanoparticles in the presence of prodrugs and its bystander effects in HCC cells of different p53 status. (a) PI staining-based FACS analysis to measure the live and apoptotic populations in HepG2 (p53+/+) and Hep3B (p53−/−) cells transfected with pcDNA, or TK–NTR, or TK–p53–NTR using SP94–PLGA–PEG–PEI nanoparticles for 24 h and treated with GCV and CB1954 combinations for different durations (24, 48, and 72 h). (b, c) Quantitative plot of HepG2 (b) and Hep3B (c) cells transfected with pcDNA, TK–NTR, and TK–p53–NTR and treated with GCV, or CB1954, or GCV + CB1954. (d) Cell cycle analysis of HepG2 cells transfected with TK–NTR and treated with GCV, or CB1954, or GCV + CB1954. (e) Evaluation of TK–p53–NTR bystander effects in HepG2 cells by PI staining-based FACS analysis at 24 and 48 h for different coculture populations with various proportions of control cells to cells pretransfected with TK–p53–NTR by SP94–PLGA–PEG–PEI nanoparticles in the presence of prodrugs GCV and CB1954 (i.e., 20, 40, 60, and 80%).
Cell viability analysis revealed that both HepG2 and Hep3B cells treated with control DNA (pcDNA)-loaded polyplexes in combination with CB1954 and GCV did not result in significant decline in cell viability as the prodrugs remained inactive in the absence of TK or NTR gene expression, whereas we observed a significant time-dependent decline (p < 0.001) in cell viability by the cells delivered with TK–NTR or TK–p53–NTR and treated with prodrugs in combinations (CB1954 and/or GCV). Although the cells delivered with TK–p53–NTR expressed both TK and NTR enzymes, in the presence of GCV, the TK gene alone had limited antiproliferative effect, which was 85.12% cell viability compared to pcDNA-treated controls at 72 h. Likewise, NTR enzyme causes cytotoxicity only in the presence of CB1954 (resulting in 84.68% cell viability), whereas the concomitant activation of CB1954 and GCV by both NTR and TK enzymes led to significantly increased decline in cell viability (69.49%) even as early as 24 h after treatment, which improved further at 48 and 72 h post treatment (Figures 2a,b and S10). Furthermore, the role of supplementing p53 to TK–NTR in augmenting suicide gene therapy was clearly established by comparing therapeutic efficacy between TK–NTR and TK–p53–NTR gene therapies in HepG2 (p53+/+) and Hep3B (p53−/−) cells under similar conditions. The cell viability at every time point of analysis (i.e., 24, 48, and 72 h) revealed that TK–p53–NTR gene therapy could achieve consistently higher antiproliferative effects as compared to the TK–NTR construct, which ultimately rose to cell viability differences of 17.6% at 72 h in the case of HepG2 cells. These results clearly indicated that the expression of functionally active exogenous p53 gene renders HCC cells much more susceptible to the cytotoxic metabolites generated by the TK and NTR enzymes/prodrug systems. As an interesting outcome of this difference in p53 status, we observed that, despite the lower transfection efficiency of PLGA–PEG–PEI nanoparticles in Hep3B cells (i.e., 41.3%), the therapeutic outcome of using TK–p53–NTR in combination with prodrugs (GCV and/or CB1954) was similar to that of HepG2 cells, i.e., 44.7% (with HepG2 cells achieving a nearly 12% higher transfection efficiency than in Hep3B cells). Thus, in spite of the relatively lower gene delivery, the Hep3B cells were as equally susceptible to TK–p53–NTR therapy as HepG2. Again, this clearly demonstrated the role of p53 in improving TK–NTR gene therapy (Figures 2a–c and S11). Flow-cytometry-based cell cycle analysis was performed to study the effects of gene therapy by estimating different phases of cell cycle status after various treatment conditions. The expression of therapeutic genes along with the cotreatment using both the prodrugs led to the suppression of both G0/G1 and G2 populations, as well as the S phase arrest of cancer cells (Figure 2d). The detailed discussion regarding the cell cycle analysis results is presented in the Supporting Results and Discussion section.10,22
TK–p53–NTR Gene Therapy Delivered Using SP94–PLGA–PEG–PEI Nanoparticles Showed Significant Bystander Effects When Treated with GCV and CB1954 Prodrug Combinations in HCC Cells.
An advantageous feature of GDEPT is the “bystander effect”, where toxic metabolites generated by the cells expressing the suicide gene(s) not only have cytotoxic effects on the tumor cells that express the gene but also permeate into nearby cells and kill them eventually.23 Thus, we evaluated TK–p53–NTR suicide gene therapy for such a bystander effect, even at lower levels of transfection in HCC cells.24 To study the bystander effects of TK–p53–NTR triple gene therapy, we cocultured control HepG2 cells with stipulated proportions of HepG2 cells pretransfected with TK–p53–NTR using SP94–PLGA–PEG–PEI nanoparticles and treated with the prodrugs GCV and CB1954. To establish this coculture, HepG2 cells treated with TK–p53–NTR-loaded nanoparticles and prodrugs were harvested at 48 h post treatment and seeded along with the control HepG2 cells at predefined cell confluence percentages (i.e., 20, 40, 60, and 80%). PI staining-based FACS analysis of cocultured cells at 24 and 48 h revealed that, even in the absence of further addition of prodrugs, HepG2 populations pretreated with TK–p53–NTR-loaded nanoparticles and prodrugs could induce significant cytotoxicity to the cocultured control HepG2 cells in an initial seeding-proportion-dependent manner (Figure 2e).
The initial seeding population of the treated cell consisted of 43.2% apoptotic cells, and upon seeding them in a 40% ratio with control HepG2 cells, 27.7% apoptotic cells were achieved at the end of 48 h in coculture. Among this apoptotic population of 27.7%, a component of these cells arose from the apoptotic population present in the initial seeding population of treated cells, i.e., 17.3% (43.2 × 40%), whereas the remaining 10.4% apoptotic cells could be accounted as a consequence of the bystander effect, mediated by the treated cells on the control cells. This trend was observed upon increasing the ratio of control to treated cells. The consistent time- and TK–p53–NTR-expression-dependent therapeutic outcome observed in these experiments clearly established the significant contribution of the bystander effect in TK–p53–NTR triple gene therapy. In addition, we measured the diffusion of CytoCy5S signal from the cells transfected to express TK–p53–NTR into nontransfected cells through the bystander effect using fluorescence microscopy (Figure S12). The result is further discussed in the Supporting Results and Discussion section.
TK–p53–NTR Transfection Efficiency Using PLGA–PEG–PEI Nanoparticles in Tumor Spheroid Culture of HCC Cells.
Systemically delivered long circulating nanoparticles accumulate in solid cancer by a combination of extravasation from leaky tumor-associated vasculature and a lack of adequate lymphatic drainage resulting in a so-called enhanced permeability and retention (EPR) effect. However, the distribution of these delivery vehicles in tumors is heterogeneous and confined mainly to areas immediately surrounding tumor blood vessels. This can reduce the potential efficacy of systemically delivered gene therapy since distances between vessels can be relatively large and prominent avascular regions may be present in the cores of tumors. To test the gene therapy transfection efficiency and depth across many cell layers, we used a spheroid culture model where the inner layers of cells were expected to have no direct physical contact with the nanoparticles for transfection. The HepG2 tumor spheroids treated with TK–p53–NTR-loaded PLGA–PEG–PEI nanoparticles surface-functionalized with SP94 targeting peptide for 48 h and subsequent CytoCy5S incubation for 4 h enabled us to identify the extent of spheroid penetration achieved under Cy5 filter and fluorescence microscopy (Figure 3). We showed that, although the majority of TK–NTR expressing cells were present on the surface of the spheroid, a significant proportion of this population were present also in deeper portions of the spheroids, indicating that the nanoparticles could penetrate significantly into tumor spheroids to effect direct gene therapy. This, along with the bystander effect, demonstrates penetration of TK–p53–NTR gene therapy deep into cancer cell spheroids.25
Figure 3.
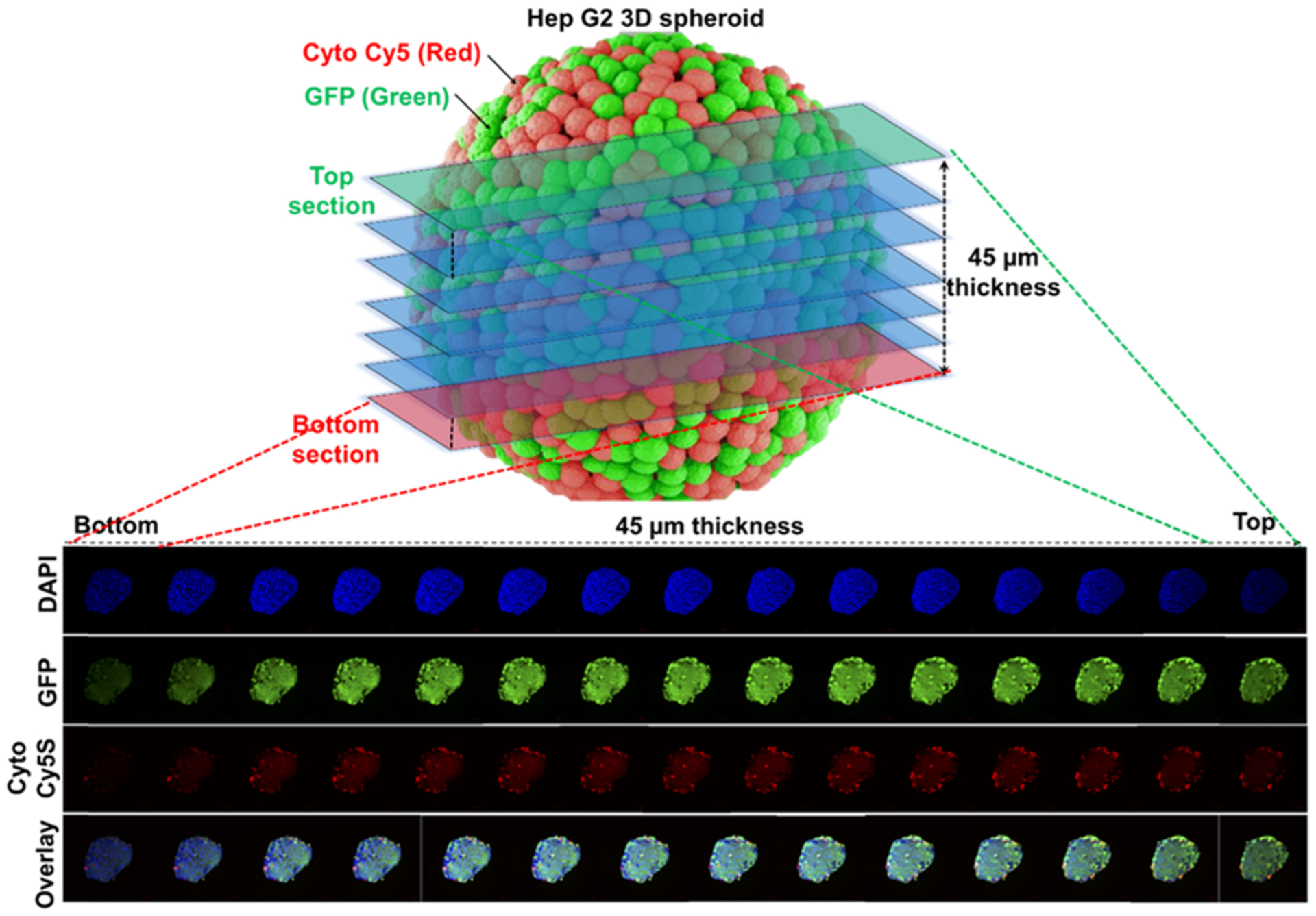
Evaluation of TK–p53–NTR transfection depth in three-dimensional (3D) spheroid culture in the presence of CytoCy5S substrate depicting the SP94-targeted PLGA–PEG–PEI nanoparticle transfection efficiency across different layers of cells in a 3D microsphere.
Immunoblot Analysis of Signaling Proteins and Validation of Apoptosis Induction Demonstrated by DNA Fragmentation Assay Showed TK–p53–NTR-Mediated Therapeutic Improvement in HCC Cells.
Since p53 plays a major role in apoptosis, we evaluated the p53 pathway proteins, and pro- and antiapoptotic proteins (p53, Bax, Bcl2, p21, p65, caspase 3) in HCC cells after different treatment conditions.26–30
As expected, the protein level significantly altered in cells treated with TK–p53–NTR with prodrug combination compared to those in control and TK–NTR-treated cells (Figure 4a–c). The results of the DNA fragmentation assay revealed strong apoptosis-mediated DNA damage in cells treated with either TK–NTR or TK–p53–NTR in combination with GCV/CB1954-mediated gene therapy compared to that in control cells (Figure 4c). The results are discussed in detail in the Supporting Results and Discussion section.
Figure 4.
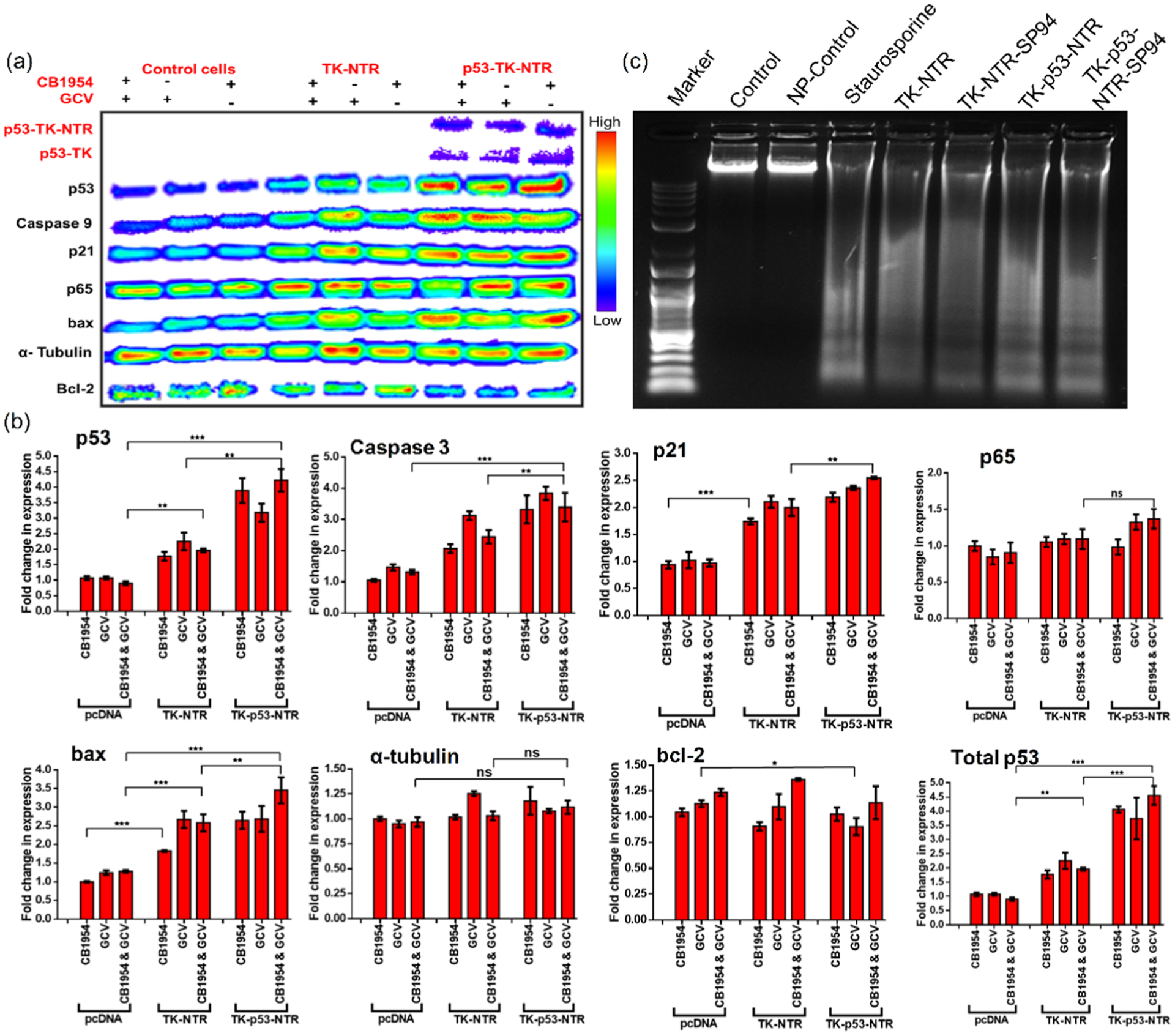
(a) Immunoblot analysis of apoptotic signaling proteins in HepG2 cells transfected with pcDNA, or pcDNA–TK–NTR, or pcDNA–TK–p53–NTR by PLGA–PEG–PEI nanoparticles with SP94 targeting peptide and treated with GCV, CB1954, or GCV plus CB1954 prodrugs for 72 h, and (b) its quantitative representation. (c) DNA fragmentation assay of cells harvested after 72 h of treatment.
Targeted Delivery of TK–p53–NTR Using PLGA–PEG–PEI Nanoparticles with and without SP94 Peptide and Following GCV/CB1954-Mediated Prodrug Therapy in Vivo in HCC Xenografts.
The in vitro results in various cell culture studies clearly established the improved antiproliferative potential of TK–p53–NTR triple gene therapy over TK–NTR delivered by PLGA–PEG–PEI NP, which further led us to evaluate the therapeutic efficacy of targeted triple gene therapy in HCC in vivo. We used nude mice bearing tumor xenografts of HepG2 cells expressing FLuc–EGFP on their lower flanks for the study. The tumor growth was assessed at different time points after the treatment by imaging the bioluminescence of tumor xenografts following intraperitoneal injection of substrate d-luciferin (3 mg/animal). The prodrugs GCV and CB1954 were administered intraperitoneally, whereas TK–p53–NTR-loaded nanoparticles with or without SP94 targeting peptide were administered intravenously at predetermined time points (Figure 5a). The study was terminated on day 29 (in spite of the observed recovery in treated mice) since, by that time, tumor volumes in the control mice reached the maximum permissible limits (>1.5 cm in diameter).
Figure 5.
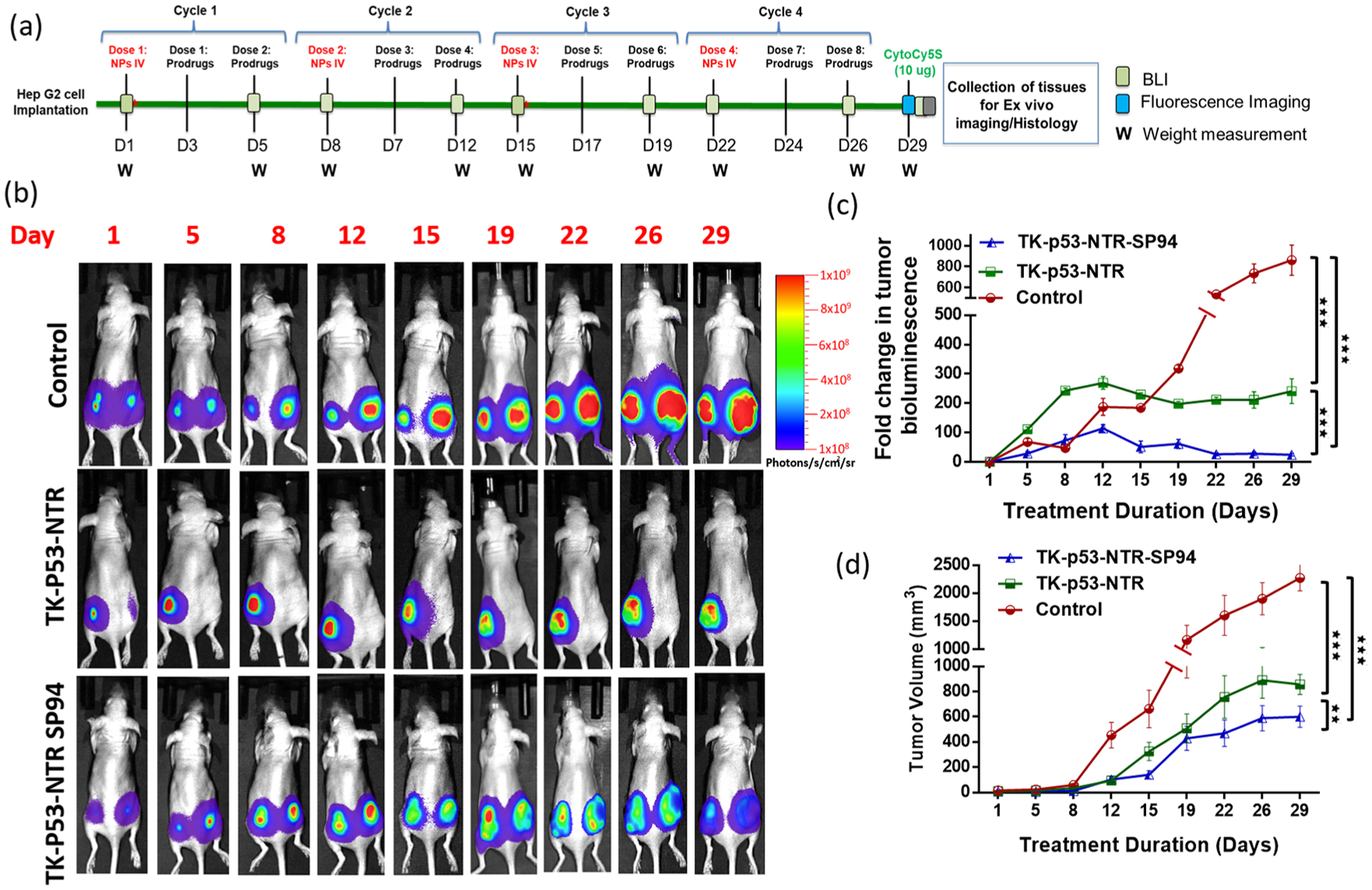
In vivo evaluation of therapeutic potential of TK–p53–NTR triple therapeutic gene in the presence of prodrugs (GCV and CB1954) in mice bearing HCC xenograft and delivered by SP94-targeted PLGA–PEG–PEI nanoparticles. (a) Schematic outline of the animal study workflow. (b) Bioluminescence imaging of nude mice bearing HepG2 tumor xenografts expressing FLuc–eGFP reporter gene at different time points (control denotes pcDNA delivered using SP94–PLGA–PEG–PEI nanoparticles, TK–p53–NTR denotes TK–p53–NTR triple gene delivered using PLGA–PEG–PEI nanoparticles, and TK–p53–NTR–SP94 denotes TK–p53–NTR triple gene delivered using SP94-targeted PLGA–PEG–PEI nanoparticles; all mice received prodrugs GCV and CB1954). (c) Its quantitative plot. (d) Estimation of tumor growth volume at different time points by caliper measurements.
Consistently, over all time points of the study, we found that SP94-targeted delivery of TK–p53–NTR gene effectively arrested the tumor growth much more efficiently than the nontargeted delivery approach, which was evident in terms of both tumor bioluminescence and tumor volume as compared to the nontargeted counterpart and control. The therapeutic expression of SP94-targeted triple gene therapy resulted in 3.8 ± 0.4-fold lower tumor bioluminescence and 35.3 ± 2.0-fold lower tumor volume with respect to controls on day 29 (Figure 5b–d). The comparison of the results of SP94-targeted delivery with the nontargeted TK–p53–NTR delivery showed ~2-fold higher tumor volume with respective bioluminescence signal, which clearly establishes the crucial implication of SP94 targeting peptide in this work.
Although control mice were also treated with equivalent amounts of prodrugs CB1954 and GCV after delivering pcDNA using SP94-targeted PLGA–PEG–PEI, the tumors continued to grow rapidly owing to the absence of the prodrug activating TK–p53–NTR therapeutic genes. Similar trends in tumor treatment response were observed in the case of TK–NTR dual gene therapy vector, with and without SP94-targeted delivery, but these were much lower than in the animals treated with the triple therapeutic gene (Figure S13). This finding further strengthens the observation of SP94-mediated targeted delivery of our triple therapeutic gene to HCC xenografts in this work. On the other hand, at the end point of the study, SP94-targeted TK–p53–NTR triple gene therapy was able to produce ~2-fold lower bioluminescence and tumor volume than obtained with SP94-targeted TK–NTR, which highlights the prominent role of p53 supplementation in improving TK–NTR therapeutic efficacy (Figures 5b–d and S13).
CytoCy5 Imaging and NTR Expression Mapping Further Support Efficient Gene Delivery in Vivo.
To establish a clear correlation between the promising therapeutic outcome and the expression levels of TK–NTR or TK–p53–NTR gene in the tumor, CytoCy5 was administered a day before study termination to trace the NTR expression level and biodistribution in the mice. In the presence of NTR, the substrate CytoCy5S is converted into its reduced form exhibiting red-shifted fluorescence, which can be traced by imaging the mice under a 640/690 nm excitation/emission filter setting in an optical imaging scanner. We did not observe any Cy5 fluorescence in the tumors of control mice upon receiving CytoCy5S, which clearly established the specificity of CytoCy5S as an NTR enzyme-specific substrate. On the other hand, in the case of mice treated with TK–NTR or TK–p53–NTR, prominent fluorescence signals were observed in the regions specifically overlapping with tumor locations indicating the localized activity of NTR enzyme in the tumor, confirming successful delivery of TK–NTR or TK–p53–NTR gene to the tumor cells and that the observed therapeutic effects are the sole outcome of the delivered therapeutic genes (Figure 6a).
Figure 6.
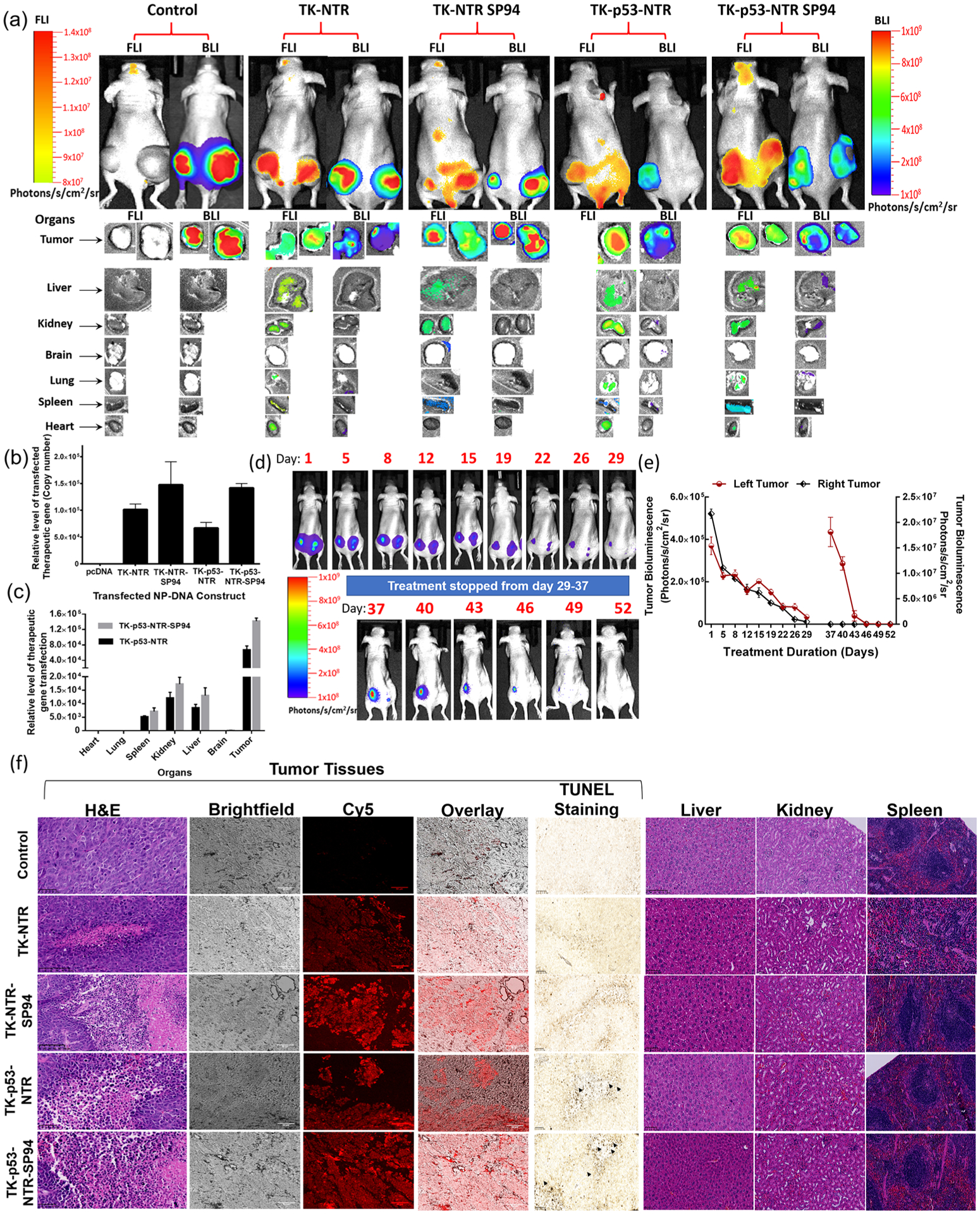
Evaluation of TK–p53–NTR therapeutic gene delivery by PLGA–PEG–PEI nanoparticles, expression in animals by CytoCy5S fluorescence imaging, biodistribution in tumor and other organs by ex vivo imaging and qPCR, and therapeutic response by bioluminescence imaging, ex vivo histology, and terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay. (a) Bioluminescence (HepG2–Fluc activity) and fluorescence 640/690 (CytoCy5 substrate conversion) images of nude mice in vivo (FLuc: 300 s, BLI: 50 s), and ex vivo (FLuc: 200 s, BLI: 50 s) imaging of tissues harvested after completion of the study. (b) Quantitative estimation of TK–NTR/TK–p53–NTR gene delivery to tumors and their biodistribution in different organs using qPCR analysis (c). (d, e) Evaluation of TK–p53–NTR therapeutic potential in complete eradication of tumor and its enhanced anticancer potential upon tumor relapse using tumor bioluminescence imaging and ROI quantitation plot of tumor BLI signal. The data are presented as mean ± standard error of the mean (SEM); * represents p < 0.05, ** represents p < 0.01, and *** represents p < 0.001. (f) Histologic analysis of tumor sections and organs of different treatment groups by H&E staining, CytoCy5S fluorescence tracking, and TUNEL assay.
Moreover, to evaluate the biodistribution of TK–NTR/TK–p53–NTR expression in treated mice, all major organs (i.e., brain, heart, lung, liver, spleen, kidney, and tumor) were harvested at study termination and imaged for the presence of the converted CytoCy5S fluorescence signal before various other ex vivo analyses. The ex vivo imaging results showed predominant CytoCy5S fluorescence in the tumors of animals that received gene delivery compared to that of controls. We also observed some fluorescence signals in the kidney, liver, and spleen but these were much lower compared to those of tumors (Figure 6a). Since the liver, kidney, and spleen are vital organs of the first-pass metabolism, the uptake of any substance introduced into the circulatory system can occur in these organs.8,31 We also observed that the topographical distribution of FLuc expression in tumors was exactly complementary to the Cy5-stained regions, which clearly indicated that TK–NTR or TK–p53–NTR expression is responsible for cell death in tumors.
In addition, to accurately evaluate SP94-mediated targeted gene delivery, we performed a real-time PCR analysis of the tumors and various organs for the presence of delivered genes in copy numbers. We isolated the DNA from the homogenized tissues by a modified plasmid extraction protocol optimized for extracting DNA from tissue homogenate. We used real-time PCR primers designed for estimating the levels of TK–NTR or TK–p53–NTR (Figure 6b,c). We compared the results with the animals that received control pcDNA and adopted the background amplification cycle threshold value as 1-fold for relatively quantifying the delivered TK–NTR and TK–p53–NTR levels in treated groups. Quantitation of TK–NTR or TK–p53–NTR delivery in tumors consistently presented a prominent increase in the tumors of animals delivered with SP94-targeted PLGA–PEG–PEI. In the case of TK–p53–NTR triple gene therapy vector, nontargeted delivery led to 66 922-fold (p < 0.001) increase in gene delivery, whereas upon targeting with SP94 peptide, the extent of gene delivery doubled (141 689-fold) (p < 0.001). This result clearly depicts the improved delivery efficiency of therapeutic genes using SP94 peptide-functionalized PLGA–PEG–PEI nanocarriers against HCC tumors. We further assessed the extent of gene delivery in major organs of mice treated with both nontargeted and SP94-targeted TK–p53–NTR genes. As observed for CytoCy5-based organ mapping of NTR enzyme activity, the PCR results also showed significant therapeutic vector levels for both nontargeted and SP94-targeted nanoparticles in kidneys, liver, and spleen (p < 0.001) but much lower compared to tumors (Figure 6b,c).
TK–p53–NTR and GCV/CB1954 Enzyme–Prodrug Gene Therapy Is Effective for Treating Recurrent Tumors in a Similar Manner as the Primary HCC Xenograft.
In cancer therapy, primary tumors generally show a better response to treatment. Recurrent tumors usually manifest with aggressive and drug-resistant phenotypes, displaying a poor response to previously used therapies. Thus, to assess the therapeutic efficacy of TK–p53–NTR gene therapy against recurrent HCC tumor, animals previously possessing HepG2 xenografts in both flanks and that had received TK–p53–NTR gene prodrug therapy resulting in complete response to therapy were then kept untreated for several days. These animals were routinely monitored for the development of recurrent tumors using bioluminescence imaging. One animal within the group developed a palpable recurrent tumor by day 37 (60-fold increase in bioluminescence signal compared to background, p < 0.001), with a concomitant increase in tumor volume. The right tumor, however, did not recur (Figure 6d,e). This animal was then re-treated using a similar regimen to that prior to day 29, as outlined in Figure 5a. The recurrent tumor responded to the treatment much faster than observed during the initial treatment phase. Complete tumor regression was observed after two cycles of gene and prodrug therapies. We acknowledge that definite conclusions cannot be drawn from this limited experiment intended as a pilot exploration of a concept. Nonetheless, this very preliminary result hints that recurrent tumors may be highly responsive to this triple gene therapy strategy. On account of this possibility, in future studies we will investigate and optimize the durations of TK–p53–NTR gene therapy relative to different stages of HCC development and progression and also study metastatic disease with chemotherapy-resistant cancer phenotypes to evaluate the potential superiority (or synergy) of suicide gene therapy over (with) standard chemotherapy.
Ex Vivo Histological Analyses.
H&E staining of tumor sections revealed that, in the control group, the tumor cells were closely aligned and possessed larger cell nuclei and higher nuclear/cytoplasm ratios than those in the other treatment groups. However, in the case of mice treated with TK–NTR or TK–p53–NTR plus prodrugs CB1954 and GCV, cancer cells exhibited sparse arrangement, cell shrinkage, fragmentation, and chromatin disappearance, indicating dispersed necrosis, apoptosis, and inflammation. As elucidated in ex vivo tissue imaging, the delivery and expression of gene therapy vectors corresponded to the distribution of fluorescent Cy5 signal generated by NTR-enzyme-catalyzed conversion of CytoCy5S, which predominantly coincided with apoptotic populations of cells. Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay of tumor sections revealed the extent of apoptosis in control and treatment groups (Figure 6f). The localized distribution pattern of CytoCy5S fluorescent substrate coincided with regions of higher apoptosis as observed in TUNEL assay, which clearly asserts the anticancer therapeutic potential of TK–NTR/TK–p53–NTR gene therapy against HCC. Furthermore, H&E staining of sections of different organs showed no sign of toxicity (Figure 6f).
DISCUSSION
GDEPT has shown great promise in cancer therapy. GDEPT involves tumor-targeted delivery of transgenes that encode enzymes capable of converting a nontoxic prodrug into an activated cytotoxic agent.32 An exclusive benefit of GDEPT over other therapeutic approaches is the bystander effect, whereby neighboring tumor cells that elude gene transfer are also killed following cell–cell diffusion of the activated cytotoxic metabolites.33 This eliminates the need for gene delivery to all of the cancer cells. Although suicide gene therapy is an efficient strategy for cancer therapy, the expression of suicide genes at therapeutically effective levels in cancer cells in vivo is a challenging task, which poses a major setback for clinical translation of gene therapy approaches. To address this limitation, the simultaneous expression of a combination of suicide genes that can target multiple cellular pathways has emerged as a potential alternative to achieve sufficient therapeutic efficiency even at low extents of gene delivery.
As such, in earlier studies we identified TK and NTR as novel therapeutic genes for combination therapy, where the toxic metabolites of these enzymes interfere with DNA synthesis and DNA damage to eliminate cancer cells independent of their cell cycle status. We evaluated this combination in various cancer types and achieved superior therapeutic effects in comparison to when each gene is expressed independently.10,34
HCC is a complex disease with a grim prognosis, and surgery can only benefit if the disease is diagnosed at an early stage. However, most HCC patients are diagnosed at very late stages, thus allowing very few treatment options. Furthermore, it has been estimated that more than 50% of HCC patients are diagnosed with a mutant p53 drug-resistant phenotype.2,11 Hence, in this study we constructed a triple therapeutic gene (TK–p53–NTR) by combining p53 with TK–NTR dual genes to rescue p53-mediated apoptotic function in cancer cells and therefore to potentially treat HCC patients independent of their p53 status. We evaluated the therapeutic efficacy of triple suicide gene therapy in HCC cells of p53-wild-type and p53-mutant phenotypes and observed a significant enhancement in anticancer effects in the presence of prodrugs in both cell types (Figure 2).
Delivery of suicide genes in in vitro conditions is often facile and efficient; the ensuing promising results can lead to false hopes of clinical translation. On the other hand, the selective delivery of therapeutic gene(s) to tumor cells in vivo presents considerable challenges in achieving full therapeutic potency of suicide gene therapy. The intricate complexities of in vivo delivery include the need to establish effective threshold levels of gene therapy vectors specifically to cancer cells, with minimal nonspecific accumulation in other tissues, as well as factoring in any potential toxicities associated with the delivery agents. Although viral-mediated gene delivery is efficient, it has limited clinical applications owing to the host immune response. Similarly, liposome-mediated delivery is restricted by toxicity.35
Therefore, to achieve targeted delivery of TK–p53–NTR triple therapeutic gene without eliciting any immunogenic response, we designed SP94 peptide-functionalized PLGA–PEG–PEI nanoparticles. These carriers exhibited efficient loading of therapeutic DNA, as well as enhanced delivery of genes in vitro in HCC cells and in vivo in HCC xenografts, all without significant toxicity. In this study, we conjugated L-PEI after PLGA–PEG nanoparticle preparation rather than grafting on top of PLGA–PEG nanoparticles,24 which allows precise control over the extent of PEI conjugation on the surface of nanoparticles while also reducing the toxicity. As such, we observed a reduction in toxicity with this approach without significant reduction in DNA loading compared to our previous study (Figures 1, S3, and S4).24 Additionally, subsequent functionalization of SP94 targeting peptide on the surface of PLGA–PEG did not affect either the PEI conjugation or the DNA loading efficiency (Figures S1 and S2).
SP94 is a HCC-specific targeting ligand identified from phage-displayed selections.36 In a recent report, glucose-regulated protein 78 (GRP78) membrane receptor, which is expressed prominently in HCC cells, has been identified as the target for SP94 peptide.37 As expected, conjugation of this peptide to PLGA–PEG–PEI nanoparticles resulted in the selective delivery of loaded therapeutic genes in vitro in HCC cells and in vivo in HCC xenografts. The evaluation of TK–p53–NTR gene prodrug therapy showed improved therapeutic efficiency in p53-mutant Hep3B cells compared to that of the TK–NTR dual gene therapy system (Figure 2). This finding corroborates a previous comparative therapeutic efficacy study, where Hep3B cells with p53-mutant phenotype were relatively more resistant to common anticancer drugs (e.g., doxorubicin and paclitaxel) when compared to HepG2 cells with wild-type-p53 phenotype.38 Independently, we also validated the delivery of TK–p53–NTR gene in HCC cells in vitro and in vivo xenografts for NTR enzyme activity by using a fluorescent substrate (CytoCy5S) conversion assay that we developed and evaluated previously (Figure S7).39,40
p53 is a tumor suppressor gene that regulates the apoptotic pathway in cells. The downstream targets of p53 include p21 (cell cycle arrest), Bax (inducer of apoptosis), and caspase 3 (inducer of apoptosis) have important roles in cell cycle arrest and apoptosis. Since we implemented the strategy of supplementing p53 to synergize the anticancer therapeutic response through enhancing apoptosis in p53-wild-type cancer cells, as well as through rescuing the p53 pathway in p53-mutant cancer cells, we studied the effects of this by scrutinizing downstream functions of p53 after TK–p53–NTR delivery into HCC cells in vitro and in vivo. We observed a prominent increase in DNA fragmentation with the concomitant increase in apoptotic protein levels in cells transfected with TK–p53–NTR triple gene compared to those of TK–NTR and controls (Figure 2). Our in vivo study also demonstrated successful targeted delivery and expression of TK–NTR and TK–p53–NTR when using SP94-functionalized PLGA–PEG–PEI nanoparticles. CytoCy5 imaging revealed that not only the expression of TK–NTR and TK–p53–NTR was higher in the case of SP94-functionalized nanoparticles, but it was also predominantly confined to tumors, in comparison to findings with the use of Sc-SP94-functionalized nanoparticles. The use of cancer cells labeled with bioluminescence reporter also facilitated the in vivo imaging and measurement of tumor growth or functional reduction.
In general, most of the suicide gene therapy studies to date have focused on improving the therapeutic gene constructs, while little attention has been given to developing a safe and targeted delivery of these genes to cancer cells in vivo; this remains a significant obstacle in clinical translation of these promising approaches. Here, we introduce important improvements to both features: first, the therapeutic efficacy of the TK–NTR dual gene is augmented by additional supplementation of the p53 gene, which effectively regresses HCC cancers independent of p53 status. Second, the SP94 peptide-functionalized PLGA–PEG–PEI nanoparticles we designed achieve targeted delivery of the triple gene construct (TK–p53–NTR) without significant off-target effects. Overall, we demonstrate the superior therapeutic efficacy of the TK–p53–NTR triple gene therapy both in vitro and in vivo studies. We believe that the described approach of cell-surface targeting of HCC using nanoparticles encapsulating this new triple therapeutic gene will be an exciting addition to cancer gene therapy options. This novel strategy could also be tailored to other solid cancers.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the Canary Center at Stanford, Department of Radiology, for facility and resources. The authors also thank SCi3 small animal imaging service center, Stanford University School of Medicine, for providing imaging facilities and data analysis support. Rayhaneh Afjei is acknowledged for her technical assistance in some of the experiments. The authors also acknowledge Dr. José G. Vilches-Moure, Veterinary pathologist, Animal Histology Services (AHS), for his advice regarding the histological analysis of animal tissues. The authors dedicate this work in memory of Dr. Juergen K. Willmann.
Funding
This research was supported by NIH R01CA209888 and NIH R21EB022298.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.9b20071.
PLGA–PEG–PEI nanoparticle synthesis and characterization procedures, pDNA loading efficiency in PLGA–PEG–PEI nanoparticles, in vitro evaluation of TK–NTR and TK–p53–NTR gene delivery by NPs, evaluation of TK–NTR and TK–p53–NTR gene expressions and therapeutic efficacy in cells by immunoblot assay, flow cytometry analysis, and fluorescence microscopy and in vivo assessment of TK–NTR therapeutic potential in HCC xenograft mice model in terms of tumor volume and bioluminescence (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsami.9b20071
The authors declare no competing financial interest.
REFERENCES
- (1).Rawla P; Sunkara T; Muralidharan P; Raj JP Update in Global Trends and Aetiology of Hepatocellular Carcinoma. Contemp. Oncol 2018, 22, 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hussain SP; Schwank J; Staib F; Wang XW; Harris CC Tp53 Mutations and Hepatocellular Carcinoma: Insights into the Etiology and Pathogenesis of Liver Cancer. Oncogene 2007, 26, 2166–2176. [DOI] [PubMed] [Google Scholar]
- (3).Kamijo T; Weber JD; Zambetti G; Zindy F; Roussel MF; Sherr CJ Functional and Physical Interactions of the Arf Tumor Suppressor with P53 and Mdm2. Proc. Natl. Acad. Sci. U.S.A 1998, 95, 8292–8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kobet E; Zeng X; Zhu Y; Keller D; Lu H Mdm2 Inhibits P300-Mediated P53 Acetylation and Activation by Forming a Ternary Complex with the Two Proteins. Proc. Natl. Acad. Sci. U.S.A 2000, 97, 12547–12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Karjoo Z; Chen X; Hatefi A Progress and Problems with the Use of Suicide Genes for Targeted Cancer Therapy. Adv. Drug Delivery Rev 2016, 99, 113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhang J; Kale V; Chen M Gene-Directed Enzyme Prodrug Therapy. AAPS J. 2015, 17, 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sukumar UK; Packirisamy G Bioactive Core-Shell Nanofiber Hybrid Scaffold for Efficient Suicide Gene Transfection and Subsequent Time Resolved Delivery of Prodrug for Anticancer Therapy. ACS Appl. Mater. Interfaces 2015, 7, 18717–18731. [DOI] [PubMed] [Google Scholar]
- (8).Bridgewater JA; Knox RJ; Pitts JD; Collins MK; Springer CJ The Bystander Effect of the Nitroreductase/Cb1954 Enzyme/Prodrug System Is Due to a Cell-Permeable Metabolite. Hum. Gene Ther 1997, 8, 709–717. [DOI] [PubMed] [Google Scholar]
- (9).Xu G; McLeod HL Strategies for Enzyme/Prodrug Cancer Therapy. Clin. Cancer Res 2001, 7, 3314–3324. [PubMed] [Google Scholar]
- (10).Sekar TV; Foygel K; Ilovich O; Paulmurugan R Noninvasive Theranostic Imaging of Hsv1-Sr39tk-Ntr/Gcv-Cb1954 Dual-Prodrug Therapy in Metastatic Lung Lesions of Mda-Mb-231 Triple Negative Breast Cancer in Mice. Theranostics 2014, 4, 460–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chan KT; Lung ML Mutant P53 Expression Enhances Drug Resistance in a Hepatocellular Carcinoma Cell Line. Cancer Chemother. Pharmacol 2004, 53, 519–526. [DOI] [PubMed] [Google Scholar]
- (12).Patil S; Gao YG; Lin X; Li Y; Dang K; Tian Y; Zhang WJ; Jiang SF; Qadir A; Qian AR The Development of Functional Non-Viral Vectors for Gene Delivery. Int J Mol Sci 2019, 20, No. 5491.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lü JM; Wang X; Marin-Muller C; Wang H; Lin PH; Yao Q; Chen C Current Advances in Research and Clinical Applications of Plga-Based Nanotechnology. Expert Rev. Mol. Diagn 2009, 9, 325–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Devulapally R; Sekar NM; Sekar TV; Foygel K; Massoud TF; Willmann JK; Paulmurugan R Polymer Nanoparticles Mediated Codelivery of Antimir-10b and Antimir-21 for Achieving Triple Negative Breast Cancer Therapy. ACS Nano 2015, 9, 2290–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zou W; Liu C; Chen Z; Zhang N Preparation and Characterization of Cationic Pla-Peg Nanoparticles for Delivery of Plasmid DNA. Nanoscale Res. Lett 2009, 4, 982–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lo A; Lin CT; Wu HC Hepatocellular Carcinoma Cell-Specific Peptide Ligand for Targeted Drug Delivery. Mol. Cancer Ther 2008, 7, 579–589. [DOI] [PubMed] [Google Scholar]
- (17).Euhus DM; Hudd C; LaRegina MC; Johnson FE Tumor Measurement in the Nude Mouse. J. Surg. Oncol 1986, 31, 229–234. [DOI] [PubMed] [Google Scholar]
- (18).Tomayko MM; Reynolds CP Determination of Subcutaneous Tumor Size in Athymic (Nude) Mice. Cancer Chemother. Pharmacol 1989, 24, 148–154. [DOI] [PubMed] [Google Scholar]
- (19).Chen XA; Zhang LJ; He ZJ; Wang WW; Xu B; Zhong Q; Shuai XT; Yang LQ; Deng YB Plasmid-Encapsulated Polyethylene Glycol-Grafted Polyethylenimine Nanoparticles for Gene Delivery into Rat Mesenchymal Stem Cells. Int. J. Nanomed 2011, 6, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Figueiredo M; Esenaliev R Plga Nanoparticles for Ultrasound-Mediated Gene Delivery to Solid Tumors. J. Drug Delivery 2012, 2012, No. 767839.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Mohammadian F; Abhari A; Dariushnejad H; Nikanfar A; Pilehvar-Soltanahmadi Y; Zarghami N Effects of Chrysin-Plga-Peg Nanoparticles on Proliferation and Gene Expression of Mirnas in Gastric Cancer Cell Line. Iran. J. Cancer Prev 2016, 9, No. e4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Teng G; Ju Y; Yang Y; Hua H; Chi J; Mu X Combined Antitumor Activity of the Nitroreductase/Cb1954 Suicide Gene System and Gamma-Rays in Hela Cells in Vitro. Mol. Med. Rep 2016, 14, 5164–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Greco O; Dachs GU Gene Directed Enzyme/Prodrug Therapy of Cancer: Historical Appraisal and Future Prospectives. J. Cell. Physiol 2001, 187, 22–36. [DOI] [PubMed] [Google Scholar]
- (24).Devulapally R; Lee T; Barghava-Shah A; Sekar TV; Foygel K; Bachawal SV; Willmann JK; Paulmurugan R Ultrasound-Guided Delivery of Thymidine Kinase-Nitroreductase Dual Therapeutic Genes by Pegylated-Plga/Pei Nanoparticles for Enhanced Triple Negative Breast Cancer Therapy. Nanomedicine 2018, 13, 1051–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Perry JL; Reuter KG; Luft JC; Pecot CV; Zamboni W; DeSimone JM Mediating Passive Tumor Accumulation through Particle Size, Tumor Type, and Location. Nano Lett. 2017, 17, 2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Fujiwara K; Daido S; Yamamoto A; Kobayashi R; Yokoyama T; Aoki H; Iwado E; Shinojima N; Kondo Y; Kondo S Pivotal Role of the Cyclin-Dependent Kinase Inhibitor P21waf1/Cip1 in Apoptosis and Autophagy. J. Biol. Chem 2008, 283, 388–397. [DOI] [PubMed] [Google Scholar]
- (27).Vaseva AV; Moll UM The Mitochondrial P53 Pathway. Biochim. Biophys. Acta, Bioenerg 2009, 1787, 414–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wu JM; Sheng H; Saxena R; Skill NJ; Bhat-Nakshatri P; Yu M; Nakshatri H; Maluccio MA Nf-Kappab Inhibition in Human Hepatocellular Carcinoma and Its Potential as Adjunct to Sorafenib Based Therapy. Cancer Lett. 2009, 278, 145–155. [DOI] [PubMed] [Google Scholar]
- (29).Zheng TS; Schlosser SF; Dao T; Hingorani R; Crispe IN; Boyer JL; Flavell RA Caspase-3 Controls Both Cytoplasmic and Nuclear Events Associated with Fas-Mediated Apoptosis in Vivo. Proc. Natl. Acad. Sci. U.S.A 1998, 95, 13618–13623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Finucane DM; Bossy-Wetzel E; Waterhouse NJ; Cotter TG; Green DR Bax-Induced Caspase Activation and Apoptosis Via Cytochrome C Release from Mitochondria Is Inhibitable by Bcl-Xl. J. Biol. Chem 1999, 274, 2225–2233. [DOI] [PubMed] [Google Scholar]
- (31).Navarro SM; Darensbourg C; Cross L; Stout R; Coulon D; Astete CE; Morgan T; Sabliov CM Biodistribution of Plga and Plga/Chitosan Nanoparticles after Repeat-Dose Oral Delivery in F344 Rats for 7 Days. Ther. Delivery 2014, 5, 1191–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Anderson WF Gene Therapy Scores against Cancer. Nat. Med 2000, 6, 862–863. [DOI] [PubMed] [Google Scholar]
- (33).Dachs GU; Hunt MA; Syddall S; Singleton DC; Patterson AV Bystander or No Bystander for Gene Directed Enzyme Prodrug Therapy. Molecules 2009, 14, 4517–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sekar TV; Foygel K; Willmann JK; Paulmurugan R Dual-Therapeutic Reporter Genes Fusion for Enhanced Cancer Gene Therapy and Imaging. Gene Ther. 2013, 20, 529–537. [DOI] [PubMed] [Google Scholar]
- (35).Yue J; Wu J; Liu D; Zhao X; Lu WW Bmp2 Gene Delivery to Bone Mesenchymal Stem Cell by Chitosan-G-Pei Nonviral Vector. Nanoscale Res. Lett 2015, 10, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lo A; Lin C-T; Wu H-C Hepatocellular Carcinoma Cell-Specific Peptide Ligand for Targeted Drug Delivery. Mol. Cancer Ther 2008, 7, 579–589. [DOI] [PubMed] [Google Scholar]
- (37).Jiang B; Zhang R; Zhang J; Hou Y; Chen X; Zhou M; Tian X; Hao C; Fan K; Yan X Grp78-Targeted Ferritin Nanocaged Ultra-High Dose of Doxorubicin for Hepatocellular Carcinoma Therapy. Theranostics 2019, 9, 2167–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chang AY; Wang M In-Vitro Growth Inhibition of Chemotherapy and Molecular Targeted Agents in Hepatocellular Carcinoma. Anticancer Drugs 2013, 24, 251–259. [DOI] [PubMed] [Google Scholar]
- (39).Bhaumik S; Sekar TV; Depuy J; Klimash J; Paulmurugan R Noninvasive Optical Imaging of Nitroreductase Gene-Directed Enzyme Prodrug Therapy System in Living Animals. Gene Ther. 2012, 19, 295–302. [DOI] [PubMed] [Google Scholar]
- (40).Sekar T; Foygel K; Willmann J; Paulmurugan R Dual-Therapeutic Reporter Genes Fusion for Enhanced Cancer Gene Therapy and Imaging. Gene Ther. 2013, 20, 529–537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.