

Abstract
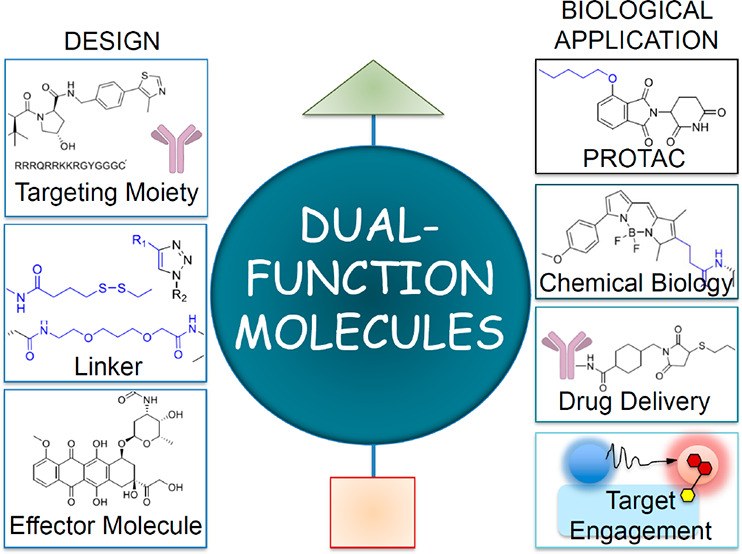
After the first seed concept introduced in the 18th century, different disciplines have attributed different names to dual-functional molecules depending on their application, including bioconjugates, bifunctional compounds, multitargeting molecules, chimeras, hybrids, engineered compounds. However, these engineered constructs share a general structure: a first component that targets a specific cell and a second component that exerts the pharmacological activity. A stable or cleavable linker connects the two modules of a chimera. Herein, we discuss the recent advances in the rapidly expanding field of chimeric molecules leveraging chemical biology concepts. This Perspective is focused on bifunctional compounds in which one component is a lead compound or a drug. In detail, we discuss chemical features of chimeric molecules and their use for targeted delivery and for target engagement studies.
1. Introduction
A chimeric molecule is an engineered construct in which two or more components are linked to form a novel biological agent. Chimeric molecules can be considered as variants of an idea proposed by Paul Ehrlich in the late 1800s. This concept describes a bifunctional molecule in which one component targets the molecule to a specific cell and the second component exerts a pharmacological activity.1,2 Different disciplines have attributed multiple names to dual-functional molecules (chimeras, hybrids, bioconjugates, bifunctional compounds, multitargeting molecules, engineered compounds) depending on the field of application, but the general structure is conserved.
Recently, the knowledge in cellular and molecular biology widely increased. The chemical biology field allowed the application of the chemistry knowledge to deliver specific biomolecules on the cell membrane and into the cells. The concepts of chemical biology were translated into drug discovery of chimeric molecules (or chimeras).3,4 These entities display (i) a targeting moiety and (ii) an effector molecule within the same chemical construct, and their individual function could be largely modulated with appropriate conjugation chemistry strategies where a linker is the bridging element (Figure 1).1,3 Recently, the exploitation of these systems for drug delivery implementation, particularly into cancer cells, has been reviewed.4
Figure 1.

Overview of chimeric compounds with a diversity of structures: (a) examples of chimeras discussed in the Perspective, where linker moiety is highlighted in blue; (b) general structures of chimeric compounds.
This Perspective discusses the recent advances in the rapidly expanding field of chimeric molecules in which one component is a lead compound or a drug. In detail we discuss chemical features of chimeric molecules, targeted delivery, and the exploitation of chimeric molecules for target engagement studies.
Section 2 is focused on linker chemistry. To develop small molecules that engage a specific cell type or protein target, a small molecule needs to be linked with another moiety that allows selective target recognition. The linker plays a pivotal role in the development of chimeric compounds and allows bridging of two pharmacophores within one molecule. The type and the length of the linker are essential parameters for the design and biological activity of chimeras, leading to a rapid expansion of the linker chemistry field.
Section 3 deals with drug delivery based on receptor-mediated endocytosis (RME). Cell membrane permeation represents the major bottleneck in achieving the sufficient drug concentration for therapeutic effect. Drug delivery systems exploiting receptor-mediated endocytosis have been proposed as a promising tool to overcome tissue barriers and have given an important contribution to medical practice, especially in the area of cancer and central nervous system (CNS) disorders. Three classes of ligands have been used to target receptors at the cell membrane and are herein discussed: (i) cell-penetrating peptides (CPPs), (ii) tumor homing peptides, and (iii) monoclonal antibodies.
Section 4 covers the recent advancements in chimeric molecules engineered to demonstrate how a drug engages its own target intracellularly. Herein, we discuss the crucial integration of chemical biology knowledge, drug discovery strategies, and medicinal chemistry to foster structure–mechanism of action studies and subsequent structural modifications.
2. Linker Features in the Modular Approach to Chimeric Compounds
2.1. Linker Chemistry
Physically connecting two chemical moieties or a small molecule with a protein occurs through a moiety called linker. A wide variety of linkers have been developed that consider if the target of the small molecule is intra- or extracellular and what type of cell or tissue the small molecule needs to target. If the desired target is intracellular, typically the linker includes a moiety that can be cleaved once the chimera is inside the cell. Linkers also play an important role in activity-based protein profiling experiments.
A commonly used linker type is hydrazone, Figure 2a. The hydrazone moiety can typically be easily installed because of its compatibility with peptide synthesis.5 The hydrazone moiety is stable at physiological pH and cleaves at an acidic pH, but additional conditions that do not require acids have been developed.6 While the hydrazone moiety has been widely used in diversity-oriented synthesis7 and as an additional handle in peptide synthesis,8 more recently it has been exploited as a reversible linker for proteomics experiments.9 Several different types have been developed, including an acyl hydrazone from the Kohn laboratory.10 Their study highlights a more efficient capture and release of the targeted protein pool as compared to standard protocols due to the mild conditions for the hydrazone release. Captured proteins do not have to be exposed to SDS or 8 M guanidine to release them for mass spectrometry experiments. The Dawson group developed a bisaryl hydrazone linker also highlighting the mild conditions that can be exploited to release captured proteins from a hydrazone-linked molecule.11
Figure 2.
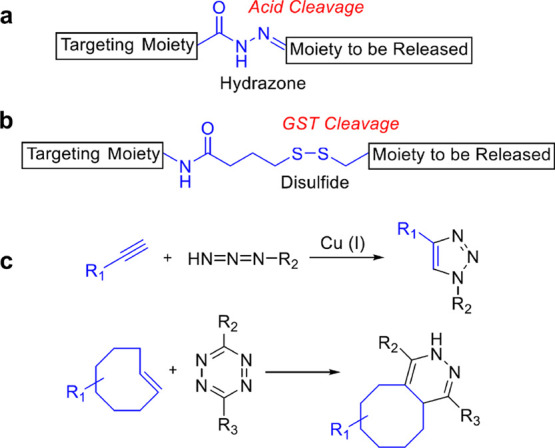
Examples of linkers and cleavage conditions: (a) hydrazone; (b) disulfide linker; (c) traditional click chemistry reaction used to easily link moieties together. The inverse Diels–Alder reaction has been used to link molecules to a solid surface for screening or for the release of a cytotoxic moiety. Linkers are highlighted in blue.
The disulfide moiety is also commonly investigated in chemical biology (Figure 2b). It has been highly utilized to cyclize peptides. The cyclization of peptides has been shown to increase their resistance to proteases and, in some cases, stabilizes the structure to boost its ability to bind to the targeted protein.12 More recent trends have promoted the concept of peptide stapling rather than disulfide bonds because of the reversibility of the disulfide bond.13 Nonetheless, disulfides in peptide therapeutics are still common, with the most well-known therapeutic peptide that incorporates disulfide bonds being insulin.14 Disulfide linkers have also been exploited in recombinant fusion proteins15 and for the synthesis of peptide libraries.16,17
Since the disulfide linker bond can be reversed under physiological conditions, it has been integrated into drug delivery approaches and in prodrug scaffolds. Cells have a high level of free −SH moieties in their cytosol. Once the disulfide-linked drug enters the cytosol, the disulfide can be reduced, releasing the drug moiety.18 The disulfide linker has been extensively used in the conjugation of small molecules to antibodies. Anticancer drugs, including doxorubicin (1), methotrexate, and mitomycin C, have been linked to antibodies and, after internalization, the disulfide linker is cleaved and the cytotoxic agent released.19 This method increases the uptake of the cytotoxic drug by cancer cells and not by the healthy ones. A linker containing a free thiol is conjugated to the small molecule of interest at a location that does not affect its activity. This entire moiety is then bound to an antibody through generation of a disulfide bond between the free thiol linked to the small molecule and a cysteine residue on the antibody.
Other linkers used in chemical biology can be generated through the reaction commonly referred to as click chemistry (Figure 2c). The term click chemistry, coined by Karl Barry Sharpless, refers to a variety of reactions that are considered simple and regiospecific and provide high yields.20 However, click chemistry has become traditionally referred to as the Huisgen 1,3-dipolar cycloaddition of azides and terminal alkynes. The most basic click reaction, with cooper as a catalyst, produces a 1,4-subsituted triazole. This reaction has been used to (i) link natural products to tags aiding in identification and detection,21,22 (ii) introduce a biotin moiety on proteins of interest for enrichment for mass spectrometry experiments,23−26 and (iii) synthesize a variety of small molecule libraries on solid-phase or polymer-like structures.27−30 A click reaction generating a releasable linker is the inverse-electron-demand Diels–Alder between a conjugated trans-cyclooctene and a tetrazine moiety. This type of cleavage linker has been demonstrated to effectively release 1 or other ligands conjugated to an antibody.31,32 The Garner laboratory has also employed this type of click reaction to develop different platforms for the screening of small molecule binders to RNA.33,34
The linkers described here are just a few of those that have been developed to help answer a variety of chemical biology questions and for therapeutic application. In the remaining subsections, we will describe more specific examples of how linkers are critical for the success of drug discovery programs and for the study of essential cellular processes.
2.2. PROTAC Linker Considerations
Proteolysis targeting chimeras or PROTACs represent a new method to target proteins of interest and degrade them to elicit a therapeutic response. This method exploits a chimeric molecule. A small molecule binder to an E3 ligase is linked to another small molecule that binds with the protein of interest. The targeted protein is then ubiquitinated after coming into close contact with the E3 ligase and degraded by the proteasome. One of the most critical decisions in designing a PROTAC is the length of the linker required to connect the small molecule binding to the protein of interest and the desired E3 ligase. PROTACs have been developed to degrade a variety of target proteins including ALK,35 the estrogen receptor,36 MDM2,37,38 tau,39 BET protein, and CDK9 protein. For these two last ones, the chimeric compounds JQ-1 and CDK9 are reported in Figure 3. Well-established PROTACs are commercially available. After selection of which E3 ligase to target, typically either cereblon (CRBN) or von Hippel–Lindau (VHL), an appropriate linker between the E3 ligase binding moiety and the molecule binding the protein to be degraded needs to be installed.
Figure 3.
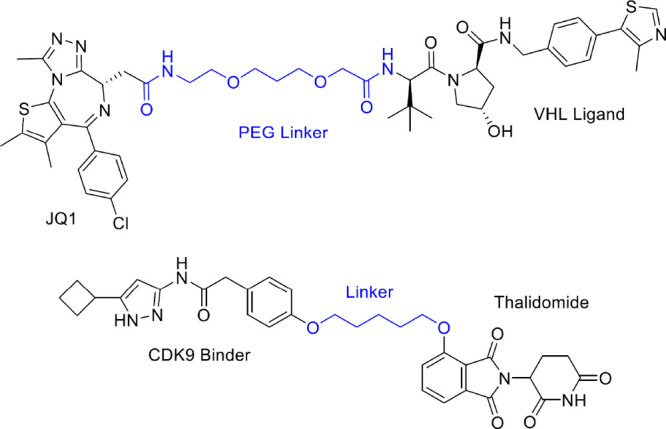
Chemical structures of the well-known, potent PROTACs including JQ1 and a CDK9 inhibitor, presenting the best linker length. Linkers are highlighted in blue.
An interesting study by the Kim group highlights how critical the linker length is in order to generate a potent degrader. They created an estrogen receptor (ER)-α-targeting PROTAC and installed a variety of linkers with different lengths. These linkers were composed of polyethylene glycol units, ranging in length from 11 to 16 atoms. Their results showed that while the 12- and 16-atom linkers had similar binding affinities to the ER, the 16-atom linker was significantly more potent in degrading the ER.40
The importance of the linker length for a PROTAC was also demonstrated by the Krönke group.41 They designed a homo-PROTAC for the degradation of the E3 ligase CRBN. If CRBN cannot ligate its cellular substrates, ubiquitinated proteins can increase, leading to cell death. They connected two thalidomide moieties with PEG linkers of various lengths and determined their abilities to degrade CRBN. In this case, the optimized linker was a short 8 atoms length PEG. These studies, along with many others, highlight that new PROTACs must be tested with a variety of different length linkers.42 Linker dynamics, such as thermodynamics, linker flexibility, and decreasing steric clash, have been studied, and all of these parameters should be considered when designing a new PROTAC.43,44
2.3. Linkers for the Discovery and Isolation of Natural Products
Natural products represent a novel pool of potential antibiotic and anticancer molecules. Traditional purification techniques are biased toward discovering natural products that have been already identified. As described in the click chemistry section, it is a method to target alkyne-containing natural products, but these are a very small pool of natural products. The biggest challenge in discovering therapeutically relevant natural products is finding small molecules that have not been previously identified. Traditional extraction methods of a crude natural product lysate followed by LC/MS analysis is biased toward discovering the most abundant molecules in the lysate. Linkers that can isolate natural products based on their functional group composition have been developed. This technique produces different pools of natural products, helping to unmask those that are too low in abundance to be detected in the crude lysate. The Carlson group has developed a family of reversible linkers to isolate hydroxyl-, phenol-, and carboxylic acid-containing natural products.45−48 These linkers contain different siloxy moieties that can selectively capture or release different molecules containing the aforementioned functional groups (Figure 4). The capture of hydroxyl-containing natural products occurs by the formation of a silyl ether bond. This links the natural product to the resin. Molecules not bound to the resin are rinsed away using a variety of solvents. The molecules linked to the resin can be released by exposing the resin to a fluoride source, such as TBAF or HF. This creates two pools of molecules, those that contain a hydroxyl moiety and those not bearing this functional group. These unique pools of molecules can be concentrated and analyzed by LC/MS or fractionated for activity-based assays.
Figure 4.

Linkers for the physical capture of natural products. Resins utilize a linker (colored in blue) with a silicon atom to capture (a) hydroxyl- or (b) carboxylic acid-containing molecules.
Linkers for the isolation of natural products containing other functional groups have also been developed. There are several examples of linkers to capture thiol containing natural products through either a disulfide bond formation or a 1,4-nucleophilic addition.49,50 Linkers to natural products containing less prevalent functional groups, including epoxides and β-lactams, have also been described.51,52
2.4. Linking Covalent Inhibitors to Fluorophores
Monitoring and visualizing essential cell processes are critical for drug development. The monitoring of enzymes critical to cell survival is an important chemical biology technique. To accomplish this, a number of covalent inhibitors have been linked to fluorophores through a variety of linkers aiming to visualize the desired cellular process. These probes can be used in confocal microscopy and/or flow cytometry to evaluate the effect of potential small molecule therapeutics. One example is the development of a fluorescent derivative of Taxol (2) (Figure 5a). This cytotoxic drug was discovered ∼50 years ago and has been used to treat a variety of cancer types. 2 targets cells that are rapidly dividing by interacting with microtubules and initiating mitotic arrest. However, it is currently unclear the mechanism by which 2 elicits its toxic effect and why some patients do not respond to the treatment.53,54 To visualize the subcellular localization of 2, the Peterson group synthesized a probe that links this microtubule-stabilizing drug to Pacific Blue.55 They tested three different linker lengths between 2 and the Pacific Blue moiety, and their results indicated that having a glycine linker, rather than a β-alanine or GABA linker, led to the best binding affinity to the tubulin heterodimer. Their probe was highly specific for tubulin binding, and they proposed that it can be used as a new tool for studying how 2 affects the proliferation rate of cancer cells.
Figure 5.
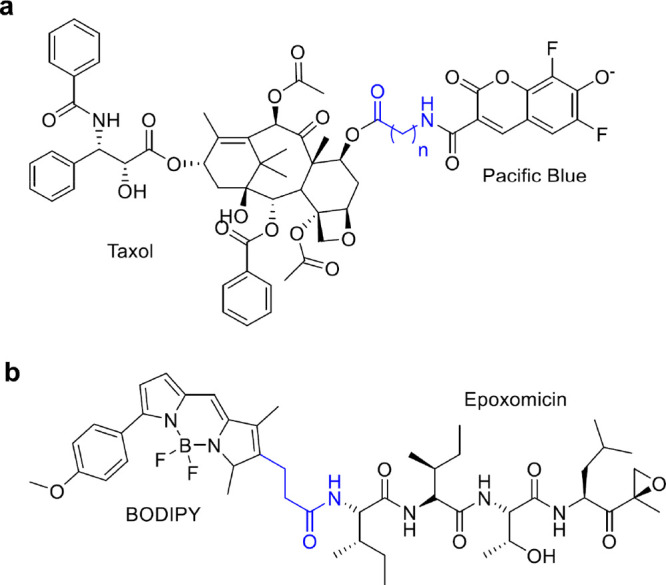
Two probes to observe (a) tubulin dynamics and (b) proteasome activity with the linker portion colored in blue. A variety of linker lengths between the small molecule binder/inhibitor and the fluorophore were evaluated to ensure that the fluorophore did not interfere with the binding to the protein of interest.
Fluorescent probes with a variety of linker types have also been developed to monitor the activity of the proteasome. The proteasome is a large protein complex in cells, responsible for proteins degradation. If unwanted proteins accumulate in cells, this can lead to endoplasmic reticulum stress and eventually apoptosis.56−58 Fluorescent probes have been developed to study the activity of the proteasome in cells.59,60 One of the major considerations when developing a proteasome activity probe is the linker length between the fluorophore and the active-site binding moiety. The fluorophore must be far enough from the binding site moiety to prevent any steric hindrance but not too bulky that it cannot enter the catalytic channel of the proteasome. The Overkleeft group has developed a number of fluorescent probes to monitor the activity of the proteasome. They have applied an activity probe that consists of the BODIPY fluorophore linked to epoxomicin, a covalent inhibitor of the proteasome (Figure 5b).61 This probe, along with others with different linker lengths, can be used to evaluate proteasome activity and determine the composition of the different types of active sites that assemble to form the full proteasome.62,63 In addition, a variety of probes with different types of linkers have been developed to monitor the activity of the immunoproteasome.64 The immunoproteasome rather than the standard proteasome is produced when cells encounter an inflammatory signal.
The recent advancements in linker chemistry suggest that in the future linkers will allow making steps forward in the design of chimeras. Moreover, the linker will play a pivotal role in the delivery and release of therapeutic agents, as well as in the investigation of biological pathways.
3. Chimeric Compounds and Receptor-Mediated Endocytosis
3.1. Receptor-Mediated Endocytosis for Drug Delivery
Lack of optimal pharmacokinetic profile is one of the main reasons why compounds fail during preclinical and clinical studies. Barrier permeability is an obstacle in achieving the therapeutic effect. Drug delivery opportunities are currently rising, and researchers are focusing their work on overcoming tissue barriers. Receptor-mediated endocytosis (RME, Figure 6) has been extensively studied as a method for boosting the transport of bioactive cargo across membranes, including the blood–brain barrier (BBB).
Figure 6.
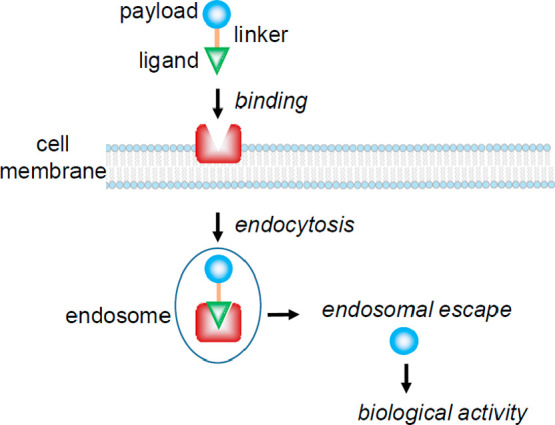
Schematic strategy of the selective delivery of biological cargos into cells exploiting receptor-mediated endocytosis.
Ligands binding to surface receptors can induce cellular uptake of therapeutics, including monoclonal antibodies, peptides, nucleic acids, small-molecule drugs, and nanoparticles. Herein, we discuss the recent advancements in the use of ligands for selectively binding to cell surface receptors. Three classes of ligands are discussed: (i) cell-penetrating peptides (CPPs), (ii) tumor homing peptides (THPs), and (iii) monoclonal antibodies. The highest ligand selectivity is displayed for antibodies, and this led to the FDA approval of five antibody–drug conjugates (ADCs): Mylotarg (3),65,66 Besponsa (4),67 Adcetris (5),68,69 Kadcyla (6),70,71 and Polivy (7).72
However, huge advancements have been shown also in the field of THPs. THPs are short peptides that have an inherent property to recognize tumor cells. Tumor necrosis factor α (hTNFα) was conjugated with a tumor homing peptide (NGR),73,74 and phase I and phase II clinical trials of NGR-hTNFα as a single agent and in combination with 1 are ongoing. In addition, THPs have a possible application in diagnostic imaging to help target radiopharmaceutical agents.75 THPs represent a step forward in cancer diagnosis and treatment.
3.2. Cell-Penetrating Peptides
CPPs are considered the least selective ligands for RME and are believed to translocate across cell membranes via a receptor-independent mechanism.76 Very recently, disclosures in cell-surface receptors responsible for cellular uptake of CPPs paved the way for the optimization and exploitation of CPPs as ligands.77
CPPs are cationic and/or amphipathic peptides of typically 8–30 amino acids and have been widely used to induce cellular uptake of bioactive cargoes.78−80 CPPs can be either covalently or noncovalently be coupled with a cargo. Identification of key amino acids to induce cellular uptake has been a pivotal parameter for the development of efficient ligands. The isolation of the active transporting peptide sequence within the HIV-TAT (TAT48–57: GRKKRRQRRR) represented a breakthrough for CPPs development. This sequence is called TAT peptide or TAT.81 Due to their high efficiency in internalization, arginine-rich CPPs such as oligoarginine and TAT facilitate the intracellular delivery of a wide range of cargoes, including peptides, antibodies, nucleic acids, nanoparticles, and small molecule drugs.82 Different studies have reported the pivotal role of arginine as a basic amino acid in CPPs, since it interacts with the guanidinium and phosphate groups at the cellular membrane. Indeed, the surface of cancer cells is known to be more negative with respect to that of normal cells. The negative charge generated on cancer cells is related to the different sugar metabolism pathways from normal cells due to the higher amount of lactic acid production.83 Positively charged CPPs bind through electrostatic interactions to the outside of cancer cells and promote RME.84 However, the widespread use of CPPs is hampered by the lack of specific selectivity. TAT has been shown to strongly enhance the intracellular delivery of 1. Due to the nonspecific cell penetrating features of TAT, CPPs have been coupled to nanocarriers. Recently, Yang et al. developed acid-sensitive micelles as delivery method for TAT protection. The luteinizing hormone modified poly(ethylene glycol)-poly(l-histidine)-1 (LHRH-PEG-PHIS-1, Figure 7a) micelles were employed to deliver 1-TAT (Figure 7b). This strategy represents a step forward in the safer use of cytotoxic agents since the micelles dissociate in response to the tumor extracellular pH. Afterward, 1-TAT can cross the cell membrane of tumor cells and elicit a cytotoxic effect.85
Figure 7.

(a) LHRH-PEG-PHIS-1. (b) 1-TAT conjugate. (c) Cleavable probe of octa-arginine peptide. Linker is colored in blue.
In 2018, an anionic cell-penetrating tetrapeptide, Glu-Thr-Trp-Trp (ETWW), with excellent potential for cell penetration, has been reported. The tetrapeptide has been coupled to liposomes to efficiently deliver 1 to the nucleus of cancer cells.86 Very recently, Dominguez-Berrocal et al. developed a chimeric trifunctional peptide composed of a CPP, a nuclear localization sequence, and a peptide blocking the interaction of the primary downstream effectors of the Hippo signaling pathway (TEAD and YAP). The novel peptide delivered the cargo specifically to the nucleus and showed an apoptotic effect in tumor cell lines. The antitumor efficacy in a breast cancer xenograft model is encouraging for the development of nuclear anticancer drugs.87
In addition to cancer treatment, nanoparticle-forming CPPs have been investigated in gene therapy approaches. However, CPP-mediated plasmid DNA (pDNA) delivery has been inefficient mostly because CPPs condense pDNA into nanoparticles that easily disintegrate, without delivering the therapeutic amount of pDNA into cells. In addition, CPPs and their cargo could be trapped into endocytic vesicles, preventing the pDNA from reaching the nucleus. These limitations can be overcome with the addition of a hydrophobic stearic acid residue since hydrophobic interactions are essential to form and stabilize the CPP/pDNA nanoparticles. Veiman et al. proposed pepFect14 (stearyl-AGYLLGKLLOOLAAAALOOLL, PF14, where O is ornithine) as a suitable non-natural peptide to form stable nanoparticles with pDNA. These nanoparticles could lead to an efficient gene delivery allowing the optimal transfection of genetic material into cells.88 The uptake of PF14 and other CPP/oligonucleotide (siRNA or pDNA) complexes is mediated by scavenger receptors (SCARA).89 These receptors bind promiscuously all negatively charged macromolecules and mediate their uptake.90
Another noteworthy application of CPPs is the delivery of neuroprotective peptides to the central nervous system for the treatment of neurological disorders. Arginine-rich CPPs show promising results in the delivery of neuroprotective peptides, especially to aid in treating cerebral ischemia and stroke. Several groups have shown that TAT and other arginine-rich cell penetrating peptides have intrinsic neuroprotective properties.91,92 Meloni et al. suggested that the neuroprotection might be related to carrier peptide endocytosis: neuronal cell surface structures such as ion channels and transporters are internalized during endocytosis, decreasing the calcium influx associated with excitotoxicity.93 In addition, endocytosis causes internalization of cell surface receptors leading to a decrease in receptor-mediated neurodamaging signaling pathways.94
Endocytosis has a crucial role in the cellular uptake of CPPs. Macropinocytosis95 and other classes of endocytosis such as clathrin-mediated96 and caveolae-mediated endocytosis97 are involved. Moreover, direct penetration of CPPs through plasma membranes has been described.98 Originally it was believed that CPPs translocated across cell membranes via a receptor-independent mechanism, leading to a not-cell-type-specific uptake.76 Very recently, Kawaguchi et al. identified syndecan-4 as a cell-surface receptor responsible for cellular uptake of octa-arginine (R8) peptide via clathrin-mediated endocytosis. A cleavable probe of the R8 peptide (Figure 7c) was used to identify syndecan-4 as an endogenous membrane-associated receptor.77 Even though this cell-surface receptor is ubiquitously expressed, it is overexpressed in breast and testicular cancer cells99,100 and in kidney cells of patients with IgA nephropathy.101
Rodriguez Plaza et al. proposed that CPPs work as cationic antibacterial peptides (CAPs) in the presence of bacterial cells. While CPPs enter eukaryotic cells without apparent toxicity, CAPs are able to make pores in the membrane and kill bacteria. Iztli peptide 1 (IP-1), showing both CPP and CAP activities, was utilized to explain this different behavior. IP-1, a hunter–killer peptide against Saccharomyces cerevisiae, makes pores only in the presence of high electric potential value at the membrane, which have been found in bacteria and mitochondria.102 Therefore, CPPs are able to switch from penetrating mammalian cells with any apparent toxicity to killing bacterial cells in the presence of large membrane potential.102,103
3.3. Tumor Homing Peptides
As described in section 3.2, the majority of CPPs lack specificity leading to reduced therapeutic efficiency and side effects. To overcome the limitations of CPPs, more specific peptides, namely, tumor homing peptides (THPs), have been developed.104 THPs are short peptides constituted by a few amino acids (3–15) and are considered a type of CPP. They have the intrinsic property to recognize oncological-specific proteins and molecular markers overexpressed on tumor cells or tumor vasculature.105 After binding to cell surface receptors, tumor homing peptides induce RME. Classical vascular-homing peptides are peptides containing the NGR motif, which binds to aminopeptidase N (CD13) or the RGD motif, which binds to αν integrins.106 Aminopeptidase N is overexpressed by endothelial cells of tumor vasculature and has been demonstrated to be involved in angiogenesis and cancer progression. Likewise, αν integrins are overexpressed in blood vessels in the tumor and represent a potential target to deliver cytokines to tumor vasculature. 1 was the first anticancer drug to be coupled to a NGR peptide. Later, phase I and phase II clinical trials of NGR-hTNFα as a single antitumor agent and in combination with 1 have been performed for a variety of cancers, including ovarian, colorectal, and small cell lung cancer (SCLC).107,108 Tumor necrosis factor α (TNFα) has demonstrated powerful antitumor activity but also severe toxicity. Conjugation of hTNFα with the tumor homing peptide NGR (Figure 8) improved safety and efficacy of TNF. Moreover, a synergism between NGR-hTNFα and chemotherapy was observed, since NGR-hTNFα has been shown to increase the intratumoral chemotherapy penetration.73 Very recently, phase II clinical results have been disclosed and NGR-hTNF plus 1 demonstrated promising activity in patients with relapsed SCLC.74 A phase III clinical trial was performed in patients with malignant pleural mesothelioma to assess the efficacy and safety of NGR-hTNF plus best investigator choice [NCT01098266]. Despite the positive results in phase II evaluation, the phase III clinical trial did not meet its end point; no significant differences in overall survival were observed between treated groups. However, further investigation is needed due to the poor prognosis of patients after first-line treatment.109
Figure 8.
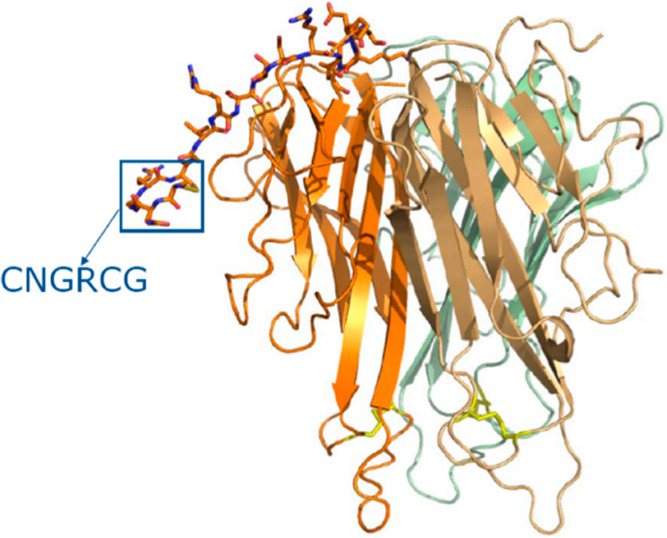
Structure of a monomer NGR-hTNF [https://www.molmed.com/node/33].
In addition to the TNF protein, the TNF gene has been employed for cancer gene therapy and has been reported to promote antitumor responses both in animal models and in patients. The plasmid DNA encoding CNGRCG-TNF and ACDCRGDCFCG-TNF (pNGR-TNF and pRGD-TNF, respectively) displayed growth inhibition of subcutaneous murine B16F1 melanomas and RMA-T lymphomas after intramuscular injection.110 RGD-TNFα was also evaluated for its ability to enhance the antitumor effect of chemotherapy; however NGR-hTNFα was mostly chosen for clinical trials. RGD has been preferentially exploited for diagnostic applications and many RGD-based radiopharmaceutical agents have been assessed for cancer imaging.75 Bispecific NGR peptides (GNGRAHA), targeting both CD13 and αvβ3 integrin in the endothelium of solid tumors, have been developed. In 2018, Seidi et al. combined the NGR peptide, GNGRAHA, with a truncated form of coagulase (tCoa) generating a bifunctional protein (tCoa-NGR) with novel anticancer properties. This strategy allowed selective targeting of the tumor neovasculature and inducing of selective thrombosis in tumor-feeding vessels. In tumor models, tCoa-NGR led to a significant reduction of tumor growth after systemic administration.111 Therefore, tCoa-NGR represents a promising anticancer strategy to induce tumor infarction and reduce systemic side effects.
Besides TNF, tumor homing peptides could facilitate distribution of other cytokines into tumor cells and enhance their therapeutic effect. In 2017, it has been shown that RGD enhances the antitumor effect of IL-24. Melanoma differentiation-associated gene-7/interleukin-24 gene (MDA-7/IL-24) is a unique tumor suppressor gene, which promotes selective apoptosis of cancer cells. RGD-coupled IL-24 construct induced apoptosis in hepatocellular carcinoma-related cell line.112 The results highlight the benefit of cytokine targeting by THPs to cancer cells. Coupling RGD to the N-terminus of IL-24 led to a stronger interaction with the receptors. On the contrary, adding RGD to the C-terminus of IL-24 disrupted native interactions and reduced the apoptosis induction properties.113 Very recently, Bina et al. confirmed these results with in silico targeting of RGD/NGR-modified IL-24 to tumor cells.114
THPs have shown potential to be versatile platforms of polymers for nonviral gene delivery.115 pDNA complexes of recombinant proteins with poly(l-lysine) and THP showed significant improvement of target specificity to cancer cells by additions of F3 and CGKRK THPs. F3 peptides are high affinity ligands for nucleolin, which is expressed on the surface of cancer angiogenic endothelial cells, and selectively bind MDA-MB-435 cells.116,117 CGKRK peptides were described to bind to heparin sulfate in cancer vessels.118,119
THP–gold nanoconjugates actively targeted MCF-7 cells in comparison to nontumor 3T3-L1 fibroblast cells.120 THPs specific for MCF-7 cells were selected from a phage display library, synthesized, and conjugated to spherical gold nanoparticles by a heterobifunctional cross-linker with an ethylene oxide spacer. This work proved the possibility of developing nanomaterials that can rely on tumor targeting potential irrespective of a specific knowledge of the target cell biology.
3.4. Monoclonal Antibodies
Over the past decade, monoclonal antibodies (mAbs) have significantly improved the clinical outcomes for cancer patients since they specifically bind tumor-associated target antigens and eventually deliver cytotoxic agents to tumor cells in a targeted manner while sparing normal cells.121 mAbs are conjugated to small-molecule chemotherapeutics, and the resulting antibody–drug conjugate (ADC) is parentally administered (intravenous or subcutaneous). After binding to their target antigens, ADCs are internalized through RME.122 The development of a procedure to produce mAbs has increased the enthusiasm of scientists for the development of precise targeted cancer therapy. Humanized and fully human antibodies have the advantage of being retained longer in circulation than their murine equivalents and led to a dramatic increase in the use of antibody-based drugs against cancer.123,124 However, many challenges have to be overcome for the development of optimized and functional antibody–drug conjugates with possible application as therapeutic agents.
One of the major challenges in the development of ADCs is to incorporate a linker able to preserve the ADC stability in systemic circulation for an extended period and to release the payload at the targeted site. Conjugation site and linker choice are key parameters in the pharmacokinetic properties of ADCs. The site of attachment to an antibody can also be engineered in different ways to incorporate a linker and subsequently a bioactive molecule.
Considering the five ADCs approved so far by FDA, gemtuzumab ozogamicin (3) and inotuzumab ozogamicin (4) have an acid-sensitive hydrazone linker (Figure 9), brentuximab vedotin (5) and polatuzumab vedotin (7) have a lysosomal protease-sensitive peptide linker (Figure 10a), and trastuzumab emtansine (6) exploits a noncleavable SMCC (N-succinimidyl-4-(maleimidomethyl)cyclohexane-1-carboxylate) linker (Figure 10b).
Figure 9.
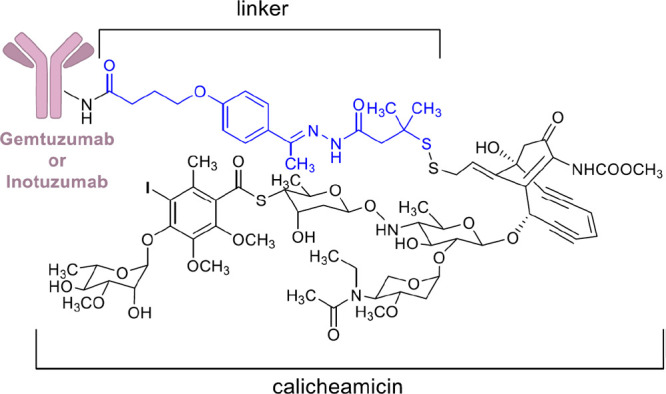
Chemical structure of 3 and 4. Linker is colored in blue.
Figure 10.

(a) Chemical structure of 5 and 7. (b) Chemical structure of 6. Linkers are colored in blue.
Compound 3 uses side chain reactive lysines of a humanized anti-CD33 mAb to attach calicheamicin, a highly cytotoxic agent that induces double-strand DNA cleavage, by a bifunctional acid sensitive hydrazone linker (Figure 9). After being launched in 2000 as therapeutic agent for relapsed acute myelogenous leukemia, this ADC was withdrawn from the market due to the limited benefit over conventional anticancer treatment and the serious hepatotoxicity.125,126 This withdrawal increased the concern on the stability of the hydrazone linker. In addition, the ADC heterogeneous nature of the drug conjugate concurred to premature release of the conjugated payload, leading to a significant toxicity compared to traditional chemotherapy. Subsequent trials using a lower dose led, in September 2017, to the FDA approval of 3 for newly diagnosed and relapsed/refractory acute myeloid leukemia.65,66
Compound 4 is another antibody–drug conjugate of calicheamicin.127 It is formed by a CD22-directed monoclonal antibody covalently bonded to N-acetyl-γ-calicheamicin (Figure 9). 4 received FDA approval in 2017 to treat relapsed or refractory CD22-positive B-cell precursor acute lymphoblastic leukemia.674 has shown excellent activity in the clinic, and ongoing trials are evaluating its value as frontline treatment.128 A phase III clinical trials is assessing the benefits of treating newly diagnosed B-cell acute lymphoblastic leukemia with 4 in combination with chemotherapy [NCT03150693].
In 2011, compound 5 received approval for Hodgkin’s lymphoma (HL) and anaplastic large-cell lymphoma (ALCL).68,695 utilizes side chain cysteines to conjugate monomethyl auristatin E (MMAE), a potent antimitotic agent, with the anti-CD30 mAb (cAC10) through an enzymatically cleavable dipeptide (valine–citrulline) linker (Figure 10a).129 A selective reduction of the disulfide bonds in the four interchain provides up to eight reactive sulfhydryl groups that facilitate drug conjugation (drug to antibody ratios are from 0 to 8).130,131 Exploiting this method to link the drug, rather than using lysine conjugation, results in ADCs that could be easily purified and pharmacokinetically characterized. Besides its application in the treatment of different types of lymphomas, the safety and antitumor activity of 5 have been demonstrated also in patients with CD30-expressing solid tumors in a phase II clinical trial.132
In 2019, compound 7, a second ADC of MMAE whose mAb targets CD79b (B-cell antigen receptor complex-associated protein β chain), was granted accelerated FDA approval for the treatment of adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) in combination with bendamustine plus rituximab (BR).72 A multicenter phase Ib/II clinical trial including a cohort of 80 patients with relapsed or refractory DLBCL [NCT02257567] granted drug approval. At the end of the therapy, the complete response rate was 40% with 7 plus BR, compared with 18% with BR alone.133
Compound 6 uses a noncleavable SMCC linker to cross-link the warhead cytotoxic agent emtansine (DM1), a microtubule inhibitor, to lysine residues of anti-HER2 mAb, trastuzumab (Figure 10b). Lysine-MCC-DM1 complex, an intercellular metabolite, turned out to be as active as the parent drug, after trastuzumab degradation by lysosomes. It is clinically employed in patients with HER2-positive metastatic breast cancer.70,71 The approval of 6 for the treatment of breast cancer highlighted the capability of ADCs to target solid malignancies in addition to hematologic tumors. With the recent approval of 7, there has been a boost in research investigating the use of ADCs in cancer treatment. ADCs are likely to become a pivotal part of future targeted cancer therapy.
Although a huge effort has been made to produce ADCs for oncology, they are also an attractive platform to deliver noncytotoxic bioactive cargos in a cell-specific way aiming to reduce potential side effects related to off-target interactions. For example, an antibody–drug conjugate that selectively recognizes immune cells through the CD11a antigen has been conjugated to a derivative of a highly potent phosphodiesterase 4 (PDE4) inhibitor (GSK256066) (Figure 11a). This strategy could limit neurological side and gastrointestinal toxicity that have hampered a broad application of PDE4 inhibitors.134 To obtain a site-specific conjugation to the anti-human CD11a antibody, the unnatural residue p-acetylphenylalanine (pAcF) was linked to the heavy chain of efalizumab (site A122). To enable conjugation of GSK256066, a linker containing a tetraethylene glycol spacer with a terminal aminooxy group was reacted under slightly acidic conditions with the pAcF ketone, resulting in stable covalent conjugates. Conjugation was performed with drug/antibody ratio of 2 (1 bioactive molecule linked to each heavy chain). Recent studies have supported the feasibility to develop mAbs-PDE4 inhibitor conjugates as promising therapeutics for treating ulcerative colitis due to the specific delivery of immune suppressants to immune compartment.135 In addition, autoimmune diseases represent a potential field for ADCs application and significant advancements have been done in the past decade. Wang et al. proposed the use of dasatinib (8), a Bcr-Ab1 tyrosine kinase inhibitor, for immune suppression and developed an immunosuppressive ADC (Figure 11b) which targets CXCR4 and delivers 8 to human T lymphocytes. Modeling and structure–activity relationship studies highlighted that the hydroxyl moiety of 8 is not required to observed pharmacological activity.89,90 Therefore, it was modified for conjugation to the antibody with a noncleavable linker by reaction with p-nitrophenyl chloroformate and carbamylation with a tetrapolyethylene glycol (PEG) linker displaying an aminooxy group. The resulting 8–antibody conjugate inhibits T cell receptor (TCR)-mediated T cell activation and cytokine expression with nanomolar EC50 and shows minimal effects on cell viability. This strategy could lead to an improved efficacy and safety of kinase inhibitors and to their exploitation in nononcological diseases.136 A phase II clinical trial is currently ongoing to determine the benefit of 5 in the treatment of systemic sclerosis, a multisystem autoimmune disease [NCT03198689].
Figure 11.

(a) Anti-inflammatory human αCD11a antibody conjugated to a PDE4 inhibitor. (b1, b2) HLCX, immunosuppressive humanized antibody that binds selectively to CXCR4, conjugated to 8 with a noncleavable linker (b1) and a disulfide-cleavable linker (b2). Linkers are colored in blue.
These results highlight that, besides cancer, ADCs have potential application in a wide range of inflammatory and autoimmune disorders. Naked therapeutic antibodies have launched a novel era of both autoimmune disease and cancer treatment, but ADCs represent the next-generation antibody therapies and will represent a breakthrough in the treatment of these illnesses.
The ability of monoclonal antibodies to selectively bind tumor-associated target antigens and release cytotoxic agents to the tumors in a targeted manner has dramatically improved the clinical practice. Further advancements in this field will lead to the success of precise targeted cancer therapy. RME is also exploited by CPPs and THPs, but they have a poor selectivity compared to antibodies. However, nanocarriers are attractive tools to be coupled to CPPs and THPs and improve their safety and selectivity. In addition, the use of nanocarrier is boosting the antibody-based delivery of biological cargos into cancer cells. In the future, these systems are expected to become essential therapeutics for the treatment of malignancies and central nervous system disorders. Moreover, THPs are essential components of radiopharmaceutical agents and will represent a step forward in cancer diagnosis.
4. Chimeric Compounds and Target Engagement
4.1. Target Engagement
One of the main failures in translating preclinical results into a positive clinical outcome is the lack of pharmacokinetic and pharmacodynamic validation of drug–target interactions in vivo with serious impact on efficiency and costs of the drug discovery process. The mechanism that small molecules adopt to engage their targets inside living cells is a crucial step in medicinal chemistry and chemical biology, since it requires the availability of appropriate assays and inhibitors/ligands. The molecular recognition event in living cells between drugs and targets is defined as “target engagement”, and the associated technologies represent a rapidly evolving field of research.137−140
This technology allows target validation in living systems: in cells, tissues, and animal models. It confirms compounds cellular entry and target binding and can suggest optimized drug delivery to enable compounds to be more effective, specific, bioavailable, and less toxic.2,141,142In vitro studies could foresee the optimization of human performance characteristics.143 To perform these studies, selected leads and drugs should be conjugated with appropriate tags to obtain chimeric compounds with a diversity of structures (see Figure 1).2,141,142
To perform a target engagement study, it is essential to (i) know the target localization into the cell, (ii) design an assay for cellular setting, (iii) ensure the detection of the observable changes on cellular surface or intracellularly, depending on targets location, and (iv) ensure the escape of off-targets and background noise of the cellular matrices.144 While quantification of compound binding to purified proteins or surface receptors (in particular to GPCR) is well established,145,146 the interaction of compounds with intracellular targets is difficult to quantify.
Regarding the fluorimetric detection, a fluorescent probe has to be covalently conjugated with the inhibitor (see section 2.4)2,141,142 and should show sufficient solubility (slightly different from the values required for a drug) and a log P of around 3, necessary for a suitable drug or inhibitor tagging. Lipophilicity may influence the amount of compound able to enter the cell and consequently available for binding. In addition, the fluorescent tag module should not mask the compound affinity for the target (see section 2). Target engagement assays might be invasive since they drive the intracellular environment away from equilibrium conditions.147−149 Orthogonal assays are usually needed to validate the results.150 Aktinson and co-workers studied the interaction of selective autophagy receptors with two conserved hydrophobic pockets (called W-site and L-site) of mATG8 (autophagy receptors to autophagy related 8) proteins through a linear residue, namely, the LC3-interacting region (LIR). Fourteen LIR-containing peptides were designed and synthesized, and their affinity for mATG8 was investigated using a competitive time-resolved FRET (TR-FRET). The assay used a GST-tagged mATG8 protein and a terbium labeled anti-GST antibody to measure the equilibrium dissociation constant, Kd, by TR-FRET. The results were confirmed by additional structural information using nuclear magnetic resonance (NMR) spectroscopy. This work points out the importance of having two assays that exploit different experimental readouts to validate the results.150
A similar approach was reported to discover inhibitors of the signal-regulatory protein (SIRP)α-CD47 interaction with a high-throughput screening approach.151 CD47 is an immune checkpoint that downregulates the functionality of both innate and adaptive anticancer immune response through its SIRPα receptor. A series of small molecule ligands that selectively target SIRPα interactions with CD47 was discovered. The assay was performed using a specific LANCE TR-FRET assay and a ∼90 000-compound library. In parallel, an AlphaScreen based on similar TR-FRET technology was adopted for validation purposes. SIRPα was biotin tagged, and an antibody with the energy donor reagent was the tagged chimeric biomolecule exploited in the assays.
In the following subsections, target engagement technologies and examples of the use of tagged compounds are described.
4.2. Strategies Based on Small Molecule and Target Protein Modification
The proximity between a bioactive small molecule and its targeted protein can be studied using spectroscopic methods such as fluorescence or bioluminescence resonance energy transfer measurements (FRET and BRET, respectively). FRET and BRET occur only when the donor and acceptor are in close proximity (2–6 nm) and are unique methods to inspect intermolecular protein interactions and protein–ligand interactions in cells.152
In FRET (fluorescence resonance energy transfer or Förster resonance energy transfer) studies, a donor fluorophore upon excitation transfers energy to a nearby acceptor fluorophore. When a suitable acceptor is present, the donor emission is quenched and emission of light occurs at a longer wavelength (Figure 12). The essential criteria to observe FRET are (i) suitable distance, (ii) appropriate donor/acceptor orientation, and (iii) large overlap of the donor emission spectrum and the acceptor absorption spectrum. FRET can be quantified determining the change in donor fluorescence lifetime through fluorescence lifetime imaging microscopy (FLIM) based on FRET readout.153−155 FRET-FLIM monitors target engagement in living cells and provides details on the temporal and spatial distribution of the ligand–protein complex.
Figure 12.
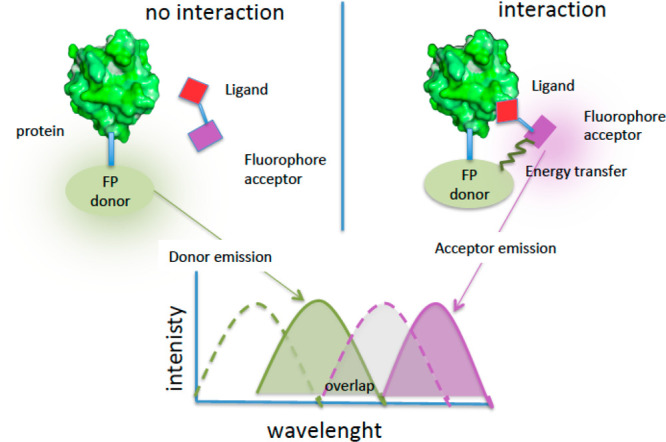
Description of FRET experiments for target engagement with no interaction (left) and interaction (right): FP, fluorescent protein; green dot line = donor excitation spectrum; green line = donor emission spectrum; violet dot line = acceptor excitation spectrum; violet line = acceptor emission spectrum.
In some cases, a fluorescent protein (FP) is fused in cells to the target protein, and a FRET signal is generated when fluorophores are in close proximity (Figure 12, left). In a different protocol, the target protein can be ectopically expressed in the same cells and modified in a specific residue in order to bind a suitable fluorescent donor (or acceptor) (Figure 12, right).156,157 The target protein could be properly engineered to allow binding detection, for example, with a tetracysteine tag.156,157 Cells expressing the target protein coupled to a FP are treated with the a small molecule labeled with a fluorescent dye; subsequently the lifetime distribution of the donor fluorophore into a cell is determined. The donor fluorescence lifetime reveals the interaction sites into a cell as well as the areas with a reduced donor lifetime.
FRET based technology has been exploited, for example, for the recognition of phosphodiesterase158 and thymidylate synthase (TS) by tagged inhibitors. TS is an obligate homodimeric enzyme, and a tetracysteine (TC4) tag is introduced at the N-terminus. The fluorescein diarsenical probe FlAsH, added to the HEK-293 cell lysate containing the ectopically expressed protein, is coordinated by the tetracysteine behaving as a fluorescence donor. The tagged substrate is an octapeptide (LR) and is conjugated with the fluorescence acceptor probe Hylite-405 (Figure 13). Titration of hTS-tetracys-Flash (acceptor) with LR-hilyte 405 (donor) in lysates of cells transfected with hTS-tetracys shows an increase in FRET signal.
Figure 13.

FRET experiment with TS dimer. The N-terminus is modified by inserting the sequence CCGPCC-tetracysteine (TC). Probe excitation at the proper wavelength causes the energy transfer and FRET signal increases upon binding of the LR-hilyte-405 ligand.
TR-FRET has been applied to the assessment of Bruton’s tyrosine kinase (BTK) occupancy in the clinical trials of tirabrutinib (9). Compound 9 (GS-4059/ONO-4059) is a second-generation, irreversible BTK inhibitor explored for the treatment of lymphoid malignancies. The inhibitor was conjugated with biotin through a carbamide–PEG mixed linker, and free and total BTK levels were measured using TR-FRET.154
4.3. BRET Experiments
BRET (bioluminescence resonance energy transfer) is a mechanism describing the energy transfer between a donor (luciferase) and an acceptor (fluorescent) molecule. The spectral separation between donor and acceptor excitation required in FRET (Figure 13) is not required in BRET since the production of light originates from a chemical reaction catalyzed by the donor enzyme (Figure 14, right). Since BRET does not require the use of excitation illumination, it has advantages over FRET. BRET is therefore more applicable to the analysis of photoresponsive cells or cells that are easily damaged by excitation light. BRET has been exploited to detect protein–protein interactions in real time in living cells.159,160 In target engagement studies, cells express the target protein fused to a luciferase, while a ligand with a fluorophore tag behaves as an acceptor (Figure 14). Different BRET techniques are known and differ for the combination of the donor/acceptor/substrate used.161 Distance, orientation, and spectral overlap are the major parameters that influence both BRET and FRET. However, external excitation of the donor is not required in BRET; therefore phenomena related to simultaneous donor/acceptor excitation, fluorescence of the background, and photobleaching are not occurring. A microplate luminescence/fluorescence reader is one of the major components of the BRET imagining microscopy system, and the acceptor fluorescence is detected as readout. BRET allows determination of the affinity of a small molecule for the target protein and the study of the intracellular residence time of inhibitors using kinetic measurements. This method was exploited to prove the isoenzyme-specific engagement of histone deacetylase inhibitors144 and ligand engagement of G-protein-coupled receptors (β2-adrenergic and adenosine receptors).145 Robers et al. exploited a Nanoluc small luciferase protein (19 kDa) as a BRET donor instead of luciferase (Luc), since it showed a higher fluorescence yield, a narrow spectrum, and a stable luminescence. As a BRET acceptor, the non-chloro-TOM dye (NCT), showing membrane permeability and significant spectral resolution, was employed. To explore the interaction of intracellular engagement of HDAC inhibitors, the hydroxamate-based inhibitor (SAHA) was conjugated with NCT and was used as displacement substrate (tracer displacement by unlabeled compounds).144
Figure 14.
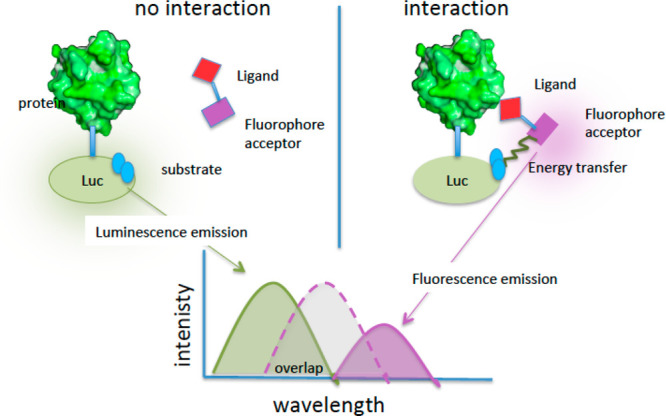
Description of BRET experiments for target engagement with no interaction (left) and interaction (right): Luc, luciferase; green line = donor emission spectrum; violet dot line = acceptor excitation spectrum; violet line = acceptor emission spectrum.
The same authors reported the quantitative aspects relevant to fully characterize the engagement. Inside living cells, a NanoLuc-tagged intracellular protein of interest achieves a dynamic equilibrium with a cell-permeable fluorescent dye (tracer). After binding of an unlabeled small molecule, complex disruption leads to a loss of BRET signal that is detected in a microplate format (Figure 14). NanoBRET tracers are often produced starting from a drug or a tool compound and allow a quantitative measurement of the apparent affinity and a real-time assessment of the residence time.162
The BRET method was also adopted for the identification of antimicrobial hits targeting the protein–protein interaction between the initiation factor σ and the β′-subunit of bacterial RNA polymerase.70 The study combined an in silico screening with an in vivo bioluminescence resonance energy transfer in yeast cells, showing the large applicability of this technology. One hit was identified and optimized using medicinal chemistry approaches.163
The description of the quantitative, real-time measurements of intracellular target engagement using energy transfer is reported, and NanoBRET tracers with optimized cell permeability have been developed and fully characterized (Figure 15a). Two main classes have been identified that represent robust chemical tools for the assay. Linkers between the dye and the reacting group such as succinimide (NanoBRET 590 SE) or reactive esters (nanoBRET 618 TFP) can influence tracer properties, including affinity and cell permeability.
Figure 15.

(a) Chemical structure of NanoBRET ester activated dyes; (b) example of linker building blocks. Dyes directly bound to the building block are known; however sometimes a linker (colored in blue) between the head tag and the fluorescent probe is necessary.
4.4. A Chemical Proteomic Approach for Covalently Binding Ligands
Affinity-based chemical proteomics (ABCP) is a method to study proteins or ligand–target interactions, based on protein isolation by an affinity reagent that can be coupled to a reporter system for detection. Affinity-based chemical proteomic has been used in target engagement studies of small-molecule drugs that covalently react with their targeted protein.164,165 The compounds have a chimeric nature since a reactive functionality such as an alkyne (Figure 16a) or azide group is introduced in a suitable position of the scaffold. After addition to the cell, the ligand reacts with the protein target bearing a reactive group exposed and regioselectively placed. Both wild type and mutant proteins can be exploited for the study. When the functional tag (or affinity tag) is added to the cell, it binds to the covalent ligand through a click chemistry reaction (alkyne with azide, Figure 16b). “Click chemistry”20 is exploited to attach in situ a functional tag, such as biotin. The functional tag allows affinity purification of the covalently bound protein of interest using, for example, streptavidin beads (Figure 16c), and protein identification is performed using tryptic digestion and nanoliquid chromatography–tandem MS analysis (Figure 16d). The functional tag presents a reactive head for the ligand and an affinity tag for the resin to allow affinity chromatography.
Figure 16.

Example of ABCP process for covalently binding ligands. (a) Different targets available in the cells for binding the covalent ligand (orange) with the alkyne reactive functionality. (b) Covalent ligand binds the target and forms a covalent or noncovalent complex. The reactive group is exposed outside the binding site. (c) The functional tag (green) is added. It reacts with the reacting group of the covalent ligand (alkyne) with a click chemistry reaction, thus forming a covalent complex with the target. The green tag has a high affinity for the resin that should sequester the chimera–target complex from the sample matrix. (d) The affinity resin is added. The complex, once detached from the resin, is analyzed through mass spectrometry and the biomolecular target identified.
This method can be applied also to probes that bind proteins in a reversible fashion by the addition of a photoreactive group for UV detection of probe–protein interactions in cells (see photoaffinity labeling).166 ABCP allows also off-targets detection in cells.167
Wong et al. investigated the specificity for a series of ATP-competitive bivalent kinase inhibitors targeting ABL1.168 They proved the affinity and selectivity of bivalent inhibitors against Abl protein kinase with respect to other off-targets using dual functional chemical proteomics probes. A bivalent inhibitor A-2 showed high affinity together with improved selectivity over the parental ATP-competitive inhibitor.
Another example of the pivotal role of chemical proteomic in chemical biology is the use of activity-based protein profiling (ABPP) to study proteins in their native environment.169 By exploitation of click chemistry, an affinity-based probe for the human adenosine A2A receptor (hA2AR) was developed to investigate the structural biology of the G-protein-coupled receptor (GPCR). Yang et al. developed compound 10 (LUF7445), a clickable affinity-based probe, with an electrophilic reactive group, as a covalent antagonist of hA2AR. LUF7445 was discovered through chemical modification of compound ZM241385 introducing a fluorosulfonyl group, and different linker lengths have been investigated. On the most potent ligand, an alkyne-click handle was introduced leading to the synthesis of probe 13 (Figure 17). The binding of the ligand to the receptor was washout-resistant. This probe allowed assessment of the presence of hA2AR in complex biological samples. The identification of the affinity probe for a GPCR is a promising tool to monitor the endogenous GPCR expression related to human diseases.
Figure 17.

Chemical structures of the hA2AR antagonists investigated by Yang et al. The selective hA2AR antagonist (ZM241385) guided the design of the covalent antagonist 10. The authors assessed the importance of the linker length (colored in blue) between the scaffold and the head on affinity and synthesized the optimized compounds 11 and 12. Starting from compound 12, the affinity-based probe 13 with an alkyne ligation-group and a fluorosulfonyl electrophilic was synthesized.
4.5. Photoaffinity Labeling
Photoaffinity labeling (PAL) is a well-known technique used to study specific protein function or inhibition.170 Photo-cross-linkers are conjugated with drugs or substrates that can bind to the target protein (protein of interest). Typically, photo-cross-linkers are (i) benzophenone (BP), (ii) aryl azide (AA), and (iii) diazirine (DA) (Figure 18). Upon photoirradiation, the photo-cross-linking functional group generates highly reactive species that react with adjacent molecules, leading to a direct covalent modification.171
Figure 18.

(a) Most relevant photoaffinity compounds used to tag protein ligands aiming to study target engagement, protein functions, and their photoreaction: benzofenone, aryl azide, and diazirine (trifluoromethylphenyl diazirine, trifluoromethylethyl diazirine). (b) General structures of scaffolds for photoaffinity tagging.
PAL can capture partners through noncovalent interactions and explore the ligand accessible protein space in a selective mode. Photo-cross-linking agents have turned out to be essential tools to study difficult targets such as protein–protein interactions. Despite the high significance and extensive application, only few photo-cross-linkers are currently available. In the 1970s, BP has been introduced as a photo-cross-linker and is the most used in PAL due to the good selectivity and affinity toward methionine. Upon irradiation by 350–365 nm wavelengths, BP is converted into an active diradical. It reacts with protein functional groups exploiting an abstraction–recombination reaction mechanism. Aryl azides cross-link through nitrene, a reactive species, that is generated by loss of N2 upon photoirradiation with 254 and 400 nm wavelengths. Nitrene reacts with nearby C–H and heteroatom–H bonds, creating a novel covalent product. AAs are known to be chemically stable and to have superior photophysical properties than the corresponding acyl and alkyl analogs. Trifluoromethyl phenyl DAs and alkyl DAs can both produce carbene as reactive species losing N2 upon photoirradiation at 350 nm. They can form covalent adducts as phenyl diazirine (Figure 18a).172 Novel functionalized scaffold to be included in the chimeric compounds can be designed starting from different precursor reagents. In Figure 18b the colored fragments are included in the final chemical photoaffinity reagent.
A typical methodology for target deconvolution in drug discovery is applied to living cells or protein complexes, including cell lysates, that are incubated with the compound. The derivatized compound has a photoaffinity linker and a reacting agent (drug, inhibitor, or ligand), and the compound–protein binding is fixed by UV irradiation.173 Affinity tag is used to isolate proteins covalently bound to the compound, which are then analyzed exploiting MS-based proteomics for proteins identification (Figure 19). T. Tomohiro identified pyruvate carboxylase and C-terminal biotin carboxyl carrier protein as biotin-binding protein from HeLa cells using a PAL-based enrichment with an isotope-coded fluorescent and photocleavable tag followed by MS.174 Relatively small functional groups for click chemistry have been recently introduced aimed at improving the photo-cross-linking yield and at gaining sensitivity of MS-based proteomics. This method could be applied also when the affinity between the target protein and the small molecule is weak.
Figure 19.

Efficient PAL method for protein identification using a bead-based multivalent probe.
The Kaori Sakurai group used PAL to detect the binding protein of benzenesulfonamide.175 They produced trifunctional probes bearing a lysine scaffold and containing a benzenesulfonamide moiety as protein-binding ligand. The photoactivatable group was BP, and biotin was selected as reporter group, allowing the detection of the protein–covalent adducts (Figure 19).
Other engineered chimeric structures have been adopted for tagging experiments. For example, a chimeric molecule to identify the target of oleanolic acid was prepared176 (Figure 20a), and a photoactivated γ-secretase inhibitor in which the tag covalently label presenilin 1 was developed177 (Figure 20b). Presenilin 1 belongs to the γ secretase complex and plays an important role in the generation of amyloid β (Aβ) from the amyloid precursor protein, and it is associated with the onset of Alzheimer disease. The novel photoaffinity labeled palmitoyl derivative (Figure 20c) is directed to the peroxisomal β-oxidation enzyme, a primary enzyme for fatty acid degradation.178
Figure 20.

(a) Chimeric molecule designed to identify the target of oleanolic acid; (b) photoactivated-γ-secretase inhibitor in which the tag covalently label presenilin 1; (c) photoaffinity labeled palmytoil derivative directed to the peroxisomal β-oxidation enzyme. Shown in green are the photoreactive groups.
Recently a high number of PAL applications have been published, underlining the increasing importance of this method in drug discovery.
Other technologies are under development within the engagement technology field such as the carbene footprinting technology.179,180 An example is given by the differential protein footprinting approach that adopted an efficient photoactivated probe and used it in mass spectrometry to map the binding cleft of lysozyme, as well as between UPS5, a deubiquitinating enzyme, and a diubiquitin substrate.179
5. Conclusion
The development of new technologies and chemical biology strategies has largely stimulated medicinal chemists’ creativity to design molecules that could meet the challenges that a drug encounters from the delivery to the patient up to target binding.
This ambitious task was initially addressed using simple structures with chemicophysical properties suitable for cell membrane penetration. However, chemistry exploration and modular approaches led to the design of engineered constructs called chimeric molecules. Chimeras have been exploited in a wide range of applications, such as drug targeting and release, drug tracking and monitoring when tagged with fluorescent probes, target engagement, and mechanism of action clarification. Recently, engineered systems in which both compounds/drugs and proteins are chemically modified to give more specific and less invasive assays have been developed. Crucial is the role of the linking fragment connecting the functional head with the tag. Starting from a disulfide and an ester, the first linkers were based on the early concept of prodrugs, in which a cleavable bond could easily release the bioactive compound. Application of this linker chemistry was promising; however, the use of these systems was hampered by the risk of low specificity. Improved engineered compounds were developed, and linkers were recognized as an essential tool for structure–activity relationship studies of chimeric compounds and for providing the requested reactivity to conjugate the head and the tag. From a linear structure, such as PEG and alkyl chains with reactive groups at the two edges, a “three-dimensional” decoration of the chain is taking place to address the biological requirements, as observed in some ADCs. Irrespective of application field, chimeric molecules and linkers are conceptually related and can be exploited also in fields different from those mentioned in the present Perspective, including biosensors, biomarkers, and molecular machine. Chimeras are being developed by teamwork of medicinal chemists and chemical biologists and represent formidable tools for targeted therapies and personalized medicine.
Acknowledgments
We thank G. Ponterini for advice and discussions. This work was supported by the Foundation for Cancer Research in Italy by Grant AIRC IG16977 (to M.P.C.).
Glossary
Abbreviations Used
- RME
receptor mediated endocytosis
- ADC
antibody–drug conjugate
- CPP
cell-penetrating peptide
- THP
tumor homing peptide
- mAbs
monoclonal antibodies
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- GST
glutathione
- PROTAC
proteolysis targeting chimera
- CRBN
cereblon
- TBAF
tetra-n-butylammonium fluoride
- TR-FRET
time-resolved Förster resonance energy transfer
- BRET
bioluminescence resonance energy transfer
- FLIM
fluorescence lifetime imaging microscopy
- BET protein
bromodomain and extraterminal domain protein family
- CDK9
cyclin dependent kinase 9.
Biographies
Chiara Borsari is a medicinal chemist, working as postdoctoral fellow at the University of Basel in the group of Prof. M. Wymann. Her research is focused on the design and synthesis of small-molecule anticancer agents. Her main goal is the development of irreversible inhibitors as a novel strategy in targeted cancer therapy. She got her Ph.D. at the University of Modena and Reggio Emilia. During her Ph.D., she has joined the NHRF in Athens and the State University of New York at Albany. She is also keen on communicating science to the public: she created a short movie “Life in Colour”, explaining the advancements in cancer research, and she is part of the European Federation for Medicinal Chemistry Communication Team.
Darci J. Trader received a Ph.D. in Chemistry under the direction of Prof. Erin E. Carlson at Indiana University in 2013. Her postdoctoral studies were at The Scripps Research Institute in the laboratory of Prof. Thomas Kodadek. She began her independent career at Purdue University in 2016. Her lab, which is in the Department of Medicinal Chemistry and Molecular Pharmacology, is focused on developing methods and probes to monitor or perturb both ubiquitin-dependent and -independent proteasome activity.
Annalisa Tait is an Associate Professor at the Department of Life Sciences of the University of Modena and Reggio Emilia. She has a broad experience in medicinal chemistry, and she is currently teaching Medicinal Chemistry/Drug Analysis in Pharmacy and Biotechnology degree courses, as well as in the Hospital Pharmacy School of Specialization. She was Director of the Doctorate School in Science and Technologies for Health Products. Her research is mainly focused on synthesis, structural characterization, and evaluation of heterocyclic derivatives as antiviral, phosphodiesterase inhibitors and ligands of α, 5-HT1A, NOP, and σ receptors.
Maria P. Costi obtained her Ph.D. in Medicinal Chemistry at the University of Modena and Reggio Emilia and was a Visiting Scientist at the University of California, San Francisco. She is Professor of Medicinal Chemistry at the Department of Life Sciences, leading the integrated laboratory of drug discovery and biotechnology. She published approximately 150 papers in international journals, deposited 20 patents, and serves as an editorial board member of different journals. Her research focus is on targeting the folate metabolism in different organisms for the discovery of anticancer, anti-infective drugs by combining medicinal chemistry and chemical biology tools. A second field of her research is the discovery of novel drugs for neglected tropical diseases. She coordinated many European and national projects.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Kuan S. L.; Bergamini F. R. G.; Weil T. Functional protein nanostructures: a chemical toolbox. Chem. Soc. Rev. 2018, 47, 9069–9105. 10.1039/C8CS00590G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Corson T. W.; Aberle N.; Crews C. M. Design and applications of bifunctional small molecules: why two heads are better than one. ACS Chem. Biol. 2008, 3, 677–692. 10.1021/cb8001792. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lautenslager G. T.; Simpson L. L. Chimeric molecules constructed with endogenous substances. Adv. Mol. Cell Biol. 1994, 9, 233–262. 10.1016/S1569-2558(08)60387-2. [DOI] [Google Scholar]
- a Maniaci C.; Ciulli A. Bifunctional chemical probes inducing protein-protein interactions. Curr. Opin. Chem. Biol. 2019, 52, 145–156. 10.1016/j.cbpa.2019.07.003. [DOI] [PubMed] [Google Scholar]; b Gilad Y.; Tuchinsky H.; Ben-David G.; Minnes R.; Gancz A.; Senderowitz H.; Luboshits G.; Firer M. A.; Gellerman G. Discovery of potent molecular chimera (CM358) to treat human metastatic melanoma. Eur. J. Med. Chem. 2017, 138, 602–615. 10.1016/j.ejmech.2017.06.066. [DOI] [PubMed] [Google Scholar]
- Tashima T. Effective cancer therapy based on selective drug delivery into cells across their membrane using receptor-mediated endocytosis. Bioorg. Med. Chem. Lett. 2018, 28, 3015–3024. 10.1016/j.bmcl.2018.07.012. [DOI] [PubMed] [Google Scholar]
- Lazny R.Hydrazone linker units. In Linker Strategies in Solid-Phase Organic Synthesis; Scott P. J. H., Ed.; John Wiley & Sons, Ltd.: Chichester, U.K., 2009; pp 303–315, 10.1002/9780470749043.ch10. [DOI] [Google Scholar]
- Enders D.; Wortmann L.; Peters R. Recovery of carbonyl compounds from N,N-dialkylhydrazones. Acc. Chem. Res. 2000, 33 (3), 157–169. 10.1021/ar990062y. [DOI] [PubMed] [Google Scholar]
- Corbett P. T.; Leclaire J.; Vial L.; West K. R.; Wietor J.-L.; Sanders J. K. M.; Otto S. Dynamic combinatorial chemistry. Chem. Rev. 2006, 106 (9), 3652–3711. 10.1021/cr020452p. [DOI] [PubMed] [Google Scholar]
- Dyniewicz J.; Lipiński P. F. J.; Kosson P.; Leśniak A.; Bochyńska-Czyż M.; Muchowska A.; Tourwé D.; Ballet S.; Misicka A.; Lipkowski A. W. Hydrazone linker as a useful tool for preparing chimeric peptide/nonpeptide bifunctional compounds. ACS Med. Chem. Lett. 2017, 8 (1), 73–77. 10.1021/acsmedchemlett.6b00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Fonović M.; Verhelst S. H. L. Cleavable linkers in chemical proteomics applications. Methods Mol. Biol. 2017, 1491, 185–203. 10.1007/978-1-4939-6439-0_14. [DOI] [PubMed] [Google Scholar]
- Park K. D.; Liu R.; Kohn H. Useful tools for biomolecule isolation, detection, and identification: acylhydrazone-based cleavable linkers. Chem. Biol. 2009, 16 (7), 763–772. 10.1016/j.chembiol.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Dirksen A.; Yegneswaran S.; Dawson P. E. Bisaryl hydrazones as exchangeable biocompatible linkers. Angew. Chem., Int. Ed. 2010, 49 (11), 2023–2027. 10.1002/anie.200906756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Góngora-Benítez M.; Tulla-Puche J.; Albericio F. Multifaceted roles of disulfide bonds. Peptides as therapeutics. Chem. Rev. 2014, 114 (2), 901–926. 10.1021/cr400031z. [DOI] [PubMed] [Google Scholar]
- Kourra C. M. B. K.; Cramer N. Converting disulfide bridges in native peptides to stable methylene thioacetals. Chem. Sci. 2016, 7 (12), 7007–7012. 10.1039/C6SC02285E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinther T. N.; Pettersson I.; Huus K.; Schlein M.; Steensgaard D. B.; Sørensen A.; Jensen K. J.; Kjeldsen T.; Hubalek F. Additional disulfide bonds in insulin: prediction, recombinant expression, receptor binding affinity, and stability. Protein Sci. Publ. Protein Soc. 2015, 24 (5), 779–788. 10.1002/pro.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Bai Y.; Zaro J. L.; Shen W.-C. Design of an in vivo cleavable disulfide linker in recombinant fusion proteins. BioTechniques 2010, 49 (1), 513–518. 10.2144/000113450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Morales-Sanfrutos J.; Angelini A.; Cutting B.; Heinis C. Structurally diverse cyclisation linkers impose different backbone conformations in bicyclic peptides. ChemBioChem 2012, 13 (7), 1032–1038. 10.1002/cbic.201200049. [DOI] [PubMed] [Google Scholar]
- Tegge W.; Bautsch W.; Frank R. Synthesis of cyclic peptides and peptide libraries on a new disulfide linker. J. Pept. Sci. 2007, 13 (10), 693–699. 10.1002/psc.879. [DOI] [PubMed] [Google Scholar]
- Arnér E. S. J.; Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase: thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267 (20), 6102–6109. 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- Garnett M. C. Targeted drug conjugates: principles and progress. Adv. Drug Delivery Rev. 2001, 53 (2), 171–216. 10.1016/S0169-409X(01)00227-7. [DOI] [PubMed] [Google Scholar]
- Hein C. D.; Liu X.-M.; Wang D. Click chemistry, a powerful tool for pharmaceutical sciences. Pharm. Res. 2008, 25 (10), 2216–2230. 10.1007/s11095-008-9616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hems E. S.; Wagstaff B. A.; Saalbach G.; Field R. A. CuAAC click chemistry for the enhanced detection of novel alkyne-based natural product toxins. Chem. Commun. 2018, 54 (86), 12234–12237. 10.1039/C8CC05113E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon H.; Lim C.; Lee J. M.; Kim S. Chemical assay-guided natural product isolation via solid-supported chemodosimetric fluorescent probe. Chem. Sci. 2015, 6 (5), 2806–2811. 10.1039/C5SC00360A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Mfuh A.; Amako Y.; Woo C. M. Small molecule interactome mapping by photoaffinity labeling reveals binding site hotspots for the NSAIDs. J. Am. Chem. Soc. 2018, 140 (12), 4259–4268. 10.1021/jacs.7b11639. [DOI] [PubMed] [Google Scholar]
- Sohn C. H.; Agnew H. D.; Lee J. E.; Sweredoski M. J.; Graham R. L. J.; Smith G. T.; Hess S.; Czerwieniec G.; Loo J. A.; Heath J. R.; Deshaies R. J.; Beauchamp J. L. Designer reagents for mass spectrometry-based proteomics: clickable cross-linkers for elucidation of protein structures and interactions. Anal. Chem. 2012, 84 (6), 2662–2669. 10.1021/ac202637n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiapparelli L. M.; McClatchy D. B.; Liu H.-H.; Sharma P.; Yates J. R.; Cline H. T. Direct detection of biotinylated proteins by mass spectrometry. J. Proteome Res. 2014, 13 (9), 3966–3978. 10.1021/pr5002862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright M. H.; Sieber S. A. Chemical proteomics approaches for identifying the cellular targets of natural products. Nat. Prod. Rep. 2016, 33 (5), 681–708. 10.1039/C6NP00001K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harju K.; Vahermo M.; Mutikainen I.; Yli-Kauhaluoma J. Solid-phase synthesis of 1,2,3-triazoles via 1,3-dipolar cycloaddition. J. Comb. Chem. 2003, 5 (6), 826–833. 10.1021/cc030110c. [DOI] [PubMed] [Google Scholar]
- Blass B. E.; Coburn K. R.; Faulkner A. L.; Hunn C. L.; Natchus M. G.; Parker M. S.; Portlock D. E.; Tullis J. S.; Wood R. Solid-phase synthesis of functionalized 1,2,3-triazoles. Tetrahedron Lett. 2002, 43 (22), 4059–4061. 10.1016/S0040-4039(02)00742-6. [DOI] [Google Scholar]
- Angelo N. G.; Arora P. S. Solution- and solid-phase synthesis of triazole oligomers that display protein-like functionality. J. Org. Chem. 2007, 72 (21), 7963–7967. 10.1021/jo701292h. [DOI] [PubMed] [Google Scholar]
- Galibert M.; Piller V.; Piller F.; Aucagne V.; Delmas A. F. Combining triazole ligation and enzymatic glycosylation on solid phase simplifies the synthesis of very long glycoprotein analogues. Chem. Sci. 2015, 6 (6), 3617–3623. 10.1039/C5SC00773A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin R.; van Duijnhoven S. M. J.; ten Hoeve W.; Janssen H. M.; Kleijn L. H. J.; Hoeben F. J. M.; Versteegen R. M.; Robillard M. S. Triggered drug release from an antibody–drug conjugate using fast “click-to-release” chemistry in mice. Bioconjugate Chem. 2016, 27 (7), 1697–1706. 10.1021/acs.bioconjchem.6b00231. [DOI] [PubMed] [Google Scholar]
- Versteegen R. M.; Rossin R.; tenHoeve W.; Janssen H. M.; Robillard M. S. Click to release: instantaneous doxorubicin elimination upon tetrazine ligation. Angew. Chem., Int. Ed. 2013, 52 (52), 14112–14116. 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- Sherman E. J.; Lorenz D. A.; Garner A. L. Click chemistry-mediated complementation assay for RNA–protein interactions. ACS Comb. Sci. 2019, 21 (7), 522–527. 10.1021/acscombsci.9b00071. [DOI] [PubMed] [Google Scholar]
- Lorenz D. A.; Garner A. L. A click chemistry-based microRNA maturation assay optimized for high-throughput screening. Chem. Commun. 2016, 52 (53), 8267–8270. 10.1039/C6CC02894B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Han X.-R.; Yang X.; Jiang B.; Liu J.; Xiong Y.; Jin J. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK). Eur. J. Med. Chem. 2018, 151, 304–314. 10.1016/j.ejmech.2018.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Guillen V. S.; Sharma N.; Flessa K.; Min J.; Carlson K. E.; Toy W.; Braqi S.; Katzenellenbogen B. S.; Katzenellenbogen J. A.; Chandarlapaty S.; Sharma A. new class of selective estrogen receptor degraders (SERDs): expanding the toolbox of PROTAC degrons. ACS Med. Chem. Lett. 2018, 9 (8), 803–808. 10.1021/acsmedchemlett.8b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines J.; Lartigue S.; Dong H.; Qian Y.; Crews C. M. MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res. 2019, 79 (1), 251–262. 10.1158/0008-5472.CAN-18-2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurz R. P.; Cee V. J. Targeted degradation of MDM2 as a new approach to improve the efficacy of MDM2-p53 inhibitors. J. Med. Chem. 2019, 62 (2), 445–447. 10.1021/acs.jmedchem.8b01945. [DOI] [PubMed] [Google Scholar]
- Silva M. C.; Ferguson F. M.; Cai Q.; Donovan K. A.; Nandi G.; Patnaik D.; Zhang T.; Huang H.-T.; Lucente D. E.; Dickerson B. C.; Mitchison T. J.; Fischer E. S.; Gray N. S.; Haggarty S. J. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. eLife 2019, 8, e45457 10.7554/eLife.45457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyrus K.; Wehenkel M.; Choi E.-Y.; Han H.-J.; Lee H.; Swanson H.; Kim K.-B. Impact of linker length on the activity of PROTACs. Mol. BioSyst. 2011, 7 (2), 359–364. 10.1039/C0MB00074D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinebach C.; Lindner S.; Udeshi N. D.; Mani D. C.; Kehm H.; Köpff S.; Carr S. A.; Gütschow M.; Krönke J. Homo-PROTACs for the chemical knockdown of cereblon. ACS Chem. Biol. 2018, 13 (9), 2771–2782. 10.1021/acschembio.8b00693. [DOI] [PubMed] [Google Scholar]
- Lai A. C.; Toure M.; Hellerschmied D.; Salami J.; Jaime-Figueroa S.; Ko E.; Hines J.; Crews C. M. Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem., Int. Ed. 2016, 55 (2), 807–810. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorba A.; Nguyen C.; Xu Y.; Starr J.; Borzilleri K.; Smith J.; Zhu H.; Farley K. A.; Ding W.; Schiemer J.; Feng X.; Chang J. S.; Uccello D. P.; Young J. A.; Garcia-Irrizary C. N.; Czabaniuk L.; Schuff B.; Oliver R.; Montgomery J.; Hayward M. M.; Coe J.; Chen J.; Niosi M.; Luthra S.; Shah J. C.; El-Kattan A.; Qiu X.; West G. M.; Noe M. C.; Shanmugasundaram V.; Gilbert A. M.; Brown M. F.; Calabrese M. F. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (31), E7285–E7292. 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B.; Tsai C.-J.; Haliloǧlu T.; Nussinov R. Dynamic allostery: linkers are not merely flexible. Structure 2011, 19 (7), 907–917. 10.1016/j.str.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odendaal A. Y.; Trader D. J.; Carlson E. E. Chemoselective enrichment for natural products discovery. Chem. Sci. 2011, 2 (4), 760–764. 10.1039/c0sc00620c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trader D. J.; Carlson E. E. Siloxyl ether functionalized resins for chemoselective enrichment of carboxylic acids. Org. Lett. 2011, 13 (20), 5652–5655. 10.1021/ol202376m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trader D. J.; Carlson E. E. Taming of a superbase for selective phenol desilylation and natural product isolation. J. Org. Chem. 2013, 78 (14), 7349–7355. 10.1021/jo4010298. [DOI] [PubMed] [Google Scholar]
- Trader D. J.; Carlson E. E. Toward the development of solid-supported reagents for separation of alcohol-containing compounds by steric environment. Tetrahedron 2014, 70 (27–28), 4191–4196. 10.1016/j.tet.2014.03.040. [DOI] [Google Scholar]
- Capehart S. L.; Carlson E. E. Mass spectrometry-based assay for the rapid detection of thiol-containing natural products. Chem. Commun. 2016, 52 (90), 13229–13232. 10.1039/C6CC07111B. [DOI] [PubMed] [Google Scholar]
- Cox C. L.; Tietz J. I.; Sokolowski K.; Melby J. O.; Doroghazi J. R.; Mitchell D. A. Nucleophilic 1,4-additions for natural product discovery. ACS Chem. Biol. 2014, 9 (9), 2014–2022. 10.1021/cb500324n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Falcón G.; Hahn D.; Reimer D.; Hughes C. C. Thiol probes to detect electrophilic natural products based on their mechanism of action. ACS Chem. Biol. 2016, 11 (8), 2328–2336. 10.1021/acschembio.5b00924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolf G. C.; Koch M. F.; Mandl F. A. M.; Sieber S. A. Subclass-specific labeling of protein-reactive natural products with customized nucleophilic probes. Chem. - Eur. J. 2015, 21 (9), 3701–3707. 10.1002/chem.201405009. [DOI] [PubMed] [Google Scholar]
- Rohena C. C.; Mooberry S. L. Recent progress with microtubule stabilizers: new compounds, binding modes and cellular activities. Nat. Prod. Rep. 2014, 31 (3), 335–355. 10.1039/C3NP70092E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlodi-Pasztor E.; Sackett D.; Wilkerson J.; Fojo T. Mitosis is not a key target of microtubule agents in patient tumors. Nat. Rev. Clin. Oncol. 2011, 8 (4), 244–250. 10.1038/nrclinonc.2010.228. [DOI] [PubMed] [Google Scholar]
- Lee M. M.; Gao Z.; Peterson B. R. Synthesis of a fluorescent analogue of paclitaxel that selectively binds microtubules and sensitively detects efflux by P-glycoprotein. Angew. Chem., Int. Ed. 2017, 56 (24), 6927–6931. 10.1002/anie.201703298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng E. A.; Carlson L. M.; Gutman D. M.; Harrington W. J.; Lee K. P.; Boise L. H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107 (12), 4907–4916. 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski R. Z.; Kuhn D. J. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin. Cancer Res. 2008, 14 (6), 1649–1657. 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- Oerlemans R.; Franke N. E.; Assaraf Y. G.; Cloos J.; van Zantwijk I.; Berkers C. R.; Scheffer G. L.; Debipersad K.; Vojtekova K.; Lemos C.; van der Heijden J. W.; Ylstra B.; Peters G. J.; Kaspers G. L.; Dijkmans B. A.; Scheper R. J.; Jansen G. Molecular basis of bortezomib resistance: proteasome subunit 5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112 (6), 2489–2499. 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- Carmony K. C.; Kim K. B. Activity-based imaging probes of the proteasome. Cell Biochem. Biophys. 2013, 67 (1), 91–101. 10.1007/s12013-013-9626-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong A.; Schuurman K. G.; Rodenko B.; Ovaa H.; Berkers C. R. Fluorescence-based proteasome activity profiling. Methods Mol. Biol. 2012, 803, 183–204. 10.1007/978-1-61779-364-6_13. [DOI] [PubMed] [Google Scholar]
- Florea B. I.; Verdoes M.; Li N.; van der Linden W. A.; Geurink P. P.; van den Elst H.; Hofmann T.; de Ru A.; van Veelen P. A.; Tanaka K.; Sasaki K.; Murata S.; den Dulk H.; Brouwer J.; Ossendorp F. A.; Kisselev A. F.; Overkleeft H. S. Activity-based profiling reveals reactivity of the murine thymoproteasome-specific subunit β5t. Chem. Biol. 2010, 17 (8), 795–801. 10.1016/j.chembiol.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovaa H.; van Swieten P. F.; Kessler B. M.; Leeuwenburgh M. A.; Fiebiger E.; van den Nieuwendijk A. M. C. H.; Galardy P. J.; van der Marel G. A.; Ploegh H. L.; Overkleeft H. S. Chemistry in living cells: detection of active proteasomes by a two-step labeling strategy. Angew. Chem., Int. Ed. 2003, 42 (31), 3626–3629. 10.1002/anie.200351314. [DOI] [PubMed] [Google Scholar]
- Verdoes M.; Hillaert U.; Florea B. I.; Sae-Heng M.; Risseeuw M. D. P.; Filippov D. V.; van der Marel G. A.; Overkleeft H. S. Acetylene functionalized BODIPY dyes and their application in the synthesis of activity based proteasome probes. Bioorg. Med. Chem. Lett. 2007, 17 (22), 6169–6171. 10.1016/j.bmcl.2007.09.025. [DOI] [PubMed] [Google Scholar]
- de Bruin G.; Xin B. T.; Kraus M.; van der Stelt M.; van der Marel G. A.; Kisselev A. F.; Driessen C.; Florea B. I.; Overkleeft H. S. A set of activity-based probes to visualize human (immuno)proteasome activities. Angew. Chem., Int. Ed. 2016, 55 (13), 4199–4203. 10.1002/anie.201509092. [DOI] [PubMed] [Google Scholar]
- Jen E. Y.; Ko C. W.; Lee J. E.; Del Valle P. L.; Aydanian A.; Jewell C.; Norsworthy K. J.; Przepiorka D.; Nie L.; Liu J.; Sheth C. M.; Shapiro M.; Farrell A. T.; Pazdur R. FDA approval: gemtuzumab ozogamicin for the treatment of adults with newly diagnosed CD33-positive acute myeloid leukemia. Clin. Cancer Res. 2018, 24, 3242–3246. 10.1158/1078-0432.CCR-17-3179. [DOI] [PubMed] [Google Scholar]
- Norsworthy K. J.; Ko C. W.; Lee J. E.; Liu J.; John C. S.; Przepiorka D.; Farrell A. T.; Pazdur R. FDA approval summary: mylotarg for treatment of patients with relapsed or refractory CD33-positive acute myeloid leukemia. Oncologist 2018, 23, 1103–1108. 10.1634/theoncologist.2017-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb Y. N. Inotuzumab ozogamicin: first global approval. Drugs 2017, 77, 1603–1610. 10.1007/s40265-017-0802-5. [DOI] [PubMed] [Google Scholar]
- Donato E. M.; Fernández-Zarzoso M.; Hueso J. A.; de la Rubia J. Brentuximab vedotin in Hodgkin lymphoma and anaplastic large-cell lymphoma: an evidence-based review. OncoTargets Ther. 2018, 11, 4583–4590. 10.2147/OTT.S141053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson N. C.; Kasamon Y. L.; Chen H.; de Claro R. A.; Ye J.; Blumenthal G. M.; Farrell A. T.; Pazdur R. FDA approval summary: brentuximab vedotin in first-line treatment of peripheral T-cell lymphoma. Oncologist 2019, 24, e180–e187. 10.1634/theoncologist.2019-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Minckwitz G.; Huang C. S.; Mano M. S.; Loibl S.; Mamounas E. P.; Untch M.; Wolmark N.; Rastogi P.; Schneeweiss A.; Redondo A.; Fischer H. H.; Jacot W.; Conlin A. K.; Arce-Salinas C.; Wapnir I. L.; Jackisch C.; DiGiovanna M. P.; Fasching P. A.; Crown J. P.; Wülfing P.; Shao Z.; Rota Caremoli E.; Wu H.; Lam L. H.; Tesarowski D.; Smitt M.; Douthwaite H.; Singel S. M.; Geyer C. E. Jr. KATHERINE Investigators. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N. Engl. J. Med. 2019, 380, 617–628. 10.1056/NEJMoa1814017. [DOI] [PubMed] [Google Scholar]
- Verma S.; Miles D.; Gianni L.; Krop I. E.; Welslau M.; Baselga J.; Pegram M.; Oh D. Y.; Diéras V.; Guardino E.; Fang L.; Lu M. W.; Olsen S.; Blackwell K. EMILIA Study Group. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks E. D. Polatuzumab vedotin: first global approval. Drugs 2019, 79 (13), 1467–1475. 10.1007/s40265-019-01175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchi A.; Gasparri A.; Curnis F.; Bellone M.; Corti A. Crucial role for interferon gamma in the synergism between tumor vasculature-targeted tumor necrosis factor alpha (NGR-TNF) and doxorubicin. Cancer Res. 2004, 64, 7150–7155. 10.1158/0008-5472.CAN-04-1445. [DOI] [PubMed] [Google Scholar]
- Gregorc V.; Cavina R.; Novello S.; Grossi F.; Lazzari C.; Capelletto E.; Genova C.; Salini G.; Lambiase A.; Santoro A. NGR-hTNF and doxorubicin as second-line treatment of patients with small cell lung cancer. Oncologist 2018, 23, 1133–e112. 10.1634/theoncologist.2018-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Niu G.; Wu H.; Chen X. Clinical application of radiolabeled RGD peptides for PET imaging of integrin αvβ3. Theranostics 2016, 6, 78–92. 10.7150/thno.13242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehnert B.; Burkhardt H.; Dübel S.; Voll R. E. The “sneaking-ligand” approach: cell-type specific inhibition of the classical NF-κB pathway. Methods Mol. Biol. 2015, 1280, 559–578. 10.1007/978-1-4939-2422-6_33. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y.; Takeuchi T.; Kuwata K.; Chiba J.; Hatanaka Y.; Nakase I.; Futaki S. Syndecan-4 is a receptor for clathrin-mediated endocytosis of arginine-rich cell-penetrating peptides. Bioconjugate Chem. 2016, 27, 1119–1130. 10.1021/acs.bioconjchem.6b00082. [DOI] [PubMed] [Google Scholar]
- Milletti F. Cell-penetrating peptides: classes, origin, and current landscape. Drug Discovery Today 2012, 17, 850–860. 10.1016/j.drudis.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Pooga M.; Langel U. Classes of cell-penetrating peptides. Methods Mol. Biol. 2015, 1324, 3–28. 10.1007/978-1-4939-2806-4_1. [DOI] [PubMed] [Google Scholar]
- Lönn P.; Dowdy S. F. Cationic PTD/CPP-mediated macromolecular delivery: charging into the cell. Expert Opin. Drug Delivery 2015, 12, 1627–1636. 10.1517/17425247.2015.1046431. [DOI] [PubMed] [Google Scholar]
- Becker-Hapak M.; McAllister S. S.; Dowdy S. F. TAT-mediated protein transduction into mammalian cells. Methods 2001, 24, 247–256. 10.1006/meth.2001.1186. [DOI] [PubMed] [Google Scholar]
- Klein M. J.; Schmidt S.; Wadhwani P.; Bürck J.; Reichert J.; Afonin S.; Berditsch M.; Schober T.; Brock R.; Kansy M.; Ulrich A. S. Lactam-stapled cell-penetrating peptides: cell uptake and membrane binding properties. J. Med. Chem. 2017, 60, 8071–8082. 10.1021/acs.jmedchem.7b00813. [DOI] [PubMed] [Google Scholar]
- Mitchell D. J.; Kim D. T.; Steinman L.; Fathman C. G.; Rothbard J. B. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56, 318–325. 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- Kalafatovic D.; Giralt E. Cell-penetrating peptides: design strategies beyond primary structure and amphipathicity. Molecules 2017, 22 (11), 1929. 10.3390/molecules22111929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T.; Li F.; Zhang H.; Fan L.; Qiao Y.; Tan G.; Zhang H.; Wu H. Multifunctional pH-sensitive micelles for tumor-specific uptake and cellular delivery. Polym. Chem. 2015, 6, 1373–1382. 10.1039/C4PY01403K. [DOI] [Google Scholar]
- Bhunia D.; Mondal P.; Das G.; Saha A.; Sengupta P.; Jana J.; Mohapatra S.; Chatterjee S.; Ghosh S. Spatial position regulates power of tryptophan: discovery of a major-groove-specific nuclear-localizing, cell-penetrating tetrapeptide. J. Am. Chem. Soc. 2018, 140, 1697–1714. 10.1021/jacs.7b10254. [DOI] [PubMed] [Google Scholar]
- Dominguez-Berrocal L.; Cirri E.; Zhang X.; Andrini L.; Marin G. H.; Lebel-Binay S.; Rebollo A. New therapeutic approach for targeting Hippo signaling pathway. Sci. Rep. 2019, 9 (1), 4771. 10.1038/s41598-019-41404-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Veiman K. L.; Mager I.; Ezzat K.; Margus H.; Lehto T.; Langel K.; Kurrikoff K.; Arukuusk P.; Suhorutsenko J.; Padari K.; Pooga M.; Lehto T.; Langel Ü. PepFect14 peptide vector for efficient gene delivery in cell cultures. Mol. Pharmaceutics 2013, 10, 199–210. 10.1021/mp3003557. [DOI] [PubMed] [Google Scholar]; b Ezzat K.; El Andaloussi S.; Zaghloul E. M.; Lehto T.; Lindberg S.; Moreno P. M.; Viola J. R.; Magdy T.; Abdo R.; Guterstam P.; Sillard R.; Hammond S. M.; Wood M. J.; Arzumanov A. A.; Gait M. J.; Smith C. I.; Hällbrink M.; Langel Ü. PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011, 39 (12), 5284–5298. 10.1093/nar/gkr072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ezzat K.; Helmfors H.; Tudoran O.; Juks C.; Lindberg S.; Padari K.; El-Andaloussi S.; Pooga M.; Langel U. Scavenger receptor-mediated uptake of cell-penetrating peptide nanocomplexes with oligonucleotides. FASEB J. 2012, 26, 1172–1180. 10.1096/fj.11-191536. [DOI] [PubMed] [Google Scholar]; b Lindberg S.; Muñoz-Alarcón A.; Helmfors H.; Mosqueira D.; Gyllborg D.; Tudoran O.; Langel U. PepFect15, a novel endosomolytic cell-penetrating peptide for oligonucleotide delivery via scavenger receptors. Int. J. Pharm. 2013, 441, 242–247. 10.1016/j.ijpharm.2012.11.037. [DOI] [PubMed] [Google Scholar]
- Lindberg S.; Regberg J.; Eriksson J.; Helmfors H.; Munoz-Alarcon A.; Srimanee A.; Figueroa R. A.; Hallberg E.; Ezzat K.; Langel U. A convergent uptake route for peptide- and polymer-based nucleotide delivery systems. J. Controlled Release 2015, 206, 58–66. 10.1016/j.jconrel.2015.03.009. [DOI] [PubMed] [Google Scholar]
- Craig A. J.; Meloni B. P.; Watt P. M.; Knuckey N. W. Attenuation of neuronal death by peptide inhibitors of AP-1 activation in acute and delayed in vitro ischaemia (oxygen/glucose deprivation) models. Int. J. Pept. Res. Ther. 2011, 17, 1–6. 10.1007/s10989-010-9234-8. [DOI] [Google Scholar]
- Vaslin A.; Puyal J.; Clarke P. G. Excitotoxicity-induced endocytosis confers drug targeting in cerebral ischemia. Ann. Neurol. 2009, 65, 337–347. 10.1002/ana.21584. [DOI] [PubMed] [Google Scholar]
- Meloni B. P.; Brookes L. M.; Clark V. W.; Cross J. L.; Edwards A. B.; Anderton R. S.; Hopkins R. M.; Hoffmann K.; Knuckey N. W. Poly-arginine and arginine-rich peptides are neuroprotective in stroke models. J. Cereb. Blood Flow Metab. 2015, 35, 993–1004. 10.1038/jcbfm.2015.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang B. T.; Cregg J. M.; DePaul M. A.; Tran A. P.; Xu K.; Dyck S. M.; Madalena K. M.; Brown B. P.; Weng Y. L.; Li S.; Karimi-Abdolrezaee S.; Busch S. A.; Shen Y.; Silver J. Modulation of the proteoglycan receptor PTPσ promotes recovery after spinal cord injury. Nature 2015, 518, 404–408. 10.1038/nature13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadia J. S.; Stan R. V.; Dowdy S. F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Melikov K.; Brooks H.; Prevot P.; Lebleu B.; Chernomordik L. V. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J. Biol. Chem. 2005, 280, 15300–15306. 10.1074/jbc.M401604200. [DOI] [PubMed] [Google Scholar]
- Ferrari A.; Pellegrini V.; Arcangeli C.; Fittipaldi A.; Giacca M.; Beltram F. Caveolae-mediated internalization of extracellular HIV-1 tat fusion proteins visualized in real time. Mol. Ther. 2003, 8, 284–294. 10.1016/S1525-0016(03)00122-9. [DOI] [PubMed] [Google Scholar]
- Kosuge M.; Takeuchi T.; Nakase I.; Jones A. T.; Futaki S. Cellular internalization and distribution of arginine-rich peptides as a function of extracellular peptide concentration, serum, and plasma membrane associated proteoglycans. Bioconjugate Chem. 2008, 19, 656–664. 10.1021/bc700289w. [DOI] [PubMed] [Google Scholar]
- Labropoulou V. T.; Skandalis S. S.; Ravazoula P.; Perimenis P.; Karamanos N. K.; Kalofonos H. P.; Theocharis A. D. Expression of syndecan-4 and correlation with metastatic potential in testicular germ cell tumours. BioMed Res. Int. 2013, 2013, 214864. 10.1155/2013/214864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba F.; Swartz K.; van Buren R.; Eickhoff J.; Zhang Y.; Wolberg W.; Friedl A. Syndecan-1 and syndecan-4 are overexpressed in an estrogen receptor-negative, highly proliferative breast carcinoma subtype. Breast Cancer Res. Treat. 2006, 98, 91–98. 10.1007/s10549-005-9135-2. [DOI] [PubMed] [Google Scholar]
- Yung S.; Woods A.; Chan T. M.; Davies M.; Williams J. D.; Couchman J. R. Syndecan-4 up-regulation in proliferative renal disease is related to microfilament organization. FASEB J. 2001, 15, 1631–1633. 10.1096/fj.00-0794fje. [DOI] [PubMed] [Google Scholar]
- Rodriguez Plaza J. G.; Morales-Nava R.; Diener C.; Schreiber G.; Gonzalez Z. D.; Ortiz M. T. L.; Blake I. O.; Pantoja O.; Volkmer R.; Klipp E.; Herrmann A.; Del Rio G. Cell penetrating peptides and cationic antibacterial peptides: two sides of the same coin. J. Biol. Chem. 2014, 289, 14448–14457. 10.1074/jbc.M113.515023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Shi Y.; Su G.; Le G. Selectivity for and destruction of Salmonella typhimurium via a membrane damage mechanism of a cell penetrating peptide ppTG20 analogue. Int. J. Antimicrob. Agents 2012, 40, 337–343. 10.1016/j.ijantimicag.2012.05.026. [DOI] [PubMed] [Google Scholar]
- Gautam A.; Kapoor P.; Chaudhary K.; Kumar R.; Raghava G. P. S. Tumor homing peptides as molecular probes for cancer therapeutics, diagnostics and theranostics. Curr. Med. Chem. 2014, 21, 2367–2391. 10.2174/0929867321666140217122100. [DOI] [PubMed] [Google Scholar]
- Fan L. Q.; Du G. X.; Li P. F.; Li M. W.; Sun Y.; Zhao L. M. Improved breast cancer cell-specific intracellular drug delivery and therapeutic efficacy by coupling decoration with cell penetrating peptide and SP90 peptide. Biomed. Pharmacother. 2016, 84, 1783–1791. 10.1016/j.biopha.2016.10.102. [DOI] [PubMed] [Google Scholar]
- Lu L.; Qi H.; Zhu J.; Sun W. X.; Zhang B.; Tang C. Y.; Cheng Q. Vascular-homing peptides for cancer therapy. Biomed. Pharmacother. 2017, 92, 187–195. 10.1016/j.biopha.2017.05.054. [DOI] [PubMed] [Google Scholar]
- Gregorc V.; Santoro A.; Bennicelli E.; Punt C. J.; Citterio G.; Timmer-Bonte J. N.; Caligaris Cappio F.; Lambiase A.; Bordignon C.; van Herpen C. M. Phase Ib study of NGR-hTNF, a selective vascular targeting agent, administered at low doses in combination with doxorubicin to patients with advanced solid tumours. Br. J. Cancer 2009, 101, 219–224. 10.1038/sj.bjc.6605162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorusso D.; Scambia G.; Amadio G.; di Legge A.; Pietragalla A.; De Vincenzo R.; Masciullo V.; Di Stefano M.; Mangili G.; Citterio G.; Mantori M.; Lambiase A.; Bordignon C. Phase II study of NGR-hTNF in combination with doxorubicin in relapsed ovarian cancer patients. Br. J. Cancer 2012, 107, 37–42. 10.1038/bjc.2012.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorc V.; Gaafar R. M.; Favaretto A.; Grossi F.; Jassem J.; Polychronis A.; Bidoli P.; Tiseo M.; Shah R.; Taylor P.; Novello S.; Muzio A.; Bearz A.; Greillier L.; Fontana F.; Salini G.; Lambiase A.; O’Brien M. NGR-hTNF in combination with best investigator choice in previously treated malignant pleural mesothelioma (NGR015): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2018, 19, 799–811. 10.1016/S1470-2045(18)30193-1. [DOI] [PubMed] [Google Scholar]
- Zarovni N.; Monaco L.; Corti A. Inhibition of tumor growth by intramuscular injection of cDNA encoding tumor necrosis factor alpha coupled to NGR and RGD tumor-homing peptides. Hum. Gene Ther. 2004, 15, 373–382. 10.1089/104303404322959524. [DOI] [PubMed] [Google Scholar]
- Seidi K.; Jahanban-Esfahlan R.; Monhemi H.; Zare P.; Minofar B.; Daei Farshchi Adli A.; Farajzadeh D.; Behzadi R.; Mesgari Abbasi M.; Neubauer H. A.; Moriggl R.; Zarghami N.; Javaheri T. NGR (Asn-Gly-Arg)-targeted delivery of coagulase to tumor vasculature arrests cancer cell growth. Oncogene 2018, 37, 3967–3980. 10.1038/s41388-018-0213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini E.; Hosseini S. Y.; Hashempour T.; Fattahi M. R.; Sadeghizadeh M. Effect of RGD coupled MDA-7/IL-24 on apoptosis induction in a hepatocellular carcinoma cell line. Mol. Med. Rep. 2017, 15, 495–501. 10.3892/mmr.2016.6009. [DOI] [PubMed] [Google Scholar]
- Zhang B. F.; Liu J. J.; Pei D. S.; Yang Z. X.; Di J. H.; Chen F. F.; Li H. Z.; Xu W.; Wu Y. P.; Zheng J. N. Potent antitumor effect elicited by RGD-mda-7, an mda-7/IL-24 mutant, via targeting the integrin receptor of tumor cells. Cancer Biother.Radiopharm. 2011, 26, 647–655. 10.1089/cbr.2011.0984. [DOI] [PubMed] [Google Scholar]
- Bina S.; Hosseini S. Y.; Shenavar F.; Hosseini E.; Mortazavi M. The effect of RGD/NGR peptide modification of melanoma differentiation-associated gene-7/interleukin-24 on its receptor attachment, an in silico analysis. Cancer Biother.Radiopharm. 2017, 32, 205–214. 10.1089/cbr.2017.2195. [DOI] [PubMed] [Google Scholar]
- Numata K.; Reagan M. R.; Goldstein R. H.; Rosenblatt M.; Kaplan D. L. Spider silk-based gene carriers for tumor cell-specific delivery. Bioconjugate Chem. 2011, 22, 1605–1610. 10.1021/bc200170u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bini E.; Foo C. W.; Huang J.; Karageorgiou V.; Kitchel B.; Kaplan D. L. RGD-functionalized bioengineered spider dragline silk biomaterial. Biomacromolecules 2006, 7, 3139–3145. 10.1021/bm0607877. [DOI] [PubMed] [Google Scholar]
- Huang J.; Valluzzi R.; Bini E.; Vernaglia B.; Kaplan D. L. Cloning, expression, and assembly of sericin-like protein. J. Biol. Chem. 2003, 278, 46117–46123. 10.1074/jbc.M307792200. [DOI] [PubMed] [Google Scholar]
- Prince J. T.; McGrath K. P.; DiGirolamo C. M.; Kaplan D. L. Construction, cloning, and expression of synthetic genes encoding spider dragline silk. Biochemistry 1995, 34, 10879–10885. 10.1021/bi00034a022. [DOI] [PubMed] [Google Scholar]
- Yan S. Z.; Beeler J. A.; Chen Y. B.; Shelton R. K.; Tang W. J. The regulation of type 7 adenylyl cyclase by its C1b region and Escherichia coli peptidylprolyl isomerase, SlyD. J. Biol. Chem. 2001, 276, 8500–8506. 10.1074/jbc.M010361200. [DOI] [PubMed] [Google Scholar]
- Galbiati E.; Gambini L.; Civitarese V.; Bellini M.; Ambrosini D.; Allevi R.; Avvakumova S.; Romeo S.; Prosperi D. “Blind” targeting in action: from phage display to breast cancer cell targeting with peptide-gold nanoconjugates. Pharmacol. Res. 2016, 111, 155–162. 10.1016/j.phrs.2016.06.007. [DOI] [PubMed] [Google Scholar]
- Peters C.; Brown S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225 10.1042/BSR20150089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura C. K. Therapeutic drug monitoring of monoclonal antibodies: applicability based on their pharmacokinetic properties. Drug Metab. Pharmacokinet. 2019, 34, 14–18. 10.1016/j.dmpk.2018.11.003. [DOI] [PubMed] [Google Scholar]
- Saeed A. F.; Wang R.; Ling S.; Wang S. Antibody engineering for pursuing a healthier future. Front. Microbiol. 2017, 8, 495. 10.3389/fmicb.2017.00495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert J. M.; Dhimolea E. The future of antibodies as cancer drugs. Drug Discovery Today 2012, 17, 954–963. 10.1016/j.drudis.2012.04.006. [DOI] [PubMed] [Google Scholar]
- Pollard J. A.; Loken M.; Gerbing R. B.; Raimondi S. C.; Hirsch B. A.; Aplenc R.; Bernstein I. D.; Gamis A. S.; Alonzo T. A.; Meshinchi S. CD33 expression and its association with gemtuzumab ozogamicin response: results from the randomized phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2016, 34, 747–755. 10.1200/JCO.2015.62.6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers E. L.; Larson R. A.; Stadtmauer E. A.; Estey E.; Löwenberg B.; Dombret H.; Karanes C.; Theobald M.; Bennett J. M.; Sherman M. L.; Berger M. S.; Eten C. B.; Loken M. R.; van Dongen J. J.; Bernstein I. D.; Appelbaum F. R. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J. Clin. Oncol. 2001, 19 (13), 3244–3254. 10.1200/JCO.2001.19.13.3244. [DOI] [PubMed] [Google Scholar]
- Ricart A. D. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin. Cancer Res. 2011, 17, 6417–6427. 10.1158/1078-0432.CCR-11-0486. [DOI] [PubMed] [Google Scholar]
- Uy N.; Nadeau M.; Stahl M.; Zeidan A. M. Inotuzumab ozogamicin in the treatment of relapsed/refractory acute B cell lymphoblastic leukemia. J. Blood Med. 2018, 9, 67–74. 10.2147/JBM.S136575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doronina S. O.; Toki B. E.; Torgov M. Y.; Mendelsohn B. A.; Cerveny C. G.; Chace D. F.; DeBlanc R. L.; Gearing R. P.; Bovee T. D.; Siegall C. B.; Francisco J. A.; Wahl A. F.; Meyer D. L.; Senter P. D. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- Hamblett K. J.; Senter P. D.; Chace D. F.; Sun M. M.; Lenox J.; Cerveny C. G.; Kissler K. M.; Bernhardt S. X.; Kopcha A. K.; Zabinski R. F.; Meyer D. L.; Francisco J. A. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- Schroeder D. D.; Tankersley D. L.; Lundblad J. L. A new preparation of modified immune serum globulin (human) suitable for intravenous administration. I. Standardization of the reduction and alkylation reaction. Vox Sang. 1981, 40, 373–382. 10.1111/j.1423-0410.1981.tb00725.x. [DOI] [PubMed] [Google Scholar]
- Sharman J. P.; Wheler J. J.; Einhorn L.; Dowlati A.; Shapiro G. I.; Hilton J.; Burke J. M.; Siddiqi T.; Whiting N.; Jalal S. I. A phase 2, open-label study of brentuximab vedotin in patients with CD30-expressing solid tumors. Invest. New Drugs 2019, 37, 738–747. 10.1007/s10637-019-00768-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehn L. H.; Kamdar M.; Herrera A. F.; McMillan A.; Flowers C.; Kim W. S.; Kim T. M.; Özcan M.; Demeter J.; Hertzberg M.; Trněný M.; Salles G. A.; Davies A.; Hirata J. H.; Cheng J.; Ku G.; Matasar M. J. Randomized phase 2 trial of polatuzumab vedotin (pola) with bendamustine and rituximab (BR) in relapsed/refractory (r/r) FL and DLBCL. J. Clin. Oncol. 2018, 36 (15), 7507. 10.1200/JCO.2018.36.15_suppl.7507. [DOI] [Google Scholar]
- Yu S.; Pearson A. D.; Lim R. K.; Rodgers D. T.; Li S.; Parker H. B.; Weglarz M.; Hampton E. N.; Bollong M. J.; Shen J.; Zambaldo C.; Wang D.; Woods A. K.; Wright T. M.; Schultz P. G.; Kazane S. A.; Young T. S.; Tremblay M. S. Targeted delivery of an anti-inflammatory PDE4 inhibitor to immune cells via an antibody-drug conjugate. Mol. Ther. 2016, 24, 2078–2089. 10.1038/mt.2016.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S.; Pearson A.; Lim R.; Rodgers D.; Li S.; Parker H.; Weglarz M.; Hampton E.; Bollong M.; Shen J.; Zumbaldo C.; Wang D.; Woods A.; Wright T.; Schultz P.; Kazane S.; Young T.; Tremblay M. P-262 A PDE4 inhibitor-antibody conjugate for treating ulcerative colitis. Inflammatory Bowel Dis. 2017, 23 (1), 262. 10.1097/01.MIB.0000512805.98510.c0. [DOI] [Google Scholar]
- Wang R. E.; Liu T.; Wang Y.; Cao Y.; Du J.; Luo X.; Deshmukh V.; Kim C. H.; Lawson B. R.; Tremblay M. S.; Young T. S.; Kazane S. A.; Wang F.; Schultz P. G. An immunosuppressive antibody-drug conjugate. J. Am. Chem. Soc. 2015, 137, 3229–3232. 10.1021/jacs.5b00620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemical Biology: From Small Molecules to Systems Biology and Drug Design; Schreiber L. S., Kapoor T., Wess G., Eds.; Wiley-VCH: Weinheim, Germany, 2007; Vol. 1-3. [Google Scholar]
- Jones L. H.; Bunnage M. E. Applications of chemogenomic library screening in drug discovery. Nat. Rev. Drug Discovery 2017, 16 (4), 285–296. 10.1038/nrd.2016.244. [DOI] [PubMed] [Google Scholar]
- Hermansonn G. T.Bioconjugate Techniques, 3rd ed.; Academic Press: San Diego, CA, U.S., 2013. [Google Scholar]
- Chen D.; Jansson A.; Sim D.; Larsson A.; Nordlund P. Structural analyses of human thymidylate synthase reveal a site that may control conformational switching between active and inactive states. J. Biol. Chem. 2017, 292, 13449–13458. 10.1074/jbc.M117.787267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham T. B.; Blanco M. J. Target engagement in lead generation. Bioorg. Med. Chem. Lett. 2015, 25, 998–1008. 10.1016/j.bmcl.2014.12.076. [DOI] [PubMed] [Google Scholar]
- Schürmann M.; Janning P.; Ziegler S.; Waldmann H. Small-molecule target engagement in cells. Cell Chem. Biol. 2016, 23, 435–441. 10.1016/j.chembiol.2016.03.008. [DOI] [PubMed] [Google Scholar]
- Rudkouskaya A.; Sinsuebphon N.; Ward J.; Tubbesing K.; Intes X.; Barroso M. Quantitative imaging of receptor-ligand engagement in intact live animals. J. Controlled Release 2018, 286, 451–459. 10.1016/j.jconrel.2018.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robers M. B.; Dart M. L.; Woodroofe C. C.; Zimprich C. A.; Kirkland T. A.; Machleidt T.; Kupcho K. R.; Levin S.; Hartnett J. R.; Zimmerman K.; Niles A. L.; Ohana R. F.; Daniels D. L.; Slater M.; Wood M. G.; Cong M.; Cheng Y. Q.; Wood K. V. Target engagement and drug residence time can be observed in living cells with BRET. Nat. Commun. 2015, 6, 10091. 10.1038/ncomms10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart L. A.; Johnstone E. K. M.; Wheal A. J.; Goulding J.; Robers M. B.; Machleidt T.; Wood K. V.; Hill S. J.; Pfleger K. D. G. Application of BRET to monitor ligand binding to GPCRs. Nat. Methods 2015, 12, 661–663. 10.1038/nmeth.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon G. M.; Niphakis M. J.; Cravatt B. F. Determining target engagement in living systems. Nat. Chem. Biol. 2013, 9, 200–205. 10.1038/nchembio.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A.; Pompliano D. L.; Meek T. D. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discovery 2006, 5, 730–739. 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Tummino P. J.; Copeland R. A. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry 2008, 47, 5481–5492. 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- Vauquelin G.; Charlton S. J. Exploring avidity: understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. Br. J. Pharmacol. 2013, 168, 1771–1785. 10.1111/bph.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson J. M.; Ye Y.; Gebru M. T.; Liu Q.; Zhou S.; Young M. M.; Takahashi Y.; Lin Q.; Tian F.; Wang H. G. Time-resolved FRET and NMR analyses reveal selective binding of peptides containing the LC3-interacting region to ATG8 family proteins. J. Biol. Chem. 2019, 294 (38), 14033–14042. 10.1074/jbc.RA119.008723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T. W.; Amason J. D.; Garcin E. D.; Lamy L.; Dranchak P. K.; Macarthur R.; Braisted J.; Rubin J. S.; Burgess T. L.; Farrell C. L.; Roberts D. D.; Inglese J. Quantitative high-throughput screening assays for the discovery and development of SIRPα-CD47 interaction inhibitors. PLoS One 2019, 14 (7), e0218897 10.1371/journal.pone.0218897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau M. J.; Morin I.; Schaeffer P. M. Quantitative determination of protein stability and ligand binding using a green fluorescent protein reporter system. Mol. BioSyst. 2010, 6, 1285–1292. 10.1039/c002001j. [DOI] [PubMed] [Google Scholar]
- Bücherl C.; Aker J.; de Vries S.; Borst J. W. Probing protein-protein Interactions with FRET-FLIM. Methods Mol. Biol. 2010, 655, 389–399. 10.1007/978-1-60761-765-5_26. [DOI] [PubMed] [Google Scholar]
- Yu H.; Truong H.; Mitchell S. A.; Liclican A.; Gosink J. J.; Li W.; Lin J.; Feng J. Y.; Jürgensmeier J. M.; Billin A.; Xu R.; Patterson S.; Pagratis N. Homogeneous BTK occupancy assay for pharmacodynamic assessment of tirabrutinib (GS-4059/ONO-4059) target engagement. SLAS Discovery 2018, 23, 919–929. 10.1177/2472555218786165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockmann H.; Todorovic V.; Richardson P. L.; Marin V.; Scott V.; Gerstein C.; Lake M.; Wang L.; Sadhukhan R.; Vasudevan A. Cell-surface receptor-ligand interaction analysis with homogeneous time-resolved FRET and metabolic glycan engineering: application to transmembrane and GPI-anchored receptors. J. Am. Chem. Soc. 2017, 139, 16822–16829. 10.1021/jacs.7b09359. [DOI] [PubMed] [Google Scholar]
- Genovese F.; Ferrari S.; Guaitoli G.; Caselli M.; Costi M. P.; Ponterini G. Dimer–monomer equilibrium of human thymidylate synthase monitored by fluorescence resonance energy transfer. Protein Sci. 2010, 19, 1023–1030. 10.1002/pro.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponterini G.; Martello A.; Pavesi G.; Lauriola A.; Luciani R.; Santucci M.; Pelà M.; Gozzi G.; Pacifico S.; Guerrini R.; Marverti G.; Costi M. P.; D’Arca D. Intracellular quantitative detection of human thymidylate synthase engagement with an unconventional inhibitor using tetracysteine-diarsenical-probe technology. Sci. Rep. 2016, 6, 27198. 10.1038/srep27198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rocco G.; Martinelli I.; Pacifico S.; Guerrini R.; Cichero E.; Fossa P.; Franchini S.; Cardarelli S.; Giorgi M.; Sola M.; Ponterini G. Fluorometric detection of protein-ligand engagement: the case of phosphodiesterase 5. J. Pharm. Biomed. Anal. 2018, 149, 335–342. 10.1016/j.jpba.2017.11.014. [DOI] [PubMed] [Google Scholar]
- Brown N. E.; Blumer J. B.; Hepler J. R. Bioluminescence resonance energy transfer to detect protein-protein interactions in live cells. Methods Mol. Biol. 2015, 1278, 457–465. 10.1007/978-1-4939-2425-7_30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koblan L. W.; Buckley D. L.; Ott C. J.; Fitzgerald M. E.; Ember S. W.; Zhu J. Y.; Liu S.; Roberts J. M.; Remillard D.; Vittori S.; Zhang W.; Schonbrunn E.; Bradner J. E. Assessment of bromodomain target engagement by a series of BI2536 analogues with miniaturized BET-BRET. ChemMedChem 2016, 11 (23), 2575–2581. 10.1002/cmdc.201600502. [DOI] [PubMed] [Google Scholar]
- Casarini L.; Riccetti L.; Limoncella S.; Lazzaretti C.; Barbagallo F.; Pacifico S.; Guerrini R.; Tagliavini S.; Trenti T.; Simoni M.; Sola M.; Di Rocco G. Probing the effect of sildenafil on progesterone and testosterone production by an intracellular FRET/BRET combined approach. Biochemistry 2019, 58, 799–808. 10.1021/acs.biochem.8b01073. [DOI] [PubMed] [Google Scholar]
- Robers M. B.; Vasta J. D.; Corona C. R.; Ohana R. F.; Hurst R.; Jhala M. A.; Comess K. M.; Wood K. V. Quantitative, real-time measurements of intracellular target engagement using energy transfer. Methods Mol. Biol. 2019, 1888, 45–71. 10.1007/978-1-4939-8891-4_3. [DOI] [PubMed] [Google Scholar]
- Sartini S.; Levati E.; Maccesi M.; Guerra M.; Spadoni G.; Bach S.; Benincasa M.; Scocchi M.; Ottonello S.; Rivara S.; Montanini B. New antimicrobials targeting bacterial RNA polymerase holoenzyme assembly identified with an in vivo BRET-based discovery platform. ACS Chem. Biol. 2019, 14 (8), 1727–1736. 10.1021/acschembio.9b00178. [DOI] [PubMed] [Google Scholar]
- McClure R. A.; Williams J. D. Impact of mass spectrometry-based technologies and strategies on chemoproteomics as a tool for drug discovery. ACS Med. Chem. Lett. 2018, 9, 785–791. 10.1021/acsmedchemlett.8b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S.; Jang S. Y.; Wang D.; Liew S. S.; Li Z.; Lee J. S.; Yao S. Q. A suite of “minimalist” photo-crosslinkers for live-cell imaging and chemical proteomics: case study with BRD4 inhibitors. Angew. Chem., Int. Ed. 2017, 56, 11816–11821. 10.1002/anie.201706076. [DOI] [PubMed] [Google Scholar]
- Parker C. G.; Galmozzi A.; Wang Y.; Correia B. E.; Sasaki K.; Joslyn C. M.; Kim A. S.; Cavallaro C. L.; Lawrence R. M.; Johnson S. R.; Narvaiza I.; Saez E.; Cravatt B. F. Ligand and target discovery by fragment-based screening in human cells. Cell 2017, 168, 527–541. 10.1016/j.cell.2016.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Guo Z.; Song T.; Zhang X.; He N.; Liu P.; Wang P.; Zhang Z. Proteome-wide identification of on- and off-targets of Bcl-2 inhibitors in native biological systems by using affinity-based probes (AfBPs). ChemBioChem 2018, 19, 2312–2320. 10.1002/cbic.201800380. [DOI] [PubMed] [Google Scholar]
- Wong M. L.; Murphy J.; Harrington E.; Gower C. M.; Jain R. K.; Schirle M.; Thomas J. R. Examining the influence of specificity ligands and ATP-competitive ligands on the overall effectiveness of bivalent kinase inhibitors. Proteome Sci. 2016, 15, 17. 10.1186/s12953-017-0125-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; van Veldhoven J. P. D.; Offringa J.; Kuiper B. J.; Lenselink E. B.; Heitman L. H.; van der Es D.; Ijzerman A. P. Development of covalent ligands for G protein-coupled receptors: a case for the human adenosine A(3) receptor. J. Med. Chem. 2019, 62 (7), 3539–3552. 10.1021/acs.jmedchem.8b02026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murale D. P.; Hong S. C.; Haque M. M.; Lee J. S. Photo-affinity labeling (PAL) in chemical proteomics: a handy tool to investigate protein-protein interactions (PPIs). Proteome Sci. 2016, 15, 14. 10.1186/s12953-017-0123-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.; Collins I. Photoaffinity labeling in target- and binding-site identification. Future Med. Chem. 2015, 7 (2), 159–183. 10.4155/fmc.14.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J. R.; Robertson A. A. B. Fishing for Drug Targets: A focus on diazirine photoaffinity probe synthesis. J. Med. Chem. 2018, 61 (16), 6945–6963. 10.1021/acs.jmedchem.7b01561. [DOI] [PubMed] [Google Scholar]
- Kubota K.; Funabashi M.; Ogura Y. Target deconvolution from phenotype-based drug discovery by using chemical proteomics approaches. Biochim. Biophys. Acta, Proteins Proteomics 2019, 1867 (1), 22–27. 10.1016/j.bbapap.2018.08.002. [DOI] [PubMed] [Google Scholar]
- Tomohiro T.Tag-Creation Approaches for Highly Efficient Profiling of Interacting Proteins and Domains. In Photoaffinity Labeling for Structural Probing Within Protein; Hatanaka Y., Hashimoto M., Eds.; Springer: Tokyo, 2017; pp 13–43. [Google Scholar]
- Sakurai K.; Tawa M.; Okada A.; Yamada R.; Sato N.; Inahara M.; Inoue M. Active/inactive dual-probe system for selective photo-affinity labeling of smallmolecule-binding proteins. Chem. - Asian J. 2012, 7, 1567–1571. 10.1002/asia.201200085. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Zhang Y.; Dong J.; Liu J.; Zhang L.; Sun H. Design and synthesis of novel photoaffinity probes for study of the target proteins of oleanolic acid. Bioorg. Med. Chem. Lett. 2012, 22 (2), 1036–1039. 10.1016/j.bmcl.2011.11.123. [DOI] [PubMed] [Google Scholar]
- Li Y. M.; Xu M.; Lai M. T.; Huang Q.; Castro J. L.; DiMuzio-Mower J.; Harrison T.; Lellis C.; Nadin A.; Neduvelil J. G.; Register R. B.; Sardana M. K.; Shearman M. S.; Smith A. L.; Shi X. P.; Yin K. C.; Shafer J. A.; Gardell S. J. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 2000, 405 (6787), 689–694. 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- Colca J. R.; McDonald W. G.; Cavey G. S.; Cole S. L.; Holewa D. D.; Brightwell-Conrad A. S.; Wolfe C. L.; Wheeler J. S.; Coulter K. R.; Kilkuskie P. M.; Gracheva E.; Korshunova Y.; Trusgnich M.; Karr R.; Wiley S. E.; Divakaruni A. S.; Murphy A. N.; Vigueira P. A.; Finck B. N.; Kletzien R. F. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT)--relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS One 2013, 8 (5), e61551 10.1371/journal.pone.0061551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzi L.; Barrow A. S.; Scott D.; Layfield R.; Wright T. G.; Moses J. E.; Oldham N. J. Carbene footprinting accurately maps binding sites in protein–ligand and protein–protein interactions. Nat. Commun. 2016, 7, 13288–13297. 10.1038/ncomms13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne J. E.; Walko M.; Calabrese A. N.; Levenstein M. A.; Brockwell D. J.; Kapur N.; Wilson A. J.; Radford S. E. Rapid mapping of protein interactions using tag-transfer photocrosslinkers. Angew. Chem., Int. Ed. 2018, 57 (51), 16688–16692. 10.1002/anie.201809149. [DOI] [PMC free article] [PubMed] [Google Scholar]