

Cryo-ET of photodamaged plasma membrane reveals shedding from protrusions that depends on F-actin dynamics, Myo1a, and Vps4B.
Abstract
Cryo–electron tomography (cryo-ET) provides structural context to molecular mechanisms underlying biological processes. Although straightforward to implement for studying stable macromolecular complexes, using it to locate short-lived structures and events can be impractical. A combination of live-cell microscopy, correlative light and electron microscopy, and cryo-ET will alleviate this issue. We developed a workflow combining the three to study the ubiquitous and dynamic process of shedding in response to plasma membrane damage in HeLa cells. We found filopodia-like protrusions enriched at damage sites and acting as scaffolds for shedding, which involves F-actin dynamics, myosin-1a, and vacuolar protein sorting 4B (a component of the ‘endosomal sorting complex required for transport’ machinery). Overall, shedding is more complex than current models of vesiculation from flat membranes. Its similarities to constitutive shedding in enterocytes argue for a conserved mechanism. Our workflow can also be adapted to study other damage response pathways and dynamic cellular events.
INTRODUCTION
Cryo–electron tomography (cryo-ET) is an imaging technique that can provide high-resolution three-dimensional (3D) structural information about biological samples preserved in a near-native frozen-hydrated state. Traditionally, cryo-ET has been used to mainly study whole bacterial cells (1) and viruses (2) owing to their small size (cryo-ET requires a thin sample) and their simplicity (annotating structures in tomograms is nontrivial). Mammalian cells, on the other hand, are thousand-fold larger and are more complex, making it difficult to reliably locate targets of interest within them. It becomes even more of a challenge when the targets of interest are transient events or structures. One solution to this is to use live-cell imaging followed by correlative light and electron microscopy (CLEM) to guide cryo-ET. Live-cell imaging can be used to determine when cells should be frozen for electron microscopy (EM); CLEM can be used to identify where the targets of interest are located within the specimen. Toward this end, we developed a workflow combining live-cell microscopy, CLEM, and cryo-ET for investigating mammalian cell biology. Although there have been technical advances in recent years to enable these workflows, including rapid freezing of samples to capture dynamic processes (3), few implementations have yet revealed previously unknown biological mechanisms, especially in higher eukaryotes (4). Here, we developed and used a novel experimental workflow to study the ultrastructural details of shedding.
Shedding is an important and ubiquitous process with diverse functions ranging from delivery of enzymes to cell-to-cell signaling. Depending on the cell type, shedding may be constitutive and/or dynamically amplified by external stimuli (5) including plasma membrane damage from mechanical injury, chemical insults, introduction of foreign pore-forming proteins such as the bacterial toxin streptolysin O (SLO) and perforin, or laser ablation (6–8). Thus far, shedding has been imaged primarily by fluorescence microscopy (FM), leading to the discovery, for instance, that when treated with SLO, damaged cells release SLO-containing vesicles (9, 10). Extracellular vesicle–like structures have also been observed at sites of laser damage by scanning EM (SEM) (7), although this technique is prone to dehydration artifacts. Unfortunately, because of resolution limitations and sample preparation challenges, FM and SEM were not able to reveal structural details directly at a molecular level. Cryo-ET, on the other hand, has the ability to provide 3D images at nanometer resolution while preserving ultrastructure and cellular context.
Using our workflow, we were able to resolve macromolecular details of plasma membrane shedding events. We report that injured cells relocate actin and membrane to sites of damage to generate F-actin–rich filopodia-like protrusions that act as scaffolds for vesicle shedding. When N-WASp (Neural Wiskott–Aldrich Syndrome protein; required for actin nucleation) or myosin-1a (Myo1a) was disrupted, cells displayed defects in generating protrusions and shedding vesicles, and disruption of vacuolar protein sorting 4B (Vps4B), an AAA adenosine triphosphatase (ATPase) in the endosomal sorting complex required for transport (ESCRT) pathway previously shown to be important for wound repair (7), led to defects in membrane scission, the final step in shedding. These results reveal that shedding in response to plasma membrane damage is notably similar to constitutive shedding from gut microvilli, suggesting a conserved underlying mechanism. We envision this kind of a workflow combining live-cell microscopy, cryogenic CLEM (cryo-CLEM), and cryo-ET to find wide applicability for studying many other dynamic processes of mammalian cell biology, including other damage responses and the shedding process.
RESULTS
Experimental setup and its rationale
As discussed earlier, shedding is constitutive in several cell types but can be amplified by environmental stimuli including plasma membrane damage. As part of our workflow, we damaged specific regions of plasma membrane in HeLa cells grown on EM grids, performed live-cell microscopy to monitor the response, and then froze cells to capture shedding events. The exact areas of interest were revisited using cryo-CLEM, and then tilt series were recorded. For the purpose of cryo-CLEM, we used HeLa cells expressing CHMP4B [charged multivesicular body protein 4B, an ESCRT-III protein that is recruited from cytosol to the site of plasma membrane injury (7)] tagged to enhanced green fluorescent protein (EGFP). Damage was induced with a 640-nm laser. Compared to other damage methods used in previous studies including detergents, toxins, and mechanical disruption, laser treatment offers reproducibility (by standardizing laser power and the size of the damaged area) and precise control over the position of damage sites. We targeted thin regions of cell periphery that can be directly imaged using cryo-ET, thus circumventing other laborious thinning methods such as cryosectioning or focused ion beam milling. Laser treatment also offers the convenience of administering the insult and monitoring the response (CHMP4B-EGFP recruitment) with the same instrument, in our case, a scanning confocal microscope. We adapted a previously published scheme for laser-based (ultraviolet wavelength) damage (7) by introducing a photosensitizer to mediate generation of reactive oxygen species (ROS) following light illumination (11–13). Photosensitizers can be used to specifically damage target cellular compartments such as mitochondria (14) and lysosomes (15). For our workflow, we briefly incubated cells with the photosensitizer disulfonated aluminum phthalocyanine (AlPcS2a) to allow it to localize to the plasma membrane [adsorptively endocytosed with its hydrophobic part intercalating into the outer leaflet of the plasma membrane (16) en route to lysosomes], thereby allowing for light-controlled, plasma membrane–localized ROS production and damage. We chose early time points (<5 min) after AlPcS2a treatment to maximize its association with plasma membrane while limiting the endocytosed pool. To standardize the recovery time after damage and to prevent cells from undergoing large morphological changes between light microscopy and plunge freezing (important for CLEM), we fixed the cells at times of interest with 4% paraformaldehyde (PFA) for 45 min before plunge-freezing them. Fixation has been shown to preserve ultrastructure to a large extent, both by single particle analysis and cryo-ET (17–19). The workflow is summarized in fig. S1.
Cryo-ET reveals F-actin–rich membrane protrusions and free vesicles at damage sites
To do membrane damage, cryo-CLEM, and cryo-ET on the periphery of cells grown on EM grids, we first standardized the parameters for laser damage using CHMP4B-EGFP recruitment to damage sites as a reporter. Consistent with previously published data (7), we observed that CHMP4B-EGFP went from being diffusely cytosolic to accumulating at damaged sites in 10 to 15 min when damaged over a circular region of 1.5 μm in diameter using 35 pulse cycles of laser (Fig. 1A and more information in Materials and Methods). Our improved method of laser damage using a photosensitizer elicits a very similar response to other established methods. We fixed cells in this time window (note that the 5-min window was to accommodate photodamage experiments on multiple cells on each EM grid). A representative experiment is illustrated in movie S1. We advise readers to watch this movie before proceeding further.
Fig. 1. Cryo-ET of plasma membrane damage sites.
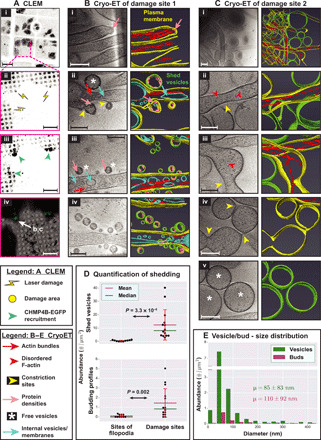
(A) CLEM. (i) Cells expressing CHMP4B-EGFP grown on an EM grid. (ii) Sites of laser damage, each 1.5 μm in diameter. (iii) Sites of laser damage showing CHMP4B-EGFP recruitment. Cells were monitored for 10 min after damage and fixed for 45 min using 4% PFA. EGFP fluorescence in (i), (ii), and (iii) is shown using inverted grayscale. (iv) Overlay of EGFP fluorescence (green) on electron micrograph. (B and C) Cryo-ET of damage sites and corresponding segmentation showing actin-filled plasma membrane protrusions, pearling/budding profiles, shed vesicles, and protein densities observed at certain sites of high membrane curvature in budding profiles and shed vesicles. (D) Quantification of shedding. Top: Abundance of shed vesicles at control sites versus sites of damage (number of vesicles per square micrometer tomogram X-Y cross-sectional area). Bottom: Abundance of budding profiles at control sites of regular filopodial clusters versus sites of damage (number of buds per square micrometer tomogram X-Y cross-sectional area). Each data point represents a tomogram. SD (±1 SD) for each distribution is shown as red error bars, and P values for the pairwise comparison of distributions are obtained using Kolmogorov-Smirnov test (KS test). (E) Size distribution of shed vesicles and budding profiles at damage sites (number of vesicles or buds in each size range per square micrometer tomogram X-Y cross-sectional area) along with the means ± SD values. A total of 14 tomograms were quantified for damage sites and 10 tomograms for sites of regular filopodial clusters in undamaged cells in (D) and (E). Scale bars, 50 μm (A, i), 10 μm (A, ii to iv), and 200 nm (B and C).
Cryo–transmission EM (cryo-TEM) of CHMP4B-EGFP accumulation sites (within an area of ~3 to 4 μm in diameter at the damage sites) pointed to the presence of numerous plasma membrane protrusions. Protrusions were more numerous at these damage sites compared to regions ~10 μm away from them or in randomly assessed regions of undamaged cells that occasionally showed protrusions in the form of filopodia (fig. S2A). These observations suggest that the protrusions are enriched in response to damage. Cryo-ET revealed that abundant F-actin longitudinally arranged in small bundles within these protrusions (Fig. 1, B to D, and movie S1). In addition, budding profiles—vesicle-like features whose bounding membrane was connected to the plasma membrane—were seen on protrusions, while abundant free vesicles were observed nearby. Budding profiles were even observed on the free vesicles. The size distribution of budding profiles matched that of the free vesicles (diameter of 85 ± 83 nm for free vesicles and 110 ± 92 nm for budding profiles; means ± SD; Fig. 1E), suggesting that the free vesicles were shed from the protrusions. Further supporting this idea, the budding profiles showed protein densities close to the cytosolic surface of plasma membrane similar to those present in the shed vesicles. Free vesicles and budding profiles were observed near/on protrusions away from damage sites as well. However, their abundance progressively reduced with their distance from the damage site. They were even found near/on filopodia of undamaged control cells but were extremely rare (Fig. 1D). A subset of damage site protrusions showed regions devoid of F-actin or with disorganized F-actin, usually at their distal tips (Fig. 1C). These regions had often lost their tubular morphology and displayed a propensity to pearl, a phenomenon by which tubular membranes spontaneously stabilize into structures with regularly spaced constrictions, resembling beads on a string (20, 21). Pearled membrane protrusions were pleomorphic, with varying numbers of constriction sites. Free vesicles of comparable sizes were abundant in the vicinity (Fig. 1C), suggesting that pearling-mediated membrane constriction contributes to vesicle shedding.
By comparison, canonical filopodia on undamaged cells showed no pearling. However, apart from pearling, damage-induced protrusions resembled filopodia (Fig. 2, A and B). In both kinds of protrusions, F-actin bundles spanned the entire length, while individual actin filaments in the bundles were shorter, as evident from the exposed free ends of individual filaments we observed within the protrusions [consistent with previous findings in Dictyostelium filopodia (22); fig. S2B]. The F-actin bundles in filopodia consisted of 10 to 35 individual filaments, while those of damage site protrusions consisted of 10 to 30 filaments (fig. S2C). Both of them displayed a lateral interfilament spacing of ~10 nm (10.2 ± 1.3 nm for filopodia and 10.1 ± 1.2 nm for damage site protrusions; means ± SD, measured from center to center). Note that F-actin was well preserved in our sample while it is typically not preserved by traditional methods (involving fixation, dehydration, heavy metal staining, and resin embedding), suggestive of the merits of our experimental scheme. Damage site protrusions displayed several other notable similarities to filopodia including (i) similar widths (mostly 50 to 150 nm for both with filopodia being marginally thinner; Fig. 2D); (ii) internal vesicles (Fig. 2E) suggesting active membrane trafficking; (iii) densities resembling linkers between filaments and between F-actin and the plasma membrane (Fig. 2, A and B, ii and iii); (iv) branch points, with F-actin bundles at the periphery (Fig. 2, A and B, iii and iv); (v) F-actin filaments derived from the cortical actin network at the base (Fig. 2, A and B, iv); and (vi) similar distribution of F-actin filament lengths, although they were slightly longer in damage-site protrusions (a mean value of 163 nm in damage site protrusions versus 122 nm in filopodia; Fig. 2F). By light microscopy, we also observed recruitment of FusionRed fused to Fimbrin/Plastin-1 (Pls1), an actin-bundling protein found in filopodia, to damage-induced protrusions (Fig. 2G and movie S2). These results suggest that the protrusions are repurposed filopodia. The abundance of protrusions at damage sites is similar to (in a few cases greater than that of filopodia) in high-density clusters (Fig. 2C); these clusters are sporadically seen around undamaged cells.
Fig. 2. Similarities between filopodia and damage site protrusions.
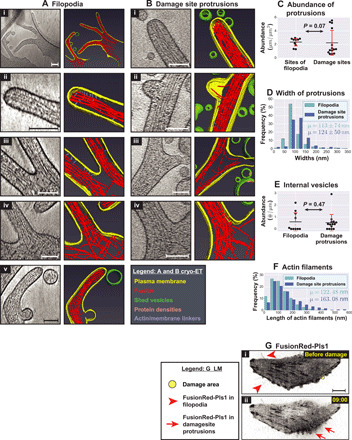
(A and B) Cryo-ET and corresponding segmentation of (A) filopodial clusters at undamaged sites and (B) plasma membrane protrusions at damage sites. (C) Quantification of the abundance of regular filopodia at sites of clustering in undamaged cells versus that of plasma membrane protrusions at damage sites (total length of protrusions in micrometers per square micrometer tomogram X-Y cross-sectional area). (D) Distribution of widths of filopodia versus damage site protrusions (percentage in each size range) along with their means ± SD values (P = 9.63 × 10−16). (E) Quantification of internal vesicles in filopodia versus damage site protrusions (number of vesicles per micrometer of protrusion length). (F) Distribution of lengths of linear actin filaments in filopodia versus damage site protrusions (percentage in each size range) along with their mean values (means ± SD values are as follows: filopodia, 122 ± 73 nm; and damage site protrusions, 163 ± 126 nm; P = 0.0001). (G) Imaging FusionRed-Pls1 in HeLa cells grown on glass (i) before and (ii) after damage. Fluorescence is shown using inverted grayscale. The damage area (yellow circle) is 3 μm in diameter, and the time points after damage are denoted in minutes:seconds. In (C) and (E), each data point represents a tomogram. Red error bars denote ±1 SD for each distribution. Sample sizes for quantifications: (C to E) 14 tomograms for damage site protrusions and 10 tomograms for filopodia in undamaged cells; (F) 450 actin filaments from filopodia and 480 filaments from damage site protrusions. P values are reported by KS tests. Scale bars, 200 nm (A, i), 10 μm (G), and 100 nm (for all other panels).
Live-cell microscopy reveals membrane and actin relocation to sites of damage
While cryo-ET delivered snapshots of local shedding events at nanometer-scale resolution, the technique is not well suited to examine the kind of large-scale membrane dynamics involving F-actin that we hypothesized were likely involved. We therefore performed live-cell microscopy using bright-field and fluorescence imaging following laser damage. To see the effects more clearly, we switched to cells grown on glass (carbon-film EM grids lower signal-to-noise) and increased the damaged area to 3 μm in diameter. We observed that prolonged laser illumination caused numerous blebs—bulging regions of plasma membrane formed by reorganization of cortical F-actin—around the cells (movie S3, cell 1). We therefore calibrated the laser illumination by reducing the number of pulse cycles (see Materials and Methods) such that blebbing was limited and cells recovered while remaining attached to the glass surface (Fig. 3 and movie S3, cells 2 to 6). One hundred and fifty illumination cycles of the laser produced more reproducible results than 100. In total, we imaged the response to laser damage in ~50 cells, with the results summarized in Table 1. Consistent with our previous experiments with cells on EM grids and a previous publication (7), we observed that CHMP4B-EGFP went from being diffusely cytosolic to accumulating at damaged sites (in 40/48 cells) at 5 to 15 min after damage, where it persisted for at least 40 more minutes (Fig. 3). This recruitment of CHMP4B-EGFP was specific to damage sites as shown by a comparison to randomly chosen control regions nearby (note that the control regions Sq2 and Sq3 show some decrease in intensity due to the relocation of cytosolic protein and/or photobleaching; Fig. 3, viii).
Fig. 3. Live-cell light microscopy of plasma membrane damage sites.
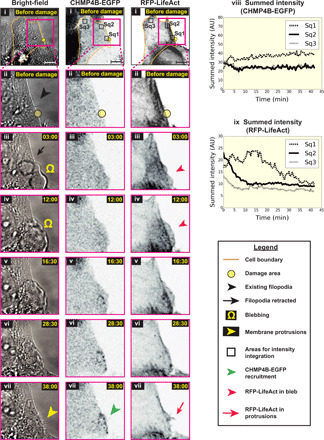
Light microscopy images of HeLa cells grown on glass (i) before and (ii to vii) at various time points after damage—(left-to-right) bright-field, CHMP4B-EGFP, and RFP-LifeAct imaging. EGFP and red fluorescent protein (RFP) fluorescence are shown using inverted grayscale. (viii and ix) Integration of pixel intensities for CHMP4B-EGFP and RFP-LifeAct over the square regions marked as Sq1, Sq2, and Sq3 in (i) at various time points after damage. Sq1 lies over the damaged area, while Sq2 and Sq3 are control sites. The damaged area (yellow circle) is 3 μm in diameter, and the time points after damage are denoted in minutes:seconds. Scale bars, 10 μm. AU, arbitrary units.
Table 1. Liver-cell microscopy of cells after laser damage.
| Laser damage |
CHMP4B-EGFP recruitment |
Blebbing |
Retraction of blebs |
Loss of filopodia |
*Appearance of at least one new protrusion |
Partial retraction of cell area |
Actin relocation |
| 150 pulse cycles of laser damage | |||||||
| Damaged | 22/26 | 21/26 | 8/21 | 22/26 | 20/26 | 3/26 | 10/11 |
| Control | 0/25 | 0/25 | – | 4/25 | 17/25 | 17/25 | 0/2 |
| 100 pulse cycles of laser damage | |||||||
| Damaged | 18/22 | 11/22 | 5/11 | 13/22 | 6/22 | 3/22 | 4/7 |
| Control | 0/7 | 0/7 | – | 0/7 | 2/7 | 0/7 | 0/3 |
| Total | |||||||
| Damaged | 40/48 | 32/48 | 13/32 | 35/48 | 26/48 | 6/48 | 14/18 |
| Control | 0/32 | 0/32 | – | 4/32 | 19/32 | 0/32 | 0/5 |
*The appearance of new protrusions was difficult to quantify by bright-field imaging owing to resolution limitations. We therefore simplified this quantification by scoring the appearance of any protrusion, which resulted in a similar number of damage sites and control sites. However, qualitative assessment suggested a much larger response to damage; numerous protrusions at damage sites outnumber those at control sites as seen in movie S3. The control sites likely reflect random rearrangements of filopodia.
Upon damaging regions near visible filopodia clusters (typically 10 to 25 μm away from the clusters), we observed large-scale remodeling of the plasma membrane by bright-field imaging (Fig. 3A and movie S3, cells 2 to 6). First, plasma membrane blebs formed (32/48 cells) and were subsequently retracted (almost completely retracted in 13/32 cells within the imaged time period). Second, dynamic ruffled membrane boundaries appeared (movie S3, cells 5 and 6). Third, existing filopodia seemed to disappear (likely retracted; 35/48 cells). And fourth, numerous new plasma membrane protrusions (which we now understood were filopodia) seemed to appear predominantly at damage sites (movie S3, cells 2 to 6). New filopodia appeared away from the damage sites as well, albeit with lesser propensity [bright-field inset (iii) shows more numerous protrusions closer to damage site compared to inset (iv), which shows a region away from damage; fig. S3A]. These light microscopy observations involving possible retraction of existing filopodia and formation of new ones are consistent with our cryo-EM experiments (fig. S2A). In addition, the same way as in the cryo-ET experiments, protrusions largely resembled filopodia but sometimes displayed pearling (note that the pearled regions observed by bright-field microscopy were an order of magnitude larger in size; fig. S3A). Damage-induced filopodia started appearing 10 to 15 min after damage, and they continued to appear/persist as CHMP4B-EGFP was recruited. The new filopodia often showed punctate CHMP4B-EGFP fluorescence along their lengths (movie S3, cell 4). The disappearance of existing filopodia and the appearance of new filopodia were sometimes simultaneous and sometimes sequential. Last, a few damaged cells retracted a portion of their area (6/48 cells; Table 1), forming retraction fibers labeled by CHMP4B-EGFP (fig. S3B). Very few control sites in undamaged cells displayed these phenomena (Table 1).
Phenomena such as blebbing, plasma membrane ruffling, and modulation of cell filopodia are suggestive of a role for F-actin as previously anticipated. We therefore imaged damage response in cells labeled with RFP-LifeAct. We observed relocation of F-actin to damage sites (Fig. 3 and movie S3, cells 2 and 3, and quantified in Fig. 3, ix). Initially, F-actin relocated to the blebs, consistent with established behavior during retraction of membrane blebs (23). Subsequently, F-actin relocated to the newly formed protrusions.
Actin-rich membrane protrusions are the source of shed vesicles
The abundance of free vesicles and intermediates around damage-induced filopodia strongly suggests that the filopodia act as scaffolds for shedding. To test this hypothesis, we analyzed the damage response in cells after disrupting the N-WASp actin-nucleation pathway with wiskostatin. Wiskostatin binds the guanosine 5′-triphosphate–binding domain of N-WASp and stabilizes it in an autoinhibited form (24), thus preventing de novo nucleation of linear chains of F-actin or activation of Arp2/3 (actin related protein 2/3) complex to form branched F-actin chains. When cells were treated with wiskostatin for 2 to 3 hours, before laser treatment, the number of filopodia visible by bright-field imaging was greatly reduced (Fig. 4A, i, iii, and v), although the cells still spread on the glass support. We observed an enrichment of vacuole-like vesicles in these cells, consistent with previous observations (25); this phenotype is possibly one of several indirect effects of wiskostatin on cellular functions (unrelated to N-WASp) due to lowering cellular adenosine 5′-triphosphate levels (26). Nonetheless, our observations pertaining to damage response, including filopodia formation and shedding, were informative. Following laser damage, CHMP4B-EGFP was recruited to the damage site as in untreated cells (Fig. 4A, ii, iv, and vi). However, no filopodia were visible by bright-field imaging. Cryo-ET similarly showed a significant reduction in the number of damage site filopodia compared to untreated cells (Fig. 4, B and C). Instead, we observed aberrant membrane structures (not found in untreated cells) that may represent accumulation of membranes that failed to form filopodia (Fig. 4B). Defect in filopodia formation was accompanied by a defect in shedding—shed vesicles were less abundant than from untreated cells. Examination of the vesicles revealed that those from wiskostatin-treated cells showed a narrower range of sizes than those from untreated cells (Fig. 4D), with fewer vesicles larger than 100 nm in diameter (Fig. 4E). This reduction is reflected in their size distribution (Fig. 4D) and their mean sizes (57 ± 44 nm for wiskostatin-treated cells compared to 85 ± 83 nm for untreated cells; means ± SD). In summary, when N-WASp–mediated actin nucleation was blocked, CHMP4B-EGFP was still recruited to damage sites, but fewer filopodia and vesicles were observed. Thus, F-actin is not only important for the formation of filopodia but also for shedding, particularly of larger membrane vesicles.
Fig. 4. Live-cell light microscopy and cryo-ET of damage sites in the presence of wiskostatin.
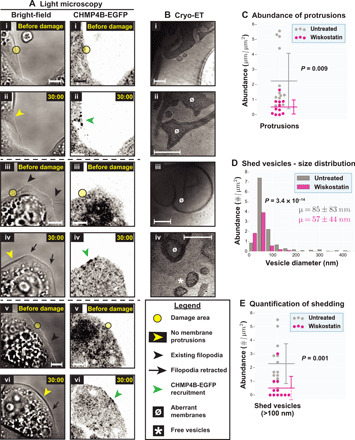
(A) Light microscopy images of three different HeLa cells treated with wiskostatin on glass, (i, iii, and v) before and (ii, iv, and vi) after damage—(left) bright-field and (right) CHMP4B-EGFP imaging. EGFP fluorescence is shown using inverted grayscale. The damage areas are 3 μm in diameter. (B) Cryo-ET of damage sites in wiskostatin-treated cells showing several aberrant membranes, a few plasma membrane protrusions and a few shed vesicles. (C) Quantification of plasma membrane protrusions at damage sites (total length of protrusions in micrometers per square micrometer tomogram X-Y cross-sectional area) in wiskostatin-treated cells versus untreated cells. (D) Size distribution of shed vesicles at damage sites in wiskostatin-treated cells versus untreated cells (number of vesicles in each size range per square micrometer tomogram X-Y cross-sectional area) along with means ± SD values. (E) Quantification of shed vesicles (>100 nm) at damage sites in wiskostatin-treated cells versus untreated cells (number of vesicles per square micrometer tomogram X-Y cross-sectional area). In (C) and (E), each data point represents a tomogram. Horizontal lines denote the mean values, while the vertical error bars denote ±1 SD for each distribution. P values for pairwise comparison of distributions are obtained using KS tests. Sample sizes for quantifications: 11 tomograms for wiskostatin-treatment (versus 10 tomograms for untreated). Scale bars, 5 μm (A) and 200 nm (B).
Myo1a is involved in the organization of filopodia and/or vesicle shedding
Actin-based membrane protrusions have been previously implicated in vesicle shedding in the brush borders of gut enterocytes (27, 28), suggesting possible similarities in the molecular machinery between the two systems. In microvilli, Myo1a forms radial densities connecting the central actin bundles to the plasma membrane that are important for stabilizing the microvilli and shedding vesicles at the distal tip (possibly by propelling membrane over actin bundles) (28). We observed similar radial densities in filopodia at both damaged and undamaged sites (Fig. 3), although they were less abundant, more irregular, and harder to quantify than those described in microvilli; they measured ~15 to 20 nm (measurements made from membrane periphery to periphery of actin filaments), while previous work estimated Myo1a linkers of microvilli to be ~12 to 16 nm (29). We decided to directly test whether Myo1a plays a role in damage-induced shedding. We knocked down Myo1a expression in cells using small interfering RNAs (siRNAs) and observed a reduction of Myo1a protein levels (although not complete; fig. S4A). We then performed damage experiments on cells that showed efficient cotransfection of BLOCK-iT Alexa Fluor Red Fluorescent Control RNA (to limit our analysis to transfected cells; fig. S4B). When these cells were laser-damaged, we observed CHMP4B-EGFP recruitment to the damage sites, with or without membrane blebbing, and loss of nearby filopodia and formation of new filopodia (Fig. 5A), the same way as in wild-type cells. Cryo-ET revealed additional similarities between Myo1a knockdown and wild-type cells: (i) similar abundance of plasma membrane filopodia (fig. S5A); (ii) similar organization of F-actin in filopodia (Fig. 5B); (iii) pearling at sites of disorganized F-actin along filopodia (Fig. 5B); (iv) similar abundance of shed vesicles with comparable size distribution (fig. S5, B and C); and (v) protein densities underneath the plasma membrane in both budding profiles and free vesicles (Fig. 5B). These observations indicate that Myo1a is not absolutely essential for the organization of filopodia, although it is also possible that the knockdown of the protein was insufficient to see an effect or that there is functional redundancy with other motor proteins. We did, however, observe some elongated budding necks (possibly scission defects; Fig. 5B, vi) and extended constrictions along filopodia (defects in organization; Fig. 5B, vii and viii). These defects, absent in wild type, were reflected in the wider distribution of widths for damage site filopodia (SD of 98 nm for Myo1a knockdown cells and 50 nm for wild type; Fig. 5C) and a significant increase in budding profiles (and constriction events) of all sizes compared to wild type (Fig. 5D and fig. S5D). However, since the increase in the abundance of budding profiles was not accompanied by a decrease in shed vesicles, it is also possible that Myo1a knockdown leads to the formation of more budding sites. Together, these results suggest that Myo1a, although probably not essential, is likely involved in damage-induced filopodia organization and shedding dynamics.
Fig. 5. Live-cell light microscopy and cryo-ET of damage sites in Myo1a knockdown cells.
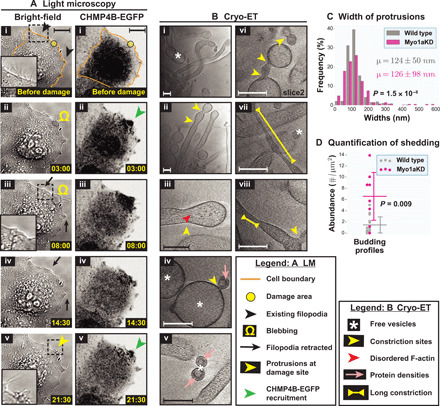
(A) Light microscopy (LM) images of Myo1a knockdown HeLa cells grown on glass (i) before and (ii to v) at various time points after damage—(left) bright-field and (right) CHMP4B-EGFP imaging. EGFP fluorescence is shown using inverted grayscale. The damage area is 3 μm in diameter. (B) Cryo-ET of damage sites in Myo1a knockdown cells showing actin-filled plasma membrane protrusions, pearling/budding profiles, shed vesicles, protein densities observed at certain sites of high membrane curvature in budding profiles and shed vesicles, defective budding profiles, and long constriction necks. (C) Width distribution of damage site protrusions in Myo1a knockdown cells versus wild type (percentage in each size range) along with means ± SD values. (D) Quantification of budding profiles at damage sites of Myo1a knockdown cells versus wild type (number of buds per square micrometer tomogram X-Y cross-sectional area). Each data point represents a tomogram. Horizontal lines denote the mean values, while the vertical error bars denote ±1 SD for each distribution. In (C) and (D), P values for pairwise comparison of distributions are obtained using KS tests. Samples sizes for quantifications, 8 tomograms for Myo1a knockdown (versus 10 tomograms for wild type). Scale bars, 10 μm (A) and 200 nm (B).
ESCRT is involved in membrane scission during shedding
ESCRT proteins are known to catalyze several membrane scission processes with the same membrane topology as shedding (30, 31). In a previous study, recruitment of ESCRT proteins was shown to directly correlate with wound closure (7). Furthermore, the authors observed a few extracellular membrane vesicles at sites of damage by SEM, leading to the hypothesis that ESCRT proteins close wounds by shedding damaged membranes. In this study, we observed punctate localization of CHMP4B-EGFP along filopodia at damage sites. These protein foci could be localized to the necks of budding profiles, thus suggesting a role for ESCRT in membrane scission. However, it is also possible that ESCRT is instead localized to endocytic compartments such as multivesicular bodies (MVBs) and other vesicles in the filopodia.
We therefore tested the role of ESCRT in membrane shedding directly by knocking down Vps4B, an essential AAA ATPase in the ESCRT pathway that was previously shown to be important for wound repair. Knockdown of Vps4B was very efficient (fig. S6A). Cells showing strong signal from cotransfected BLOCK-iT Alexa Fluor Red Fluorescent Control RNA were imaged (fig. S6B). Again, we saw that the plasma membrane exhibited blebbing at the site of damage, existing filopodia were retracted away from the damage site, new filopodia were formed at (and close) to the damage site, and CHMP4B-EGFP was recruited to the damage site (Fig. 6A). Cryo-ET of damage sites showed (i) numerous filopodia, (ii) F-actin bundles in filopodia, and (iii) pearling (Fig. 6B and fig. S7A), all similar to wild type. Although the abundance of these damage site filopodia seemed to show a different distribution than in wild-type cells [as indicated by the P value from a Kolmogorov-Smirnov test (KS test)], the mean was quite similar to that of wild-type cells (~2.5 μm of protrusions per square micrometer of tomogram X-Y cross-sectional area for both samples; fig. S7A). These observations indicate that Vps4B is not essential for the organization of filopodia. On the other hand, we observed numerous long chains of budding profiles (Fig. 6B, v) reminiscent of failed HIV-1 budding profiles from the plasma membrane upon disruption of membrane scission via Vps4 function (32). These budding profiles tended to be <100 nm and were not seen in wild type, therefore pointing to a defect in membrane scission during shedding of smaller vesicles. Consistent with this idea, there was a significant change in the distribution of vesicle and bud sizes; there was a decrease in the number of shed vesicles smaller than 200 nm in diameter compared to wild type (fig. S7B), as indicated by the mean vesicle diameter (141 ± 141 nm for Vps4B knockdown cells compared to 85 ± 83 nm for wild type; means ± SD) and a corresponding increase in the abundance of budding profiles (or constriction events) smaller than 200 nm in diameter (fig. S7C). These smaller budding profiles and shed vesicles displayed protein densities underneath their membrane (as seen in wild type), and the densities were enriched in the chain of budding profiles (Fig. 6B, v). The effect of Vps4B knockdown on shedding of larger vesicles was less pronounced and therefore unclear.
Fig. 6. Live-cell light microscopy and cryo-ET of damage sites in Vps4B knockdown cells.
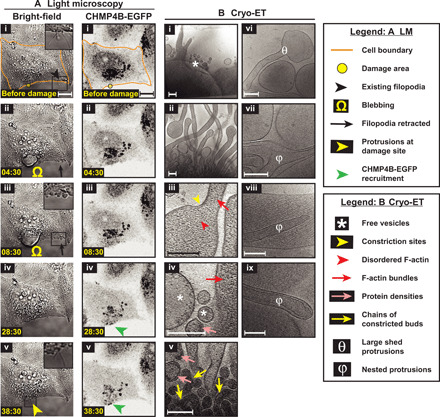
(A) Light microscopy images of Vps4B knockdown HeLa cells grown on glass (i) before and (ii to v) at various time points after damage—(left) bright-field and (right) CHMP4B-EGFP imaging. EGFP fluorescence is shown using inverted grayscale. The damage area is 3 μm in diameter. (B) Cryo-ET of damage sites of Vps4B knockdown cells showing actin-filled plasma membrane protrusions, pearling/budding profiles, shed vesicles, protein densities observed at certain sites of high membrane curvature in budding profiles and shed vesicles, chains of budding profiles, shed membrane protrusions devoid of F-actin, and nested protrusions. Samples sizes for quantifications: 11 tomograms for Vps4B knockdown (versus 10 tomograms for wild type). Scale bars, 10 μm (A) and 200 nm (B).
In addition to wild type–like filopodia, we occasionally observed shed filopodia devoid of F-actin (Fig. 6B, vi) and a few nested filopodia with or without F-actin (Fig. 6B, vii to ix). These defects, not seen in wild type, are likely due to accumulation of unshed membrane upon disruption of Vps4 function and are quantitatively reflected in the greater spread of filopodia widths (SD of 98 nm for Vps4B knockdown cells compared to 50 nm for wild type; fig. S7D). Together, VPS4B is likely involved in membrane scission during damage-induced shedding of smaller vesicles, consistent with its published role in closure of small wounds on plasma membrane (7) and the role of Vps4, in general, in other membrane scission events such as retroviral budding, MVB formation, and abscission during cell division (30).
DISCUSSION
Our workflow combining live-cell microscopy, CLEM, and cryo-ET illustrates the strength of these methods to study dynamic mechanisms in mammalian cell biology in general and plasma membrane damage in particular. The same experimental approach could be used to investigate the role of other molecular players in shedding, for instance, or to study other damage responses in eukaryotic cells. For example, photodamage protocols similar to the one administered in our study exist for lysosomes (15) and mitochondria (14), and our experimental approach could be adapted to study their dynamic repair pathways including autophagy. We could further extend the reach of cryo-ET beyond the thin periphery of a eukaryotic cell to its entire volume by incorporating cryogenic focused ion beam (cryo-FIB) milling (33) to cut a site-specific window into a thick sample. Imaging site-specific dynamic processes deeper in eukaryotic cells with precision will, however, likely require cryogenic FM (of an already-plunge-frozen sample) to precisely locate the fluorescence signal for subsequent cryo-FIB milling and cryo-ET (34).
Our experiments here imaging plasma membrane damage have yielded several important findings that directly lead to a possible model for membrane shedding summarized in Fig. 7: (i) Actin and membrane from other regions of the cell could be relocated to the site of damage; (ii) in a process dependent on F-actin nucleation and Myo1a, relocated membrane and actin could be used to construct new filopodia (predominantly at the damage site and to a lesser extent at nearby sites) to act as scaffolds for vesicle shedding; and (iii) F-actin dynamics, Myo1a, and the ESCRT machinery mediate membrane remodeling and scission to shed damaged membrane. Damage-induced plasma membrane shedding is thus more complex than current models depicting simple vesiculation from flat plasma membrane domains (5, 35). Tufts of microvilli-like plasma membrane protrusions were previously reported in bovine retinal microvascular endothelial cells in response to wounding (36), and filopodia-like protrusions were observed in epithelial cells of Drosophila embryos upon wounding and were demonstrated to be important for healing (37). The possible function of these structures in membrane shedding remained unknown, however, until now.
Fig. 7. Model for damage-induced plasma membrane shedding.
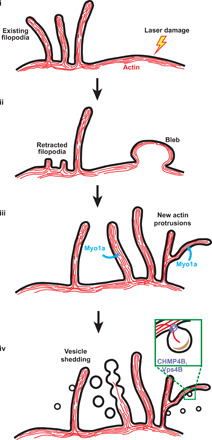
(i and ii) F-actin and membrane are redirected to the site of plasma membrane damage from other regions of the cell (possibly from existing filopodia as well) during the formation of membrane blebs at this site. (iii) Membrane blebs are withdrawn into the cell and reorganized into membrane protrusions using F-actin. There is an overall enrichment of protrusions at sites of damage. (iv) F-actin depolymerization could cause membrane deformation and pearling. Proteins that sense or induce high negative membrane curvature could provide additional force necessary for membrane deformation (especially for smaller vesicles). Myo1a is involved in building protrusions and/or shedding dynamics, while Vps4B is involved in membrane scission during shedding of smaller vesicles. Overall, cells likely form new protrusions in response to damage and shed membrane vesicles from these protrusions, both processes occurring predominantly at the site of damage.
In the first step of shedding, membrane could be transferred to sites of damage by lateral diffusion or by a more complex process involving endocytosis at the source, followed by exocytosis at the sites of damage. We observed numerous internal vesicles near damage sites in the cytosol and inside protrusions, supporting the endocytosis-exocytosis model. Retraction of existing filopodia at other sites suggests that they contribute actin and membrane for remodeling at and around damage sites. To our knowledge, a function for filopodia as a reservoir for membrane and actin during plasma membrane repair has not been suggested previously.
Because the damage site protrusions we imaged share so many features with previously described filopodia (summarized in Results), we concluded that they are filopodia, distinguished mainly by the increased shedding. Notably, we found that damage site and regular filopodia even share the molecular marker Pls1 (an actin-bundling protein). Consistent with this model, even regular filopodia exhibit basal levels of shedding, although this function has not been well studied. Filopodia-like protrusions could offer an advantage over a flat patch of plasma membrane by providing a higher membrane curvature more suitable for shedding. As already noted, shedding from brush-border enterocytes occurs from microvilli, another form of plasma membrane protrusions (27, 28). Another study reported shedding from mesenchymal cells in response to progesterone treatment via putative vesicle-budding events on plasma membrane protrusions (38). Thus, shedding from actin-rich membrane protrusions could be a universal mechanism.
Damage-induced filopodia are likely built by F-actin along with Myo1a and other motor proteins. As reported for previously described filopodia (22), we found several free ends of F-actin throughout the length of all filopodia (including ones that were induced by damage), supporting a de novo filament nucleation model that describes filopodia growth through multiple nucleation events along its length. Growing F-actin at the tip could provide the force to propel membrane forward, while the polymerized F-actin bundles dictate the shape of the filopodia; for example, in curved filopodia, we always saw F-actin filaments/bundles of a similar contour closely associated with their plasma membrane (Fig. 2A, v). F-actin growth in filopodia could be accompanied by Myo1a or other Myo1 isoforms [Myo1b and Myo1f are expressed in HeLa cells (39, 40)] moving membranes on these filament tracks toward the tip. Myo1b was reported to localize to filopodia in HeLa cells (39). Consistent with a role for motor proteins in filopodia formation, an engineered form of Myo6, Myo6+ (the mutant version is directed toward the F-actin plus end), was shown to induce formation of filopodia (41). Moreover, Myo1 isoforms bind phosphoinositides (PIs) enriched in filopodia (39), suggesting that membrane-binding properties of motor proteins are important for filopodia formation. In addition to plasma membrane moved along F-actin by myosin motors, cytosolic vesicles trafficked internally along F-actin within filopodia could be used to meet the membrane demands of a growing filopodium. These internal vesicles, which we observed in damage site filopodia, could also help sort damaged membrane domains to the sites of shedding.
The final steps of shedding involve membrane deformation and scission to release free vesicles to the exterior. Note that the free vesicles observed in this study showed a very broad size distribution. This size variation and the presence of budding profiles on damage site filopodia argue against the possibility that these vesicles are exosomes (derived from fusion of MVBs with the plasma membrane): exosomes are smaller (<80 nm in diameter) and more or less uniform in size (5). We postulate two different mechanisms, not mutually exclusive, that could cause deformation of membrane domains for shedding: (i) Rapid depolymerization of F-actin could cause unsupported membrane to vesiculate or pearl; and (ii) membrane-binding proteins could sense/induce high negative curvature, particularly for smaller vesicles. Several previous reports support our first model: (i) Destruction of the F-actin cortex with latrunculin A induces pearling of plasma membrane (42); (ii) actin polymerization (for subsequent destabilization), direct destabilization of the actin cytoskeleton, and disruption of actin-membrane interactions induce shedding from neutrophils and platelets (43–45); and (iii) microvilli of enterocytes that shed vesicles from their tip show a similar lack of tubular morphology at their tips that correlates with a lack of F-actin bundles in these regions (27, 28). In support of our second model, we observed protein densities underneath membrane regions of high curvature in budding profiles and smaller vesicles. Several inverse BAR (Bin/amphiphysin/Rvs) domain containing proteins such as IRSp53 (insulin receptor tyrosine kinase substrate p53) have been reported to have high affinity for PIs (enriched in filopodia) and induce negative curvature during various kinds of cellular morphogenesis including filopodia formation (46, 47). Moreover, they are known to couple membrane deformation to actin dynamics, an important feature of shedding in our experiments. Involvement of these BAR-domain proteins would not come as an unexpected since one such protein, Angiomotin, is known to function in another process involving a membrane curvature of similar topology, namely, HIV-1 budding from the host cell (48). Subsequent to membrane deformation, the ESCRT machinery is involved in membrane scission during shedding. Previous work hypothesized this function for ESCRT due to their involvement in wound closure and several other membrane scission processes of similar topology (7). Here, we show direct evidence for this function by showing a defect in membrane scission (accumulated budding profiles and a reduced number of free vesicles <200 nm in diameter) when Vps4B function is disrupted. This defect was not complete, so there is likely functional redundancy in the ESCRT system, perhaps with Vps4A. Together, we propose a model for damage-induced shedding that involves multiple molecular mechanisms working in concert. Consistent with this model, wiskostatin-mediated disruption of F-actin and disruption of ESCRT function have complex and distinct effects on shedding—the former results in reduction of larger shed vesicles, while the latter affects smaller shed vesicles. The current study addresses the structural mechanisms of shedding during plasma membrane damage repair, but its purpose remains at least partly elusive. Shedding seems to occur even 10 to 15 min after damage, but membrane resealing is thought to occur within 2 to 4 min (7, 49). Therefore, it is possible that shedding is required for remodeling plasma membrane, or even for extracellular signaling, after the resealing process is complete.
Our findings on shedding could have wider implications as shed vesicles perform several important functions in the eukaryotic world. To name a few, (i) they deliver enzymes to the intestinal lumen from enterocytes for digestion (50) and detoxification of bacterial lipopolysaccharides (51) to control bacterial population (52); (ii) they regulate inflammation in the case of neutrophils and other immune response cells (53–55); (iii) they contain signaling growth factors from astrocytes and neurons (56, 57); and (iv) they play important roles in angiogenesis, metastasis, atherothrombosis, and other diseases (58–60). Although shedding is so ubiquitous, a mechanistic view of the process had been missing except in the case of intestinal enterocytes. Our experimental workflow could be adapted to deliver structural mechanisms in several of the above-listed scenarios.
MATERIALS AND METHODS
Cell growth
A HeLa Kyoto cell line stably expressing CHMP4B-EGFP (gift from A. A. Hyman, Max Planck Institute) was grown in a humidified 37°C incubator with a constant supply of 5% CO2. Cells were cultured in high glucose (l-glutamine+) Dulbecco’s modified Eagle’s medium (DML09, Caisson Labs, Smithfield, UT) supplemented with 10% fetal bovine serum (catalog no. 10437028, Thermo Fisher Scientific), 1 mM sodium pyruvate (catalog no. 11360070, Thermo Fisher Scientific), penicillin (100 U/ml), and streptomycin (100 μg/ml). The CHMP4B-EGFP plasmid was maintained in these cells using G-418 disulfate (400 μg/ml; catalog no. G64500, Research Products International). For experiments involving confocal microscopy, cells were grown on poly-d-lysine–coated 35-mm coverslip bottom dishes (P35GC-1.5-14-C, MatTek Corporation). For experiments further involving CLEM and cryo-ET, cells were grown on 200-mesh gold R2/2 London Finder Quantifoil grids (Quantifoil Micro Tools GmbH, Jena, Germany) added to the bottom of MatTek dishes. Before addition of these grids to the MatTek dishes, they were coated with human fibronectin (0.1 mg/ml; catalog no. C-43060, PromoCell) by floating them on fibronectin droplets on parafilm for approximately 15 to 30 min. In addition, they were coated with 10-nm Au fiducials to be later used for tomography. Roughly 4 μl of 15× diluted Au fiducials (catalog no. 15703, Ted Pella) in 0.01% bovine serum albumin were dried onto the grids. Cells were grown to a density of approximately two to three per grid-square over a period of 1 to 3 days depending on the experiment.
Gene silencing, expression of fluorescent proteins, and drug treatments
Knockdown experiments for Myo1a and Vps4B were performed in CHMP4B-EGFP–expressing HeLa cells using Lipofectamine RNAiMax (catalog no. 13778075, Thermo Fisher Scientific). Cells were grown on 35-mm MatTek dishes or on grids placed at the bottom of these dishes overnight. They were transfected with 50 pmol of siMyo1a (SMARTpool ON-TARGETplus MYO1A siRNA, catalog no. L-008765-01, GE Dharmacon) or with 50 pmol of siVps4B (ON-TARGETplus Human VPS4B siRNA, catalog no. L-013119-00, GE Dharmacon). Transfections were performed for two rounds, each lasting for 24 hours. Cells were cotransfected with BLOCK-iT Alexa Fluor Red Fluorescent Control (catalog no. 14750100, Thermo Fisher Scientific) to ensure transfection efficiency and to identify transfected cells for photodamage experiments. Transfection experiments with RFP-LifeAct and FusionRed-Fimbrin/Pls1 were conducted similar to other transfections before live-cell microscopy. For experiments involving wiskostatin (catalog no. 4434, Tocris Bioscience), the drug was administered to the cells at 25 μM for 2 to 3 hours before photodamage experiments.
Confocal microscopy and laser damage
Imaging was performed at the Caltech Biological Imaging Facility on a Zeiss LSM800 microscope equipped with a large environmental chamber to maintain the temperature at 37°C and a smaller insert module that helped maintain both the temperature and a CO2 level of 5%. Before confocal microscopy and laser damage, AlPcS2a (catalog no. P40632, Frontier Scientific) was added to the cell medium at ~1.3 μM final concentration. Laser damage experiments were performed within the next 10 min to prevent any large interference from endocytosed photosensitizer. No washes were performed after incubation with the photosensitizer. The cell media also contained 50 mM Hepes (catalog no. 15630080, Thermo Fisher Scientific) to prevent pH fluctuations during cell transport and handling. Both bright-field and fluorescence imaging were performed using an LD C-Apochromat 40× water-immersion objective with a numerical aperture of 1.1, and images were recorded using photomultiplier tubes (PMTs; for bright-field image) and GaAsP-PMT (for fluorescence). Green fluorescence imaging was performed using a diode laser at 488 nm at ~1.5 to 2% of its maximum power. Photodamage was administered using a diode laser at 640 nm operated at 100% of its maximum power. The maximum power for the laser lines was 500 mW at the source but measured to be ~750 μW at the level of the objective lenses for the 488-nm laser and ~400 μW for the 640-nm laser. We observed that cells grown on EM grids were more susceptible to laser damage than ones grown on glass. Therefore, we reduced the number of laser pulse cycles and the damaged area for cells on EM grids accordingly. For photodamaging cells grown on glass, a circular area of 3 μm in diameter was chosen close to the cell periphery and scanned for 100 or 150 cycles. For cells on grids, a circular area of 1.5 μm in diameter was scanned for 35 cycles. Damage response and recovery were monitored intermittently (~every 1 to 2 min) for up to 1 hour after photodamage. A scan speed of 7 was used for both photodamaging and imaging cells. The pixel size for imaging was set at 0.312 μm (0.156 μm at a zoom factor of 2), while the image sizes were fixed at 512 by 512 pixels. For CLEM and cryo-ET experiments, cells were fixed at 10 to 15 min after damage for 45 min with 4% PFA (catalog no. RT-15710, Electron Microscopy Sciences) in phosphate-buffered saline (PBS). Cells were washed three times with PBS before plunge-freezing for CLEM and cryo-ET. Using this fixation procedure, we observed by cryo-ET that filopodia and actin filaments contained inside were well preserved (comparable to those of unfixed HeLa cells and other cell types), although 4% PFA has been historically found to be insufficient for preserving the same structures in traditional EM. We believe that the discrepancy arises due to other, accompanying procedures during sample preparation for traditional EM. In traditional EM, a stronger fixative such as glutaraldehyde is required to preserve actin during dehydration, resin embedding, heavy metal staining, and, occasionally, detergent treatment. In contrast, in our workflow, the samples were preserved in a frozen-hydrated state without the need for these additional procedures. Note that cryo-ET does not require any additional, chemical fixation procedures to preserve actin; we used PFA only to conveniently arrest the cellular response to laser damage at a particular time point before plunge-freezing. While studying damage response in cells transfected with siRNAs, candidate cells were chosen on the basis of cytosolic levels of a cotransfected fluorescent BLOCK-iT RNA.
Plunge freezing
EM grids containing photodamaged and fixed cells were plunge-frozen in a liquid ethane/propane mixture using a Vitrobot Mark IV (FEI, Hillsboro, OR) (61). The Vitrobot was set to 95 to 100% relative humidity at 37°C, and blotting was performed manually from the back side of the grids using Whatman filter paper strips. Plunge-frozen grids were subsequently loaded into Polara EM cartridges (FEI) or Krios autogrid cartidges (Thermo Fisher Scientific). EM cartridges containing frozen grids were stored in liquid nitrogen and maintained at ≤−170°C throughout storage, transfer, and cryo-EM imaging.
CLEM and cryo-ET
Cells previously photodamaged and imaged by confocal microscopy were imaged by cryo-EM using either an FEI G2 Polara 300-kV FEG cryo-TEM or a Thermo Fisher Scientific Krios G3i 300-kV FEG cryo-TEM at the Caltech Cryo-EM Facility. Both these microscopes were equipped with a 4k × 4k K2 Summit direct detector (Gatan Inc.) operated in electron counting mode. An energy filter was used to increase the contrast at both medium and higher magnifications with a slit width of 50 and 20 eV, respectively. In addition, defocus values of close to –100 and –8 μm were used to boost the contrast (in the lower spatial resolution range) at the medium and higher magnifications, respectively. Magnifications typically used on the Polara were 3000× and 22,500× (in the medium and higher ranges), corresponding to pixel sizes of 3.7 nm and 4.8 Å, respectively. On the Krios, 3600×/4800× and 26,000× were used in the two magnification ranges that correspond to pixel sizes of 4.2 nm/3.1 nm and 5.38 Å, respectively. A Volta phase plate was optionally used on the Krios to further improve contrast at higher magnifications in certain cases. SerialEM software (62) was used for all imaging.
Photodamaged cells were located in the electron microscope using the markers on the Finder grids. The markers were clearly visible by transmission light microscopy but only partially identifiable by cryo-EM after freezing. However, a full grid montage at a low magnification of close to 100× is sufficient to positively identify these markers based on their overall arrangement on the grid. The photodamaged locations in the cells were located by roughly correlating the light microscopy images with the EM maps based on the shape of the cells or using the image registration protocol in SerialEM. Cracks, regularly spaced 2-μm holes in the carbon film, ice contamination, and other features visible by both light and electron microscopy, were sufficient to obtain an accurate enough correlation (<500-nm precision) for the purpose of tomography. Once the areas of interest were identified and marked, anchor maps were used to revisit these locations and collect tilt series in an automated fashion. Each tilt series was collected from –60° to +60° with an increment of 1° or 2° in an automated fashion using the low-dose functions of tracking and focusing. The cumulative dose of each tilt series ranged between 80 and 150 e−/Å2. Once acquired, tilt series were binned into 1k × 1k arrays before alignment and reconstruction into 3D tomograms with the IMOD software package (63) and tomo3D (64). Tilt series were aligned using 10-nm Au fiducials or patch tracking in IMOD, while reconstructions were performed using simultaneous iterative reconstructive technique (SIRT) in tomo3D. In addition to tilt series, projection images were saved at other magnifications such as 360× for correlation after data acquisition.
For data processing, analysis, and generating figures, more precise correlations were performed using custom Python scripts in a semiautomated fashion. Features used as control points here were similar to the ones used for correlation during data acquisition. However, the number of control points used was larger, and a robust best-fit method was used to increase the precision for correlation. Control points that gave the most accurate correlation (based on the overall error in the fit) were selected from the set provided by the user for accurate correlation. Precision for correlation at lower magnifications is particularly important because of the large pixel sizes involved.
Segmentation
Segmentations of tomograms were manually performed using Amira (Thermo Fisher Scientific). Animations of segmented tomograms were created using Amira and Adobe Photoshop CC (Adobe Inc., San Jose, USA). Segmentations were performed to the best of our abilities bearing in mind the limitation of the missing wedge of information in cryo-ET. Distinctions between plasma membrane and shed vesicles were based only on unambiguously segmented data.
Quantification
Live-cell light microscopy
Cells were observed for ~45 min after damage (or without damage). Damage sites showing any newly visible filopodia were counted positive for appearance of new protrusions (these protrusions were much more abundant at damage sites than control sites). Integrated intensities over a square area of pixels were plotted over time for damage sites and two other sites from the same cell as control. Intensity data were extracted from live-cell microscopy movies over these square areas using Fiji (ImageJ) before plotting them in Python 3.5 using Numpy and Matplotlib.
Actin
F-actin was analyzed after segmentation in Amira using the filament module. Actin filaments that were unambiguously linear (no branching, showing free ends on either side) were used for length measurements. A total of ~450 filaments were measured for filopodia, and ~480 filaments were measured for damage site protrusions. Interfilament spacing was measured manually at multiple positions between parallel F-actin filaments using IMOD. A total of ~150 measurements were made for filopodia and ~100 for damage-site protrusions.
Other measurements from cryo-EM
Quantifications were performed on a per-tomogram basis and displayed as beeswarm plots or alternatively, combined from several tomograms and displayed as histograms. Quantifications for damage sites, sites ~10 μm away from damage, and control sites in undamaged cells were all made at the same distance from the cell edge; the field of view was <1 μm from the cell edge. For each damage site, one to two tomograms were randomly selected close to the site of CHMP4B-EGFP recruitment (within a region of 3 to 4 μm in diameter centered around the damage site). The following numbers of tomograms were selected for each sample—14 tomograms from wild-type damage sites, 10 from control sites with no damage, 11 from damage sites of wiskostatin-treated cells, 8 from damage sites of Myo1a knockdown cells, and 11 from damage sites of Vps4B knockdown cells. All measurements were made using IMOD. Density of protrusions was measured as total lengths of protrusions in a tomogram (in micrometers) divided by tomogram X-Y cross-sectional area (in square micrometers). Widths of plasma membrane protrusions were measured at regular intervals at local maxima, minima, and anywhere in between. Density of shed vesicles was measured as number of vesicles per square micrometer cross-sectional per tomogram. The vesicle sizes were measured as cross-sectional diameters. Similar quantifications were performed for budding profiles as well. New budding profiles in shed vesicles were included in the analysis. Internal vesicles were measured as number of vesicles per micrometer length of protrusion per tomogram. In the case of fig. S2A, projection images were used to quantify abundance of plasma membrane protrusions. All model files were exported as text before plotting using Numpy and Matplotlib libraries in Python 3.5. KS tests were performed using the ks_2samp method from the Scipy package.
Acknowledgments
We thank A. A. Hyman and I. Poser for providing the HeLa cell line stably expressing CHMP4B-EGFP. We thank A. Collazo and S. Wilbert for technical assistance with confocal microscopy. We also thank S. Chen and A. Malyutin for technical assistance with cryo-EM. The bulk of the confocal imaging was performed at the Biological Imaging Facility, and EM was performed at the Beckman Institute Resource Center for Transmission Electron Microscopy, both at Caltech. Funding: This work was supported by funding from the NIH (P50 AI150464 awarded to G.J.J.). Author contributions: W.Y.Y. performed light microscopy experiments and prepared sample for cryo-ET with help from S.K.M. S.K.M. performed CLEM, cryo-ET, and data analyses. S.K.M. prepared the manuscript with help from W.Y.Y., C.M.O., and G.J.J. Y.C. performed the Western blot assays for Myo1a and Vps4B knockdowns. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the first and corresponding authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabc6345/DC1
REFERENCES AND NOTES
- 1.Oikonomou C. M., Chang Y. W., Jensen G. J., A new view into prokaryotic cell biology from electron cryotomography. Nat. Rev. Microbiol. 14, 205–220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luque D., Caston J. R., Cryo-electron microscopy for the study of virus assembly. Nat. Chem. Biol. 16, 231–239 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Fuest M., Nocera G. M., Modena M. M., Riedel D., Mejia Y. X., Burg T. P., Cryofixation during live-imaging enables millisecond time-correlated light and electron microscopy. J. Microsc. 272, 87–95 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Jun S., Ro H.-J., Bharda A., Kim S. I., Jeoung D., Jung H. S., Advances in cryo-correlative light and electron microscopy: Applications for studying molecular and cellular events. Protein J. 38, 609–615 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Cocucci E., Racchetti G., Meldolesi J., Shedding microvesicles: Artefacts no more. Trends Cell Biol. 19, 43–51 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Babiychuk E. B., Monastyrskaya K., Potez S., Draeger A., Intracellular Ca2+ operates a switch between repair and lysis of streptolysin O-perforated cells. Cell Death Differ. 16, 1126–1134 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Jimenez A. J., Maiuri P., Lafaurie-Janvore J., Divoux S., Piel M., Perez F., ESCRT machinery is required for plasma membrane repair. Science 343, 1247136 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Keefe D., Shi L., Feske S., Massol R., Navarro F., Kirchhausen T., Lieberman J., Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity 23, 249–262 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Keyel P. A., Loultcheva L., Roth R., Salter R. D., Watkins S. C., Yokoyama W. M., Heuser J. E., Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci. 124, 2414–2423 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Potez S., Luginbühl M., Monastyrskaya K., Hostettler A., Draeger A., Babiychuk E. B., Tailored protection against plasmalemmal injury by annexins with different Ca2+ sensitivities. J. Biol. Chem. 286, 17982–17991 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsieh C.-W., Chu C. H., Lee H.-M., Yuan Yang W., Triggering mitophagy with far-red fluorescent photosensitizers. Sci. Rep. 5, 10376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kochevar I. E., Lambert C. R., Lynch M. C., Tedesco A. C., Comparison of photosensitized plasma membrane damage caused by singlet oxygen and free radicals. Biochim. Biophys. Acta 1280, 223–230 (1996). [DOI] [PubMed] [Google Scholar]
- 13.Yang W. Y., Optogenetic probing of mitochondrial damage responses. Ann. N. Y. Acad. Sci. 1350, 48–51 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Yang J. Y., Yang W. Y., Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat. Commun. 4, 2428 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Hung Y. H., Chen L. M., Yang J. Y., Yang W. Y., Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat. Commun. 4, 2111 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Vikdal M., Generalov R., Berg K., The photosensitizer disulfonated aluminum phthalocyanine reduces uptake and alters trafficking of fluid phase endocytosed drugs in vascular endothelial cells—Impact on efficacy of photochemical internalization. Biochem. Pharmacol. 86, 748–758 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Kastner B., Fischer N., Golas M. M., Sander B., Dube P., Boehringer D., Hartmuth K., Deckert J., Hauer F., Wolf E., Uchtenhagen H., Urlaub H., Herzog F., Peters J. M., Poerschke D., Lührmann R., Stark H., GraFix: Sample preparation for single-particle electron cryomicroscopy. Nat. Methods 5, 53–55 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Shukla A. K., Westfield G. H., Xiao K., Reis R. I., Huang L. Y., Tripathi-Shukla P., Qian J., Li S., Blanc A., Oleskie A. N., Dosey A. M., Su M., Liang C. R., Gu L. L., Shan J. M., Chen X., Hanna R., Choi M., Yao X. J., Klink B. U., Kahsai A. W., Sidhu S. S., Koide S., Penczek P. A., Kossiakoff A. A., Woods Jr V. L., Kobilka B. K., Skiniotis G., Lefkowitz R. J., Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512, 218–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramanian P., Pirbadian S., El-Naggar M. Y., Jensen G. J., Ultrastructure of Shewanella oneidensis MR-1 nanowires revealed by electron cryotomography. Proc. Natl. Acad. Sci. U.S.A. 115, E3246–E3255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bar-Ziv R., Tlusty T., Moses E., Safran S. A., Bershadsky A., Pearling in cells: A clue to understanding cell shape. Proc. Natl. Acad. Sci. U.S.A. 96, 10140–10145 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campelo F., Hernandez-Machado A., Model for curvature-driven pearling instability in membranes. Phys. Rev. Lett. 99, 088101 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Medalia O., Beck M., Ecke M., Weber I., Neujahr R., Baumeister W., Gerisch G., Organization of actin networks in intact filopodia. Curr. Biol. 17, 79–84 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Charras G. T., Hu C. K., Coughlin M., Mitchison T. J., Reassembly of contractile actin cortex in cell blebs. J. Cell Biol. 175, 477–490 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peterson J. R., Bickford L. C., Morgan D., Kim A. S., Ouerfelli O., Kirschner M. W., Rosen M. K., Chemical inhibition of N-WASP by stabilization of a native autoinhibited conformation. Nat. Struct. Mol. Biol. 11, 747–755 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Inoue T., Pattabiraman P. P., Epstein D. L., Vasantha Rao P., Effects of chemical inhibition of N-WASP, a critical regulator of actin polymerization on aqueous humor outflow through the conventional pathway. Exp. Eye Res. 90, 360–367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guerriero C. J., Weisz O. A., N-WASP inhibitor wiskostatin nonselectively perturbs membrane transport by decreasing cellular ATP levels. Am. J. Physiol. Cell Physiol. 292, C1562–C1566 (2007). [DOI] [PubMed] [Google Scholar]
- 27.McConnell R. E., Higginbotham J. N., Shifrin D. A. Jr., Tabb D. L., Coffey R. J., Tyska M. J., The enterocyte microvillus is a vesicle-generating organelle. J. Cell Biol. 185, 1285–1298 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McConnell R. E., Tyska M. J., Myosin-1a powers the sliding of apical membrane along microvillar actin bundles. J. Cell Biol. 177, 671–681 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsudaira P. T., Burgess D. R., Identification and organization of the components in the isolated microvillus cytoskeleton. J. Cell Biol. 83, 667–673 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christ L., Raiborg C., Wenzel E. M., Campsteijn C., Stenmark H., Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem. Sci. 42, 42–56 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Hurley J. H., ESCRTs are everywhere. EMBO J. 34, 2398–2407 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garrus J. E., von Schwedler U. K., Pornillos O. W., Morham S. G., Zavitz K. H., Wang H. E., Wettstein D. A., Stray K. M., Côté M., Rich R. L., Myszka D. G., Sundquist W. I., Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107, 55–65 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Rigort A., Bauerlein F. J. B., Villa E., Eibauer M., Laugks T., Baumeister W., Plitzko J. M., Focused ion beam micromachining of eukaryotic cells for cryoelectron tomography. Proc. Natl. Acad. Sci. U.S.A. 109, 4449–4454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnold J., Mahamid J., Lucic V., de Marco A., Fernandez J. J., Laugks T., Mayer T., Hyman A. A., Baumeister W., Plitzko J. M., Site-specific cryo-focused ion beam sample preparation guided by 3D correlative microscopy. Biophys. J. 110, 860–869 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pollet H., Conrard L., Cloos A. S., Tyteca D., Plasma membrane lipid domains as platforms for vesicle biogenesis and shedding? Biomolecules 8, 94 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyake K., McNeil P. L., Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J. Cell Biol. 131, 1737–1745 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wood W., Jacinto A., Grose R., Woolner S., Gale J., Wilson C., Martin P., Wound healing recapitulates morphogenesis in Drosophila embryos. Nat. Cell Biol. 4, 907–912 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Casado S., Lobo M., Paino C. L., Dynamics of plasma membrane surface related to the release of extracellular vesicles by mesenchymal stem cells in culture. Sci. Rep. 7, 6767 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Komaba S., Coluccio L. M., Localization of myosin 1b to actin protrusions requires phosphoinositide binding. J. Biol. Chem. 285, 27686–27693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kulak N. A., Pichler G., Paron I., Nagaraj N., Mann M., Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Masters T. A., Buss F., Filopodia formation and endosome clustering induced by mutant plus-end–directed myosin VI. Proc. Natl. Acad. Sci. U.S.A. 114, 1595–1600 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heinrich D., Ecke M., Jasnin M., Engel U., Gerisch G., Reversible membrane pearling in live cells upon destruction of the actin cortex. Biophys. J. 106, 1079–1091 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cauwenberghs S., Feijge M. A. H., Harper A. G. S., Sage S. O., Curvers J., Heemskerk J. W. M., Shedding of procoagulant microparticles from unstimulated platelets by integrin-mediated destabilization of actin cytoskeleton. FEBS Lett. 580, 5313–5320 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Fox J. E., Austin C. D., Boyles J. K., Steffen P. K., Role of the membrane skeleton in preventing the shedding of procoagulant-rich microvesicles from the platelet plasma membrane. J. Cell Biol. 111, 483–493 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Middelhoven P. J., van Buul J. D., Kleijer M., Roos D., Hordijk P. L., Actin polymerization induces shedding of FcgammaRIIIb (CD16) from human neutrophils. Biochem. Biophys. Res. Commun. 255, 568–574 (1999). [DOI] [PubMed] [Google Scholar]
- 46.Mattila P. K., Lappalainen P., Filopodia: Molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 9, 446–454 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Zhao H., Pykäläinen A., Lappalainen P., I-BAR domain proteins: Linking actin and plasma membrane dynamics. Curr. Opin. Cell Biol. 23, 14–21 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Mercenne G., Alam S. L., Arii J., Lalonde M. S., Sundquist W. I., Angiomotin functions in HIV-1 assembly and budding. eLife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinhardt R. A., Bi G., Alderton J. M., Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 263, 390–393 (1994). [DOI] [PubMed] [Google Scholar]
- 50.Hooton D., Lentle R., Monro J., Wickham M., Simpson R., The secretion and action of brush border enzymes in the mammalian small intestine. Rev. Physiol. Biochem. Pharmacol. 168, 59–118 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Bates J. M., Akerlund J., Mittge E., Guillemin K., Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2, 371–382 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shifrin D. A. Jr., McConnell R. E., Nambiar R., Higginbotham J. N., Coffey R. J., Tyska M. J., Enterocyte microvillus-derived vesicles detoxify bacterial products and regulate epithelial-microbial interactions. Curr. Biol. 22, 627–631 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Distler J. H., Pisetsky D. S., Huber L. C., Kalden J. R., Gay S., Distler O., Microparticles as regulators of inflammation: Novel players of cellular crosstalk in the rheumatic diseases. Arthritis Rheum. 52, 3337–3348 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Gasser O., Schifferli J. A., Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood 104, 2543–2548 (2004). [DOI] [PubMed] [Google Scholar]
- 55.MacKenzie A., Wilson H. L., Kiss-Toth E., Dower S. K., North R. A., Surprenant A., Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity 15, 825–835 (2001). [DOI] [PubMed] [Google Scholar]
- 56.Proia P., Schiera G., Mineo M., Ingrassia A. M., Santoro G., Savettieri G., di Liegro I., Astrocytes shed extracellular vesicles that contain fibroblast growth factor-2 and vascular endothelial growth factor. Int. J. Mol. Med. 21, 63–67 (2008). [PubMed] [Google Scholar]
- 57.Schiera G., Proia P., Alberti C., Mineo M., Savettieri G., di Liegro I., Neurons produce FGF2 and VEGF and secrete them at least in part by shedding extracellular vesicles. J. Cell. Mol. Med. 11, 1384–1394 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.George F. D., Microparticles in vascular diseases. Thromb. Res. 122 ( suppl. 1), S55–S59 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Janowska-Wieczorek A., Wysoczynski M., Kijowski J., Marquez-Curtis L., Machalinski B., Ratajczak J., Ratajczak M. Z., Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int. J. Cancer 113, 752–760 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Leroyer A. S., Tedgui A., Boulanger C. M., Role of microparticles in atherothrombosis. J. Intern. Med. 263, 528–537 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Iancu C. V., Tivol W. F., Schooler J. B., Dias D. P., Henderson G. P., Murphy G. E., Wright E. R., Li Z., Yu Z., Briegel A., Gan L., He Y., Jensen G. J., Electron cryotomography sample preparation using the Vitrobot. Nat. Protoc. 1, 2813–2819 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Mastronarde D. N., Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 63.Kremer J. R., Mastronarde D. N., McIntosh J. R., Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996). [DOI] [PubMed] [Google Scholar]
- 64.Agulleiro J. I., Fernandez J. J., Fast tomographic reconstruction on multicore computers. Bioinformatics 27, 582–583 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabc6345/DC1