

IFNAR1 signaling in NK cells compromises cellular and humoral immune responses to hamper virus control.
Abstract
Inhibition of type 1 interferon (IFN-I) signaling promotes the control of persistent virus infection, but the underlying mechanisms remain poorly understood. Here, we report that genetic ablation of Ifnar1 specifically in natural killer (NK) cells led to elevated numbers of T follicular helper cells, germinal center B cells, and plasma cells and improved antiviral T cell function, resulting in hastened virus clearance that was comparable to IFNAR1 neutralizing antibody treatment. Antigen-specific B cells and antiviral antibodies were essential for the accelerated control of LCMV Cl13 infection following IFNAR1 blockade. IFNAR1 signaling in NK cells promoted NK cell function and general killing of antigen-specific CD4 and CD8 T cells. Therefore, inhibition of IFN-I signaling in NK cells enhances CD4 and CD8 T cell responses, promotes humoral immune responses, and thereby facilitates the control of persistent virus infection.
INTRODUCTION
Persistent viral infections represent significant global health problems. Hyperimmune activation is a common feature of persistent virus infection and is characterized by prolonged activation of T, B, and NK cells; elevated proinflammatory mediators; and sustained type 1 interferon (IFN-I) gene signatures (1–3). Persistent viruses take advantage of this inflammatory state to promote sustained expression of negative immunoregulatory molecules that act to restrain antiviral CD4 and CD8 T cell responses (4–7) and to facilitate long-term virus persistence. We and another group reported that IFN-I signaling during persistent viral infection promoted immune system activation, increased expression of immunoregulatory molecules, and potentiated the dissolution of lymphoid architecture. Blockade of IFN-I signaling using an IFNAR1 neutralizing antibody reduced immune system activation, decreased expression of interleukin-10 (IL-10) and programmed death-ligand 1 (PD-L1), and restored lymphoid architecture in mice persistently infected with lymphocytic choriomeningitis virus (LCMV) Cl13. Blockade of IFNAR1 both before and after established persistent LCMV infection promoted faster virus clearance, which required an intact CD4 T cell compartment (8, 9). Additional studies suggest that IFNAR1 blockade might help control chronic HIV infection in humanized mouse models (10, 11), extending these findings to persistent virus infection in humans. Despite these reports, the specific mechanisms through which IFNAR1 blockade promotes control of persistent viral infection remain incompletely understood.
The early failure of antiviral CD8 T cells to control viral replication results in T cell exhaustion and long-term LCMV Cl13 persistence (7, 12, 13). Following the establishment of virus persistence, the host relies on CD4 T cell–mediated immunity to eventually terminate Cl13 infection (14). The switch in antiviral immunity correlates with increased differentiation of T follicular helper cells (TFH) and germinal center (GC) B cells in spleen 30 days after Cl13 infection and is followed by eventual elimination of Cl13 viruses from most tissues (15, 16). Moreover, B cell–deficient mice fail to control LCMV Cl13 infection, suggesting that B cells and antiviral antibodies contribute to the eventual control of persistent viral replication (17, 18). While blockade of IFN-I signaling had modest effects on virus-specific CD8 T cell function, α-IFNAR1 antibody treatment resulted in significantly enhanced CD4 T cell function (8, 9, 19). Moreover, depletion of CD4 T cells before Cl13 infection completely abrogated α-IFNAR1 treatment–mediated enhancement of virus control, suggesting that IFN-I signaling during persistent virus infection restrains optimal antiviral CD4 T cell responses. Given the importance of antiviral CD4 T cells in promoting accelerated control of Cl13 infection, it is reasonable to infer that IFN-I blockade might enhance the ability of CD4 T cells to promote humoral immune responses during persistent viral infection.
Many studies have highlighted multiple mechanisms by which persistent LCMV infection induces the pan-immunosuppressive environment that prevents initial viral control (20). In addition to the roles immunoregulatory cytokines and cell surface receptors play in suppressing the adaptive immune response during Cl13 infection, several studies reported that nature killer (NK) cells can restrain T and/or B cell responses through lysis of antiviral T cells (21–26). Moreover, it was reported that NK cell depletion can accelerate clearance of Cl13 infection (27), demonstrating that NK cells play a causal role in Cl13 persistence. Expression of IFNAR1 on T cells has been reported to protect them from NK cell–mediated lysis (28, 29), and NK cell–intrinsic IFN-I signaling promotes NK cell expansion and effector functions (30, 31). Thus, IFNAR1 expression and signaling on NK cells following Cl13 infection may be essential to restrain antiviral T cell responses and promote virus persistence.
Here, we took a genetic approach to assess the role of IFNAR1 signaling in individual immune cell compartments during Cl13 infection. Deletion of Ifnar1 from NCR1+ cells, which include NK cells and some additional innate lymphocytes, resulted in accelerated control of Cl13 infection. We determined that NCR1+ cell–intrinsic deletion of Ifnar1 resulted in enhanced antiviral TFH, GCB, and plasma cell responses following Cl13 infection. The increase in TFH, GCB, and plasma cell responses in mice lacking IFNAR1 expression on NCR1+ cells was similar to that observed following IFN-I blockade. We further demonstrate that optimal humoral immune responses are essential for controlling persistent LCMV infection and that passive transfer of LCMV immune serum or purified immunoglobulin G (IgG) significantly reduces viral loads similar to α-IFNAR1 treatment. CD4 T cell–specific deletion of the Bcl6 transcription factor, which controls TFH development, GC formation, and antibody isotype switching/hypermutation, completely abrogated enhanced control of LCMV Cl13 following blockade of IFN-I signaling. Moreover, we show that hastened control of Cl13 infection following IFN-I blockade requires LCMV-specific B cells, as MD4 mice that lack LCMV-specific B cells abolished accelerated virus clearance following α-IFNAR1 treatment. Last, we demonstrate that α-IFNAR1 treatment inhibits the maturation and effector differentiation of NK cells and that NK cell deletion or IFNAR1 blockade inhibited NK cell ability to lyse activated CD4 and CD8 T cells. Our results suggest that early IFN-I signaling during persistent virus infection inhibits the generation of optimal TFH, GCB, and plasma cell responses by promoting optimal NK cell function and its killing of T cells, thereby facilitating virus persistence.
RESULTS
IFNAR1 signaling in NCR1+ cells supports T cell exhaustion and virus persistence
To mechanistically investigate how in vivo IFN-I signaling promotes virus persistence at the cellular level, we crossed Ifnar1-floxed (Ifnar1fl/fl) mice with various cell-specific Cre recombinase mouse lines to selectively delete Ifnar1 from specific cellular populations. We began by deleting Ifnar1 from B cells (Ifnar1fl/fl;Mb1Cre), T cells (Ifnar1fl/fl;CD4Cre), dendritic cells (Ifnar1fl/fl;CD11cCre), or NK cells and innate lymphocytes (Ifnar1fl/fl;NCR1Cre) and infected these strains with a persistent dose of LCMV Cl13. Ifnar1 deletion in these Cre strains was confirmed by flow cytometry (fig. S1A). We observed no significant changes in clearance of Cl13 infection in mice that lacked IFNAR1 expression in B cells (Fig. 1A). Further, deletion of Ifnar1 from CD11c+ or CD4/CD8+ T cells resulted in elevated viral titers in the plasma throughout the course of infection (Fig. 1A). However, Cl13 infection of mice that lack IFNAR1 specifically in NCR1+ cells resulted in significant reductions in plasma viral titers starting at 20 days post-infection (d.p.i.), with 50 and 90% of Ifnar1fl/fl;NCR1Cre mice clearing virus below detection limits by 40 and 50 d.p.i., respectively, compared to Ifnar1fl/fl littermate controls (Fig. 1A). NK cell differentiation was not affected by the absence of IFNAR1 on NK cells, as indicated by comparable numbers of NK cells (fig. S1B) and similar frequency of CD27+CD11b−, CD27+CD11b+, CD27−CD11b+, and CD27−CD11b− NK cells in various tissues between naïve Ifnar1fl/fl and Ifnar1fl/fl;NCR1Cre mice (fig. S1C). Upon LCMV Cl13 infection, the frequency and number of NK cells are still comparable between Ifnar1fl/fl and Ifnar1fl/fl;NCR1Cre mice (fig. S2A). The effector functions of NK cells were impaired in the absence of IFNAR1, indicated by reduced IFN-γ and granzyme B expression (fig. S2B). We next analyzed the antiviral adaptive immune response in Ifnar1fl/fl;NCR1Cre mice following Cl13 infection. In agreement with the hastened clearance of virus, we observed increases in virus-specific CD8 T cells in Ifnar1fl/fl;NCR1Cre mice compared to Ifnar1fl/fl littermate controls (Fig. 1B). Moreover, we detected significant increases in the frequency of IFN-γ+ and IFN-γ+ TNF-α+ (tumor necrosis factor–α–positive) virus–specific CD4 and CD8 T cells in Ifnar1fl/fl;NCR1Cre mice compared to Ifnar1fl/fl littermate controls (Fig. 1, C and D), suggesting that IFN-I signaling in NCR1+ cells suppresses T cell function during Cl13 infection. We also assessed TFH and B cell responses in Ifnar1fl/fl;NCR1Cre and Ifnar1fl/fl mice. Deletion of Ifnar1 from NCR1+ cells increased frequencies and numbers of CXCR5+Bcl6+ TFH, Fas+GL7+ GCB, and CD138+ plasma cells following Cl13 infection (Fig. 1E). However, while the frequency of TFC CD8 T cells was slightly reduced in Ifnar1fl/fl;NCR1Cre mice, we observed similar numbers of those cells in Ifnar1fl/fl;NCR1Cre and Ifnar1fl/fl mice (fig. S2C). Increased GC reaction and T cell response were also observed in Ifnar1fl/fl;NCR1Cre mice when compared to Ifnar1fl/+;NCR1Cre mice, which expressed NCR1Cre but retained one intact allele of the Ifnar1 gene (fig. S2D), further confirming that the increased T cell and B cell responses were driven by the loss of IFNAR1 on NK cells but not reduced NCR1 expression caused by NCR1Cre or possible genetic background variance of our NCR1Cre mice. Together, our data suggest that deletion of Ifnar1 in NCR1+ cells improves antiviral T cell and humoral immune responses following Cl13 infection.
Fig. 1. IFNAR1 signaling in NCR1+ cells promotes T cell exhaustion and virus persistence.
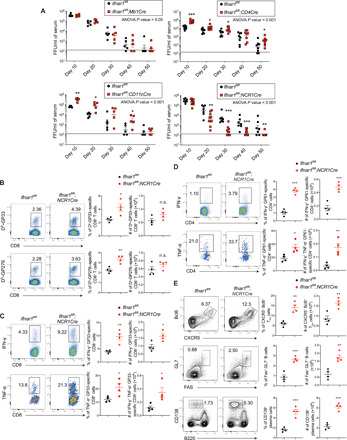
(A) Ifnar1fl/fl;Mb1Cre, Ifnar1fl/fl;CD4Cre, Ifnar1fl/fl;CD11cCre, Ifnar1fl/fl;NCR1Cre, and Ifnar1fl/fl littermate control mice were infected with LCMV Cl13. Viral titers in serum were measured by focus-forming assay (FFU) at indicated time points. (B to E) Ifnar1fl/fl;NCR1Cre and littermate control mice were infected with LCMV Cl13. Frequency and number of GP33- or GP276-specific CD8 T cells (B), GP33-specific cytokine-producing CD8 T cells (C), GP61-specific cytokine-producing CD4 T cells (D), and TFH, GCB, and plasma cells (E) in the spleen were analyzed at day 6 after infection. Statistical comparisons were performed using two-way ANOVA followed by Mann-Whitney U test with Holm-Šídák multiple testing correction (A) or two-tailed Student’s t test (B to E).
IFNAR1 blockade enhances TFH, GC, and plasma cell responses following Cl13 infection
Increased TFH, GCB, and plasma cell responses in Ifnar1fl/fl;NCR1Cre mice prompted us to ask whether α-IFNAR1 treatment also facilitated TFH, GCB, and plasma cell generation during Cl13 infection. Blockade of IFN-I signaling induced elevated GC response from day 9 to 20 after LCMV Cl13 infection as assessed by the numbers of GCB and plasma cells (Fig. 2, B to D) and preserved splenic GC architecture (Fig. 2E). We also observed a significant increase in TFH as early as 5 d.p.i., a time when there were few GCB cells (Fig. 2, A and B). The increase in TFH, GCB, and plasma cells following α-IFNAR1 treatment was accompanied by increased numbers of LCMV Cl13–specific IgG+ antibody-secreting cells (ASCs) (Fig. 2F). Although α-LCMV Cl13 IgG antibody levels were comparable between isotype and α-IFNAR1–treated mice at 9 d.p.i., LCMV-specific IgG antibody was significantly higher in α-IFNAR1–treated mice at 40 d.p.i. (fig. S3A). Next, we examined whether blockade of IFN-I signaling expedites the generation of pre-TFH. SMARTA CD4+ T cells displayed an increased capacity to differentiate into pre-TFH following α-IFNAR1 antibody treatment compared to isotype treatment at 3 d.p.i. (Fig. 2, G and H). These data suggest that IFN-I signaling restrains antiviral humoral immune responses following Cl13 infection.
Fig. 2. IFNAR1 blockade expedites GC reaction during LCMV C13 infection.
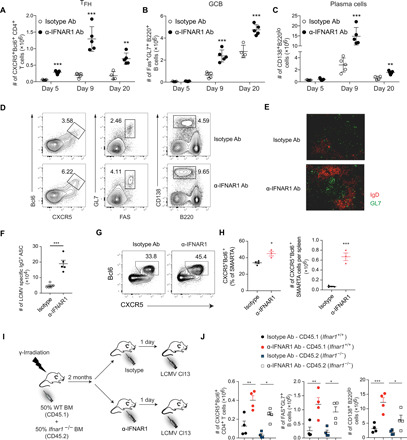
(A to F) Wild-type (WT) mice were treated with isotype or α-IFNAR1 antibodies (Ab) 1 day before LCMV Cl13 infection. Scatter plots of total numbers of TFH (A), GCB cells (B), and plasma cells (C) in the spleen at indicated time points. Representative flow cytometry plots of TFH, GCB, and plasma cells (D) and immunohistochemistry analysis of GC reaction in spleens at 9 d.p.i. (E). (F) Total numbers of LCMV-specific IgG+ ASCs in the spleen at 9 d.p.i. measured by enzyme-linked immunospot (ELISPOT). (G and H) CD45.1+ naïve SAMRTA CD4+ T cells (1.0 × 106) were adoptively transferred into CD45.2+ WT mice and treated with isotype or α-IFNAR1 antibodies 1 day before LCMV Cl13 infection. CXCR5+Bcl6hi pre-TFH among SMARTA CD4+ T cells were analyzed by flow cytometry at 3 d.p.i. (I) Chimera were generated by reconstitution of irradiated CD45.1 mice with bone marrow (BM) cells from CD45.1 Ifnar1+/+ and CD45.2 Ifnar1−/− mice at a 1:1 ratio. The chimera mice were divided into two groups—one treated with isotype control, and the other with α-IFNAR1—and then infected with LCMV Cl13 the next day. (J) Frequency and total numbers of TFH, GCB cell, and plasma cell in the chimera mice were analyzed at day 9 after infection. n = 4 to 5 mice per group in each experiment. Statistical comparisons were performed using two-way ANOVA and Holm-Šídák post hoc test (A to C), two-tailed Student’s t test (F and G), or one-way ANOVA with Tukey’s post hoc test (J). Data are representative of more than three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001.
To better understand the mechanism by which IFNAR blockade promotes the GC reaction, we generated mixed bone marrow chimeras by reconstituting irradiated CD45.1 B6 mice with CD45.2 Ifnar1−/− and CD45.1 Ifnar1+/+ bone marrow cells at a 1:1 ratio. These chimeras were pretreated with isotype or α-IFNAR1 antibody, infected with LCMV Cl13, and analyzed at 9 d.p.i. (Fig. 2I). Consistent with previous findings, we found significant increases in the numbers of CD45.1 IFNAR+/+ TFH, GCB, and plasma cells in α-IFNAR1–treated mice compared to isotype-treated mice (Fig. 2J). Significant increases in the numbers of CD45.2 Ifnar1−/− TFH, GCB, and plasma cells were also found in α-IFNAR1–treated mice (Fig. 2J). These data suggest that IFNAR1 blockade enhances the humoral immune response through TFH-, GCB cell–, and plasma cell–extrinsic pathways.
The humoral immune response is essential for enhanced control of LCMV Cl13 following IFNAR1 blockade
Increased numbers of TFH, GCB, and plasma cells in α-IFNAR1–treated mice and in Ifnar1fl/fl;NCR1Cre mice led us to examine the importance of B cell–mediated immunity in resolving persistent viral infection. To explore the importance of antibody secretion, a unique feature of B lymphocytes, we used Prdm1fl/fl;CD19Cre mice in which the generation of antibody-secreting plasma cells is abolished due to the absence of Blimp-1 (encoded by the Prdm1 gene), the transcription factor essential for plasma cell differentiation. Prdm1fl/fl;CD19Cre mice were unable to control persistent viral infection, as indicated by sustained plasma viral titers following LCMV Cl13 infection (fig. S3B). Also, α-LCMV IgG antibody production was severely compromised in those mutant mice (fig. S3C). These data demonstrate a critical role for plasma cells and α-LCMV Cl13 antibodies in controlling Cl13 infection.
Blimp-1 deficiency in B cells abolishes both GC-independent and GC-dependent plasma cell differentiation. The latter depends on TFH help that enables the production of class-switched and high-affinity antibodies. Because Bcl6 is a key transcription factor in TFH generation and is required for the formation of GCs, we generated Bcl6 conditional knockout (KO) mice (fig. S4A) to address the role of GC-dependent plasma cell differentiation in controlling persistent viral infection. We first examined the quantity and quality of antibodies produced against LCMV Cl13 in Bcl6fl/fl;CD4Cre mice. The amount of α-LCMV Cl13 IgM and IgG antibodies in Bcl6fl/fl;CD4Cre mice was comparable to that in wild-type (WT) mice at 10 d.p.i. when GCs were only beginning to form (fig. S4, B and C). However, by 40 d.p.i., the production of class-switched α-LCMV IgG antibodies was compromised in Bcl6fl/fl;CD4Cre mice, while IgM antibodies were produced at levels similar to Bcl6fl/fl littermate control mice (fig. S4, B and C). These data confirmed that Bcl6fl/fl;CD4Cre mice are unable to induce the GC-dependent plasma cell differentiation program but retained the capacity to generate GC-independent plasma cells. To elucidate roles of the GC-dependent program in controlling persistent viral infection, Bcl6fl/fl;CD4Cre mice were challenged with LCMV Cl13 and viral titers were determined. The Bcl6fl/fl;CD4Cre mice were unable to control LCMV Cl13 infection, as indicated by high viral loads in serum and organs at 80 d.p.i (fig. S4, D and E).
We reasoned that enhanced generation of TFH, GCB, and plasma cells in Ifnar1fl/fl;NCR1Cre mice and in α-IFNAR1–treated mice may be necessary for hastened LCMV Cl13 clearance. Because loss of Bcl6 in T cells results in compromised α-LCMV IgG antibody production, we initially tested this hypothesis with Bcl6fl/fl;CD4Cre mice, in which Bcl6 was deleted in both CD4 and CD8 T cells. As expected, deletion of Bcl6 in T cells resulted in a significant decrease in TFH, GC, and plasma cells compared to littermate controls following LCMV Cl13 infection (Fig. 3, A and B). Moreover, α-IFNAR1 treatment increased the frequencies and numbers of TFH, GCB, and plasma cells in Bcl6fl/fl control mice (Fig. 3, A and B) and T cell–specific Bcl6 deletion abrogated α-IFNAR1 treatment’s ability to increase TFH, GCB, and plasma cell frequencies or total numbers (Fig. 3, A and B). α-IFNAR1 treatment was unable to accelerate virus control in Bcl6fl/fl;CD4Cre mice compared to Bcl6fl/fl littermate controls (Fig. 3E). Further, 40% of α-IFNAR1–treated Bcl6fl/fl littermate control mice cleared LCMV Cl13 in serum and several organs at 40 d.p.i., while α-IFNAR1–treated Bcl6fl/fl;CD4Cre mice harbored ~4 to 7 logs of virus in serum and tissues regardless of α-IFNAR1 treatment (Fig. 3E and fig. S4F). These results demonstrate that Bcl6 expression in T cells is necessary for accelerated control of persistent LCMV Cl13 infection following IFNAR1 blockade.
Fig. 3. GC reaction is essential for IFNAR1 blockade–mediated hastened virus control.
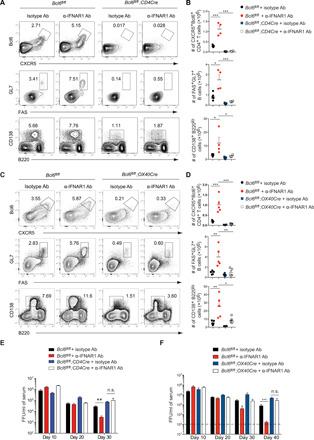
(A to D) Bcl6fl/fl;CD4Cre (A and B) and Bcl6fl/fl;OX40Cre (C and D) mice were treated with isotype or α-IFNAR1 antibodies 1 day before LCMV Cl13 infection. Frequencies and numbers of TFH (upper), GCB (middle), and plasma cells (lower) in spleens at 9 d.p.i. were analyzed by flow cytometry (A and C) and summarized in scatter plots (B and D). Bcl6fl/fl littermates were used as control. (E and F) Viral titers in serum of Bcl6fl/fl;CD4Cre (E) and Bcl6fl/fl;OX40Cre (F) mice were measured at indicated time points. n = 5 mice per group in each experiment. Data are representative of two independent experiments. Statistical comparisons were performed using one-way ANOVA with Tukey’s post hoc test (B and D) or two-way ANOVA followed by Mann-Whitney U test with Holm-Šídák multiple testing correction (E and F). *P < 0.05, **P < 0.01, and ***P < 0.001. n.s., not significant.
The absence of Bcl6 can have profound effects on CD8 T cell differentiation and function (32). The Bcl6 and Blimp-1 transcription factors are known to antagonistically regulate the genetic switch of T cell memory versus effector cell fate decisions (33, 34). Bcl6 expression in CD8 T cells has also been reported to play a critical role in the generation of CXCR5+ CD8 T cells (35–37). Given the importance of Blimp-1 in promoting terminal CD8 T cell differentiation and lytic capacity (38, 39), as well as the potential function of CXCR5+ CD8 T cells in controlling Cl13 infection, we reasoned that the reduced ability of Bcl6fl/fl;CD4Cre mice to control LCMV Cl13 infection could be due to altered CD8 T cell differentiation and function. The frequencies and numbers of IFN-γ+ GP33-specific CD8 T cells were significantly reduced in Bcl6fl/fl;CD4Cre mice compared to Bcl6fl/fl littermate controls (fig. S5A), suggesting that CD8 T cell differentiation/function is altered in mice in which T cells are deficient in Bcl6. To exclude the possibility that CD8 T cell–intrinsic Bcl6 functions might promote control of LCMV Cl13 infection, we generated Bcl6fl/fl;OX40Cre mice in which Cre expression is induced following CD4 T cell activation (40). Similar to Bcl6fl/fl;CD4Cre mice, the generation of TFH, GCB and plasma cells was significantly reduced in Bcl6fl/fl;OX40Cre mice compared to Bcl6fl/fl controls (Fig. 3, C and D). Analysis of virus-specific T cell function revealed that the numbers of IFN-γ+ GP33-specific as well as IFN-γ+TNF-α+ and IFN-γ+TNF-α+IL-2+ polyfunctional cytokine-producing CD8 T cells were comparable between littermate Bcl6fl/fl and Bcl6fl/fl;OX40Cre mice (fig. S5, B and C), demonstrating normal generation of antiviral CD8 T cell function upon CD4 T cell–intrinsic deletion of Bcl6. With respect to antiviral CD4 T cells, we observed similar numbers of GP61-specific IFN-γ+ as well as IFN-γ+TNF-α+ and IFN-γ+TNF-α+IL-2+ cytokine-producing CD4 T cells in Bcl6fl/fl;OX40Cre mice compared to Bcl6fl/fl littermate controls (fig. S5D), suggesting that deleting Bcl6 following CD4 T cell activation has minimal effects on antiviral CD4 T cell responses during Cl13 infection. However, the ability of α-IFNAR1 treatment to increase the numbers of GP61-specific IFN-γ+, IFN-γ+TNF-α+, and IFN-γ+TNF-α+IL-2+ cytokine-producing CD4 T cells was lost in Bcl6fl/fl;OX40Cre mice (fig. S5D), suggesting that CD4 T cell–intrinsic Bcl6 expression is required for enhanced expansion of T helper 1 cell (TH1) antiviral CD4 T cells following IFN-I signaling blockade. In agreement with reduced TFH, GCB, and plasma cells, the production of class-switched α-LCMV IgG antibodies was modestly diminished at 10 d.p.i. and severely compromised at 40 d.p.i. in Bcl6fl/fl;OX40Cre mice (fig. S5E). Moreover, similar to Bcl6fl/fl;CD4Cre mice, Bcl6fl/fl;OX40Cre mice displayed a reduced ability to control LCMV Cl13 infection (Fig. 3F). Further, α-IFNAR1 treatment was unable to accelerate virus clearance in Bcl6fl/fl;OX40Cre mice as they retained ~5 logs of virus following IFN-I blockade, while α-IFNAR1–treated littermate control Bcl6fl/fl mice were approaching complete clearance in plasma at 40 d.p.i. (Fig. 3F).
LCMV-specific antibodies facilitate Cl13 clearance following IFNAR1 blockade
To further assess the role of α-LCMV Cl13 antibodies in reducing viral loads, we treated LCMV Cl13–infected mice with serum collected from WT mice 120 days after LCMV Cl13 infection (LCMV serum). We assessed the antibody content in donor serum and determined that it harbored notable levels of α-LCMV Cl13 IgG antibodies, particularly IgG2a (IgG2c), as the presence of this antibody isotype predicts control of LCMV Cl13 (41). Serum collected from mice immunized with ovalbumin (OVA)/alum/lipopolysaccharide 30 days after immunization (OVA serum) was used as control serum. Administration of LCMV, but not OVA serum, significantly decreased viral loads in tissues and serum (fig. S6, A and B). Moreover, injection of purified antibodies from LCMV serum lowered viral loads to a degree similar to LCMV serum (fig. S6, C and D), suggesting that antiviral antibody and not some unidentified serum factor was responsible for viral load reduction. These data confirm previous studies and further support the importance of α-LCMV Cl13 antibodies in resolving persistent viral infection in serum and tissues.
To further confirm the critical role of B cells and antiviral antibodies in promoting α-IFNAR1 treatment–mediated hastened virus clearance, MD4 mice, in which the majority of B cells carry a fixed B cell receptor specific for hen egg lysozyme, were pretreated with isotype control or α-IFNAR1 antibody before Cl13 infection. Because of the lack of LCMV-specific B cells, MD4 mice generated less TFH, GCB cells, and plasma cells after Cl13 infection compared to littermate controls (Fig. 4, A and B). Following α-IFNAR1 treatment, TFH in MD4 mice showed no measurable increase in frequencies or numbers (Fig. 4, A and B). However, α-IFNAR1 treatment resulted in slightly elevated GCB and plasma cell frequency and number in MD4 mice, although the increase was significantly less than observed in WT littermate controls (Fig. 4, A and B). Similar to Bcl6fl/fl;OX40Cre mice, the increase in cytokine-producing CD4 T cells following α-IFNAR1 treatment was abrogated in MD4 mice (Fig. 4C), suggesting that enhanced CD4 T cell function following IFNAR1 blockade requires an antigen-specific B cell response. Enzyme-linked immunosorbent assay (ELISA) assay showed that these mice produced low levels of LCMV-specific IgG antibody (Fig. 4D). Moreover, virus titers remained elevated in MD4 mice, and the accelerated virus clearance following α-IFNAR1 treatment was lost compared to littermate control animals (Fig. 4E). Together, our data show that antigen-specific B cell responses are indispensable for control of Cl13 infection and the ability of α-IFNAR1 treatment to accelerate Cl13 clearance.
Fig. 4. Antigen-specific B cell response is required for IFNAR1 blockade–mediated hastened virus control.
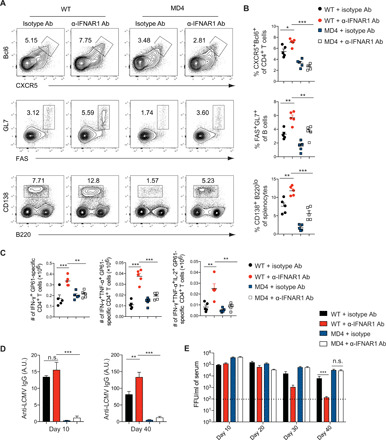
MD4 transgenic mice and WT littermates were treated with isotype or α-IFNAR1 antibodies 1 day before LCMV Cl13 infection. Frequencies (A and B) of TFH, GCB, and plasma cells and (C) numbers of cytokine-producing GP61 LCMV-specific CD4 T cells in spleens at 9 d.p.i. were analyzed by flow cytometry (A) and summarized in scatter plots (B and C). (D) LCMV-specific IgG antibody in serum at days 10 and 40 was quantified by ELISA. A.U., arbitrary units. (E) Viral titers in serum were measured at indicated time points. n = 5 mice per group in each experiment. Data are representative of two independent experiments. Statistical comparisons were performed using one-way ANOVA and Tukey’s post hoc test (B to D) or two-way ANOVA followed by Mann-Whitney U test with Holm-Šídák multiple testing correction (E). *P < 0.05, **P < 0.01, and ***P < 0.001.
NK cells are major cellular mediators of elevated humoral immune responses caused by IFNAR1 blockade
It was recently reported that CD8 T cells suppress humoral responses through direct killing of antigen-specific B cells during LCMV Cl13 infection (42, 43). To assess whether enhanced GC and plasma cell responses following α-IFNAR1 treatment are caused by suppression of CD8 T cell function, CD8 T cell–depleted or α-IFNAR1–treated mice were infected with Cl13 and analyzed at 9 d.p.i. We did observe that depletion of CD8 T cells resulted in slightly increased numbers of GCB cells; however, the effect was much weaker than IFNAR1 blockade (Fig. 5B). Moreover, TFH and plasma cells were reduced in CD8 T cell–depleted mice (Fig. 5, A and C), which contrasted the significant increases in TFH and plasma cells observed following α-IFNAR1 treatment (Fig. 5, A and C), demonstrating that CD8 T cell depletion does not mimic α-IFNAR1 treatment during Cl13 infection.
Fig. 5. NK cells are major cellular mediators of IFNAR1 blockade.
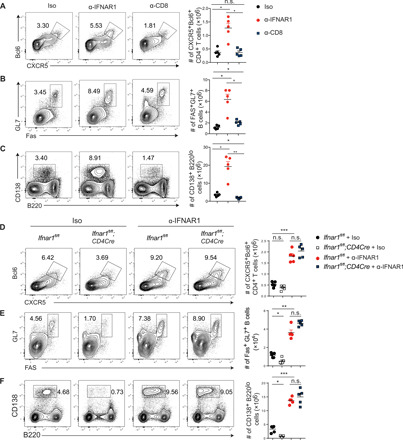
(A to C) Isotype (Iso) control–, α-IFNAR1–, or α-CD8–treated mice were infected with LCMV Cl13. Frequencies (left) and numbers (right) of TFH (A), GCB (B), and plasma cells (C) were analyzed by flow cytometry at 9 d.p.i. (D to F) Ifnar1fl/fl;CD4Cre and Ifnar1fl/fl littermate control mice were treated with isotype control or α-IFNAR1 antibodies and infected with LCMV Cl13. Frequencies (left) and numbers (right) of TFH (D), GCB (E), and plasma cells (F) were analyzed by flow cytometry at 9 d.p.i. Statistical comparisons were performed using Dunnett’s T3 test. **P < 0.01 and ***P < 0.001.
Signaling through IFNAR1 promotes activation and proper effector generation of CD8 T cells, which may impede antiviral humoral responses (44–47). To further assess the role of T cell–intrinsic IFNAR1 signaling in regulating humoral immune responses during Cl13 infection, we treated Ifnar1fl/fl;CD4Cre mice with either isotype or α-IFNAR1 antibody and assessed TFH, GCB, and plasma cell responses. Deletion of Ifnar1 from both CD4 and CD8 T cells resulted in a modest but significant reduction in the frequencies and total numbers of TFH, GCB, and plasma cells compared to Ifnar1fl/fl littermate controls (Fig. 5, D to F). Moreover, α-IFNAR1 treatment of Ifnar1fl/fl;CD4Cre animals before Cl13 infection resulted in expansion of TFH, GCB, or plasma cell frequencies and total numbers to a similar magnitude as observed in α-IFNAR1–treated Ifnar1fl/fl littermates (Fig. 5, D to F), further confirming that IFNAR1 signaling in a cell type other than CD4 or CD8 T cells contributes to restraining the expansion of TFH, GCB, and plasma cells during Cl13 infection.
Several studies demonstrated that NK cells eliminate activated CD4 and CD8 T cells following LCMV Cl13 infection (21–26). Further, IFN-I signaling has been reported to promote NK cell activation and killing ability (30, 31). We observed significant decreases in the expression of both IFN-γ and granzyme B in NK cells from Ifnar1fl/fl;NCR1Cre mice (fig. S2B). NK cell depletion before adoptive transfer of virus-specific T cell receptor transgenic CD4 and CD8 T cells resulted in substantial increases in the frequencies and numbers of these cells (Fig. 6A), supporting previous findings that NK cells regulate the numbers of antigen-specific T cells.
Fig. 6. IFNAR1 blockade suppresses NK cell killing of T cells.
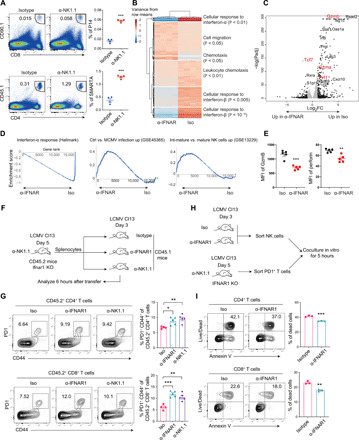
(A) CD90.1+ P14 CD8 T cells or CD45.1+ SMARTA CD4 T cells were adoptively transferred into isotype control– or α-NK1.1–treated B6 mice, which were subsequently infected with LCMV Cl13. P14 and SMARTA cells in the spleen were analyzed at 3.d.p.i. (B to D) NK cells from isotype control– or α-IFNAR1–treated mice were sorted at 3 days after LCMV Cl13 infection and analyzed by RNA sequencing. (B) Heatmap of hierarchically clustered differentially expressed genes [adjusted P < 0.05, absolute fold change (FC) > 2.0]. Top Gene Ontology terms significantly enriched are shown on the right. (C) Volcano plot of differentially expressed genes (adjusted P < 0.05). Black dots mark genes with absolute FC > 2, and gray dots mark genes with absolute FC < 2. Selected genes of interest are marked in red. (D) Select gene sets significantly enriched (adjusted P < 0.05) in α-IFNAR– or isotype-treated groups. (E) Mean florescence intensity (MFI) of granzyme B (GzmB) and perforin in NK cells of isotype- or α-IFNAR1–treated mice at 3 d.p.i. (F) Experimental scheme of in vivo killing assay. (G) Recovery of CD44+PD1+ CD4+ or CD8+ donor cells in the spleen at 6 hours after transfer. (H) Experimental scheme of in vitro killing assay. (I) Death of CD4 (top) and CD8 (bottom) T cells after 5 hours of coculture with sorted NK cells. Statistical comparisons were performed using two-tailed Student’s t test (A, E, and I), the differential expression DESeq2 test (C), or Dunnett’s test (G). *P < 0.05, **P < 0.01, and ***P < 0.001.
Transcriptome analysis of NK cells isolated from α-IFNAR1– or isotype-treated, Cl13-infected mice identified genes and pathways differentially expressed between the groups. As expected, IFN-stimulated gene expression levels were drastically reduced in the α-IFNAR1–treated group (Fig. 6B). Genes enriched in chemotaxis and cell migration were also significantly different between the groups, with expression of chemokines Ccl4, Cxcl10, and the lectin Cd69 decreased, whereas the lymphoid-homing S1P receptor S1pr1 increased in NK cells from α-IFNAR1–treated mice (Fig. 6C). The most significantly increased gene in the α-IFNAR1 group was the TCF1-ecoding gene Tcf7, which limits terminal differentiation of NK cells (48). Conversely, effector molecules Gzmb, Gzma, and Prf1 were lower in the α-IFNAR1 group. Gene set enrichment analysis found that expression of gene sets correlating to NK cell maturation and activation was lower in the α-IFNAR1 group (Fig. 6, C and D). Flow cytometry analysis further confirmed that expression of NK cell effector molecules, including granzyme B and perforin, was suppressed upon α-IFNAR1 treatment (Fig. 6E). These results suggest that IFNAR1 blockade limits the terminal maturation and activation of NK cells.
We next assessed how α-IFNAR1 treatment and NK cell depletion affected the direct killing of activated CD4 and CD8 T cells in vitro and in vivo. We treated Ifnar1-deficient mice with anti-NK1.1 antibody to deplete NK cells, infected them with Cl13, isolated splenocytes at 5 d.p.i., and adoptively transferred them into mice infected with Cl13 3 days before. Those recipient mice were treated with isotype, α-IFNAR1, or α-NK1.1 antibody 1 day before Cl13 infection and analyzed 6 hours after transfer (Fig. 6F). Both NK cell depletion and α-IFNAR1 treatment resulted in similar increases in the frequency of CD44+PD1+ Ifnar1-deficient CD4+ and CD8+ T cells compared to isotype control–treated mice (Fig. 6G). Ifnar1-deficient T cells were used as target cells for NK cell killing in this and following experiments because it has been shown that Ifnar1-deficient T cells are more sensitive to NK cell killing than their WT counterparts (28, 29). To further demonstrate that NK cells were the likely source of killing, we performed an in vitro NK cell killing experiment. We treated WT mice with either isotype or α-IFNAR1 antibody, infected them with Cl13, purified NK cells at 3 d.p.i., and cocultured them with PD1+ T cells sorted from Ifnar1-deficient mice, which underwent NK cell depletion and Cl13 infection 5 days before. We then analyzed the death of Ifnar1-deficient T cells 5 hours after coculture (Fig. 6H). In agreement with our previous results, NK cells from α-IFNAR1–treated mice killed PD-1+ CD4+ and CD8+ T cells less efficiently than isotype control–treated NK cells (Fig. 6I). To study whether NK cells specifically kill TFH, PD1+CXCR5+ TFH CD4 T cells or PD1+CXCR5− non-TFH CD4+ T cells were used as target cells. We observed similar reduction of killing for NK cells from Ifnar1fl/fl;NCR1Cre mice against these different CD4 T cell subsets (fig. S7A). We next sought to understand where in lymphoid tissue do NK cells interact with and kill activated T cells. Spleen sections from isotype- or α-IFNAR1–treated mice were stained with B220, CD3, and NK1.1. The spleen architecture of isotype-treated mice is less intact than that of α-IFNAR1–treated mice, indicated by the diffused distribution of T cells across the section, while T cells in the spleen of α-IFNAR1–treated mice form a distinct T cell zone (fig. S7B).
In general, we found that NK cells localize outside of B cell zones (fig. S7B). In the spleens of isotype-treated mice, there was no clear separation of NK cells and T cells, while in the spleen of α-IFNAR1–treated mice, we did observe some T cells in the vicinity of NK cells; however, most of the T cells were not located in close proximity to NK cells (fig. S7B). From these results, we conclude that NK cells do not appear to penetrate compact T cell zones that could partially explain the reduced T cell killing observed in α-IFNAR1–treated mice. Together, our results demonstrated that IFNAR1 blockade diminished NK cell killing of activated CD4+ and CD8+ T cells by reducing NK cell effector functions and possibly by preventing T-NK interactions in vivo.
DISCUSSION
The mechanisms by which inhibition of IFN-I signaling promotes control of persistent LCMV infection are incompletely understood. In this study, we provide mechanistic insights into this phenomenon by demonstrating that IFN-I blockade results in elevated numbers of TFH, GCB, and plasma cells. This translated into increased numbers of LCMV-specific antibody-forming cells and elevated levels of isotype-switched LCMV-specific antibodies. It has been recently shown in an LCMV-specific B cell receptor transgenic mouse model that CD8 T cells kill antigen-specific B cells to suppress antigen-specific B cell and antibody responses (42, 43). However, the cumulative effects of IFNAR1 blockade on controlling Cl13 infection appear to be more complex. Notably, deletion of Ifnar1 from T cells resulted in elevations in viral titers (Fig. 1A) and consistent decreases in TFH, GCB, and plasma cells compared to mice with Ifnar1-sufficient T cells (Fig. 5, D to F), demonstrating that deletion of Ifnar1 specifically in T cells is not sufficient for enhanced humoral responses and viral control. Moreover, in our studies, increased TFH differentiation precedes the increase in GCB cells (Fig. 2, A and B), suggesting that accelerated TFH differentiation may indirectly regulate GCB and plasma cell responses following IFNAR1 blockade. IFNAR1 blockade promotes TFH differentiation as early as day 3 after infection, at which time GCs have not yet formed (Fig. 2, G and H). Moreover, CD8 depletion modestly increased GCB cells; however, the overall effect was much weaker than IFNAR1 blockade in enhancing the GC reaction (Fig. 5, A to C), suggesting that both CD8 T cell–dependent and T cell–independent pathways contribute to enhanced humoral responses following IFNAR1 blockade. CD8 T cell–depleted mice generate less TFH and plasma cells, which runs counter to the increases in frequencies and total numbers observed in TFH and plasma cell populations following IFNAR1 blockade (Fig. 5, A and C). Instead, our data show that IFNAR1 signaling in NK cells promotes differentiation and activation of NK cells and NK cell killing of activated CD4 and CD8 T cells, thereby suppressing antiviral cellular and humoral immune responses (Figs. 1 and 6). Our data agree with the literature, as NK cells have been reported to regulate antiviral T and B cell responses during Cl13 infection (21–25) and IFNAR1 signaling is essential in NK cell activation and differentiation (25, 30, 31). Thus, the gene signature in NK cells observed following α-IFNAR1 treatment coupled to the reduced CD4 and CD8 T cell killing ability of NK cells from α-IFNAR1–treated mice demonstrates reduced NK cell function at both the molecular and cellular levels (Fig. 6). Overall, our study highlights the complexity and multicellular modulation of IFNAR1 blockade in persistent viral infection where IFNAR1 signaling on various cellular subsets cumulatively contributes to the accelerated viral control.
The contribution of humoral immunity in controlling ongoing persistent viral infections is well supported in the literature. The existence of broadly neutralizing antibodies to HIV long after establishment of viral chronicity and their ability to protect against virus challenge in animal models is encouraging (49, 50). More recently, therapeutic antibody delivery has been demonstrated to control infection in a humanized mouse model of HIV infection (51). During LCMV Cl13 infection, B cell–deficient mice or mice expressing an unrelated transgenic B cell receptor are unable to terminate Cl13 persistence (17, 18). A more recent study further demonstrated that the absence of the transcription factor T-bet in B cells resulted in ablation of antiviral IgG2a/c antibody production and led to prolonged virus persistence (41), suggesting that IgG2a/c antibody and/or T-bet–expressing B cells are necessary for termination of Cl13 persistence. Our results demonstrate that mice lacking Blimp-1 in B cells, a transcription factor necessary for plasma cell generation and antibody production, are unable to purge Cl13 infection (fig. S3B), supporting an essential role for plasma cells and their production of antiviral antibody in controlling Cl13 infection. In addition, the ability of passive serum or anti-LCMV antibody transfer to lower viral loads demonstrates that antibody is effective in virus control in persistently infected mice (fig. S6). It should be noted that therapeutic treatment of LCMV Cl13–infected mice with LCMV serum or antibodies reduced viral loads in serum and tissues but was insufficient to completely purge mice of Cl13. This could be a result of suboptimal concentrations or exposure duration of antiviral antibody. However, it is possible that production of α-LCMV antibodies is essential, but not sufficient, to terminate persistent viral infection. This may suggest that other effector mechanisms are needed to terminate persistent viral infection in conjunction with the production of antiviral antibodies. Notably, how antiviral antibodies act to terminate Cl13 persistence given that α-LCMV antibodies have poor neutralizing capacity and their neutralization potential does not correlate with protective efficacy (52) remains an open question. Moreover, large concentrations of immune complexes generated during persistent virus infection can also suppress Fc-receptor effector functions in animal models (53, 54), suggesting that antibody responses can also promote immune suppression. Further exploration into what antibody effector functions are essential for purging LCMV infection may point the way to the generation of specific antibody properties that will stimulate optimal control of ongoing persistent virus infections.
The redirection of antiviral CD4 T cell responses away from TH1 and toward TFH generation before the host gaining control of the infection suggests that this antiviral program may be important outside of directing humoral immunity (15, 16) and act to preserve antiviral immune potential. The recent demonstration that virus-specific CXCR5+Bcl6+ CD8 T cells are generated during and promote control of persistent LCMV infection highlights the importance of Bcl6 in preserving immune responses exposed to suppressive environments (35–37). The α-IFNAR1–mediated enhanced generation of TFH and GCB cells following Cl13 infection (Figs. 2 to 5) correlates with differentiation of CXCR5+Bcl6+ CD8 T cells (55, 56). Moreover, it is interesting that Bcl6 expression in CD4 T cells was essential for enhanced generation of polyfunctional GP61-specific CD4 T cells following IFN-I blockade during Cl13 infection given that Bcl6 has not been previously linked to TH1 generation. One possibility is that Bcl6 may act as a general pro-proliferative or pro-survival factor in antiviral CD4 and CD8 T cells, which would be consistent with recent studies in antiviral CD8 T cells (35–37) and our data presented herein. Further, data generated in this study suggest that NK cell–mediated killing of virus-specific CD4 and CD8 T cells contributes to the early restraint of TFH, GCB, plasma cell, and functional CD4 and CD8 T cell responses and, in turn, promotes Cl13 persistence.
Together, our study uncovers a previously unappreciated role of IFNAR1 signaling in NK cells in the establishment of Cl13 persistence. Notably, the increases in TFH, GCB, and plasma cells in mice that lacked IFNAR1 in NCR1+ cells were similar to the magnitude of TFH, GCB, and plasma cell increases observed in α-IFNAR1–treated mice following Cl13 infection, suggesting that IFNAR1 signaling in NK cells restrains antiviral humoral responses. We further demonstrate that B cells and antiviral antibodies are essential for the accelerated control of Cl13 infection following α-IFNAR1 treatment. Last, IFNAR1 signaling in NK cells promoted NK cell maturation and lytic gene expression and enhanced cytokine and chemokine production, thereby enhancing NK killing of antiviral CD4 and CD8 T cells, compromising humoral immune responses, and hampering virus control.
MATERIALS AND METHODS
Mice and viruses
Ifnar1-floxed mice were provided by U. Kalinke (TWINCORE Center of Experimental and Clinical Infection Research, Hannover, Germany). NCR1-Cre mice were a gift from V. Sexl (Pharmacology and Toxicology, Veterinary University of Vienna). MD4 (Jax stock no. 002595) mice were from The Jackson Laboratory. Prdm1fl/fl mice were a gift from Dr. M. McHeyzer-Williams. Bcl6fl/fl mice were generated at TSRI Mouse Genetics Core following established protocol using an embryonic stem-cell clone obtained from the European Conditional Mouse Mutagenesis Program (EUCOMM) (clone ID: EPD0745_4_C04). All mice were bred and housed under specific pathogen–free conditions. For virus infection, LCMV clone 13 was injected by the intravenous route at a dose of 2 × 106 plaque-forming units per mouse. For quantification of viremia, blood was drawn from the retro-orbital sinus under isoflurane anesthesia. Serum was isolated by low-speed centrifugation to remove red and white blood cells. Ten microliters of serum was used to perform 10-fold serial dilutions and quantified by focus-forming assay on VeroE6 cells as previously described (57). Tissues (spleen, lung, liver, and kidney) were harvested from euthanized mice and frozen at −80°C. At the time of the assay, tissues were thawed, weighed, and homogenized in medium to 10% (w/v) and clarified by low-speed centrifugation, and 10 μl of the supernatant was used to quantify titers by focus-forming assay. All mice were used in accordance with guidelines from the Institutional Animal Care and Use Committee of The Scripps Research Institute.
Mixed bone marrow chimeras
CD45.1 mice were irradiated with two rounds of 500 cGy with a 3-hour interval. Bone marrow from Ifnar1+/+ (CD45.1+) and Ifnar1−/− (CD45.2+) was subjected to lysis of red blood cells and depleted of T cells by magnetic separation. Bone marrow cells were mixed at a ratio of 1:1 (2 × 106 cells for each genotype) and transferred by injection intravenously. Two months later, recipient mice were treated with isotype control or α-IFNAR1 antibody and infected with LCMV Cl13.
Antibody treatments
Mice were treated with 1 mg of anti-IFNAR1 antibody intraperitoneally (clone MAR1-5A3) or a mouse IgG1 isotype control (clone MOPC21) 1 day before virus infection. For CD8 T cell depletion, mice were treated on day −1 and day 4 with 0.5 mg of the CD8 antibody (clone YTS 169.4.2.1). For NK cell depletion, mice were treated with 0.25 mg of anti-NK1.1 (clone PK136) or isotype control 2 days before virus infection.
Flow cytometry and intracellular staining
Single-cell suspensions were prepared from spleen. Antibody for cell surface markers was added, followed by incubation for 30 min at 4°C. For staining of CXCR5 of TFH, single cells were stained with purified antibody to mouse CXCR5 (2G8; BD Biosciences), followed by biotin-conjugated antibody to rat IgG2a (MRG2a-83; BioLegend) and streptavidin-allophycocyanin. For intracellular cytokine staining, splenocytes were stimulated with major histocompatibility complex (MHC) class I–restricted LCMV-GP33–41 (2 μg/ml) or MHC class II–restricted LCMV-GP61–80 peptide (10 μg/ml) for 1 hour in the absence of brefeldin A and then 5 hours in the presence of brefeldin A (4 μg/ml; Sigma-Aldrich, St. Louis, MO). Cells were fixed and permeabilized with 2% saponin, and intracellular staining was performed with antibodies to IFN-γ (XMG1.2), TNF-α (MP6-XT22), and IL-2 (JES6-5H4). Flow cytometric analysis was performed using BD LSR II (Becton Dickinson), and data were analyzed using FlowJo (Tree Star Inc., Ashland, OR). Absolute cell numbers were determined by multiplying the frequency of specific cell populations by the total number of viable cells.
Enzyme-linked immunosorbent assay
Microtiter plates were coated with lysate of LCMV Cl13–infected baby-hamster kidney (BHK) cells. Nonspecific binding was blocked with 3% bovine serum albumin (BSA) in phosphate-buffered saline (PBS). Serum samples were serially diluted in PBS with 1% BSA and incubated in blocked plates at 4°C overnight. Plates were incubated with biotin-conjugated anti-IgG (1030-08; Southern Biotech) for 2 hours, with streptavidin–alkaline phosphatase (Roche) for 1 hour, and then with alkaline phosphatase substrate solution containing 4-nitro-phenyl phosphate (Sigma-Aldrich) for color development, followed by quantification on a VERSAmax microplate reader (Molecular Devices).
Histology and sectioning
To obtain tissues for immunofluorescent staining, spleens were harvested at day 9 after LCMV C13 infection and frozen in optimal cutting temperature compound. Six-micrometer frozen sections were cut, fixed with 4% paraformaldehyde, blocked with 10% fetal bovine serum (FBS), stained at room temperature for 2 hours with Alexa Fluor 488–anti-mouse-GL7 and Alexa Fluor 647–anti-mouse–IgD, and then washed and mounted using mounting medium.
Enzyme-linked immunospot
Nitrocellulose-bottom 96-well multiscreen plates (Millipore Corporation, CA) were coated with 100 μl per well of lysate of LCMV Cl13–infected BHK cells and incubated at 4°C overnight. Plates were washed with PBS containing 0.1% Tween 20 (PBS-T) and then washed with PBS. The plates were blocked with 100 μl of RPMI medium with 10% FBS for 1 hour at room temperature. Single-cell suspensions were serially diluted in complete medium and incubated in blocked plates for 15 hours at 37°C, 5% CO2 incubator. Plates were washed with PBS-T and then incubated with biotinylated anti-mouse IgGγ (Jackson ImmunoResearch) overnight at 4°C. The plates were washed with PBS-T and incubated with horseradish peroxidase–conjugated avidin-D (Vector) followed by incubation with a 3-amino-9-ethylcarbazole (AEC) substrate. The plates were then washed and dried, and red spots were counted.
In vivo killing assay
Ifnar1 KO mice (CD45.2) were treated with anti-NK1.1 antibody and infected with LCMV Cl13. Splenocytes were prepared from those mice at 5 d.p.i. and adoptively transferred into CD45.1 mice infected with Cl13 3 days before. Those CD45.1 recipient mice were treated with isotype, α-IFNAR1, or α-NK1.1 antibodies 1 day before Cl13 infection, and their splenocytes were harvested 6 hours after transfer and analyzed for CD44+PD1+ T cell recovery by flow cytometry (21).
In vitro killing assay
Ifnar1 KO mice were treated with anti-NK1.1 antibody and infected with LCMV Cl13. PD1+ CD4 and CD8 T cells were sorted from these mice at day 5 after infection. NK1.1+CD3− NK cells were sorted from WT mice treated with either isotype or anti-IFNAR1 antibody and subsequently infected with LCMV Cl13 at day 3 after infection. NK cells (3 × 105) were mixed with 1 × 105 T cells, cultured in RPMI medium for 5 hours in round bottom 96-well plates, and then stained with Live/Dead stain and annexin V to determine T cell killing by NK cells (28, 29).
Statistical analysis
Statistical comparisons of viral titer data in Figs. 1A, 3 (E and F), and 4E and figs. S3B and S4 (D and E) were performed using two-way analysis of variance (ANOVA) followed by Mann-Whitney U test with Holm-Šídák multiple testing correction for overall and time point– or organ-specific comparison of experimental groups. Log-transformed viral titer values were used. Statistical comparison of sample groups in fig. S4F was performed using the Kruskal-Wallis one-way ANOVA test on log-transformed titer data and Dunn multiple comparisons test for all pairwise comparisons. P values in Fig. 2 (A to C) were determined using two-way ANOVA and Holm-Šídák post hoc test for comparing the isotype-treated group with the α-IFNAR1–treated group. Statistical analysis of fig. S1 (B and C) was performed using two-way ANOVA, and P value for the experimental group comparison (Cre+ versus Cre− mice) is reported. P values in fig. S3C were determined using two-way ANOVA and Holm-Šídák post hoc test for comparing cre− groups and cre+ groups at each time point. P values in Figs. 2J, 3 (B and D), and 4 (B to D) and figs. S2D and S5 (C and D) were determined by one-way ANOVA and Tukey’s post hoc multiple comparisons test for all pairwise comparisons. P values in Fig. 5 (A to F) were determined by Dunnett’s T3 multiple comparisons test for all pairwise comparisons. P values in Fig. 6G were determined by Dunnett’s method with the isotype group as control. The remaining P values were determined using two-tailed Student’s t test. Statistical significance is displayed as *P < 0.05, **P < 0.01, and ***P < 0.001.
RNA sequencing and analysis
Total RNA from purified cells was isolated and subjected to mRNA library preparation and sequencing using single-end 75–base pair reads at a depth of >20 million reads per sample. Demultiplexed FASTQ files were aligned to the mouse genome (Ensembl mm10 with transcriptome annotations version 95) using STAR version 2.7.1a (58). Statistical normalization and differential expression analysis were performed using R version 3.6.2 (59) and R package DESeq2 version 1.26.0 (60). Gene set enrichment analysis was performed using GSEA version 4.0.3 (61). Enrichment analysis of heatmap clusters was performed using AmiGO2. Raw data were deposited in Gene Expression Omnibus under the accession no. GSE147330.
Acknowledgments
We thank S. Paust (TSRI) and members of Xiao and Teijaro laboratories for discussion and critical reading of the manuscript. Funding: This study is supported by NIH (R01AI121155, R01AI137252, and R21AI143529 to C.X. and AI123210 and AI118862 to J.R.T.). J.Z. is the recipient of a Cancer Research Institute/Irvington Postdoctoral Fellowship. Author contributions: C.X., J.R.T., Z.H., and S.G.K. conceived and designed this study. J.Z. performed bioinformatic analysis of RNA-sequencing data. Z.H., S.G.K., and Y.L. performed all experiments, with assistance from N.S., K.D., J.S., and R.B. Z.H., S.G.K., J.R.T., and C.X. wrote the manuscript, with input from all authors. Competing interests: The authors declare that they have no competing financial interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Correspondence and requests for materials should be addressed to C.X. (cxiao@scripps.edu) and J.R.T. (teijaro@scripps.edu).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabb8087/DC1
REFERENCES AND NOTES
- 1.Appay V., Sauce D., Immune activation and inflammation in HIV-1 infection: Causes and consequences. J. Pathol. 214, 231–241 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Chang J. J., Altfeld M., Innate immune activation in primary HIV-1 infection. J. Infect. Dis. 202 ( Suppl. 2), S297–S301 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.d’Ettorre G., Paiardini M., Ceccarelli G., Silvestri G., Vullo V., HIV-associated immune activation: From bench to bedside. AIDS Res. Hum. Retroviruses 27, 355–364 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Brooks D. G., Trifilo M. J., Edelmann K. H., Teyton L., McGavern D. B., Oldstone M. B. A., Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 12, 1301–1309 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barber D. L., Wherry E. J., Masopust D., Zhu B., Allison J. P., Sharpe A. H., Freeman G. J., Ahmed R., Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Yi J. S., Cox M. A., Zajac A. J., T-cell exhaustion: Characteristics, causes and conversion. Immunology 129, 474–481 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zajac A. J., Blattman J. N., Murali-Krishna K., Sourdive D. J. D., Suresh M., Altman J. D., Ahmed R., Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 188, 2205–2213 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teijaro J. R., Ng C., Lee A. M., Sullivan B. M., Sheehan K. C. F., Welch M., Schreiber R. D., de la Torre J. C., Oldstone M. B. A., Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340, 207–211 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson E. B., Yamada D. H., Elsaesser H., Herskovitz J., Deng J., Cheng G., Aronow B. J., Karp C. L., Brooks D. G., Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340, 202–207 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhen A., Rezek V., Youn C., Lam B., Chang N., Rick J., Carrillo M., Martin H., Kasparian S., Syed P., Rice N., Brooks D. G., Kitchen S. G., Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J. Clin. Invest. 127, 260–268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng L., Ma J., Li J., Li D., Li G., Li F., Zhang Q., Yu H., Yasui F., Ye C., Tsao L.-C., Hu Z., Su L., Zhang L., Blocking type I interferon signaling enhances T cell recovery and reduces HIV-1 reservoirs. J. Clin. Invest. 127, 269–279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wherry E. J., T cell exhaustion. Nat. Immunol. 12, 492–499 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Wherry E. J., Blattman J. N., Murali-Krishna K., van der Most R., Ahmed R., Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77, 4911–4927 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matloubian M., Concepcion R. J., Ahmed R., CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol. 68, 8056–8063 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fahey L. M., Wilson E. B., Elsaesser H., Fistonich C. D., McGavern D. B., Brooks D. G., Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J. Exp. Med. 208, 987–999 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harker J. A., Lewis G. M., Mack L., Zuniga E. I., Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science 334, 825–829 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergthaler A., Flatz L., Verschoor A., Hegazy A. N., Holdener M., Fink K., Eschli B., Merkler D., Sommerstein R., Horvath E., Fernandez M., Fitsche A., Senn B. M., Verbeek J. S., Odermatt B., Siegrist C.-A., Pinschewer D. D., Impaired antibody response causes persistence of prototypic T cell-contained virus. PLOS Biol. 7, e1000080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitmire J. K., Asano M. S., Kaech S. M., Sarkar S., Hannum L. G., Shlomchik M. J., Ahmed R., Requirement of B cells for generating CD4+ T cell memory. J. Immunol. 182, 1868–1876 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osokine I., Snell L. M., Cunningham C. R., Yamada D. H., Wilson E. B., Elsaesser H. J., de la Torre J. C., Brooks D., Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc. Natl. Acad. Sci. U.S.A. 111, 7409–7414 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wherry E. J., Kurachi M., Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waggoner S. N., Cornberg M., Selin L. K., Welsh R. M., Natural killer cells act as rheostats modulating antiviral T cells. Nature 481, 394–398 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waggoner S. N., Taniguchi R. T., Mathew P. A., Kumar V., Welsh R. M., Absence of mouse 2B4 promotes NK cell-mediated killing of activated CD8+ T cells, leading to prolonged viral persistence and altered pathogenesis. J. Clin. Invest. 120, 1925–1938 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pallmer K., Barnstorf I., Baumann N. S., Borsa M., Jonjic S., Oxenius A., NK cells negatively regulate CD8 T cells via natural cytotoxicity receptor (NCR) 1 during LCMV infection. PLOS Pathog. 15, e1007725 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rydyznski C., Daniels K. A., Karmele E. P., Brooks T. R., Mahl S. E., Moran M. T., Li C., Sutiwisesak R., Welsh R. M., Waggoner S. N., Generation of cellular immune memory and B-cell immunity is impaired by natural killer cells. Nat. Commun. 6, 6375 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cook K. D., Kline H. C., Whitmire J. K., NK cells inhibit humoral immunity by reducing the abundance of CD4+ T follicular helper cells during a chronic virus infection. J. Leukoc. Biol. 98, 153–162 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lang P. A., Lang K. S., Xu H. C., Grusdat M., Parish I. A., Recher M., Elford A. R., Dhanji S., Shaabani N., Tran C. W., Dissanayake D., Rahbar R., Ghazarian M., Brustle A., Fine J., Chen P., Weaver C. T., Klose C., Diefenbach A., Haussinger D., Carlyle J. R., Kaech S. M., Mak T. W., Ohashi P. S., Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc. Natl. Acad. Sci. U.S.A. 109, 1210–1215 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waggoner S. N., Daniels K. A., Welsh R. M., Therapeutic depletion of natural killer cells controls persistent infection. J. Virol. 88, 1953–1960 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu H. C., Grusdat M., Pandyra A. A., Polz R., Huang J., Sharma P., Deenen R., Köhrer K., Rahbar R., Diefenbach A., Gibbert K., Löhning M., Höcker L., Waibler Z., Häussinger D., Mak T. W., Ohashi P. S., Lang K. S., Lang P. A., Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity 40, 949–960 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Crouse J., Bedenikovic G., Wiesel M., Ibberson M., Xenarios I., von Laer D., Kalinke U., Vivier E., Jonjic S., Oxenius A., Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity 40, 961–973 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Mack E. A., Kallal L. E., Demers D. A., Biron C. A., Type 1 interferon induction of natural killer cell gamma interferon production for defense during lymphocytic choriomeningitis virus infection. MBio 2, e00169-11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madera S., Rapp M., Firth M. A., Beilke J. N., Lanier L. L., Sun J. C., Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J. Exp. Med. 213, 225–233 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ichii H., Sakamoto A., Kuroda Y., Tokuhisa T., Bcl6 acts as an amplifier for the generation and proliferative capacity of central memory CD8+ T cells. J. Immunol. 173, 883–891 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Johnston R. J., Poholek A. C., DiToro D., Yusuf I., Eto D., Barnett B., Dent A. L., Craft J., Crotty S., Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325, 1006–1010 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crotty S., Johnston R. J., Schoenberger S. P., Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat. Immunol. 11, 114–120 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He R., Hou S., Liu C., Zhang A., Bai Q., Han M., Yang Y., Wei G., Shen T., Yang X., Xu L., Chen X., Hao Y., Wang P., Zhu C., Ou J., Liang H., Ni T., Zhang X., Zhou X., Deng K., Chen Y., Luo Y., Xu J., Qi H., Wu Y., Ye L., Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature 540, 470 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Leong Y. A., Chen Y., Ong H. S., Wu D., Man K., Deleage C., Minnich M., Meckiff B. J., Wei Y., Hou Z., Zotos D., Fenix K. A., Atnerkar A., Preston S., Chipman J. G., Beilman G. J., Allison C. C., Sun L., Wang P., Xu J., Toe J. G., Lu H. K., Tao Y., Palendira U., Dent A. L., Landay A. L., Pellegrini M., Comerford I., McColl S. R., Schacker T. W., Long H. M., Estes J. D., Busslinger M., Belz G. T., Lewin S. R., Kallies A., Yu D., CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat. Immunol. 17, 1187–1196 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Im S. J., Hashimoto M., Gerner M. Y., Lee J., Kissick H. T., Burger M. C., Shan Q., Hale J. S., Lee J., Nasti T. H., Sharpe A. H., Freeman G. J., Germain R. N., Nakaya H. I., Xue H.-H., Ahmed R., Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rutishauser R. L., Martins G. A., Kalachikov S., Chandele A., Parish I. A., Meffre E., Jacob J., Calame K., Kaech S. M., Transcriptional repressor Blimp-1 promotes CD8+ T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 31, 296–308 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shin H., Blackburn S. D., Intlekofer A. M., Kao C., Angelosanto J. M., Reiner S. L., Wherry E. J., A role for the transcriptional repressor Blimp-1 in CD8+ T cell exhaustion during chronic viral infection. Immunity 31, 309–320 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klinger M., Kim J. K., Chmura S. A., Barczak A., Erle D. J., Killeen N., Thymic OX40 expression discriminates cells undergoing strong responses to selection ligands. J. Immunol. 182, 4581–4589 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barnett B. E., Staupe R. P., Odorizzi P. M., Palko O., Tomov V. T., Mahan A. E., Gunn B., Chen D., Paley M. A., Alter G., Reiner S. L., Lauer G. M., Teijaro J. R., Wherry E. J., Cutting Edge: B cell-intrinsic T-bet expression is required to control chronic viral infection. J. Immunol. 197, 1017–1022 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moseman E. A., Wu T., de la Torre J. C., Schwartzberg P. L., McGavern D. B., Type I interferon suppresses virus-specific B cell responses by modulating CD8+ T cell differentiation. Sci. Immunol. 1, eaah3565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fallet B., Narr K., Ertuna Y. I., Remy M., Sommerstein R., Cornille K., Kreutzfeldt M., Page N., Zimmer G., Geier F., Straub T., Pircher H., Larimore K., Greenberg P. D., Merkler D., Pinschewer D. D., Interferon-driven deletion of antiviral B cells at the onset of chronic infection. Sci. Immunol. 1, eaah6817 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jennings R. N., Grayson J. M., Barton E. S., Type I interferon signaling enhances CD8+ T cell effector function and differentiation during murine gammaherpesvirus 68 infection. J. Virol. 88, 14040–14049 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curtsinger J. M., Valenzuela J. O., Agarwal P., Lins D., Mescher M. F., Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 174, 4465–4469 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Pinto A. K., Daffis S., Brien J. D., Gainey M. D., Yokoyama W. M., Sheehan K. C. F., Murphy K. M., Schreiber R. D., Diamond M. S., A temporal role of type I interferon signaling in CD8+ T cell maturation during acute West Nile virus infection. PLOS Pathog. 7, e1002407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Welsh R. M., Bahl K., Marshall H. D., Urban S. L., Type 1 interferons and antiviral CD8 T-cell responses. PLOS Pathog. 8, e1002352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jeevan-Raj B., Gehrig J., Charmoy M., Chennupati V., Grandclément C., Angelino P., Delorenzi M., Held W., The transcription factor Tcf1 contributes to normal NK cell development and function by limiting the expression of granzymes. Cell Rep. 20, 613–626 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Hessell A. J., Rakasz E. G., Tehrani D. M., Huber M., Weisgrau K. L., Landucci G., Forthal D. N., Koff W. C., Poignard P., Watkins D. I., Burton D. R., Broadly neutralizing monoclonal antibodies 2F5 and 4E10 directed against the human immunodeficiency virus type 1 gp41 membrane-proximal external region protect against mucosal challenge by simian-human immunodeficiency virus SHIVBa-L. J. Virol. 84, 1302–1313 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mascola J. R., Lewis M. G., Stiegler G., Harris D., Van Cott T. C., Hayes D., Louder M. K., Brown C. R., Sapan C. V., Frankel S. S., Lu Y., Robb M. L., Katinger H., Birx D. L., Protection of macaques against pathogenic simian/human immunodeficiency virus 89.6PD by passive transfer of neutralizing antibodies. J. Virol. 73, 4009–4018 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klein F., Halper-Stromberg A., Horwitz J. A., Gruell H., Scheid J. F., Bournazos S., Mouquet H., Spatz L. A., Diskin R., Abadir A., Zang T., Dorner M., Billerbeck E., Labitt R. N., Gaebler C., Marcovecchio P. M., Incesu R.-B., Eisenreich T. R., Bieniasz P. D., Seaman M. S., Bjorkman P. J., Ravetch J. V., Ploss A., Nussenzweig M. C., HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature 492, 118–122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baldridge J. R., Buchmeier M. J., Mechanisms of antibody-mediated protection against lymphocytic choriomeningitis virus infection: Mother-to-baby transfer of humoral protection. J. Virol. 66, 4252–4257 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamada D. H., Elsaesser H., Lux A., Timmerman J. M., Morrison S. L., de la Torre J. C., Nimmerjahn F., Brooks D. G., Suppression of Fcγ-receptor-mediated antibody effector function during persistent viral infection. Immunity 42, 379–390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wieland A., Shashidharamurthy R., Kamphorst A. O., Han J. H., Aubert R. D., Choudhury B. P., Stowell S. R., Lee J., Punkosdy G. A., Shlomchik M. J., Selvaraj P., Ahmed R., Antibody effector functions mediated by Fcγ-receptors are compromised during persistent viral infection. Immunity 42, 367–378 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu T., Ji Y., Moseman E. A., Xu H. C., Manglani M., Kirby M., Anderson S. M., Handon R., Kenyon E., Elkahloun A., Wu W., Lang P. A., Gattinoni L., McGavern D. B., Schwartzberg P. L., The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 1, eaai8593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang Z., Zak J., Pratumchai I., Shaabani N., Vartabedian V. F., Nguyen N., Wu T., Xiao C., Teijaro J. R., IL-27 promotes the expansion of self-renewing CD8+ T cells in persistent viral infection. J. Exp. Med. 216, 1791–1808 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Battegay M., Cooper S., Althage A., Bänziger J., Hengartner H., Zinkernagel R. M., Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24- or 96-well plates. J. Virol. Methods 33, 191–198 (1991). [DOI] [PubMed] [Google Scholar]
- 58.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.R Core Team, R: A language and environment for statistical computing. v. 3.6.2. (R Foundation for Statistical Computing, Vienna, Austria, 2019); https://www.R-project.org.
- 60.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabb8087/DC1