

Abstract
Proxima is a molecular perception library designed with a double purpose: to be used with immersive molecular viewers (thus providing any required feature not supported by third party libraries) and to be integrated in workflow managers thus providing the functionalities needed for the first steps of molecular modeling studies. It thus stands at the boundary between visualization and computation. The purpose of the present article is to provide a general introduction to the first release of Proxima, describe its most significant features, and highlight its performance by means of some case studies. The current version of Proxima is available for evaluation purposes at https://bitbucket.org/sns-smartlab/proxima/src/master/.
Introduction
The goal of molecular perception algorithms is to build topological models of molecular systems starting from the minimal information conveyed by atomic symbols and coordinates, to be next used for scientific visualization purposes, or for building initial guesses for quantum mechanics (QM) and/or molecular mechanics (MM) simulations. As a matter of fact, molecular perception has been (quite restrictively) defined as “the process of distinguishing between different chemical environments in order to assign force field parameters”.1
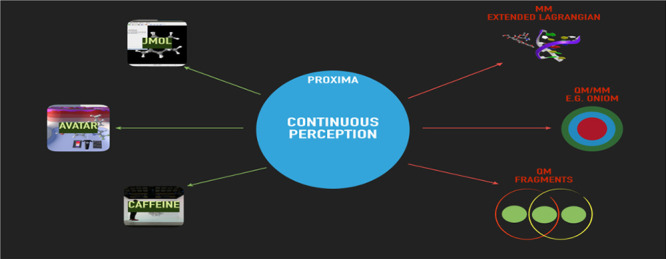
In this work, we present Proxima, the molecular perception library under development at the SMART laboratory of Scuola Normale Superiore.2 The purpose of Proxima is (i) to provide information about bonds, rings, atom types, and related data to imersive visualization software (e.g., Caffeine3), (ii) to compute molecular properties such as atomic charges, atomic hybridization, or hydrogen bonds, (iii) to be connected to workflow managers in order to exploit property calculations to prepare input files, and (iv) to allow user-friendly use also by nonexperts. In other words, our aim is to build a tool standing in the middle between computation and visualization (see Figure 1).
Figure 1.
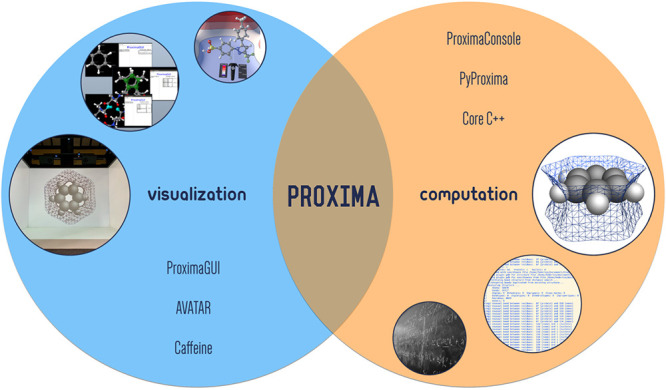
Proxima is a molecular perception library aimed at computational chemistry but also employable by molecular visualization software.
We tried to explore some innovative strategies for molecular perception like, e.g., the computation of continuous bond orders by setting sensible limits to atomic valence and thus taking into account uncertainty in the starting atom coordinates.
As an example of application of Proxima for computational purposes, the unsupervised computation of biaryl torsional parameters in complex systems is analyzed. For visualization tasks we demonstrate the remarkable flexibility of the library by showing its use in conjunction with Caffeine, the stand alone ProximaGUI, and the AVATAR Virtual Reality application4 for exploring potential energy surfaces computed by Proxima itself.
Methodology
Perception
Starting from Cartesian coordinates and atom numbers (see the section about the Proxima I/O in the Supporting Information) we proceed with chemical perception. The first step is the perception of different kinds of bonds (currently, covalent and hydrogen bonds are perceived).
Bond Perception
Several characteristics (e.g., strength, polarity, etc.) of chemical bonds are tuned by the nature of the involved atoms and by their environment. We decided to use the same mathematical form not only for detecting the presence of a bond, but also for evaluating its strength. In other words, Proxima is based on the concept of continuous chemical perception. In particular, the strength of a bond can be written as
| 1 |
Where B(θ) is an angular term required for three-body interactions (e.g., hydrogen bonds). Both the A and the B functions have the same mathematical form shown below for the radial component:
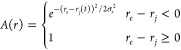 |
2 |
This function is continuous also with its first derivative. The nature of the parameters (re and σr in the example) depends on the type of bond considered.
Covalent Bonds
Traditionally, the computation of the covalent bond connectivity is performed by evaluating the distance between each pair of atoms without further corrections related to atom types. If the distance between two atoms is longer than a small threshold (here 0.4 Å) and shorter than the sum of their respective covalent radii, plus some “tolerance” factor (usually 0.4 Å7), then a covalent bond is assigned to the given pair. We increased the sensitivity of this formulation by including the electronegativity in the theoretical bond length expression, in a way that has been proposed for the first time by Schomacker and Stevenson8 and then improved by Porterfield:9
| 3 |
This is the lower bound to the single covalent bond length (the tolerance is taken as 0.4).
Here, re is the distance parameter used in eq 2 (in Angstroms), Rcova and Rcov are the covalent radii for the atoms “a” and “b” respectively, Δχ is the electronegativity difference between the two species (values are taken from a work of Little and Jones10), whose contribution is significant especially when dealing with ionic interactions. Since the covalent bond is a two-body interaction, only the radial component of eq 1 is considered, and the σr of eq 2 is taken as
| 4 |
A new covalent bond is assigned only if the strength of the new bond is above 0.4. An example is the potassium channel of Figure 2. Here, OpenBabel 2.3.2 overestimates the number of covalent bonds with the potassium atoms, whereas Proxima correctly describes such system thanks to a proper account of the electronegativity contribution (see eq 3). Proxima also checks the consistence of bond assignments by removing excess bonds that make the coordination of the atom exceed its valence. Once bonds are detected, valence and dihedral angles can be straightforwardly determined.
Figure 2.
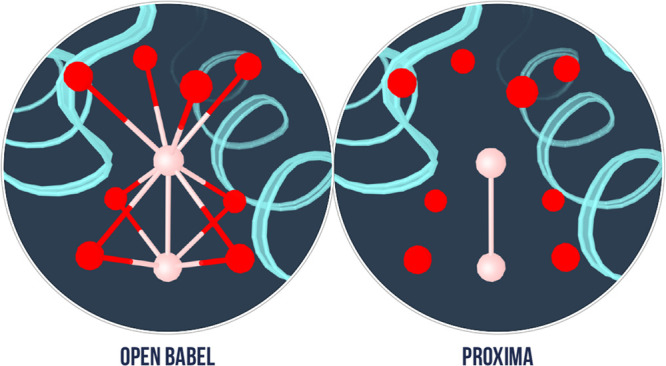
Looking at a difficult case: the potassium atoms within the potassium channel (PDB ID: 1BL8). (left) Bond perception performed by the Avogadro software5 (version 1.1.1) (using the OpenBabel library6 (version 2.3.2)): number of bonds perceived, 2918. (right) Bond perception performed by Proxima: number of bonds perceived, 2897.
Hydrogen Bonds
Following a simple geometric approach widely used in MM, if the donor–hydrogen–acceptor geometry is compatible with a reference geometry, then an hydrogen bond is assigned to such an assembly of atoms. Since this is a three-body interaction, we use the general expression of eq 1 including also the angular contribution (θ is the donor–H–acceptor angle).
Proxima employs the parameters proposed by Pagliai11−13 and supports nitrogen and oxygen atoms both as donors and acceptors.
Ring Perception
Proxima implements the Horton’s algorithm for the perception of chemical rings.14 Despite its worst-case complexity of O(n7), the algorithm can be applied only to the cyclic portions of the system (called blocks in the following), which represent a very small part of the molecular systems of current interest for Proxima applications (e.g., proteins). To this end, we have implemented the block partitioning scheme suggested by Fan, Doucet, and Barbu.15 Some benchmarks are shown in the Supporting Information.
Hybridization Perception
The perception of hybridization is performed by looking at the bond angles around each atom. This is similar to the OpenBabel approach, with some additional tweaks like, e.g. the hybridization of the most common transition elements. After computing the minimum and the maximum bond angles for each atom, Proxima assigns the hybridization in relation to an heuristic, provided that the considered atom has a sufficient number of bonds. Thus, terminal atoms must be processed separately. Moreover, it is important to note that in very small ring structures the values of the angles are not really representative of the hybridization state. Thus, there are two categories of atoms that Proxima processes separately: (i) cyclic planar atoms and (ii) terminal atoms. The hybridization of all atoms belonging to planar cycles is taken as an sp2. Next, the average torsion angle of those atoms is computed. Typically, the hybridization of terminal atoms is left unknown. There are some special cases, though, that are explicitly treated such as oxygen, nitrogen, and carbon atoms. For cycles that have four or five bonds, the cycle is considered to be planar only if the mean torsion angle is below 7.5°, whereas this threshold is increased to 15° for larger cycles. Further details are included in the Supporting Information.
Charges and Bond Orders
Electrostatic interactions are the longest ranged contributions in MM force fields and usually account for the largest fraction of the total energy. Therefore, their computation is a critical component of any FF and the simplest and most widespread approach is the computation of fixed point charges assigned to atoms. These can be either perceived directly on the basis of tabulated quantities (e.g., electronegativity) and coordinates or be the results of a quantum chemistry calculation (e.g., RESP16 or Hirshfeld17 charges). The possibility of computing charges with a cheap perception model can be a useful feature in the initial stages of a study, so that several perception libraries and molecular editors offer such feature. Along these lines Proxima allows computation of the point charges using the Gasteiger18 (popular in the medicinal chemistry domain) and the QeQ19,20 (which is the default method of assigning charges in UFF) methods. Both methods depend on tabulated atomic parameters and possibly on more refined atom types (the CHARMM ones21 for QeQ).
Concerning bond orders, the goal of Proxima is to compute continuous quantities instead of the discrete ones usually employed by other software. We have already implemented a method based on bond lengths, which is, however quite error prone for structures with low spatial resolution. As a consequence, we have also developed a custom filtering procedure, which enforces correct boundaries. It is important to notice how we decoupled the problem of finding bond orders from the problem of finding the strength of a bond.
Distance Based Bond Order
The custom Proxima method for computing bond orders is based on the knowledge of the covalent radii for single, double and triple bonds for all the elements of the periodic table, which were tabulated by Pyykkö.22
In order to move from a discrete to a continuous approach, we have fitted the three covalent bond radii of each element (rcov,s, rcov,d, and rcov,t) with the corresponding bond order values (1, 2, and 3). The function used to fit such quantities is an exponential:
| 5 |
Here, rcov is the analogous of the covalent bond radius in a continuous formulation and represents the contribution of the atom to the total bond length. BO is the overall bond order for the bond. By simple algebraic manipulations, one obtains:
| 6 |
Proxima Filtering Procedure
As mentioned above, distance-based bond orders show a huge dependence on the reference molecular geometry. However, it is possible to check the consistency of a given bond order assignment by looking at the sum of bond orders around each atom. If this value is not compatible with the valence of the atom itself, then the given bond orders are not the correct ones. Minimization of the squared distance between the new bond orders and the original values with the constraint of correct atom valences leads to a Lagrange multiplier problem, which is solved in Proxima by standard routines of the Eigen linear algebra library.23
Application to Biaryl Systems
As a case study, we consider the problem of finding torsional parameters for biaryl groups in molecules. A recent work by Liu, Barigye, Shahamat, Labute, and Moitesser24 has shown that it is possible to relate one torsional parameter to the difference in electronegativity between the atoms of the two aromatic rings involved in the biaryl. The software Liu24 developed (called HTEQ) is a Java application that takes a mol2 file containing the biaryl molecule and a text file containing the serial numbers of the atoms of the inter-ring bond as inputs. The goal was to use the perception algorithms of Proxima for
Finding bridge bonds
Extracting the local geometry of the biaryl group from the overall system
Finding Bridge Bonds
In order to find bridge bonds, Proxima iterates over each couple of planar rings found in the system, tagging as bridge bond any bond connecting one atom of the first ring with one atom of the second ring. Next, the serial numbers of the atoms involved in each bridge bond are stored in the format required by the HTEQ software.
Extracting Local Geometry
Once bridge bonds have been found, Proxima starts “cutting” the molecular system around each of them in order to isolate the biaryl group from the rest of the molecule. This system is then stored in the mol2 file required as input by the software that computes the torsional parameters. The combination of this cutting system with the serial numbers of the bridge atoms (retrieved in the previous step) provides all the information needed by the HTEQ software.
As an example, in Figure 3 a celecoxib molecule is shown. Here, the highlighted atoms are the bridge atoms of this system automatically detected by Proxima. Selecting a cutting distance of four bonds, one obtains the “cut” system around the red bridge shown on the right side of Figure 3, where the cut atoms are replaced by hydrogen atoms placed at the correct distance (that is the sum of the respective covalent radii). In real applications, higher cutting distances are recommended. In this specific case, the HTEQ software predicts a V2 torsional parameter of −1.142 kcal/mol for the bridge bond highlighted in red, and a V2 torsional parameter of −0.942 kcal/mol for the bridge bond highlighted in green.
Figure 3.
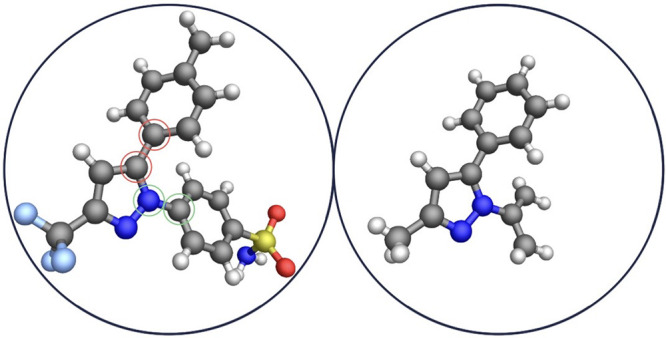
Celecoxib molecule on the left. On the right, the molecular system cut at a distance of four bonds around the red atoms.
Immersive Virtual Reality (IVR) Viewers
Proxima can be used as perception tool by our in-house IVR molecular viewer Caffeine.3 Moreover, the computation of molecular electrostatic potential surfaces is currently implemented in Proxima. This method uses the atomic charges (either Gasteiger or QeQ charges) to compute the electrostatic potential in each voxel of the bounding box (whose dimensions are chosen by the user) for the given molecular system. These electrostatic potential data can be saved in a cube file,25 which can be visualized by several visualization tools such as VMD,26 JMol,27 etc.
Computation and Virtual Exploration of Potential Energy Surfaces
To show the versatility of Proxima, we present a test case for the computation of potential energy surfaces with Proxima and their visualization in interactive VR environments. In particular, we are working on a virtual reality application (called AVATAR4), developed using the popular Unity game engine,28 for the interactive visualization of three-dimensional energy surfaces with the HTC Vive VR headset.29 The interesting aspect of such application is not only the visualization of the energy surface in virtual reality but also the possibility for the user to walk onto the surface and see how the molecular geometry changes along the path. For this test case we used Proxima to compute the potential energy surface (PES) of the celecoxib molecule associated with the rotation of the two inter-ring torsion angles (see Figure 3), while keeping the other coordinates frozen.
We have employed the Python version of Proxima (PyProxima) to rotate the molecule along the torsion angles (from 0° to 360°), to compute the V2 torsional parameter, the charges and the bond connectivity of the molecule and to output the results in a custom version of the multimodel xyz file format compatible with the VR application. The calculation of the van der Waals energy for each conformation has been performed in an external Python script, based on PyProxima, by employing the original parameters of Rappe, Casewit, Colwell, Goddard, and Skiff.30Figure 4a and b shows the resulting energy surface and two screenshots of the AVATAR application.4 As can be noted, the molecular conformation associated with the underlying point of the surface is displayed on the right controller with the corresponding energy value and updated in real time. In particular, the surface presents some divergent points where the energy rises drastically due to the overlapping of two hydrogen atoms of the two different rings (e.g., where both the torsions are equal to 0°). Moreover, the point with the lowest energy shows a geometry compatible with the one resulting from a UFF geometry optimization.
Figure 4.

(a) Screenshot of the configuration panel of the AVATAR application4 for the exploration of the potential energy surfaces. The computation of the shown PES has been performed by taking into account the van der Waals, electrostatic, and V2 torsional components of the energy and relies on Proxima for the computation of the V2 parameters, the charges, the input, and the output of files. (b) Celecoxib molecule on its potential energy surface.
The examples described in the preceding sections show how Proxima can be exploited in tandem with other software for the interactive visualization of molecular systems and related properties.
Conclusions and Perspectives
In this paper, we have introduced a new molecular perception library called Proxima. This library is provided with parsers for pdb, xyz, and mol (v3000) file formats. Concerning molecular perception, Proxima can detect or compute the covalent bond connectivity, chemical rings within molecular systems, atomic hybridizations, and atomic charges (Gasteiger or QeQ).
It is worth noting that, in analogy with many other available perception methods, the information detected or computed by Proxima is sufficient to perform atom type assignment; in fact, this is what many packages do, linking the perception to a domain (e.g., Antechamber with AMBER31 or GAFF32) or general (e.g., UFF33) FF.
From the visualization point of view most of the outputs of Proxima can be visualized in Caffeine.3 In addition to the Proxima C++ core library, we have also developed additional tools, allowing a greater flexibility and ease of use: PyProxima, a Python module that enables the use of Proxima in scripts or in the python interpreter, and ProximaConsole, a console application for using Proxima in the direct computation of the desired molecular properties.
Finally, we have analyzed in some detail a test case in which PyProxima has been exploited for the computation of a potential energy surface for the conformational analysis of the Celecoxib molecule, to be provided as input to the AVATAR4 VR application.
Acknowledgments
This work has been supported by the Italian MIUR (PRIN 2017, project Physico-chemical Heuristic Approaches: Nanoscale Theory Of Molecular Spectroscopy, PHANTOMS, prot. 2017A4XRCA).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.0c00076.
Further details about the parsing of PDB files highlighting the way residues and alternate locations are treated. Moreover, some benchmarks for the computation of the Horton’s rings are reported. Also, the complete set of rules used for the computation of the hybridization of the atoms is shown. Lastly, Caffeine, PyProxima, ProximaConsole, and ProximaGUI are discussed more in detail by also showing some pictures of the software (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Zanette C.; Bannan C. C.; Bayly C. I.; Fass J.; Gilson M. K.; Shirts M. R.; Chodera J. D.; Mobley D. L. Toward Learned Chemical Perception of Force Field Typing Rules. J. Chem. Theory Comput. 2019, 15, 402–423. 10.1021/acs.jctc.8b00821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMART SNS–Theoretical and Computational Chemistry at SNS. http://smart.sns.it (accessed 2020-02-28).
- Salvadori A.; Del Frate G.; Pagliai M.; Mancini G.; Barone V. Immersive virtual reality in computational chemistry: Applications to the analysis of QM and MM data. Int. J. Quantum Chem. 2016, 116, 1731–1746. 10.1002/qua.25207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino M.; Salvadori A.; Lazzari F.; Paoloni L.; Nandi S.; Mancini G.; Barone V.; Rampino S. Chemical promenades: Exploring potential-energy surfaces with immersive virtual reality. J. Comput. Chem. 2020, 41, 1310. 10.1002/jcc.26172. [DOI] [PubMed] [Google Scholar]
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeersch T.; Zurek E.; Hutchison G. R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4, 17. 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Cheng T.; Wang R. Automatic Perception of Organic Molecules Based on Essential Structural Information. J. Chem. Inf. Model. 2007, 47, 1379–1385. 10.1021/ci700028w. [DOI] [PubMed] [Google Scholar]; PMID: 17530839
- Schomaker V.; Stevenson D. P. Some Revisions of the Covalent Radii and the Additivity Rule for the Lengths of Partially Ionic Single Covalent Bonds. J. Am. Chem. Soc. 1941, 63, 37–40. 10.1021/ja01846a007. [DOI] [Google Scholar]
- Porterfield W. W.Inorganic Chemistry; Academic Press, 1993. [Google Scholar]
- Little E. J.; Jones M. M. A complete table of electronegativities. J. Chem. Educ. 1960, 37, 231. 10.1021/ed037p231. [DOI] [Google Scholar]
- Pagliai M.; Muniz-Miranda F.; Cardini G.; Righini R.; Schettino V. Hydrogen Bond Dynamics of Methyl Acetate in Methanol. J. Phys. Chem. Lett. 2010, 1, 2951–2955. 10.1021/jz1010994. [DOI] [Google Scholar]
- Pagliai M.; Cardini G.; Righini R.; Schettino V. Hydrogen bond dynamics in liquid methanol. J. Chem. Phys. 2003, 119, 6655–6662. 10.1063/1.1605093. [DOI] [Google Scholar]
- Macchiagodena M.; Mancini G.; Pagliai M.; Barone V. Accurate prediction of bulk properties in hydrogen bonded liquids: amides as case studies. Phys. Chem. Chem. Phys. 2016, 18, 25342–25354. 10.1039/C6CP04666E. [DOI] [PubMed] [Google Scholar]
- Horton J. A Polynomial-Time Algorithm to Find the Shortest Cycle Basis of a Graph. SIAM Journal on Computing 1987, 16, 358–366. 10.1137/0216026. [DOI] [Google Scholar]
- Fan B. T.; Panaye A.; Doucet J. P.; Barbu A. Ring perception. A new algorithm for directly finding the smallest set of smallest rings from a connection table. J. Chem. Inf. Model. 1993, 33, 657–662. 10.1021/ci00015a002. [DOI] [Google Scholar]
- Bayly C. I.; Cieplak P.; Cornell W.; Kollman P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993, 97, 10269–10280. 10.1021/j100142a004. [DOI] [Google Scholar]
- Marenich A. V.; Jerome S. V.; Cramer C. J.; Truhlar D. G. Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases. J. Chem. Theory Comput. 2012, 8, 527–541. 10.1021/ct200866d. [DOI] [PubMed] [Google Scholar]
- Gasteiger J.; Marsili M. Iterative partial equalization of orbital electronegativity – a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. 10.1016/0040-4020(80)80168-2. [DOI] [Google Scholar]
- Patel S.; Brooks C. L. CHARMM fluctuating charge force field for proteins: I parameterization and application to bulk organic liquid simulations. J. Comput. Chem. 2004, 25, 1–16. 10.1002/jcc.10355. [DOI] [PubMed] [Google Scholar]
- Rappe A. K.; Goddard W. A. Charge equilibration for molecular dynamics simulations. J. Phys. Chem. 1991, 95, 3358–3363. 10.1021/j100161a070. [DOI] [Google Scholar]
- CHARM–Chemistry at Harvard Macromolecular Mechanics. https://www.charmm.org/charmm/ (accessed 2020-02-28).
- Pyykkö P. Additive Covalent Radii for Single-, Double-, and Triple-Bonded Molecules and Tetrahedrally Bonded Crystals: A Summary. J. Phys. Chem. A 2015, 119, 2326–2337. 10.1021/jp5065819. [DOI] [PubMed] [Google Scholar]
- Eigen, the C++ template library for linear algebra: matrices, vectors, numerical solvers, and related algorithms. http://eigen.tuxfamily.org/ (accessed 2020-02-28).
- Liu Z.; Barigye S. J.; Shahamat M.; Labute P.; Moitessier N. Atom Types Independent Molecular Mechanics Method for Predicting the Conformational Energy of Small Molecules. J. Chem. Inf. Model. 2018, 58, 194–205. 10.1021/acs.jcim.7b00645. [DOI] [PubMed] [Google Scholar]
- Gaussian–Cube File. http://gaussian.com/cubegen/ (accessed 2020-02-28).
- VMD: Theoretical and Computational Biophysics Group, NIH Center for Macromolecular Modeling and Bioinformatics, at the Beckman Institute, University of Illinois, Urbana-Champaign. https://www.ks.uiuc.edu/Research/vmd/ (accessed 2020-02-28).
- Jmol: an open-source Java viewer for chemical structures in 3D. http://jmol.sourceforge.net/ (accessed 2020-02-28).
- Unity 3D Engine. https://unity.com/ (accessed 2020-02-28).
- VIVE. https://www.vive.com/eu/ (accessed 2020-02-28).
- Rappe A. K.; Casewit C. J.; Colwell K. S.; Goddard W. A.; Skiff W. M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. 10.1021/ja00051a040. [DOI] [Google Scholar]
- Case D., et al. AMBER 2018; University of California, San Francisco, 2018. [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Artemova S.; Jaillet L.; Redon S. Automatic molecular structure perception for the universal force field: Automatic Molecular Structure Perception for the UFF. J. Comput. Chem. 2016, 37, 1191. 10.1002/jcc.24309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.