

Abstract

Sphingolipids (SphLs) are a diverse class of molecules that are regulated by a complex network of enzymatic pathways. A disturbance in these pathways leads to lipid accumulation and initiation of several SphL-related disorders. Acid ceramidase is one of the key enzymes that regulate the metabolism of ceramides and glycosphingolipids, which are important members of the SphL family. Herein, we describe the lead optimization studies of benzoxazolone carboxamides resulting in piperidine 22m, where we demonstrated target engagement in two animal models of neuropathic lysosomal storage diseases (LSDs), Gaucher’s and Krabbe’s diseases. After daily intraperitoneal administration at 90 mg kg–1, 22m significantly reduced the brain levels of the toxic lipids glucosylsphingosine (GluSph) in 4L;C* mice and galactosylsphingosine (GalSph) in Twitcher mice. We believe that 22m is a lead molecule that can be further developed for the correction of severe neurological LSDs where GluSph or GalSph play a significant role in disease pathogenesis.
Introduction
Sphingolipids (SphLs) are a large class of diverse amphipathic molecules found in abundance in plasma membranes.1,2 Besides being important as structural cellular components, SphLs play a central role in different biological processes, which are essential to maintain the homeostasis and the development of eukaryotic cells. These processes include signaling, angiogenesis, cell growth, proliferation, and death, senescence, inflammation, immune responses, metabolism, autophagy, and brain development and functions.2 Aided by recent technological advances, much has been accomplished in terms of the identification of the basic biological components of the complex network in dynamic and interconnected enzymatic pathways that regulate the biosynthesis of SphLs and the formation of a variety of bioactive metabolites in distinct cellular compartments.1
In recent years, both academia and industry have shown growing interest in advancing our understanding of the multifaceted roles of SphL species under physiological and pathological conditions.2 Collected evidence suggests that a disturbance between the synthesis and catabolism of SphLs leads to their accumulation in specific cellular compartments, such as the lysosomes, and the initiation of several SphL-related disorders. Lysosomes are critical organelles responsible for cellular homeostasis.3 They contain different degradative enzymes that can hydrolyze proteins, DNA, RNA, polysaccharides, and lipids.4
Acid ceramidase (AC, also known as N-acylsphingosine amidohydrolase-1, ASAH-1) is a lysosomal cysteine amidase that catalyzes the hydrolysis of ceramides (Cer) into fatty acids and sphingosine, which is then converted into sphingosine 1-phosphate (Sph1P) by sphingosine kinase.5−7 Cer and Sph1P are important members of the SphL class and have opposing actions in the control of the cellular fate;8−10 while Cer mediates cellular senescence11 and apoptosis,12,13 Sph1P promotes cell survival and proliferation.14−17 Recent studies have shown that AC is abnormally expressed in various types of human cancer (for example, prostate, head and neck, colon, and glioblastoma), and serum AC levels are elevated in patients with melanoma relative to control subjects.18 Therefore, inhibition of AC has been envisaged as a potential cancer drug target (Figure 1). Aberrant AC activity has also been described in several other common diseases, including inflammation, pain, and various pulmonary disorders.19,20
Figure 1.
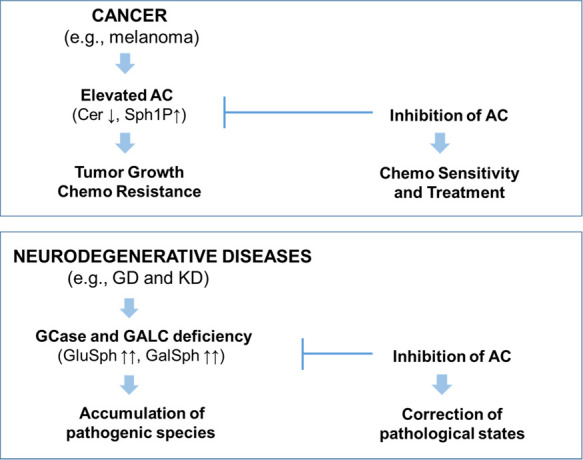
Some potential applications of AC inhibition therapy.
Over the recent years, the multifaceted catabolic role of AC has attracted much attention for its potential therapeutic applications in many other altered conditions. Important genetic studies have identified specific mutations in several genes that encode defective expressions of some lysosomal enzymes as the causes of the onset and progression of severe pathological conditions, called lysosomal storage diseases (LSDs).21−24 For example, Gaucher’s disease (GD) is caused by a defective function of acid β-glucocerebrosidase (GCase), a lysosomal membrane-associated protein responsible for the hydrolysis of glucosylceramide (GluCer) to glucose and ceramides.25−27 Krabbe’s disease (KD) is associated with defective β-galactosyl-ceramidase (GALC) activity, a lysosomal enzyme responsible for the hydrolysis of galactosylceramide (GalCer).28
As a result of either enzyme absences or deficiencies, these metabolic lysosomal disorders are characterized by an abnormal storage of substrates or metabolites to concentration levels that are toxic or otherwise detrimental to the cells in various compartments, including the skeleton, skin, liver, spleen, lung, heart, and central nervous system (CNS). The substrate or metabolite accumulations are believed to be responsible for the disease progression.24 In GD patients, for example, the accumulation of GluCer (3-fold) and/or glucosylsphingosine (GluSph) (200-fold) has been related to the brain pathogenesis of neuronopathic GD patients due to neuronal death, which is propagated by the toxic effects of GluCer and/or GluSph.29,30 Recent evidence demonstrates an active role of AC in an alternative catabolic pathway, which causes GluSph accumulation through the deacylation of the lysosomal GluCer.31,32 In KD patients, deficiency of GALC activity leads to accumulation of neurotoxic galactosylsphingosine (GalSph or psychosine) in tissues, especially in the brain. It is possible that accumulation of GalSph mediates pathology of KD. A very recent report suggests that genetic ablation of AC or pharmacological inhibition of AC could eliminate psychosine accumulation and prolong the life span of Twitcher mice, a model of KD.33,34 No approved therapeutic approaches are available to treat neuropathic GD and KD; inhibiting AC may provide an efficacious strategy for treating these two devastating diseases.
Efforts over the last decade to develop potent AC inhibitors have resulted in limited success. The first structural analysis of mammalian AC has recently been solved by Gebai and co-workers,35 which may aid future medicinal chemistry programs. In 2013, Realini et al. reported the discovery of carmofur 1 and some close uracil analogs as the first class of single-digit nanomolar inhibitors of intracellular AC activity and studied their potential use as chemosensitizing agents (Figure 2).36,37 Despite being potent AC inhibitors, the uracil derivatives suffered from low chemical and metabolic stability. Subsequently, Diamanti et al. selected compound 1 as a ligand template for a computational-assisted virtual screening approach, leading to the identification of a new class of potent AC inhibitors, the pyrazole carboxamides.38 However, although very potent against AC activity, as exemplified by pyrazole 3, this class of molecules suffered from low metabolic stability, limiting their therapeutic potential (Figure 2).
Figure 2.

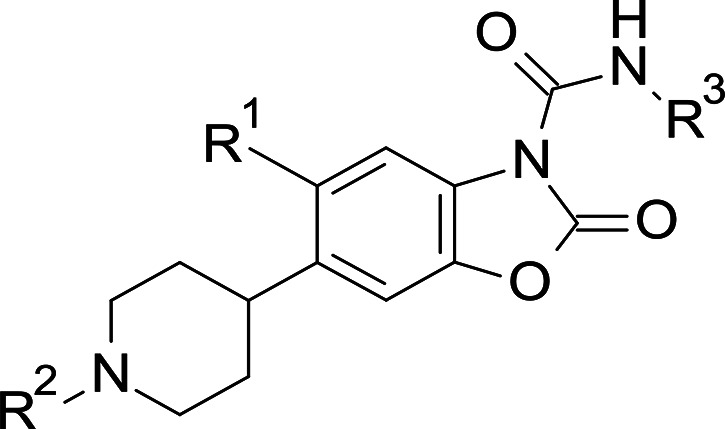
Structures of representative known AC inhibitors (1–4) (top) and general structure of the benzoxazolone carboxamide series 5 explored in this study (bottom).
An alternative approach, consisting of a screening campaign of a small compound library, led to the identification of a novel and very promising class of covalent AC inhibitors, the benzoxazolone carboxamides, exemplified by the hit 2a (Figure 2).39 Preliminary chemical exploration of this series led to the identification of 2b(39) and 2c(40) as more advanced and systematically active analogs. More recently, Ortega et al. reported a systematic computational investigation of the general pharmacophore model for AC inhibition, comprising a 6 + 5 fused ring heterocycle linked to an aliphatic substituent via a urea moiety. These studies resulted in the identification of the novel class of benzimidazole derivatives 4a–d with promising activity in different melanoma cell lines (Figure 2).41
Although some of the molecules discussed above exhibited potent inhibitory effects toward AC, they generally suffer from low aqueous solubility and moderate chemical or metabolic stability, which hamper their further development. As part of our continued efforts to optimize the class of benzoxazolone carboxamides, we further extended the preliminary studies around 2b(39) (and 2c)40 and performed a focused structure–activity relationship (SAR) study around this scaffold (compound 5, Figure 2), with the aim of identifying an optimal compound with improved physicochemical and pharmacokinetic profiles favoring oral administration. The subject of this manuscript describes the lead optimization and medicinal chemistry strategies that led to the discovery of 22m as a lead candidate with improved oral bioavailability and excellent distribution to the CNS.
Chemistry
All target compounds were prepared by synthetic routes outlined in Schemes 1–11. Compounds 8a–d were synthesized under standard conditions by reacting 7a–d with 4-phenylbutyl isocyanate (Scheme 1). The novel core scaffold of 12a was prepared in three steps from the α-bromo ketone 9 (Scheme 2A). Reaction with TZD gave compound 10, which was then converted in moderate yield to the fused bicyclic derivative 11 via an intramolecular cyclization under basic conditions in anhydrous THF. Subsequent coupling of 11 to 4-phenylbutyl isocyanate gave 12a, which upon removal of the N-Boc protecting group gave the key intermediate 12b, which was subsequently transformed to 12c–e via standard reductive amination and acetylation reactions. Alternatively, the isomeric key intermediate 17b was prepared in four steps from the commercially available epoxide 13 (Scheme 2B). Ring opening42 and subsequent oxidation of the corresponding alcohol 14a followed by intramolecular cyclization of 15 afforded compound 16. Finally, as discussed for the synthesis of 12b, standard reactions transformed 16 to 17b.
Scheme 1. Synthesis of 8a–d.

Reagents and conditions: (a) CDI, MeCN, rt, 2 h; (b) 4-phenylbutyl isocyanate, DMAP, toluene/DMF, rt, 2 h (20–60% over two steps for 8a and 8b); 4-phenylbutyl isocyanate, Et3N, MeCN, rt, 2 h (20–26% for 8c and 8d).
Scheme 11. Synthesis of 23j and 23k.

Reagents and conditions: (a) 1-methylpiperazin-2-one (for 52a), Et3N, MeCN, 80 °C, 16 h (60%); 4-methylpiperazin-2-one (for 52b), CuI, K3PO4, N,N-dimethyl-1,2-ethanediamine, dioxane, reflux, 24 h (50%); (b) 10% Pd/C, cyclohexene, EtOH, 65 °C, 16 h; (c) CDI, MeCN, rt, 1 h (70–80% over two steps); (d) 4-phenylbutyl isocyanate, DMAP, MeCN, rt, 16 h (10–20%).
Scheme 2. Synthesis of fused bicyclic piperidine-oxazolone derivatives 12a–e, 17a, and 17b.

Reagents and conditions: for the synthesis of 12a–e: (a) TZD, K2CO3, DMF, rt, 2 h (85%); (b) tBuOK, THF, rt, 30 min (60%); (c) 4-phenylbutyl isocyanate, DMAP, MeCN, rt, 16 h (68%); (d) 4 M HCl, dioxane, rt, 1 h (60%); (e) HCHO (for 12c) or PhCHO (for 12d), NaBH(OAc)3, AcOH, MeCN, rt, 3 h (56–90%); (f) AcCl, Et3N, DCM, rt, 3 h (62%). For the synthesis of 17a and 17b: (a) TZD, Mg(ClO4)2, DMF, 115 °C, 5 h (50%); (b) Dess–Martin reagent, DCM, 0 °C to rt, 12 h (70%); (c) tBuOK, THF, rt, 30 min; (d) 4-phenylbutyl isocyanate, DMAP, MeCN, rt, 30 min (25% over two steps); (e) 4 M HCl, dioxane, rt, 1 h (60%).
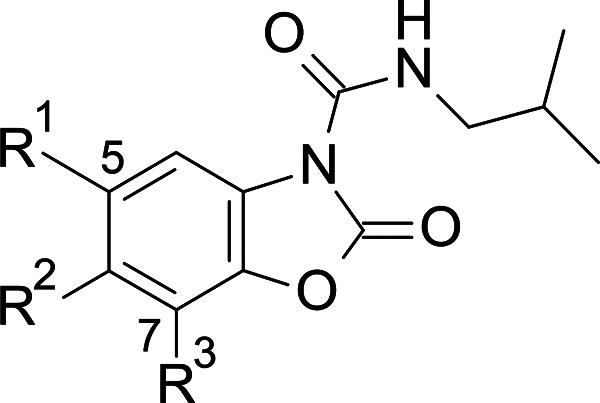
We introduced cyclic and heterocyclic groups at C(5)-, C(6)-, and C(7)-positions of the benzoxazolone cores by exploring different synthetic pathways (Schemes 3–11). The exploration at the C(4)-position of the benzoxazolone scaffold was abandoned because, in accordance with previously reported results on the 4-Me and 4-Ph derivatives of 2a (Figure 2),40 we experienced a pronounced chemical instability of our targeted C(4)-derivatives.
Scheme 3. Synthesis of C(6)-substituted benzoxazolone carboxamides 22a–d and 22i.

Reagents and conditions: (a) 5-bromo-2-nitrophenol (for 19a–c), 5-bromo-4-fluoro-2-nitrophenol (for 19d), Pd(PPh3)4, 2 M Na2CO3, dioxane, reflux, 18 h (56–90%); (b) H-Cube, Pd/C, EtOAc, rt, 1–2 h; (c) CDI, MeCN, 60 °C, 2 h (60–84% over two steps); (d) 4-phenylbutyl isocyanate, DMAP, MeCN, rt, 16 h (50–98%); (e) 4 M HCl, dioxane, rt, 3 h (86%); (f) AcCl, Et3N, THF, rt, 4 h (90%).
The C(6)-substituted benzoxazolones 21a–d were prepared in three steps starting from boronic esters 18a–c, using Pd-catalyzed cross coupling reactions with the corresponding bromo-nitrophenols followed by hydrogenation and intramolecular cyclization in the presence of CDI (Scheme 3). An additional step consisting of the in situ formation of boronic ester 29b, from ketone 28 via enol triflate 29a, was necessary for the preparation of the benzoxazolone 21l (Scheme 5). A Pd-catalyzed cross coupling procedure was also used for the synthesis of the C(5)-substituted benzoxazolone 33 (Scheme 6) and other C(6)-substituted benzoxazolones, such as 37a and 41a (Schemes 7 and 8). In contrast, to overcome some synthetic problems in the Pd-catalyzed cross coupling reaction, we performed an alternative synthetic approach for the preparation of the benzoxazolone 21m (Scheme 9A). Lithium-halogen exchange of 6-bromo-3H-1,3-benzoxazol-2-one43 followed by the addition of the ketone 42a afforded the alcohol 43, which upon dehydration and hydrogenation led to the key intermediate 21m. A similar synthetic procedure was applied to insert the functionalization at the C(7)-position of the benzoxazolone, as in 47 (Scheme 9B).
Scheme 5. Synthesis of 22q.

Reagents and conditions: (a) N,N-bis(trifluoromethanesulfonyl)aniline, Et3N, THF, 0 °C, 16 h (74%); (b) Pd(dppf)Cl2, [B2(pin)2], KOAc, dioxane, 70 °C, 3 h; (c) 2-(benzyloxy)-4-bromo-1-nitrobenzene, Na2CO3 2 M, 70 °C, 1 h (93% over two steps) (d) NaH, MeI, THF, 0 °C, 20 h (45%); (e) H2, 10% Pd/C, EtOH, rt, 1 h; (f) CDI, MeCN, rt, 1 h (70% over two steps); (g) 4-phenylbutyl isocyanate, DMAP, pyridine, rt, 16 h (90%).
Scheme 6. Synthesis of 24c.

Reagents and conditions: (a) 4-bromo-2-nitrophenol, PdCl2(PPh3)2, 2 M Na2CO3, dioxane, reflux, 2 h (95%); (b) 10% Pd/C, cyclohexene, MeOH, reflux, 4 h; (c) CDI, MeCN, rt, 3 h (60% over two steps); (d) isobutylamine, triphosgene, Et3N, DCM (70%); (e) 4 M HCl, dioxane (95%); (f) HCHO, NaBH(OAc)3, AcOH, MeCN, rt, 2 h (83%).
Scheme 7. Synthesis of 26.

Reagents and conditions: (a) 2-benzyloxy-4-bromo-1-nitrobenzene, Pd(dppf)Cl2, Na2CO3, dioxane, reflux, 16 h (40%); (b) cyclohexene, Pd/C, MeOH, 70 °C, 2 h; (c) CDI, MeCN, rt, 16 h; (d) HCHO, NaBH(OAc)3, AcOH, MeCN, rt, 2 h (43% over three steps); (e) isobutylamine, triphosgene, Et3N, DCM, rt, 4 h (30%).
Scheme 8. Synthesis of 27.

Reagents and conditions: (a) 5-bromo-2-nitrophenol, Pd(dppf)Cl2, Na2CO3, dioxane, reflux, 2 h (53%); (b) 10% Pd/C, cyclohexene, EtOH, 65 °C, 4 h; (c) CDI, MeCN, rt, 1 h (90% over two steps) (d) 4 M HCl, dioxane, rt, 30 min; (e) HCHO, NaBH(OAc)3, AcOH, MeCN, rt, 30 min (70% over two steps); (f) isobutylamine, triphosgene, Et3N, DCM, rt, 3 h (70%).
Scheme 9. Synthesis of 22r and 25.

Reagents and conditions: for the synthesis of 22r: (a) 6-bromo-3H-1,3-benzoxazol-2-one, MeMgBr, n-BuLi, THF, −78 °C, 2 h (30%); (b) p-TsOH, toluene, reflux, 1 h (quant.); (c) H2, 10% Pd/C, MeOH, 60 °C, 2 h; (d) 4-phenylbutyl isocyanate, DMAP, pyridine (73% over two steps). For the synthesis of 25: (a) 7-bromo-3H-1,3-benzoxazol-2-one, MeMgBr, n-BuLi, THF, −78 °C 1.5 h (44%); (b) p-TsOH, toluene, 90 °C, 3 h (quant.); (c) HCHO, NaBH(OAc)3, MeCN, rt, 16 h; (d) H2, Pd/C, MeOH, 40 °C, 2 h; (e) isobutylamine, triphosgene, Et3N, DCM (30% over three steps).
Other C(6)-substituted benzoxazolones, for example, 23a–d and 50j (Schemes 10 and 11), were prepared in three steps and in satisfactory yields using a nucleophilic aromatic substitution (SNAr)44 reaction of activated fluoro-phenyls with a set of heterocyclic amines followed by hydrogenation and intramolecular cyclization reaction with CDI. An alternative approach was used for the synthesis of 50k (Scheme 11). In this case, a Cu-catalyzed cross coupling N-arylation of O-Bn-protected bromo-nitrophenol 51b with 4-methylpiperazin-2-one afforded 52b in acceptable yield,45 which upon standard reactions led to the benzoxazolone 50k.
Scheme 10. Synthesis of C(6)-substituted benzoxazolone carboxamides 23a–i.

Reagents and conditions: (a) 5-fluoro-2-nitrophenol, DIPEA, MeCN, 60–80 °C, 15 h (40% for 49c); (b) 10% Pd/C, cyclohexene, MeOH, reflux, 2–16 h; (c) CDI, MeCN, rt (or 50 °C for 49g), 2 h (75–80% over three steps for 50a and 50d; 45–60% over two steps for 50b and 50c); (d) 4-phenylbutyl isocyanate, DMAP, MeCN, rt, 16 h (20–85%); (e) 4 M HCl, dioxane, rt, 3 h (quant.); (f) RCHO, AcOH, NaBH(OAc)3, DCE, THF or MeCN, rt, 2 h (60–90%); (g) 4-phenylbutyl isocyanate, DMAP, MeCN, rt, 16 h (30–75%).
Finally, the carboxamide functionalities were introduced under standard conditions, which involved the reaction of the benzoxazolone intermediates with the corresponding commercially available isocyanates, as in the preparation of 22a–c (Scheme 3) and 23a–d (Scheme 10). Alternatively, the isocyanates were prepared in situ, upon activation of the corresponding amines by reaction with Boc2O in the presence of DMAP in MeCN,46 as in the synthesis of 22j, 22l, 22n, and 22o (Scheme 4), or by reaction with triphosgene in the presence of Et3N in DCM,47 as in the synthesis of 22m (Scheme 4).
Scheme 4. Synthesis of C(6)-substituted benzoxazolone carboxamides 22e–h and 22j–p.

Reagents and conditions: (a) 4 M HCl, dioxane, rt, 3 h; (b) RCHO, AcOH, NaBH(OAc)3, DCE, THF or MeCN, rt, 1–3 h (40% over two steps for 21i, quant. For 21h); (c) RNCO, DMAP, MeCN, rt, 2–16 h (50–73% for 22e–h, 22k, and 22p); or RNH2, triphosgene, Et3N, DCM, 0 °C to rt, 2 h (45% for 22m), or RNH2, Boc2O, DMAP, MeCN, rt, 1 h (24–40% for 22j, 22l, 22n, and 22o).
Results and Discussion
The four most potent classes of AC inhibitors described to date are illustrated in Figure 2. Each class is defined by the presence of a common chemical warhead—the urea-like functionality—that can covalently react with the catalytic cysteine (Cys-143) of AC to form a thioester bond.35 It has been reported that carboxamides 2a(39) and 4a–b41 form, upon incubation experiments with the protein, the corresponding cysteine adducts. This has recently been confirmed by Dementiev and co-workers, who described the crystal structural analysis of the uracil 1 covalently bound to Cys-143 at 2.7 Å resolution.48
While potent and, in some cases, systemically active,39,40 these molecules share two features that limit their use as oral drugs. First, the presence of a reactive warhead on the molecular scaffolds described to date contributes to their chemical and metabolic instability (e.g., uracil 1),37 and second, the hydrophobic linear side chain that ensures target recognition and some degree of specificity negatively affects their drug-likeness (e.g., benzoxazolone 2a).39 Thus, the need for optimized AC inhibitors remains an important issue to be addressed.49
As previously reported, preliminary structural modifications of 2a by variation of the lateral side chain of the urea functionality (Region A) and substitution of the benzoxazolone moiety (Region B) led to the identification of 2b(39) and 2c(40) (Figure 3). Despite good potency and enhanced drug-likeness compared to the previous uracil37 series, compounds 2b and 2c suffer from low solubility in aqueous media and moderate chemical and metabolic stability that limit their utility as oral drugs.39,40 To address these issues, our lead optimization strategy focused on designing additional structural modifications on Regions A and B (compound 5, Figure 2) with the aim of improving the physicochemical and metabolic properties while maintaining inhibitory potency.
Figure 3.

Inhibitory potencies (IC50 in μM) of compounds 2d–g, 8a–d, 12b–e, and 17b on the activity of hAC expressed in HEK-293 cells.
We initially investigated modifications of the lateral side chain (Region A) of 2c confirming that, as previously reported with 2a analogs,40 this region is involved in lipophilic interactions important for target recognition (Figure 3). In fact, different attempts to improve solubility and metabolic stability by reducing lipophilicity of the side chain were detrimental regarding potency (Figure 3). Although the removal of the phenyl ring was tolerated, as for the n-pentyl analog 2e (hAC IC50 = 60 nM), no enhancement of solubility was observed (<1 μM, PBS, pH 7.4). Replacement of one methylene unit with an oxygen (e.g., ethers 2d, 2f, and 2g) to increase the hydrophilicity significantly reduced the inhibitory potency to the μM range. A similar trend was observed for the corresponding analogs in the 2b series (data not shown), indicating that the lipophilic side chain of the urea was very likely occupying a hydrophobic pocket.
We then shifted our attention to the left-hand side (Region B) of the scaffold by evaluating the replacement of the benzoxazolone moiety with some bioisosteric 6 + 5 fused ring heterocyclic systems (Figure 3), alternative to those already reported by Ortega et al.41
However, both the isatin analog 8c and the oxindole analog 8d were inactive at concentrations up to 10 μM. We then investigated the bioisosteric insertion of an aza-group in the phenyl ring of the benzoxazolone moiety, and this change resulted in very potent compounds. For example, compounds 8a and 8b gave hAC IC50’s of 6 and 3 nM, respectively, compared to the earlier compound 2a,39 which has an hAC IC50 of 64 nM. We envisaged that the insertion of a polar group on the left-hand side of the scaffold (Region B) could have an impact on the solubility of this series in aqueous buffer (PBS, pH 7.4), but, unfortunately, both 8a and 8b had very poor chemical stability in these conditions (t1/2 < 15 min).
These findings prompted us to evaluate the inhibitory potency of the fused bicyclic derivatives 12b and 17b (Figure 3). We speculated that changing the left-hand-side leaving group at the urea functionality could have an effect on the chemical stability of the scaffold. Although we generally observed a loss in potency to the sub-μM range, regardless of the substituent (12b–e) or the position of the nitrogen atom (17b), we were pleased to notice that, as for 12b and 17b, this novel class of hAC inhibitors showed improved chemical stability in PBS at pH 7.4 (t1/2 > 8 h) and improved aqueous solubility (82 and 230 μM, respectively). Despite the novel chemotype of these AC inhibitors with promising physicochemical properties, our attempts to improve the potency of this series were unsuccessful (data not shown). In addition, although these compounds exhibited high mouse plasma stability (e.g., t1/2 > 2 h, for 12b and 17b), this class of molecules also suffered from poor mouse liver microsomal stability (t1/2 < 15 min).
Overall, these results confirmed the benzoxazolone moiety as a “privileged scaffold”, thus focusing our SAR strategy on Region B with the intention of reducing the lipophilicity by replacing the phenyl ring of 2c at the C(6)-position with aliphatic heterocyclic rings (5, Figure 2). We envisaged that the reduction of the number of sp2-hybridized carbon atoms and the insertion of heteroatoms in this region could improve the overall physicochemical and metabolic stability of this class of inhibitors.50,51
We were pleased to observe that both the cyclohexyl analog 22a and the tetrahydropyrane analog 22b resulted in equipotent inhibition (hAC IC50 = 0.089 and 0.068 μM, respectively) compared to the corresponding phenyl derivative 2c(39) (Table 1 and Figure 2). With these results in hand, we were then interested in evaluating the effect of increasing the hydrophilicity by the addition of a polar basic amine, as in the piperidine analogs 22d and 22e. With this modification, we observed that both compounds showed only a slight loss of potency compared to the aliphatic analog 22a (hAC IC50 = 0.134 and 0.129 μM, respectively). Encouraged by these results, we explored the effect of other N-containing heterocyclic systems, such as the piperidine 23a, the morpholine 23b, the 1,1-dioxothiomorpholine 23c, and the piperazines 23e and 23f. Overall, this set of compounds showed similar potency to the initial cyclohexyl analog 22a. Notably, the piperidine 23a and the piperazine 23f were the most potent compounds, showing IC50 values of 0.080 and 0.116 μM, respectively (Table 1). Based on these promising results, we selected the piperidine and piperazine series, exemplified by 22d and 23e, respectively, as novel scaffolds for further studies, exploiting the presence of a distal nitrogen atom as an anchor point for additional structural modifications (Table 2). First, we evaluated the SAR exploration around the piperidine series by introducing both linear and branched alkyl chains on the nitrogen atom, such as the ethyl 22f, isopropyl 22g, and isobutyl 22h analogs (Table 2). In general, no significant differences were observed on the inhibitory potency of these derivatives, with 22f–h almost being equipotent to the unsubstituted 22d. Moreover, the removal of the basic center by the introduction of either an exocyclic (22i) or endocyclic N-acyl group (22q) was tolerated, showing IC50 values of 0.064 and 0.105 μM, respectively. These results further confirmed that different polar groups were tolerated in this region of the scaffold.
Table 1. Inhibitory Potencies of Compounds 22a, 22b, 22d, 22e, 23a–c, 23e, and 23f on the Activity of hAC.

IC50 values are the mean of at least three independent experiments, performed in three technical replicates.
Table 2. Inhibitory Potencies of Piperidines 22f–i and 22q and Piperazines 23g–k on the Activity of hAC.

IC50 values are the mean of at least three independent experiments performed in three technical replicates.
The same strategy was applied to the piperazine series (Table 2). Specifically, both the N-alkyl derivatives 23g–i and the piperazinones 23j and 23k resulted in more potent AC inhibition than the parent 23e. For example, the N-ethyl piperazine 23g and the piperazinone 23k were almost 7-fold more potent than 23e (hAC IC50 = 0.363 μM), showing IC50 values of 0.056 and 0.052 μM, respectively.
However, a comparison of the piperazine and piperidine series in terms of aqueous kinetic solubility (PBS, pH 7.4) in vitro metabolism highlighted some significant differences (Table 3). Interestingly, the piperidine analogs, bearing small linear alkyl groups (22d–f), were highly soluble (kinetic solubility >100 μM) and, in some cases (22d), had acceptable stability profiles both in mouse plasma and in liver microsomes. On the other hand, the piperidine derivatives, bearing more sterically hindered lipophilic alkyl groups, such as the isopropyl 22g and isobutyl 22h, or the acyls 22i and 22q suffered from low solubility and, with the exception of 22h, poor stability in mouse plasma (Table 3). Conversely, all the piperazine derivatives generally suffered from poor aqueous solubility and poor microsomal and plasma stability (Table 3). As illustrative examples, the piperazines 23f and 23g showed poor solubility in water (<1 μM), rapid metabolism in liver microsomes, and poor plasma stability (m-plasma and m-liver microsomes, t1/2 < 5 min). Some improvement in microsomal stability was observed with the des-methylated 23e and with 23k, which bears a heterocyclic ring at a higher oxidative state compared to 23f.
Table 3. Aqueous Kinetic Solubility and In Vitro Metabolism of Some Selected Compounds in the Piperidines 22d–i, 22q and Piperazines 23e–g, 23j, and 23k Series.
| compound | solubility (μM)a (PBS, pH 7.4) | m-plasmabt1/2 (min) | m-LMc t1/2 (min)[% at 60 min] |
|---|---|---|---|
| piperidine series | |||
| 22d | 150 | 60 | >60 [70%] |
| 22e | 120 | 50 | 40 |
| 22f | 198 | 50 | 60 |
| 22g | 50 | 40 | 60 |
| 22h | <1 | 60 | 60 |
| 22i | <1 | 30 | 60 |
| 22q | 20 | 36 | 35 |
| piperazine series | |||
| 23e | 20 | 30 | 45 |
| 23f | <1 | <5 | <5 |
| 23g | <1 | <5 | <5 |
| 23j | <1 | 20 | 30 |
| 23k | 20 | 20 | <5 |
Aqueous kinetic solubility in phosphate-buffered saline. Values are the mean of at least two independent experiments performed in two technical replicates.
Mouse plasma. Values are the mean of at least two independent experiments performed in two technical replicates.
Mouse liver microsomes. Values are the mean of at least two independent experiments performed in two technical replicates.
With these results in hand, we focused our efforts on exploring Regions A and B of the N-methyl piperidine 22e, as reported in Table 4. In order to reduce the lipophilicity and improve metabolic stability of this scaffold, we followed different strategies: (a) insertion of a heteroatom, removal of the phenyl ring, and reduction of the side chain length (Region A); and (b) removal of potential metabolic soft spots (Regions A and B).
Table 4. Inhibitory Potencies of Piperidines 22j–p and 22r on the Activity of hAC and Aqueous Kinetic Solubility and In Vitro Metabolism of Some Selected Compounds.

IC50 values are the mean of at least three independent experiments performed in three technical replicates.
Aqueous kinetic solubility in phosphate-buffered saline. Values are the mean of at least two independent experiments performed in two technical replicates.
Mouse plasma. Values are the mean of at least two independent experiments performed in two technical replicates.
Mouse liver microsomes. Values are the mean of at least two independent experiments performed in two technical replicates.
An immediate loss in potency was observed with the removal of the lipophilic phenyl ring (22 k) or the insertion of an oxygen on the lateral chain (22j and 22l), while the bioisosteric replacement of a fluorine on the distal phenyl ring resulted in 22o, being almost equipotent to 22e (Table 4). Nonetheless, exploration of Region A continued with the insertion of branched alkyl groups. We were pleased that the isobutyl analog 22m (hAC IC50 = 0.166 μM) was equipotent to the corresponding butyl phenyl 22e, demonstrating that it was possible to remove the phenyl group and reduce the overall lipophilicity without compromising potency. On the other hand, a methyl group adjacent to the urea functionality, such as the sec-butyl analog 22n, was detrimental for potency, with an IC50 of 2.1 μM. Moving the SAR exploration back to Region B, insertion of a fluorine on the benzoxazolone ring 22p boosted the inhibitory potency (IC50 = 0.024 μM), while the difluoroethyl analog 22r showed similar potency (IC50 = 0.095 μM) to 22e. The kinetic aqueous solubility and in vitro metabolic stability of a selection of compounds in the piperidine series are summarized in Table 4. Notably, while the insertion of an oxygen did not affect either the solubility or metabolic stability in microsomes of 22j compared to 22e, reducing lipophilicity with small aliphatic groups (22k and 22m) was particularly beneficial. For example, 22k and 22m showed high aqueous solubility (240 μM) and improved plasma and liver microsomal stabilities (t1/2 > 60 min). On the other hand, attempts to improve the liver microsomal stability of 22e by inserting a fluorine atom at different potential metabolic soft spots of both Regions A and B (compounds 22o, 22p, and 22r) were not successful. Not surprisingly, these bioisosteric replacements negatively affected the aqueous solubilities of 22o, 22p, and 22r, without a substantial improvement of the metabolic stability in microsomes.
With these results in hand, SAR studies continued on the scaffold of compound 22m (Table 5). Specifically, we evaluated the effect of the location of both the N-methyl piperidine ring, at C(5)- and C(7)-positions of the benzoxazolone moiety (compounds 24c and 25), and the N-methylated nitrogen atom, within the piperidine nucleus (compounds 26 and 27). Overall, we generally observed a loss in the inhibitory potency of these targeted analogs compared to 22m, which was even more pronounced with compounds 26 and 27, showing IC50 values in the μM range. Finally, the evaluation of the kinetic aqueous solubility and in vitro metabolism of 22m and close analogs was completed (Table 5). In general, all the targeted compounds showed high solubility values in aqueous media, except for 24c, which bears the piperidine ring at the C(5)-position of the benzoxazolone system. On the other hand, major differences were observed comparing their metabolic stability properties. In particular, we observed that substitution at the C(6)-position was critical to maintaining acceptable mouse plasma and liver microsomal stabilities (compound 22m, m-plasma t1/2 = 80 min and m-liver microsomes t1/2 > 60 min (76% remaining at 1 h)). On the other hand, both derivatives with the piperidine ring at the C(5)- and C(7)-positions, 24c and 25 showed reduced mouse plasma and liver microsomal stability. A similar trend was observed by moving the nitrogen atom to a different position on the piperidine ring, except for 27, which showed a similar liver microsomal stability to 22m. Due to its inhibitory potency and improved overall drug-likeness profile, the piperidine 22m was selected for further biological and pharmacological investigations.
Table 5. Inhibitory Potencies of Compounds 24c and 25–27 on hAC and Aqueous Kinetic Solubility and In Vitro Metabolism.

IC50 values are the mean of at least three independent experiments performed in three technical replicates.
Aqueous kinetic solubility in phosphate-buffered saline. Values are the mean of at least two independent experiments performed in two technical replicates.
Mouse plasma. Values are the mean of at least two independent experiments performed in two technical replicates.
Mouse liver microsomes. Values are the mean of at least two independent experiments performed in two technical replicates.
We first envisaged that the inhibition of 22m, belonging to the same class of the benzoxazolone carboxamide 2c,39 should occur through the same covalent AC modification. According to our hypothesis, the corresponding benzoxazolone 21g (Scheme 4), tested at 1 and 10 μM, was not able to inhibit hAC due to the lack of the reactive urea-like functionality. Moreover, kinetic studies on hAC-enriched lysates showed that 22m causes a concentration-dependent reduction in the maximal catalytic velocity of AC (Vmax) without influencing the Michaelis–Menten constant (KM) (Figure 4B and Table S1) and time-dependent inhibition at different 22m concentrations with ki/KI = 0.02 μM–1 min–1 and ki = 0.15 min–1 (Figure 4C,D), suggesting a very fast covalent bond formation to the enzyme.52,53
Figure 4.

(A) Concentration–response curve for inhibition of hAC activity by 22m; (B) Michaelis–Menten analysis of the reaction of hAC in the presence of vehicle (DMSO 1%, ●) or 22m (100 nM, ▲; 400 nM, ■). Rbm 14–12: fluorogenic substrate of hAC; (C) time-dependent inhibition of hAC by 22m (two independent experiments, each performed in two technical replicates); (D) determination of kinetic parameter ki/KI of 22m (two independent experiments, each performed in two technical replicates).
The selectivity of 22m was evaluated against a set of related lysosomal enzymes. The compound showed only a weak inhibitory effect (IC50 = 8.0 μM) on human N-acylethanolamine acid amidase (hNAAA), a lysosomal cysteine amidase that shares 33–34% sequence identity and a very similar reactive site to AC.5422m had no effect at the concentrations tested (1 and 10 μM) on the activity of either acid sphingomyelinase (ASM) and GCase. We next assessed the selectivity of 22m against two of the most representative members of serine hydrolases, human fatty acid amide hydrolyase (FAAH)55 and monoacylglycerol lipase (MAGL):5622m showed inhibitory activity on FAAH with an IC50 of 0.070 μM and no effect on monoacylglycerol lipase (MAGL) at the concentrations tested (1 and 10 μM). Although off-target activity of 22m against FAAH is observed, to our knowledge, no evidence for biological cross-talk between the sphingolipid-signaling pathways2 and the FAAH-signaling pathway55,57 has been reported that could preclude further development of 22m.
The favorable overall profile of 22m prompted us to test its ability to inhibit AC in intact cells. Human neuroblastoma SH-SY5Y cells were incubated in the presence of 22m at different doses (1, 2.5, 5, and 10 μM). AC activity was measured with a liquid chromatography/mass spectrometry (LC/MS)-based activity assay after different incubation times (30 min, 1 h, 3 h, and 6 h), and SphL levels were identified and quantified by LC/MS, showing that 22m effectively engages AC in these cells leading to the expected variations in the SphL levels, as reported in Figures 5 and 6. Treatment of cultures of human neuroblastoma SH-SY5Y cells with 22m caused a concentration- (Figure 5A) and time-dependent reduction of AC activity (Figure 6A). After 3 h of incubation, this effect resulted in an intracellular accumulation of various ceramide species, including Cer (d18:0/16:0) and Cer (d18:1/16:0) (Figure 5B,C) and a corresponding decrease in the levels of sphingosine (Figure 5D) in a concentration-dependent manner. The effect of 22m (10 μM) on AC activity inhibition and SphL persisted for up to 6 h under our experimental conditions (Figure 6B–D). The results indicated that 22m inhibits AC in the complex cellular environment leading to an increased Cer (d18:0/16:0) and Cer (d18:1/16:0) (Figure 6B,C) and decreased sphingosine levels with a partial recovery of sphingosine levels after 3–6 h (Figure 6D). Conversely, as expected, no major variations were observed in the levels of sphingomyelin (SM) (d18:1/16:0) (Figures 5E and 6E) and hexosylceramide (HexCer) (d18:1/16:0) (Figures 5F and 6F).
Figure 5.

Effects of 22m in SH-SY5Y cells after a 3 h of incubation. Concentration dependence of the effects on AC activity (A) and sphingolipid levels (B–F). GraphPad Prism software (GraphPad Software, Inc., USA) was used for statistical analysis. Data were analyzed using the Student t test or one-way ANOVA followed by the Bonferroni post hoc test for multiple comparisons. Differences between groups were considered statistically significant at values of p < 0.05. Values are expressed as means ± S.E.M of at least six determinations. Experiments were repeated twice with similar results.
Figure 6.

Time course of the effects of 22m (10 μM) in SH-SY5Y cells on AC activity (A) and sphingolipid levels (B–F). GraphPad Prism software (GraphPad Software, Inc., USA) was used for statistical analysis. Data were analyzed using the Student t test or one-way ANOVA followed by the Bonferroni post hoc test for multiple comparisons. Differences between groups were considered statistically significant at values of p < 0.05. Values are expressed as means ± S.E.M of at least six determinations. Experiments were repeated twice with similar results.
Pharmacokinetic studies of 22m were determined in CD1 mice, and relevant pharmacokinetic parameters are reported in Table 6. Values of plasma clearance (Clp), volume of distribution (Vdss), and plasma half-life (t1/2) were calculated after intravenous administration of 22m at 3 mg kg–1. Clearance was moderately high (14.1 L h–1 kg–1), with a relatively short plasma half-life (1 h) and high Vdss (12.5 L kg–1) indicating that 22m well distributed out of the circulating plasma compartment. Good oral bioavailability was observed dosing 22m at 10 mg kg–1 (F = 58%), with significant exposures in plasma, brain, and cerebrospinal fluid (CSF) (AUC values = 412, 14648, and 119 (h × ng mL–1), respectively). A maximum tolerated dose (MTD) study in mice was also conducted in the same background as the pharmacodynamic model using C57BL/6 mice at intraperitoneal dose escalation of 20, 40, 80, and 120 mg kg–1 in the time range of 4 days, and no clinical abnormalities were observed in any animals within the doses and time range used.
Table 6. Pharmacokinetic Properties of 22m after Intravenous (A, 3 mg kg–1, N = 18) and Oral Administration (B, 10 mg kg–1, N = 18) in Male CD1 Mice.
| A | |||
|---|---|---|---|
| parameter (3mpk, i.v.) | plasma | brain | CSF |
| tmax (h) | - | 0.250 | 0.250 |
| Cmax (ng mL–1) | - | 6443 | 71.6 |
| t1/2 (h) | 1.26 | 1.01 | 0.661 |
| Cl (L h–1 kg–1) | 14.1 | - | - |
| Vdss (L kg–1) | 12.5 | - | - |
| AUC (h × ng mL–1) | 212 | 10128 | 77.8 |
| B | |||
|---|---|---|---|
| parameter (10mpk, p.o.) | plasma | brain | CSF |
| tmax (h) | 0.5 | 1.00 | 1.00 |
| Cmax (ng mL–1) | 216 | 6900 | 52.2 |
| t1/2 (h) | 1.03 | 1.18 | 1.23 |
| AUC (h × ng mL–1) | 412 | 14648 | 119 |
| F (%) | 58.3 | - | - |
Based on these results, we decided to study the effect of dosing 22m in 4L;C* mice, a validated genetic mutated animal model for neuropathic GD.58 4L;C* mice have a marked increase (20- to 30-fold) of GluSph and moderate elevation (1.5- to 3-fold) of GluCer in the brain; therefore, they are a unique model suitable for testing GluSph reduction therapy. 22m was administered at selected doses of 30 and 90 mg kg–1 by intraperitoneal injection (i.p.) once a day for 14 days starting at postnatal day 5. Preliminary results showed that compound 22m significantly reduces GluSph (d18:1) in the brain of 4L;C* mice in a dose-dependent manner (Figure 7). Target engagement was demonstrated at a high dose of 90 mg kg–1 with 54% reduction of the GluSph levels relative to control.
Figure 7.

Dose response reduction of brain levels of GluSph (d18:1) after intraperitoneal injection of 22m at 30 and 90 mg kg–1 in 4L;C* mice (N = 4–8 with mixed males and females for each group).
Next, we evaluated 22m in the Twitcher mouse, an animal model of Krabbe’s disease. The Twitcher mice naturally carry a GALC mutation that contains a premature stop codon in GALC and leads to a complete loss of GALC activity. As a result, a dramatic increase of the extremely toxic lipid GalSph is observed in Twitcher mouse brains. After i.p. administration at 30 and 90 mg kg–1 once daily for a treatment period of 20 days starting at postnatal day 10, 22m showed dose-dependent reduction of the toxic lipid GalSph (d18:1) levels in the brains of Twitcher mice by 72 and 41% at high and low doses, respectively (Figure 8).
Figure 8.

Dose response reduction of brain levels of GalSph (d18:1) after intraperitoneal injection of 22m at 30 and 90 mg kg–1 in Twitcher (Twi) mice (N = 3 males + N = 3 females for each group).
In the group of 4L;C* mice at 90 mg kg–1 doses, the unbound drug level in the brain 1 h post last dose (day 14) is 2.6 μM (6.4-fold higher than the EC50 value) (Table 7), while at a lower dose of 30 mg kg–1, the unbound drug level is 0.77 μM (1.9-fold higher than the EC50 value). In the group of Twitcher mice at 90 mg kg–1 doses, the unbound drug level in the brain 1 h post last dose (day 20) is 2.15 μM (5.2-fold higher than the EC50 value), while at a lower dose of 30 mg kg–1, the unbound drug level is 0.80 μM (2.0-fold higher than the EC50 value). Overall, these data support the observed dose responses in the two animal models.
Table 7. Plasma and Brain Concentrations of 22m in 4L;C* and Twitcher Mice.
| EC50 (μM)a | Fp, u (%)b | Fb, u (%)c | mouse model | dose (mg kg–1) | Cp (μM)d | Cp,u (μM)e | Cb (μM)f | Cb, u (μM)g |
|---|---|---|---|---|---|---|---|---|
| 0.410 ± 0.100 | 13.8 | 0.70 | 4L;C*h | 90 | 16.81 | 2.32 | 373.34 | 2.61 |
| 30 | 3.08 | 0.42 | 110.12 | 0.77 | ||||
| Twitcheri | 90 | 3.85 | 0.52 | 307.92 | 2.15 | |||
| 30 | 0.85 | 0.11 | 114.50 | 0.80 |
EC50 value as a mean of two independent experiments, each performed in two technical replicates. Primary fibroblast cells from Krabbe’s disease patients were incubated with 22m for 2 h at different concentrations.
Fp, u: plasma fraction unbounded. Values are the mean of two technical replicates.
Fb, u: brain fraction unbounded. Values are the mean of two technical replicates.
Cp: plasma concentration.
Cp, u: plasma unbounded concentration.
Cb: brain concentration.
Cb, u: brain unbounded concentration.
4L;C* mice were sacrificed 1 h after the last doses (day 14), and the compound 22m levels were measured in plasma and brain (N = 4–8 with mixed males and females).
Twitcher mice were sacrificed 1 h after the last doses (day 20), and the compound 22m levels were measured in plasma and brain (N = 3 males + N = 3 females for each group).
To our knowledge, this is the first report showing the efficacy of inhibiting AC on reducing the neurotoxic lipids GluSph in the brains of 4L;C* mice. Our result that inhibiting AC reduces neurotoxic lipid GalSph levels in the brains of Twitcher mice is consistent with the recent report.34
Further pharmacological studies of 22m will be reported in due course.
Conclusions
The present work outlines the lead optimization studies of a class of benzoxazolone carboxamides as AC inhibitors. We further extended the preliminary studies around 2b (and 2c)39,40 and performed a focused structure–activity relationship (SAR) study on Regions A and B of this scaffold with the aim of improving the physicochemical and metabolic properties of the series while maintaining the inhibitory potency. Introduction of different heterocyclic groups on the benzoxazolone moiety was tolerated regarding inhibitory potency, as for the tetrahydropyrane 22b, the piperidines 22d and 23a, and the piperazines 23e and 23f. A more focused exploration around 22d and 23e by changing the nature of substitution on the distal nitrogen atom led to the identification of novel potent analogs with improved solubility, for example, the piperidines 22e and 22f. Targeted modifications on different positions of Regions A and B of the N-methylated piperidine series led to compound 22m as a potent and oral bioavailable AC inhibitor with excellent brain penetration in mice. Preliminary results demonstrated target engagement of 22m both in the 4L;C* and Twitcher mouse models where dose-dependent reductions in GluSph and GalSph were observed, supporting that further optimized AC inhibitors may be used in the correction of severe pathological neurological states of LSD where these toxic lipids may play a significant role in the pathology, such as GD and KD.
Experimental Section
Chemicals, Materials, and Methods
Solvents and reagents were obtained from commercial suppliers and were used without further purification. Automated column chromatography purifications were done using a Teledyne ISCO apparatus (CombiFlash Rf) with prepacked SiO2 columns of different sizes (from 4 to 40 g). Mixtures of increasing polarity of Cy and EtOAc or DCM and MeOH were used as eluents. TLC analyses were performed using a Supelco on TLC Al foils 0.2 mm with a fluorescence indicator at 254 nm. Purifications of basic compounds were done using an IST ISOLUTE SCX packed into SPE cartridges (SCX). Hydrogenation reactions were performed using H-Cube continuous hydrogenation equipment (SS-reaction line version), employing disposable catalyst cartridges (CatCart) preloaded with the required heterogeneous catalyst. Microwave heating was performed using an Explorer-48 positions instrument (CEM). NMR experiments of all the intermediates and final compounds were run on a Bruker Avance III 400 system (400.13 MHz for 1H and 100.62 MHz for 13C) equipped with a BBI probe and Z-gradients. Spectra were acquired at 300 K using deuterated dimethylsulfoxide (DMSO-d6) or deuterated chloroform (CDCl3) as solvent. Chemical shifts for 1H and 13C spectra were recorded in parts per million using the residual non-deuterated solvent as the internal standard (for DMSO-d6: 2.50 ppm, 1H; 39.52 ppm, 13C; for CDCl3: 7.26 ppm, 1H and 77.16 ppm, 13C). Data are reported as follows: chemical shift (ppm), multiplicity (indicated as bs, broad singlet; s, singlet; d, doublet; t, triplet; q, quartet; p, quintet, sx, sextet; m, multiplet, and combinations thereof), coupling constants (J) in hertz (Hz), and integrated intensity. Quantitative 1H-NMR analyses of the freshly prepared 10 mM DMSO-d6 stock solutions (used for biological screenings) of the final compounds were performed using the PULCON method (PUlse Length based CONcentration determination, Bruker software, topspin 3.0. References: (a) Wider G., Reires L. J. Am. Chem. Soc. 2006, 128 (8), 2571–2576; (b) Burton I. W., Quilliam M. A., Valter J. A., Anal. Chem. 2005, 77, 3123–3131). UPLC/MS analyses of all the intermediates and final compounds were performed on a Waters ACQUITY UPLC/MS system consisting of a Single Quadrupole Detector (SQD) Mass Spectrometer (MS) equipped with an Electrospray Ionization (ESI) interface and a Photodiode Array Detector (PDA). The PDA range was 210–400 nm. Analyses were performed on an ACQUITY UPLC BEH C18 column (50 × 2.1 mm ID, particle size 1.7 μm) with a VanGuard BEH C18 precolumn (5 × 2.1 mm ID, particle size 1.7 μm). The mobile phase was 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN/H2O (95:5) at pH 5 (B). ESI in both positive and negative modes was used in the mass scan range of 100–650 Da. Analyses were performed with method A, B, C, or D. Method A: gradient 5 to 95% B over 2.5 min. Flow rate 0.5 mL min–1. Temperature 40 °C. Method B: gradient 50 to 100% B over 2.5 min. Flow rate 0.5 mL min–1. Temperature 40 °C. Method C: gradient 0 to 100% B over 2.5 min. Flow rate 0.5 mL min–1. Temperature 40 °C. Method D: isocratic 55% B over 5 min. Flow rate 0.5 mL min–1. Temperature 40 °C. UPLC/MS analyses of the final compounds were performed with method E or F using freshly prepared 10 mM DMSO-d6 stock solutions (used for biological screenings), diluted 20-fold or 100 fold in MeCN/H2O (1:1), and directly analyzed. An ACQUITY UPLC BEH C18 (100 × 2.1 mm ID, particle size 1.7 μm) with a VanGuard BEH C18 precolumn (5 × 2.1 mm ID, particle size 1.7 μm) was used. The mobile phase was 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN/H2O (95:5) at pH 5 (B). ESI in both positive and negative modes was used in the mass scan range of 100–650 Da. Method E: gradient: 10 to 90% B over 6 min. Flow rate 0.5 mL min–1. Temperature 40 °C. Method F: gradient: 50 to 100% B over 6 min. Flow rate 0.5 mL min–1. Temperature 40 °C. The detection wavelength (λ) was set at 215 nm for relative purity determination. Rt of the final compounds are reported in Table S2. Accurate mass measurements were performed on a Synapt G2 Quadrupole-ToF Instrument (Waters, USA) equipped with an ESI ion source; compounds were diluted to 50 μM in H2O/MeCN and analyzed. Leucine enkephalin (2 ng mL–1) was used as a lock mass reference compound for spectral recalibration. All final compounds displayed ≥95% purity as determined by NMR and UPLC/MS analysis.
General Procedure for Palladium-Catalyzed Cross Coupling Reaction (Procedure A)
To a solution of the appropriate phenyl bromide (1.0 equiv.) in dry 1,4-dioxane (0.5 M, previously degassed under a nitrogen atmosphere) was added the appropriate boronic acid or its corresponding boronic ester (1.1 equiv.) followed by the addition of Pd(PPh3)4 or Pd(dppf)Cl2 (0.05–0.2 equiv.) and 2 M Na2CO3 (2.5 equiv.). The dark reaction mixture was stirred at reflux for 15 h, then diluted with EtOAc, and filtered through a pad of Celite. The filtrate was concentrated under reduced pressure, and the crude was purified by column chromatography, eluting with Cy/EtOAc as indicated in each case.
General Procedure for Catalytic Hydrogenation Reaction (Procedure B)
Method A
To a suspension of the appropriate 2-nitrophenol (1.0 equiv.) in MeOH, EtOH, or EtOAc (0.4 M) were added 10% Pd/C (0.25 equiv.) and cyclohexene (30 equiv.), and the reaction mixture was stirred at reflux until the disappearance of the starting material, as indicated by UPLC/MS analysis. The suspension was filtered through a pad of Celite, and the filtrate was quickly evaporated under reduced pressure. The crude was used in the next step without further purification.
Method B
A suspension of the appropriate 2-nitrophenol (1.0 equiv.) in MeOH (0.4 M) was hydrogenated with the H-Cube apparatus using 10% Pd/C at 60 °C and full H2 mode. After complete conversion (UPLC/MS analysis monitoring), the solvent was evaporated under reduced pressure. The crude was used in the next step without further purification.
General Procedure for Intramolecular Cyclization Using CDI (Procedure C)
To a solution of the appropriate 2-aminophenol (1.0 equiv.) in MeCN (0.1 M) was added CDI (1.0–1.5 equiv.). The reaction mixture was stirred at rt for 2 h. Then the solvent was evaporated under reduced pressure, and the crude was redissolved in EtOAc, washed with H2O and brine, and dried over Na2SO4. After evaporation of the solvent, the crude was purified by column chromatography, eluting with Cy/EtOAc or DCM/MeOH, or used in the next step without further purification, as indicated in each case.
General Procedure for Carboxamide Synthesis (Procedure D)
Method A
To a stirred solution of the appropriate oxazolone (1.0 equiv.) and DMAP (1.1 equiv.) in dry MeCN was added the appropriate isocyanate (1.1–3.0 equiv.). The reaction mixture was stirred at rt for 30 min under a nitrogen atmosphere. After evaporation of the solvent, the crude was purified by column chromatography, eluting with Cy/EtOAc or DCM/MeOH as indicated in each case.
Method B
To a stirred solution of triphosgene (0.33 equiv.) in dry DCM (0.2 M) were added the appropriate amine (1.5–3.0 equiv.) and dry Et3N (3.0 equiv.) at −15 °C. The resulting mixture was stirred at rt for 30 min under a nitrogen atmosphere and then added to a solution of the appropriate oxazolone (1.0 equiv.) and Et3N (1.0 equiv.) in dry DCM. The reaction mixture was stirred at rt for 30 min under nitrogen and then diluted with DCM. The organic phase was washed with saturated aqueous NH4Cl solution and brine and dried over Na2SO4. After evaporation of the solvent, the crude was purified by column chromatography, eluting with Cy/EtOAc or DCM/MeOH, as indicated in each case.
Method C
To a stirred solution of Boc2O (2.0 equiv.) in MeCN (0.4 M) were added DMAP (2.0 equiv.) and the appropriate amine (2.0 equiv.). The resulting solution was stirred at rt for 10 min, then the appropriate oxazolone derivative (1.0 equiv.) was added, and the mixture was stirred at rt for 1 h. After evaporation of the solvent, the crude was purified by flash column chromatography, eluting with Cy/EtOAc or DCM/MeOH, as indicated in each case.
General Procedure for N-Boc Removal (Procedure E)
To a suspension of the appropriate N-Boc-protected derivative (1.0 equiv.) in 1,4-dioxane or DCM (0.1 M) was added HCl (30 equiv., 4 M in 1,4-dioxane), and the reaction mixture was stirred at rt for 2 h. After evaporation of the solvent, the crude was triturated with Et2O or used in the next step without further purification, as indicated in each case.
General Procedure for Reductive Amination Reaction (Procedure F)
To a solution of the appropriate secondary amine (1.0 equiv.) in MeCN or THF (0.1 M) were added the appropriate aldehyde or ketone (1.6–5.0 equiv.), AcOH (1.6–5.0 equiv.), and NaBH(OAc)3 (1.6–3.0 equiv.). The mixture was stirred at rt for 2–16 h under a nitrogen atmosphere. Then the reaction mixture was poured into saturated aqueous NaHCO3 solution and extracted with EtOAc. The organic phase was washed with brine and dried over Na2SO4. After evaporation of the solvent, the crude was purified by SCX.
General Procedure for Nucleophilic Aromatic Substitution Reaction (SNAr) (Procedure G)
To a solution of the appropriate 4-fluoronitrobenzene (1.0 equiv.) in MeCN were added the appropriate amine (2.0 equiv.) and DIPEA (2.0 equiv.). The reaction mixture was refluxed (or stirred under MW irradiation, 90 °C, power 200 W) until the disappearance of the starting material, as indicated by UPLC/MS analysis. After evaporation of the solvent, the crude was purified by flash column chromatography, eluting with Cy/EtOAc or DCM/MeOH, as indicated in each case.
General Procedure for Intramolecular Cyclization under Basic Conditions (Procedure H)
To a solution of the appropriate thiazolidinedione derivative (1.0 equiv.) in dry THF (0.1 M) was added tBuOK (2.0–4.0 equiv.) at rt under a nitrogen atmosphere. After 30 min, the reaction mixture was diluted with EtOAc, washed with saturated aqueous NH4Cl solution and brine, and dried over Na2SO4. After evaporation of the solvent, the crude was used in the next step without further purification.
General Procedure for Lithium/Halogen Exchange - Addition Reaction (Procedure I)
To a solution of the appropriate bromobenzoxazolone (1.0 equiv.) in dry THF (0.1 M) was added MeMgBr (1.5 equiv., 3.0 M in Et2O) at −78 °C under a nitrogen atmosphere for 30 min followed by the addition of n-BuLi (1.2 equiv., 2.5 M in hexanes). After 30 min, a solution of the appropriate piperidone (1.7 equiv.) in dry THF (0.7 M) was added dropwise at −78 °C under a nitrogen atmosphere, and then the reaction mixture was allowed to warm to rt. After 30 min, the reaction was quenched by addition of saturated aqueous NH4Cl solution, diluted with EtOAc, washed with brine, and dried over Na2SO4. After evaporation of the solvent, the crude was purified by column chromatography, eluting with Cy/EtOAc or DCM/MeOH, as indicated in each case.
General Procedure for Dehydration Reaction of Tertiary Alcohols (Procedure L)
To a suspension of the appropriate tertiary alcohol (1.0 equiv.) in dry toluene (0.1 M) was added p-TsOH (3.0 equiv.), and the reaction mixture was stirred at reflux for 2 h. After evaporation of the solvent, the crude was purified by SCX or used in the next step without further purification, as indicated in each case.
Synthesis of N-(2-Benzyloxyethyl)-6-(4-fluorophenyl)-2-oxo-1,3-benzoxazole-3-carboxamide (2d)
Compound 2d was prepared according to general procedure D (method B) using 6-(4-fluorophenyl)-3H-1,3-benzoxazol-2-one39 (0.060 g, 0.26 mmol), 2-(benzyloxy)-1-ethanamine hydrochloride (0.073 g, 0.39 mmol), and Et3N (0.11 mL, 0.079 g, 0.78 mmol) in dry DCM (3 mL). The crude was purified by column chromatography (Cy/EtOAc, 80:20) to afford 2d as a white solid (0.06 g, 57%). 1H NMR (600 MHz, CDCl3) δ 8.35 (bs, 1H), 8.08 (d, J = 8.3 Hz, 1H), 7.52 (dd, J = 8.8, 5.2 Hz, 2H), 7.43 (dd, J = 8.3, 1.7 Hz, 1H), 7.40 (d, J = 1.7 Hz, 2H), 7.39–7.33 (m, 4H), 7.28 (tt, J = 7.0, 1.6 Hz, 1H), 7.14 (t, J = 8.6 Hz, 2H), 4.59 (s, 2H), 3.74–3.65 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 162.82 (d, JC–F = 247.5 Hz), 153.12, 149.94, 142.43, 137.91, 137.51, 136.18, 136.16, 128.86 (d, JC–F = 8.2 Hz), 128.62, 127.96, 127.95, 127.28, 123.87, 116.03 (d, JC–F = 21.4 Hz), 115.78, 108.57, 73.40, 68.26, 40.37. UPLC/MS (method A): Rt 2.78 min. MS (ES) C23H19FN2O4 requires 406, found 407 [M + H]+. HRMS C23H20FN2O4 [M + H]+: calculated 407.1407, measured: 407.1424, Δppm 4.2.
Synthesis of 6-(4-Fluorophenyl)-2-oxo-N-pentyl-1,3-benzoxazole-3-carboxamide (2e)
Compound 2e was prepared according to general procedure D (method A) using 6-(4-fluorophenyl)-3H-1,3-benzoxazol-2-one (0.080 g, 0.35 mmol) and 1-pentyl isocyanate (0.05 mL, 0.040 g, 0.39 mmol) in dry MeCN (3 mL). The crude was purified by column chromatography (Cy/EtOAc, 80:20) to afford 2e as a white solid (0.100 g, 82%). 1H NMR (400 MHz, CDCl3) δ. 8.10 (d, J = 8.3 Hz, 1H), 8.04 (bs, 1H), 7.52 (dd, J = 8.6, 5.3 Hz, 2H), 7.47–7.42 (m, 1H), 7.41–7.38 (m, 1H), 7.14 (t, J = 8.6 Hz, 2H), 3.44 (q, J = 6.9 Hz, 2H), 1.66 (p, J = 7.2 Hz, 2H), 1.45–1.32 (m, 4H), 0.93 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 162.70 (d, JC–F = 247.6 Hz), 153.23, 149.71, 142.28, 137.38, 136.06, 128.73 (d, JC–F = 8.8 Hz), 123.78, 116.00 (d, JC–F = 25.3 Hz), 115.75, 108.42, 99.96, 40.34, 29.12, 28.96, 22.30, 13.94. UPLC/MS (method A): Rt 2.43 min. MS (ES) C19H19FN2O3 requires 342, found 343 [M + H]+. HRMS C19H20FN2O3 [M + H]+: calculated 343.1458, measured: 343.1449, Δppm −2.6.
Synthesis of N-(2-Ethoxyethyl)-6-(4-fluorophenyl)-2-oxo-1,3-benzoxazole-3-carboxamide (2f)
Compound 2f was prepared according to general procedure D (method B) using 6-(4-fluorophenyl)-3H-1,3-benzoxazol-2-one (0.130 g, 0.57 mmol), 2-ethoxyethylamine (0.09 mL, 0.080 g, 0.85 mmol), and Et3N (0.20 mL, 0.140 g, 1.42 mmol) in dry DCM (15 mL). The crude was purified by column chromatography (Cy/EtOAc, 80:20) to afford 2f as a white solid (0.03 g, 13%). 1H NMR (400 MHz, CDCl3) δ 8.32 (bs, 1H), 8.10 (d, J = 8.3 Hz, 1H), 7.57–7.49 (m, 2H), 7.46–7.38 (m, 2H), 7.18–7.10 (m, 2H), 3.68–3.61 (m, 4H), 3.56 (q, J = 7.0 Hz, 2H), 1.24 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 162.69 (d, JC–F = 247.7 Hz), 153.16, 149.98, 142.47, 137.54, 136.20, 128.88 (d, JC–F = 8.2 Hz), 127.34, 123.89, 116.04 (d, JC–F = 21.7 Hz), 115.81, 108.60, 68.63, 66.81, 40.43, 15.25. UPLC/MS (method A): Rt 2.56 min. MS (ES) C18H17FN2O4 requires 344, found 345 [M + H]+. HRMS C18H18FN2O4 [M + H]+: calculated 345.1251, measured: 345.1258, Δppm 2.
Synthesis of 6-(4-Fluorophenyl)-N-(3-methoxypropyl)-2-oxo-1,3-benzoxazole-3-carboxamide (2g)
Compound 2g was prepared according to general procedure D (method B) using 6-(4-fluorophenyl)-3H-1,3-benzoxazol-2-one (0.08 g, 0.35 mmol), 3-methoxypropylamine (0.06 mL, 0.050 g, 0.52 mmol), and Et3N (0.12 mL, 0.09 g, 0.88 mmol) in dry DCM (15 mL). The crude was purified by column chromatography (Cy/EtOAc, 80:20) to afford 2g as a white solid (0.020 g, 18%). 1H NMR (400 MHz, CDCl3) δ 8.35 (bs, 1H), 8.10 (d, J = 8.3 Hz, 1H), 7.52 (dd, J = 8.6, 5.3 Hz, 2H), 7.43 (dd, J = 8.4, 1.4 Hz, 1H), 7.41–7.39 (m, 1H), 7.14 (t, J = 8.6 Hz, 2H), 3.55 (dt, J = 16.4, 6.0 Hz, 4H), 3.39 (s, 3H), 1.92 (p, J = 6.1 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 162.67 (d, JC–F = 247.4 Hz), 153.16, 142.84, 142.43, 137.44, 136.23, 128.70 (d, JC–F = 8.1 Hz, 2C), 127.40, 123.84, 116.12, 115.88 (d, JC–F = 11.3 Hz, 2C), 108.53, 70.95, 58.95, 38.60, 29.28. UPLC/MS (method A): Rt 2.49 min. MS (ES) C18H17FN2O4 requires 344, found 345 [M + H]+. HRMS C18H18FN2O4 [M + H]+: calculated 345.1251, measured: 345.1258, Δppm 2.
Synthesis of 3H-Oxazolo[4,5-c]pyridin-2-one (7a)
Compound 7a was prepared according to general procedure C using 6a (0.10 g, 0.91 mmol) and CDI (0.290 g, 1.82 mmol, 2.0 equiv.) in a mixture of MeCN/DMF (9 mL, 2:1). The crude was triturated with DCM to afford 7a as a whitish solid (0.100 g, 80%). 1H NMR (400 MHz, DMSO-d6) δ 8.34 (s, 1H), 8.32 (d, J = 5.3 Hz, 1H), 7.38 (d, J = 5.3 Hz, 1H). UPLC/MS (method A): Rt 0.61 min. MS (ES) C6H4N2O2 requires 136, found 137 [M + H]+, 135 [M–H]−.
Synthesis of 1H-Oxazolo[5,4-c]pyridin-2-one (7b)
Compound 7b was prepared according to general procedure C using 6b (0.100 g, 0.91 mmol) and CDI (0.441 g, 2.72 mmol) in a mixture of MeCN/DMF (9 mL, 1:4). The crude was triturated with Et2O to afford 7b as a brown solid (0.123 g, quant.). 1H NMR (400 MHz, DMSO-d6) δ 12.51 (bs, 1H), 8.38 (s, 1H), 8.23 (d, J = 5.5 Hz, 1H), 7.16 (d, J = 5.5 Hz, 1H). UPLC/MS (method C): Rt 1.06 min. MS (ES) C6H4N2O2 requires 136, found 137 [M + H]+, 135 [M–H]−.
Synthesis of 2-Oxo-N-(4-phenylbutyl)oxazolo[4,5-c]pyridine-3-carboxamide (8a)
Compound 8a was prepared according to general procedure D (method A) using 7a (0.03 g, 0.22 mmol) and 4-phenylbutyl isocyanate (0.045 mL, 0.046 g, 0.26 mmol) in a mixture of DMF/toluene (3 mL, 2:1). The crude was purified by column chromatography (Cy/EtOAc, from 95:5 to 70:30) to afford 8a as a white solid (0.015 g, 41%). 1H NMR (400 MHz, CDCl3) δ 9.28 (s, 1H), 8.55 (d, J = 5.1 Hz, 1H), 7.85 (bs, 1H), 7.31–7.21 (m, overlapped with CDCl3 signal, 3H), 7.21–7.13 (m, 3H), 3.47 (q, J = 6.5 Hz, 2H), 2.68 (t, J = 7.1 Hz, 2H), 1.79–1.64 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 151.67, 148.95, 148.06, 146.45, 141.91, 136.75, 136.72, 128.53 (4C), 126.06, 105.75, 40.48, 35.54, 29.11, 28.63. UPLC/MS (method A): Rt 2.23 min. MS (ES) C17H17N3O3 requires 311, found 312 [M + H]+. HRMS C17H18N3O3 [M + H]+: calculated 312.1348, measured: 312.134, Δppm −2.6.
Synthesis of 2-Oxo-N-(4-phenylbutyl)oxazolo[5,4-c]pyridine-1-carboxamide (8b)
Compound 8b was prepared according to general procedure D (method A) using 7b (0.08 g, 0.59 mmol) and 4-phenylbutyl isocyanate (0.11 mL, 0.113 g, 0.65 mmol) in a mixture of DMF/MeCN (12 mL, 4:1). The crude was purified by column chromatography (Cy/EtOAc, 80:20) to afford 8b as a white solid (0.107 g, 59%). 1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 8.54 (d, J = 5.3 Hz, 1H), 8.04 (d, J = 5.3 Hz, 1H), 7.91 (bs, 1H), 7.33–7.23 (m, overlapped with CDCl3 signal, 2H), 7.23–7.13 (m, 3H), 3.46 (q, J = 6.4 Hz, 2H), 2.68 (t, J = 7.1 Hz, 2H), 1.80–1.61 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 151.99, 148.79, 146.15, 141.86, 139.86, 130.48, 128.55 (4C), 128.53, 126.10, 110.80, 40.52, 35.54, 29.04, 28.62. UPLC/MS (method A): Rt 2.26 min. MS (ES) C17H17N3O3 requires 311, found 312 [M + H]+. HRMS C17H18N3O3 [M + H]+: calculated 312.1348, measured: 312.1341, Δppm −2.2.
Synthesis of 2,3-Dioxo-N-(4-phenylbutyl)indoline-1-carboxamide (8c)
Compound 8c was prepared according to general procedure D (method A) using 7c (0.074 g, 0.50 mmol) and 4-phenylbutyl isocyanate (0.097 mL, 0.100 g, 0.55 mmol). The crude was purified by column chromatography (Cy/EtOAc, 85:15) to afford 8c as a yellow solid (0.029 g, 21%). 1H NMR (400 MHz, DMSO-d6) δ 8.22 (t, J = 5.7 Hz, 1H), 8.16 (d, J = 8.2 Hz, 1H), 7.75–7.64 (m, 2H), 7.31–7.23 (m, 3H), 7.23–7.13 (m, 3H), 3.39–3.24 (m, overlapped with H2O signal, 2H), 2.62 (t, J = 7.3 Hz, 2H), 1.68–1.51 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 180.90, 159.48, 151.08, 148.69, 142.51, 138.04, 128.70 (4C), 126.14, 125.16, 124.81, 119.47, 116.97, 39.70, 35.23, 29.14, 28.66. UPLC/MS (method A): Rt 1.41 min. MS (ES) C19H18N2O3 requires 322, found 323 [M + H]+. HRMS C19H19N2O3 [M + H]+: calculated 323.1396, measured: 323.1391, Δppm −1.5.
Synthesis of 2-Oxo-N-(4-phenylbutyl)indoline-1-carboxamide (8d)
Compound 8d was prepared according to general procedure D (method A) using 7d (0.066 g, 0.50 mmol) and 4-phenylbutyl isocyanate (0.094 mL, 0.096 g, 0.55 mmol). The crude was purified by column chromatography (Cy/EtOAc, 90:10) to afford 8d as a white solid (0.04 g, 26%). 1H NMR (400 MHz, CDCl3) δ 8.60 (bs, 1H), 8.25 (d, J = 8.2 Hz, 1H), 7.35–7.22 (m, overlapped with CDCl3 signal, 4H), 7.21–7.11 (m, 4H), 3.71 (s, 2H), 3.42 (q, J = 6.7 Hz, 2H), 2.67 (t, J = 7.3 Hz, 2H), 1.79–1.62 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 177.17, 152.03, 141.89, 141.69, 128.22, 128.15, 128.11, 125.62, 124.11, 123.61, 122.71, 116.28, 39.52, 36.80, 35.32, 28.96, 28.50. UPLC/MS (method A): Rt 2.61 min. MS (ES) C19H20N2O2 requires 308, found 309 [M + H]+. HRMS C19H21N2O2 [M + H]+: calculated 309.1603, measured 309.1598, Δppm −1.6.
Synthesis of tert-Butyl 3-(2,4-dioxothiazolidin-3-yl)-4-oxo-piperidine-1-carboxylate (10)
To a solution of 9 (0.782 g, 1.00 mmol, 1.0 equiv.) in dry DMF (5 mL) were added TZD (0.141 g, 1.20 mmol, 1.2 equiv.) and K2CO3 (0.207 g, 1.50 mmol, 1.5 eq.). The reaction was stirred at rt for 2 h and then diluted with EtOAc. The organic phase was washed with brine, dried over Na2SO4, and concentrated under reduced pressure to afford 10 as an orange oil (0.247 g, 79%). 1H NMR (400 MHz, CDCl3) δ 4.80–4.67 (m, 1H), 4.57–4.19 (m, 2H), 4.02 (s, 2H), 3.79–3.58 (m, 1H), 3.30–3.10 (m, 1H), 2.68–2.47 (m, 2H), 1.49 (s, 9H). UPLC/MS (method A): Rt 1.88 min. MS (ES) C13H18N2O5S requires 314, found 313[M–H]−.
Synthesis of tert-Butyl 2-oxo-3,4,6,7-tetrahydrooxazolo[4,5-c]pyridine-5-carboxylate (11)
Compound 11 was prepared according to general procedure H using 10 (0.247 g, 0.79 mmol, 1.0 equiv.) and tBuOK (0.176 g, 1.57 mmol, 2.0 equiv.) in dry THF (8 mL). The crude was purified by column chromatography (Cy/EtOAc, 80:20) to afford 11 as yellow oil (0.055 g, 29%). UPLC/MS (method A): Rt 1.64 min. MS (ES) C11H16N2O4 requires 240, found 241 [M + H]+.
Synthesis of tert-Butyl 2-oxo-3-(4-phenylbutylcarbamoyl)-6,7-dihydro-4H-oxazolo[4,5-c]pyridine-5-carboxylate (12a)
Compound 12a was prepared according to general procedure D (method A) using 11 (0.055 g, 0.23 mmol) and 4-phenylbutyl isocyanate (0.079 mL, 0.081 g, 0.46 mmol, 2.0 equiv.) in dry MeCN (1 mL). The crude was purified by column chromatography (Cy/EtOAc, 90:10) to afford 12a as an off-white solid (0.070 g, 68%). 1H NMR (400 MHz, CDCl3) δ 7.98 (bs, 1H), 7.31–7.23 (m, overlapped signals with CDCl3, 2H), 7.21–7.11 (m, 3H), 4.64–4.58 (m, 2H), 3.74–3.66 (m, 2H), 3.35 (q, J = 6.7 Hz, 2H), 2.64 (q, J = 7.6 Hz, 2H), 2.56–2.46 (m, 2H), 1.78–1.58 (m, overlapped with H2O signal, 4H), 1.48 (s, 9H). UPLC/MS (method A): Rt 2.81 min. MS (ES) C22H29N3O5 requires 415, found 416 [M + H]+.
Synthesis of 2-Oxo-N-(4-phenylbutyl)-4,5,6,7-tetrahydrooxazolo[4,5-c]pyridine-3-carboxamide Hydrochloride (12b)
Compound 12b was prepared according to general procedure E using compound 12a (0.065 g, 0.16 mmol). The crude was triturated with Et2O to afford 12b as a yellow solid (0.030 g, 60%). 1H NMR (400 MHz, CDCl3) δ 10.33 (bs, 2H), 7.82 (t, J = 5.6 Hz, 1H), 7.31–7.24 (m, overlapped with CDCl3 signal, 2H), 7.21–7.13 (m, 3H), 4.58–4.38 (m, 2H), 3.63–3.45 (m, 2H), 3.33 (q, J = 6.5 Hz, 2H), 2.99–2.87 (m, 2H), 2.65 (t, J = 7.3 Hz, 2H), 1.74–1.53 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 152.81, 149.01, 141.92, 133.04, 128.54 (4C), 126.04, 114.29, 40.31, 35.53, 29.06, 28.63, 19.24. MS UPLC/MS (method A): Rt 1.83 min. MS (ES) C17H21N3O3 requires 315, found 316 [M + H]+. HRMS C17H22N3O3 [M + H]+: calculated 316.1661, measured: 316.1661, Δppm 0.0.
Synthesis of 5-Methyl-2-oxo-N-(4-phenylbutyl)-6,7-dihydro-4H-oxazolo[4,5-c]pyridine-3-carboxamide Hydrochloride (12c)
Compound 12c was prepared according to general procedure F using compound 12b (0.030 g, 0.09 mmol), 37% aqueous solution of formaldehyde (0.005 mL, 0.18 mmol), NaBH(OAc)3 (0.381 g, 1.80 mmol), and AcOH (0.008 mL, 0.008 g, 0.14 mmol) in dry MeCN (1.0 mL). The crude was dissolved in DCM (1 mL) followed by the addition of HCl (0.68 mL, 2.70 mmol, 4 M in 1,4-dioxane). After evaporation of the solvent, the residue was triturated with Et2O to afford 12c as a white solid (0.026 g, 90%). 1H NMR (400 MHz, CDCl3) δ 13.62 (bs, 1H), 7.80 (t, J = 5.5 Hz, 1H), 7.31–7.25 (m, overlapped with CDCl3 signal, 2H), 7.21–7.14 (m, 3H), 4.71 (d, J = 16.0 Hz, 1H), 4.14–4.01 (m, 1H), 3.75–3.60 (m, 1H), 3.54–3.39 (m, 1H), 3.34 (p, J = 6.3 Hz, 2H), 3.28–3.14 (m, 1H), 2.96 (s, 3H), 2.64 (t, J = 7.3 Hz, 2H), 1.74–1.51 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 152.71, 149.00, 141.89, 132.64, 128.52 (4C), 126.04, 113.48, 50.56 (2C), 49.25 (2C), 43.26, 40.32, 35.52, 29.03 (2C), 28.60 (2C), 19.49. UPLC/MS (method A): Rt 2.13 min. MS (ES) C18H23N3O3 requires 329, found 330[M + H]+. HRMS C18H24N3O3 [M + H]+: calculated 330.1818, measured: 330.182, Δppm 0.6.
Synthesis of 5-Benzyl-2-oxo-N-(4-phenylbutyl)-6,7-dihydro-4H-oxazolo[4,5-c]pyridine-3-carboxamide (12d)
Compound 12d was prepared according to general procedure F using compound 12b (0.050 g, 0.16 mmol), benzaldehyde (0.033 mL, 0.32 mmol), NaBH(OAc)3 (0.054 g, 0.26 mmol), and AcOH (0.015 mL, 0.015 g, 0.26 mmol) in dry MeCN (2 mL). The crude was purified by column chromatography (Cy/EtOAc, 85:15) to afford 12d as a white solid (0.043 g, 68%). 1H NMR (400 MHz, CDCl3) δ 8.01 (bs, 1H), 7.41–7.23 (overlapped with CDCl3 signal, m, 7H), 7.21–7.13 (m, 3H), 3.85–3.64 (m, 4H), 3.32 (q, J = 6.8 Hz, 2H), 2.89–2.76 (m, 2H), 2.64 (t, J = 7.4 Hz, 2H), 2.58–2.43 (m, 2H), 1.82–1.52 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 149.72, 142.06, 128.68 (2C), 128.53 (3C), 128.49 (4C), 125.99, 61.29, 48.43 (2C), 40.07, 35.58, 29.18, 28.67, 21.84. UPLC/MS (method A): Rt 1.98 min. MS (ES) C24H27N3O3 requires 405, found 406 [M + H]+. HRMS C24H28N3O3 [M + H]+: calculated 406.2131, measured: 406.2126, Δppm −1.2.
Synthesis of 5-Acetyl-2-oxo-N-(4-phenylbutyl)-6,7-dihydro-4H-oxazolo[4,5-c]pyridine-3-carboxamide (12e)
To a solution of 12b (0.030 g, 0.09 mmol) in dry DCM (0.9 mL) were added Et3N (0.025 mL, 0.018 g, 0.18 mmol, 2.0 equiv.) and acetyl chloride (0.008 g, 0.10 mmol, 1.1 equiv.) at 0 °C. The reaction mixture was stirred at rt for 3 h and then was diluted with EtOAc, washed with saturated aqueous NH4Cl solution and brine, and dried over Na2SO4. After evaporation of the solvent, the crude was triturated with Et2O to afford 12e as a white solid (0.018 g, 56%). 1H NMR (400 MHz, CDCl3) δ 8.07–7.85 (m, 1H), 7.31–7.24 (m, overlapped with CDCl3 signal, 2H), 7.21–7.13 (m, 3H), 4.87–4.58 (m, 2H), 3.96–3.64 (m, 2H), 3.36 (q, J = 6.6 Hz, 2H), 2.65 (t, J = 7.2 Hz, 2H), 2.61–2.49 (m, 2H), 2.17 (s, 3H), 1.76–1.58 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 128.52 (4C), 126.04, 43.33, 40.19, 38.26, 35.56, 29.16, 28.64, 22.23, 21.66. UPLC/MS (method A): Rt 2.12 min. MS (ES) C19H23N3O4 requires 357, found 358 [M + H]+.
Synthesis of tert-Butyl 4-(2,4-Dioxothiazolidin-3-yl)-3-hydroxy-piperidine-1-carboxylate (14a)
To a solution of 13 (0.220 g, 1.10 mmol, 1.2 equiv.) in dry DMF (2 mL) were added TZD (0.100 g, 0.89 mmol, 1 equiv.) and magnesium perchlorate (0.040 g, 0.18 mmol, 0.2 equiv.). The reaction mixture was stirred at rt for 20 min and then gradually heated to 115 °C over 2 h. After 3 h, the reaction was cooled, diluted with EtOAc, washed with H2O, brine, and 15% LiCl in H2O, and dried over Na2SO4. After evaporation of the solvent, the crude was purified by column chromatography (Cy/EtOAc, 60:40) to afford 14a as a white solid (0.139 g, 50%). 1H NMR (400 MHz, DMSO-d6) δ 5.37 (d, J = 3.3 Hz, 1H), 4.13 (d, J = 4.7 Hz, 2H), 4.11–3.86 (m, 4H), 2.88–2.61 (m, 1H), 2.06 (qd, J = 12.7, 4.7 Hz, 1H), 1.65–1.53 (m, 1H), 1.40 (s, 9H). UPLC/MS (method A): Rt 1.77 min, MS (ES) C13H20N2O5S requires 316, found 315 [M–H]−.
Synthesis of 3-(1-Methyl-3-oxo-4-piperidyl)thiazolidine-2,4-dione (15)
To a solution of 14a (0.100 g, 0.32 mmol) in dry DCM (3 mL) was added portionwise Dess–Martin periodinane (0.300 g, 0.70 mmol, 2.2 equiv.) under an argon atmosphere. The reaction was stirred at rt for 16 h, and then saturated aqueous NaHCO3 solution was added followed by the addition of 10% Na2SO3 in H2O. The mixture was stirred at rt for 30 min, and then the organic phase was separated, and the aqueous layer was extracted with DCM (3 times). The collected organic phases were dried over Na2SO4, and the solvent was removed under reduced pressure. The crude was purified by column chromatography (Cy/EtOAc, 75:15) to afford 15 as a white solid (0.070 g, 70%). 1H NMR (400 MHz, CDCl3) δ 4.80 (dd, J = 12.3, 6.6 Hz, 1H), 4.40 (d, J = 18.5 Hz, 1H), 4.24–3.91 (m, 3H), 3.29–3.49 (m, 1H), 2.63 (qd, J = 12.4, 5.1 Hz, 1H), 2.16–2.04 (m, 1H), 1.51–1.49 (m, 1H), 1.48 (s, 9H). UPLC/MS (method A): Rt 1.83 min, MS (ES) C13H18N2O5S requires 314, found 315 [M + H]+.
Synthesis of tert-Butyl 2-Oxo-1,4,6,7-tetrahydrooxazolo[5,4-c]pyridine-5-carboxylate (16)
Compound 16 was prepared according to general procedure H using 15 (0.070 g, 0.22 mmol) and tBuOK (0.190 g, 0.89 mmol, 4.0 equiv.) in dry THF (2 mL). The crude was used in the next step without further purification. UPLC/MS (method A): Rt 1.64 min. MS (ES) C11H16N2O4 requires 240, found 239 [M–H]−.
Synthesis of tert-Butyl 2-Oxo-1-(4-phenylbutylcarbamoyl)-6,7-dihydro-4H-oxazolo[5,4-c]pyridine-5-carboxylate (17a)
Compound 17a was prepared according to general procedure D (method A) using 16 (0.052 g, 0.22 mmol) and 4-phenylbutyl isocyanate (0.039 mL, 0.040 g, 0.22 mmol, 1.0 equiv.) in dry MeCN (2 mL). The crude was purified by column chromatography (Cy/EtOAc, 90:10) to afford 17a as a white solid (0.023 g, 25% over two steps). 1H NMR (400 MHz, CDCl3) δ 8.01 (bs, 1H), 7.31–7.24 (m, overlapped with CDCl3 signal, 2H), 7.22–7.14 (m, 3H), 4.32–4.19 (m, 2H), 3.72–3.59 (m, 2H), 3.34 (q, J = 6.7 Hz, 2H), 2.99–2.86 (m, 2H), 2.64 (t, J = 7.3 Hz, 2H), 1.75–1.59 (m, 4H), 1.48 (s, 9H). UPLC/MS (method B): Rt 1.91 min. MS (ES) C22H29N3O5 requires 415, found 416 [M + H]+.
Synthesis of 2-Oxo-N-(4-phenylbutyl)-4,5,6,7-tetrahydrooxazolo[5,4-c]pyridine-1-carboxamide Hydrochloride (17b)
Compound 17b was prepared according to general procedure E using 17a (0.023 g, 0.055 mmol). After evaporation of the solvent, the crude was triturated with Et2O to obtain 17b as a white solid (0.012 g, 60%). 1H NMR (400 MHz, DMSO-d6) δ 9.96 (bs, 2H), 8.05 (bs, 1H), 7.31–7.23 (m, 2H), 7.22–7.12 (m, 3H), 4.08–3.99 (m, 2H), 4.04 (s, 2H), 3.26 (q, J = 6.4 Hz, 2H), 3.01–2.93 (m, 2H), 2.59 (t, J = 6.9 Hz, 2H), 1.66–1.44 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 153.08, 149.11, 141.97, 128.54 (2C), 126.02, 40.28 (2C), 38.16, 36.03, 35.56, 29.09, 28.66. UPLC/MS (method A): Rt 1.88 min. MS (ES) C17H21N3O3 requires 315, found 316 [M + H]+. HRMS C17H22N3O3 [M + H]+: calculated 316.1661, measured: 316.1669, Δppm 2.5.
Synthesis of 5-(Cyclohexen-1-yl)-2-nitrophenol (19a)
Compound 19a was prepared according to general procedure A using 5-bromo-2-nitrophenol (0.218 g, 1.0 mmol), 18a (0.229 g, 1.10 mmol), Pd(PPh3)4 (0.058 g, 0.05 mmol), and 2 M Na2CO3 (1.30 mL, 2.50 mmol) in degassed 1,4-dioxane (20 mL). The crude was purified by column chromatography (Cy) to afford 19a as colorless oil (0.200 g, 90%). 1H NMR (400 MHz, CDCl3) δ 10.66 (s, 1H), 8.01 (d, J = 9.0 Hz, 1H), 7.09 (d, J = 1.9 Hz, 1H), 7.03 (dd, J = 9.0, 2.0 Hz, 1H), 6.39 (tt, J = 4.0, 1.6 Hz, 1H), 2.43–2.34 (m, 2H), 2.31–2.22 (m, 2H), 1.85–1.74 (m, 2H), 1.73–1.62 (m, 2H). UPLC/MS (method A): Rt 1.35 min. MS (ES) C12H13NO3 requires 219, found 220 [M + H]+.
Synthesis of 5-(3,6-Dihydro-2H-pyran-4-yl)-2-nitrophenol (19b)
Compound 19b was prepared according to general procedure A using 5-bromo-2-nitrophenol (0.218 g, 1.00 mmol), 18b (0.231 g, 1.10 mmol), Pd(PPh3)4 (0.058 g, 0.05 mmol), and 2 M Na2CO3 (1.30 mL, 2.50 mmol) in degassed 1,4-dioxane (20 mL). The crude was purified by column chromatography (Cy/EtOAc, 85:15) to afford 19b as a white powder (0.123 g, 56%). 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 7.95–7.85 (m, 1H), 7.15–7.10 (m, 2H), 6.50–6.45 (m, 1H), 4.30–4.20 (m, 2H), 3.82 (t, J = 5.4 Hz, 2H), 2.44–2.35 (m, 2H). UPLC/MS (method A): Rt 0.51 min. MS (ES) C11H11NO4 requires 221, found 220 [M–H]−.
Synthesis of tert-Butyl 4-(3-Hydroxy-4-nitrophenyl)-3,6-dihydro-2H-pyridine-1-carboxylate (19c)
Compound 19c was prepared according to general procedure A using 5-bromo-2-nitrophenol (1.60 g, 7.35 mmol), 18c (2.50 g, 8.09 mmol), Pd(PPh3)4 (0.424 g, 0.36 mmol), and 2 M Na2CO3 (9.2 mL, 18.38 mmol) in degassed 1,4-dioxane (15 mL). The crude was purified by column chromatography (Cy/EtOAc, 80:20) to afford 19c as a white powder (1.88 g, 80%). 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 7.91 (d, J = 8.7 Hz, 1H), 7.19–7.02 (m, 2H), 6.44–6.27 (m, 1H), 4.09–3.97 (m, 2H), 3.54 (t, J = 5.7 Hz, 2H), 2.48–2.41 (m, 2H), 1.43 (s, 9H). UPLC/MS (method A): Rt 2.54 min. MS (ES) C16H20N2O5 requires 320, found 319 [M–H]−.
Synthesis of tert-Butyl 4-(2-Fluoro-5-hydroxy-4-nitrophenyl)-3,6-dihydro-2H-pyridine-1-carboxylate (19d)
Compound 19d was prepared according to general procedure A using 5-bromo-4-fluoro-2-nitrophenol (0.280 g, 1.18 mmol), 18c (0.402 g, 1.3 mmol), Pd(PPh3)4 (0.07 g, 0.06 mmol), and 2 M Na2CO3 (1.48 mL, 2.95 mmol) in degassed 1,4-dioxane (12 mL). The crude was purified by column chromatography (Cy/EtOAc, 80:20) to afford 19d as a yellow solid (0.267 g, 67%). UPLC/MS (method A): Rt 2.45 min. MS (ES) C16H19FN2O5 requires 338, found 339 [M + H]+.
Synthesis of 2-Amino-5-cyclohexylphenol (20a)
Compound 20a was prepared according to general procedure B (method B) using 19a (0.200 g, 0.91 mmol). 1H NMR (400 MHz, CDCl3) δ 6.74–6.68 (m, 1H), 6.67–6.60 (m, 2H), 2.43–2.30 (m, 1H), 1.93–1.76 (m, 4H), 1.76–1.67 (m, 1H), 1.43–1.29 (m, 4H), 1.29–1.15 (m, 1H). UPLC/MS (method A): Rt 0.98 min. MS (ES) C12H17NO requires 191, found 192 [M + H]+.
Synthesis of 2-Amino-5-tetrahydropyran-4-ylphenol (20b)
Compound 20b was prepared according to general procedure B (method B) using 19b (0.123 g, 0.56 mmol). UPLC/MS (method A): Rt 0.60 min. MS (ES) C11H15NO2 requires 193, found 194 [M + H]+.
Synthesis of tert-Butyl 4-(4-Amino-3-hydroxyphenyl)piperidine-1-carboxylate (20c)
Compound 20c was prepared according to general procedure B (method B) using 19c (0.239 g, 0.75 mmol). UPLC/MS (method A): Rt 0.98 min. MS (ES) C16H24N2O3 requires 292, found 293 [M + H]+.
Synthesis of tert-Butyl 4-(4-Amino-2-fluoro-5-hydroxyphenyl)piperidine-1-carboxylate (20d)
Compound 20d was prepared according to general procedure B (method B) using 19d (0.265 g, 0.78 mmol). UPLC/MS (method D): Rt 1.51 min. MS (ES) C16H23FN2O3 requires 310, found 311 [M + H]+.
Synthesis of 6-Cyclohexyl-3H-1,3-benzoxazol-2-one (21a)
Compound 21a was prepared according to general procedure C using 20a (0.174 g, 0.91 mmol) and CDI (0.295 g, 1.82 mmol) in dry MeCN (9 mL). The crude was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 8.07 (bs, 1H), 7.09–7.06 (m, 1H), 7.04–6.89 (m, 2H), 2.61–2.44 (m, 1H), 1.96–1.80 (m, 4H), 1.81–1.62 (m, 1H), 1.47–1.31 (m, 4H), 1.31–1.16 (m, 1H). UPLC/MS (method A): Rt 2.38 min. MS (ES) C13H15NO2 requires 217, found 218 [M + H]+.
Synthesis of 6-Tetrahydropyran-4-yl-3H-1,3-benzoxazol-2-one (21b)
Compound 21b was prepared according to general procedure C using 20b (0.108 g, 0.56 mmol) and CDI (0.136 g, 0.84 mmol) in dry MeCN (6 mL). The crude was purified by column chromatography (Cy/EtOAc, 70:30) to afford 21b as a white powder (0.089 g, 72% over two steps). 1H NMR (400 MHz, DMSO-d6) δ 11.48 (s, 1H), 7.23–7.18 (m, 1H), 7.05–6.95 (m, 2H), 4.00–3.85 (m, 2H), 3.45–3.35 (m, 2H), 2.80–2.70 (m, 1H), 1.70–1.60 (m, 4H). UPLC/MS (method A): Rt 1.59 min. MS (ES) C12H13NO3 requires 219, found 220 [M + H]+.
Synthesis of tert-Butyl 4-(2-Oxo-3H-1,3-benzoxazol-6-yl)piperidine-1-carboxylate (21c)
Compound 21c was prepared according to general procedure D using 20c (0.219 g, 0.75 mmol) and CDI (0.183 g, 1.13 mmol) in dry MeCN (8 mL). The crude was purified by column chromatography (Cy/EtOAc, 45:55) to afford 21c as a white powder (0.195 g, 82% over two steps). 1H NMR (400 MHz, DMSO-d6) δ 11.49 (bs, 1H), 7.24–7.16 (m, 1H), 7.06–6.96 (m, 2H), 4.18–3.94 (m, 2H), 2.94–2.73 (m, 2H), 2.68 (tt, J = 11.9, 3.4 Hz, 1H), 1.78–1.70 (m, 2H), 1.55–1.43 (m, 2H), 1.42 (s, 9H). UPLC/MS (method A): Rt 2.16 min. MS (ES) C17H22N2O4 requires 318, found 317 [M–H]−.
Synthesis of tert-Butyl 4-(5-Fluoro-2-oxo-3H-1,3-benzoxazol-6-yl)piperidine-1-carboxylate (21d)
Compound 21d was prepared according to general procedure C using 20d (0.242 g, 0.78 mmol) and CDI (0.19 g, 1.17 mmol) in dry MeCN (8 mL). The crude was purified by column chromatography (Cy/EtOAc, 70:30) to afford 21d as a white solid (0.157 g, 60% over two steps). 1H NMR (400 MHz, DMSO-d6) δ 11.68 (bs, 1H), 7.29 (d, J = 6.0 Hz, 1H), 6.95 (d, J = 9.7 Hz, 1H), 4.18–3.97 (m, 2H), 2.95 (ddd, J = 12.0, 8.7, 3.4 Hz, 1H), 2.90–2.67 (m, 2H), 1.74–1.63 (m, 2H), 1.54 (qd, J = 12.5, 4.1 Hz, 2H), 1.41 (s, 9H). UPLC/MS (method D): Rt 2.21 min. MS (ES) C17H21FN2O4 requires 336, found 337 [M + H]+.
Synthesis of 6-(4-Piperidyl)-3H-1,3-benzoxazol-2-one; Hydrochloric Salt (21e)
Compound 21e was prepared according to general procedure E using 21c (0.193 g, 0.61 mmol). The crude was used in the next step without further purification. UPLC/MS (method A): Rt 0.91 min. MS (ES) C12H14N2O2 requires 218, found 219 [M + H]+.
Synthesis of 5-Fluoro-6-(4-piperidyl)-3H-1,3-benzoxazol-2-one; Hydrochloric Salt (21f)
Compound 21f was prepared according to general procedure E using 21d (0.157 g, 0.47 mmol). The crude used in the next step without further purification. UPLC/MS (method D): Rt 0.37 min. MS (ES) C12H13FN2O2 requires 236, found 237 [M + H]+.
Synthesis of 6-(1-Methyl-4-piperidyl)-3H-1,3-benzoxazol-2-one (21g)
Compound 21g was prepared according to general procedure F using 21e (0.155 g, 0.61 mmol), 37% aqueous solution of formaldehyde (0.03 mL, 1.22 mmol), NaBH(OAc)3 (0.386 g, 1.83 mmol), and AcOH (0.07 mL, 0.073 g, 1.22 mmol) in dry MeCN (6 mL). The crude was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ 7.19–7.15 (m, 1H), 7.03–6.96 (m, 2H), 2.90–2.80 (m, 2H), 2.53–2.40 (m, overlapped with DMSO signal, 1H), 2.18 (s, 3H), 1.95 (td, J = 11.5, 2.7 Hz, 2H), 1.76–1.57 (m, 4H). UPLC/MS (method A): Rt 0.91 min. MS (ES) C13H16N2O2 requires 232, found 233 [M + H]+.
Synthesis of 6-(1-Ethyl-4-piperidyl)-3H-1,3-benzoxazol-2-one (21h)
Compound 21h was prepared according to general procedure F using 21e (0.254 g, 1.00 mmol), acetaldehyde (0.21 mL, 1.05 mmol, 5 M in THF), NaBH(OAc)3 (0.318 g, 1.6 mmol), and AcOH (0.150 g, 2.5 mmol) in dry THF (10 mL). The crude was purified by SCX to afford 21h as a yellow powder (0.245 g, quant.). 1H NMR (400 MHz, DMSO-d6) δ 6.82 (s, 1H), 6.80 (d, J = 6.0 Hz, 1H), 6.59 (dd, J = 8.5, 2.3 Hz, 1H), 3.04–2.97 (m, 4H), 2.52–2.43 (m, overlapped with DMSO signal, 5H), 2.35 (q, J = 7.2 Hz, 2H), 1.02 (t, J = 7.2 Hz, 3H). UPLC/MS (method A): Rt 0.98 min. MS (ES) C14H18N2O2 requires 246, found 247[M + H]+.
Synthesis of 6-(1-Isopropyl-4-piperidyl)-3H-1,3-benzoxazol-2-one (21i)
Compound 21i was prepared according to general procedure F using 21e (0.254 g, 1.00 mmol), NaBH(OAc)3 (0.318 g, 1.6 mmol), and AcOH (0.29 mL, 0.30 g, 5.0 mmol) in acetone (10 mL). The residue was purified by column chromatography (DCM/MeOH, 80:20) to afford 21i as a pink powder (0.104 g, 40%). UPLC/MS (method A): Rt 0.99 min. MS (ES) C15H20N2O2 requires 261, found 262 [M + H]+.
Synthesis of 6-(1-Isobutyl-4-piperidyl)-3H-1,3-benzoxazol-2-one (21j)
Compound 21j was prepared according to general procedure F using 21e (0.254 g, 1.00 mmol), NaBH(OAc)3 (0.318 g, 1.5 mmol), AcOH (0.29 mL, 0.30 g, 5.0 mmol), and isobutyraldehyde (0.36 g, 5.0 mmol) in dry MeCN (10 mL). The crude was purified by SCX to afford 21j as a white solid (0.236 g, 86%). 1H NMR (400 MHz, DMSO-d6) δ 7.16–7.13 (m, 1H), 7.02–6.93 (m, 2H), 2.95–2.85 (m, 2H), 2.53–2.43 (m, overlapped with DMSO signal, 1H), 2.05 (d, J = 7.4 Hz, 2H), 1.93 (td, J = 11.6, 2.5 Hz, 2H), 1.84–1.69 (m, 3H), 1.69–1.56 (m, 2H), 0.86 (d, J = 6.5 Hz, 6H). UPLC/MS (method A): Rt 1.23 min. MS (ES) C16H22N2O2 requires 274, found 275 [M + H]+.
Synthesis of 5-Fluoro-6-(1-methyl-4-piperidyl)-3H-1,3-benzoxazol-2-one (21k)
Compound 21k was prepared according to general procedure F using 21f (0.123 g, 0.47 mmol) 37% aqueous solution of formaldehyde (0.03 mL, 0.94 mmol), NaBH(OAc)3 (0.386 g, 1.83 mmol), and AcOH (0.054 mL, 0.056 g, 0.94 mmol) in dry MeCN (5 mL). The crude was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ 7.24 (d, J = 6.03 Hz, 1H), 6.94 (d, J = 9.7 Hz, 1H), 2.93–2.84 (m, 2H), 2.73 (ddd, J = 15.5, 10.1, 4.4 Hz, 1H), 2.22 (s, 3H), 2.02 (td, J = 11.3, 3.1 Hz, 2H), 1.79–1.60 (m, 4H). UPLC/MS (method A): Rt 0.99 min. MS (ES) C13H15FN2O2 requires 250, found 251 [M + H]+.
Synthesis of 6-Cyclohexyl-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (22a)
Compound 22a was prepared following general procedure D (method A) using 21a (0.169 g, 0.78 mmol) and 4-phenylbutyl isocyanate (0.15 mL, 0.86 mmol) in dry pyridine (8 mL). The crude was purified by column chromatography (Cy/EtOAc, 80:20) to afford 22a as a white solid (0.300 g, 98%). 1H NMR (400 MHz, CDCl3) δ 8.04 (t, J = 5.4 Hz, 1H), 7.93 (d, J = 8.2 Hz, 1H), 7.32–7.24 (m, overlapped with CDCl3 signal, 2H), 7.22–7.14 (m, 3H), 7.13–7.06 (m, 2H), 3.44 (q, J = 6.8 Hz, 2H), 2.67 (t, J = 7.2 Hz, 2H), 2.61–2.48 (m, 1H), 1.93–1.80 (m, 4H), 1.80–1.59 (m, 5H), 1.41 (q, J = 10.9, 9.8 Hz, 4H), 1.32–1.19 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 153.60, 150.06, 145.57, 142.06, 128.56, 128.52, 126.01, 125.90, 123.62, 115.31, 108.28, 44.64, 40.21, 35.60, 34.78, 29.22, 28.71, 26.91, 26.16. UPLC/MS (method A): Rt 2.45 min. MS (ES) C24H28N2O3 requires 392, found 393 [M + H]+. HRMS C24H29N2O3 [M + H]+: calculated 393.2178, measured: 393.218, Δppm 0.5.
Synthesis of 2-Oxo-N-(4-phenylbutyl)-6-tetrahydropyran-4-yl-1,3-benzoxazole-3-carboxamide (22b)
Compound 22b was prepared according to general procedure D (method A) using 21b (0.080 g, 0.37 mmol) and 4-phenylbutyl isocyanate (0.07 mL, 0.072 g, 0.41 mmol) in dry MeCN (2 mL). The crude was purified by column chromatography (Cy/EtOAc, 90:10) to afford 22b as a white solid (0.075 g, 51%). 1H NMR (400 MHz, CDCl3) δ 8.03 (t, J = 5.4 Hz, 1H), 7.97 (d, J = 8.2 Hz, 1H), 7.35–7.25 (m, overlapped with H2O signal, 2H), 7.25–7.17 (m, 3H), 7.17–7.10 (m, 2H), 4.20–4.05 (m, 2H), 3.61–3.51 (m, 2H), 3.44 (q, J = 6.7 Hz, 2H), 2.88–2.76 (m, 1H), 2.67 (t, J = 7.2 Hz, 2H), 1.88–1.68 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 153.45, 149.96, 143.24, 142.14, 142.03, 128.53, 128.50, 126.01, 123.52, 115.56, 108.24, 68.33, 41.61, 40.23, 35.58, 34.20, 29.19, 28.68. UPLC/MS (method A): Rt 2.83 min. MS (ES) C23H26N2O4 requires 394, found 218 [M-H-CONH(CH2)4Ph]−. HRMS C23H27N2O4 [M + H]+: calculated 395.1955, measured: 395.1971, Δppm −4.0.
Synthesis of tert-Butyl 4-[2-Oxo-3-(4-phenylbutylcarbamoyl)-1,3-benzoxazol-6-yl]piperidine-1-carboxylate (22c)
Compound 22c was prepared according to general procedure D (method A) using 21c (0.087 g, 0.27 mmol), DMAP (0.037 g, 0.30 mmol), and 4-phenylbutyl isocyanate (0.05 mL, 0.053, 0.30 mmol) in dry MeCN (3 mL). The crude was purified by column chromatography (DCM/MeOH, 90:10) to afford 22c as a white solid (0.112 g, 84%). 1H NMR (400 MHz, CDCl3) δ 8.02 (t, J = 5.6 Hz, 1H), 7.96 (d, J = 8.2 Hz, 1H), 7.31–7.24 (m, overlapped signals with CDCl3, 2H), 7.24–7.14 (m, 3H), 7.14–7.05 (m, 2H), 4.39–4.17 (m, 2H), 3.44 (q, J = 6.7 Hz, 2H), 2.80 (t, J = 12.3 Hz, 2H), 2.74–2.62 (m, 3H), 1.89–1.78 (m, 2H), 1.78–1.54 (m, overlapped with H2O signal, 6H), 1.48 (s, 9H). UPLC/MS (method B): Rt 2.42 min. MS (ES) C28H35N3O5 requires 493, found 494 [M + H]+.
Synthesis of 2-Oxo-N-(4-phenylbutyl)-6-(4-piperidyl)-1,3-benzoxazole-3-carboxamide Hydrochloride (22d)
Compound 22d was prepared according to general procedure E using 22c (0.112 g, 0.23 mmol). The crude was triturated with Et2O to afford 22d as a white solid (0.085 g, 86%). 1H NMR (400 MHz, DMSO-d6) δ 9.00 (bs, 2H), 8.11 (t, J = 5.8 Hz, 1H), 7.83 (d, J = 8.3 Hz, 1H), 7.32–7.23 (m, 3H), 7.23–7.08 (m, 4H), 3.40–3.28 (m, overlapped with H2O signal, 3H), 3.05–2.82 (m, 3H), 2.61 (t, J = 7.2 Hz, 2H), 2.02–1.77 (m, 4H), 1.68–1.49 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 152.25, 149.26, 142.00, 141.70, 141.21, 128.27, 128.21, 126.64, 125.65, 122.52, 114.48, 108.08, 43.36, 38.67, 34.73, 29.38, 28.59, 28.12. UPLC/MS (method A): Rt 2.40 min. MS (ES) C23H27N3O3 requires 393, found 394 [M + H]+. HRMS C23H28N3O3 [M + H]+: calculated 394.213, measured: 394.2131, Δppm −0.3.
Synthesis of 6-(1-Methyl-4-piperidyl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (22e)
Compound 22e was prepared according to general procedure D using 21g (0.142 g, 0.61 mmol) and 4-phenylbutyl isocyanate (0.11 mL, 0.118 g, 0.67 mmol) in dry MeCN (3 mL). The crude was purified by column chromatography (DCM/MeOH, 70:30) to afford 22e as a white solid (0.181 g, 73% over three steps). 1H NMR (400 MHz, DMSO-d6) δ 8.10 (t, J = 5.8 Hz, 1H), 7.78 (d, J = 8.2 Hz, 1H), 7.32 (d, J = 1.5 Hz, 1H), 7.30–7.24 (m, 2H), 7.23–7.13 (m, 4H), 3.38–3.25 (m, overlapped with H2O signal, 2H), 2.85 (d, J = 11.4 Hz, 2H), 2.61 (t, J = 7.3 Hz, 2H), 2.54–2.46 (m, overlapped with DMSO signal, 1H), 2.19 (s, 3H), 1.95 (dd, J = 11.4 , 2.8 Hz, 2H), 1.77–1.49 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 154.40, 149.98, 143.59, 142.05, 140.24, 128.55, 128.51, 126.30, 126.01, 123.69, 115.50, 108.32, 56.28, 46.43, 42.04, 40.23, 35.60, 33.65, 29.21, 28.69. UPLC/MS (method A): Rt 2.21 min. MS (ES) C24H29N3O3 requires 407, found 408 [M + H]+. HRMS C24H30N3O3 [M + H]+: calculated 408.2287, measured: 408.2291, Δppm 1.0.
Synthesis of 6-(1-Ethyl-4-piperidyl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (22f)
Compound 22f was prepared according to general procedure D (method A) using 21h (0.100 g, 0.41 mmol) and 4-phenylbutyl isocyanate (0.08 mL, 0.079 g, 0.45 mmol). The crude was purified by column chromatography (DCM/MeOH, 70:30) to afford 22f as a yellow solid (0.105 g, 61%). 1H NMR (400 MHz, CDCl3) δ 8.03 (t, J = 5.7 Hz, 1H), 7.95 (d, J = 8.9 Hz, 1H), 7.32–7.23 (m, overlapped with CDCl3 signal, 2H), 7.21–7.15 (m, 3H), 7.15–7.10 (m, 2H), 3.44 (q, J = 6.6 Hz, 2H), 3.09 (d, J = 12.0 Hz, 2H), 2.67 (t, J = 7.2 Hz, 2H), 2.61–2.51 (m, 1H), 2.47 (q, J = 7.2 Hz, 2H), 2.04 (td, J = 11.7, 2.9 Hz, 2H), 1.92–1.76 (m, 4H), 1.76–1.60 (m, 4H), 1.13 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 153.49, 149.97, 143.81, 142.09, 142.04, 128.54 (2C), 128.50, 126.00, 123.69, 115.44, 108.30, 53.89, 52.74, 42.81, 40.21, 35.59, 33.73, 29.20, 28.69, 12.25. UPLC/MS (method A): Rt 2.22 min, MS (ES) C25H31N3O3 requires 421, found 422 [M + H]+, 245 [M–CONH(CH2)4Ph)]−. HRMS C25H32N3O3 [M + H]+: calculated 422.2444, measured 422.2449, Δppm 1.2.
Synthesis of 6-(1-Isopropyl-4-piperidyl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (22g)
Compound 22g was prepared according to general procedure D (method A) using 21i (0.100 g, 0.38 mmol) and 4-phenylbutyl isocyanate (0.074 g, 0.42 mmol) in dry MeCN (2 mL). The crude was purified by column chromatography (DCM/MeOH, 70:30) to afford 22g as a white solid (0.113 g, 68%). 1H NMR (400 MHz, CDCl3) δ 8.03 (t, J = 5.7 Hz, 1H), 7.94 (d, J = 8.84 Hz, 1H), 7.31–7.24 (m, overlapped with CDCl3 signal, 2H), 7.21–7.15 (m, 3H), 7.15–7.10 (m, 2H), 3.44 (q, J = 6.59 Hz, 2H), 3.09–2.96 (m, 2H), 2.85–2.72 (m, 1H), 2.67 (t, J = 7.2 Hz, 2H), 2.53 (tt, J = 12.1, 4.0 Hz, 1H), 2.32–2.20 (m, 2H), 1.92–1.61 (m, 8H), 1.09 (d, J = 6.5 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 153.50, 149.98, 144.01, 142.08, 128.53 (2C), 128.50, 126.15, 126.00, 123.70, 115.40, 108.32, 54.85, 49.44, 43.11, 40.21, 35.59, 34.12, 29.20, 28.69, 18.55. UPLC/MS (method A): Rt 2.25 min, MS (ES) C26H33N3O3 requires 435, found 436 [M + H]+. HRMS C26H34N3O3 [M + H]+: calculated 436.2603, measured 436.26, Δppm 0.6.
Synthesis of 6-(1-Isobutyl-4-piperidyl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (22h)
Compound 22h was prepared according to general procedure D (method A) using 21j (0.10 g, 0.36 mmol) and 4-phenylbutyl isocyanate (0.07 mL, 0.07 g, 0.4 mmol). The residue was purified by column chromatography (Cy/EtOAc, 75:25) to afford 22h as a white powder (0.115 g, 72%). 1H NMR (400 MHz, CDCl3) δ 8.04 (t, J = 5.7 Hz, 1H), 7.94 (d, J = 8.86 Hz, 1H), 7.32–7.24 (m, overlapped with CDCl3 signal, 2H), 7.22–7.15 (m, 3H), 7.15–7.07 (m, 2H), 3.44 (q, J = 6.5 Hz, 2H), 3.08–2.92 (m, 2H), 2.67 (t, J = 7.2 Hz, 2H), 2.60–2.46 (m, 1H), 2.19–2.07 (m, 2H), 2.07–1.93 (m, 2H), 1.90–1.50 (m, 9H), 0.92 (d, J = 6.5 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 153.54, 150.01, 142.66, 142.09, 128.55 (2C), 128.52, 126.01, 123.70, 115.40, 108.35, 67.38, 54.75, 42.87, 40.23, 35.60, 33.83, 29.22, 28.71, 25.79, 21.21 (2C). UPLC/MS (method A): Rt 2.43 min, MS (ES) C27H35N3O3 requires 449, found 450 [M + H]+. HRMS C27H36N3O3 [M + H]+: calculated 450.2756, measured 450.2757, Δppm −0.2.
Synthesis of 6-(1-Acetyl-4-piperidyl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (22i)
To a solution of 22d (0.184 g, 0.47 mmol, 1.0 equiv.) in dry THF (5 mL) was added Et3N (0.10 g, 0.98 mmol, 2.0 equiv.) dropwise at 0 °C followed by the addition of AcCl (0.039 g, 0.49 mmol, 1.05 equiv.). The reaction mixture was stirred at rt for 4 h, then diluted with EtOAc, washed with saturated aqueous NH4Cl solution and brine, and dried over NaSO4. After evaporation of the solvent, the crude was purified by column chromatography (DCM/MeOH, 90:10) to afford 22i as a white powder (0.181 g, 89%). 1H NMR (400 MHz, CDCl3) δ 8.02 (t, J = 5.6 Hz, 1H), 7.97 (d, J = 8.2 Hz, 1H), 7.31–7.23 (m, overlapped with CDCl3 signal, 2H), 7.21–7.14 (m, 3H), 7.12–7.05 (m, 2H), 4.90–4.70 (m, 1H), 4.05–3.84 (m, 1H), 3.43 (q, J = 6.6 Hz, 2H), 3.28–3.07 (m, 1H), 2.79 (tt, J = 12.1, 3.6 Hz, 1H), 2.67 (t, J = 7.2 Hz, 2H), 2.65–2.55 (m,1H), 2.13 (s, 3H), 1.99–1.83 (m, 2H), 1.80–1.49 (m, overlapped with H2O signal, 6H). 13C NMR (101 MHz, CDCl3) δ 169.02, 149.90, 144.12, 142.54, 142.02, 128.53 (2C), 128.45, 126.56, 126.02, 123.53, 115.66, 108.24, 47.01, 42.78, 42.17, 40.24, 35.58, 29.19, 28.69, 21.64. UPLC/MS (method A): Rt 2.48 min, MS (ES) C25H29N3O4 requires 435, found 436 [M + H]+. HRMS C25H30N3O4 [M + H]+: calculated 436.2244, measured 436.2236, Δppm 1.8.
Synthesis of N-(2-Benzyloxyethyl)-6-(1-methyl-4-piperidyl)-2-oxo-1,3-benzoxazole-3-carboxamide (22j)
Compound 22j was prepared according to general procedure D (method C) using 21g (0.080 g, 0.34 mmol) and 2-(benzyloxy)-1-ethanamine (0.056 g, 0.37 mmol) in dry MeCN (3 mL). The crude was purified by column chromatography (DCM/MeOH, 94:6) to afford 22j as a white solid (0.033 g, 24%). 1H NMR (400 MHz, CDCl3) δ 8.32 (bs, 1H), 7.93 (d, J = 8.8 Hz, 1H), 7.40–7.30 (m, 4H), 7.29–7.23 (m, overlapped with CDCl3 signal, 1H), 7.16–7.08 (m, 2H), 4.57 (s, 2H), 3.71–3.60 (m, 4H), 3.06–2.94 (m, 2H), 2.59–2.48 (m, 1H), 2.35 (s, 3H), 2.09 (td, J = 11.4, 3.5 Hz, 2H), 1.92–1.74 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 153.27, 150.07, 143.59, 128.61 (2C), 127.93 (2C), 127.90, 126.26, 123.63, 115.42, 108.33, 73.39, 68.32, 56.29 46.46, 42.05, 40.32, 33.68. UPLC/MS (method A): Rt 1.92 min, MS (ES) C23H27N3O4 requires 409, found 410 [M + H]+. HRMS C23H28N3O4 [M + H]+: calculated 410.208, measured 410.2087, Δppm 1.7.
Synthesis of 6-(1-Methyl-4-piperidyl)-2-oxo-N-pentyl-1,3-benzoxazole-3-carboxamide (22k)
Compound 22k was prepared according to general procedure D (method A) using 21g (0.050 g, 0.22 mmol) and pentyl isocyanate (0.031 mL, 0.027 g, 0.24 mmol) in dry MeCN (1 mL). The crude was purified by column chromatography (DCM/MeOH, 95:5) to afford 22k as a white solid (0.039 g, 51%). 1H NMR (400 MHz, CDCl3) δ 8.02 (t, J = 5.8 Hz, 1H), 7.95 (d, J = 8.2 Hz, 1H), 7.16–7.07 (m, 2H), 3.41 (q, J = 7.0 Hz, 2H), 3.08–2.92 (m, 2H), 2.62–2.45 (m, 1H), 2.35 (s, 3H), 2.09 (td, J = 11.3, 3.8 Hz, 2H), 1.91–1.74 (m, 4H), 1.69–1.57 (m, 2H), 1.44–1.29 (m, 4H), 0.98–0.85 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 153.50, 149.96, 143.55, 126.32, 123.68, 115.50, 108.30, 56.28, 46.44, 42.03, 40.40, 33.66, 29.27, 29.11, 22.45, 14.09. UPLC/MS (method A): Rt 2.00 min, MS (ES) C19H27N3O3 requires 345, found 346 [M + H]+. HRMS C19H28N3O3 [M + H]+: calculated 346.2131, measured 346.2116, Δppm −4.3.
Synthesis of N-(2-Ethoxyethyl)-6-(1-methyl-4-piperidyl)-2-oxo-1,3-benzoxazole-3-carboxamide (22l)
Compound 22l was prepared according to general procedure D (method C) using 21g (0.050 g, 0.22 mmol) and 2-ethoxyethylamine (0.021 g, 0.24 mmol) in dry MeCN (2 mL). The crude was purified by column chromatography (DCM/MeOH, 92:8) to afford 22l as a white solid (0.030 g, 39%). 1H NMR (600 MHz, CDCl3) δ 8.29 (bs, 1H), 7.94 (d, J = 9.0 Hz, 1H), 7.14–7.08 (m, 2H), 3.63–3.59 (m, 4H), 3.54 (q, J = 7.0 Hz, 2H), 3.04–2.94 (m, 2H), 2.59–2.47 (m, 1H), 2.33 (s, 3H), 2.07 (td, J = 11.5, 3.3 Hz, 2H), 1.90–1.72 (m, 4H), 1.23 (t, J = 7.0 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 153.26, 150.06, 143.10, 142.13, 126.36, 123.63, 115.46, 108.35, 68.64, 66.77, 56.11, 46.13, 41.78, 40.33, 33.24, 15.23. UPLC/MS (method A): Rt 1.57 min, MS (ES) C18H25N3O4 requires 347, found 348 [M + H]+. HRMS C18H26N3O4 [M + H]+: calculated 348.1923, measured 348.1921, Δppm 0.6.
Synthesis of N-Isobutyl-6-(1-methyl-4-piperidyl)-2-oxo-1,3-benzoxazole-3-carboxamide (22m)
Compound 22m was prepared according to general procedure D (method B) using 21g (0.404 g, 1.74 mmol) and isobutylamine (0.382 g, 5.22 mmol) in dry DCM (20 mL). The crude was purified by column chromatography (DCM/MeOH, 92:8) to afford 22m as a white solid (0.259 g, 45%). 1H NMR (400 MHz, CDCl3) δ 8.08 (t, J = 5.9 Hz, 1H), 7.94 (d, J = 8.9 Hz, 1H), 7.16–7.05 (m, 2H), 3.24 (t, J = 6.4 Hz, 2H), 3.05–2.93 (m, 2H), 2.59–2.45 (m, 1H), 2.33 (s, 3H), 2.07 (td, J = 11.4, 3.4 Hz, 2H), 1.97–1.86 (m, 1H), 1.86–1.72 (m, 4H), 0.96 (d, J = 6.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 153.51, 150.06, 143.58, 142.08, 126.29, 123.65, 115.47, 108.27, 56.27, 47.66, 46.45, 42.03, 33.68, 28.58, 20.14. UPLC/MS (method A): Rt 1.82 min, MS (ES) C18H25N3O3 requires 331, found 332 [M + H]+. HRMS C18H26N3O3 [M + H]+: calculated 332.1974, measured 332.1969, Δppm −1.5.
Synthesis of 6-(1-Methyl-4-piperidyl)-2-oxo-N-sec-butyl-1,3-benzoxazole-3-carboxamide (22n)
Compound 22n was prepared according to general procedure D (method C) using 21g (0.08 g, 0.34 mmol) and sec-butylamine (0.027 g, 0.37 mmol) in dry MeCN (1 mL). The crude was purified by column chromatography (DCM/MeOH, 95:5) to afford 22n as a white solid (0.029 g, 26%). 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.0 Hz, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.14–7.08 (m, 2H), 4.07–3.83 (m, 1H), 3.07–2.94 (m, 2H), 2.60–2.46 (m, 1H), 2.35 (s, 3H), 2.09 (td, J = 11.4, 3.3 Hz, 2H), 1.89–1.75 (m, 4H), 1.61 (p, J = 7.3, 6.9 Hz, 2H), 1.26 (d, J = 6.6 Hz, 3H), 0.97 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 159.92, 154.18, 149.39, 143.50, 126.36, 123.65, 115.51, 108.27, 56.28, 48.18, 46.45, 42.02, 33.67, 29.63, 20.49, 10.41. UPLC/MS (method A): Rt 1.83 min. MS (ES) C18H25N3O3 requires 331, found 332 [M + H]+. HRMS C18H26N3O3 [M + H]+: calculated 332.1974, measured: 332.1967, Δppm −2.1.
Synthesis of N-[4-(4-Fluorophenyl)butyl]-6-(1-methyl-4-piperidyl)-2-oxo-1,3-benzoxazole-3-carboxamide (22o)
Compound 22o was prepared according to general procedure D (method C) using 21g (0.085 g, 0.51 mmol) and 4-fluorobenzenebutanamine (0.085 g, 0.51 mmol) in dry MeCN (2 mL). The crude was purified by column chromatography (DCM/MeOH, 87:13) to afford 22o as a white solid (0.042 g, 29%). 1H NMR (600 MHz, CDCl3) δ 8.03 (t, J = 5.5 Hz, 1H), 7.94 (d, J = 8.2 Hz, 1H), 7.19–7.06 (m, 4H), 7.01–6.89 (m, 2H), 3.43 (q, J = 6.5 Hz, 2H), 3.06–2.95 (m, 2H), 2.63 (t, J = 7.0 Hz, 2H), 2.59–2.47 (m, 1H), 2.34 (s, 3H), 2.08 (td, J = 11.4, 3.2 Hz, 2H), 1.90–1.76 (m, 4H), 1.74–1.56 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 161.41 (d, JC–F = 243.5 Hz), 153.45, 149.96, 143.29, 142.08, 137.61 (d, JC–F = 3.2 Hz), 129.82 (d, JC–F = 7.7 Hz), 126.30, 123.70, 115.48, 115.22 (d, JC–F = 21.0 Hz), 108.33, 56.14, 46.21, 41.84, 40.14, 34.75, 33.36, 29.10, 28.81. UPLC/MS (method A): Rt 2.20 min. MS (ES) C24H28FN3O3 requires 425, found 426 [M + H]+. HRMS C24H29FN3O3 [M + H]+: calculated 426.2193, measured: 426.2188, Δppm −1.2.
Synthesis of 5-Fluoro-6-(1-methyl-4-piperidyl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (22p)
Compound 22p was prepared according to general procedure D (method A) using 21k (0.117 g, 0.47 mmol) and 4-phenylbutyl isocyanate (0.088 mL, 0.091 g, 0.52 mmol) in dry MeCN (5 mL). The crude was purified by column chromatography (DCM/MeOH, 92:8) to afford 22p as a white solid (0.099 g, 50% over three steps). 1H NMR (600 MHz, CDCl3) δ 7.98 (t, J = 5.3 Hz, 1H), 7.77 (d, J = 9.9 Hz, 1H), 7.31–7.23 (m, overlapped with CDCl3 signal, 2H), 7.22–7.14 (m, 3H), 7.11 (d, J = 5.8 Hz, 1H), 3.43 (q, J = 6.6 Hz, 2H), 3.05–2.97 (m, 2H), 2.95–2.84 (m, 1H), 2.66 (t, J = 7.4 Hz, 2H), 2.35 (s, 3H), 2.17 (td, J = 11.5, 2.9 Hz, 2H), 1.89–1.78 (m, 4H), 1.75–1.58 (m, overlapped with H2O signal, 4H). 13C NMR (151 MHz, CDCl3) δ 157.38 (d, JC–F = 241.2 Hz), 153.43, 149.58, 141.98, 138.08 (d, JC–F = 2.2 Hz), 129.37 (d, J = 17.7 Hz), 128.53, 128.51, 126.42 (d, J = 14.5 Hz), 126.02, 108.44 (d, JC–F = 5.6 Hz), 103.97 (d, JC–F = 33.4 Hz), 56.08, 46.27, 40.26, 35.56, 34.29, 32.05, 29.13, 28.66. UPLC/MS (method A): Rt 2.20 min. MS (ES) C24H28FN3O3 requires 425, found 426 [M + H]+. HRMS C24H29FN3O3 [M + H]+: calculated 426.2193, measured: 426.2191, Δppm −0.5.
Synthesis of (6-Oxo-2,3-dihydro-1H-pyridin-4-yl) trifluoromethanesulfonate (29a)
To a solution of 28 (0.400 g, 3.54 mmol, 1.0 equiv.) in dry THF (30 mL) were added at 0 °C under stirring Et3N (0.716 g, 7.08 mmol, 2.0 equiv.) and N,N-bis(trifluoromethylsulfonyl)aniline (1.388 g, 4.24 mmol, 1.2 equiv.) dissolved in dry THF (6 mL). The reaction mixture was slowly warmed to rt and stirred for 16 h. The mixture was diluted with EtOAc, washed with a saturated aqueous NH4Cl solution, and dried over Na2SO4. After evaporation of the solvent, the crude was purified by column chromatography (DCM/MeOH, 94:6) to afford 29a as a white solid (0.640 g, 74%). 1H NMR (400 MHz, DMSO-d6) δ 7.79 (s, 1H), 5.95–5.93 (m, 1H), 3.37 (td, J = 7.1, 2.7 Hz, 2H), 2.72 (td, J = 7.2, 1.4 Hz, 2H). UPLC/MS (method A): Rt 1.47 min, MS (ES) C6H6F3NO4S requires 245, found 246 [M + H]+.
Synthesis of 4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3-dihydro-1H-pyridin-6-one (29b)
To a stirred solution of compound 29a (0.100 g, 0.41 mmol, 1.0 equiv.) in degassed dioxane (4 mL) were added ([B2(pin)2]) (0.124 g, 0.49 mmol, 1.2 equiv.), Pd(dppf)Cl2 (0.058 g, 0.08 mmol, 0.2 equiv.), and KOAc (0.080 g, 0.82 mmol, 2.0 equiv.). The reaction mixture was stirred at 70 °C for 90 min, then cooled to rt, and used directly in the next step. UPLC/MS (method A): Rt 0.44 min, MS (ES) C11H18BNO3 requires 223, found 141 [M–(CH3)2CC(CH3)2]+.
Synthesis of 4-(3-Benzyloxy-4-nitrophenyl)-2,5-dihydro-1H-pyridin-6-ne (30a)
Compound 30a was prepared according to general procedure A using 29b (0.091 g, 0.41 mmol), 2-benzyloxy-4-bromo-1-nitrobenzene (0.138 g, 0.45 mmol), Pd(PPh3)4 (0.023 g, 0.02 mmol), and 2 M Na2CO3 (0.51 mL, 1.025 mmol) in degassed 1,4-dioxane (10 mL). The crude was purified by column chromatography (DCM/MeOH, 90:10) to afford 30a as a brown powder (0.124 g, 93% over two steps). 1H NMR (400 MHz, DMSO-d6) δ 7.93 (d, J = 8.5 Hz, 1H), 7.61 (d, J = 1.7 Hz, 1H), 7.60 (bs, 1H), 7.50–7.47 (m, 2H), 7.46–7.40 (m, 2H), 7.39–7.33 (m, 2H), 6.34 (q, J = 1.5 Hz, 1H), 5.42 (s, 2H), 3.39 (td, J = 7.0, 2.6 Hz, 2H), 2.74 (td, J = 7.0, 1.5 Hz, 2H). UPLC/MS (method A): Rt 1.80 min, MS (ES) C18H16N2O4 requires 324, found 325 [M + H]+.
Synthesis of 4-(3-Benzyloxy-4-nitrophenyl)-1-methyl-2,3-dihydropyridin-6-one (30b)
To a stirred solution of 30a (0.124 g, 0.38 mmol, 1.0 equiv.) in dry THF (4 mL) was added NaH (0.018 g, 60% in mineral oil, 0.46 mmol, 1.2 equiv.) at 0 °C under stirring. After 30 min, CH3I (0.047 mL, 0.76 mmol, 2.0 equiv.) was added, and the mixture was slowly warmed to rt. After 5 h, saturated aqueous NH4Cl solution was added, and the mixture extracted with EtOAc. The combined organic phases were washed with brine, dried over MgSO4, and concentrated. The crude was purified by column chromatography (DCM/EtOAc, 70:30) to afford 30b as a brown solid (0.058 g, 45%). 1H NMR (400 MHz, DMSO-d6) δ 7.94 (d, J = 8.5 Hz, 1H), 7.63 (d, J = 1.8 Hz, 1H), 7.51–7.46 (m, 2H), 7.46–7.39 (m, 2H), 7.40–7.32 (m, 2H), 6.41 (t, J = 1.4 Hz, 1H), 5.42 (s, 2H), 3.53 (t, J = 7.1 Hz, 2H), 2.92 (s, 3H), 2.83 (td, J = 7.2, 1.4 Hz, 2H). UPLC/MS (method A): Rt 1.89 min, MS (ES) C19H18N2O4 requires 338, found 339 [M + H]+.
Synthesis of 4-(4-Amino-3-hydroxyphenyl)-1-methyl-piperidin-2-one (31)
Compound 31 was prepared according to general procedure B (method B) using 30b (0.056 g, 0.16 mmol). UPLC/MS (method A): Rt 1.05 min, MS (ES) C12H16N2O2 requires 220, found 221 [M + H]+.
Synthesis of 6-(1-Methyl-2-oxo-4-piperidyl)-3H-1,3-benzoxazol-2-one (21l)
Compound 21l was prepared according to general procedure C using 31 (0.035 g, 0.16 mmol) and CDI (0.039 g, 0.24 mmol) in dry MeCN (2 mL). The crude was purified by column chromatography (DCM/MeOH, 95:5) to afford 21l as a white powder (0.028 g, 70% over two steps). 1H NMR (400 MHz, DMSO-d6) δ 7.26–7.23 (m, 1H), 7.08–6.96 (m, 2H), 3.45–3.23 (m, 2H), 3.14–3.03 (m, 1H), 2.85 (s, 3H), 2.49–2.28 (m, 2H), 2.06–1.76 (m, 2H). UPLC/MS (method A): Rt 1.17 min, MS (ES) C13H14N2O3 requires 246, found 247 [M + H]+.
Synthesis of 6-(1-Methyl-2-oxo-4-piperidyl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (22q)
Compound 22q was prepared according to general procedure D (method A) using 21l (0.025 g, 0.10 mmol) and 4-phenylbutyl isocyanate (0.019 mL, 0.019 g, 0.11 mmol) in dry MeCN (1 mL). The crude was purified by column chromatography (DCM/EtOAc, 70:30) to afford 22q as a white solid (0.038 g, 90%). 1H NMR (400 MHz, CDCl3) δ 8.07–7.95 (m, 2H), 7.33–7.23 (m, overlapped with CDCl3 signal, 2H), 7.22–7.13 (m, 3H), 7.13–7.05 (m, 2H), 3.51–3.38 (m, 3H), 3.38–3.28 (m, 1H), 3.21–3.08 (m, 1H), 2.99 (s, 3H), 2.79–2.68 (m, 1H), 2.67 (t, J = 7.2 Hz, 2H), 2.46 (dd, J = 17.3, 11.1 Hz, 1H), 2.19–2.08 (m, 1H), 2.07–1.91 (m, 1H), 1.80–1.67 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 168.96, 149.85, 142.26, 142.01, 140.73, 128.53 (2C), 126.87, 126.02, 123.31, 115.86, 108.10, 100.13, 49.07, 40.27, 39.61, 38.89, 35.59, 34.65, 30.45, 29.19, 28.68. UPLC/MS (method A): Rt 2.09 min, MS (ES) C24H27N3O4 requires 421, found 422 [M + H]+. HRMS C24H28N3O4 [M + H]+: calculated 422.208, measured 422.2074, Δppm −1.4.
Synthesis of 6-[1-(2,2-Difluoroethyl)-4-hydroxy-4-piperidyl]-3H-1,3-benzoxazol-2-one (43)
Compound 43 was prepared according to general procedure I using 6-bromo-3H-1,3-benzoxazol-2-one (0.150 g, 0.7 mmol), 42a (0.171 g, 1.05 mmol), MeMgBr (0.35 mL, 0.125 g, 1.05 mmol, 3 M in Et2O), and n-BuLi (0.336 mL, 0.84 mmol, 2.5 M in hexanes) in dry THF (7 mL). The crude was purified by column chromatography (DCM/MeOH, 98:4) to afford 43 as a white solid (0.063 g, 30%). 1H NMR (400 MHz, DMSO-d6) δ 7.37 (d, J = 1.4 Hz, 1H), 7.26 (dd, J = 8.2, 1.7 Hz, 1H), 7.01 (d, J = 8.1 Hz, 1H), 6.14 (tt, J = 55.9, 4.3 Hz, 1H), 4.88 (bs, 1H), 2.82–2.68 (m, 4H), 2.63 (td, J = 11.6, 2.4 Hz, 2H), 1.93 (td, J = 12.8, 4.7 Hz, 2H), 1.57 (dd, J = 13.8, 2.5 Hz, 2H). UPLC/MS (method A): Rt 1.11 min, MS (ES) C14H16F2N2O3 requires 298, found 299 [M + H]+.
Synthesis of 6-[1-(2,2-Difluoroethyl)-3,6-dihydro-2H-pyridin-4-yl]-3H-1,3-benzoxazol-2-one (44)
Compound 44 was prepared according to general procedure L using 43 (0.033 g, 0.11 mmol). The crude was purified by SCX to afford 44 as a pale brown solid (0.031 g, quant.). 1H NMR (400 MHz, DMSO-d6) δ 11.57 (bs, 1H), 7.38 (d, J = 1.6 Hz, 1H), 7.22 (dd, J = 8.2, 1.7 Hz, 1H), 7.03 (d, J = 8.1 Hz, 1H), 6.18 (tt, J = 55.8, 4.3 Hz, 1H), 6.10 (td, J = 3.5, 1.7 Hz, 1H), 3.27–3.19 (m, 2H), 2.92–2.74 (m, 4H), 2.49–2.41 (m, 2H). UPLC/MS (method A): Rt 1.56 min, MS (ES) C14H14F2N2O2 requires 280, found 281 [M + H]+.
Synthesis of 6-[1-(2,2-Difluoroethyl)-4-piperidyl]-3H-1,3-benzoxazol-2-one (21m)
Compound 21m was prepared according to general procedure B (method B) using 44 (0.028 g, 0.1 mmol). The crude was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ 11.46 (bs, 1H), 7.20 (d, J = 1.4 Hz, 1H), 7.08–6.95 (m, 2H), 6.14 (tt, J = 55.8, 4.4 Hz, 1H), 3.04–2.95 (m, 2H), 2.75 (td, J = 15.7, 4.4 Hz, 2H), 2.26 (td, J = 11.6, 2.8 Hz, 2H), 1.80–1.56 (m, 4H). UPLC/MS (method A): Rt 1.50 min, MS (ES) C14H16F2N2O2 requires 282, found 283 [M + H]+.
Synthesis of 6-[1-(2,2-Difluoroethyl)-4-piperidyl]-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (22r)
Compound 22r was prepared according to general procedure D (method A) using 21m (0.028 g, 0.1 mmol) and 4-phenylbutyl isocyanate (0.019 mL, 0.019 g, 0.11 mmol) in dry MeCN (2.0 mL). The crude was purified by column chromatography (DCM/EtOAc, 90:10) to afford 22r as a white solid (0.033 g, 73% over two steps). 1H NMR (400 MHz, CDCl3) δ 8.02 (t, J = 5.7 Hz, 1H), 7.95 (d, J = 8.3 Hz, 1H), 7.32–7.24 (m, overlapped with CDCl3 signal, 2H), 7.22–7.15 (m, 3H), 7.15–7.08 (m, 2H), 5.92 (tt, J = 56.0, 4.3 Hz, 1H), 3.44 (q, J = 6.6 Hz, 2H), 3.07 (d, J = 11.6 Hz, 2H), 2.79 (td, J = 15.0, 4.3 Hz, 2H), 2.67 (t, J = 7.2 Hz, 2H), 2.61–2.49 (m, 1H), 2.34 (td, J = 11.3, 3.5 Hz, 2H), 1.91–1.78 (m, 4H), 1.78–1.66 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 153.47, 150.61, 149.97, 143.39, 142.04, 128.54 (2C), 128.49, 126.02, 123.62, 115.52 (2C), 108.28, 60.57 (t, JC–F = 24.9 Hz), 55.07, 42.14, 40.24, 35.59, 33.63, 29.20, 28.69. UPLC/MS (method A): Rt 1.82 min, MS (ES) C25H29F2N3O3 requires 457, found 458 [M + H]+. HRMS C25H30F2N3O3 [M + H]+: calculated 458.2255, measured 458.2258, Δppm 0.7.
Synthesis of 2-Nitro-5-(1-piperidyl)phenol (49a)
Compound 49a was prepared according to general procedure G using 5-fluoro-2-nitrophenol (0.150 g, 0.95 mmol), 48a (0.265 g, 1.43 mmol), and DIPEA (0.33 mL, 0.25 g, 1.90 mmol) in dry MeCN (8 mL) under MW irradiation. Saturated aqueous NH4Cl solution was added, and the aqueous phase was extracted with DCM. The organic layers were collected, dried over Na2SO4, and concentrated under reduced pressure. The crude was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 11.31 (s, 1H), 7.93 (d, J = 9.7 Hz, 1H), 6.45 (dd, J = 9.7, 2.7 Hz, 1H), 6.31 (d, J = 2.7 Hz, 1H), 3.47 (d, J = 5.8 Hz, 4H), 1.70 (s, 6H). UPLC/MS (method A): Rt 2.45 min. MS (ES) C11H14N2O3 requires 222, found 223 [M + H]+.
Synthesis of 5-(1,1-Dioxo-1,4-thiazinan-4-yl)-2-nitrophenol (49c)
Compound 49c was prepared according to general procedure G using 5-fluoro-2-nitrophenol (0.470 g, 3.00 mmol), 48c (0.811 g, 6.00 mmol), and DIPEA (1.05 mL, 0.775 g, 6.00 mmol) in MeCN (15 mL), heating at reflux for 16 h. The crude was purified by column chromatography (EtOAc) to afford 49c as a yellow powder (0.33 g, 40%). 1H NMR (400 MHz, DMSO-d6) δ 10.85 (bs, 1H), 7.91 (d, J = 9.6 Hz, 1H), 6.73 (dd, J = 9.6, 2.8 Hz, 1H), 6.59 (d, J = 2.8 Hz, 1H), 4.02–3.94 (m, 4H), 3.20–3.14 (m, 4H). UPLC/MS (method A): Rt 1.49 min. MS (ES) C10H12N2O5S requires 272, found 273 [M + H]+.
Synthesis of tert-Butyl 4-(3-Hydroxy-4-nitrophenyl)piperazine-1-carboxylate (49d)
Compound 49d was prepared according to general procedure G using 5-fluoro-2-nitrophenol (2.00 g, 12.73 mmol), 48d (3.56 g, 19.09 mmol), and DIPEA (2.47 g, 19.1 mmol) in MeCN (25 mL), heating at reflux for 16 h. The crude was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ 10.94 (bs, 1H), 7.88 (d, J = 9.7 Hz, 1H), 6.64 (dd, J = 9.7, 2.7 Hz, 1H), 6.42 (d, J = 2.7 Hz, 1H), 3.56–3.40 (m, 8H), 1.42 (s, 9H). UPLC/MS (method A): Rt 2.36 min, MS (ES) C15H21N3O5 requires 323, found 324 [M + H]+.
Synthesis of 2-Amino-5-(1-piperidyl)phenol (49e)
Compound 49e was prepared according to general procedure B (method A) using 49a (0.130 g, 0.59 mmol). After evaporation of the solvent, the crude was used in the next step without purification. UPLC/MS (method A): Rt 0.94 min. MS (ES) C11H16N2O requires 192, found 193 [M + H]+.
Synthesis of 2-Amino-5-morpholinophenol (49f)
Compound 49f was prepared according to general procedure B (method A) using the commercially available 49b (0.224 g, 1.0 mmol). After evaporation of the solvent, the crude was used in the next step without purification. UPLC/MS (method A): Rt 1.18 min. MS (ES) C10H14N2O2 requires 194, found 195 [M + H]+.
Synthesis of 2-Amino-5-(1,1-dioxo-1,4-thiazinan-4-yl)phenol (49g)
Compound 49g was prepared according to general procedure B (method A) using 49c (0.272 g, 1.00 mmol). After evaporation of the solvent, the crude was used in the next step without purification. UPLC/MS (method A): Rt 0.52 min. MS (ES) C10H14N2O3S requires 242, found 243 [M + H]+.
Synthesis of tert-Butyl 4-(4-Amino-3-hydroxyphenyl)piperazine-1-carboxylate (49h)
Compound 49h was prepared according to general procedure B (method B) using 49d (4.11 g, 12.73 mmol). After evaporation of the solvent, the crude was used in the next step without purification. UPLC/MS (method A): Rt 1.77 min. MS (ES) C15H23N3O3 requires 293, found 294 [M + H]+.
Synthesis of 6-(1-Piperidyl)-3H-1,3-benzoxazol-2-one (50a)
Compound 50a was prepared according to general procedure C using 49e (0.110 g, 0.58 mmol) and CDI (0.141 g, 0.87 mmol) in dry MeCN (6 mL). The pink solid was triturated with Et2O to afford 50a as a pinkish solid (0.165 g, 80% over three steps). 1H NMR (400 MHz, DMSO-d6) δ 11.26 (bs, 1H), 6.93 (d, J = 2.2 Hz, 1H), 6.90 (d, J = 8.5 Hz, 1H), 6.70 (dd, J = 2.3, 8.5 Hz, 1H), 3.08–2.93 (m, 4H), 1.61 (p, J = 5.7 Hz, 4H), 1.50 (p, J = 5.7 Hz, 2H). UPLC/MS (method A): Rt 2.38 min. MS (ES) C12H14N2O2 requires 218, found 219 [M + H]+.
Synthesis of 6-Morpholino-3H-1,3-benzoxazol-2-one (50b)
Compound 50b was prepared according to general procedure C using 49f (0.194 g, 1.00 mmol) and CDI (0.243 g, 1.50 mmol) in dry MeCN (10 mL). The crude was purified by column chromatography (Cy/EtOAc, 70:30) to afford 50b as a pink powder (0.132 g, 60%, over two steps). 1H NMR (400 MHz, DMSO-d6) δ 11.31 (bs, 1H), 6.98 (d, J = 2.3 Hz, 1H), 6.94 (d, J = 8.5 Hz, 1H), 6.72 (dd, J = 8.6, 2.3 Hz, 1H), 3.81–3.65 (m, 4H), 3.10–2.93 (m, 4H). UPLC/MS (method A): Rt 1.32 min. MS (ES) C11H12N2O3 requires 220, found 221 [M + H]+.
Synthesis of 6-(1,1-Dioxo-1,4-thiazinan-4-yl)-3H-1,3-benzoxazol-2-one (50c)
Compound 50c was prepared according to general procedure C using 49g (0.242 g, 1.0 mmol) and CDI (0.162 g, 1.0 mmol) in dry MeCN (10 mL). The crude was purified by column chromatography (Cy/EtOAc 70:30) to afford 50c as a yellow solid (0.120 g, 45% over two steps). 1H NMR (400 MHz, DMSO-d6) δ 6.94 (d, J = 2.4 Hz, 1H), 6.87 (d, J = 8.4 Hz, 1H), 6.72 (dd, J = 8.4, 2.4 Hz, 1H), 3.70–3.65 (m, 4H), 3.17–3.12 (m, 4H). UPLC/MS (method A): Rt 1.15 min. MS (ES) C11H12N2O4S requires 268, found 267 [M-H]−.
Synthesis of tert-Butyl 4-(2-Oxo-3H-1,3-benzoxazol-6-yl)piperazine-1-carboxylate (50d)
Compound 50d was prepared according to general procedure C using 49h (3.73 g, 12.73 mmol) and CDI (3.096 g, 19.09 mmol) in dry MeCN (25 mL). The crude was purified by column chromatography (Cy/EtOAc, 80:20) to afford 50d as a pink powder (3.046 g, 75% over three steps). 1H NMR (400 MHz, DMSO-d6) δ 11.34 (bs, 1H), 7.00 (d, J = 2.2 Hz, 1H), 6.94 (d, J = 8.5 Hz, 1H), 6.74 (dd, J = 8.5, 2.3 Hz, 1H), 3.50–3.40 (m, 4H), 3.05–2.96 (m, 4H), 1.42 (s, 9H). UPLC/MS (method A): Rt 2.14 min, MS (ES) C16H21N3O4 requires 319, found 320 [M + H]+.
Synthesis of 6-Piperazin-1-yl-3H-1,3-benzoxazol-2-one Hydrochloride (50e)
Compound 50e was prepared according to general procedure E using 50d (1.50 g, 4.70 mmol). The reaction mixture was concentrated under reduced pressure to afford 50e as a gray solid (1.198 g, quant.). 1H NMR (400 MHz, DMSO-d6) δ 11.41 (bs, 1H), 8.80 (bs, 2H), 7.05 (d, J = 2.2 Hz, 1H), 6.97 (d, J = 8.5 Hz, 1H), 6.77 (dd, J = 8.5, 2.3 Hz, 1H), 3.31–3.18 (m, 8H). UPLC/MS (method A): Rt 0.55 min. MS (ES) C11H13N3O2 requires 219, found 220 [M + H]+.
Synthesis of 6-(4-Methylpiperazin-1-yl)-3H-1,3-benzoxazol-2-one (50f)
Compound 50f was prepared according to general procedure F using 50e (0.388 g, 1.52 mmol), 37% aqueous solution of formaldehyde (0.17 mL, 6.08 mmol), NaBH(OAc)3 (0.21 g, 1.0 mmol), and AcOH (0.096 mL, 0.101 g, 1.68 mmol) in dry MeCN (5 mL). The crude was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 6.97–6.90 (m, 2H), 6.73–6.69 (m, 1H), 3.09–3.02 (m, 4H), 2.47–2.41 (m, 4H) 2.22 (s, 3H). UPLC/MS (method A): Rt 0.85 min. MS (ES) C12H15N3O2 requires 233, found 234 [M + H]+.
Synthesis of 6-(4-Ethylpiperazin-1-yl)-3H-1,3-benzoxazol-2-one (50g)
Compound 50g was prepared according to general procedure F using 50e (0.171 g, 0.67 mmol), NaBH(OAc)3 (0.21 g, 1.0 mmol), AcOH (0.096 mL, 0.101 g, 1.68 mmol), and acetaldehyde (0.14 mL, 0.70 mmol, 5 M in THF) in dry THF (7 mL). The crude was purified by SCX to afford 50g as a white solid (0.150 g, 90%). UPLC/MS (method A): Rt 0.88 min. MS (ES) C13H17N3O2 requires 247, found 248 [M + H]+.
Synthesis of 6-(4-Isopropylpiperazin-1-yl)-3H-1,3-benzoxazol-2-one (50h)
Compound 50h was prepared according to general procedure F using 50e (0.256 g, 1.00 mmol), NaBH(OAc)3 (0.636 g, 3.0 mmol), and AcOH (0.286 mL, 0.300 g, 5.00 mmol) in acetone (10 mL). The crude was purified by SCX to afford 50h as a white solid (0.158 g, 60%). 1H NMR (400 MHz, DMSO-d6) δ 11.40 (bs, 1H), 7.10–7.03 (m, 1H), 6.97 (d, J = 8.5 Hz, 1H), 6.78 (dd, J = 8.5, 2.2 Hz, 1H), 3.94–2.79 (m, overlapped with H2O signal, 9H), 1.25 (s, 6H). UPLC/MS (method A): Rt 0.99 min, MS (ES) C14H19N3O2 requires 261, found 262 [M + H]+.
Synthesis of 6-(4-Isobutylpiperazin-1-yl)-3H-1,3-benzoxazol-2-one (50i)
Compound 50i was prepared according to general procedure F using 50e (0.256 g, 1.0 mmol), isobutyraldehyde (0.361 g, 5.0 mmol), NaBH(OAc)3 (0.318 g, 1.50 mmol), and AcOH (0.286 mL, 0.300 g, 5.0 mmol) in dry MeCN (10 mL). The crude was purified by SCX to afford 50i as a violet solid (0.220 g, 80%). 1H NMR (400 MHz, DMSO-d6) δ 6.88–6.80 (m, 2H), 6.63 (dd, J = 8.5, 2.3 Hz, 1H), 3.09–2.83 (m, 4H), 2.47–2.39 (m, 4H), 2.07 (d, J = 7.4 Hz, 2H), 1.78 (dt, J = 13.6, 6.8 Hz, 1H), 0.87 (d, J = 6.6 Hz, 6H). UPLC/MS (method A): Rt 1.21 min. MS (ES) C15H21N3O2 requires 275, found 276 [M + H]+.
Synthesis of 2-Oxo-N-(4-phenylbutyl)-6-piperidin-1-ium-1-yl-1,3-benzoxazole-3-carboxamide Hydrochloride (23a)
Compound 23a was prepared according to general procedure D (method A) using 50a (0.055 g, 0.25 mmol) and 4-phenylbutyl isocyanate (0.047 mL, 0.049 g, 0.86 mmol) in a mixture of toluene/DMF (3 mL, 9:1). The crude was purified by column chromatography (Cy) (0.029 g, 30%). The free base of 23a was dissolved in DCM (0.7 mL, 0.1 M) followed by the addition of HCl (0.55 mL, 0.08 g, 2.14 mmol, 4 M in 1,4-dioxane). After evaporation of the solvent, the residue was triturated with Et2O to afford 23a as a white solid (0.026 g, 86%). 1H NMR (400 MHz, DMSO-d6) δ 8.10 (t, J = 5.8 Hz, 1H), 8.06–7.83 (m, 2H), 7.82–7.46 (m, 1H), 7.32–7.24 (m, 2H), 7.23–7.13 (m, 3H), 4.63 (bs, 1H), 3.58–3.40 (m, 4H), 3.34 (q, J = 6.4 Hz, 2H), 2.61 (t, J = 7.2 Hz, 2H), 2.22–1.79 (m 4H), 1.78–1.51 (m, 6H). 13C NMR (151 MHz, CDCl3) δ 152.59, 149.20, 141.91, 141.89, 139.51, 129.29, 128.52, 128.50, 126.05, 118.19, 116.86, 105.34, 58.14, 40.39, 35.52, 29.06, 28.63, 23.14, 21.80. UPLC/MS (method A): Rt 2.33 min. MS (ES) C23H27N3O3 requires 393, found 394 [M + H]+. HRMS C23H28N3O3 [M + H]+: calculated 394.2131, measured: 394.2122, Δppm −2.3.
Synthesis of 6-Morpholino-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (23b)
Compound 23b was prepared according to general procedure D (method A) using 50b (0.130 g, 0.6 mmol) and 4-phenylbutyl isocyanate (0.157 g, 0.9 mmol) in dry MeCN (6 mL). The crude was purified by column chromatography (Cy/EtOAc, 90:10) to afford 23b as a white powder (0.142 g, 60%). 1H NMR (400 MHz, CDCl3) δ 7.99 (t, J = 5.7 Hz, 1H), 7.91 (d, J = 8.6 Hz, 1H), 7.32–7.24 (m, overlapped with CDCl3 signal, 2H), 7.20–7.07 (m, 3H), 6.90–6.78 (m, 2H), 3.95–3.82 (m, 4H), 3.44 (q, J = 6.6 Hz, 2H), 3.20–3.10 (m, 4H), 2.67 (t, J = 7.2 Hz, 2H), 1.78–1.60 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 153.48, 150.00, 142.91, 142.05, 128.54, 128.51, 126.01, 115.97, 112.77, 98.66, 66.76, 50.27, 40.22, 35.60, 29.22, 28.70. UPLC/MS (method A): Rt 2.63 min. MS (ES) C22H25N3O4 requires 395, found 396 [M + H]+. HRMS C22H26N3O4 [M + H]+: calculated 396.1923, measured 396.1925, Δppm 0.5.
Synthesis of 6-(1,1-Dioxo-1,4-thiazinan-4-yl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (23c)
Compound 23c was prepared according to general procedure D (method A) using 50c (0.100 g, 0.37 mmol) and 4-phenylbutyl isocyanate (0.20 g, 1.12 mmol) in dry MeCN (4 mL). The crude was purified by column chromatography (Cy/EtOAc, 80:20) to afford 23c as a white powder (0.050 g, 20%). 1H NMR (400 MHz, DMSO-d6) δ 8.06 (t, J = 5.8 Hz, 1H), 7.71 (d, J = 8.8 Hz, 1H), 7.32–7.24 (m, 2H), 7.24–7.12 (m, 4H), 6.94 (dd, J = 8.9, 2.5 Hz, 1H), 3.87–3.67 (m, 4H), 3.42–3.26 (m, 2H), 3.21–3.06 (m, 4H), 2.61 (t, J = 7.2 Hz, 2H), 1.73–1.43 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 152.82, 149.82, 145.77, 143.21, 142.51, 128.76, 128.70, 126.14, 121.16, 115.39, 112.38, 99.29, 50.27, 47.85, 39.90, 35.22, 29.10, 28.63. UPLC/MS (method A): Rt 2.49 min. MS (ES) C22H25N3O5S requires 443, found 267 [M–CO(CH2)4Ph)]−. HRMS C22H26N3O5S [M + H]+: calculated 444.1593, measured 444.1588, Δppm −1.1.
Synthesis of tert-Butyl 4-[2-Oxo-3-(4-phenylbutylcarbamoyl)-1,3-benzoxazol-6-yl]piperazine-1-carboxylate (23d)
Compound 23d was prepared according to general procedure D (method A) using 50d (0.10 g, 0.31 mmol), 4-phenylbutyl isocyanate (0.060 g, 0.34 mmol), and DMAP (0.042 g, 0.34 mmol) in dry MeCN (3 mL). The crude was purified by column chromatography (DCM/MeOH, 97:3) to afford 23d as a white solid (0.130 g, 85%). UPLC/MS (method B): Rt 2.21 min, MS (ES) C27H34N4O5 requires 494, found 495 [M + H]+.
Synthesis of 2-Oxo-N-(4-phenylbutyl)-6-piperazin-1-yl-1,3-benzoxazole-3-carboxamide Hydrochloride (23e)
Compound 23e was prepared according to general procedure E using 23d (0.120 g, 0.24 mmol). The reaction mixture was concentrated under reduced pressure to afford 23e as a white solid (0.103 g, quant.). 1H NMR (400 MHz, DMSO-d6) δ 9.53 (bs, 2H), 8.06 (t, J = 5.8 Hz, 1H), 7.72 (d, J = 8.8 Hz, 1H), 7.33–7.24 (m, 2H), 7.24–7.11 (m, 4H), 6.90 (dd, J = 8.9, 2.4 Hz, 1H), 3.44–3.35 (m, 4H), 3.32 (q, J = 6.4 Hz, 2H), 3.27–3.15 (m, 4H), 2.62 (t, J = 7.2 Hz, 2H), 1.76–1.49 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 152.35, 149.33, 147.49, 142.50, 142.02, 128.28, 128.22, 125.66, 121.16, 114.71, 112.07, 98.98, 45.99, 42.35, 39.42, 34.74, 28.62, 28.14. UPLC/MS (method A): Rt 2.12 min, MS (ES) C22H26N4O3 requires 394, found 395 [M + H]+. HRMS C22H27N4O3 [M + H]+: calculated 395.2083, measured 395.2086, Δppm 1.4.
Synthesis of 6-(4-Methylpiperazin-1-yl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (23f)
Compound 23f was prepared according to general procedure D (method A) using 50f (0.060 g, 0.26 mmol) and 4-phenylbutyl isocyanate (0.088 mL, 0.090 g, 0.51 mmol) in dry MeCN (3 mL). The crude was purified by column chromatography (DCM/MeOH, 98:2) to afford 23f as a white powder (0.080 g, 75%). 1H NMR (400 MHz, CDCl3) δ 7.99 (t, J = 5.8 Hz, 1H), 7.93–7.85 (m, 1H), 7.33–7.23 (m, overlapped with CDCl3 signal, 2H), 7.22–7.14 (m, 3H), 6.86–6.77 (m, 2H), 3.43 (q, J = 6.4 Hz, 2H), 3.37–3.18 (m, 4H), 2.83–2.70 (m, 4H), 2.67 (t, J = 7.2 Hz, 2H) 2.47 (s, 3H), 1.80–1.61 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 153.52, 150.03, 142.90, 142.06, 128.55, 128.52, 126.01, 115.93, 113.26, 99.01, 54.79, 49.32, 45.66, 40.21, 35.60, 29.23, 28.71. UPLC/MS (method A): Rt 2.23 min. MS (ES) C23H28N4O3 requires 408, found 409 [M + H]+. HRMS C23H29N4O3 [M + H]+: calculated 409.224, measured 409.224, Δppm 0.0.
Synthesis of 6-(4-Ethylpiperazin-1-yl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (23g)
Compound 23g was prepared according to general procedure D (method A) using 50g (0.100 g, 0.40 mmol) and 4-phenylbutyl isocyanate (0.077 g, 0.44 mmol) in dry MeCN (4 mL). The crude was purified by column chromatography (DCM/MeOH, 95:5) to afford 23g as a yellow solid (0.109 g, 64%). 1H NMR (400 MHz, CDCl3) δ 8.00 (t, J = 5.8 Hz, 1H), 7.87 (d, J = 9.5 Hz, 1H), 7.32–7.23 (m, overlapped with CDCl3 signal, 2H), 7.22–7.14 (m, 3H), 6.83–6.77 (m, 2H), 3.43 (q, J = 6.6 Hz, 2H), 3.24–3.16 (m, 4H), 2.66 (t, J = 7.3 Hz, 2H), 2.64–2.59 (m, 4H), 2.49 (q, J = 7.2 Hz, 2H), 1.78–1.60 (m, overlapped with H2O signal, 4H), 1.14 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 153.57, 150.07, 149.47, 142.92, 142.06, 128.54, 128.50, 125.99, 120.71, 115.82, 112.81, 98.52, 52.79, 52.44, 49.78, 40.18, 35.59, 29.23, 28.70, 12.10. UPLC/MS (method A): Rt 2.24 min, MS (ES) C24H30N4O3 requires 422, found 423 [M + H]+. HRMS C24H31N4O3 [M + H]+: calculated 423.2396, measured 423.2397, Δppm 0.2.
Synthesis of 6-(4-Isopropylpiperazin-1-yl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (23h)
Compound 23h was prepared according to general procedure D (method A) using 50h (0.100 g, 0.38 mmol) and 4-phenylbutyl isocyanate (0.074 g, 0.42 mmol) in dry MeCN (4 mL). The crude was purified by column chromatography (DCM/MeOH, 95:5) to afford 23h as a pink solid (0.050 g, 30%). 1H NMR (400 MHz, , CDCl3) δ 8.00 (t, J = 5.7 Hz, 1H), 7.87 (d, J = 9.5 Hz, 1H), 7.32–7.23 (m, overlapped with CDCl3 signal, 2H), 7.22–7.14 (m, 3H), 6.83–6.77 (m, 2H), 3.43 (q, J = 6.7 Hz, 2H), 3.27–3.11 (m, 4H), 2.79–2.69 (m, 5H), 2.67 (t, J = 7.3 Hz, 2H), 1.80–1.60 (m, 4H), 1.10 (d, J = 6.5 Hz, 6H). 13C NMR (101 MHz, , CDCl3) δ 153.56, 150.07, 149.43, 142.90, 142.06, 128.53, 128.49, 125.99, 120.77, 115.82, 112.89, 98.57, 54.86, 49.96, 48.65, 40.17, 35.59, 29.22, 28.69, 18.59. UPLC/MS (method A): Rt 2.31 min, MS (ES) C25H32N4O3 requires 436, found 437 [M + H]+. HRMS C25H33N4O3 [M + H]+: calculated 437.2553, measured 437.2557, Δppm 0.9.
Synthesis of 6-(4-Isobutylpiperazin-1-Yl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (23i)
Compound 23i was prepared according to general procedure D (method A) using 50i (0.100 g, 0.36 mmol) and 4-phenylbutyl isocyanate (0.07 mL, 0.07 g, 0.4 mmol) in dry MeCN (4 mL). The crude was purified by column chromatography (DCM/MeOH, 95:5) to afford 23i as a white solid (0.116 g, 72%). 1H NMR (400 MHz, CDCl3) δ 8.00 (t, J = 5.7 Hz, 1H), 7.87 (d, J = 9.5 Hz, 1H), 7.32–7.23 (m, overlapped with CDCl3 signal, 2H), 7.22–7.14 (m, 3H), 6.83–6.77 (m, 2H), 3.43 (q, J = 6.7 Hz, 2H), 3.26–3.10 (m, 4H), 2.67 (t, J = 7.2 Hz, 2H), 2.61–2.46 (m, 4H), 2.22–2.06 (m, 2H), 1.90–1.78 (m, 1H), 1.77–1.62 (m, 4H), 0.93 (d, J = 6.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 150.11, 149.62, 142.93, 142.08, 128.55, 128.51, 126.00, 115.79, 112.76, 98.45, 66.91, 53.51, 49.79, 40.19, 35.60, 29.24, 28.71, 25.56, 21.06. UPLC/MS (method B): Rt 2.08 min. MS (ES) C26H34N4O3 requires 450, found 451 [M + H]+. HRMS C26H35N4O3 [M + H]+: calculated 451.2719, measured: 451.2716, Δppm 1.6.
Synthesis of 4-(3-Benzyloxy-4-nitrophenyl)-1-methylpiperazin-2-one (52a)
Compound 52a was prepared according to general procedure G using 2-benzyloxy-4-fluoro-1-nitrobenzene (0.490 g, 2.00 mmol), 51a (0.300 g, 2.00 mmol), and Et3N (0.55 mL, 0.405 g, 4.00 mmol) in dry MeCN (20 mL), heating at reflux for 16 h. The crude was purified by column chromatography (EtOAc) to afford 52a as a yellow solid (0.350 g, 60%). 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 9.3 Hz, 1H), 7.51 (d, J = 7.2 Hz, 2H), 7.40 (t, J = 7.4 Hz, 2H), 7.33 (dd, J = 8.4, 6.3 Hz, 1H), 6.38 (dd, J = 9.3, 2.6 Hz, 1H), 6.29 (d, J = 2.5 Hz, 1H), 5.23 (s, 2H), 3.98 (s, 2H), 3.61 (dd, J = 6.5, 4.2 Hz, 2H), 3.50 (dd, J = 6.5, 4.2 Hz, 2H), 3.07 (s, 3H). UPLC/MS (method A): Rt 1.98 min, MS (ES) C18H19N3O4 requires 341, found 342 [M + H]+.
Synthesis of 1-(3-Benzyloxy-4-nitrophenyl)-4-methylpiperazin-2-one (52b)
To a solution of 2-benzyloxy-4-bromo-1-nitrobenzene (1.00 g, 3.25 mmol, 1.0 equiv.) in degassed 1,4-dioxane (26 mL) were added 51b (0.410 g, 3.6 mmol, 1.1 equiv.), K3PO4 (1.38 g, 6.50 mmol, 2.0 equiv.), and N,N′-dimethylethylenediamine (0.06 g, 0.07 mL, 0.65 mmol, 0.2 equiv.). The reaction mixture was degassed for another 15 min, and then copper(I) iodide (0.06 g, 0.33 mmol, 0.1 equiv.) was added. The reaction was refluxed for 24 h, then diluted with EtOAc, and filtered through a pad of Celite. The filtrate was concentrated under reduced pressure, and the crude was purified by column chromatography (EtOAc) to afford 52b as an orange solid (0.554 g, 50%). 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8.8 Hz, 1H), 7.46 (d, J = 7.3 Hz, 2H), 7.43–7.36 (m, 2H), 7.36–7.30 (m, 2H), 6.97 (dd, J = 8.8, 2.1 Hz, 1H), 5.22 (s, 2H), 3.72 (t, J = 5.1 Hz, 2H), 3.33 (s, 2H), 2.83 (s, 2H), 2.43 (s, 3H). UPLC/MS (method A): Rt 1.74 min. MS (ES) C18H19N3O4 requires 341, found 342 [M + H]+.
Synthesis of 4-(4-Amino-3-hydroxyphenyl)-1-methylpiperazin-2-one (49i)
Compound 49i was prepared according to general procedure B (method A) using 52a (0.341 g, 1.00 mmol). UPLC/MS (method A): Rt 0.89 min. MS (ES) C11H15N3O2 requires 221, found 222 [M + H]+.
Synthesis of 1-(4-Amino-3-hydroxyphenyl)-4-methylpiperazin-2-one (49j)
Compound 49j was prepared according to general procedure B (method A) using 52b (0.50 g, 1.46 mmol). UPLC/MS (method C): Rt 1.42 min. MS (ES) C11H15N3O2 requires 221, found 222 [M + H]+.
Synthesis of 6-(4-Methyl-3-oxo-piperazin-1-yl)-3H-1,3-benzoxazol-2-one (50j)
Compound 50j was prepared according to general procedure C using 49i (0.221 g, 1.00 mmol) and CDI (0.162 g, 1.00 mmol) in dry MeCN (10 mL). The crude was purified by column chromatography (DCM/MeOH, 90:10) to afford 50j as a violet solid (0.197 g, 80% over two steps). 1H NMR (400 MHz, CDCl3) δ 6.95 (d, J = 8.5 Hz, 1H), 6.82 (d, J = 2.3 Hz, 1H), 6.67 (dd, J = 2.3, 8.5 Hz, 1H), 3.81 (s, 2H), 3.48 (dd, J = 3.4, 5.8 Hz, 2H), 3.42 (dd, J = 4.0, 6.2 Hz, 2H), 3.04 (s, 3H). UPLC/MS (method A): Rt 1.16 min. MS (ES) C12H13N3O3 requires 247, found 248 [M + H]+.
Synthesis of 6-(4-Methyl-2-oxo-piperazin-1-yl)-3H-1,3-benzoxazol-2-one (50k)
Compound 50k was prepared according to general procedure C using 49j (0.323 g, 1.46 mmol) and CDI (0.240 g, 1.46 mmol) in dry MeCN (15 mL). The crude was purified by column chromatography (DCM/MeOH, 80:20) to afford 50k as orange oil (0.270 g, 70% over two steps). 1H NMR (400 MHz, CDCl3) δ 7.73–7.65 (m, 1H), 7.03–6.86 (m, 2H), 3.76–3.66 (m, 2H), 3.29 (s, 2H), 2.87–2.68 (m, 2H), 2.42 (s, 4H). UPLC/MS (method A): Rt 1.16 min. MS (ES) C12H13N3O3 requires 247, found 248 [M + H]+.
Synthesis of 6-(4-Methyl-3-oxo-piperazin-1-yl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (23j)
Compound 23j was prepared according to general procedure D (method A) using 50j (0.170 g, 0.69 mmol) and 4-phenylbutyl isocyanate (0.13 mL, 0.130 g, 0.76 mmol) in dry MeCN (7 mL). The crude was purified by column chromatography (DCM/MeOH, 90:10) to afford 23j as a white solid (0.060 g, 20%). 1H NMR (400 MHz, CDCl3) δ 7.98 (t, J = 5.3 Hz, 1H), 7.93 (d, J = 8.7 Hz, 1H), 7.32–7.23 (m, overlapped with CDCl3 signal, 2H), 7.21–7.14 (m, 3H), 6.82–6.73 (m, 2H), 3.85 (s, 2H), 3.54–3.46 (m, 4H), 3.44 (q, J = 6.7 Hz, 2H), 3.05 (s, 3H), 2.67 (t, J = 7.1 Hz, 2H), 1.77–1.62 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 128.54 (2C), 126.05, 120.78, 115.97, 107.97, 47.76, 46.79, 40.34, 35.58, 35.35, 29.15, 28.67. UPLC/MS (method A): Rt 2.32 min. MS (ES) C23H26N4O4 requires 422, found 423 [M + H]+. HRMS C23H27N4O4 [M + H]+: calculated 423.2032, measured: 423.202, Δppm −2.8.
Synthesis of 6-(4-Methyl-2-oxo-piperazin-1-yl)-2-oxo-N-(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (23k)
Compound 23k was prepared according to general procedure D (method A) using 50k (0.288 g, 1.17 mmol) and 4-phenylbutyl isocyanate (0.22 mL, 0.225 g, 1.28 mmol) in dry MeCN (8 mL). The crude was purified by column chromatography (DCM/MeOH, 90:10) to afford 23k as a white solid (0.049 g, 10%). 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 8.6 Hz, 1H), 7.98 (t, J = 5.5 Hz, 1H), 7.31–7.26 (m, overlapped with CDCl3 signal, 3H), 7.24–7.13 (m, 4H), 3.76 (s, 2H), 3.44 (q, J = 6.7 Hz, 2H), 3.33 (s, 2H), 2.93–2.80 (m, 2H), 2.67 (t, J = 7.2 Hz, 2H), 2.45 (s, 3H), 1.79–1.63 (m, 4H).13C NMR (101 MHz, CDCl3) δ 153.26, 149.71, 142.03, 141.92, 128.56, 128.55, 126.92, 126.05, 122.56, 116.07, 109.05, 59.54, 52.08, 50.30, 45.11, 40.32, 35.60, 29.18, 28.70. UPLC/MS (method A): Rt 1.94 min. MS (ES) C23H26N4O4 requires 422, found 423 [M + H]+. HRMS C23H27N4O4 [M + H]+: calculated 423.2032, measured: 423.2026, Δppm −1.4.
Synthesis of tert-Butyl 4-(4-Hydroxy-3-nitrophenyl)-3,6-dihydro-2H-pyridine-1-carboxylate (32a)
Compound 32a was prepared according to general procedure A using 4-bromo-2-nitrophenol (0.500 g, 2.29 mmol), 18c (0.92 g, 2.98 mmol), PdCl2(PPh3)4 (0.016 g, 0.023 mmol), and 2 M Na2CO3 (2.87 mL, 5.73 mmol) in degassed 1,4-dioxane (25 mL). The crude was purified by column chromatography (heptane/EtOAc, 90:10) to afford 32a as yellow oil (0.700 g, 95%). 1H NMR (400 MHz, CDCl3) δ 10.54 (s, 1H), 8.07 (d, J = 2.2 Hz, 1H), 7.65 (dd, J = 8.8, 2.3 Hz, 1H), 7.14 (d, J = 8.8 Hz, 1H), 6.07 (s, 1H), 4.09 (d, J = 2.8 Hz, 2H), 3.65 (t, J = 5.7 Hz, 2H), 3.53 (t, J = 5.8 Hz, 2H), 1.47 (s, 9H). UPLC/MS (method B): Rt 1.48 min. MS (ES) C16H20N2O5 requires 320, found 319 [M–H]−.
Synthesis of tert-Butyl 4-(3-Amino-4-hydroxyphenyl)piperidine-1-carboxylate (32b)
Compound 32b was prepared according to general procedure B (method A) using 32a (0.078 g, 0.24 mmol). UPLC/MS (method A): Rt 1.51 min. MS (ES) C16H24N2O3 requires 292, found 291 [M-H]−.
Synthesis of tert-Butyl 4-(2-Oxo-3H-1,3-benzoxazol-5-yl)piperidine-1-carboxylate (33)
Compound 33 was prepared according to general procedure C using 32b (0.070 g, 0.24 mmol) and CDI (0.058 g, 0.36 mmol) in dry MeCN (2.5 mL). The crude was purified by column chromatography (Cy/EtOAc, 70:30) to afford 33 as a white solid (0.046 g, 60% over two steps). 1H NMR (400 MHz, DMSO-d6) δ 11.55 (bs, 1H), 7.22–7.14 (m, 1H), 6.99–6.90 (m, 2H), 4.07 (d, J = 13.0 Hz, 2H), 2.98–2.56 (m, 3H), 1.84–1.65 (m, 2H), 1.54–1.44 (m, 2H), 1.42 (s, 9H). UPLC/MS (method A): Rt 2.20 min. MS (ES) C17H22N2O4 requires 318, found 319 [M + H]+.
Synthesis of tert-Butyl 4-[3-(Isobutylcarbamoyl)-2-oxo-1,3-benzoxazol-5-yl]piperidine-1-carboxylate (24a)
Compound 24a was prepared according to general procedure D (method B) using 33 (0.200 g, 0.63 mmol), isobutylamine (0.094 mL, 0.069 g, 0.94 mmol), and Et3N (0.44 mL, 0.318 g, 3.14 mmol) in dry DCM (7 mL). The crude was purified by column chromatography (Cy/EtOAc, 90:10) to afford 24a as a white solid (0.186 g, 70%). 1H NMR (400 MHz, CDCl3) δ 8.16 (t, J = 5.8 Hz, 1H), 7.98 (d, J = 1.8 Hz, 1H), 7.18 (d, J = 8.3 Hz, 1H), 7.08 (dd, J = 8.4, 1.8 Hz, 1H), 4.27 (d, J = 13.2 Hz, 2H), 3.28 (dd, J = 6.8, 5.9 Hz, 2H), 2.89–2.65 (m, 3H), 2.02–1.76 (m, 3H), 1.72–1.59 (m, 2H), 1.50 (s, 9H), 1.02 (d, J = 6.7 Hz, 6H). UPLC/MS (method B): Rt 2.07 min. MS (ES) C22H31N3O5 requires 417, found 418 [M + H]+.
Synthesis of N-Isobutyl-2-oxo-5-(4-piperidyl)-1,3-benzoxazole-3-carboxamide Hydrochloride (24b)
Compound 24b was prepared according to general procedure E using 24a (0.160 g, 0.38 mmol). The crude was triturated with Et2O to afford 24b as a white solid (0.128 g, 95%). 1H NMR (400 MHz, DMSO-d6) δ 9.16 (bs, 1H), 8.96 (bs, 1H), 8.14 (t, J = 5.9 Hz, 1H), 7.83 (d, J = 1.8 Hz, 1H), 7.39 (d, J = 8.3 Hz, 1H), 7.13 (dd, J = 8.4, 1.9 Hz, 1H), 3.37 (s, 1H), 3.17 (t, J = 6.4 Hz, 2H), 3.08–2.81 (m, 3H), 2.00–1.77 (m, 5H), 0.92 (d, J = 6.7 Hz, 6H). UPLC/MS (method A): Rt 1.78 min. MS (ES) C17H23N3O3 requires 317, found 318 [M + H]+.
Synthesis of N-Isobutyl-5-(1-methyl-4-piperidyl)-2-oxo-1,3-benzoxazole-3-carboxamide (24c)
Compound 24c was prepared according to general procedure F using 24b (0.088 g, 0.25 mmol), 37% aqueous solution of formaldehyde (0.038 mL, 1.25 mmol), NaBH(OAc)3 (0.106 g, 0.5 mmol), and AcOH (0.03 mL, 0.024 g, 0.4 mmol) in dry MeCN (3 mL). The crude was triturated with Et2O to afford 24c as a white solid (0.07 g, 83%). 1H NMR (400 MHz, DMSO-d6) δ 8.14 (t, J = 5.1 Hz, 1H), 7.80 (s, 1H), 7.33 (d, J = 8.3 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 3.15–3.07 (t, J = 6.4 Hz, 2H), 2.94–2.79 (m, 2H), 2.59–2.43 (m, overlapped with DMSO signal, 1H), 2.20 (s, 3H), 2.06–1.91 (m, 2H), 1.91–1.79 (m, 1H), 1.79–1.54 (m, 4H), 0.92 (d, J = 6.7 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 152.56, 149.50, 142.88, 139.85, 128.14, 122.36, 112.74, 109.60, 55.68, 46.79, 46.12, 41.27, 33.32, 27.90, 19.77. UPLC/MS (method A): Rt 1.81 min. MS (ES) C18H25N3O3 requires 331, found 332 [M + H]+. HRMS C18H26N3O3 [M + H]+: calculated 332.1974, measured: 332.1964, Δppm −3.0.
Synthesis of tert-Butyl 4-Hydroxy-4-(2-oxo-3H-1,3-benzoxazol-7-yl)piperidine-1-carboxylate (45)
Compound 45 was prepared according to general procedure I using 42b (0.793 g, 3.98 mmol), 7-bromo-3H-1,3-benzoxazol-2-one (0.500 g, 2.34 mmol), MeMgBr (1.17 mL, 0.418 g, 3.51 mmol, 3 M in Et2O), and n-BuLi (1.12 mL, 2.81 mmol, 2.5 M in hexanes) in dry THF (25 mL). The crude was purified by column chromatography (Cy/EtOAc, 35:65) to afford 45 as a white solid (0.345 g, 44%). 1H NMR (400 MHz, DMSO-d6) δ 11.60 (bs, 1H), 7.26 (dd, J = 8.1, 1.3 Hz, 1H), 7.12 (t, J = 7.9 Hz, 1H), 6.97 (dd, J = 7.7, 1.3 Hz, 1H), 5.34 (s, 1H), 3.97–3.78 (m, 2H), 3.25–2.99 (m, 2H), 2.09 (td, J = 13.1, 4.7 Hz, 2H), 1.63–1.52 (m, 2H), 1.43 (s, 9H). UPLC/MS (method A): Rt 1.76 min. MS (ES) C17H22N2O5 requires 334, found 335 [M + H]+.
Synthesis of 7-(1,2,3,6-Tetrahydropyridin-4-yl)-3H-1,3-benzoxazol-2-one (46a)
Compound 46a was prepared according to general procedure L using 45 (0.345 g, 1.03 mmol). The crude was purified by SCX to afford 46a as a brown solid (quant.). UPLC/MS (method A): Rt 0.95 min. MS (ES) C12H12N2O2 requires 216, found 217 [M + H]+.
Synthesis of 7-(1-Methyl-3,6-dihydro-2H-pyridin-4-yl)-3H-1,3-benzoxazol-2-one (46b)
Compound 46b was prepared according to general procedure F using 46a (0.108 g, 0.5 mmol), NaBH(OAc)3 (0.318 g, 1.50 mmol), AcOH (0.03 mL, 0.030 g, 0.50 mmol), and 37% aqueous solution of formaldehyde (0.080 mL, 2.5 mmol) in dry MeCN (5 mL). The crude was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ 6.87 (d, J = 7.76 Hz, 1H), 6.80–6.74 (m, 2H), 6.36 (t, J = 3.49 Hz, 1H), 3.07–3.00 (m, 2H), 2.58–2.47 (m, overlapped with DMSO signal, 4H), 2.27 (s, 3H). UPLC/MS (method A): Rt 0.98 min. MS (ES) C13H14N2O2 requires 230, found 231 [M + H]+.
Synthesis of 7-(1-Methyl-4-piperidyl)-3H-1,3-benzoxazol-2-one (47)
Compound 47 was prepared according to general procedure B (method B) using 46b (0.115 g, 0.50 mmol). The crude was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ 7.12–7.06 (m, 1H), 6.94 (t, J = 7.4, 1.2 Hz, 2H), 2.98 (d, J = 11.8, 3.4 Hz, 2H), 2.82–2.69 (m, 1H), 2.31 (s, 3H), 2.25–2.11 (m, 2H), 1.88–1.72 (m, 4H). UPLC/MS (method A): Rt 0.98 min, MS (ES) C13H16N2O2 requires 232, found 233 [M + H]+.
Synthesis of N-Isobutyl-7-(1-methyl-4-piperidyl)-2-oxo-1,3-benzoxazole-3-carboxamide (25)
Compound 25 was prepared according to general procedure D (method B) using 47 (0.116 g, 0.5 mmol), isobutylamine (0.014 g, 0.19 mmol), and Et3N (0.15 mL, 0.111 g, 1.10 mmol) in dry DCM (7 mL). The crude was purified by column chromatography (DCM/MeOH, 92:8) to afford 25 as a pink solid (0.050 g, 30% over three steps). 1H NMR (400 MHz, CDCl3) δ 8.14 (t, J = 5.0 Hz, 1H), 7.91 (dd, J = 8.0, 1.3 Hz, 1H), 7.21 (t, J = 8.0 Hz, 1H), 7.11 (dd, J = 8.1, 1.3 Hz, 1H), 3.30–3.21 (m, 2H), 3.07–2.96 (m, 2H), 2.96–2.82 (m, 1H), 2.35 (s, 3H), 2.13 (td, J = 11.5, 3.2 Hz, 2H), 2.03–1.81 (m, 5H), 0.99 (d, J = 6.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 153.43, 150.10, 139.67, 128.76, 127.90, 125.28, 122.48, 113.52, 56.18, 47.71, 46.50, 35.66, 31.80, 28.61, 20.18. UPLC/MS (method A): Rt 1.79 min, MS (ES) C18H25N3O3 requires 331, found 332 [M + H]+. HRMS C18H26N3O3 [M + H]+: calculated 332.1974, measured 332.1967, Δppm −2.1.
Synthesis of 2-(3-Benzyloxy-4-nitrophenyl)pyridine (35)
Compound 35 was prepared according to general procedure A using 2-benzyloxy-4-bromo-1-nitrobenzene (0.309 g, 1.00 mmol), 34 (0.174 g, 1.10 mmol), Pd(dppf)Cl2 (0.146 g, 0.2 mmol), KOAc (0.196 g, 2 mmol), and 2 M Na2CO3 (1.30 mL, 2.50 mmol) in degassed 1,4-dioxane (15 mL). The crude was purified by column chromatography (Cy/EtOAc, 80:20) to afford 35 as a yellow solid (0.121 g, 40%). 1H NMR (400 MHz, DMSO-d6) δ 8.75 (ddd, J = 4.8, 1.7, 0.9, 1H), 8.14 (d, J = 8.0 Hz, 1H), 8.12 (d, J = 1.6 Hz, 1H), 8.03 (d, J = 8.5 Hz, 1H), 7.98 (td, J = 7.8, 1.8 Hz, 1H), 7.85 (dd, J = 8.5, 1.7 Hz, 1H), 7.54–7.32 (m, 6H), 5.45 (s, 2H). UPLC/MS (method A): Rt 2.47 min, MS (ES) C18H14N2O3 requires 306, found 307 [M + H]+.
Synthesis of 2-Amino-5-(2-piperidyl)phenol (36)
Compound 36 was prepared according to general procedure B (method B) using 35 (0.520 g, 1.69 mmol). UPLC/MS (method A): Rt 0.93 min. MS (ES) C11H16N2O requires 192, found 193 [M + H]+.
Synthesis of 6-(2-Piperidyl)-3H-1,3-benzoxazol-2-one (37a)
Compound 37a was prepared according to general procedure C using 36 (0.390 g, 1.69 mmol) and CDI (0.274 g, 1.69 mmol) in dry MeCN (17 mL). The crude was used in the next step without further purification. UPLC/MS (method A): Rt 1.05 min. MS (ES) C12H14N2O2 requires 218, found 219 [M + H]+.
Synthesis of 6-(1-Methyl-2-piperidyl)-3H-1,3-benzoxazol-2-one (37b)
Compound 37b was prepared according to general procedure F using 37a (0.368 g, 1.69 mmol), 37% aqueous solution of formaldehyde (0.09 mL, 3.38 mmol), NaBH(OAc)3 (1.075 g, 5.07 mmol), and AcOH (0.15 mL, 0.162 g, 2.70 mmol) in dry MeCN (9 mL). The crude was purified by SCX to afford 37b as a white solid (0.169 g, 43% over three steps). 1H NMR (400 MHz, DMSO-d6) δ 7.64 (bs, 1H), 7.14 (d, J = 1.2 Hz, 1H), 7.06–6.95 (m, 2H), 3.01–2.85 (m, 1H), 2.74 (dd, J = 10.8, 2.7 Hz, 1H), 2.01 (td, J = 11.6, 3.2 Hz, 1H), 1.88 (s, 3H), 1.78–1.66 (m, 1H), 1.67–1.50 (m, 3H), 1.50–1.36 (m, 1H), 1.37–1.22 (m, 1H). UPLC/MS (method A): Rt 1.04 min. MS (ES) C13H16N2O2 requires 232, found 233 [M + H]+.
Synthesis of (±)-N-Isobutyl-6-(1-methyl-2-piperidyl)-2-oxo-1,3-benzoxazole-3-carboxamide (26)
Compound 26 was prepared according to general procedure D (method B) using 37b (0.170 g, 0.73 mmol) and isobutylamine (0.160 g, 2.19 mmol) in dry DCM (10 mL). The crude was purified by column chromatography (DCM/MeOH, 92:8) to afford 26 as a white solid (0.069 g, 29%). 1H NMR (400 MHz, CDCl3) δ 8.10 (t, J = 5.3 Hz, 1H), 7.94 (d, J = 8.2 Hz, 1H), 7.30–7.23 (m, overlapped with CDCl3 signal, 1H), 7.20 (dd, J = 8.3, 1.6 Hz, 1H), 3.25 (dd, J = 6.8, 5.8 Hz, 2H), 3.09–3.00 (m, 1H), 2.95 (dd, J = 11.1, 2.8 Hz, 1H), 2.80 (dd, J = 11.1, 2.8 Hz, 1H), 2.18–2.05 (m, 1H), 1.98 (s, 3H), 1.96–1.85 (m, 1H), 1.85–1.76 (m, 1H), 1.76–1.65 (m, 2H), 1.61–1.47 (m, 1H), 1.42–1.28 (m, 1H), 0.98 (d, J = 6.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 153.56, 150.07, 142.34, 142.16, 126.95, 124.20, 115.37, 108.82, 70.79, 57.48, 47.68, 44.59, 36.37, 28.60, 26.12, 24.94, 20.15. UPLC/MS (method A): Rt 1.88 min, MS (ES) C18H25N3O3 requires 331, found 332 [M + H]+. HRMS C18H26N3O3 [M + H]+: calculated 332.1971, measured 332.1974, Δppm −0.9.
Synthesis of tert-Butyl 5-(3-Hydroxy-4-nitrophenyl)-3,4-dihydro-2H-pyridine-1-carboxylate (39)
Compound 39 was prepared according to general procedure A using 38 (0.834 g, 2.7 mmol), 5-bromo-2-nitrophenol (0.530 g, 2.43 mmol), Pd(PPh3)4 (0.156 g, 0.135 mmol), and 2 M Na2CO3 (3.04 mL, 6.075 mmol) in degassed 1,4-dioxane (27 mL). The crude was purified by column chromatography (Cy/EtOAc, 90:10) to afford 39 as a yellow solid (0.460 g, 53%). 1H NMR (400 MHz, CDCl3) δ 10.79 (s, 1H), 7.99 (d, J = 8.8 Hz, 1H), 7.82–7.48 (m, 1H), 7.11–6.94 (m, 2H), 3.62 (s, 2H), 2.42 (t, J = 6.0 Hz, 2H), 1.98 (p, J = 6.1 Hz, 2H), 1.54 (s, 9H). UPLC/MS (method B): Rt 1.86 min. MS (ES) C16H20N2O5 requires 320, found 321 [M + H]+.
Synthesis of tert-Butyl 3-(4-Amino-3-hydroxyphenyl)piperidine-1-carboxylate (40)
Compound 40 was prepared according to general procedure B (method A) using 39 (0.450 g, 1.41 mmol). UPLC/MS (method B): Rt 0.73 min. MS (ES) C16H24N2O3 requires 292, found 291 [M-H]−.
Synthesis of tert-Butyl 3-(2-Oxo-3H-1,3-benzoxazol-6-yl)piperidine-1-carboxylate (49a)
Compound 49a was prepared according to general procedure C using 40 (0.412 g, 1.41 mmol) and CDI (0.229 g, 1.41 mmol) in dry MeCN (14 mL). The crude was purified by column chromatography (Cy/EtOAc, 70:30) to afford 41a as brown oil (0.403 g, 90% over two steps). 1H NMR (400 MHz, CDCl3) δ 10.04 (bs, 1H), 7.07–6.88 (m, 3H), 4.20–4.06 (m, 2H), 2.80–2.43 (m, 3H), 2.03–1.93 (m, 1H), 1.72 (dd, J = 3.2, 6.4 Hz, 1H), 1.66–1.52 (m, 2H), 1.44 (s, 9H). UPLC/MS (method A): Rt 2.19 min. MS (ES) C17H22N2O4 requires 318, found 319 [M + H]+.
Synthesis of 6-(3-Piperidyl)-3H-1,3-benzoxazol-2-one Hydrochloride (41b)
Compound 41b was prepared according to general procedure E using 41a (0.185 g, 0.58 mmol). The crude was used in the next step without further purification. UPLC/MS (method A): Rt 0.99 min. MS (ES) C12H14N2O2 requires 218, found 219 [M + H]+.
Synthesis of 6-(1-Methyl-3-piperidyl)-3H-1,3-benzoxazol-2-one (41c)
Compound 41 was prepared according to general procedure F using 41b (0.147 g, 0.58 mmol), 37% aqueous solution of formaldehyde (0.03 mL, 1.16 mmol), NaBH(OAc)3 (0.370 g, 1.74 mmol), and AcOH (0.07 mL, 0.070 g, 1.16 mmol) in dry MeCN (3 mL). The crude was purified by SCX to afford 41c as a white solid (0.094 g, 70% over two steps). 1H NMR (400 MHz, CDCl3) δ 7.09 (s, 1H), 7.02 (d, J = 8.2 Hz, 1H), 6.95 (d, J = 8.1 Hz, 1H), 3.01–2.81 (m, 3H), 2.31 (s, 3H), 2.03–1.87 (m, 3H), 1.87–1.67 (m, 2H), 1.39 (qd, J = 12.6, 4.2 Hz, 1H). UPLC/MS (method A): Rt 1.81 min. MS (ES) C13H16N2O2 requires 232, found 233 [M + H]+.
Synthesis of (±)-N-Isobutyl-6-(1-methyl-3-piperidyl)-2-oxo-1,3-benzoxazole-3-carboxamide (27)
Compound 27 was prepared according to general procedure D (method B) using 41c (0.088 g, 0.28 mmol) and isobutylamine (0.06 mL, 0.06 g, 0.84 mmol) in dry DCM (4 mL). The crude was purified by column chromatography (DCM/MeOH, 90:10) to afford 27 as a white solid (0.08 g, 68%). 1H NMR (400 MHz, CDCl3) δ 8.09 (bs, 1H), 7.96 (d, J = 8.9 Hz, 1H), 7.17–7.08 (m, 2H), 3.25 (d, J = 6.59, 2H), 3.03–2.87 (m, 3H), 2.34 (s, 3H), 2.13–1.68 (m, 6H), 1.41 (qd, J = 12.1, 5.5 Hz, 1H), 0.99 (d, J = 6.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 153.50, 150.05, 142.02, 141.71, 126.47, 123.99, 115.48, 108.69, 63.12, 55.85, 47.69, 46.54, 42.82, 31.21, 28.60, 25.64, 20.16. UPLC/MS (method A): Rt 1.87 min. MS (ES) C18H25N3O3 requires 331, found 332 [M + H]+. HRMS C18H26N3O3 [M + H]+: calculated 332.1974, measured 332.1972, Δppm −0.9.
In Vitro Pharmacological Assay
In Vitro hAC Fluorescence Assay
Cell Culture Conditions and Preparation of hAC-Enriched Lysate
HEK293 cells stably expressing hAC were grown in Dulbecco’s modified Eagle medium (DMEM) containing 10% FBS, 1% glutamine, 1 mM sodium pyruvate, and 500 μg mL–1 G418. Cells were harvested, and pellets were stored at −80 °C until lysosomal-enriched lysate preparation. Cells were suspended in 20 mM Tris HCl (pH 7.5) with 0.32 M sucrose, sonicated, and centrifuged at 800 × g for 30 min at 4 °C. Supernatants were then centrifuged at 12000 × g for 30 min at 4 °C. Pellets were resuspended in PBS (pH 7.4) and subjected to three freeze–thaw cycles at −80 °C. The suspension was finally centrifuged at 105000 × g for 1 h at 4 °C, and protein concentration was measured in the supernatant with the bicinchoninic acid based protein assay. This hAC-enriched preparation allowed us to further optimize the enzymatic assay and to use small amounts of lysate (2 μg per well) at a 5 μM substrate (Rbm14–12) around its KM (KM = 5.0 μM).
Fluorogenic hAC Assay
The assay was performed in Optiplate 96-well black plates, with each reaction well containing a mixture of 25 mM NaOAc buffer (pH 4.5) and a fixed amount of protein (2 μg) in a volume of 85 μL. After 10 min of preincubation with test compounds (diluted 20× from DMSO stock solutions at different concentrations), the fluorogenic probe was added (diluted 40× from EtOH stock solution, final concentration 5 μM). After 3 h of incubation at 37 °C, reactions were stopped with 50 μL of MeOH and 100 μL of a 2.5 mg mL–1 NaIO4 fresh solution in 100 mM glycine/NaOH buffer (pH 10.6). The plates were further incubated for 2 h at 37 °C in the dark, and fluorescence intensities were measured at excitation/emission wavelengths of 355/460 nm. Negative control samples consisted of the same incubation mixture in the absence of protein-enriched extracts. Data were plotted as a function of compound concentrations. IC50 values were calculated by nonlinear regression analysis using GraphPad Prism 5 (GraphPad Software Inc., CA, USA) applying a standard slope curve fitting. The reported IC50 values are the mean of at least three independent experiments performed in three technical replicates.
Kinetic Studies
Michaelis–Menten Analysis
Assay conditions for the kinetic studies were the same as those described for the fluorogenic hAC assay. Enzyme-enriched lysate (2 μg) was incubated with the following concentrations of substrate Rbm14–12: 0.25, 0.5, 1, 2.5, 5, 10, 12.5, and 15 μM. Compound 22m was used at final concentrations of 100 and 400 nM. Initial velocities (V0) were determined and automatically fitted to the Michaelis–Menten equation to obtain the kinetic parameters (KM and Vmax). The graph is representative of two independent experiments, each performed in three technical replicates. Graphs and data analysis were performed using GraphPad Prism 5 software (GraphPad Software Inc., CA, USA).
Determination of Kinetic Parameter ki/KI
hAC activity was measured as a function of reaction time in the presence of different concentrations of 22m. The apparent inactivation rate constant of hAC (kobs) was analyzed by nonlinear square fitting each data set to the pseudo-first-order rate equation Y = vi(1 – exp(−kobst))/kobs. Replotting of calculated kobs vs [22m] was made, and the kinetic parameter ki/KI was calculated by nonlinear square fitting data to the equation Y = kiI/(KI + I). The graphs are representative of two independent experiments, each performed in two technical replicates.
In Vitro hASM Assay
hASM activity measurement was conducted using the fluorogenic substrate 6-hexadecanoylamino-4-methylumbelliferylphosphorylcholine (HMU-PC, Toronto Research Chemicals) at 0.5 μM and 1.3 nM purified human full-length ASM enzyme (purified in house) in a buffer containing 50 mM citrate, 150 mM NaCl, 5 mM ZnCl2, and 0.43 mM Triton X-100 at pH 4.7 in a final volume of 50 μL. The reaction mixtures were incubated for 45 min at rt and stopped by the addition of 150 μL of 1 M glycine at pH 12.5. The formation of the fluorescent product was monitored by a plate reader at excitation/emission wavelengths of 385/450 nm. The average hASM activity was calculated from two independent experiments, each performed in two technical replicates.
In Vitro hGCase Assay
hGCase activity measurement was conducted using the fluorogenic substrate 4-methylumbelliferyl-β-d-glucuronide hydrate (4-MUG, Merck) at 1 mM and 5 nM purified human full-length GCase enzyme and 50 nM of its natural activator SapC (both GCase and SapC were purified in house) in a buffer containing 50 mM citric acid, 174 mM K2HPO4, 15 μM phosphatidylserine, and 0.32 mM Triton X-100 at pH 4.7 in a final volume of 50 μL. The reaction mixtures were incubated for 15 min at rt and stopped by the addition of 150 μL of 1 M glycine at pH 12.5. The cleavage of 4-MUG was monitored by a plate reader at excitation/emission wavelengths of 365/440 nm. The average hGCase activity was calculated from two independent experiments, each performed in two technical replicates.
In Vitro hNAAA Fluorescence Assay
Cell Culture and Preparation of hNAAA-Enriched Lysate
HEK-293 cells stably transfected with the hNAAA coding sequence cloned from a human spleen cDNA library (catalog no. 639124, Clontech, Mountain View, CA, USA) were used as the enzyme source. Cells were grown in Dulbecco’s modified Eagle medium (DMEM) containing 10% FBS, 1% glutamine, 1 mM sodium pyruvate, and 500 μg mL–1 G418. Cells were harvested, and pellets were stored at −80 °C until lysosomal-enriched lysate preparation. Cells were suspended in 20 mM Tris HCl (pH 7.4) with 0.32 M sucrose, sonicated, and centrifuged at 800 × g for 30 min at 4 °C. Supernatants were then ultracentrifuged at 12000 × g for 30 min at 4 °C. Pellets were resuspended in PBS buffer (pH 7.4) and subjected to three freeze–thaw cycles at −80 °C. The suspension was finally ultracentrifuged at 105000 × g for 1 h at 4 °C, supernatants were collected, protein concentration was measured, and samples were aliquoted and stored at −80 °C until use.
Fluorogenic hNAAA Assay
The assay was run in 96-well microplates (Black OptiPlate-96F; PerkinElmer, Massachusetts, USA) in a total reaction volume of 200 μL. hNAAA protein preparation (4.0 μg) was preincubated for 30 min with various concentrations of test compounds or vehicle control (DMSO 5%) in 100 mM citrate/phosphate buffer (pH 4.5) containing 3.0 mM DTT, 0.1% NP40 0.1%, 0.05% BSA, 150 mM NaCl. N-(4-Methyl-2-oxo-chromen-7-yl)-hexadecanamide (PAMCA) was used as a substrate (2.0 μM), and the reaction was carried out for 50 min at 37 °C. Fluorescence was measured with an EnVision 2014 Multilabel Reader (PerkinElmer, Massachusetts, USA) using an excitation wavelength of 355 nm and emission of 460 nm. IC50 values were calculated by nonlinear regression analysis of log[concentration]/inhibition curves using GraphPad Prism 5 (GraphPad Software Inc., CA, USA) applying a standard slope curve fitting. The reported IC50 values are the mean of at least three independent experiments performed in three technical replicates.
In Vitro hFAAH Fluorescence Assay
Cell Culture and Preparation of hFAAH-Enriched Lysate
hFAAH was obtained from a HEK-293 FAAH-1 overexpressing stable cell line. Cells were grown in Dulbecco’s modified Eagle medium (DMEM) containing 10% FBS, 1% penicillin/streptomycin, 1% glutamine, 1 mM sodium pyruvate, and 500 μg mL–1 G418. Cells were harvested, and pellets were stored at −80 °C until membrane-enriched lysate preparation. The cell pellet was resuspended in 20 mM Tris HCl (pH 7.4, 0.32 M sucrose), sonicated, and centrifuged at 1000 × g (10 min, 4 °C). The collected supernatant was centrifuged at 12000 × g for 10 min at 4 °C, and the supernatants were further centrifuged at 100000 × g for 1 h at 4 °C. Membrane pellets were resuspended in PBS, protein concentration was measured, and samples were aliquoted and stored at −80 °C until use.
Fluorogenic hFAAH Assay
The fluorescence assay to measure FAAH activity was performed in 96-well black plates (Black OptiPlate-96F; PerkinElmer, Massachusetts, USA): 2.5 μg of hFAAH membrane preparation was preincubated for 50 min at 37 °C in 190 μL of assay buffer (50 mM Tris HCl pH 7.4, 0.05% fatty acid free BSA), with 5 μL of inhibitor or 5 μL of DMSO to measure FAAH total activity. The background (no activity) samples were prepared using 190 μL of assay buffer without hFAAH and 5 μL of DMSO. The reaction was then started by the addition of 5 μL of substrate (AMC Arachidonyl Amide, A6855, Merck) dissolved in DMSO and used at a final concentration of 800 nM. The reaction was carried out for 45 min at 37 °C, and fluorescence was measured with an EnVision 2014 Multilabel Reader (PerkinElmer, Massachusetts, USA) (excitation wavelength 355 nm/emission wavelength 460 nm). The concentration causing half-maximal inhibition (IC50) was determined by nonlinear regression analysis of the log[concentration]/response curves generated with mean replicate values using a four-parameter Hill equation curve fitting with GraphPad Prism 5 (GraphPad Software Inc., CA, USA). The reported IC50 values are the mean of at least three independent experiments performed in three technical replicates.
In Vitro hMAGL Colorimetric Assay
The colorimetric assay to measure hMAGL activity was performed using an assay kit provided by Cayman Scientific (item. 705192), according to the manufacturer’s instructions. Briefly, in vitro activity was measured in 96-well plates, and DMSO was used as solvent. 10 μL of DMSO (100% initial activity wells: 100% IA) or compounds at two concentrations (1 and 10 μM) were preincubated for 5 min at rt with 150 μL of diluted assay buffer (10 mM Tris HCl, pH 7.2, containing 1 mM EDTA) containing hMAGL. In blank wells, 160 μL of the diluted assay buffer and 10 μL of DMSO were added. The reactions were initiated by adding 10 μL of MAGL substrate to all the wells, and plates were incubated for 10 min at rt. Absorbance values were measured at 405 nm, and percent inhibition was calculated by the following method: 100 – (Inhibitor/100% IA) × 100. The reported percentages of inhibition are the mean of at least three independent experiments performed in three technical replicates.
Cell Culture and Treatments
SH-SY5Y cells were purchased from Sigma Aldrich (Italy) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum at 37 °C and 5% CO2. Drugs were dissolved in DMSO (10 mM) and diluted in the cell culture medium with reduced serum (1%) for cell treatments.
hAC LC/MS-Based Activity Assay
hAC activity measurement was performed as previously described.36,37 Total lysates from cells were diluted in assay buffer (100 mM sodium phosphate, 0.1% Nonidet P-40, 150 mM NaCl, 3 mM DTT, 100 mM sodium citrate, pH 4.5). Reactions were started by the addition of 50 μM N-lauroyl ceramide (Nu-Chek Prep, Elysian, MN) and carried out for 1 h at 37 °C. Reactions were stopped by addition of a mixture of CHCl3/MeOH (2:1) containing 1 nmol of 11-lauroleic acid (Nu-Chek Prep). The organic phases were collected, dried under nitrogen, and analyzed by UPLC/MS (ACQUITY, Waters) in the negative-ion mode monitoring the reaction product (lauric acid, m/z: 199) using 11-lauroleic acid as internal standard. Lipids were eluted on an ACQUITY UPLC BEH C18 column (50 mm length, 2.1 mm ID, 1.7 μm pore size, Waters) at 0.5 mL min–1 for 1.5 min with a gradient of MeCN and H2O, both containing 0.25% acetic acid and 5 mM ammonium acetate (70 to 100% MeCN in 0.5 min, 100% MeCN for 0.5 min, 70% MeCN for 0.4 min). The column temperature was 40 °C. Electrospray ionization (ESI) was in the negative mode, capillary voltage was 1 kV, and cone voltage was 50 V. N2 was used as drying gas at a flow rate of 500 L h–1 and at a temperature of 400 °C. The [M–H]− ion was monitored in the selected-ion monitoring mode (m/z values: lauric acid 199, 11-lauroleic acid 197.35). Calibration curves were generated with authentic lauric acid (Nu-Chec Prep).
Lipid Extraction and Ceramide Analysis
Lipid extraction and sphingolipid measurements were performed as previously described.36,37 Lipids were extracted with a CHCl3/MeOH mixture (2:1, 3 mL) containing internal standards. The organic phase was collected, dried under nitrogen, and dissolved in CHCl3/MeOH (1:3) for LC/MS analyses. Ceramides and sphingosine were analyzed by LC/MS/MS, using a Waters ACQUITY UPLC coupled to a Waters Xevo TQMS and interfaced with an ESI ion source. Separation was performed on a Waters ACQUITY BEH C18 1.7 μm column (2.1 × 50 mm) at 60 °C. A linear gradient of 0.1% formic acid in MeCN/isopropyl alcohol (20:80) as solvent B in 0.1% formic acid in MeCN/H2O (20:80) as solvent A was applied at a flow rate of 0.4 mL min–1. Detection of sphingolipids was performed in positive-ion mode. Capillary voltage was 3.5 kV, and cone voltage was 25 V. The source temperature and desolvation temperatures were set at 120 and 600 °C, respectively. Desolvation gas and cone gas (N2) flows were 800 and 20 L h–1, respectively. Ceramides were identified by comparison of their LC retention times and MS/MS fragmentation patterns with those of authentic standards (Avanti Polar Lipids). Multiple Reaction Monitoring (MRM) ion chromatograms were used to quantify myristoyl ceramide (C14:0, m/z: 492.5 > 264.3), palmitoyl ceramide (C16:0, m/z 520.3 > 264.3), stearoyl ceramide (C18:0, m/z: 548.3 > 264.3), lignoceroyl ceramide (C24:0, m/z: 632.3 > 264.3), and nervonoyl ceramide (C24:1 m/z: 630.3 > 264.3) using lauroyl ceramide standard (m/z: 464.5 > 264.3). Detection and analysis were controlled by Waters MassLynx software version 4.1. Sphingosine was identified by comparison of its LC retention times and MS2 fragmentation patterns with those of authentic standards (Avanti Polar Lipids). Extracted ion chromatograms were used to quantify sphingosine standard (d18:1, m/z: 300.5 > 282.5). Detection and analysis were controlled by Waters MassLynx software version 4.1. Calibration curves were prepared for every experiment.
Statistics
GraphPad Prism software (GraphPad Software, Inc., USA) was used for statistical analysis. Data were analyzed using the Student t test or one-way ANOVA followed by the Bonferroni post hoc test for multiple comparisons. Differences between groups were considered statistically significant at values of p < 0.05. Results are expressed as mean ± S.E.M.
EC50 Determination in Primary Fibroblast Cells from Krabbe’s Disease Patients
Cells were plated in a 6-well plate. After 24 h, cells were treated with 22m at different concentrations for 2 h. Next, cells were washed with PBS, and cell pellets were washed, collected, and stored at −80 °C. Finally, cell pellets were lysed, and hAC activity in cell lysates was analyzed using the same biochemical fluorogenic assay as described for compound IC50 determination. Using this methodology, EC50 was determined to be 0.41 ± 0.1 μM. The EC50 value is a mean of two independent experiments, each performed in two technical replicates.
In Vitro Physicochemical and Metabolic Stability Assays
Aqueous Kinetic Solubility Assay
The aqueous kinetic solubility was determined from a 10 mM MeCN stock solution of test compound in Phosphate-Buffered Saline (PBS) at pH 7.4. The study was performed by incubation of an aliquot of 10 mM MeCN stock solution in PBS (pH 7.4) at a target concentration of 250 μM. The incubation was carried out under shaking at 25 °C for 1 h followed by centrifugation at 21100 × g for 30 min. The supernatant was analyzed by UPLC/MS for the quantification of the dissolved compound (in μM) by UV at a specific wavelength (215 nm). The aqueous kinetic solubility (in μM) was calculated by dividing the peak area of the dissolved test compound (supernatant) by the peak area of the test compound in the reference (250 μM in MeCN) and further multiplied by the target concentration and dilution factor. The UPLC/MS analyses were performed on a Waters ACQUITY UPLC/MS system consisting of a single quadrupole detector (SQD) Mass Spectrometer (MS) equipped with an Electrospray Ionization (ESI) interface and a Photodiode Array Detector (PDA). The PDA range was 210–400 nm. ESI in positive mode was used in the mass scan range of 100–650 Da. The analyses were run on an ACQUITY UPLC BEH C18 column (50 × 2.1 mm ID, particle size 1.7 μm) with a VanGuard BEH C18 precolumn (5 × 2.1 mm ID, particle size 1.7 μm), using 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN/H2O (95:5) at pH 5 (B) as the mobile phase. Values are reported as mean values of ≥2 experiments performed.
Chemical Stability Assay
Chemical stability of selected compounds was evaluated under physiological pH conditions (0.01 M phosphate-buffered saline, pH 7.4) for up to 8 h. The buffer was added with 10% MeCN. Stock solutions of each compound (10 mM) were freshly prepared in MeCN. Each compound was incubated at a final concentration of 1 μM in preheated buffer (37 °C). The sample solutions were divided into aliquots in glass vials (preheated at 37 °C) for each time point. The samples were maintained at 37 °C in the UPLC/MS autosampler during the study (no shaking). A reference solution of each compound (final concentration: 1 μM) in preheated MeCN was prepared from the stock solutions and maintained at 37 °C in the UPLC/MS autosampler during the study. For each time point, the samples were analyzed directly by LC/MS without any further sample preparation. The samples were analyzed by integrating the corresponding MRM peak areas. The relative compound concentration was calculated by dividing the peak area at each time point by the peak area at t = 0 min. The reference solution was analyzed at the beginning (t = 0 min) and at the end of the study (t = 8 h). The apparent half-life (t1/2) of the disappearance of the compound was calculated using the best fitting equation by GraphPad Prism (GraphPad Software, Inc., USA). The analyses were performed on a Waters ACQUITY UPLC/MS TQD system consisting of a triple quadrupole detector (TQD) MS equipped with an ESI interface and a photodiode array detector. The analyses were run on an ACQUITY UPLC BEH C18 1.7 μm 2.1 × 50 mm column with a VanGuard BEH C18 1.7 μm preolumn at 40 °C. For each compound, the appropriate mobile phase was chosen. ESI was applied in positive mode. Values are the mean of at least two independent experiments performed in two technical replicates.
In Vitro Plasma Stability Study
Freshly prepared 10 mM MeCN stock solution of test compound was diluted 50-fold with DMSO/H2O (1:1) and incubated at 37 °C for 2 h with mouse plasma added in 5% DMSO (preheated at 37 °C for 10 min). The final concentration was 2 μM. At each time point (0, 5, 15, 30, 60, and 120 min), 50 μL of incubation mixture was diluted with 200 μL of cold MeCN spiked with 200 nM internal standard followed by centrifugation at 3300 × g for 20 min. The supernatant was further diluted with H2O (1:1) for analysis. The concentration of test compound was quantified by LC/MS/MS on a Waters ACQUITY UPLC/MS TQD system consisting of a TQD MS equipped with an ESI interface. The analyses were run on an ACQUITY UPLC BEH C18 (50 × 2.1 mm ID, particle size 1.7 μm) with a VanGuard BEH C18 precolumn (5 × 2.1 mm ID, particle size 1.7 μm) at 40 °C. For each compound, the appropriate mobile phase was chosen. ESI was applied in positive mode. The response factors, calculated on the basis of the internal standard peak area, were plotted over time. When possible, response vs time profiles were fitted with Prism (GraphPad Software, Inc., USA) to estimate compound t1/2 in plasma. Values are the mean of at least two independent experiments performed in two technical replicates.
In Vitro Microsomal Stability Study
Freshly prepared 10 mM MeCN stock solution of test compound was preincubated at 37 °C for 15 min with mouse liver microsomes added in 0.1 M Tris HCl buffer (pH 7.4). The final concentration was 4.6 μM. After preincubation, the cofactors (NADPH, G6P, G6PDH, and MgCl2 predissolved in 0.1 M Tris HCl) were added to the incubation mixture, and the incubation was continued at 37 °C for 1 h. At each time point (0, 5, 15, 30, and 60 min), 30 μL of incubation mixture was diluted with 200 μL of cold MeCN spiked with 200 nM internal standard followed by centrifugation at 3300 × g for 15 min. The supernatant was further diluted with H2O (1:1) for analysis. The concentration of the test compound was quantified by LC/MS/MS on a Waters ACQUITY UPLC/MS TQD system consisting of a TQD MS equipped with an ESI interface. The analyses were run on an ACQUITY UPLC BEH C18 (50 × 2.1 mm ID, particle size 1.7 μm) with a VanGuard BEH C18 precolumn (5 × 2.1 mm ID, particle size 1.7 μm) at 40 °C. For each compound, the appropriate mobile phase was chosen. ESI was applied in positive mode. The percentage of test compound remaining at each time point relative to t = 0 was calculated. t1/2 was determined by a one-phase decay equation using a nonlinear regression of compound concentration vs time. Values are the mean of at least two independent experiments performed in two technical replicates.
In Vitro Mouse Plasma and Mouse Brain Tissue Protein Binding
Studies were performed by the DMPK Group at Shanghai ChemPartner Co., Ltd., using the equilibrium dialysis method. Values are the mean of two technical replicates.
Animal Models
In Vivo Pharmacokinetic Study
Male CD1 mice (22–24 g, 6 weeks old, SLAC Laboratory Animal Co. Ltd.) were group-housed in ventilated cages and had free access to water and food. They were maintained under a 24 h light/dark cycle at controlled temperature and relative humidity. All efforts were made to minimize animal suffering and to use the minimal number of animals required to produce reliable results. All procedures were performed in accordance with the Ethical Guidelines on the Protection of Animals Used for Scientific Purposes at the DMPK Group at Shanghai ChemPartner Co., Ltd. 22m was administrated intravenously (i.v.) at 3 mg kg–1 (vehicle: 100% saline at 0.6 mg mL–1) via tail vein injection (N = 18) and via oral administration (p.o.) at 10 mg kg–1 (vehicle: 100% saline at 2.0 mg mL–1) by oral gavage (N = 18). Sample collection. Samples were collected at 0.25, 0.5, 1, 4, 8, and 24 h. Animals were sacrificed 24 h after 22m administration; plasma, brain, and CSF samples were collected and stored at −80 °C. Blood collection: The animal was restrained manually, and approximately 150 μL of blood/time point was collected into the K2EDTA tube via retro orbital puncture under anesthesia with isoflurane. The blood sample was put on ice and centrifuged to obtain the plasma sample (2000 × g, 5 min under 4 °C) within 15 min and then acidified following 100 μL of plasma + 1.0 μL of formic acid. An aliquot of 20 μL sample (pretreatment with 1% formic acid) was added with 200 μL of IS (propranolol, 40 ng mL–1) in MeCN. The mixture was vortexed for 5 min and centrifuged at 6000 rpm for 10 min. The 0.5 μL mixture was injected into LC/MS/MS. Brain collection: Brain was removed and immediately homogenized immediately for 2 min with three volumes (v/w) of homogenizing solution (PBS:formic acid = 100:1), and then the solution was stored in tubes under −70 °C until analysis. An aliquot of 20 μL sample was added with 200 μL of MeCN, which contains IS (propranolol, 40 ng mL–1) for protein precipitation. The mixture was vortexed for 5 min and centrifuged at 6000 rpm for 10 min. The 0.5 μL mixture was injected into LC/MS/MS. CSF collection: A midline incision was made on the neck. The muscle under the skin was cut to expose the cisterna magna. The CSF was collected by capillary. An aliquot of 3 μL sample was added with 90 μL of IS (propranolol, 40 ng mL–1) in MeCN:H2O = 2:1 (added 1% formic acid). The mixture was vortexed for 5 min and centrifuged at 6000 rpm for 10 min. The 1.5 μL mixture was injected into LC/MS/MS. 22m sample levels were monitored on an LC/MS/MS-19 (API5500, Qtriple) system, using the calibration curve and propanol as internal standard. Chromatography was carried out on a Waters BEH C18 column (2.1 × 50 mm, 1.7 μm) at 60 °C, setting a flow rate of 0.60 mL min–1. Mobile phases were as follows: A = H2O/0.025% formic acid/1 mM NH4OAc and B = MeOH/0.025% formic acid/1 mM NH4OAc. After the initial 0.20 min at 10% of mobile phase B, the percentage of mobile phase B increased at 70% at 0.50 min, reaching steadily 90% in the range of 0.80–1.30 min. Then the system returned to the initial conditions in a single step until 1.80 min. The following parent (m/z)/daughter (m/z) transitions were monitored: 22m: m/z = 332.20/333.20 Da; propanol (IS): m/z: 260.30/116.10 Da.
Maximum Tolerated Dose (MTD) Study
An MTD study was conducted on male C57BL/6 mice (15–19 g, 5 weeks old, SLAC Laboratory Animal Co. Ltd.). Animals were injected via intraperitoneal (i.p.) injection with single administration at 20 mg kg–1 (N = 18, vehicle: 100% saline at 0.6 mg mL–1) and multiple administrations for a duration of 4 days at 20 (N = 18, day 1), 40 (N = 18, day 2), 80 (N = 18, day 3), and 120 mg kg–1 (N = 18, day 4). Clinical observations/samples of plasma, CSF, and brain were collected at 0.25, 0.5, 1, 4, 8, and 24 h. All procedures were performed in accordance with the Ethical Guidelines on the Protection of Animals Used for Scientific Purposes at the DMPK Group at Shanghai ChemPartner Co., Ltd.
In Vivo Mouse Model Studies
4L;C* mice (C57BL/6 J/129SvEV) were randomly assigned to three treatment groups (N = 4–8 with mixed males and females) and dosed once a day i.p. with 90 or 30 mg kg–1 of 22m or vehicle. Treatment started at 5 days of age for a duration of 14 days. The application volume was set to 10 μL per gram of body weight, and dosage was adjusted accordingly. Animals of all groups were sacrificed 1 h after the last dose, and the brain tissues and plasma were collected. The left brain containing cortex, cerebella, thalamus, and brainstem was analyzed for SphL levels by MS. Data were analyzed using the Student t test. The right brain containing cortex, cerebella, thalamus, and brainstem and plasma were analyzed for drug levels of 22m. All mice were housed under pathogen-free conditions in the animal facility, and animal experiment was performed according to the IACUC approved protocol (2018-0056) at Cincinnati Children’s Hospital Research Foundation. Wild Type (WT) (GALC+/+) and Twitcher (Twi) (GALC–/−) mice were genotyped by PCR as previously described.58 Twi mice were randomly assigned to three treatment groups (N = 3 males + N = 3 females for each group) and dosed once a day i.p. with 90 or 30 mg kg–122m or vehicle for a duration of 20 days. Treatment started at 10 days of age. The application volume was set to 10 μL per gram of body weight, and dosage was adjusted accordingly. Two groups (N = 3 males + N = 3 females for each group) of WT controls treated with vehicle or high dose (90 mg kg–1) of 22m were also included in the study. Animals of all groups were sacrificed 1 h after the last dose, and the brain tissues and plasma were collected. The left brain containing cortex, cerebella, thalamus, and brainstem was analyzed for SphL levels by MS. Data were analyzed using the Student t test. The right brain containing cortex, cerebella, thalamus, and brainstem and plasma were analyzed for drug levels of 22m. All animal work in this study was performed in accordance with approved animal protocols from the Animal Care and Use Committee at the University of Illinois at Chicago.
Acknowledgments
The authors thank Silvia Venzano, Luca Goldoni, and Marina Veronesi for their technical support.
Glossary
Abbreviations Used
- AC
acid ceramidase
- Cer
ceramide
- GALC
β-galactosyl-ceramidase
- GalCer
galactosylceramide
- GalSph
galactosylsphingosine
- GD
Gaucher’s disease
- GCase
β-glucocerebrosidase
- GluCer
glucosylceramide
- GluSph
glucosylsphingosine
- LSDs
lysosomal storage diseases
- KD
Krabbe’s disease
- SphLs
sphingolipids
- Sph1P
sphingosine 1-phosphate
- Twi
Twitcher
- AcOH
glacial acetic acid
- MeCN
acetonitrile
- NH4OAc
ammonium acetate
- NH4Cl
ammonium chloride
- n-BuLi
n-butyllithium
- CDI
1′-carbonyldiimidazole
- CHCl3
chloroform
- Cy
cyclohexane
- Celite
diatomaceous earth
- Et2O
diethyl ether
- DIPEA
N,N-diisopropylethylamine
- (Pd(dppf)Cl2)
[1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II)
- EtOH
ethanol
- EtOAc
ethyl acetate
- equiv.
equivalent
- HCl
hydrochloric acid
- LiCl
lithium chloride
- MeOH
methanol
- MeMgBr
methylmagnesium bromide
- Pd/C
palladium on carbon
- [B2(pin)2]
bis(pinacolato)diboron
- KOAc
potassium acetate
- K2CO3
potassium carbonate
- K3PO4
potassium phosphate tribasic
- Rt
retention time
- SiO2
silica gel
- NaOAc
sodium acetate
- NaHCO3
sodium bicarbonate
- Na2CO3
sodium carbonate
- Na2SO4
sodium sulfate
- NaBH(OAc)3
sodium triacetoxyborohydride
- Pd(PPh3)4
tetrakis(triphenylphosphine)palladium(0)
- Dess–Martin periodinane
1,1,1-tris(acetyloxy)-1,1-dihydro-1,2-benziodoxol-3-(1H)-one
- Et3N
trimethylamine
- Boc2O
di-tert-butyl dicarbonate
- TZD
2,4-thiazolidinedione
- p-TsOH
p-toluenesulfonic acid monohydrate
- H2O
water
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b02004.
Vmax and KM determinations; concentration–response curve of 22m in primary fibroblast cells of Krabbe’s patients; 1H NMR and 13C NMR spectra of the final compounds; retention times and UPLC analytical methods of the final compounds; UPLC traces of the final compounds (PDF)
A csv file containing molecular formula strings (CSV)
Author Present Address
◆ X4 Pharmaceuticals, 955 Massachusetts Avenue, 4th Floor, Cambridge, MA 02139, United States (Y.S.).
Author Contributions
○ S.D.M. and P.T. contributed equally. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): All the work in the manuscript was funded by Lysosomal Therapeutics Inc. A Sponsored Research Agreement was signed between Fondazione Istituto Italiano di Tecnologia, Drug Discovery and Development (D3)-Validation and Lysosomal Therapeutics Inc.
Supplementary Material
References
- Gault C. R.; Obeid L. M.; Hannun Y. A. An overview of sphingolipid metabolism: from synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. 10.1007/978-1-4419-6741-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun Y. A.; Obeid L. M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. 10.1038/nrm.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts B.; Johnson A.; Lewis J.; Raff M.; Roberts K.; Walter P. In Molecular Biology of the Cell; 5th ed. th ed.; Garland Science: New York, USA, Ed. 2008, 779–787. [Google Scholar]
- Saftig P.; Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- Bernardo K.; Hurwitz R.; Zenk T.; Desnick R. J.; Ferlinz K.; Schuchman E. H.; Sandhoff K. Purification, characterization, and biosynthesis of human acid ceramidase. J. Biol. Chem. 1995, 270, 11098–11102. 10.1074/jbc.270.19.11098. [DOI] [PubMed] [Google Scholar]
- Mao C.; Obeid L. M. Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2008, 1781, 424–434. 10.1016/j.bbalip.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coant N.; Sakamoto W.; Mao C.; Hannun Y. A. Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv. Biol. Regul. 2017, 63, 122–131. 10.1016/j.jbior.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun Y. A. Functions of ceramide in coordinating cellular responses to stress. Science 1996, 274, 1855–1859. 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- Ballou L. R.; Laulederkind S. J. F.; Rosloniec E. F.; Raghow R. Ceramide signalling and the immune response. Biochim. Biophys. Acta 1996, 1301, 273–287. 10.1016/0005-2760(96)00004-5. [DOI] [PubMed] [Google Scholar]
- Ruvolo P. P. Intracellular signal transduction pathways activated by ceramide and its metabolites. Pharmacol. Res. 2003, 47, 383–392. 10.1016/S1043-6618(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Huang X.; Withers B. R.; Dickson R. C. Sphingolipids and lifespan regulation. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2014, 1841, 657–664. 10.1016/j.bbalip.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettus B. J.; Chalfant C. E.; Hannun Y. A. Ceramide in apoptosis: an overview and current perspectives. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2002, 1585, 114–125. 10.1016/S1388-1981(02)00331-1. [DOI] [PubMed] [Google Scholar]
- Morales A.; Lee H.; Goñi F. M.; Kolesnick R.; Fernandez-Checa J. C. Sphingolipids and cell death. Apoptosis 2007, 12, 923–939. 10.1007/s10495-007-0721-0. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Desai N. N.; Olivera A.; Seki T.; Brooker G.; Spiegel S. Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J. Cell Biol. 1991, 114, 155–167. 10.1083/jcb.114.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne S. G.; Milstien S.; Spiegel S. Sphingosine-1-phosphate: dual messenger functions. FEBS Lett. 2002, 531, 54–57. 10.1016/S0014-5793(02)03480-4. [DOI] [PubMed] [Google Scholar]
- Spiegel S.; Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- Takabe K.; Spiegel S. Export of sphingosine-1-phosphate and cancer progression. J. Lipid Res. 2014, 55, 1839–1846. 10.1194/jlr.R046656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Realini N.; Palese F.; Pizzirani D.; Pontis S.; Basit A.; Bach A.; Ganesan A.; Piomelli D. Acid ceramidase in melanoma: expression, localization, and effects of pharmacological inhibition. J. Biol. Chem. 2016, 291, 2422–2434. 10.1074/jbc.M115.666909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuchman E. H. Acid ceramidase and the treatment of ceramide diseases: The expanding role of enzyme replacement therapy. Biochim. Biophys. Acta, Mol. Basis Dis. 2016, 1862, 1459–1471. 10.1016/j.bbadis.2016.05.001. [DOI] [PubMed] [Google Scholar]
- Iwabuchi K.; Nakayama H.; Oizumi A.; Suga Y.; Ogawa H.; Takamori K. Role of ceramide from glycosphingolipids and its metabolites in immunological and inflammatory responses in humans. Mediators Inflammation 2015, 2015, 1–10. 10.1155/2015/120748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieselmann V. Lysosomal storage diseases. Biochim. Biophys. Acta, Mol. Basis Dis. 1995, 1270, 103–136. 10.1016/0925-4439(94)00075-2. [DOI] [PubMed] [Google Scholar]
- Vanier M.-T.Disorders of Sphingolipid Metabolism. In Inborn Metabolic Diseases: Diagnosis and Treatment; Fernandes J.; Saudubray J.-M.; van den Berghe G.; Walter J. H., Eds. Springer: Berlin, Heidelberg, 2006, 479–494. [Google Scholar]
- Ballabio A.; Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim. Biophys. Acta, Mol. Cell Res. 2009, 1793, 684–696. 10.1016/j.bbamcr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Cox T. M.; Cachón-González M. B. The cellular pathology of lysosomal diseases. J. Pathol. 2012, 226, 241–254. 10.1002/path.3021. [DOI] [PubMed] [Google Scholar]
- Brady R. O.; Kanfer J.; Shapiro D. The metabolism of glucocerebrosides. I. Purification and properties of a glucocerebroside-cleaving enzyme from spleen tissue. J. Biol. Chem. 1965, 240, 39–43. [PubMed] [Google Scholar]
- Sorbera L. A.; Sundaravinayagam D.; Dulsat C.; Rosa E. Therapeutic targets for Gaucher′s disease. Drugs Future 2009, 34, 1001. 10.1358/dof.2009.034.12.1444443. [DOI] [Google Scholar]
- Stirnemann J.; Belmatoug N.; Camou F.; Serratrice C.; Froissart R.; Caillaud C.; Levade T.; Astudillo L.; Serratrice J.; Brassier A.; Rose C.; de Villemeur T. B.; Berger M. G. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int. J. Mol. Sci. 2017, 18, 441–471. 10.3390/ijms18020441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won J.-S.; Singh A. K.; Singh I. Biochemical, cell biological, pathological, and therapeutic aspects of Krabbe’s disease. J. Neurosci. Res. 2016, 94, 990–1006. 10.1002/jnr.23873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson O.; Svennerholm L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J. Neurochem. 1982, 39, 709–718. 10.1111/j.1471-4159.1982.tb07950.x. [DOI] [PubMed] [Google Scholar]
- Sidransky E. Gaucher disease: complexity in a ″simple″ disorder. Mol. Genet. Metab. 2004, 83, 6–15. 10.1016/j.ymgme.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Ferraz M. J.; Marques A. R. A.; Appelman M. D.; Verhoek M.; Strijland A.; Mirzaian M.; Scheij S.; Ouairy C. M.; Lahav D.; Wisse P.; Overkleeft H. S.; Boot R. G.; Aerts J. M. Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. 10.1002/1873-3468.12104. [DOI] [PubMed] [Google Scholar]
- Murugesan V.; Chuang W.-L.; Liu J.; Lischuk A.; Kacena K.; Lin H.; Pastores G. M.; Yang R.; Keutzer J.; Zhang K.; Mistry P. K. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 2016, 91, 1082–1089. 10.1002/ajh.24491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K.; Suzuki K. The Twitcher Mouse: A model for Krabbe disease and for experimental therapies. Brain Pathol. 1995, 5, 249–258. 10.1111/j.1750-3639.1995.tb00601.x. [DOI] [PubMed] [Google Scholar]
- Li Y.; Xu Y.; Benitez B. A.; Nagree M. S.; Dearborn J. T.; Jiang X.; Guzman M. A.; Woloszynek J. C.; Giaramita A.; Yip B. K.; Elsbernd J.; Babcock M. C.; Lo M.; Fowler S. C.; Wozniak D. F.; Vogler C. A.; Medin J. A.; Crawford B. E.; Sands M. S. Genetic ablation of acid ceramidase in Krabbe disease confirms the psychosine hypothesis and identifies a new therapeutic target. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 20097–20103. 10.1073/pnas.1912108116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebai A.; Gorelik A.; Li Z.; Illes K.; Nagar B. Structural basis for the activation of acid ceramidase. Nat. Commun. 2018, 9, 1621. 10.1038/s41467-018-03844-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Realini N.; Solorzano C.; Pagliuca C.; Pizzirani D.; Armirotti A.; Luciani R.; Costi M. P.; Bandiera T.; Piomelli D. Discovery of highly potent acid ceramidase inhibitors with in vitro tumor chemosensitizing activity. Sci. Rep. 2013, 3, 1035. 10.1038/srep01035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzirani D.; Pagliuca C.; Realini N.; Branduardi D.; Bottegoni G.; Mor M.; Bertozzi F.; Scarpelli R.; Piomelli D.; Bandiera T. Discovery of a new class of highly potent inhibitors of acid ceramidase: synthesis and structure-activity relationship (SAR). J. Med. Chem. 2013, 56, 3518–3530. 10.1021/jm301879g. [DOI] [PubMed] [Google Scholar]
- Diamanti E.; Bottegoni G.; Goldoni L.; Realini N.; Pagliuca C.; Bertozzi F.; Piomelli D.; Pizzirani D. Pyrazole-based acid ceramidase inhibitors: design, synthesis, and structure-activity relationships. Synthesis 2016, 48, 2739–2756. 10.1055/s-0035-1561456. [DOI] [Google Scholar]
- Pizzirani D.; Bach A.; Realini N.; Armirotti A.; Mengatto L.; Bauer I.; Girotto S.; Pagliuca C.; De Vivo M.; Summa M.; Ribeiro A.; Piomelli D. Benzoxazolone carboxamides: potent and systemically active inhibitors of intracellular acid ceramidase. Angew. Chem., Int. Ed. 2014, 54, 485–489. 10.1002/anie.201409042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach A.; Pizzirani D.; Realini N.; Vozella V.; Russo D.; Penna I.; Melzig L.; Scarpelli R.; Piomelli D. Benzoxazolone carboxamides as potent acid ceramidase inhibitors: synthesis and structure-activity relationship (SAR) studies. J. Med. Chem. 2015, 58, 9258–9272. 10.1021/acs.jmedchem.5b01188. [DOI] [PubMed] [Google Scholar]
- Ortega J. A.; Arencibia J. M.; La Sala G.; Borgogno M.; Bauer I.; Bono L.; Braccia C.; Armirotti A.; Girotto S.; Ganesan A.; De Vivo M. Pharmacophore identification and scaffold exploration to discover novel, potent, and chemically stable inhibitors of acid ceramidase in melanoma cells. J. Med. Chem. 2017, 60, 5800–5815. 10.1021/acs.jmedchem.7b00472. [DOI] [PubMed] [Google Scholar]
- Li X.; Russell R. K.; Spink J.; Ballentine S.; Teleha C.; Branum S.; Wells K.; Beauchamp D.; Patch R.; Huang H.; Player M.; Murray W. Process development for scale-up of a novel 3,5-substituted thiazolidine-2,4-dione compound as a potent inhibitor for estrogen-related receptor 1. Org. Process Res. Dev. 2014, 18, 321–330. 10.1021/op400325r. [DOI] [Google Scholar]
- DeOrazio R. J.; Maeng J.-H.; Manning D. D.; Sherer B. A.; Scott I. L.; Nikam S. S. A simple strategy for the preparation of 6-substituted 3H-benzoxazol-2-ones and 3H-benzothiazol-2-ones. Synth. Commun. 2011, 41, 3551–3555. 10.1080/00397911.2010.519093. [DOI] [Google Scholar]
- Rynearson K. D.; Charrette B.; Gabriel C.; Moreno J.; Boerneke M. A.; Dibrov S. M.; Hermann T. 2-Aminobenzoxazole ligands of the hepatitis C virus internal ribosome entry site. Bioorg. Med. Chem. Lett. 2014, 24, 3521–3525. 10.1016/j.bmcl.2014.05.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toto P.; Gesquière J.-C.; Deprez B.; Willand N. Synthesis of N-(iodophenyl)-amides via an unprecedented Ullmann-Finkelstein tandem reaction. Tetrahedron Lett. 2006, 47, 1181–1186. 10.1016/j.tetlet.2005.12.022. [DOI] [Google Scholar]
- Knöelker H.-J.; Braxmeier T.; Schlechtingen G. A novel method for the synthesis of isocyanates under mild conditions. Angew. Chem., Int. Ed. Engl. 1995, 34, 2497–2500. 10.1002/anie.199524971. [DOI] [Google Scholar]
- Eckert H.; Forster B. Triphosgene, a crystalline phosgene substitute. Angew. Chem., Int. Ed. Engl. 1987, 26, 894–895. 10.1002/anie.198708941. [DOI] [Google Scholar]
- Dementiev A.; Joachimiak A.; Nguyen H.; Gorelik A.; Illes K.; Shabani S.; Gelsomino M.; Ahn E.-Y. E.; Nagar B.; Doan N. Molecular mechanism of inhibition of acid ceramidase by carmofur. J. Med. Chem. 2019, 62, 987–992. 10.1021/acs.jmedchem.8b01723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S. M.; Kwon H. J. Acid ceramidase, an emerging target for anti-cancer and anti-angiogenesis. Arch. Pharmacal Res. 2019, 42, 232–243. 10.1007/s12272-019-01114-3. [DOI] [PubMed] [Google Scholar]
- Ishikawa M.; Hashimoto Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 2011, 54, 1539–1554. 10.1021/jm101356p. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. Improving drug design: an update on recent applications of efficiency metrics, strategies for replacing problematic elements, and compounds in nontraditional drug space. Chem. Res. Toxicol. 2016, 29, 564–616. 10.1021/acs.chemrestox.6b00043. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists; Wiley-Interscience: Hoboken, New Jersey, USA, Ed. 2005, 48–248. [PubMed] [Google Scholar]
- Strelow J. D. W.; Iversen P. W.; Brooks H. B.; Radding J. A.; McGee J.; Weidner J., In Assay Guidance Manual; Sittampalam G. S. G. A.; Brimacombe K.; Arkin M.; Auld D.; Austin C. P.; Baell J.; Bejcek B.; Caaveiro J. M. M.; Chung T. D. Y.; Coussens N. P.; Dahlin J. L.; Devanaryan V.; Foley T. L.; Glicksman M.; Hall M. D.; Haas J. V.; Hoare S. R. J.; Inglese J.; Iversen P. W.; Kahl S. D.; Kales S. C.; Kirshner S.; Lal-Nag M.; Li Z.; McGee J.; McManus O.; Riss T.; Saradjian P.; Trask O. J.; Weidner J. R. Jr.; Wildey M. J.; Xia M.; Xu X., Ed. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences, 2004, 65–86. [PubMed] [Google Scholar]
- Gorelik A.; Gebai A.; Illes K.; Piomelli D.; Nagar B. Molecular mechanism of activation of the immunoregulatory amidase NAAA. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E10032–E10040. 10.1073/pnas.1811759115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K.; McKinney M. K.; Cravatt B. F. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 2008, 108, 1687–1707. 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman J. L.; Simon G. M.; Cravatt B. F. A Comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discovery 2018, 17, 623–639. 10.1038/nrd.2018.115. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Liou B.; Ran H.; Skelton M. R.; Williams M. T.; Vorhees C. V.; Kitatani K.; Hannun Y. A.; Witte D. P.; Xu Y.-H.; Grabowski G. A. Neuronopathic Gaucher disease in the mouse: viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010, 19, 1088–1097. 10.1093/hmg/ddp580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.