

Abstract
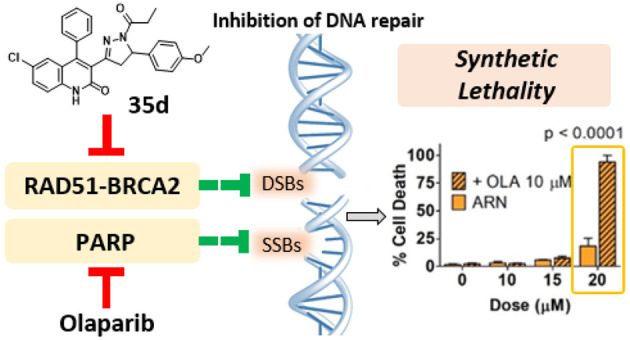
Synthetic lethality is an innovative framework for discovering novel anticancer drug candidates. One example is the use of PARP inhibitors (PARPi) in oncology patients with BRCA mutations. Here, we exploit a new paradigm based on the possibility of triggering synthetic lethality using only small organic molecules (dubbed “fully small-molecule-induced synthetic lethality”). We exploited this paradigm to target pancreatic cancer, one of the major unmet needs in oncology. We discovered a dihydroquinolone pyrazoline-based molecule (35d) that disrupts the RAD51-BRCA2 protein–protein interaction, thus mimicking the effect of BRCA2 mutation. 35d inhibits the homologous recombination in a human pancreatic adenocarcinoma cell line. In addition, it synergizes with olaparib (a PARPi) to trigger synthetic lethality. This strategy aims to widen the use of PARPi in BRCA-competent and olaparib-resistant cancers, making fully small-molecule-induced synthetic lethality an innovative approach toward unmet oncological needs.
Introduction
Synthetic lethality is a new opportunity for discovering new anticancer molecules for personalized targeted therapies. The concept derives from genetic studies in model organisms.1−5 Two genes are synthetically lethal if the perturbation of either gene alone has no effect on cell viability, but the simultaneous impairment of both genes results in cell death. In principle, small organic molecules can target the synthetically lethal partner of an altered gene in cancer cells but not in normal cells. This creates opportunities to selectively kill cancer cells while sparing normal cells.6−12
The DNA repair and DNA damage response (DDR) pathways are suitable for the application of synthetic lethality as a novel anticancer therapeutic strategy.13−15 Genome instability is a hallmark of cancer.16 DNA damage occurs constantly in cells due to the continuous exposition to endogenous and exogenous stressors. Consequently, cells have evolved a complex coordinated DDR, which orchestrates a network of cellular processes to repair DNA damage and preserve genome integrity. DDR thus prevents the transmission of altered genetic material to daughter cells and acts as a tumor-suppressive barrier. Defects in DDR are associated with the accumulation of oncogenic mutations and genome instability, and they contribute to cancer initiation and progression. However, cancer cells with defects in one DDR pathway can become reliant on other pathways for their survival. Targeting these other DDR pathways can potentially cause selective cancer cell death through synthetic lethality. The classic example is the clinical application of poly (ADP-ribose)polymerase (PARP) inhibitors in oncology patients with BRCA1/2 mutations. PARP is crucial for repairing DNA single-strand breaks (SSBs), whereas BRCA1/2 are important for repairing DNA double-strand breaks (DSBs) by homologous recombination (HR). The simultaneous impairment of both repair mechanisms results in cell-cycle arrest and apoptosis of cancer cells through synthetic lethality. In 2014, olaparib was the first PARP inhibitor (PARPi) approved to treat advanced ovarian cancer associated with defective BRCA genes.17 In 2018, olaparib was approved to treat metastatic breast tumors associated with germline BRCA mutations.18 In 2019, olaparib gained the FDA approval as first-line maintenance treatment of germline BRCA-mutated metastatic pancreatic cancer. It appears to be a new treatment option for this disease, which is one of the major unmet needs in oncology.19
One of BRCA2’s key mechanisms in DDR is to recruit RAD51, an evolutionarily conserved recombinase, at the site of DSBs where it performs DSB repair through HR.20 Additionally, the expression of RAD51 and the rate of RAD51-mediated HR are both elevated in a wide variety of cancers (e.g., breast, pancreatic).21 Moreover, the cellular amount of RAD51 is positively correlated with resistance to radiotherapies or chemotherapies that induce DNA damage.22,23
The RAD51-BRCA2 interaction is mediated by eight well-conserved motifs, known as BRC repeats.24−27 The X-ray crystallographic structure of the fourth BRC repeat (BRC4) is available in complex with the catalytic domain of RAD51, making the RAD51-BRCA2 interaction suitable for the structure-based design of small molecule inhibitors of protein–protein interactions (PPIs). Indeed, Lee et al. recently identified a small molecule inhibitor of RAD51-BRCA2 for potential cancer treatment.28,29
In this context, we recently proposed a new anticancer drug discovery concept, dubbed “fully small-molecules-induced synthetic lethality”.30,31 This concept combines RAD51-BRCA2 disruptors with olaparib to simultaneously impair two DNA repair pathways, thus mimicking the synthetic lethality described above. We carried out a successful virtual screening campaign at the FxxA pocket (i.e., zone I), one of the two RAD51 pockets responsible for BRC4 binding. This allowed us to discover a series of triazole-based compounds. Compounds 1 and 2 were selected as initial hit candidates (Figure 1). In line with our hypothesis, 2 increased the sensitivity to olaparib in pancreatic cancer cells (BxPC-3) with fully functional BRCA2. Notably, this synergistic effect was not observed in Capan-1, pancreas adenocarcinoma cells that lack BRCA2.30 Furthermore, to discover more effective compounds, we conducted a chemical modification campaign around the triazole moiety. We also improved the biological screening cascade with experiments to characterize how the new compounds disrupt the RAD51-BRCA2 interaction and inhibit DSB repair. We obtained compound 3 (Figure 1) with an improved profile relative to the initial hits (according to a biochemical ELISA assay) and a clear mechanism of action, allowing synergy with olaparib in cancer cells BxPC3, where olaparib is normally inactive. However, with 3, we could not fully reproduce the paradigm of synthetic lethality.31 This could be due to its low-level potency, which did not cause HR inhibition greater than 40%. Additionally, the inherent resistance to apoptosis of BxPC3 cells, which bear a mutant p53, could have further prevented apoptosis. Therefore, further biological experiments and new classes of RAD51-BRCA2 are needed to confirm this paradigm and to assess its potential as an innovative anticancer strategy.
Figure 1.

Structures of the previously identified triazoles 1–3.
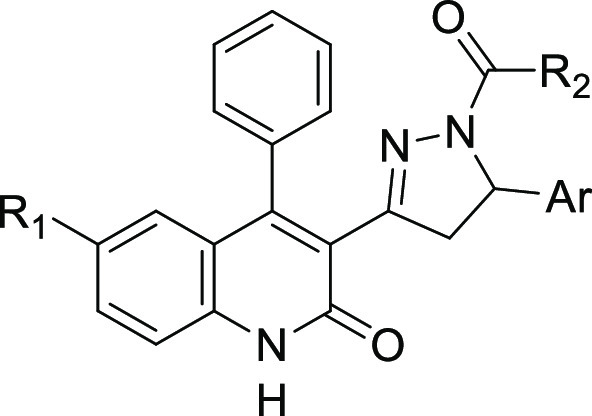
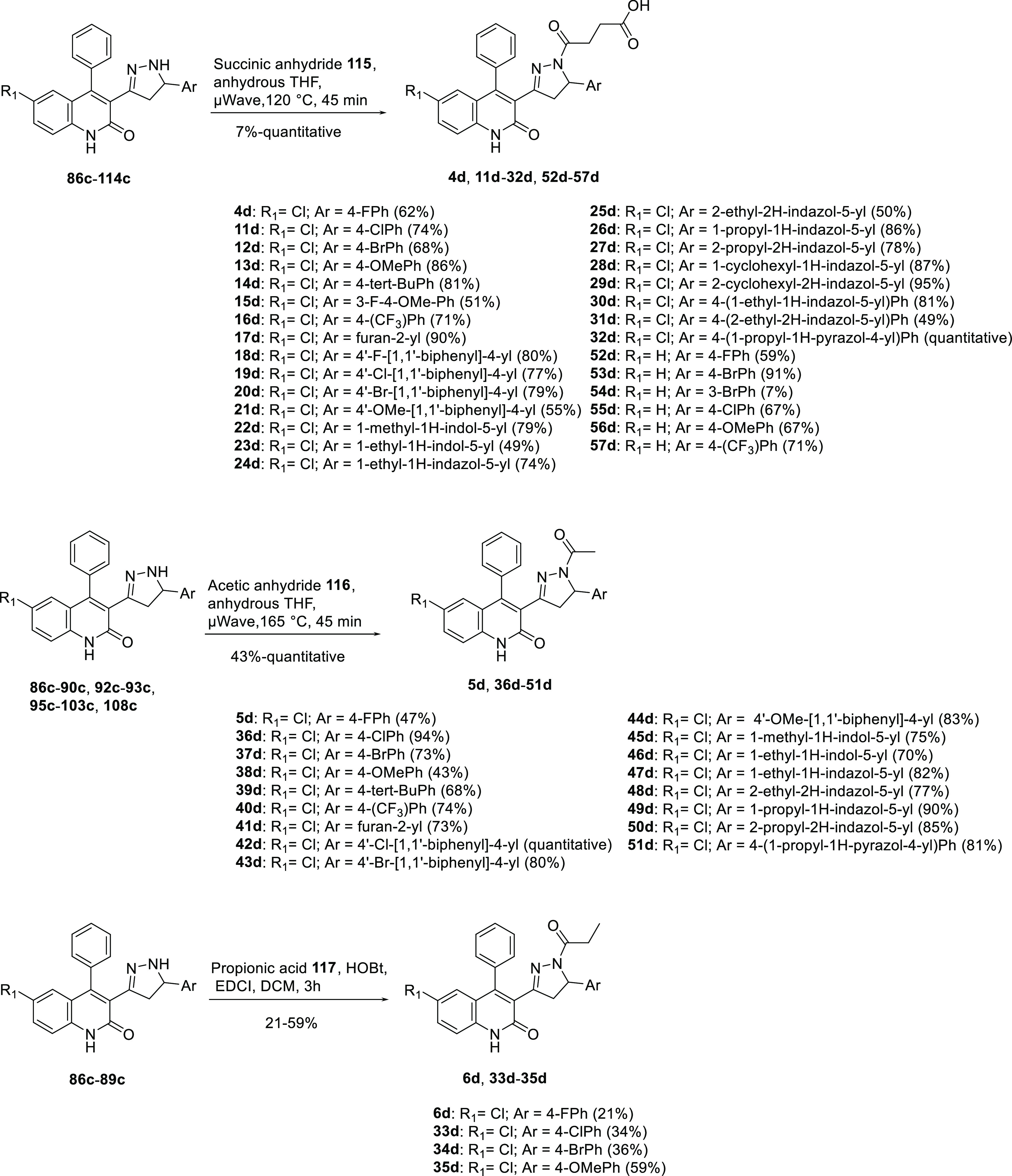
To this end, we report here on the identification of a new class of RAD51-BRCA2 disruptors based on the dihydroquinolone pyrazoline moiety (4d–57d, Table 1, Scheme 2). We attempt to depict general structure–activity relationships (SARs) of this new class of compounds as RAD51-BRCA2 inhibitors and outline the biological profile of the most promising derivative 35d (Table 1, Scheme 2).
Table 1. Structures and EC50 of Compounds 4d–57d on ELISA Assaya.


Footnotes: *All points were tested in triplicate with error bars indicating the standard deviation. **NA: not active.
Scheme 2. Synthesis of Final Dihydroquinolone Pyrazolines 4d–57d.
Results and Discussion
Hit Identification and Optimization
Targeting protein–protein interactions (PPIs) is an attractive strategy for designing innovative drugs for complex diseases such as cancer. Indeed, the first PPI inhibitors for cancer are now in clinical development.32
In this context, to identify RAD51-BRCA2 disruptors, we used the available X-ray crystallographic structure of the fourth BRC repeat (BRC4) in complex with the catalytic domain of RAD51.24 BRC4 binds RAD51 in two different hydrophobic pockets (zone I and zone II, respectively). One pocket (named zone I) can lodge BRC4’s FxxA motif (residues 1524–1527 of BRCA2) and is critical for RAD51 multimerization. The other pocket (named zone II) can lodge the BRC4’s LFDE motif (residues 1545–1548 of BRCA2) far from the oligomerization interface (Figure 2).24,33−35 Recently, we ran a successful virtual screening campaign based on high-throughput docking at the FxxA pocket to identify the first RAD51-BRCA2 disruptors.30 To increase the chemical diversity and identify a novel class of RAD51-BRCA2 disruptors, we performed a second virtual screening campaign targeting the LFDE binding pocket (see the Supporting Information). This binding pocket is more evolutionarily conserved than the FxxA. Furthermore, mutation at the LFDE causes cellular lethality and failure of RAD51 assembly in nuclear foci at the site of DNA breaks in vivo. This further suggests this pocket as a critical site for RAD51’s mechanism of action.33 To the best of our knowledge, no inhibitor that binds the LFDE binding pocket has been reported so far in the literature. This may open up new possibilities for combining molecules targeting zone I and zone II toward a more in depth understanding of the mechanism of inhibition of RAD51-BRCA2 interaction.
Figure 2.

(A) RAD51-BRCA2 BRC repeat complex (PDB code 1N0W). RAD51 is represented as a surface, BRC4 as a cartoon. The two hot spots of the interaction between the proteins (Phe1524 and Phe1546) are highlighted in sticks. (B) Zone II magnification showing the interacting residues of BRC4 (yellow) and RAD51 (white).
Here, 42 small molecules were selected, purchased, and tested for their inhibitory activity using a competitive biochemical ELISA assay, as previously described by Rajendra et al.33 Among the tested compounds, the commercially available dihydroquinolone pyrazoline derivative 4d (Figures 3 and 4) was the best candidate in terms of EC50 and chemical tractability. Its activity was confirmed by retesting the newly synthesized compound 4d (Scheme 2). Indeed, the dihydroquinolone pyrazoline moiety is a core structure of compounds with different biological targets.36,37 The binding mode to RAD51 of both enantiomers of 4d (Figure 3), as obtained by induced-fit docking simulations, displays some points of interaction similar to those of the crystallographic BRC4-RAD51 complex. Specifically, the docking model suggests that (i) the fluorophenyl ring in position 5 of the pyrazoline lies (similar to the Phe1546 of BRC4) in a hydrophobic pocket outlined by the side chains of Leu204, Tyr205, Met251, Leu255, and Phe259 of RAD51 and (ii) the carboxyl group of the pyrazoline side chain forms an ionic interaction with the Arg250 (or Arg247) of RAD51, as does the side chain Glu1548 of BRC4. In addition, the model suggests that the carbonyl and the nitrogen of the dihydroquinolone moiety, together with the carbonyl group of the pyrazoline side chain, establish hydrogen bonds with Arg254 and Glu258. Notably, both enantiomers show the same global pattern of interactions.
Figure 3.

Both enantiomers of compound 4d docked into the LFDE binding site (site II) of RAD51 (PDB code 1N0W).
Figure 4.

Overview of the optimization strategy of 4d for SAR exploration.
To improve the RAD51-BRCA2 inhibitory activity of 4d, we conducted a chemical modification campaign around the dihydroquinolone pyrazoline core. We synthesized a chemical library that contained a variety of aromatic substitutions (red and blue regions) in combination with modifications of the acyl chain moiety (green region) (Figure 4). All compounds were synthesized and tested as racemic mixtures, after verifying that the enantiomers of the hit compound 4d showed the same biochemical activity and the same binding mode (Figure 3, details in the Biological Evaluation section). First, a series of different acyl chains (namely, acetyl, propionyl, 3-aminopropionyl, 4-amino-4-oxobutanoyl, 4-methoxy-4-oxobutanoyl, 3-(methylsulfonamido)propanoyl) was introduced on the pyrazoline nitrogen (5d–10d, Table 1). Next, the aromatic ring A was modified by replacing the fluorine atom with different substituents, including chlorine and bromine atoms and methoxy, tert-butyl, and trifluoromethyl groups, leaving the succinate acyl chain unchanged (11d–16d, Table 1). In addition, the aromatic ring A was replaced by different substituted biphenyl or heterocycle groups in order to probe its role (17d–32d, Table 1). The ring A was also modified in combination with the propionyl (33d–35d, Table 1) or acetyl substitution on the pyrazoline nitrogen (36d–51d, Table 1). Regarding the dihydroquinolone core, the chlorine atom in the C-ring was removed, leaving the acyl chain unchanged and introducing some different substituents in the phenyl ring A (52d–57d, Table 1).
Chemistry
The desired dihydroquinolone pyrazoline derivatives 4d–6d and 11d–57d were achieved, taking advantage of a common synthetic strategy previously reported by Acker et al.36 The commercially available 2-aminobenzophenones 58 and 59 treated with ethyl acetoacetate afforded the corresponding methyl ketones 60a and 61a, which underwent base-catalyzed condensation with the appropriate aryl aldehydes 62–85 yielding the α,β-unsaturated aryl ketones 86b–114b. In turn, 86b–114b were treated with hydrazine monohydrate to yield the pyrazoline derivatives 86c–114c (Scheme 1). The pyrazoline amines 86c–114c functionalized with succinic anhydride 115, acetic anhydride 116, or propionic acid 117 afforded the corresponding desired dihydroquinolone pyrazoline derivatives 4d–6d and 11d–57d (Scheme 2). 7d (Scheme 3) was obtained by coupling 86c with the commercially available 3-((tert-butoxycarbonyl)amino)propanoic acid 118 to achieve the Boc-aminopyrazoline derivative 119. In turn, 119 was Boc-deprotected to afford the desired 7d. 8d (Scheme 4) was obtained by HATU-mediated coupling of 4d with ammonium chloride. 9d (Scheme 5) was afforded by coupling 86c with the commercially available 4-methoxy-4-oxobutanoic acid 120. The synthesis of 10d (Scheme 6) began with the commercially available methyl 3-aminopropanoate 121, which was treated with methanesulfonyl chloride 122 to afford the corresponding methyl 3-(methylsulfonamido)propanoate 123, which underwent basic hydrolysis to give 3-(methylsulfonamido)propanoic acid 124. The coupling reaction of 86c with 124 afforded the desired 10d.
Scheme 1. Synthesis of Dihydroquinolone Pyrazoline Intermediates 86c–114c.
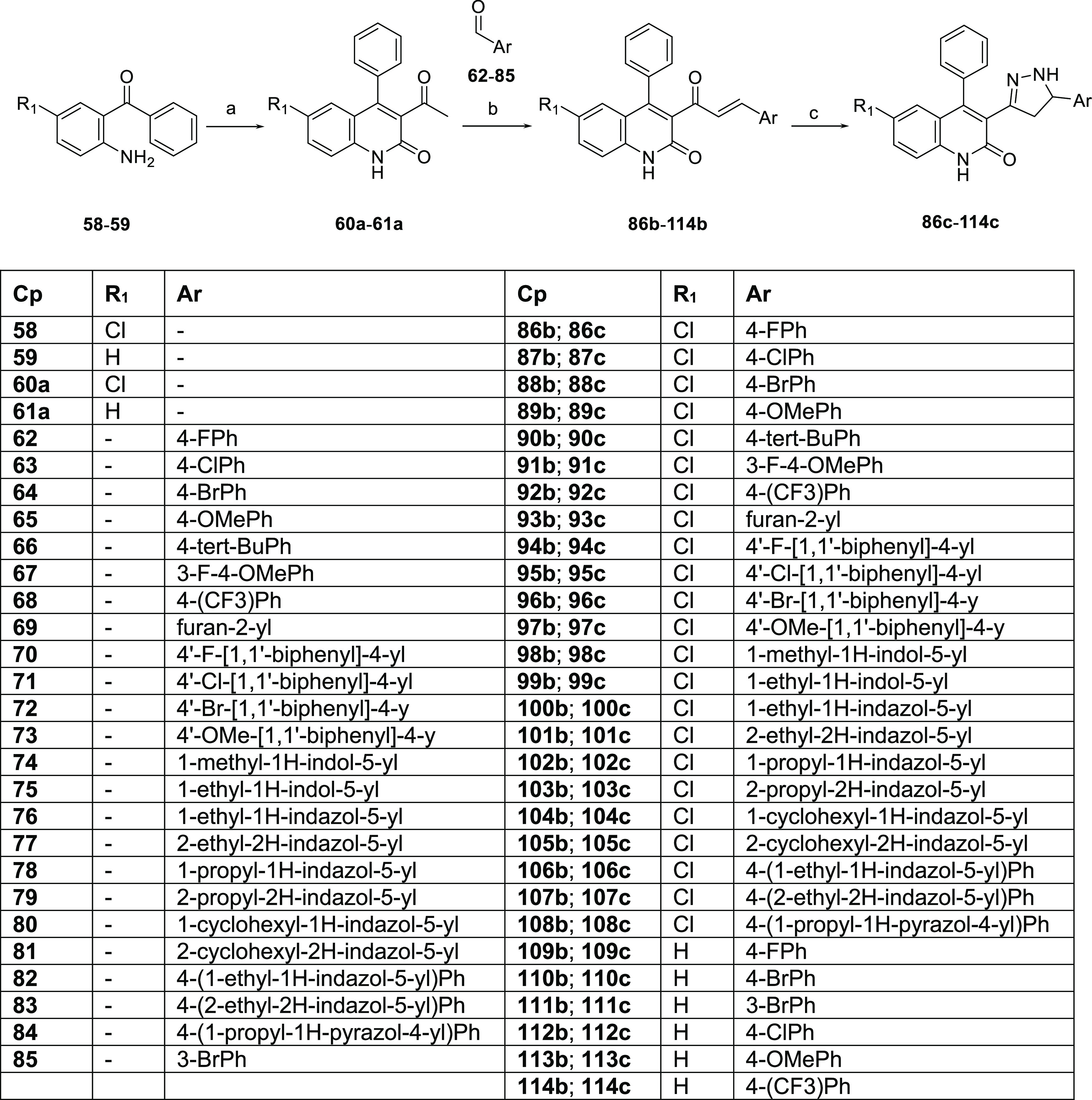
Reagents and conditions: (a) ethyl acetoacetate, DMF, 120 °C μWave or reflux, 60a quantitative, 61a 53%; (b) KOH, EtOH/H2O 4:3 v/v (0.05 M), 0 °C to rt, 53%, quantitative; (c) hydrazine monohydrate, EtOH, 110 °C μWave, 45 min, 64%, quantitative.
Scheme 3. Synthesis of Compound 7d.

Reagents and conditions: (a) HOBt, EDCI, DCM, overnight, yield 25%; (b) HCl 4 M in dioxane, rt, 15 min; (c) NaOH 0.5 M in EtOAc, rt, 15 min, yield 50%.
Scheme 4. Synthesis of Compound 8d.

Reagents and conditions: (a) HATU (1.5 equiv), EDC (1.5 equiv), DCM, DMF, ammonium chloride (5.0 equiv), DIPEA (4.0 equiv), rt, 26 h, yield 46%.
Scheme 5. Synthesis of Compound 9d.

Reagents and conditions: (a) HOBt (1.1 equiv), EDC (1.1 equiv), TEA (2.2. equiv) DCM, rt, 16 h, yield 42%.
Scheme 6. Synthesis of Compound 10d.

Reagents and conditions: (a) TEA (5.0 equiv), anhydrous DCM, methanesulfonyl chloride 122 (2.0 equiv), rt, 48 h, yield 82%; (b) MeOH/THF (1:1 v/v), 2 M LiOH, rt, 16 h, yield 82%; (c) HOBt (1.1 equiv), EDC (1.1 equiv), TEA (2.2 equiv), DCM, rt, 16 h, yield 15%.
The aldehydes 67, 70, 74–84, not commercially available, were prepared following standard procedures as reported in Supporting Information (Schemes S1–S9).
Biological Evaluation
To investigate the mechanism of action of the new dihydroquinolone pyrazoline derivatives, different biological assays were performed. As a primary screening, the ability of compounds 5d–57d to inhibit RAD51-BRCA2 interaction was investigated with a competitive biochemical ELISA assay against the parent compound 4d (Table 1). This assay is effective in evaluating the ability of new molecules to compete with BRC4 to bind to RAD51.30 Replacing the acyl chain of the pyrazoline nitrogen yielded compounds (5d–10d, Table 1) that had reduced inhibitory activity (5d, EC50 = 50 ± 10 μM; 6d, EC50 = 34 ± 3 μM) or were completely inactive (7d–10d). These data indicate that no improvement in RAD51-BRCA2 inhibitory activity was achieved with this subset of compounds relative to the parent 4d (EC50 = 16 ± 4 μM). Replacing the fluorine atom on ring A with bromine led to 12d (EC50 = 16 ± 2 μM), which shows the same activity as the initial hit 4d. The activity was affected when the fluorine was replaced with chlorine (11d, EC50 = 59 ± 8 μM) and trifluoromethyl groups (16d, EC50 = 29 ± 2 μM). The replacement of fluorine with electron-donating groups yielded the inactive compounds (13d–15d). For the subset of dihydroquinolone pyrazolines, in which the aromatic ring A was replaced by different substituted biphenyl or heterocycle groups (17d–32d, Table 1), the N-methylindole derivative 22d showed a fairly good potency with an EC50 = 2 ± 0.5 μM, 8 times higher than that of the parent 4d (EC50 = 16 ± 4 μM). 18d–21d, 23d, 25d, 30d, and 31d were active at micromolar range, all very similar to the initial hit. A drop in activity was observed with compound 32d, while derivatives 24d, 26d–29d were inactive. The replacement of the aromatic ring A in combination with the substitution of the pyrazoline nitrogen with either propionyl or acetyl chain yielded compounds 33d–51d. The 1-N-acetyl-5-(1-N-propyl)indazolylpyrazoline 49d showed the best activity of the series with EC50 = 0.95 ± 0.05 μM, while 33d–36d, 39d, 45d showed an activity very similar to that of the initial hit. A drop in potency was observed with compounds 37d, 42d–44d, 46d–47d, and 51d, while 38d, 40d–41d, 48d, and 50d were inactive. Finally, removing the chlorine atom on the dihydroquinolone core led to the completely inactive compounds 52d–57d, suggesting an active role for the halogen. The enantiomers (4d-I and 4d-II) of the racemic hit compound 4d, separated via reverse phase chiral chromatography, showed a very similar inhibitory activity (4d-I, EC50 = 4 ± 0.5 μM; 4d-II, 10 ± 1 μM) to that of the parent 4d, suggesting no stereochemical preference of these compounds for the hypothesized molecular target RAD51 (see Experimental Section). As expected for PPI disruptors, the SARs of the new series of dihydroquinolone pyrazoline were rather complex to rationalize, with many cliffs and spikes that were difficult to understand. Nonetheless, the SAR campaign allowed us to identify several compounds with interesting EC50 values ranging from 0.95 to 20 μM. Accordingly, 4d, 12d, 18d, 20d–23d, 30d, 31d, 33d–36d, 39d, 45d, and 49d were submitted to cell-based study (Table 2).
Table 2. Preliminary Biological Screening of Compounds Showing EC50 on ELISA Assay Ranging from 0.95 to 20 μM.
| compd | EC50 ELISA (μM) | preliminary biological screening |
|---|---|---|
| 4d | 16 ± 4 | HR inhibition = 10%, 40 μM |
| 12d | 16 ± 2 | HR inhibition = not present |
| olaparib association = not present | ||
| 18d | 13 ± 1 | olaparib association = not present |
| 20d | 20 ± 2 | olaparib association = not present |
| 21d | 20 ± 4 | NEa |
| 22d | 2 ± 0.5 | HR inhibition = not present |
| olaparib association = not present at 5 μM | ||
| 23d | 10 ± 2 | olaparib association = not present |
| 30d | 15 ± 3 | NEa |
| 31d | 17 ± 4 | HR inhibition = NDDb |
| olaparib association = present at 20 μM | ||
| 33d | 8 ± 2 | HR inhibition = not present |
| olaparib association = not present | ||
| 34d | 19 ± 1 | HR inhibition = not present |
| olaparib association = NEa | ||
| 35d | 19 ± 1 | HR inhibition = 54%, 20 μM |
| olaparib association = present at 15 μM | ||
| 36d | 10 ± 0.7 | HR inhibition = 24%, 10 μM |
| olaparib association = not present | ||
| 39d | 15 ± 4 | HR inhibition = NDDb |
| 45d | 18 ± 1 | HR inhibition = not present |
| 49d | 0.95 ± 0.05 | HR inhibition = not present |
| olaparib association = not present |
NE, not evaluable.
NDD, not dose-dependent.
Our working hypothesis is that compounds disrupting RAD51-BRCA2 interaction should affect HR repair and increase the efficacy of PARPi in treating breast, ovarian, and pancreatic cancer. For its clinical relevance, pancreatic adenocarcinoma was selected as our final model for cell-based experiments, and BxPC3 cells were selected for a straightforward comparison with the previously reported triazole derivatives.30,31 BxPC3 is derived from a human adenocarcinoma that expresses fully functional BRCA2.38 As shown in Table 2 and according to the rationale of our hypothesis, our preliminary screening consisted of verifying the efficacy of compounds in inhibiting cell HR and/or in increasing the antiproliferative effect of olaparib. For each compound, both parameters were verified using only one or two doses, in the range of the EC50 obtained with the ELISA test. HR activity was assessed by evaluating the recombination rate between two transfected plasmids, using a commercially available assay. This preliminary investigation allowed us to rapidly exclude molecules showing (i) low or no activity (4d, 12d, 18d, 20d, 22d, 23d, 33d, 45d, 49d), (ii) poor solubility in cell culture media (21d, 30d, 34d), (iii) discrepancy between the data obtained in the two different screening procedures such as HR inhibition not confirmed by increased olaparib efficacy in the viability assay or, on the contrary, increased olaparib efficacy without HR inhibition (31d, 36d), (iv) a non-dose-dependent effect (39d). As for the emissucinic acid-containing compounds (4d, 12d, 18d, 20d–23d, 30d, 31d), one of the reasons for their lower potency in cells might be the general poor permeability, likely related to the ionizable acid moiety. The data reported in Table 2 show that 35d was the most promising compound in the HR activity test. We then characterized 35d in additional biophysical and cell-based experiments.
To further assess the physical interaction between 35d and RAD51, a microscale thermophoresis (MST) assay was performed on the recombinant human RAD51 (see Experimental Section). The binding assay allowed us to determine the dissociation constant (Kd) for the RAD51-35d interaction and binding. The final binding curve (Figure 5) shows that 35d binds to RAD51 with a Kd value of 11 ± 6 μM. This is in agreement with the ELISA assay, supporting the initial hypothesis that 35d could act as a RAD51-BRCA2 disruptor.
Figure 5.

MST analysis of His-hRAD51-35d binding. Titration curve of (RED-tris-NTA 2nd Generation)-His-hRAD51 (80 nM) with increasing concentrations of 35d. Sigmoidal fitting curve was obtained using the Affinity Analysis software of NanoTemper Technologies. MST data are the average of three replicates.
To further characterize the effect of 35d on HR, the compound was administered at different doses to BxPC3 cells for 5 h simultaneously with plasmid transfection; as shown in Figure 6A, it produced a statistically significant dose–response effect in reducing cell HR. The compound was tested up to 40 μM (an upper limit in terms of solubility), and we estimated the dose causing a 50% inhibition of HR by applying the polynomial regression to the collected data. The EC50 was 18.4 μM.
Figure 6.
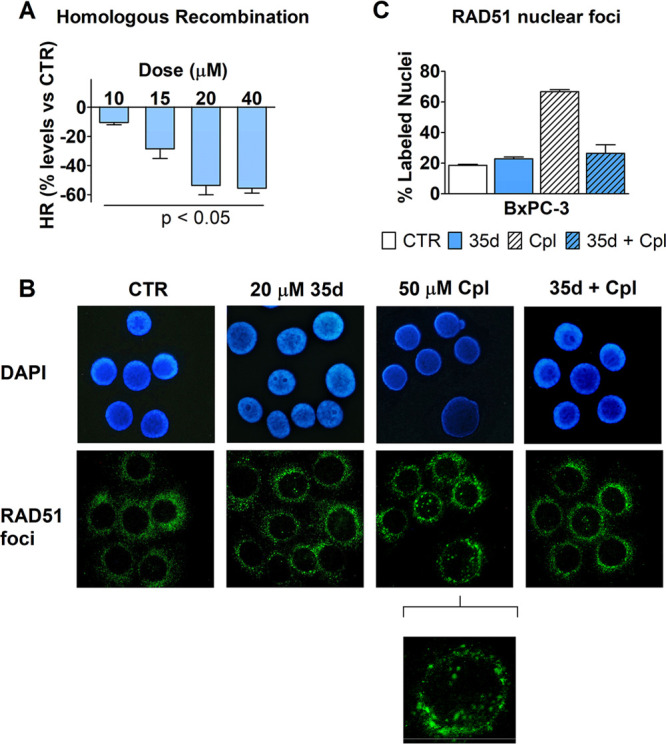
(A) Effect on HR caused by 35d administered to BxPC3 cells during plasmid transfection (5 h). HR was evaluated by real-time PCR, as described in the Experimental Section. Data were statistically analyzed using the column statistics of Prism 5 software, which applies the inferences analysis and the one-sample t test. The observed inhibitory effect was significantly different from 0 (the level of untreated cultures) for all tested doses, with p < 0.05. (B, C) Immunofluorescence detection of RAD51 in BxPC-3 nuclei after a treatment with 50 μM cisplatin (Cpl) given separately or in combination with 20 μM 35d. Experimental details are reported in the Experimental Section. (B) Representative pictures showing DAPI-stained cell nuclei and the corresponding immune-labeling of RAD51 localization. In untreated cells (CTR), RAD51 labeling is clearly evident in cytoplasm and does not appear in cell nuclei. In the pictures of Cpl-exposed cells, nuclear localization of the protein is clearly evident in 3 out of the 6 shown nuclei. A higher magnification detail was included for this sample. (C) The bar graph shows the percentage of RAD51-labeled nuclei counted by two independent observers who analyzed the treated cultures. Data were statistically evaluated by applying the one-way ANOVA, which indicated a significantly increased nuclear RAD51 labeling caused by Cpl (p < 0.05) and no statistically significant difference between cells treated with 35d and those exposed to 35d + Cpl.
An additional evidence of compromised HR was obtained by assessing the localization of RAD51 in BxPC-3 nuclei after DNA damage. The results of this experiment are reported in Figure 6B,C. To obtain massive DNA damage, BxPC-3 cultures were exposed for 1 h to 50 μM cisplatin. The immunohistochemical staining of RAD51 shown in Figure 6B revealed evident nuclear foci in cisplatin-treated cells, which appeared significantly reduced when the drug was administered in association with 20 μM 35d. Furthermore, the percentage of RAD51-labeled nuclei measured in cultures exposed to 20 μM 35d was superimposable to that observed in cells exposed to the association of cisplatin with 20 μM 35d (Figure 6C).
These results were in good agreement with the ELISA and MST outcomes, ultimately pointing to 35d as a novel RAD51-BRCA2 disruptor with a clear capability to interfere with HR.
The sustained inhibition of HR in cells should result in increased DNA damage, ultimately leading to mutations and chromosome aberrations; these effects are expected to be further amplified by PARP inhibition. The extent of DNA damage produced in cells treated for 48 h with 20 μM 35d, administered singularly or in association with 10 μM olaparib, was studied by evidencing nuclear γ-H2AX foci by immunofluorescence. The experiment was performed on both BxPC-3 and Capan-1 cultures. Capan-1 cells are derived from a human pancreatic adenocarcinoma (very similar to BxPC3cells) and are BRCA2-defective.38 As a consequence, they do not operate RAD51-BRCA2-dependent HR. The olaparib dose was selected on the basis of previously obtained results with the same cell cultures.30,31 Results are reported in Figure 7A,B.
Figure 7.

(A, B) Evaluation of DNA damage through immune detection of nuclear γ-H2AX foci in BxPC-3 and Capan-1 cells exposed for 48 h to olaparib (10 μM) or 35d (20 μM), given alone or in combination. (A) Representative pictures showing DAPI-stained cell nuclei and the corresponding immune-labeling of γ-H2AX. In BxPC-3 cells, coadministration of 35d and olaparib produced increased γ-H2AX labeling. A higher magnification detail was included for this sample. As expected, Capan-1 cells showed a constitutive γ-H2AX labeling that was highly increased by olaparib but was unaffected by 35d coadministration. (B) The bar graph shows the percentage of γ-H2AX -labeled nuclei counted by two independent observers who analyzed the treated cultures. Data obtained in bxPC-3 cells were statistically evaluated by applying the one-way ANOVA, which indicated a statistically significant difference between the cultures treated with olaparib and those exposed to olaparib + 35d. (C, D) Evaluation of micronuclei generation in BxPC3 cells treated (72 h) with 35d and olaparib, given alone or in combination. (C) Representative pictures showing DAPI-stained cell nuclei. White asterisks indicate the presence of micronuclei. (D) The percentage of cells bearing micronuclei was estimated by two independent observers, by analyzing 100–250 cells for each treatment sample. The obtained results were statistically analyzed by applying the one-way ANOVA, which indicated a p value of <0.01.
The microscope pictures shown in Figure 7A showed increased evidence of γ-H2AX labeling in nuclei of BxPC-3 cells exposed to the compounds’ association, compared to the labeling observed in cultures treated with olaparib. In Capan-1 cultures, the constitutive γ-H2AX labeling in nuclei appeared more evident than that observed in BxPC-3 cells.
Moreover, in agreement with data showing higher PARPi sensitivity for cells lacking functional HR,17 these cultures showed increased nuclear labeling when exposed to olaparib. Notably, this labeling was not further enhanced by 35d coadministration. The percentage of γ-H2AX-labeled nuclei measured in all treated cultures is shown in the bar graphs of Figure 7B.
The sustained and increased DNA damage produced in BxPC-3 nuclei generates chromosomal aberrations which can be visualized through the presence of small DNA-staining bodies outside the main nucleus (micronuclei). The microscope pictures of Figure 7C show the appearance of this feature in BxPC3 cells exposed for 72 h to 20 μM 35d, administered alone or in combination with 10 μM olaparib. The percentage of cells bearing micronuclei is reported in the graph of Figure 7D. Notably, cells bearing micronuclei were markedly more frequent in cultures treated with the 35d/olaparib combination.
Taken together, the results reported in Figures 6 and 7 significantly support the requested mechanism of action for 35d. Therefore, we conducted further experiments to test whether the 35d/olaparib combination would induce synthetic lethality.
We simultaneously evaluated cell viability and cell death, measured at 72 h in BxPC3 cells exposed to 35d alone or in combination with 10 μM olaparib (Figure 8). The statistical analysis (see legend of Figure 8) compared the results for cultures treated with different doses of 35d or the 35d/olaparib combination (Figure 8A, upper panel). When applied to the data of the cell viability experiment, this analysis indicated a statistically significant difference produced by 35d in combination with olaparib in all the treated BxPC3 cultures, with p values ranging from 0.01 (10 μM 35d) to 0.0001 (15 and 20 μM 35d). When we applied the same analysis to the data of the cell death experiment, we found no statistically significant increase in cell death for olaparib when coadministered with 10–15 μM 35d. Notably, in cultures exposed to olaparib + 20 μM 35d (the concentration producing the highest inhibition of HR), the evidence for cell death was markedly increased and statistically significant, with p < 0.0001 (Figure 8A, lower panel).
Figure 8.

(A) BxPC3 cell viability and death measured after 72 h exposure to 35d and 10 μM olaparib, given alone or in combination. Data were analyzed by two-way ANOVA using the two treatments (35d and olaparib) as variables. In the cell viability experiment, Bonferroni post-test indicated a statistically significant difference produced by olaparib coadministration in all BxPC3 cultures treated with the different 35d doses, with p values ranging from 0.01 (10 μM 35d) to 0.0001 (15 and 20 μM 35d). The same analysis was applied to the data from the cell death experiment and indicated that no statistically significant increase in cell death was produced by olaparib when coadministered with 10–15 μM 35d. In cultures exposed to olaparib + 20 μM 35d the evidence for cell death was markedly increased and statistically significant, with p < 0.0001. (B) After the 72 h treatment, BxPC3 cells were stained with vital dyes. As shown in the microscope pictures, the only culture displaying sharp evidence of cell death was that exposed to the combination of olaparib/20 μM 35d, as demonstrated by PI nuclear staining.
When cell death is a consequence of progressive DNA damage accumulation induced by simultaneous PARP and HR inhibition, we would expect it to emerge gradually over time. To confirm and better characterize the lethality observed in cultures treated with the 35d/olaparib combination, we therefore considered it inappropriate to conduct a simple evaluation of the commonly used markers (e.g., caspase activation), since these can show very transient changes. Instead, we observed cell morphology and reaction to vital dyes after the 72 h treatment, when the experiment of Figure 8A indicated a significant level of cell death.
Figure 8B shows microscope pictures of BxPC3 cells stained with mixed DAPI and PI. The simultaneous use of these two dyes can demonstrate cell death and indicate the death pattern.39 DAPI is cell-permeable and shows nuclear morphology; healthy cells appear to display normal nuclear morphology in the absence of PI staining, since this dye is not cell-permeable. Cells undergoing apoptosis display nuclear condensation, which is indicated by increased DAPI staining. PI staining indicates compromised membrane integrity, which characterizes necrotic cells and late-apoptotic cells maintained in culture.
The microscope pictures show that untreated BxPC3 cells display only a moderate DAPI staining of their nucleus. Nuclear DAPI staining is slightly increased in cells treated with 35d alone but is strikingly bright only in cells treated with the 35d/olaparib combination. Furthermore, PI staining appeared only in these cultures, confirming the manifestation of synthetic lethality. The simultaneous marked staining of these cells with both dyes could indicate an apoptotic phenomenon followed by compromised membrane integrity because of cell persistence in culture.
As expected, the sustained and increased DNA damage observed in BxPC3 cells treated with the 35d/olaparib combination (Figure 7) reproduced the desired mechanism of synthetic lethality (Figure 8). These results are also relevant given the mutated p53 status of BxPC3 cells, which should make them more resistant to mechanisms that induce cell death.
Finally, the antiproliferative effect of the 35d/olaparib combination was also studied on the HR-defective Capan-1 culture and on a non-neoplastic cell line derived from human kidney (HK-2). Moreover, to evaluate the antineoplastic potency of the 35d/olaparib association, we also calculated the combination index (CI) of the two compounds, using the procedure previously described30,31 (Figure 9).
Figure 9.
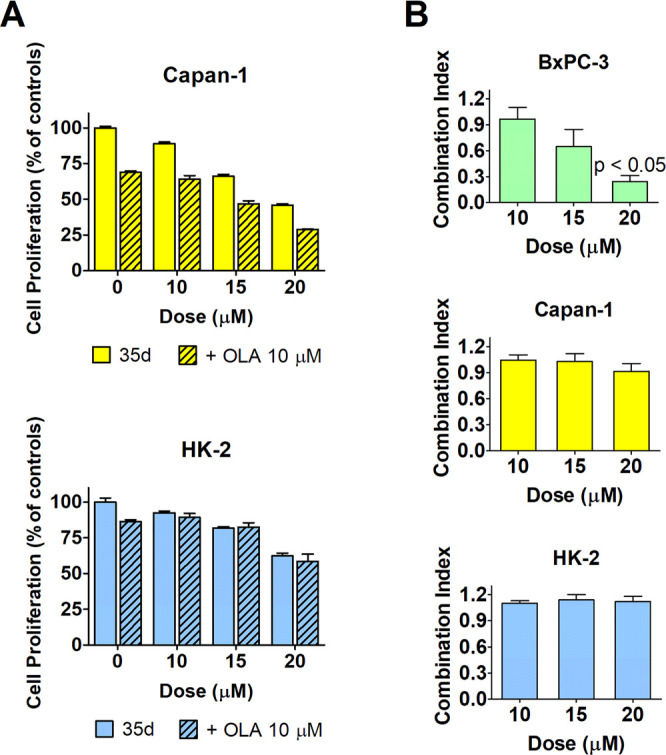
(A) Antiproliferative effect caused by 35d and its association with olaparib, measured in Capan-1 cells (which do not operate RAD51-BRCA2-dependent HR) and in normal immortalized human renal cells (HK-2). The same procedures described for BxPC3 cells were used here. (B) Combination indexes of the olaparib/35d association measured in the three used cell lines, calculated according to the method previously described.30,31 For BxPC-3 cells, the data reported in Figure 8A (cell proliferation) were used. Data were analyzed using the column statistics and the one-sample t test of Prism 5 software. For BxPC-3 cells, this test showed a statistically significant difference from 0.8 for the combination of olaparib and 20 μΜ 35d. Values of <0.8 indicate synergism between the two compounds.
According to this method, CI < 0.8 indicates synergism while a result ranging from 0.8 to 1.2 indicates additive effects. This evaluation (Figure 9) was performed for all the three studied cell cultures; for BxPC-3 cells, the data reported in Figure 8A (cell viability experiment) were used for the calculation of CI. Interestingly, in these cells the potency of the compound combination increased in parallel with the 35d dose, reaching a statistically significant difference from 0.8 at the 20 μM concentration. The effects observed when 20 μM 35d is combined with 10 μM olaparib may thus indicate a synergism between the two compounds.
In HK-2 cultures, no dose of 35d appeared to significantly increase the antiproliferative power of olaparib (two-way ANOVA). In Capan-1 cells, the same statistical evaluation showed significantly increased antiproliferative effects for 35d/olaparib. However, in these cells, the CI did not show increasing potency of the compound combination with dose escalation. Moreover, at all tested doses, the value of CI was not significantly different from 1, which is the level measured in BxPC3 cells exposed to 10 μM 35d, a dose that did not relevantly affect (≈10%) HR (Figure 6A). This result further supports the idea that the overadditive effects in the 35d/olaparib combination arise from and are strictly related to HR inhibition.
Overall, these data supported our working hypothesis that combining a RAD51-BRCA2 small molecule disruptor with olaparib could be a valuable strategy for inducing synthetic lethality in cancer cell lines with fully functional BRCA genes and homologous recombination. This includes pancreatic cancer, which is a major unmet need in oncology. We believe that this paradigm could be used to discover innovative anticancer therapies based on other lethal gene pairs using a similar medicinal chemistry strategy.
Conclusions
Continuing our research line, we described a series of dihydroquinolone pyrazoline derivatives as a new class of RAD51-BRCA2 disruptors. Compound 4d was identified as a promising hit, and subsequent SAR efforts yielded 35d with the desired biological profile. As expected, 35d bound to its target (RAD51) and inhibited the protein–protein interaction between RAD51 and BRCA2. Importantly, it synergized and reproduced the paradigm of synthetic lethality in combination with olaparib in pancreatic cancer cells (BxPC3). These effects were strictly related to the extent of HR inhibition in a dose-dependent trend. This is the most promising achievement of the current investigation and supports our working hypothesis that one can trigger synthetic lethality using only small organic molecules. Interestingly, the observed synthetic lethality was triggered by tackling two biochemically different mechanisms: enzyme inhibition (PARP) and protein–protein disruption (RAD51-BRCA2). This highlights how complex and diverse mechanisms of action can synergistically contribute to the same physiological and, in turn, pharmacological activity. We note, however, that 35d’s low solubility may affect its metabolic and pharmacokinetic profile (DM/PK), preventing it from being studied further in in vivo cancer models. Structural tuning is therefore required (and currently ongoing) to discover more drug-like dihydroquinolone pyrazoline derivatives.
In conclusion, we have further shown that synthetic lethality may be a suitable framework for discovering innovative anticancer therapies. We are confident that this novel concept will open up several new avenues based on other lethal gene pairs to meet the medical needs in oncology.
Experimental Section
Chemistry. General Chemical Methods
Solvents and reagents were obtained from commercial suppliers and used without further purification. If required, solvents were distilled prior to use. Automated column chromatography purifications were conducted using a Teledyne ISCO apparatus (CombiFlash Rf) with prepacked silica gel columns of different sizes (from 4 to 120 g). Mixtures of increasing polarity of cyclohexane and ethyl acetate or dichloromethane and methanol/ethanol were used as eluents. Preparative TLCs were performed using Macherey-Nagel precoated 0.05 mm TLC plates (SIL G-50 UV254). Microwave heating was performed using Explorer-48 positions instrument (CEM). NMR experiments were run on a Bruker Avance III 400 MHz spectrometer (400.13 MHz for 1H and 100.62 MHz for 13C), equipped with a BBI probe and Z-gradients, or on a Bruker FT NMR Avance III 600-MHz spectrometer (600.130 MHz for 1H and 150.903 MHz for 13C) equipped with a 5 mm CryoProbe QCI quadruple resonance, a shielded Z-gradient coil, and the automatic sample changer SampleJet NMR system. Spectra were acquired at 300 K, using deuterated dimethylsulfoxide (DMSO-d6) or deuterated chloroform (CDCl3) as solvents. Chemical shifts for 1H and 13C spectra were recorded in parts per million using the residual nondeuterated solvent as the internal standard (for CDCl3, 1H 7.26 ppm, 13C 77.16 ppm; for DMSO-d6, 1H 2.50 ppm, 13C 39.52 ppm). UPLC–MS analyses were run on a Waters ACQUITY UPLC/MS system consisting of an SQD (single quadrupole detector) mass spectrometer equipped with an electrospray ionization interface and a photodiode array detector. The PDA range was 210–400 nm. The analyses were performed on either an ACQUITY UPLC HSS T3 C18 column (50 × 2.1 mm i.d., particle size 1.8 μm) with a VanGuard HSS T3 C18 precolumn (5 mm × 2.1 mm i.d., particle size 1.8 μm) (log D < 1) or an ACQUITY UPLC BEH C18 column (50 mm × 2.1 mm i.d., particle size 1.7 μm) with a VanGuard BEH C18 precolumn (5 mm × 2.1 mm i.d., particle size 1.7 μm) (log D > 1). The mobile phase was 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN–H2O (95:5) at pH 5 (B). Electrospray ionization in positive and negative mode was applied in the mass scan range 100–500 Da. Methods and gradients used were the following: Polar method. Column: Waters ACQUITY UPLC HSS T3 C18, 1.8 μm, 50 mm × 2.1 mm i.d. Precolumn: VanGuard HSS T3 C18, 1.8 μm, 5 mm × 2.1 mm i.d. Linear gradient: 0–0.2 min, 0% B; 0.2–2.7 min, 0–50% B; 2.7–2.8 min, 50–100% B; 2.8–3.0 min, 100% B. Flow rate: 0.5 mL/min. Generic method. Column: Waters ACQUITY UPLC BEH C18, 1.7 μm, 50 mm × 2.1 mm i.d. Linear gradient: 0–0.2 min, 5% B; 0.2–2.7 min, 5–95% B; 2.7–2.8 min, 95–100% B; 2.8–3.0 min, 100% B. Flow rate: 0.5 mL/min. Apolar method. Column: Waters ACQUITY UPLC BEH C18, 1.7 μm, 50 mm × 2.1 mm i.d. Precolumn: VanGuard BEH C18, 1.7 μm, 5 mm × 2.1 mm i.d. Gradient: 0–0.2 min, 50% B; 0.2–2.7 min, 50–100% B; 2.7–3.0 min, 100% B. Flow rate: 0.5 mL/min. Compounds were named using the naming algorithm developed by CambridgeSoft Corporation and used in ChemBioDraw Ultra 15.0. All final compounds displayed ≥95% purity as determined by UPLC/MS analysis. All final synthesized compounds were checked for PAINS compliance.
General Procedure A for the Synthesis of Quinolin-2(1H)-one Acrolyl Intermediates
This procedure has been applied to the preparation of 86b–114b (Scheme 1). In a round-bottomed flask, the appropriate quinolin-2(1H)-one (60a–61a, 1.00 equiv) and potassium hydroxide (25.00 equiv) were stirred in EtOH/H2O (4:3 v/v, 0.05 M) at 0 °C for 45 min prior to the addition of an appropriately substituted aryl aldehyde (62–85, 1.00 equiv). The reaction mixture was stirred overnight as it gradually reached room temperature. The reaction was quenched by slow addition of acetic acid (25.00 equiv). The crude was extracted with DCM/H2O (3 × 50 mL), the organic layer was then dried over Na2SO4, and the solvent was removed under reduced pressure. The desired compound was obtained after purification over silica gel unless otherwise noted.
General Procedure B for Synthesis of Pyrazol-3-yl-quinolin-2(1H)-one Intermediates (Scheme 1)
This procedure has been applied to the preparation of 86c–114c. In a microwaveable vessel, the appropriate quinolin-2(1H)-one acrolyl intermediate (86b–114b, 1.00 equiv) was dissolved in EtOH absolute (0.2 M), and hydrazine monohydrate (2.00 equiv) was added. The mixture was microwaved with stirring for 45 min at 110 °C (200 W). The EtOH was removed under reduced pressure. Crude was purified over silica gel, unless otherwise noted, to afford desired compounds.
General Procedures C1 to Obtain the Final Desired Dihydroquinolone Pyrazoline Derivatives (Scheme 2)
This procedure has been applied to synthesize 4d, 11d–32d, 52d–57d. In a microwaveable vessel, the appropriate pyrazol-3-ylquinolin-2(1H)-one intermediate (86c−90c, 92c−93c, 95c−103c, 108c, 1.00 equiv) was dissolved in anhydrous THF (0.5 M). The succinic anhydride 115 (2.00 equiv) was added. The solution was microwaved (200 W) with stirring for 45 min at the appropriate temperature. The THF was removed under reduced pressure, and the organic layers were dissolved in DCM, washed with HClaq pH 2 (3 × 30 mL), and dried over Na2SO4. The solvent was removed under reduced pressure, and the crude was purified over silica gel.
General Procedures C2 to Obtain the Final Desired Dihydroquinolone Pyrazoline Derivatives (Scheme 2)
This procedure has been applied to synthesize 5d, 36d–51d. In a microwaveable vessel, the appropriate pyrazol-3-ylquinolin-2(1H)-one intermediate (86c−90c, 92c−93c, 95c−103c, 108c, 1.00 equiv) was dissolved in anhydrous THF (0.5 M). The acetic anhydride 116 (2.00 equiv) was added. The solution was microwaved (200 W) with stirring for 45 min at the appropriate temperature. The THF was removed under reduced pressure, and the organic layers were dissolved in DCM, washed with HClaq pH 2 (3 × 30 mL), and dried over Na2SO4. The solvent was removed under reduced pressure, and the crude was purified over silica gel.
General Procedures C3 to Obtain the Final Dihydroquinolone Pyrazoline Derivatives (Scheme 2)
This procedure has been applied to synthesize 6d, 33d–35d. In a round-bottomed flask, propionic acid 117 (1.80 equiv), HOBt (1.80 equiv), and EDCI (1.80 equiv) were stirred in DCM (0.50 M) at room temperature for 1 h. Then a solution of the pyrazol-3-ylquinolin-2(1H)-one intermediate 86c–89c (1.00 equiv) and Et3N (2.50 equiv) in DCM (0.50 M) was added. The reaction was stirred at room temperature for 3 h. The organic layer was washed with NaHCO3(aq) 1 M (1 × 50 mL), citric acid 10% (1 × 50 mL), and H2O (1 × 50 mL), dried over Na2SO4, and the solvent was removed under reduced pressure. The desired compound was obtained after purification over silica gel unless otherwise noted.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (4d)
6-Chloro-3-(5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one 86c (270 mg, 0.65 mmol) and succinic anhydride 115 (130 mg, 1.30 mmol) were microwaved (120 °C, 200 W) according to general procedure C1. The crude was purified by flash column chromatography (SiO2 gold 24 g; 0–50% EtOH/DCM) to afford the desired 4d (208 mg, 62% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.02 (s, 1H), 7.64 (dd, J = 8.8, 2.4 Hz, 1H), 7.60–7.37 (m, 5H), 7.28 (dt, J = 6.8, 2.0 Hz, 1H), 7.04 (t, J = 8.9 Hz, 2H), 6.94 (d, J = 2.3 Hz, 1H), 6.88–6.76 (m, 2H), 5.32 (dd, J = 12.0, 4.5 Hz, 1H), 3.73 (dd, J = 18.5, 12.0 Hz, 1H), 2.79 (dd, J = 18.4, 4.5 Hz, 1H), 2.48–2.40 (m, 2H), 2.32–2.26 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.95, 169.09, 160.56, 152.85, 150.38, 137.78, 135.00, 131.68, 129.88, 128.97, 128.88, 128.81, 127.99, 127.90, 126.57, 126.51, 125.07, 121.14, 118.09, 115.67, 115.46, 95.62, 58.69, 45.66, 29.04, 28.69. In agreement with that previously reported by Acker et al.36tR = 2.04 min (generic method). ESI-MS for C28H21ClFN3O4: calculated 517.1, found m/z 518.4, 520.4 [M + H] +; 516.4, 518.4 [M – H]−. UPLC–MS purity (UV at 215 nm) 98%.
3-(1-Acetyl-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (5d)
6-Chloro-3-(5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one 86c (200 mg, 0.48 mmol) and acetic anhydride 116 (91 μL, 0.96 mmol) were microwaved (120 °C, 200 W) according to general procedure C2. The crude was purified by direct phase flash column chromatography (SiO2 gold 24 g, 0–20% MeOH/DCM) to afford the desired 5d (207 mg, yield 47%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 7.64 (dd, J = 8.8, 2.4 Hz, 1H), 7.61–7.50 (m, 3H), 7.46 (d, J = 8.8 Hz, 1H), 7.41 (dt, J = 6.6, 1.9 Hz, 1H), 7.29 (dt, J = 6.4, 1.9 Hz, 1H), 7.06 (t, J = 8.9 Hz, 2H), 6.94 (d, J = 2.3 Hz, 1H), 6.84 (dd, J = 8.6, 5.6 Hz, 2H), 5.32 (dd, J = 12.0, 4.5 Hz, 1H), 3.73 (dd, J = 18.4, 12.1 Hz, 1H), 2.84 (dd, J = 18.5, 4.5 Hz, 1H), 1.88 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.53, 162.80, 160.57, 160.39, 152.67, 150.35, 138.87, 138.84, 137.79, 135.12, 131.64, 129.84, 129.04, 128.96, 128.80, 128.76, 128.00, 127.93, 126.53, 126.49, 125.02, 121.11, 118.09, 115.67, 115.45, 58.49, 45.71, 21.85. tR = 2.38 min (generic method). ESI-MS for C26H19ClFN3O2: calculated 459.1, found m/z 460.1, 462.1 [M + H]+; 458.1, 460.1 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
6-Chloro-3-(5-(4-fluorophenyl)-1-propionyl-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (6d)
Compound 6d was synthesized via general procedure C3 using 86c (250 mg, 0.60 mmol) and propionic acid 117 (80.8 μL, 1.08 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–1% MeOH/DCM) to afford 6d (60 mg, yield 21%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 7.64 (dd, J = 2.0, 8.0 Hz, 1H), 7.58–7.50 (m, 3H), 7.45 (d, J = 8.8 Hz, 1H), 7.41–7.39 (m, 1H), 7.28 (d, J = 5.6 Hz, 1H), 7.06 (t, J = 8.8 Hz, 2H), 6.93 (d, J = 2.0 Hz, 1H), 6.84 (dd, J = 4.0, 8.0, Hz, 2H), 5.32 (dd, J = 8.0, 12.0 Hz, 1H), 3.73 (dd, J = 12.0, 16.0 Hz, 1H), 2.81 (dd, J = 4.0, 16.0 Hz, 1H), 2.30–2.16 (m, 2H), 0.83 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 206.89, 170.93, 162.79, 160.57, 160.38, 152.63, 150.30, 139.00, 138.98, 137.75, 135.12, 131.63, 129.79, 128.97, 128.95, 128.86, 128.80, 127.96, 127.88, 126.53, 126.47, 125.10, 121.11, 118.07, 115.67, 115.46, 58.54, 45.54, 29.44, 9.27. ESI-MS for C27H21ClFN3O2: calculated 473.1, found m/z 474.2, 476.2 [M + H]+; 472.3, 474.3 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
3-(1-(3-Aminopropanoyl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (7d) (Scheme 3)
In round-bottom flask 119 (200 mg, 0.34 mmol) was treated with 4 M HCl in dioxane (4 mL) and stirred at rt for 15 min. The solvent was removed under reduced pressure. In a round-bottom flask the solid residue (110 mg, 0.21 mmol) was treated with NaOH 0.5 M (420 μL) in EtOAc (5 mL) stirring at rt for 30 min. The mixture was then diluted with further EtOAc and washed twice with H2O. The organic layer was dried over Na2SO4, filtered, and evaporated to dryness. Purification was performed by direct phase flash chromatography (0–10% MeOH/DCM, 0–0.1% NH4OH). Yield 50 mg, 50%. 1H NMR (400 MHz, DMSO-d6) δ 7.63 (dd, J = 12.0, 4.0 Hz, 1H), 7.57–7.50 (m, 3H), 7.54 (d, J = 4.8 Hz, 1H), 7.42–7.40 (m, 1H), 7.26 (d, J = 7.2 Hz, 1H), 7.03 (t, J = 8.8 Hz, 2H), 6.92 (d, J = 2.0 Hz, 1H), 6.81 (dd, J = 2.0, 8.0 Hz, 2H), 5.32 (dd, J = 2.0, 12.0 Hz, 1H), 3.72 (dd, J = 12.0, 20.0 Hz, 1H), 2.76 (dd, J = 4.0, 16.0 Hz, 1H), 2.57 (t, J = 6.6 Hz, 2H), 2.42–2.28 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 169.42, 162.77, 160.56, 160.36, 152.76, 150.29, 138.96, 138.93, 137.78, 135.02, 131.63, 129.86, 128.99, 128.92, 128.89, 128.80, 127.95, 127.86, 126.52, 126.45, 125.09, 121.11, 118.10, 115.66, 115.45, 110.00, 58.53, 45.61, 38.06, 38.04. ESI-MS for C27H22ClFN4O2: calculated 488.1, found m/z 489.4, 491.4 [M + H]+, 487.3, 489.4 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanamide (8d) (Scheme 4)
In a round bottomed flask 4-(3-(6-chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic acid 4d (258 mg, 0.50 mmol) was dissolved with 3.2 mL of anhydrous DCM, then HATU (285 mg, 0.75 mmol), EDC (144 mg, 0.75 mmol), and 1.8 mL of anhydrous DMF were added. The mixture was stirred at rt for 10 min. Ammonium chloride (144 mg 2.50 mmol) and soon after DIPEA (348 μL, 2.00 mmol) were added. The reaction mixture was thus stirred at rt for 26 h. Purification was performed by direct phase chromatography (SiO2 gold 24 g, 2.5–50% EtOH/DCM). Yield 120 mg, 46%. 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 7.64 (dd, J = 8.8, 2.4 Hz, 1H), 7.60–7.38 (m, 5H), 7.28 (d, J = 7.4 Hz, 1H), 7.19 (s, 1H), 7.04 (dd, J = 9.9, 7.7 Hz, 2H), 6.93 (d, J = 2.3 Hz, 1H), 6.84–6.78 (m, 2H), 6.68 (s, 1H), 5.31 (dd, J = 12.0, 4.5 Hz, 1H), 3.71 (dd, J = 18.5, 12.1 Hz, 1H), 2.76 (dd, J = 18.4, 4.5 Hz, 1H), 2.54–2.45 (m, 2H), 2.15 (t, J = 7.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.51, 169.56, 160.65, 160.38, 152.73, 150.28, 138.90, 137.94, 135.00, 131.61, 129.94, 129.94, 129.02, 128.99, 128.87, 128.79, 127.99, 127.90, 126.48, 125.10, 121.14, 118.19, 115.66, 115.45, 58.66, 45.64, 29.82, 29.38. tR = 2.09 min (generic method). ESI-MS for C28H22ClFN4O3: calculated 516.1, found m/z 517.4, 519.3 [M + H]+; 515.4, 517.4 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
Methyl 4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoate (9d) (Scheme 5)
In a round-bottomed flask, commercially available 4-methoxy-4-oxobutanoic acid 120 (153 mg, 1.16 mmol), 6-chloro-3-(5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one 86c (486 mg, 1.16 mmol), HOBT (173 mg, 1.28 mmol) were dissolved in DCM. Then TEA (355.8 μL, 2.55 mmol) was added, followed by EDCI (245 mg, 1.28 mmol) suspended in DCM. The mixture was stirred overnight at rt. The solvent was removed under vacuum, the residue was dissolved in ethyl acetate and washed with H2O, NaHCO3 sat. solution and 5% citric acid. The organic phase was dried over Na2SO4 and evaporated to dryness. The title compound was obtained after purification by direct phase flash column chromatography (0–30% EtOAc/DCM). Yield 256 mg, 42%. 1H NMR (400 MHz, DMSO-d6) δ 12.36 (s, 1H), 7.60 (dd, J = 2.2, 9.0 Hz, 1H), 7.53–7.48 (m, 3 H), 7.41 (d, J = 8.8 Hz, 1H), 7.37 (dd, J = 2.2, 4.6 Hz, 1H), 7.23 (d, J = 6.8 Hz, 1H), 7.01 (t, J = 8.8 Hz, 2H), 6.89 (d, J = 2.0, 1H), 6.77 (dd, J = 5.4, 8.6 Hz, 2H), 5.28 (dd, J = 12.0, 4.0 Hz, 1H), 3.50 (s, 3H), 3.67 (dd, J = 12.0, 18.0 Hz, 1H), 2.75 (dd, J = 4.0, 18.0 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) 172.9, 168.8, 160.0, 153.1, 150.1, 139.0, 135.0, 131.9, 129.8, 128.9, 127.9, 126.5, 125.0, 120.9, 118.0, 115.6, 115.4, 58.7, 21.7, 45.0, 28.9, 28.4. UPLC–MS purity (UV at 215 nm) 98%.
N-(3-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-3-oxopropyl)methanesulfonamide (10d) (Scheme 6)
In a round-bottom flask, 6-chloro-3-(5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one 86c (500 mg, 1.20 mmol), 3-(methylsulfonamido)propanoic acid 124 (200 mg, 1.20 mmol), HOBT (178 mg, 1.32 mmol) were stirred in DCM prior to the addition of triethylamine (367.7 mL, 2.64 mmol) and EDCI (253 mg, 1.32 mmol) at 0 °C. The reaction was stirred overnight while the mixture gradually reached rt. The solvent was removed under vacuum, and the residue was dissolved in DCM. The organic layer was washed with H2O, NaHCO3 sat. solution, and 5% citric acid, dried over Na2SO4, filtered, and evaporated to dryness. The title compound was obtained after purification over direct phase flash column chromatography (0–60% EtOAc/DCM). Yield 100 mg, 15%). 1H NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 7.60 (dd, J = 2.0, 8.0 Hz, 1H), 7.55–7.49 (m, 3H), 7.41 (d, J = 8.0 Hz, 1H), 7.38 (d, J = 6.0 Hz, 1H), 7.24 (d, J = 6.0 Hz, 1H), 7.00 (t, J = 8.0 Hz, 2H), 6.89 (d, J = 2.0 Hz, 2H), 6.77 (dd, J = 4.0, 8.0 Hz, 2H), 5.29 (dd, J = 4.0, 12.0 Hz, 1H), 3.69 (dd, J = 12.0, 20.0 Hz, 1H), 3.03–2.98 (m, 2H), 2.80 (s, 3H), 2.72 (dd, J = 4.0, 20.0 Hz, 1H), 2.51–2.47 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.01, 162.81, 160.49, 160.40, 153.25, 150.39, 138.71, 137.78, 134.86, 131.68, 129.94, 129.10, 128.94, 128.91, 128.83, 128.02, 127.94, 126.57, 125.01, 121.07, 118.09, 115.69, 115.48, 58.63, 45.67, 38.61, 34.73. UPLC–MS purity (UV at 215 nm) 99%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (11d)
Compound 11d was synthesized via general procedure C1 using 87c (253 mg, 0.58 mmol) with succinic anhydride 115 (116 mg, 1.16 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–60% EtOH/DCM) to afford 11d (231 mg, yield 74%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.03 (s, 1H), 7.64 (dd, J = 8.8, 2.4 Hz, 1H), 7.59–7.48 (m, 3H), 7.45 (d, J = 8.8 Hz, 1H), 7.41 (dt, J = 6.3, 2.0 Hz, 1H), 7.31–7.23 (m, 3H), 6.93 (d, J = 2.4 Hz, 1H), 6.83–6.76 (m, 2H), 5.32 (dd, J = 12.0, 4.6 Hz, 1H), 3.74 (dd, J = 18.5, 12.1 Hz, 1H), 2.78 (dd, J = 18.5, 4.6 Hz, 1H), 2.48–2.41 (m, 2H), 2.29 (t, J = 7.1 Hz, 2H). In agreement with that previously reported by Acker et al.36tR = 2.16 min (generic method). ESI-MS for C28H21Cl2N3O4: calculated 533.1, found m/z 534.4, 536.4, 538.3 [M + H]+; 532.4, 534.4, 536.4 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
4-(5-(4-Bromophenyl)-3-(6-chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (12d)
Compound 12d was synthesized via general procedure C1 using 88c (330 mg, 0.69 mmol) with succinic anhydride 115 (138 mg, 1.38 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–50% EtOH/DCM) to afford 12d (275 mg, yield 68%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.03 (s, 1H), 7.63 (dd, J = 8.8, 2.3 Hz, 1H), 7.59–7.47 (m, 3H), 7.45 (d, J = 8.8 Hz, 1H), 7.40 (d, J = 8.4 Hz, 2H), 7.27 (dd, J = 6.5, 1.8 Hz, 1H), 6.93 (d, J = 2.3 Hz, 1H), 6.73 (d, J = 8.5 Hz, 2H), 5.30 (dd, J = 12.0, 4.6 Hz, 1H), 3.74 (dd, J = 18.5, 12.1 Hz, 1H), 2.77 (dd, J = 18.5, 4.6 Hz, 1H), 2.48–2.36 (m, 2H), 2.28 (t, J = 7.1 Hz, 2H). In agreement with that previously reported by Acker et al.36tR = 2.18 min (generic method). ESI-MS for C28H21BrClN3O4: calculated 577.0, found m/z 578.2, 580.2, 582.3 [M + H]+; 576.1, 578.1, 580.0 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (13d)
Compound 13d was synthesized via general procedure C1 using 89c (320 mg, 0.74 mmol) with succinic anhydride 115 (148 mg, 1.48 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–15% EtOH/DCM) to afford 13d (339 mg, yield 86%). 1H NMR (400 MHz, DMSO-d6) δ 12.19 (s, 1H), 7.57 (dd, J = 8.8, 2.3 Hz, 1H), 7.51 (m, 3H), 7.42 (d, J = 8.8 Hz, 1H), 7.36–7.31 (m, 1H), 7.27–7.21 (m, 1H), 7.02 (dd, J = 8.9, 5.9 Hz, 3H), 6.90 (d, J = 2.3 Hz, 1H), 6.80 (d, J = 8.7 Hz, 2H), 4.52 (td, J = 10.4, 2.9 Hz, 1H), 3.73 (s, 3H), 3.18 (dd, J = 16.4, 10.9 Hz, 1H), 2.58–2.52 (m, 5H). In agreement with that previously reported by Acker et al.36tR = 1.99 min (generic method). ESI-MS for C29H24ClN3O5: calculated 529.1, found m/z 530.5, 532.4 [M + H]+; 528.4, 530.4 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(5-(4-(tert-Butyl)phenyl)-3-(6-chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (14d)
Compound 14d was synthesized via general procedure C1 using 90c (124 mg, 0.27 mmol) with succinic anhydride 115 (54 mg, 0.54 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–20% EtOH/DCM) to afford 14d (121 mg, yield 81%). 1H NMR (400 MHz, DMSO-d6) δ 12.27 (d, J = 78.0 Hz, 1H), 12.08 (s, 1H), 7.64 (dd, J = 8.8, 2.3 Hz, 1H), 7.59–7.47 (m, 3H), 7.47–7.43 (m, 1H), 7.40 (dd, J = 4.6, 2.1 Hz, 1H), 7.28 (d, J = 7.5 Hz, 1H), 7.23 (d, J = 8.3 Hz, 2H), 6.93 (d, J = 2.3 Hz, 1H), 6.73 (d, J = 8.3 Hz, 2H), 5.26 (dd, J = 11.9, 4.5 Hz, 1H), 3.69 (dt, J = 24.9, 12.5 Hz, 1H), 2.85 (ddd, J = 23.0, 18.2, 4.3 Hz, 1H), 2.47 (dd, J = 6.7, 4.0 Hz, 2H), 2.36–2.22 (m, 2H), 1.27 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 173.52, 168.56, 160.09, 152.41, 149.87, 149.19, 139.24, 137.30, 134.59, 131.15, 129.30, 128.57, 128.47, 128.33, 126.06, 126.03, 125.13, 125.09, 124.67, 120.69, 117.60, 58.62, 45.26, 34.12, 31.11, 28.57, 28.21, 26.32. tR = 2.39 min (generic method). ESI-MS for C32H30ClN3O4: calculated 555.2, found m/z 556.5, 558.4 [M + H]+; 554.4, 556.4 [M – H]−. UPLC–MS purity (UV at 215 nm) 97%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(3-fluoro-4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (15d)
Compound 15d was synthesized via general procedure C1 using 91c (184 mg, 0.41 mmol) with succinic anhydride 115 (82 mg, 0.82 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–10% EtOH/DCM) to afford 15d (115 mg, yield 51%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.06 (s, 1H), 7.64 (dd, J = 8.8, 2.4 Hz, 1H), 7.59–7.43 (m, 4H), 7.43–7.38 (m, 1H), 7.29 (dd, J = 7.1, 1.9 Hz, 1H), 7.01 (t, J = 8.8 Hz, 1H), 6.94 (d, J = 2.4 Hz, 1H), 6.69–6.61 (m, 2H), 5.27 (dd, J = 12.0, 4.5 Hz, 1H), 3.82 (s, 3H), 3.70 (dd, J = 18.5, 12.0 Hz, 1H), 2.82 (dd, J = 18.5, 4.5 Hz, 1H), 2.46 (dd, J = 14.3, 7.0 Hz, 2H), 2.29 (t, J = 7.2 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.96, 169.09, 160.59, 152.89, 150.37, 146.62, 137.79, 135.43, 135.05, 131.68, 129.76, 129.01, 128.94, 128.84, 128.74, 126.57, 125.06, 123.56, 122.25, 121.13, 118.10, 114.22, 113.68, 113.50, 58.52, 56.48, 45.52, 29.02, 28.67. tR = 2.00 min (generic method). ESI-MS for C29H23ClFN3O5: calculated 547.1, found m/z 548.3, 550.3 [M + H]+; 546.3, 548.2 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-(trifluoromethyl)phenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (16d)
Compound 16d was synthesized via general procedure C1 using 92c (309 mg, 0.66 mmol) with succinic anhydride 115 (132 mg, 1.32 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 40 g, 0–10% EtOH/DCM) to afford 16d (266 mg, yield 71%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.05 (s, 1H), 7.63 (dd, J = 8.8, 2.4 Hz, 1H), 7.60–7.39 (m, 7H), 7.31–7.19 (m, 1H), 7.00 (d, J = 8.1 Hz, 2H), 6.93 (d, J = 2.3 Hz, 1H), 5.42 (dd, J = 12.1, 4.7 Hz, 1H), 3.78 (dd, J = 18.5, 12.2 Hz, 1H), 2.80 (dd, J = 18.5, 4.7 Hz, 1H), 2.59–2.38 (m, 2H), 2.36–2.26 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.45, 168.78, 160.05, 152.42, 150.01, 146.65, 137.33, 134.55, 131.24, 129.42, 128.54, 128.44, 128.38, 128.35, 127.85, 127.53, 127.22, 126.20, 126.12, 126.04, 125.54, 125.39, 125.35, 124.45, 122.83, 120.63, 117.63, 58.51, 45.05, 28.49, 28.18, 26.32. In agreement with that previously reported by Acker et al.36tR = 2.22 min (generic method). ESI-MS for C29H21ClF3N3O4: calculated 567.1, found m/z 568.5, 570.4, [M + H]+; 566.4, 568.4 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(furan-2-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (17d)
Compound 17d was synthesized via general procedure C1 using 93c (201 mg, 0.51 mmol) with succinic anhydride 115 (102 mg, 1.02 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–10% EtOH/DCM) to afford 17d (226 mg, yield 90%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.06 (s, 1H), 7.64 (dd, J = 8.8, 2.3 Hz, 1H), 7.54–7.43 (m, 5H), 7.32 (t, J = 6.4 Hz, 2H), 6.96 (d, J = 2.3 Hz, 1H), 6.33 (dd, J = 3.1, 1.9 Hz, 1H), 5.99 (d, J = 3.2 Hz, 1H), 5.39 (dd, J = 11.8, 4.5 Hz, 1H), 3.58 (dd, J = 18.2, 11.9 Hz, 1H), 3.17 (dd, J = 18.2, 4.6 Hz, 1H), 2.41–2.18 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 173.58, 173.49, 168.82, 160.10, 152.84, 152.49, 150.04, 142.05, 137.37, 134.77, 131.19, 128.81, 128.78, 128.29, 128.26, 128.24, 126.12, 126.07, 124.33, 120.63, 117.60, 110.33, 106.27, 56.01, 52.62, 41.72, 28.84, 28.52, 28.15. In agreement with that previously reported by Acker et al.36tR = 1.90 min (generic method). ESI-MS for C26H20ClN3O5: calculated 489.1, found m/z 490.4, 492.4 [M + H]+; 488.4, 490.4 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4′-fluoro-[1,1′-biphenyl]-4-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (18d)
Compound 18d was synthesized via general procedure C1 using 94c (171 mg, 0.35 mmol) with succinic anhydride 115 (70 mg, 0.70 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–10% EtOH/DCM) to afford 18d (167 mg, yield 80%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.08 (s, 1H), 7.71–7.61 (m, 3H), 7.55 (ddt, J = 10.6, 9.5, 4.0 Hz, 3H), 7.50–7.40 (m, 4H), 7.33–7.25 (m, 3H), 6.94 (d, J = 2.3 Hz, 1H), 6.87 (d, J = 8.3 Hz, 2H), 5.35 (dd, J = 12.0, 4.6 Hz, 1H), 3.77 (dd, J = 18.5, 12.1 Hz, 1H), 2.84 (dd, J = 18.4, 4.7 Hz, 1H), 2.57–2.44 (m, 2H), 2.30 (t, J = 6.8 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.63, 173.50, 168.63, 163.05, 160.62, 160.11, 152.46, 149.94, 141.37, 137.96, 137.31, 136.34, 136.31, 134.60, 131.19, 129.41, 128.64, 128.56, 128.47, 128.33, 126.73, 126.08, 124.64, 120.69, 117.62, 115.78, 115.57, 58.66, 45.22, 28.96, 28.60, 28.23. tR = 2.36 min (generic method). ESI-MS for C34H25ClFN3O4: calculated 593.1, found m/z 594.1, 596.1 [M + H]+, 592.2, 594.2 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4′-chloro-[1,1′-biphenyl]-4-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (19d)
Compound 19d was synthesized via general procedure C1 using 95c (227 mg, 0.44 mmol) with succinic anhydride 115 (88 mg, 0.88 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–10% EtOH/DCM) to afford 19d (205 mg, yield 77%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.04 (s, 1H), 7.67 (d, J = 8.6 Hz, 2H), 7.63 (dd, J = 8.8, 2.4 Hz, 1H), 7.59–7.48 (m, 7H), 7.47–7.39 (m, 2H), 7.29 (dd, J = 6.5, 2.0 Hz, 1H), 6.94 (d, J = 2.4 Hz, 1H), 6.88 (d, J = 8.3 Hz, 2H), 5.35 (dd, J = 12.0, 4.6 Hz, 1H), 3.77 (dd, J = 18.5, 12.1 Hz, 1H), 2.84 (dd, J = 18.5, 4.6 Hz, 1H), 2.50 (m, 2H), 2.29 (t, J = 6.8 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.58, 173.49, 168.64, 160.10, 152.44, 149.94, 141.77, 138.63, 137.62, 137.31, 134.60, 132.26, 131.18, 129.40, 128.84, 128.52, 128.45, 128.37, 128.32, 126.72, 126.14, 126.10, 126.05, 124.62, 120.68, 117.61, 58.66, 45.21, 28.60, 28.23. tR = 2.51 min (generic method). ESI-MS for C34H25Cl2N3O4: calculated 609.1, found m/z 610.0, 612.0, 614.2 [M + H]+; 608.1, 610.1, 612.0 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-[3-[4-(4-4-(5-(4′-Bromo-[1,1′-biphenyl]-4-yl)-3-(6-chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (20d)
Compound 20d was synthesized via general procedure C1 using 96c (214 mg, 0.38 mmol) with succinic anhydride 115 (76 mg, 0.76 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–20% EtOH/DCM) to afford 20d (197 mg, yield 79%). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 12.08 (s, 1H), 7.68–7.38 (m, 12H), 7.30–7.26 (m, 1H), 6.94 (d, J = 2.4 Hz, 1H), 6.89 (d, J = 8.4 Hz, 2H), 5.35 (dd, J = 12.0, 4.6 Hz, 1H), 3.78 (dd, J = 18.5, 12.1 Hz, 1H), 2.84 (dd, J = 18.5, 4.7 Hz, 1H), 2.58–2.43 (m, J = 3.7 Hz, 2H), 2.30 (t, J = 6.9 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.50, 168.66, 160.11, 152.44, 149.95, 141.81, 139.00, 137.67, 137.31, 134.60, 131.76, 131.18, 129.40, 128.71, 128.52, 128.45, 128.33, 126.68, 126.16, 126.11, 126.05, 124.62, 120.84, 120.68, 117.61, 58.67, 45.20, 28.61, 28.24. tR = 2.52 min (generic method). ESI-MS for C34H25BrClN3O4: calculated 653.1, found m/z 653.9, 655.9, 657.9 [M + H]+; 652.0, 654.0, 655.8 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4′-methoxy-[1,1′-biphenyl]-4-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (21d)
Compound 21d was synthesized via general procedure C1 using 97c (545 mg, 1.10 mmol) with succinic anhydride 115 (220 mg, 2.20 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 40 g, 0–10% EtOH/DCM) to afford 21d (359 mg, yield 55%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.03 (s, 1H), 7.67–7.50 (m, 6H), 7.44 (dt, J = 7.7, 5.9 Hz, 4H), 7.29 (dd, J = 6.7, 2.1 Hz, 1H), 7.02 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 2.3 Hz, 1H), 6.84 (d, J = 8.3 Hz, 2H), 5.33 (dd, J = 12.0, 4.6 Hz, 1H), 3.79 (s, 4H), 2.84 (dd, J = 18.4, 4.6 Hz, 1H), 2.30 (t, J = 6.9 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.51, 168.62, 160.12, 158.85, 152.44, 149.92, 140.63, 138.67, 137.31, 134.60, 132.23, 131.17, 129.40, 128.54, 128.44, 128.32, 127.68, 126.23, 126.10, 126.00, 124.66, 120.69, 117.61, 114.34, 58.70, 55.14, 45.24, 28.62, 28.25. tR = 2.30 min (generic method). ESI-MS for C35H28ClN3O5: calculated 605.2, found m/z 606.0, 608.0 [M + H]+; 604.0, 606.0 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(1-methyl-1H-indol-5-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (22d)
Compound 22d was synthesized via general procedure C1 using 98c (153 mg, 0.34 mmol) with succinic anhydride 115 (68 mg, 0.68 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–20% EtOH/DCM) to afford 22d (150 mg, yield 79%). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 12.18 (s, 1H), 7.63 (dd, J = 8.8, 2.3 Hz, 1H), 7.60–7.55 (m, 2H), 7.55–7.49 (m, 1H), 7.44 (dd, J = 7.7, 4.3 Hz, 2H), 7.31–7.20 (m, 3H), 6.98 (d, J = 1.7 Hz, 1H), 6.93 (d, J = 2.3 Hz, 1H), 6.61 (dd, J = 8.5, 1.7 Hz, 1H), 6.34 (dd, J = 3.0, 0.8 Hz, 1H), 5.35 (dd, J = 12.0, 4.6 Hz, 1H), 3.84–3.69 (m, 4H), 2.85 (dd, J = 18.4, 4.6 Hz, 1H), 2.53–2.45 (m, 2H), 2.26 (t, J = 6.9 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.72, 173.52, 168.43, 160.17, 152.27, 149.80, 137.29, 135.60, 134.66, 133.04, 131.11, 129.90, 129.47, 128.52, 128.43, 128.25, 127.73, 126.06, 124.84, 120.75, 118.98, 117.58, 117.22, 109.64, 100.28, 59.51, 45.76, 32.46, 29.17, 28.70, 28.27. tR = 2.09 min (generic method). ESI-MS for C31H25ClN4O4: calculated 552.2, found m/z 553.1, 555.2 [M + H]+; 551.2, 553.2 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(1-ethyl-1H-indol-5-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (23d)
Compound 23d was synthesized via general procedure C1 using 99c (153 mg, 0.33 mmol) with succinic anhydride 115 (66 mg, 0.66 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–10% EtOH/DCM) to afford 23d (92 mg, yield 49%). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 12.02 (s, 1H), 7.63 (dd, J = 8.8, 2.4 Hz, 1H), 7.58 (dd, J = 5.5, 3.3 Hz, 2H), 7.52 (ddd, J = 8.9, 5.2, 3.1 Hz, 1H), 7.44 (dd, J = 8.7, 4.4 Hz, 2H), 7.35 (d, J = 3.1 Hz, 1H), 7.29 (dd, J = 8.5, 2.2 Hz, 2H), 6.95 (dd, J = 15.5, 2.0 Hz, 2H), 6.60 (dd, J = 8.5, 1.7 Hz, 1H), 6.34 (d, J = 3.1 Hz, 1H), 5.34 (dd, J = 12.0, 4.7 Hz, 1H), 4.17 (q, J = 7.2 Hz, 2H), 3.77 (dd, J = 18.4, 12.1 Hz, 1H), 2.86 (dd, J = 18.4, 4.7 Hz, 1H), 2.48–2.42 (m, 2H), 2.26 (t, J = 6.9 Hz, 2H), 1.34 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.53, 168.45, 160.17, 152.28, 149.80, 137.29, 134.66, 134.56, 133.01, 131.11, 129.45, 128.53, 128.42, 128.28, 127.89, 126.05, 124.84, 120.74, 118.93, 117.58, 117.32, 109.62, 100.49, 59.51, 45.75, 40.26, 28.72, 28.30, 15.53. tR = 2.20 min (generic method). ESI-MS for C32H27ClN4O4: calculated 566.2, found m/z 567.2, 569.2 [M + H]+; 565.3, 567.3 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(1-ethyl-1H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (24d)
Compound 24d was synthesized via general procedure C1 using 100c (196 mg, 0.42 mmol) with succinic anhydride 115 (84 mg, 0.84 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–10% EtOH/DCM) to afford 24d (177 mg, yield 74%). 1H NMR (400 MHz, DMSO-d6) δ 12.39 (s, 1H), 12.03 (s, 1H), 7.98 (s, 1H), 7.64 (dd, J = 8.8, 2.4 Hz, 1H), 7.59 (dd, J = 5.4, 3.5 Hz, 2H), 7.52 (dd, J = 10.0, 6.6 Hz, 2H), 7.48–7.43 (m, 2H), 7.29 (d, J = 7.5 Hz, 1H), 7.14 (d, J = 1.5 Hz, 1H), 6.94 (d, J = 2.3 Hz, 1H), 6.83 (dd, J = 8.7, 1.6 Hz, 1H), 5.42 (dd, J = 12.0, 4.6 Hz, 1H), 4.42 (q, J = 7.2 Hz, 2H), 3.80 (dd, J = 18.5, 12.1 Hz, 1H), 2.85 (dd, J = 18.5, 4.6 Hz, 1H), 2.54–2.48 (m, 2H), 2.28 (t, J = 6.9 Hz, 2H), 1.38 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.99, 169.05, 160.62, 152.86, 150.36, 138.51, 137.79, 135.07, 134.85, 132.81, 131.65, 130.00, 129.09, 128.97, 128.93, 128.78, 126.57, 126.54, 125.22, 124.57, 123.78, 121.19, 118.09, 117.79, 110.22, 59.57, 45.96, 43.53, 29.13, 28.72, 15.43. tR = 1.95 min (generic method). ESI-MS for C31H26ClN5O4: calculated 567.2, found m/z 568.5, 570.4 [M + H]+, 566.4, 568.4 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(2-ethyl-2H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (25d)
Compound 25d was synthesized via general procedure C1 using 101c (198 mg, 0.42 mmol) with succinic anhydride 115 (84 mg, 0.84 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–20% EtOH/DCM) to afford 25d (120 mg, yield 50%). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 12.04 (s, 1H), 8.29 (s, 1H), 7.66–7.55 (m, 3H), 7.51 (td, J = 6.6, 5.5, 3.3 Hz, 1H), 7.44 (dd, J = 10.0, 6.8 Hz, 3H), 7.28 (d, J = 7.5 Hz, 1H), 7.07 (d, J = 1.6 Hz, 1H), 6.93 (d, J = 2.4 Hz, 1H), 6.61 (dd, J = 8.9, 1.7 Hz, 1H), 5.35 (dd, J = 12.0, 4.6 Hz, 1H), 4.43 (q, J = 7.3 Hz, 2H), 3.77 (dd, J = 18.4, 12.1 Hz, 1H), 2.82 (dd, J = 18.4, 4.6 Hz, 1H), 2.54–2.44 (m, 2H), 2.28 (t, J = 6.9 Hz, 2H), 1.49 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.50, 168.55, 160.12, 152.37, 149.84, 147.32, 137.29, 134.57, 134.39, 131.15, 129.53, 128.57, 128.49, 128.42, 128.28, 126.07, 126.04, 124.75, 123.57, 123.00, 120.94, 120.72, 117.60, 117.32, 116.68, 59.31, 47.68, 45.24, 28.67, 28.26, 15.84. tR = 1.85 min (generic method). ESI-MS for C31H26ClN5O4: calculated 567.2, found m/z 568.4, 570.4 [M + H]+, 566.5, 568.4 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(1-propyl-1H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (26d)
Compound 26d was synthesized via general procedure C1 using 102c (176 mg, 0.36 mmol) with succinic anhydride 115 (72 mg, 0.72 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–25% EtOH/DCM) to afford 26d (178 mg, yield 86%). 1H NMR (400 MHz, DMSO-d6) δ 12.36 (s, 1H), 12.05 (s, 1H), 7.97 (d, J = 0.9 Hz, 1H), 7.62 (dd, J = 8.8, 2.4 Hz, 1H), 7.57 (qd, J = 4.0, 1.0 Hz, 2H), 7.53–7.47 (m, 2H), 7.47–7.41 (m, 2H), 7.30–7.25 (m, 1H), 7.16–7.11 (m, 1H), 6.94 (d, J = 2.3 Hz, 1H), 6.84 (dd, J = 8.8, 1.6 Hz, 1H), 5.42 (dd, J = 12.0, 4.6 Hz, 1H), 4.33 (t, J = 6.9 Hz, 2H), 3.78 (dd, J = 18.5, 12.0 Hz, 1H), 2.85 (dd, J = 18.5, 4.6 Hz, 1H), 2.54–2.48 (m, 2H), 2.28 (t, J = 6.9 Hz, 2H), 1.83 (h, J = 7.2 Hz, 2H), 0.83 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.53, 168.61, 160.15, 152.41, 149.89, 138.59, 137.31, 134.60, 134.31, 132.32, 131.17, 129.50, 128.60, 128.49, 128.43, 128.32, 126.11, 126.06, 124.73, 124.13, 123.19, 120.71, 117.62, 117.28, 109.80, 59.09, 49.64, 45.49, 28.67, 28.26, 22.83, 11.17. tR = 2.06 min (generic method). ESI-MS for C32H28ClN5O4: calculated 581.2, found m/z 582.1, 584.1 [M + H]+, 580.4, 582.0 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(2-propyl-2H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (27d)
Compound 27d was synthesized via general procedure C1 using 103c (173 mg, 0.36 mmol) with succinic anhydride 115 (72 mg, 0.72 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–20% EtOH/DCM) to afford 27d (162 mg, yield 78%). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 2H), 8.28 (d, J = 0.9 Hz, 1H), 7.63–7.54 (m, 3H), 7.50 (td, J = 6.6, 5.5, 3.3 Hz, 1H), 7.47–7.40 (m, 3H), 7.30–7.24 (m, 1H), 7.08 (t, J = 1.2 Hz, 1H), 6.93 (d, J = 2.4 Hz, 1H), 6.62 (dd, J = 8.9, 1.7 Hz, 1H), 5.36 (dd, J = 12.0, 4.6 Hz, 1H), 4.35 (t, J = 6.9 Hz, 2H), 3.77 (dd, J = 18.5, 12.0 Hz, 1H), 2.83 (dd, J = 18.5, 4.6 Hz, 1H), 2.54–2.50 (m, 2H), 2.29 (t, J = 6.9 Hz, 2H), 1.96–1.87 (m, 2H), 0.83 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.57, 168.64, 160.16, 152.44, 149.89, 147.41, 137.31, 134.59, 134.40, 131.16, 129.56, 128.60, 128.52, 128.43, 128.31, 126.13, 126.07, 124.77, 123.75, 123.60, 120.86, 120.74, 117.62, 117.39, 116.71, 59.36, 54.27, 45.27, 28.73, 28.32, 23.44, 10.91. tR = 2.00 min (generic method). ESI-MS for C32H28ClN5O4: calculated 581.2, found m/z 582.2, 584.3 [M + H]+, 580.3, 582.4 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(1-cyclohexyl-1H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (28d)
Compound 28d was synthesized via general procedure C1 using 104c (164 mg, 0.31 mmol) with succinic anhydride 115 (62 mg, 0.62 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–12% EtOH/DCM) to afford 28d (169 mg, yield 87%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.07 (s, 1H), 7.96 (s, 1H), 7.65–7.49 (m, 5H), 7.48–7.41 (m, 2H), 7.28 (dd, J = 7.5, 1.8 Hz, 1H), 7.11 (d, J = 1.6 Hz, 1H), 6.94 (d, J = 2.3 Hz, 1H), 6.84 (dd, J = 8.8, 1.6 Hz, 1H), 5.41 (dd, J = 12.0, 4.6 Hz, 1H), 4.54 (tt, J = 10.0, 5.5 Hz, 1H), 3.80 (dd, J = 18.5, 12.1 Hz, 1H), 2.85 (dd, J = 18.5, 4.7 Hz, 1H), 2.53–2.46 (m, 2H), 2.28 (t, J = 6.9 Hz, 2H), 2.01–1.81 (m, 6H), 1.70 (dt, J = 12.8, 3.4 Hz, 1H), 1.58–1.43 (m, 2H), 1.26 (qt, J = 12.7, 3.5 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 173.74, 173.51, 168.59, 160.16, 152.36, 149.90, 137.70, 137.31, 134.64, 134.41, 132.08, 131.16, 129.51, 128.59, 128.48, 128.45, 128.30, 126.12, 126.07, 124.73, 123.92, 123.18, 120.71, 117.61, 117.24, 109.77, 59.12, 56.63, 45.50, 32.34, 28.67, 28.26, 25.08. tR = 2.33 min (generic method). ESI-MS for C35H32ClN5O4: calculated 621.2, found m/z 622.3, 624.2 [M + H]+, 620.3, 622.4 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(2-cyclohexyl-2H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (29d)
Compound 29d was synthesized via general procedure C1 using 105c (100 mg, 0.19 mmol) with succinic anhydride 115 (38 mg, 0.38 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–20% EtOH/DCM) to afford 29d (113 mg, yield 95%). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 12.10 (s, 1H), 8.29 (d, J = 1.0 Hz, 1H), 7.65–7.55 (m, 3H), 7.53–7.48 (m, 1H), 7.44 (t, J = 8.7 Hz, 3H), 7.30–7.24 (m, 1H), 7.10–7.05 (m, 1H), 6.93 (d, J = 2.4 Hz, 1H), 6.60 (dd, J = 9.0, 1.7 Hz, 1H), 5.35 (dd, J = 12.0, 4.6 Hz, 1H), 4.44 (tt, J = 11.2, 3.8 Hz, 1H), 3.77 (dd, J = 18.4, 12.1 Hz, 1H), 2.82 (dd, J = 18.4, 4.7 Hz, 1H), 2.55–2.46 (m, 2H), 2.28 (t, J = 6.9 Hz, 2H), 2.14–2.02 (m, 2H), 1.95–1.81 (m, 4H), 1.70 (dd, J = 12.6, 3.7 Hz, 1H), 1.45 (qt, J = 12.5, 3.4 Hz, 2H), 1.25 (tdd, J = 15.6, 11.7, 7.8 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 173.72, 173.52, 168.56, 160.14, 152.38, 149.87, 146.88, 137.30, 134.58, 134.35, 131.15, 129.52, 128.59, 128.51, 128.44, 128.30, 126.09, 126.05, 124.76, 123.49, 121.57, 120.73, 120.62, 117.61, 117.47, 116.76, 61.63, 59.36, 45.25, 33.29, 29.13, 28.69, 28.28, 24.92, 24.89. tR = 2.20 min (generic method). ESI-MS for C35H32ClN5O4: calculated 621.2, found m/z 622.2, 624.2 [M + H]+, 620.3, 622.3 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-(1-ethyl-1H-indazol-5-yl)phenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (30d)
Compound 30d was synthesized via general procedure C1 using 106c (170 mg, 0.31 mmol) with succinic anhydride 115 (62 mg, 0.62 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–20% EtOH/DCM) to afford 30d (163 mg, yield 81%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.07 (s, 1H), 8.10 (d, J = 0.8 Hz, 1H), 7.99 (d, J = 1.6 Hz, 1H), 7.74 (d, J = 8.8 Hz, 1H), 7.67 (dd, J = 8.8, 1.7 Hz, 1H), 7.63 (dd, J = 8.8, 2.4 Hz, 1H), 7.60–7.51 (m, 5H), 7.48–7.40 (m, 2H), 7.30 (dd, J = 5.9, 1.8 Hz, 1H), 6.94 (d, J = 2.3 Hz, 1H), 6.89 (d, J = 8.3 Hz, 2H), 5.35 (dd, J = 12.0, 4.6 Hz, 1H), 4.46 (q, J = 7.2 Hz, 2H), 3.78 (dd, J = 18.5, 12.1 Hz, 1H), 2.86 (dd, J = 18.5, 4.6 Hz, 1H), 2.59–2.44 (m, 2H), 2.31 (t, J = 6.8 Hz, 2H), 1.41 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.53, 168.65, 160.13, 152.47, 149.95, 140.77, 139.50, 138.23, 137.32, 134.63, 132.92, 132.54, 131.18, 129.43, 128.55, 128.47, 128.34, 126.90, 126.11, 126.07, 125.52, 124.67, 124.25, 120.70, 118.46, 117.62, 110.02, 58.72, 45.26, 43.15, 28.64, 28.26, 14.94. tR = 2.24 min (generic method). ESI-MS for C37H30ClN5O4: calculated 643.2, found m/z 644.1, 646.1 [M + H]+, 642.2, 644.1 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-(2-ethyl-2H-indazol-5-yl)phenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (31d)
Compound 31d was synthesized via general procedure C1 using 107c (117 mg, 0.21 mmol) with succinic anhydride 115 (42 mg, 0.42 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–20% EtOH/DCM) to afford 31d (66 mg, yield 49%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.15 (s, 1H), 8.42 (d, J = 0.9 Hz, 1H), 7.92 (t, J = 1.3 Hz, 1H), 7.68 (d, J = 8.9 Hz, 1H), 7.63 (dd, J = 8.8, 2.4 Hz, 1H), 7.60–7.50 (m, 6H), 7.48–7.41 (m, 2H), 7.30 (dd, J = 6.1, 1.9 Hz, 1H), 6.94 (d, J = 2.3 Hz, 1H), 6.87 (d, J = 8.4 Hz, 2H), 5.35 (dd, J = 12.0, 4.6 Hz, 1H), 4.47 (q, J = 7.3 Hz, 2H), 3.78 (dd, J = 18.5, 12.1 Hz, 1H), 2.86 (dd, J = 18.5, 4.7 Hz, 1H), 2.57–2.43 (m, 2H), 2.30 (t, J = 6.9 Hz, 2H), 1.52 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.63, 173.52, 168.64, 160.13, 152.47, 149.95, 147.41, 140.72, 137.33, 132.68, 128.56, 128.47, 128.35, 126.74, 126.11, 126.06, 126.02, 125.23, 124.68, 123.68, 121.91, 120.71, 117.89, 117.63, 117.45, 58.72, 47.80, 28.93, 28.63, 28.26, 15.76. tR = 2.10 min (generic method). ESI-MS for C37H30ClN5O4: calculated 643.2, found m/z 644.2, 646.2 [M + H]+, 642.3, 644.3 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-(1-propyl-1H-pyrazol-4-yl)phenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (32d)
Compound 32d was synthesized via general procedure C1 using 108c (155 mg, 0.30 mmol) with succinic anhydride 115 (60 mg, 0.60 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–30% EtOH/DCM) to afford 32d (189 mg, quantitative yield). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 12.10 (s, 1H), 8.14 (s, 1H), 7.84 (s, 1H), 7.62 (dd, J = 8.8, 2.4 Hz, 1H), 7.59–7.50 (m, 3H), 7.48–7.36 (m, 4H), 7.31–7.25 (m, 1H), 6.94 (d, J = 2.3 Hz, 1H), 6.77 (d, J = 8.0 Hz, 2H), 5.28 (dd, J = 12.0, 4.6 Hz, 1H), 4.07 (t, J = 7.0 Hz, 2H), 3.75 (dd, J = 18.4, 12.1 Hz, 1H), 2.82 (dd, J = 18.5, 4.6 Hz, 1H), 2.48–2.41 (m, 2H), 2.28 (t, J = 6.9 Hz, 2H), 1.96–1.87 (m, 2H), 0.85 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.50, 168.56, 160.12, 152.39, 149.89, 139.75, 137.30, 135.88, 134.62, 131.56, 131.16, 129.42, 128.53, 128.44, 128.29, 126.88, 126.09, 125.98, 124.96, 124.68, 121.24, 120.70, 117.60, 58.75, 53.00, 45.20, 28.63, 28.26, 23.17, 10.93. tR = 2.09 min (generic method). ESI-MS for C34H30ClN5O4: calculated 607.2, found m/z 608.0, 610.0 [M + H]+, 606.1, 608.0 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
6-Chloro-3-(5-(4-chlorophenyl)-1-propionyl-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (33d)
Compound 33d was synthesized via general procedure C3 using 87c (200 mg, 0.46 mmol) and propionic acid 117 (60.5 μL, 0.83 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–2% MeOH/DCM) to afford 33d (76 mg, yield 34%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 7.63 (dd, J = 9.0, 2.4 Hz, 1H), 7.54–7.49 (m, 3H), 7.44 (d, J = 8.8 Hz, 1H), 7.41–7.39 (m, 1H), 7.30–7.27 (m, 3H), 6.92 (d, J = 2.4 Hz, 1H), 6.80 (d, J = 8.8 Hz, 2H), 5.31 (dd, J = 12.2, 4.6 Hz, 1H), 3.74 (dd, J = 18.4, 12.0 Hz, 1H), 2.80 (dd, J = 18.4, 4.0 Hz, 1H), 2.31–2.14 (m, 2H), 0.82 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 206.88, 170.96, 160.55, 152.65, 150.33, 141.76, 137.75, 135.13, 132.03, 131.64, 129.80, 128.95, 128.87, 128.82, 128.79, 127.81, 126.55, 126.48, 125.04, 121.10, 118.07, 110.00, 58.61, 45.45, 31.11, 27.06, 9.26. ESI-MS for C27H21Cl2N3O2: calculated 489.1, found m/z 490.2, 492.1, 494.1 [M + H]+; 488.3, 490.3, 492.1 [M – H]−. UPLC–MS purity (UV at 215 nm) 98%.
3-(5-(4-Bromophenyl)-1-propionyl-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (34d)
Compound 34d was synthesized via general procedure C3 using 88c (150 mg, 0.31 mmol) and propionic acid 117 (41.8 μL, 0.56 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–0.5% MeOH/DCM) to afford 34d (60 mg, yield 36%). 1H NMR (400 MHz, DMSO-d6) δ 12.41 (s, 1H), 7.64 (dd, J = 12.8, 1.6 Hz, 1H), 7.55–7.50 (m, 3H), 7.46–7.41 (m, 4H), 7.28 (d, J = 6.8 Hz, 1H), 6.93 (d, J = 2.0 Hz, 1H), 6.75 (d, J = 8.0 Hz, 2H), 5.30 (dd, J = 12.4, 4.0 Hz, 1H), 3.75 (dd, J = 18.4, 12.0 Hz, 1H), 2.80 (dd, J = 18.2, 4.6 Hz, 1H), 2.31–2.14 (m, 2H), 0.83 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 206.78, 171.02, 160.54, 152.60, 150.33, 142.18, 137.77, 135.16, 131.72, 131.61, 129.79, 128.97, 128.92, 128.84, 128.78, 128.18, 126.56, 126.49, 125.03, 121.13, 120.51, 118.06, 58.73, 45.40, 31.08, 27.05, 9.23. ESI-MS for C27H21BrClN3O2: calculated 533.0, found m/z 534.2, 536.1, 538.1 [M + H]+; 532.2, 534.1, 536.2 [M – H]−. UPLC–MS purity (UV at 215 nm) 97%.
6-Chloro-3-(5-(4-methoxyphenyl)-1-propionyl-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (35d)
Compound 35d was synthesized via general procedure C3 using 89c (150 mg, 0.36 mmol) and propionic acid 117 (48.5 μL, 0.65 mmol). The crude was purified by direct phase flash column chromatography (SiO2 gold 24 g; 0–2% MeOH/DCM), to afford 35d (100 mg, yield 59%). 1H NMR (400 MHz, DMSO-d6) δ 12.36 (s, 1H), 7.64 (dd, J = 8.6, 2.4 Hz, 1H), 7.56–7.52 (m, 3H), 7.45 (d, J = 8.4 Hz, 1H), 7.40–7.38 (m, 1H), 7.29–7.27 (m, 1H), 6.92 (d, J = 2.0 Hz, 1H), 6.75 (dd, J = 15.2, 9.2 Hz, 4H), 5.24 (dd, J = 12.0, 4.8 Hz, 1H), 3.72 (s, 3H), 3.70 (dd, J = 16.0, 12.0 Hz, 1H), 2.81 (dd, J = 20.0, 4.4 Hz, 1H), 2.29–2.12 (m, 2H), 0.82 (t, J = 8.0 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.80, 160.60, 158.67, 152.56, 150.23, 137.74, 135.16, 134.94, 131.58, 129.76, 129.03, 128.89, 128.84, 128.77, 127.20, 126.51, 126.47, 125.22, 121.15, 118.05, 114.15, 58.73, 55.49, 45.59, 27.12, 9.31. ESI-MS for C28H24ClN3O3: calculated 485.1, found m/z 486.4, 488.4 [M + H]+; 484.3, 486.3 [M – H]-. UPLC–MS purity (UV at 215 nm) 98%.
3-(1-Acetyl-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (36d)
Compound 36d was synthesized via general procedure C2 using 87c (210 mg, 0.48 mmol) with acetic anhydride 116 (888 μL, 0.96 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–1.5% MeOH/DCM) to afford 36d (200 mg, yield 94%). 1H NMR (400 MHz, DMSO-d6) δ 12.41 (s, 1H), 7.64 (dt, J = 8.0, 1.2 Hz, 1H), 7.58–7.53 (m, 3H), 7.44 (d, J = 8.0 Hz, 1H), 7.41–7.39 (m, 1H), 7.30–7.28 (m, 3H), 6.93 (d, J = 0.8 Hz, 1H), 6.80 (d, J = 7.6 Hz, 2H), 5.32 (dd, J = 12.0, 8.0 Hz, 1H), 3.74 (dd, J = 20.0, 12.0 Hz, 1H), 2.82 (dd, J = 16.0, 4.0 Hz, 1H), 1.87 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.56, 160.54, 152.67, 150.39, 141.60, 137.75, 135.11, 132.05, 131.65, 129.86, 129.01, 128.96, 128.81, 128.75, 127.83, 126.55, 126.49, 124.96, 121.10, 118.06, 58.56, 45.62, 21.82. ESI-MS for C26H19Cl2N3O2: calculated 475.1, found m/z 476.3, 478.4, 480.4 [M + H]+; 474.3, 476.3, 478.1 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
3-(1-Acetyl-5-(4-bromophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (37d)
Compound 37d was synthesized via general procedure C2 using 88c (100 mg, 0.21 mmol) with acetic anhydride 116 (40 μL, 0.42 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 12 g, 0–2% MeOH/DCM) to afford 37d (80 mg, yield 73%). 1H NMR (400 MHz, DMSO-d6) δ 12.39 (s, 1H), 7.64 (dd, J = 8.0, 4.0 Hz, 1H), 7.57–7.53 (m, 3H), 7.45–7.39 (m, 4H), 7.28 (dd, J = 6.0, 1.6 Hz, 1H), 6.93 (d, J = 2.0 Hz, 1H), 6.75 (d, J = 8.8 Hz, 2H), 5.30 (dd, J = 12.0, 4.4 Hz, 1H), 3.74 (dd, J = 18.5, 12.0 Hz, 1H), 2.82 (dd, J = 4.0, 20.0 Hz, 1H), 1.86 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.58, 160.54, 152.64, 150.39, 142.02, 137.78, 135.15, 131.72, 131.63, 129.83, 129.03, 128.94, 128.78, 128.74, 128.20, 126.56, 126.50, 124.96, 121.13, 120.54, 118.06, 58.67, 45.58, 21.78. ESI-MS for C26H19BrClN3O2: calculated 519.0, found m/z 520.1, 522.1, 524.1 [M + H]+; 518.1, 520.1, 522.2 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
3-(1-Acetyl-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (38d)
Compound 38d was synthesized via general procedure C2 using 89c (65 mg, 0.15 mmol) with acetic anhydride 116 (28 μL, 0.30 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 12 g, 0–1.5% MeOH/DCM) to afford 38d (30 mg, yield 43%). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 7.63 (dd, J = 8.8, 2.0 Hz, 1H), 7.55–7.52 (m, 3H), 7.45 (d, J = 8.8 Hz, 1H), 7.40–7.39 (m, 1H), 7.29 (d, J = 6.8 Hz, 1H), 6.93 (d, J = 2.0 Hz, 1H), 6.75 (dd, J = 8.8, 16.0 Hz, 4H), 5.24 (dd, J = 12.0, 4.0 Hz, 1H), 3.72 (s, 3H), 3.70–3.65 (m,1H), 2.83 (dd, J = 4.4, 18.4 Hz, 2H), 1.84 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.42, 160.60, 158.70, 152.63, 150.29, 137.75, 135.14, 134.79, 131.60, 129.80, 129.09, 128.92, 128.78, 128.75, 127.24, 126.52, 126.49, 125.13, 121.14, 118.07, 114.15, 58.67, 55.49, 45.74, 21.90. ESI-MS for C27H22ClN3O3: calculated 471.1, found m/z 472.2, 474.2, [M + H]+; 470.3, 472.3 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
3-(1-Acetyl-5-(4-(tert-butyl)phenyl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (39d)
Compound 39d was synthesized via general procedure C2 using 90c (141 mg, 0.31 mmol) with acetic anhydride 116 (59 μL, 0.62 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–5% EtOH/DCM) to afford 39d (105 mg, yield 68%). 1H NMR (400 MHz, DMSO-d6) δ 12.36 (s, 1H), 7.63 (dt, J = 9.8, 4.9 Hz, 1H), 7.60–7.48 (m, 3H), 7.44 (t, J = 9.5 Hz, 1H), 7.41–7.36 (m, 1H), 7.30 (d, J = 7.1 Hz, 1H), 7.24 (d, J = 8.3 Hz, 2H), 6.94 (d, J = 2.3 Hz, 1H), 6.75 (d, J = 8.3 Hz, 2H), 5.32–5.17 (m, 1H), 3.69 (dt, J = 28.5, 14.3 Hz, 1H), 2.86 (dd, J = 18.4, 4.6 Hz, 1H), 1.86 (s, 3H), 1.27 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 167.01, 160.12, 152.20, 149.87, 149.25, 139.30, 137.30, 134.72, 131.14, 129.30, 128.66, 128.44, 128.31, 128.29, 126.07, 126.04, 125.16, 125.13, 124.65, 120.69, 117.60, 58.45, 45.30, 34.12, 31.10, 21.42. tR = 1.83 min (apolar method). ESI-MS for C30H28ClN3O2: calculated 497.2, found m/z 498.5, 500.5 [M + H]+; 494.4, 496.4 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
3-(1-Acetyl-5-(4-(trifluoromethyl)phenyl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (40d)
Compound 40d was synthesized via general procedure C2 using 92c (326 mg, 0.70 mmol) with acetic anhydride 116 (132 μL, 1.40 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–5% EtOH/DCM) to afford 40d (268 mg, yield 74%). 1H NMR (400 MHz, DMSO-d6) δ 12.39 (s, 1H), 7.67–7.49 (m, 6H), 7.46 (d, J = 8.8 Hz, 1H), 7.44–7.39 (m, 1H), 7.32–7.25 (m, 1H), 7.03 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 2.4 Hz, 1H), 5.42 (dd, J = 12.1, 4.7 Hz, 1H), 3.79 (dd, J = 18.5, 12.1 Hz, 1H), 2.87 (dd, J = 18.5, 4.8 Hz, 1H), 1.89 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.21, 160.08, 152.27, 150.01, 146.70, 137.34, 134.69, 131.23, 129.39, 128.54, 128.51, 128.33, 127.90, 127.58, 126.24, 126.11, 126.05, 125.52, 125.43, 125.39, 125.35, 124.41, 122.82, 120.62, 117.63, 58.36, 45.14, 21.30. tR = 2.55 min (generic method). ESI-MS for C27H19ClF3N3O2: calculated 509.1, found m/z 510.5, 513.5 [M + H]+; 508.4, 510.4 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
3-(1-Acetyl-5-(furan-2-yl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (41d)
Compound 41d was synthesized via general procedure C2 using 93c (202 mg, 0.52 mmol) with acetic anhydride 116 (98 μL, 1.04 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–5% EtOH/DCM) to afford 41d (162 mg, yield 73%). 1H NMR (400 MHz, DMSO-d6) δ 12.36 (s, 1H), 7.63 (dd, J = 8.8, 2.3 Hz, 1H), 7.52–7.42 (m, 5H), 7.35–7.29 (m, 2H), 6.96 (d, J = 2.3 Hz, 1H), 6.33 (dd, J = 3.3, 1.9 Hz, 1H), 6.03 (d, J = 3.2 Hz, 1H), 5.38 (dd, J = 11.9, 4.6 Hz, 1H), 3.57 (dd, J = 18.2, 11.9 Hz, 1H), 3.20 (dd, J = 18.2, 4.7 Hz, 1H), 1.75 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.26, 160.12, 152.98, 152.28, 150.04, 142.02, 137.37, 134.92, 131.18, 128.87, 128.80, 128.25, 128.19, 126.12, 126.07, 124.31, 120.62, 117.59, 110.33, 106.33, 52.35, 41.84, 21.33. tR = 0.95 min (apolar method). ESI-MS for C24H18ClN3O3: calculated 431.1, found m/z 432.4, 434.4 [M + H]+; 430.4, 432.5 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
3-(1-Acetyl-5-(4′-chloro-[1,1′-biphenyl]-4-yl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (42d)
Compound 42d was synthesized via general procedure C2 using 95c (224 mg, 0.44 mmol) with acetic anhydride 116 (83 μL, 0.88 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–10% EtOH/DCM) to afford 42d (245 mg, quantitative yield). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 7.71–7.48 (m, 10H), 7.48–7.39 (m, 2H), 7.29 (dt, J = 5.2, 2.0 Hz, 1H), 6.94 (d, J = 2.4 Hz, 1H), 6.90 (d, J = 8.3 Hz, 2H), 5.35 (dd, J = 12.1, 4.6 Hz, 1H), 3.78 (dd, J = 18.4, 12.1 Hz, 1H), 2.88 (dd, J = 18.5, 4.6 Hz, 1H), 1.88 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.09, 160.12, 152.23, 149.93, 141.80, 138.63, 137.66, 137.30, 134.73, 132.26, 131.16, 129.39, 128.82, 128.59, 128.49, 128.40, 128.37, 128.29, 126.74, 126.15, 126.10, 126.05, 124.60, 120.67, 117.60, 58.48, 45.29, 21.41. tR = 2.00 min (apolar method). ESI-MS for C32H23Cl2N3O2: calculated 551.1, found m/z 552.0, 554.0, 556.1 [M + H]+; 550.1, 552.1, 554.0 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
3-(1-Acetyl-5-(4′-bromo-[1,1′-biphenyl]-4-yl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (43d)
Compound 43d was synthesized via general procedure C2 using 96c (203 mg, 0.36 mmol) with acetic anhydride 116 (68 μL, 0.72 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–10% EtOH/DCM) to afford 43d (175 mg, yield 80%). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 7.66–7.36 (m, 12H), 7.32–7.24 (m, 1H), 6.98–6.85 (m, 3H), 5.35 (dd, J = 12.0, 4.6 Hz, 1H), 3.78 (dd, J = 18.4, 12.1 Hz, 1H), 2.90 (dd, J = 18.4, 4.6 Hz, 1H), 1.88 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.10, 160.12, 152.20, 149.92, 141.81, 138.97, 137.70, 137.30, 134.73, 131.71, 131.12, 129.36, 128.67, 128.58, 128.46, 128.36, 128.27, 126.67, 126.17, 126.12, 126.04, 124.57, 120.84, 120.65, 117.58, 79.15, 58.51, 45.28, 21.39. tR = 2.52 min (apolar method). ESI-MS for C32H23BrClN3O2: calculated 595.1, found m/z 595.9, 597.9, 600.0 [M + H]+; 594.0, 596.0, 597.9 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
3-(1-Acetyl-5-(4′-methoxy-[1,1′-biphenyl]-4-yl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (44d)
Compound 44d was synthesized via general procedure C2 using 97c (179 mg, 0.35 mmol) with acetic anhydride 116 (66 μL, 0.70 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–8% EtOH/DCM) to afford 44d (158 mg, yield 83%). 1H NMR (600 MHz, DMSO-d6) δ 12.39 (s, 1H), 7.63 (dd, J = 8.8, 2.4 Hz, 1H), 7.60–7.53 (m, 5H), 7.45 (d, J = 8.5 Hz, 3H), 7.43–7.39 (m, 1H), 7.31–7.27 (m, 1H), 7.03–6.99 (m, 2H), 6.94 (d, J = 2.4 Hz, 1H), 6.86 (d, J = 8.3 Hz, 2H), 5.33 (dd, J = 12.1, 4.6 Hz, 1H), 3.79 (s, 4H), 2.88 (dd, J = 18.5, 4.6 Hz, 1H), 1.88 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 167.09, 160.17, 158.87, 152.28, 149.95, 140.70, 138.74, 137.33, 134.75, 132.24, 131.20, 129.43, 128.64, 128.53, 128.43, 128.33, 127.72, 126.28, 126.12, 126.08, 126.04, 124.66, 120.71, 117.64, 114.35, 58.53, 55.16, 45.34, 21.47. tR = 1.65 min (apolar method). ESI-MS for C33H26ClN3O3: calculated 547.2, found m/z 548.0, 550.0 [M + H]+; 546.0, 548.0 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
3-(1-Acetyl-5-(1-methyl-1H-indol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (45d)
Compound 45d was synthesized via general procedure C2 using 98c (127 mg, 0.28 mmol) with acetic anhydride 116 (53 μL, 0.56 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–10% EtOH/DCM) to afford 45d (107 mg, yield 75%). 1H NMR (400 MHz, DMSO-d6) δ 12.36 (s, 1H), 7.64–7.56 (m, 3H), 7.53 (ddd, J = 8.9, 5.6, 3.3 Hz, 1H), 7.47–7.40 (m, 2H), 7.31–7.23 (m, 3H), 6.99 (d, J = 1.7 Hz, 1H), 6.94 (d, J = 2.3 Hz, 1H), 6.65 (dd, J = 8.5, 1.7 Hz, 1H), 6.35 (dd, J = 3.1, 0.8 Hz, 1H), 5.36 (dd, J = 12.0, 4.5 Hz, 1H), 3.75 (s, 4H), 2.90 (dd, J = 18.4, 4.6 Hz, 1H), 1.86 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.93, 160.20, 152.06, 149.80, 137.30, 135.61, 134.79, 133.10, 131.09, 129.92, 129.46, 128.59, 128.47, 128.37, 128.23, 127.74, 126.06, 124.81, 120.74, 119.02, 117.58, 117.16, 109.63, 100.26, 59.32, 45.86, 32.45, 21.54. tR = 2.41 min (generic method). ESI-MS for C29H23ClN4O2: calculated 494.1, found m/z 495.2, 497.2 [M + H]+; 493.3, 495.3 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
3-(1-Acetyl-5-(1-ethyl-1H-indol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (46d)
Compound 46d was synthesized via general procedure C2 using 99c (168 mg, 0.36 mmol) with acetic anhydride 116 (68 μL, 0.72 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–10% EtOH/DCM) to afford 46d (128 mg, yield 70%). 1H NMR (400 MHz, DMSO-d6) δ 12.36 (s, 1H), 7.64–7.50 (m, 4H), 7.47–7.40 (m, 2H), 7.35 (d, J = 3.1 Hz, 1H), 7.32–7.27 (m, 2H), 6.97 (dd, J = 22.0, 2.0 Hz, 2H), 6.64 (dd, J = 8.5, 1.7 Hz, 1H), 6.35 (dd, J = 3.1, 0.8 Hz, 1H), 5.35 (dd, J = 12.0, 4.6 Hz, 1H), 4.16 (q, J = 7.2 Hz, 2H), 3.78 (dd, J = 18.4, 12.0 Hz, 1H), 2.91 (dd, J = 18.4, 4.6 Hz, 1H), 1.86 (s, 3H), 1.33 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.95, 160.20, 152.07, 149.81, 137.29, 134.80, 134.57, 133.07, 131.09, 129.43, 128.61, 128.47, 128.36, 128.31, 128.24, 127.90, 126.07, 124.82, 120.74, 118.97, 117.58, 117.29, 109.62, 100.48, 59.33, 45.85, 40.28, 21.55, 21.18, 15.52. tR = 2.51 min (generic method). ESI-MS for C30H25ClN4O2: calculated 508.2, found m/z 509.3, 511.2 [M + H]+; 507.3, 509.3 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
3-(1-Acetyl-5-(1-ethyl-1H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (47d)
Compound 47d was synthesized via general procedure C2 using 100c (131 mg, 0.28 mmol) with acetic anhydride 116 (53 μL, 0.56 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 12 g, 0–5% EtOH/DCM) to afford 47d (118 mg, yield 82%). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 7.98 (d, J = 0.9 Hz, 1H), 7.66–7.57 (m, 3H), 7.57–7.49 (m, 2H), 7.48–7.40 (m, 2H), 7.29 (ddt, J = 7.6, 2.0, 1.0 Hz, 1H), 7.15 (t, J = 1.1 Hz, 1H), 6.94 (d, J = 2.3 Hz, 1H), 6.84 (dd, J = 8.7, 1.6 Hz, 1H), 5.41 (dd, J = 12.0, 4.5 Hz, 1H), 4.41 (q, J = 7.2 Hz, 2H), 3.79 (dd, J = 18.5, 12.1 Hz, 1H), 2.88 (dd, J = 18.4, 4.6 Hz, 1H), 1.88 (s, 3H), 1.37 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.05, 160.18, 152.21, 149.88, 138.03, 137.33, 134.72, 134.44, 132.33, 131.16, 129.51, 128.57, 128.41, 128.29, 126.08, 124.71, 124.11, 123.28, 120.72, 117.63, 117.30, 109.78, 58.90, 45.57, 43.06, 21.50, 14.95. tR = 2.29 min (generic method). ESI-MS for C29H24ClN5O2: calculated 509.2, found m/z 510.2, 512.2 [M + H]+, 508.3, 510.4 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
3-(1-Acetyl-5-(2-ethyl-2H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (48d)
Compound 48d was synthesized via general procedure C2 using 101c (80 mg, 0.17 mmol) with acetic anhydride 116 (32 μL, 0.34 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–10% EtOH/DCM) to afford 48d (67 mg, yield 77%). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 8.31 (d, J = 0.9 Hz, 1H), 7.67–7.50 (m, 4H), 7.48–7.42 (m, 3H), 7.36–7.26 (m, 1H), 7.13–7.05 (m, 1H), 6.94 (d, J = 2.4 Hz, 1H), 6.65 (dd, J = 9.0, 1.7 Hz, 1H), 5.36 (dd, J = 12.0, 4.6 Hz, 1H), 4.44 (q, J = 7.3 Hz, 2H), 3.78 (dd, J = 18.4, 12.0 Hz, 1H), 2.87 (dd, J = 18.4, 4.6 Hz, 1H), 1.89 (s, 3H), 1.50 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.04, 160.18, 152.20, 149.83, 147.32, 137.35, 134.71, 134.46, 131.13, 129.52, 128.57, 128.54, 128.37, 128.27, 126.04, 124.73, 123.59, 123.01, 120.93, 120.72, 117.63, 117.34, 116.65, 59.13, 47.69, 45.35, 21.52, 15.84. tR = 2.14 min (generic method). ESI-MS for C29H24ClN5O2: calculated 509.2, found m/z 510.2, 512.2 [M + H]+, 508.2, 510.3 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
3-(1-Acetyl-5-(1-propyl-1H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (49d)
Compound 49d was synthesized via general procedure C2 using 102c (188 mg, 0.39 mmol) with acetic anhydride 116 (74 μL, 0.78 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–10% EtOH/DCM) to afford 49d (184 mg, yield 90%. 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 7.99 (d, J = 0.9 Hz, 1H), 7.65–7.57 (m, 3H), 7.56–7.50 (m, 2H), 7.47–7.41 (m, 2H), 7.36–7.25 (m, 1H), 7.17–7.09 (m, 1H), 6.94 (d, J = 2.3 Hz, 1H), 6.85 (dd, J = 8.8, 1.7 Hz, 1H), 5.41 (dd, J = 12.0, 4.6 Hz, 1H), 4.33 (t, J = 6.9 Hz, 2H), 3.78 (dd, J = 18.5, 12.0 Hz, 1H), 2.89 (dd, J = 18.5, 4.6 Hz, 1H), 1.90–1.77 (m, 5H), 0.82 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.06, 160.14, 152.20, 149.85, 147.37, 137.29, 134.69, 134.43, 131.13, 129.52, 128.53, 128.35, 128.26, 126.07, 124.73, 123.73, 123.58, 120.81, 120.71, 117.59, 117.37, 116.65, 59.13, 54.23, 45.34, 23.40, 21.51, 10.86. tR = 1.21 min (generic method). ESI-MS for C30H26ClN5O2: calculated 523.2, found m/z 524.1, 526.2 [M + H]+, 522.3, 524.2 [M – H]−. UPLC–MS purity (UV at 215 nm) 94%.
3-(1-Acetyl-5-(2-propyl-2H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (50d)
Compound 50d was synthesized via general procedure C2 using 103c (132 mg, 0.27 mmol) with acetic anhydride 116 (51 μL, 0.54 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–10% EtOH/DCM) to afford 50d (120 mg, yield 85%). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 8.29 (d, J = 1.0 Hz, 1H), 7.65–7.56 (m, 3H), 7.56–7.49 (m, 1H), 7.48–7.40 (m, 3H), 7.32–7.26 (m, 1H), 7.08 (t, J = 1.2 Hz, 1H), 6.93 (d, J = 2.3 Hz, 1H), 6.64 (dd, J = 8.9, 1.7 Hz, 1H), 5.35 (dd, J = 12.0, 4.5 Hz, 1H), 4.36 (t, J = 6.9 Hz, 2H), 3.77 (dd, J = 18.4, 12.0 Hz, 1H), 2.87 (dd, J = 18.4, 4.6 Hz, 1H), 1.97–1.83 (m, 5H), 0.83 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.07, 160.15, 152.21, 149.85, 147.38, 137.31, 134.69, 134.43, 131.13, 129.52, 128.57, 128.53, 128.36, 128.26, 126.07, 126.04, 124.74, 123.74, 123.58, 120.81, 120.72, 117.61, 117.38, 116.65, 59.13, 54.24, 45.35, 23.41, 21.52, 10.87. tR = 1.01 min (apolar method). ESI-MS for C30H26ClN5O2: calculated 523.2, found m/z 524.2, 526.2 [M + H]+, 522.3, 524.3 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
3-(1-Acetyl-5-(4-(1-propyl-1H-pyrazol-4-yl)phenyl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (51d)
Compound 51d was synthesized via general procedure C2 using 108c (146 mg, 0.31 mmol) with acetic anhydride 116 (59 μL, 0.62 mmol) (165 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 0–8% EtOH/DCM) to afford 51d (138 mg, yield 81%). 1H NMR (400 MHz, DMSO-d6) δ 12.36 (s, 1H), 8.13 (s, 1H), 7.83 (d, J = 0.7 Hz, 1H), 7.62 (dd, J = 8.8, 2.4 Hz, 1H), 7.60–7.52 (m, 3H), 7.45 (d, J = 8.8 Hz, 1H), 7.43–7.38 (m, 3H), 7.30 (dt, J = 6.2, 1.7 Hz, 1H), 6.94 (d, J = 2.3 Hz, 1H), 6.79 (d, J = 8.3 Hz, 2H), 5.28 (dd, J = 12.0, 4.5 Hz, 1H), 4.07 (t, J = 7.0 Hz, 2H), 3.75 (dd, J = 18.4, 12.0 Hz, 1H), 2.86 (dd, J = 18.4, 4.6 Hz, 1H), 1.91–1.75 (m, 5H), 0.85 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.01, 160.14, 152.19, 149.89, 139.79, 137.30, 135.89, 134.74, 131.59, 131.15, 129.41, 128.60, 128.49, 128.38, 128.26, 126.88, 126.08, 126.00, 124.96, 124.66, 121.22, 120.69, 117.59, 58.57, 52.99, 45.27, 23.17, 21.42, 10.92. tR = 1.22 min (apolar method). ESI-MS for C32H28ClN5O2: calculated 549.2, found m/z 550.0, 552.0 [M + H]+, 548.1, 550.0 [M – H]−. UPLC–MS purity (UV at 215 nm) 99%.
4-(5-(4-Fluorophenyl)-3-(2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (52d)
Compound 52d was synthesized via general procedure C1 using 109c (150 mg, 0.39 mmol) with succinic anhydride 115 (78 mg, 0.78 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 20–50% EtOH/DCM) to afford 52d (114 mg, yield 59%). 1H NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 12.01 (s, 1H), 7.63–7.34 (m, 6H), 7.25 (dt, J = 6.8, 1.9 Hz, 1H), 7.14 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.07–6.98 (m, 3H), 6.86–6.78 (m, 2H), 5.31 (dd, J = 12.0, 4.5 Hz, 1H), 3.72 (dd, J = 18.4, 12.0 Hz, 1H), 2.78 (dd, J = 18.4, 4.6 Hz, 1H), 2.47 (m, 2H), 2.32–2.25 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.48, 168.56, 160.25, 152.77, 151.05, 141.11, 138.53, 138.42, 135.16, 131.27, 129.38, 128.53, 128.22, 128.20, 128.13, 127.54, 127.45, 127.38, 123.30, 122.25, 119.35, 115.51, 115.16, 114.95, 58.16, 45.27, 28.58, 28.23. tR = 1.90 min (generic method). ESI-MS for C28H22FN3O4: calculated 483.2, found m/z 484.5 [M + H]+, 482.5 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(5-(4-Bromophenyl)-3-(2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydropyrazol-1-yl)-4-oxobutanoic Acid (53d)
Compound 53d was synthesized via general procedure C1 using 110c (41 mg, 0.09 mmol) with succinic anhydride 115 (18 mg, 0.18 mmol) (120 °C, 200 W). Title compound 53d was obtained after the acidic workup (46 mg, yield 91%). 1H NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 12.01 (s, 1H), 7.61–7.45 (m, 4H), 7.45–7.36 (m, 4H), 7.25 (dt, J = 6.8, 1.9 Hz, 1H), 7.14 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.04 (dd, J = 8.2, 1.4 Hz, 1H), 6.74 (d, J = 8.4 Hz, 2H), 5.29 (dd, J = 12.0, 4.6 Hz, 1H), 3.74 (dd, J = 18.4, 12.1 Hz, 1H), 2.77 (dd, J = 18.5, 4.6 Hz, 1H), 2.50 (m, 2H), 2.28 (t, J = 7.1 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.46, 168.60, 160.25, 152.78, 151.09, 141.60, 138.54, 135.17, 131.29, 131.23, 129.41, 128.51, 128.21, 128.14, 127.75, 127.39, 123.23, 122.27, 121.42, 120.03, 119.35, 115.53, 58.30, 45.12, 28.55, 28.22. tR = 2.03 min (generic method). ESI-MS for C28H22BrN3O4: calculated 543.1, found m/z 544.5, 546.5 [M + H]+; 542.5, 544.4. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(5-(3-Bromophenyl)-3-(2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (54d)
Compound 54d was synthesized via general procedure C1 using 111c (121 mg, 0.27 mmol) with succinic anhydride 115 (54 mg, 0.54 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 20–90% EtOH/DCM) to afford 54d (11 mg, yield 7%). 1H NMR (400 MHz, DMSO-d6) δ 12.24 (s, 1H), 12.13 (s, 1H), 7.61–7.34 (m, 7H), 7.26 (dt, J = 7.8, 1.8 Hz, 1H), 7.23–7.11 (m, 3H), 7.06 (dd, J = 8.2, 1.4 Hz, 1H), 6.76 (dt, J = 7.8, 1.3 Hz, 1H), 5.32 (dd, J = 12.1, 4.8 Hz, 1H), 3.74 (dd, J = 18.5, 12.1 Hz, 1H), 2.84 (dd, J = 18.5, 4.8 Hz, 1H), 2.51 (m, 2H), 2.33–2.26 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 174.08, 173.94, 169.19, 160.76, 153.36, 151.60, 145.30, 139.04, 135.72, 131.79, 131.18, 130.45, 129.75, 128.92, 128.71, 128.56, 127.90, 124.94, 123.65, 122.75, 122.09, 119.80, 116.01, 58.80, 45.64, 29.39, 29.01, 28.67. tR = 2.00 min (generic method). ESI-MS for C28H22BrN3O4: calculated 543.1, found m/z 544.4, 546.4 [M + H]+; 542.4, 544.4 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(5-(4-Chlorophenyl)-3-(2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (55d)
Compound 55d was synthesized via general procedure C1 using 112c (121 mg, 0.27 mmol) with succinic anhydride 115 (54 mg, 054 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 4–20% EtOH/DCM) to afford 55d (130 mg, yield 67%). 1H NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 12.05 (s, 1H), 7.61–7.46 (m, 4H), 7.43 (dd, J = 8.3, 1.1 Hz, 1H), 7.39 (dt, J = 6.0, 2.0 Hz, 1H), 7.30–7.23 (m, 3H), 7.15 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.05 (dd, J = 8.2, 1.4 Hz, 1H), 6.81 (d, J = 8.5 Hz, 2H), 5.31 (dd, J = 12.0, 4.6 Hz, 1H), 3.75 (dd, J = 18.5, 12.1 Hz, 1H), 2.78 (dd, J = 18.5, 4.6 Hz, 1H), 2.58–2.44 (m, 2H), 2.30 (t, J = 7.1 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.95, 169.08, 160.73, 153.27, 151.57, 141.65, 139.02, 135.65, 132.01, 131.77, 129.89, 129.00, 128.79, 128.69, 128.62, 127.88, 123.73, 122.74, 119.83, 116.01, 58.72, 45.66, 29.04, 28.70. tR = 1.97 min (generic method). ESI-MS for C28H22ClN3O4: calculated 499.1, found m/z 500.4, 502.4 [M + H]+; 498.4, 500.3 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-(5-(4-Methoxyphenyl)-3-(2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)-4-oxobutanoic Acid (56d)
Compound 56d was synthesized via general procedure C1 using 113c (120 mg, 0.30 mmol) with succinic anhydride 115 (60 mg, 0.60 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 5–35% EtOH/DCM) to afford 56d (99 mg, yield 67%). 1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 12.00 (s, 1H), 7.61–7.34 (m, 6H), 7.29–7.23 (m, 1H), 7.14 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.04 (dd, J = 8.2, 1.4 Hz, 1H), 6.79–6.66 (m, 4H), 5.23 (dd, J = 11.9, 4.5 Hz, 1H), 3.72 (s, 4H), 2.79 (dd, J = 18.4, 4.5 Hz, 1H), 2.46 (dd, J = 6.8, 2.4 Hz, 2H), 2.27 (t, J = 6.9 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.99, 168.91, 160.78, 158.68, 153.22, 151.47, 139.00, 135.70, 134.85, 131.71, 129.84, 129.08, 128.66, 128.60, 127.87, 127.27, 123.91, 122.71, 119.88, 115.99, 114.16, 58.85, 55.51, 45.80, 29.11, 28.74. tR = 1.83 min (generic method). ESI-MS for C29H25N3O5: calculated 495.1, found m/z 496.5 [M + H]+; 494.5 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
4-Oxo-4-(3-(2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-(trifluoromethyl)phenyl)-4,5-dihydro-1H-pyrazol-1-yl)butanoic Acid (57d)
Compound 57d was synthesized via general procedure C1 using 114c (189 mg, 0.45 mmol) with succinic anhydride 115 (90 mg, 0.90 mmol) (120 °C, 200 W). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g, 2–50% EtOH/DCM) to afford 57d (173 mg, yield 71%). 1H NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 12.02 (s, 1H), 7.64–7.36 (m, 8H), 7.25 (d, J = 7.3 Hz, 1H), 7.15 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H), 7.08–6.98 (m, 3H), 5.42 (dd, J = 12.1, 4.7 Hz, 1H), 3.79 (dd, J = 18.5, 12.1 Hz, 1H), 2.81 (dd, J = 18.5, 4.7 Hz, 1H), 2.59–2.48 (m, 2H), 2.36–2.26 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.94, 169.21, 160.71, 153.30, 151.65, 147.20, 139.03, 135.67, 131.80, 129.88, 128.96, 128.70, 128.65, 128.29, 127.88, 126.70, 126.02, 125.84, 125.80, 123.63, 123.31, 122.76, 119.81, 116.02, 58.95, 45.63, 28.99, 28.68. tR = 2.06 min (generic method). ESI-MS for C29H22F3N3O4: calculated 533.2, found m/z 534.5 [M + H]+; 532.5 [M – H]−. UPLC–MS purity (UV at 215 nm) >99.5%.
3-Acetyl-6-chloro-4-phenylquinolin-2(1H)-one (60a) (Scheme 1)
2-Amino-5-chlorophenyl)phenylmethanone 58 (1.460 g, 6.3 mmol) and ethyl acetoacetate (2.4 mL, 18.9 mmol) were dissolved in DMF (1.00 M) in an appropriately sized screw-capped pressure tube. The mixture was heated at 153 °C and stirred for 19 h. The solvent was removed under reduced pressure to afford 60a (1.875 g, quantitative yield). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 7.64 (dd, J = 8.8, 2.4 Hz, 1H), 7.59–7.48 (m, 3H), 7.44 (d, J = 8.8 Hz, 1H), 7.39–7.28 (m, 2H), 6.95 (d, J = 2.3 Hz, 1H), 2.22 (s, 3H). tR = 2.06 min (generic method). ESI-MS for C17H12ClNO2: calculated 297.1, found m/z 298.3, 300.3 [M + H]+; 296.3, 298.3 [M – H]−.
3-Acetyl-4-phenylquinolin-2(1H)-one (61a) (Scheme 1)
(2-Aminophenyl)phenylmethanone 59 (2.00 g, 10.1 mmol) and ethyl acetoacetate (1.9 mL, 15.2 mmol) were dissolved in DMF (1.00 M) in an appropriately sized microwaveable vessel and microwaved at 120 °C (200 W) for 1.5 h. The solvent was concentrated under reduced pressure, and the residue was diluted with DCM and washed (3 × 100 mL) with H2O. The organic layer was dried over Na2SO4, filtered and the solvent removed under reduced pressure. 61a was obtained after precipitation from EtOAc (1.44 g, 53% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 7.56 (ddd, J = 8.4, 7.1, 1.5 Hz, 1H), 7.54–7.47 (m, 3H), 7.41 (dd, J = 8.4, 1.2 Hz, 1H), 7.34–7.28 (m, 2H), 7.14 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H), 7.05 (dd, J = 8.2, 1.4 Hz, 1H), 2.21 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 201.60, 159.26, 146.96, 138.45, 134.19, 133.29, 131.20, 128.71, 128.64, 128.46, 127.03, 122.33, 118.89, 115.64, 31.47. tR = 1.86 min (generic method). ESI-MS for C17H13NO2: calculated 263.1, found m/z 264.5 [M + H]+; 262.4 [M – H]−.
(E)-6-Chloro-3-(3-(4-fluorophenyl)acryloyl)-4-phenylquinolin-2(1H)-one (86b)
3-Acetyl-6-chloro-4-phenylquinolin-2(1H)-one 60a (2.517 g, 8.45 mmol) and 4-fluorobenzaldehyde 62 (906 μL, 8.45 mmol) were allowed to react overnight according to general procedure A. Purification was performed by direct phase flash chromatography (SiO2 gold 40 g; 0–20% EtOH/DCM) to afford 86b (2.548 g, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 7.81–7.70 (m, 2H), 7.66 (dd, J = 8.8, 2.4 Hz, 1H), 7.54–7.41 (m, 5H), 7.36–7.30 (m, 2H), 7.28–7.19 (m, 2H), 6.98 (d, J = 2.4 Hz, 1H), 6.73 (d, J = 16.4 Hz, 1H). tR = 2.42 min (generic method). ESI-MS for C24H15ClFNO2: calculated 403.1, found m/z 404.1, 406.1 [M + H]+; 402.1, 404.1 [M – H]−.
6-Chloro-3-(5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (86c)
(E)-6-Chloro-3-(3-(4-fluorophenyl)acryloyl)-4-phenylquinolin-2(1H)-one 86b (317 mg, 0.78 mmol) and hydrazine monohydrate (76 μL, 1.56 mmol) were allowed to react according to general procedure B. Crude compound was purified by flash column chromatography (SiO2 gold 40 g; 0–20% EtOH/DCM) to afford 86c (303 mg, yield 92%). 1H NMR (400 MHz, DMSO-d6) δ 12.19 (s, 1H), 7.57 (dd, J = 8.8, 2.4 Hz, 1H), 7.50 (m, 2H), 7.41 (d, J = 8.8 Hz, 1H), 7.33 (dt, J = 6.9, 1.5 Hz, 1H), 7.23 (dt, J = 6.8, 2.0 Hz, 1H), 7.14 (d, J = 3.2 Hz, 1H), 7.13–7.02 (m, 4H), 6.89 (d, J = 2.3 Hz, 1H), 4.59 (td, J = 10.9, 10.2, 3.1 Hz, 1H), 3.23 (dd, J = 16.5, 11.0 Hz, 1H), 2.59–2.52 (m, 1H). tR = 2.31 min (generic method). ESI-MS for C24H17ClFN3O: calculated 417.1, found m/z 418.4, 420.4 [M + H]+; 416.4, 418.3 [M – H]−.
(E)-6-Chloro-3-(3-(4-chlorophenyl)acryloyl)-4-phenylquinolin-2(1H)-one (87b)
Compound 87b was synthesized via general procedure A using 60a (304 mg, 1.00 mmol) and 4-chlorobenzaldehyde 63 (140 mg, 1.00 mmol). Title compound 87b was obtained by precipitation and filtration from the reaction crude (429 mg, quantitative yield). 1H NMR (400 MHz, DMSO-d6) δ 7.70 (d, J = 8.6 Hz, 2H), 7.63 (dd, J = 8.8, 2.4 Hz, 1H), 7.55–7.37 (m, 7H), 7.32 (dd, J = 7.6, 1.9 Hz, 2H), 6.96 (d, J = 2.4 Hz, 1H), 6.78 (d, J = 16.4 Hz, 1H). tR = 2.55 min (generic method). ESI-MS for C24H15Cl2NO2: calculated 419.0, found m/z 420.4, 422.4, 424.4 [M + H]+; 418.5, 420.4, 422.3 [M – H]−.
6-Chloro-3-(5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (87c)
Compound 87c was synthesized via general procedure B using 87b (429 mg, 1.00 mmol) with hydrazine hydrate (97 μL, 2.00 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–10% EtOH/DCM) to afford 87c (301 mg, yield 68%). 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 7.58 (dd, J = 8.8, 2.4 Hz, 1H), 7.50 (tt, J = 7.5, 2.9 Hz, 3H), 7.42 (d, J = 8.8 Hz, 1H), 7.34 (dt, J = 7.0, 1.7 Hz, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.23 (dt, J = 7.3, 2.0 Hz, 1H), 7.18 (d, J = 3.2 Hz, 1H), 7.09 (d, J = 8.4 Hz, 2H), 6.90 (d, J = 2.3 Hz, 1H), 4.59 (td, J = 11.6, 9.3, 3.1 Hz, 1H), 3.25 (dd, J = 16.5, 11.1 Hz, 1H), 2.58–2.53 (m, 1H). tR = 2.45 min (generic method). ESI-MS for C24H17Cl2N3O: calculated 433.1, found m/z 434.4, 436.4, 438.5 [M + H]+; 432.4, 434.4, 436.4 [M – H]−.
(E)-3-(3-(4-Bromophenyl)acryloyl)-6-chloro-4-phenylquinolin-2(1H)-one (88b)
Compound 88b was synthesized via general procedure A using 60a (328 mg, 1.10 mmol) and 4-bromobenzaldehyde 64 (204 mg, 1.10 mmol). Title compound 88b was obtained by precipitation and filtration from the reaction crude (511 mg, quantitative yield). 1H NMR (400 MHz, DMSO-d6) δ 7.67–7.47 (m, 6H), 7.46–7.35 (m, 4H), 7.30 (dd, J = 7.7, 1.8 Hz, 2H), 6.93 (d, J = 2.3 Hz, 1H), 6.80 (d, J = 16.4 Hz, 1H). tR = 2.59 min (generic method). ESI-MS for C24H15BrClNO2: calculated 463.0, found m/z 464.3, 466.2, 468.2 [M + H]+; 462.2, 464.2, 466.2 [M – H]−.
3-(5-(4-Bromophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (88c)
Compound 88c was synthesized via general procedure B using 88b (510 mg, 1.10 mmol) with hydrazine hydrate (110 μL, 2.20 mmol). Purification was performed by direct phase flash chromatography (0–8% EtOH/DCM) to afford 88c (349 mg, yield 64%). 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 7.58 (dd, J = 8.8, 2.4 Hz, 1H), 7.56–7.45 (m, 3H), 7.42 (dd, J = 8.6, 5.5 Hz, 3H), 7.34 (dt, J = 7.2, 1.6 Hz, 1H), 7.23 (dt, J = 7.2, 2.0 Hz, 1H), 7.18 (d, J = 3.2 Hz, 1H), 7.07–6.99 (m, 2H), 6.89 (d, J = 2.3 Hz, 1H), 4.58 (td, J = 11.6, 9.3, 3.2 Hz, 1H), 3.25 (dd, J = 16.5, 11.1 Hz, 1H), 2.57–2.53 (m, 1H). tR = 2.50 min (generic method). ESI-MS for C24H17BrClN3O: calculated 477.0, found m/z 478.2, 480.2, 482.2 [M + H]+; 476.3, 478.2, 480.3 [M – H]−.
(E)-6-Chloro-3-(3-(4-methoxyphenyl)acryloyl)-4-phenylquinolin-2(1H)-one (89b)
3-Acetyl-6-chloro-4-phenylquinolin-2(1H)-one 60a (611 mg, 2.00 mmol) and p-anisaldehyde 65 (243 μL, 2.00 mmol) were allowed to react overnight according to general procedure A. Purification was performed by direct phase flash chromatography (SiO2 gold 40 g; 0–5% EtOH/DCM), affording the desired 89b (867 mg, quantitative yield). 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 7.68–7.58 (m, 3H), 7.49–7.38 (m, 5H), 7.34–7.29 (m, 2H), 6.99–6.89 (m, 3H), 6.60 (d, J = 16.3 Hz, 1H), 3.78 (s, 3H). tR = 1.19 min (apolar method). ESI-MS for C25H18ClNO3: calculated 415.1, found m/z 416.4, 418.4 [M + H]+; 414.4, 416.4 [M – H]−.
6-Chloro-3-(5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (89c)
(E)-6-Chloro-3-(3-(4-methoxyphenyl)acryloyl)-4-phenylquinolin-2(1H)-one 89b (780 mg, 1.9 mmol) and hydrazine monohydrate (185 μL, 3.8 mmol) were allowed to react according to general procedure B. Crude compound was purified by flash column chromatography (SiO2 gold 40 g; 0–10% EtOH/DCM) to afford 89c (611 mg, yield 75%). 1H NMR (400 MHz, DMSO-d6) δ 12.19 (s, 1H), 7.57 (dd, J = 8.8, 2.3 Hz, 1H), 7.53–7.47 (m, 3H), 7.42 (d, J = 8.8 Hz, 1H), 7.34 (dd, J = 10.3, 3.8 Hz, 1H), 7.27–7.19 (m, 1H), 7.02 (dd, J = 8.9, 5.9 Hz, 3H), 6.90 (d, J = 2.3 Hz, 1H), 6.80 (d, J = 8.7 Hz, 2H), 4.52 (td, J = 10.4, 2.9 Hz, 1H), 3.73 (s, 3H), 3.18 (dd, J = 16.4, 10.9 Hz, 1H), 2.58–2.53 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 166.84, 160.17, 152.10, 149.77, 149.57, 137.28, 134.76, 131.10, 129.96, 129.28, 128.71, 128.42, 128.30, 128.28, 126.41, 126.04, 124.80, 120.72, 117.58, 112.23, 58.37, 45.22, 40.19, 21.49. tR = 2.24 min (generic method). ESI-MS for C25H20ClN3O2: calculated 429.1, found m/z 430.4, 432.4 [M + H]+; 428.4, 430.5 [M – H]−.
(E)-3-(3-(4-(tert-Butyl)phenyl)acryloyl)-6-chloro-4-phenylquinolin-2(1H)-one (90b)
Compound 90b was synthesized via general procedure A using 60a (543 mg, 1.80 mmol) and 4-tert-butylbenzaldeyde 66 (301 μL, 1.80 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 40 g; 0–10% EtOH/DCM) to afford 90b (441 mg, yield 55%). 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 7.64 (dd, J = 8.8, 2.4 Hz, 1H), 7.58 (d, J = 8.5 Hz, 2H), 7.50–7.37 (m, 7H), 7.32 (dd, J = 7.7, 1.9 Hz, 2H), 6.97 (d, J = 2.4 Hz, 1H), 6.69 (d, J = 16.4 Hz, 1H), 1.26 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 193.82, 159.40, 153.81, 146.61, 146.19, 137.50, 133.60, 132.56, 131.52, 130.89, 128.86, 128.80, 128.50, 128.43, 126.64, 126.05, 125.71, 125.56, 120.56, 117.68, 79.15, 34.62, 30.97, 30.82. Rt 2.18 min (generic method). ESI-MS for C28H24ClNO2: calculated 441.1, found m/z 442.4, 444.4 [M + H]+; 440.4, 442.4 [M – H]-.
3-[5-(4-tert-Butylphenyl)-4,5-dihydro-1H-pyrazol-3-yl]-6-chloro-4-phenylquinolin-2(1H)-one (90c)
Compound 90c was synthesized via general procedure B using 90b (369 mg, 0.83 mmol) with hydrazine hydrate (81 μL, 1.66 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–90% EtOAc/CHX) to afford 90c (351 mg, yield 93%). 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 7.56 (dd, J = 8.8, 2.4 Hz, 1H), 7.49 (dtd, J = 9.6, 7.2, 5.3 Hz, 3H), 7.42 (d, J = 8.8 Hz, 1H), 7.33 (dt, J = 7.2, 1.7 Hz, 1H), 7.25 (d, J = 8.3 Hz, 3H), 7.06 (d, J = 3.0 Hz, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.90 (d, J = 2.3 Hz, 1H), 4.54 (t, J = 10.4 Hz, 1H), 3.19 (dd, J = 16.4, 10.9 Hz, 1H), 2.58 (dd, J = 16.4, 9.6 Hz, 1H), 1.26 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 160.54, 149.21, 148.30, 145.49, 140.31, 136.97, 135.21, 130.32, 129.19, 128.72, 128.26, 128.21, 128.14, 127.79, 127.18, 126.88, 126.33, 126.18, 125.75, 125.62, 124.92, 120.92, 117.33, 79.16, 62.69, 44.34, 34.09, 31.13, 31.05, 30.74. tR = 1.82 min (generic method). ESI-MS for C28H26ClN3O: calculated 455.2, found m/z 456.5, 458.4, [M + H]+; 454.5 456.5 [M – H]−.
(E)-6-Chloro-3-(3-(3-fluoro-4-methoxyphenyl)acryloyl)-4-phenylquinolin-2(1H)-one (91b)
Compound 91b was synthesized via general procedure A using 60a (473 mg, 1.60 mmol) and 3-fluoro-4-methoxybenzaldehyde 67 (247 mg, 1.60 mmol). Title compound 91b was obtained after precipitation and filtration from the reaction crude (547 mg, yield 79%). 1H NMR (600 MHz, DMSO-d6) δ 12.38 (s, 1H), 7.70–7.62 (m, 2H), 7.52–7.40 (m, 6H), 7.35–7.30 (m, 2H), 7.18 (t, J = 8.7 Hz, 1H), 6.97 (d, J = 2.4 Hz, 1H), 6.69 (d, J = 16.3 Hz, 1H), 3.88 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 194.19, 159.89, 153.13, 150.69, 149.82, 149.71, 147.06, 145.83, 138.00, 134.10, 133.01, 131.35, 129.34, 129.27, 128.90, 127.92, 127.85, 127.10, 126.87, 126.52, 126.02, 121.07, 118.17, 115.70, 115.52, 114.26, 56.62. tR = 2.35 min (generic method). ESI-MS for C25H17ClFNO3: calculated 433.1, found m/z 434.4, 436.4 [M + H]+; 432.3, 434.3 [M – H]−.
6-Chloro-3-(5-(3-fluoro-4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (91c)
Compound 91c was synthesized via general procedure B using 91b (203 mg, 0.50 mmol) with hydrazine hydrate (49 μL, 1.00 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–5% EtOH/DCM) to afford 91c (213 mg, quantitative yield). 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 7.63–7.39 (m, 5H), 7.35–7.29 (m, 1H), 7.23 (dt, J = 7.0, 2.0 Hz, 1H), 7.12 (d, J = 3.1 Hz, 1H), 7.03 (t, J = 8.6 Hz, 1H), 6.97–6.85 (m, 3H), 4.55 (ddd, J = 11.5, 9.0, 2.9 Hz, 1H), 3.81 (s, 3H), 3.19 (dd, J = 16.5, 11.0 Hz, 1H), 2.60–2.53 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 161.01, 152.92, 150.50, 148.84, 146.52, 145.91, 137.45, 137.04, 135.64, 130.85, 129.61, 129.14, 128.70, 127.20, 126.24, 126.10, 123.00, 121.36, 117.82, 114.42, 114.23, 114.02, 62.31, 56.48, 44.83. tR = 2.27 min (generic method). ESI-MS for C25H19ClFN3O2: calculated 447.1, found m/z 448.4, 450.3 [M + H]+; 446.3, 448.2 [M – H]−.
(E)-6-Chloro-4-phenyl-3-(3-(4-(trifluoromethyl)phenyl)acryloyl)quinolin-2(1H)-one (92b)
Compound 92b was synthesized via general procedure A using 60a (760 mg, 2.50 mmol) and 4-(trifluoromethyl)benzaldehyde 68 (341 μL, 2.50 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 40 g; 0–5% EtOH/DCM) to afford 92b (820 mg, yield 71%). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 7.89 (d, J = 8.2 Hz, 2H), 7.74 (d, J = 8.4 Hz, 2H), 7.67 (dd, J = 8.8, 2.4 Hz, 1H), 7.62–7.57 (m, 1H), 7.53–7.40 (m, 4H), 7.36–7.31 (m, 2H), 6.99 (d, J = 2.3 Hz, 1H), 6.91 (d, J = 16.5 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 193.90, 159.39, 147.09, 144.03, 138.29, 137.58, 133.52, 132.23, 131.05, 130.34, 130.07, 130.02, 129.59, 129.19, 128.88, 128.48, 126.12, 125.68, 125.65, 125.62, 125.30, 122.59, 120.55, 117.74. tR = 1.65 min (apolar method). ESI-MS for C25H15ClF3NO2: calculated 453.1, found m/z 454.4, 456.4 [M + H]+; 452.4, 454.4 [M – H]−.
6-Chloro-4-phenyl-3-(5-(4-(trifluoromethyl)phenyl)-4,5-dihydro-1H-pyrazol-3-yl)quinolin-2(1H)-one (92c)
Compound 92c was synthesized via general procedure B using 92b (752 mg, 1.70 mmol) with hydrazine hydrate (165 μL, 3.40 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 40 g; 0–5% EtOH/DCM) to afford 92c (696 mg, yield 90%). 1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 7.63–7.56 (m, 3H), 7.56–7.44 (m, 3H), 7.42 (d, J = 8.8 Hz, 1H), 7.37–7.32 (m, 1H), 7.29 (d, J = 8.5 Hz, 3H), 7.25–7.20 (m, 1H), 6.90 (d, J = 2.3 Hz, 1H), 4.75–4.64 (m, 1H), 3.39–3.25 (m, 1H), 2.63–2.54 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 160.49, 148.49, 148.37, 145.45, 136.99, 135.13, 130.43, 129.25, 128.63, 128.29, 128.23, 128.20, 127.68, 127.22, 126.58, 125.79, 125.64, 125.11, 125.08, 122.95, 120.88, 117.37, 62.16, 44.50. tR = 2.52 min (generic method). ESI-MS for C25H17ClF3N3O: calculated 467.1, found m/z 468.4, 469.5 [M + H]+, 466.4, 468.4 [M – H]−.
(E)-6-Chloro-3-(3-(furan-2-yl)acryloyl)-4-phenylquinolin-2(1H)-one (93b)
Compound 93b was synthesized via general procedure A using 60a (503 mg, 1.70 mmol) and furan-2-carbaldehyde 69 (147 μL, 1.70 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–5% EtOH/DCM) to afford 93b (471 mg, yield 74%). 1H NMR (400 MHz, DMSO-d6) δ 12.34 (s, 1H), 7.85 (d, J = 1.7 Hz, 1H), 7.64 (dd, J = 8.8, 2.4 Hz, 1H), 7.51–7.40 (m, 4H), 7.35 (s, 1H), 7.33–7.26 (m, 2H), 6.96 (dd, J = 6.9, 2.9 Hz, 2H), 6.63 (dd, J = 3.5, 1.8 Hz, 1H), 6.37 (d, J = 16.1 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 193.04, 159.41, 150.27, 146.93, 146.60, 137.52, 133.55, 132.45, 132.30, 130.98, 128.84, 128.46, 126.08, 125.58, 123.96, 120.51, 117.69, 117.67, 113.14, 40.19, 39.98. tR = 2.24 min (generic method). ESI-MS for C22H14ClNO3: calculated 375.1, found m/z 376.3, 378.3 [M + H]+; 374.3, 376.4 [M – H]−.
6-Chloro-3-(5-(furan-2-yl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (93c)
Compound 93c was synthesized via general procedure B using 93b (442 mg, 1.20 mmol) with hydrazine hydrate (117 μL, 2.40 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 40 g; 0–7.5% EtOH/DCM) to afford 93c (422 mg, yield 91%). 1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 7.57 (dd, J = 8.8, 2.3 Hz, 1H), 7.54–7.51 (m, 1H), 7.50–7.38 (m, 4H), 7.31–7.23 (m, 2H), 7.07 (d, J = 2.8 Hz, 1H), 6.90 (d, J = 2.3 Hz, 1H), 6.34 (dd, J = 3.1, 1.9 Hz, 1H), 6.17 (d, J = 3.2 Hz, 1H), 4.61–4.53 (m, 1H), 3.04 (dd, J = 16.2, 10.6 Hz, 1H), 2.87 (dd, J = 16.2, 9.2 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 160.55, 155.04, 148.48, 146.34, 142.11, 137.05, 135.23, 130.50, 128.95, 128.90, 128.34, 128.26, 128.18, 126.54, 125.83, 125.74, 120.91, 117.42, 110.33, 105.94, 40.63, 40.06. tR = 2.14 min (generic method). ESI-MS for C22H16ClN3O2: calculated 389.1, found m/z 390.4, 392.3 [M + H]+; 388.4, 390.4 [M – H]−.
(E)-6-Chloro-3-(3-(4′-fluoro-[1,1′-biphenyl]-4-yl)acryloyl)-4-phenylquinolin-2(1H)-one (94b)
Compound 94b was synthesized via general procedure A using 60a (214 mg, 0.72 mmol) and 4′-fluoro-[1,1′-biphenyl]-4-carbaldehyde 70 (144 mg, 0.72 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–7% EtOH/CHCl3) to afford 94b (305 mg, yield 89%). 1H NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 7.82–7.62 (m, 7H), 7.57–7.40 (m, 5H), 7.36–7.26 (m, 4H), 6.98 (d, J = 2.3 Hz, 1H), 6.80 (d, J = 16.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 193.83, 163.40, 160.96, 159.45, 146.77, 145.64, 141.15, 137.55, 135.57, 135.54, 133.63, 133.34, 132.54, 131.52, 131.42, 130.93, 129.33, 128.90, 128.84, 128.80, 128.72, 128.46, 127.29, 126.98, 126.10, 125.60, 120.59, 117.71, 115.91, 115.70. tR = 1.90 min (apolar method). ESI-MS for C30H19ClFNO2: calculated 479.1, found m/z 480.2, 482.2 [M + H]+, 478.2, 480.5 [M – H]−.
6-Chloro-3-(5-(4′-fluoro-[1,1′-biphenyl]-4-yl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (94c)
Compound 94c was synthesized via general procedure B using 94b (273 mg, 0.57 mmol) with hydrazine hydrate (55 μL, 1.14 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–10% EtOH/DCM) to afford 94c (188 mg, yield 67%). 1H NMR (400 MHz, DMSO-d6) δ 12.18 (s, 1H), 7.71–7.64 (m, 2H), 7.60–7.46 (m, 6H), 7.42 (d, J = 8.8 Hz, 1H), 7.37–7.32 (m, 1H), 7.31–7.21 (m, 3H), 7.19–7.13 (m, 3H), 6.90 (d, J = 2.3 Hz, 1H), 4.67–4.57 (m, 1H), 3.26 (dd, J = 16.6, 11.1 Hz, 1H), 2.60 (dd, J = 16.4, 9.5 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 162.98, 160.55, 160.52, 148.35, 145.48, 142.66, 137.78, 136.96, 136.44, 136.41, 135.19, 130.33, 129.22, 128.69, 128.51, 128.43, 128.25, 128.17, 127.06, 126.78, 126.48, 125.75, 125.62, 120.91, 117.32, 115.73, 115.52, 62.49, 44.43. tR = 1.72 min (apolar method). ESI-MS for C30H21ClFN3O: calculated 493.1, found m/z 494.2, 496.1 [M + H]+, 492.2, 494.2 [M – H]−.
(E)-6-Chloro-3-(3-(4′-chloro-[1,1′-biphenyl]-4-yl)acryloyl)-4-phenylquinolin-2(1H)-one (95b)
Compound 95b was synthesized via general procedure A using 60a (339 mg, 1.10 mmol) and 4′-chloro-[1,1′-biphenyl]-4-carbaldehyde 71 (238 mg, 1.10 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–5% EtOH/DCM) to afford 95b (567 mg, quantitative yield). 1H NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 7.78–7.68 (m, 6H), 7.65 (dd, J = 8.8, 2.4 Hz, 1H), 7.57–7.48 (m, 3H), 7.48–7.40 (m, 4H), 7.34 (dd, J = 7.8, 1.8 Hz, 2H), 6.99 (d, J = 2.3 Hz, 1H), 6.81 (d, J = 16.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 193.80, 159.41, 146.75, 145.51, 140.78, 137.87, 137.53, 133.70, 133.61, 132.90, 132.51, 130.92, 129.34, 128.93, 128.88, 128.82, 128.45, 127.42, 126.98, 126.07, 125.58, 120.57, 117.69, 79.15. tR = 2.16 min (apolar method). ESI-MS for C30H19Cl2NO2: calculated 495.1, found m/z 496.0, 497.9, 499.9, [M + H]+; 494.1, 496.0, 498.0 [M – H]−.
6-Chloro-3-(5-(4′-chloro-[1,1′-biphenyl]-4-yl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (95c)
Compound 95c was synthesized via general procedure B using 95b (558 mg, 1.10 mmol) with hydrazine hydrate (107 μL, 2.20 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 40 g; 0–15% EtOH/DCM) to afford 95c (501 mg, yield 87%). 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 7.69–7.63 (m, 2H), 7.59–7.46 (m, 8H), 7.41 (d, J = 8.7 Hz, 1H), 7.37–7.31 (m, 1H), 7.26–7.20 (m, 1H), 7.17 (d, J = 8.3 Hz, 2H), 6.90 (d, J = 2.3 Hz, 1H), 4.63 (ddd, J = 11.3, 9.5, 3.0 Hz, 1H), 3.27 (dd, J = 16.4, 11.1 Hz, 1H), 2.60 (dd, J = 16.4, 9.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 160.53, 148.37, 145.49, 143.11, 138.74, 137.45, 136.97, 135.20, 132.16, 130.35, 129.23, 128.82, 128.69, 128.28, 128.18, 127.14, 126.77, 126.49, 125.77, 125.63, 120.91, 117.34, 62.49, 44.45. tR = 1.99 min (apolar method). ESI-MS for C30H21Cl2N3O: calculated 509.1, found m/z 510.0, 511.9, 514.0 [M + H]+; 508.0, 510.0, 511.9 [M – H]−.
(E)-3-(3-(4′-Bromo-[1,1′-biphenyl]-4-yl)acryloyl)-6-chloro-4-phenylquinolin-2(1H)-one (96b)
Compound 96b was synthesized via general procedure A using 60a (337 mg, 1.10 mmol) and 4′-bromo-[1,1′-biphenyl]-4-carbaldehyde 72 (287 mg, 1.10 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–5% EtOH/DCM) to afford 96b (576 mg, yield 89%). 1H NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 7.80–7.68 (m, 4H), 7.68–7.62 (m, 5H), 7.57–7.39 (m, 5H), 7.34 (dd, J = 7.7, 1.8 Hz, 2H), 6.99 (d, J = 2.3 Hz, 1H), 6.81 (d, J = 16.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 193.81, 159.41, 146.76, 145.52, 140.83, 138.24, 137.53, 133.74, 133.60, 132.51, 131.85, 130.92, 129.36, 128.88, 128.82, 128.77, 128.44, 127.43, 126.94, 126.07, 125.58, 121.53, 120.57, 117.69, 79.15. tR = 2.20 min (apolar method). ESI-MS for C30H19BrClNO2: calculated 539.0, found m/z 539.9, 541.9, 543.9 [M + H]+; 538.0, 540.0, 542.0 [M – H]−.
3-(5-(4′-Bromo-[1,1′-biphenyl]-4-yl)-4,5-dihydro-1H-pyrazol-3-yl)-6-chloro-4-phenylquinolin-2(1H)-one (96c)
Compound 96c was synthesized via general procedure B using 96b (570 mg, 1.00 mmol) with hydrazine hydrate (97 μL, 2.00 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–10% EtOH/DCM) to afford 96c (472 mg, yield 81%). 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 7.68–7.46 (m, 10H), 7.41 (d, J = 8.8 Hz, 1H), 7.37–7.33 (m, 1H), 7.26–7.21 (m, 1H), 7.19–7.15 (m, 3H), 6.90 (d, J = 2.4 Hz, 1H), 4.63 (ddd, J = 11.3, 9.5, 3.1 Hz, 1H), 3.27 (dd, J = 16.4, 11.0 Hz, 1H), 2.60 (dd, J = 16.4, 9.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 160.53, 148.37, 145.48, 143.17, 139.11, 137.48, 136.97, 135.20, 131.74, 130.36, 129.23, 128.69, 128.63, 128.27, 128.18, 127.16, 126.77, 126.45, 125.76, 125.63, 120.91, 120.72, 117.34, 79.15, 62.49, 44.45, 40.15, 39.99, 39.94, 39.73, 39.52, 39.31, 39.10, 38.89. tR = 2.05 min (apolar method). ESI-MS for C30H21BrClN3O: calculated 553.0, found m/z 553.9, 555.9, 557.9 [M + H]+, 551.9, 554.0, 556.0 [M – H]−.
(E)-6-Chloro-3-(3-(4′-methoxy-[1,1′-biphenyl]-4-yl)acryloyl)-4-phenylquinolin-2(1H)-one (97b)
Compound 97b was synthesized via general procedure A using 60a (430 mg, 1.40 mmol) and 4′-methoxy-[1,1′-biphenyl]-4-carbaldehyde 73 (297 mg, 1.40 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–10% EtOH/DCM) to afford 97b (528 mg, yield 74%). 1H NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 7.75–7.62 (m, 6H), 7.55–7.40 (m, 6H), 7.33 (ddt, J = 6.7, 3.2, 1.8 Hz, 2H), 7.05–7.00 (m, 2H), 6.98 (d, J = 2.4 Hz, 1H), 6.77 (d, J = 16.4 Hz, 1H), 3.79 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.75, 159.42, 159.38, 159.07, 146.68, 145.85, 141.92, 137.52, 133.63, 132.58, 131.32, 131.13, 130.90, 129.30, 128.88, 128.81, 128.69, 128.65, 128.43, 127.84, 126.86, 126.37, 126.22, 126.06, 125.69, 125.57, 120.58, 117.69, 114.43, 55.19, 31.34. tR = 1.81 min (apolar method). ESI-MS for C31H22ClNO3: calculated 491.1, found m/z 492.0, 494.0 [M + H]+; 490.0, 492.1 [M – H]−.
6-Chloro-3-(5-(4′-methoxy-[1,1′-biphenyl]-4-yl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (97c)
Compound 97c was synthesized via general procedure B using 97b (522 mg, 1.10 mmol) with hydrazine hydrate (107 μL, 2.20 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 40 g; 0–10% EtOH/DCM) to afford 97c (412 mg, yield 77%). 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 7.57 (dd, J = 8.9, 2.4 Hz, 3H), 7.54–7.44 (m, 5H), 7.41 (d, J = 8.8 Hz, 1H), 7.38–7.31 (m, 1H), 7.24 (td, J = 4.2, 3.7, 1.8 Hz, 1H), 7.16–7.11 (m, 3H), 7.01 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 2.3 Hz, 1H), 4.60 (ddd, J = 11.0, 9.5, 3.1 Hz, 1H), 3.79 (s, 3H), 3.25 (dd, J = 16.4, 11.0 Hz, 1H), 2.60 (dd, J = 16.4, 9.5 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 160.54, 158.79, 148.34, 145.49, 141.88, 138.51, 136.97, 135.21, 132.34, 130.35, 129.24, 128.71, 128.27, 128.18, 127.60, 126.99, 126.82, 126.01, 125.75, 125.63, 120.93, 117.33, 114.32, 62.58, 55.14, 44.44. tR = 2.60 min (generic method). ESI-MS for C31H24ClN3O2: calculated 505.2, found m/z 506.0, 508.0 [M + H]+; 504.1, 506.1 [M – H]−.
(E)-6-Chloro-3-(3-(1-methyl-1H-indol-5-yl)acryloyl)-4-phenylquinolin-2(1H)-one (98b)
Compound 98b was synthesized via general procedure A using 60a (545 mg, 1.80 mmol) and 1-methyl-1H-indole-5-carbaldehyde 74 (287 mg, 1.80 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–10% EtOH/DCM) to afford 98b (427 mg, yield 53%). 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 7.86 (d, J = 1.5 Hz, 1H), 7.65 (dd, J = 8.8, 2.3 Hz, 1H), 7.56 (d, J = 16.3 Hz, 1H), 7.51–7.30 (m, 9H), 6.97 (d, J = 2.3 Hz, 1H), 6.65 (d, J = 16.3 Hz, 1H), 6.44 (dd, J = 3.2, 0.7 Hz, 1H), 3.79 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.46, 159.48, 148.78, 146.31, 137.77, 137.46, 133.77, 132.92, 131.03, 130.76, 128.89, 128.72, 128.37, 128.15, 125.99, 125.53, 125.36, 124.31, 123.22, 121.09, 120.65, 117.63, 110.33, 101.50, 32.61. tR = 2.45 min (generic method). ESI-MS for C27H19ClN2O2: calculated 438.1, found m/z 439.2, 441.1 [M + H]+; 437.3, 439.3 [M – H]−.
6-Chloro-3-(5-(1-methyl-1H-indol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (98c)
Compound 98c was synthesized via general procedure B using 98b (370 mg, 0.84 mmol) with hydrazine hydrate (82 μL, 1.68 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–5% EtOH/DCM) to afford 98c (325 mg, yield 85%). 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 7.57–7.46 (m, 4H), 7.41 (d, J = 8.8 Hz, 1H), 7.35 (dt, J = 7.2, 1.7 Hz, 1H), 7.31–7.21 (m, 4H), 7.02 (s, 1H), 6.91 (td, J = 3.9, 1.7 Hz, 2H), 6.34 (dd, J = 3.0, 0.7 Hz, 1H), 4.66 (t, J = 10.5 Hz, 1H), 3.75 (s, 3H), 3.22 (dd, J = 16.4, 11.0 Hz, 1H), 2.63 (dd, J = 16.4, 9.9 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 160.60, 148.21, 145.51, 136.97, 135.70, 135.30, 133.87, 130.28, 129.72, 129.24, 128.74, 128.27, 128.17, 127.79, 127.03, 125.76, 125.62, 120.96, 120.01, 118.09, 117.32, 109.44, 100.21, 63.57, 44.92, 32.45. tR = 2.38 min (generic method). ESI-MS for C27H21ClN4O: calculated 452.1, found m/z 453.2, 455.2 [M + H]+; 451.3, 453.3 [M – H]−.
(E)-6-Chloro-3-(3-(1-ethyl-1H-indol-5-yl)acryloyl)-4-phenylquinolin-2(1H)-one (99b)
Compound 99b was synthesized via general procedure A using 60a (402 mg, 1.30 mmol) and 1-ethyl-1H-indole-5-carbaldehyde 75 (225 mg, 1.30 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 40 g; 0–5% EtOH/DCM) to afford 99b (395 mg, yield 65%). 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 7.85 (d, J = 1.4 Hz, 1H), 7.65 (dd, J = 8.8, 2.4 Hz, 1H), 7.56 (d, J = 16.3 Hz, 1H), 7.50–7.45 (m, 3H), 7.45–7.38 (m, 4H), 7.33 (dd, J = 7.7, 1.8 Hz, 2H), 6.97 (d, J = 2.3 Hz, 1H), 6.65 (d, J = 16.2 Hz, 1H), 6.45 (d, J = 3.1 Hz, 1H), 4.20 (q, J = 7.2 Hz, 2H), 1.34 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.47, 159.48, 148.79, 146.30, 137.46, 136.80, 133.77, 132.92, 130.76, 129.42, 128.98, 128.89, 128.72, 128.66, 128.37, 128.30, 125.99, 125.53, 125.35, 124.27, 123.35, 121.02, 120.65, 117.63, 110.32, 101.75, 79.15, 15.44. tR = 2.56 min (generic method). ESI-MS for C28H21ClN2O2: calculated 452.1, found m/z 453.2, 455.2 [M + H]+; 451.3, 453.3 [M – H]−.
6-Chloro-3-(5-(1-ethyl-1H-indol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (99c)
Compound 99c was synthesized via general procedure B using 99b (336 mg, 0.79 mmol) with hydrazine hydrate (77 μL, 1.58 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–10% EtOH/DCM) to afford 99c (322 mg, yield 87%). 1H NMR (400 MHz, DMSO-d6) δ 12.18 (s, 1H), 7.60–7.48 (m, 4H), 7.41 (d, J = 8.8 Hz, 1H), 7.38–7.30 (m, 3H), 7.25 (dt, J = 6.6, 2.1 Hz, 2H), 7.00 (d, J = 3.2 Hz, 1H), 6.89 (dd, J = 9.7, 2.0 Hz, 2H), 6.34 (dd, J = 3.2, 0.8 Hz, 1H), 4.64 (td, J = 10.4, 3.2 Hz, 1H), 4.17 (q, J = 7.2 Hz, 2H), 3.21 (dd, J = 16.4, 11.0 Hz, 1H), 2.63 (dd, J = 16.4, 9.9 Hz, 1H), 1.33 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 160.59, 148.22, 145.47, 136.97, 135.29, 134.66, 133.82, 130.30, 129.23, 128.75, 128.27, 128.18, 128.12, 127.94, 127.04, 125.74, 125.63, 120.97, 119.94, 118.22, 117.33, 109.48, 100.42, 63.56, 56.02, 44.87, 18.54, 15.49. tR = 2.51 min (generic method). ESI-MS for C28H23ClN4O: calculated 466.2, found m/z 467.2, 469.2 [M + H]+; 465.3, 467.4 [M – H]−.
(E)-6-Chloro-3-(3-(1-ethyl-1H-indazol-5-yl)acryloyl)-4-phenylquinolin-2(1H)-one (100b)
Compound 100b was synthesized via general procedure A using 60a (292 mg, 0.98 mmol) and 1-ethyl-1H-indazole-5-carbaldehyde 76 (171 mg, 0.98 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–5% EtOH/DCM) to afford 100b (275 mg, yield 61%). 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 8.09 (s, 1H), 8.07 (d, J = 1.4 Hz, 1H), 7.72 (dd, J = 8.9, 1.6 Hz, 1H), 7.69–7.57 (m, 3H), 7.48 (d, J = 8.8 Hz, 1H), 7.46–7.36 (m, 3H), 7.34 (dd, J = 7.8, 1.8 Hz, 2H), 6.98 (d, J = 2.3 Hz, 1H), 6.74 (d, J = 16.3 Hz, 1H), 4.43 (q, J = 7.2 Hz, 2H), 1.38 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.64, 159.45, 147.35, 146.51, 139.48, 137.50, 133.76, 133.70, 132.72, 130.84, 128.89, 128.76, 128.40, 126.97, 126.03, 125.64, 125.55, 125.18, 123.97, 123.80, 120.63, 117.66, 110.26, 43.20, 14.85. tR = 2.35 min (generic method). ESI-MS for C27H20ClN3O2: calculated 453.1, found m/z 454.4, 456.4 [M + H]+, 452.4, 454.4 [M – H]−.
6-Chloro-3-(5-(1-ethyl-1H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (100c)
Compound 100c was synthesized via general procedure B using 100b (260 mg, 0.57 mmol) with hydrazine hydrate (55 μL, 1.14 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–10% EtOH/DCM) to afford 100c (228 mg, yield 86%). 1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 7.97 (d, J = 0.9 Hz, 1H), 7.59–7.46 (m, 5H), 7.42 (d, J = 8.8 Hz, 2H), 7.38–7.34 (m, 1H), 7.27–7.21 (m, 1H), 7.17–7.08 (m, 2H), 6.91 (d, J = 2.4 Hz, 1H), 4.71 (ddd, J = 11.3, 9.3, 2.5 Hz, 1H), 4.41 (q, J = 7.2 Hz, 2H), 3.27 (dd, J = 16.5, 11.1 Hz, 1H), 2.62 (dd, J = 16.5, 9.4 Hz, 1H), 1.38 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 161.06, 148.81, 145.93, 138.63, 137.46, 136.04, 135.71, 134.85, 132.74, 130.82, 129.75, 129.17, 128.76, 128.70, 127.36, 126.93, 126.25, 126.11, 125.62, 124.49, 123.84, 121.42, 118.50, 117.82, 110.00, 109.89, 63.43, 45.22, 43.52, 15.40. tR = 2.19 min (generic method). ESI-MS for C27H22ClN5O: calculated 467.1, found m/z 468.5, 470.5 [M + H]+, 466.5, 468.5 [M – H]−.
(E)-6-Chloro-3-(3-(2-ethyl-2H-indazol-5-yl)acryloyl)-4-phenylquinolin-2(1H)-one (101b)
Compound 101b was synthesized via general procedure A using 60a (307 mg, 1.00 mmol) and 2-ethyl-2H-indazole-5-carbaldehyde 77 (174 mg, 1.00 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–5% EtOH/DCM) to afford 101b (386 mg, yield 82%). 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 8.48 (s, 1H), 8.02 (s, 1H), 7.65 (dd, J = 8.8, 2.4 Hz, 1H), 7.61–7.51 (m, 3H), 7.51–7.38 (m, 4H), 7.34 (dd, J = 7.7, 1.9 Hz, 2H), 6.98 (d, J = 2.3 Hz, 1H), 6.68 (d, J = 16.3 Hz, 1H), 4.44 (q, J = 7.3 Hz, 2H), 1.49 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.61, 159.46, 148.47, 147.59, 146.48, 137.49, 133.71, 132.78, 130.83, 128.88, 128.76, 128.40, 127.38, 126.02, 125.55, 125.30, 125.16, 123.56, 121.42, 120.63, 117.65, 117.56, 47.87, 15.57. tR = 2.22 min (generic method). ESI-MS for C27H20ClN3O2: calculated 453.1, found m/z 454.4, 456.4 [M + H]+, 452.4, 454.4 [M – H]−.
6-Chloro-3-(5-(2-ethyl-2H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (101c)
Compound 101c was synthesized via general procedure B using 101b (302 mg, 0.60 mmol) with hydrazine hydrate (58 μL, 1.20 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–10% EtOH/DCM) to afford 101c (236 mg, yield 76%). 1H NMR (400 MHz, DMSO-d6) δ 12.19 (s, 1H), 8.27 (d, J = 0.9 Hz, 1H), 7.59–7.38 (m, 6H), 7.35 (td, J = 3.1, 1.5 Hz, 2H), 7.28–7.20 (m, 1H), 7.08 (d, J = 3.2 Hz, 1H), 6.98–6.86 (m, 2H), 4.65 (td, J = 10.3, 3.1 Hz, 1H), 4.42 (q, J = 7.3 Hz, 2H), 3.24 (dd, J = 16.5, 11.1 Hz, 1H), 2.60 (dd, J = 16.5, 9.6 Hz, 1H), 1.48 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 160.56, 148.29, 147.50, 145.53, 136.97, 135.54, 135.22, 130.32, 129.28, 128.68, 128.27, 128.20, 126.91, 125.76, 125.62, 124.73, 122.79, 121.02, 120.94, 117.33, 117.28, 117.07, 63.27, 47.64, 44.41, 15.77. tR = 2.06 min (generic method). ESI-MS for C27H22ClN5O: calculated 467.1, found m/z 468.5, 470.4 [M + H]+, 466.4, 468.5 [M – H]−.
(E)-6-Chloro-4-phenyl-3-(3-(1-propyl-1H-indazol-5-yl)acryloyl)quinolin-2(1H)-one (102b)
Compound 102b was synthesized via general procedure A using 60a (402 mg, 1.35 mmol) and 1-propyl-1H-indazole-5-carbaldehyde 78 (254 mg, 1.35 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–7% EtOH/DCM) to afford 102b (401 mg, yield 64%). 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 8.10 (d, J = 0.8 Hz, 1H), 8.07 (d, J = 1.4 Hz, 1H), 7.72 (dd, J = 9.0, 1.6 Hz, 1H), 7.70–7.57 (m, 3H), 7.48 (d, J = 8.8 Hz, 1H), 7.44–7.38 (m, 3H), 7.34 (dd, J = 7.7, 1.8 Hz, 2H), 6.98 (d, J = 2.4 Hz, 1H), 6.74 (d, J = 16.3 Hz, 1H), 4.36 (t, J = 6.9 Hz, 2H), 1.82 (h, J = 7.2 Hz, 2H), 0.80 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.66, 159.46, 147.36, 146.52, 140.12, 137.50, 133.80, 133.70, 132.74, 130.85, 128.89, 128.77, 128.41, 126.94, 126.03, 125.65, 125.55, 125.20, 123.98, 123.65, 120.64, 117.67, 110.36, 49.71, 22.82, 11.04. tR = 2.51 min (generic method). ESI-MS for C28H22ClN3O2: calculated 467.1, found m/z 468.2, 470.2 [M + H]+, 466.3, 468.3 [M – H]−.
6-Chloro-4-phenyl-3-(5-(1-propyl-1H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)quinolin-2(1H)-one (102c)
Compound 102c was synthesized via general procedure B using 102b (395 mg, 0.84 mmol) with hydrazine hydrate (82 μL, 1.68 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–10% EtOH/DCM) to afford 102c (381 mg, yield 94%). 1H NMR (400 MHz, DMSO-d6) δ 12.19 (s, 1H), 8.00–7.91 (m, 1H), 7.59–7.45 (m, 6H), 7.42 (d, J = 8.7 Hz, 2H), 7.38–7.31 (m, 1H), 7.24 (dd, J = 6.8, 1.9 Hz, 1H), 7.12 (dd, J = 8.7, 1.6 Hz, 1H), 6.90 (d, J = 2.3 Hz, 1H), 4.70 (t, J = 10.3 Hz, 1H), 4.33 (t, J = 6.9 Hz, 2H), 3.25 (dd, J = 16.5, 11.1 Hz, 1H), 2.62 (dd, J = 16.5, 9.5 Hz, 1H), 1.88–1.77 (m, 2H), 0.81 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 160.54, 148.29, 145.42, 138.73, 136.96, 135.44, 135.21, 132.22, 130.31, 129.23, 128.68, 128.25, 128.22, 128.18, 126.87, 125.74, 125.60, 125.13, 123.18, 120.92, 117.98, 117.32, 109.56, 62.94, 49.59, 44.66, 22.81, 11.09. tR = 2.34 min (generic method). ESI-MS for C28H24ClN5O: calculated 481.2, found m/z 482.1, 484.2 [M + H]+, 480.2, 482.3 [M – H]−.
(E)-6-Chloro-4-phenyl-3-(3-(2-propyl-2H-indazol-5-yl)acryloyl)quinolin-2(1H)-one (103b)
Compound 103b was synthesized via general procedure A using 60a (292 mg, 0.98 mmol) and 2-propyl-2H-indazole-5-carbaldehyde 79 (184 mg, 0.98 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–7% EtOH/DCM) to afford 103b (372 mg, yield 81%). 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 8.47 (s, 1H), 8.04–8.00 (m, 1H), 7.65 (dd, J = 8.8, 2.3 Hz, 1H), 7.60–7.51 (m, 3H), 7.47 (d, J = 8.8 Hz, 1H), 7.44–7.38 (m, 3H), 7.36–7.32 (m, 2H), 6.97 (d, J = 2.3 Hz, 1H), 6.68 (d, J = 16.2 Hz, 1H), 4.36 (t, J = 6.9 Hz, 2H), 1.96–1.87 (m, 2H), 0.83 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.63, 159.46, 148.52, 147.59, 146.47, 137.50, 133.71, 132.79, 130.83, 128.88, 128.77, 128.41, 127.39, 126.02, 125.87, 125.55, 125.31, 123.56, 121.32, 120.63, 117.66, 117.59, 54.36, 23.22, 10.84. tR = 2.37 min (generic method). ESI-MS for C28H22ClN3O2: calculated 467.1, found m/z 468.2, 470.2 [M + H]+, 466.3, 468.3 [M – H]−.
6-Chloro-4-phenyl-3-(5-(2-propyl-2H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)quinolin-2(1H)-one (103c)
Compound 103c was synthesized via general procedure B using 103b (342 mg, 0.73 mmol) with hydrazine hydrate (71 μL, 1.46 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–10% EtOH/DCM) to afford 103c (316 mg, yield 89%). 1H NMR (400 MHz, DMSO-d6) δ 12.19 (s, 1H), 8.27 (d, J = 0.9 Hz, 1H), 7.57 (dd, J = 8.8, 2.3 Hz, 1H), 7.55–7.44 (m, 4H), 7.41 (d, J = 8.8 Hz, 1H), 7.38–7.33 (m, 2H), 7.24 (dt, J = 6.6, 1.9 Hz, 1H), 7.08 (d, J = 3.2 Hz, 1H), 6.92 (dd, J = 8.9, 1.6 Hz, 1H), 6.90 (d, J = 2.4 Hz, 1H), 4.70–4.59 (m, 1H), 4.35 (t, J = 6.9 Hz, 2H), 3.24 (dd, J = 16.5, 11.1 Hz, 1H), 2.60 (dd, J = 16.4, 9.6 Hz, 1H), 1.96–1.87 (m, 2H), 0.82 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 160.58, 148.31, 147.56, 145.50, 136.98, 135.55, 135.23, 130.37, 129.31, 128.70, 128.30, 128.28, 128.23, 126.93, 125.76, 125.64, 124.75, 123.59, 120.95, 120.89, 117.36, 117.31, 117.11, 63.26, 54.20, 44.40, 23.40, 10.89. tR = 2.23 min (generic method). ESI-MS for C28H24ClN5O: calculated 481.2, found m/z 482.2, 484.2 [M + H]+, 480.3, 482.3 [M – H]−.
(E)-6-Chloro-3-(3-(1-cyclohexyl-1H-indazol-5-yl)acryloyl)-4-phenylquinolin-2(1H)-one (104b)
Compound 104b was synthesized via general procedure A using 60a (134 mg, 0.46 mmol) and 1-cyclohexyl-1H-indazole-5-carbaldehyde 80 (105 mg, 0.46 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–5% EtOH/DCM) to afford 104b (193 mg, yield 83%). 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 8.08 (s, 1H), 8.06 (d, J = 1.4 Hz, 1H), 7.72–7.69 (m, 2H), 7.65 (dd, J = 8.8, 2.4 Hz, 1H), 7.60 (d, J = 16.3 Hz, 1H), 7.48 (d, J = 8.8 Hz, 1H), 7.45–7.37 (m, 3H), 7.33 (dd, J = 7.7, 1.8 Hz, 2H), 6.98 (d, J = 2.4 Hz, 1H), 6.73 (d, J = 16.3 Hz, 1H), 4.60 (tt, J = 9.6, 4.8 Hz, 1H), 1.98–1.80 (m, 6H), 1.70 (d, J = 13.0 Hz, 1H), 1.48 (td, J = 12.4, 11.1, 4.6 Hz, 2H), 1.25 (dtd, J = 12.9, 9.7, 3.6 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 193.66, 159.47, 147.44, 146.53, 139.15, 137.51, 133.72, 133.57, 132.73, 130.86, 128.90, 128.79, 128.42, 127.00, 126.05, 125.59, 125.56, 125.00, 123.98, 123.66, 120.65, 117.68, 110.37, 56.77, 32.26, 24.98. tR = 1.94 min (apolar method). ESI-MS for C31H26ClN3O2: calculated 507.2, found m/z 508.2, 510.2 [M + H]+, 506.3, 508.3 [M – H]−.
6-Chloro-3-(5-(1-cyclohexyl-1H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (104c)
Compound 104c was synthesized via general procedure B using 104b (187 mg, 0.37 mmol) with hydrazine hydrate (36 μL, 0.74 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–10% EtOH/DCM) to afford 104c (172 mg, yield 86%). 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 7.95 (d, J = 0.8 Hz, 1H), 7.62–7.55 (m, 2H), 7.50 (dddd, J = 13.9, 7.1, 5.1, 4.0 Hz, 3H), 7.44–7.39 (m, 2H), 7.38–7.32 (m, 1H), 7.28–7.20 (m, 1H), 7.15–7.08 (m, 2H), 6.90 (d, J = 2.3 Hz, 1H), 4.69 (ddd, J = 11.0, 9.5, 3.2 Hz, 1H), 4.55 (tt, J = 10.0, 5.3 Hz, 1H), 3.25 (dd, J = 16.5, 11.1 Hz, 1H), 2.61 (dd, J = 16.5, 9.5 Hz, 1H), 1.88 (ddd, J = 20.4, 10.2, 3.5 Hz, 6H), 1.71 (dt, J = 13.0, 3.2 Hz, 1H), 1.51 (qd, J = 12.1, 11.7, 5.9 Hz, 2H), 1.26 (tdd, J = 12.8, 9.5, 4.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 160.56, 148.32, 145.45, 137.82, 136.98, 135.55, 135.24, 132.00, 130.35, 129.24, 128.70, 128.29, 128.26, 128.21, 126.89, 125.76, 125.63, 124.92, 123.21, 120.93, 118.01, 117.35, 109.60, 62.97, 56.62, 44.74, 32.30, 25.06. tR = 2.65 min (generic method). ESI-MS for C31H28ClN5O: calculated 521.2, found m/z 522.3, 524.3 [M + H]+, 520.3, 522.4 [M – H]−.
(E)-6-Chloro-3-(3-(2-cyclohexyl-2H-indazol-5-yl)acryloyl)-4-phenylquinolin-2(1H)-one (105b)
Compound 105b was synthesized via general procedure A using 60a (89 mg, 0.30 mmol) and 2-cyclohexyl-2H-indazole-5-carbaldehyde 81 (68 mg, 0.30 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 12 g; 0–5% EtOH/DCM) to afford 105b (136 mg, yield 90%). 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 8.49 (s, 1H), 8.00 (d, J = 1.5 Hz, 1H), 7.65 (dd, J = 8.8, 2.3 Hz, 1H), 7.59–7.52 (m, 3H), 7.47 (d, J = 8.8 Hz, 1H), 7.45–7.38 (m, 3H), 7.35–7.30 (m, 2H), 6.97 (d, J = 2.3 Hz, 1H), 6.66 (d, J = 16.3 Hz, 1H), 4.45 (tt, J = 11.3, 3.8 Hz, 1H), 2.09 (dd, J = 13.0, 4.0 Hz, 2H), 1.86 (ddt, J = 14.4, 9.8, 5.6 Hz, 4H), 1.76–1.63 (m, 1H), 1.51–1.37 (m, 2H), 1.25 (qt, J = 13.1, 3.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 193.63, 159.47, 148.04, 147.67, 146.47, 137.49, 133.72, 132.78, 130.84, 128.89, 128.78, 128.41, 127.34, 126.03, 125.55, 125.42, 125.24, 123.78, 123.45, 121.13, 120.65, 117.70, 61.84, 33.12, 24.85, 24.82. tR = 2.60 min (generic method). ESI-MS for C31H26ClN3O2: calculated 507.2, found m/z 508.3, 510.2 [M + H]+, 506.3, 508.4 [M – H]−.
6-Chloro-3-(5-(2-cyclohexyl-2H-indazol-5-yl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (105c)
Compound 105c was synthesized via general procedure B using 105b (130 mg, 0.26 mmol) with hydrazine hydrate (25 μL, 0.52 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–10% EtOH/DCM) to afford 105c (117 mg, yield 86%). 1H NMR (400 MHz, DMSO-d6) δ 12.19 (s, 1H), 8.28 (d, J = 0.9 Hz, 1H), 7.59–7.44 (m, 5H), 7.41 (d, J = 8.8 Hz, 1H), 7.38–7.31 (m, 2H), 7.23 (dt, J = 6.5, 2.0 Hz, 1H), 7.07 (d, J = 3.2 Hz, 1H), 6.94–6.87 (m, 2H), 4.64 (td, J = 9.9, 3.1 Hz, 1H), 4.43 (tt, J = 11.3, 3.8 Hz, 1H), 3.23 (dd, J = 16.5, 11.1 Hz, 1H), 2.59 (dd, J = 16.4, 9.6 Hz, 1H), 2.15–2.03 (m, 2H), 1.90–1.80 (m, 4H), 1.69 (dt, J = 12.5, 3.3 Hz, 1H), 1.44 (qt, J = 13.0, 3.5 Hz, 2H), 1.35–1.17 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 160.58, 148.32, 147.05, 145.53, 136.99, 135.52, 135.24, 130.37, 129.30, 128.70, 128.31, 128.29, 128.24, 126.93, 125.77, 125.65, 124.65, 121.38, 120.96, 120.70, 117.37, 117.34, 117.22, 63.29, 61.58, 44.46, 33.30, 24.93, 24.90. tR = 2.47 min (generic method). ESI-MS for C31H28ClN5O: calculated 521.2, found m/z 522.2, 524.2 [M + H]+, 520.2, 522.3 [M – H]−.
(E)-6-Chloro-3-(3-(4-(1-ethyl-1H-indazol-5-yl)phenyl)acryloyl)-4-phenylquinolin-2(1H)-one (106b)
Compound 106b was synthesized via general procedure A using 60a (137 mg, 0.46 mmol) and 4-(1-ethyl-1H-indazol-5-yl)benzaldehyde 82 (115 mg, 0.46 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–2% EtOH/DCM) to afford 106b (244 mg, quantitative yield). 1H NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 8.10 (d, J = 7.3 Hz, 2H), 7.74 (s, 5H), 7.65 (dd, J = 8.8, 2.4 Hz, 1H), 7.59–7.39 (m, 6H), 7.34 (dd, J = 7.8, 1.8 Hz, 2H), 6.99 (d, J = 2.4 Hz, 1H), 6.80 (d, J = 16.3 Hz, 1H), 4.46 (q, J = 7.2 Hz, 2H), 1.40 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.80, 159.46, 146.73, 145.88, 142.84, 138.45, 137.54, 133.65, 133.16, 132.74, 132.59, 131.71, 130.93, 129.35, 128.91, 128.47, 127.09, 126.97, 126.09, 125.60, 125.35, 124.25, 120.61, 118.93, 117.71, 110.15, 43.17, 14.92. tR = 2.64 min (generic method). ESI-MS for C33H24ClN3O2: calculated 529.2, found m/z 530.2, 532.2 [M + H]+, 528.3, 530.3 [M – H]−.
6-Chloro-3-(5-(4-(1-ethyl-1H-indazol-5-yl)phenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (106c)
Compound 106c was synthesized via general procedure B using 106b (233 mg, 0.44 mmol) with hydrazine hydrate (43 μL, 0.88 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–8% EtOH/DCM) to afford 106c (181 mg, yield 75%). 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 8.10 (d, J = 0.9 Hz, 1H), 7.98 (t, J = 1.2 Hz, 1H), 7.74 (d, J = 8.8 Hz, 1H), 7.67 (dd, J = 8.8, 1.7 Hz, 1H), 7.60–7.47 (m, 6H), 7.42 (d, J = 8.8 Hz, 1H), 7.36 (dt, J = 6.4, 1.8 Hz, 1H), 7.28–7.24 (m, 1H), 7.20–7.14 (m, 3H), 6.90 (d, J = 2.4 Hz, 1H), 4.63 (td, J = 10.9, 10.3, 3.2 Hz, 1H), 4.46 (q, J = 7.2 Hz, 2H), 3.29–3.20 (m, 1H), 2.62 (dd, J = 16.4, 9.4 Hz, 1H), 1.41 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 160.55, 148.36, 145.50, 142.03, 139.35, 138.19, 136.98, 135.22, 132.90, 132.66, 130.37, 129.25, 128.73, 128.29, 128.20, 127.06, 126.83, 126.69, 125.76, 125.64, 125.48, 124.24, 120.94, 118.34, 117.35, 110.00, 62.58, 44.46, 43.14, 14.93. tR = 1.49 min (apolar method). ESI-MS for C33H26ClN5O: calculated 543.2, found m/z 544.1, 546.1 [M + H]+, 542.2, 544.1 [M – H]−.
(E)-6-Chloro-3-(3-(4-(2-ethyl-2H-indazol-5-yl)phenyl)acryloyl)-4-phenylquinolin-2(1H)-one (107b)
Compound 107b was synthesized via general procedure A using 60a (92 mg, 0.31 mmol) and 4-(2-ethyl-2H-indazol-5-yl)benzaldehyde 83 (78 mg, 0.31 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 12 g; 0–10% EtOH/DCM) to afford 107b (150 mg, yield 91%). 1H NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 8.44 (d, J = 0.8 Hz, 1H), 8.04 (t, J = 1.3 Hz, 1H), 7.73 (s, 4H), 7.70–7.63 (m, 2H), 7.59 (dd, J = 9.1, 1.8 Hz, 1H), 7.57–7.42 (m, 5H), 7.34 (dd, J = 7.8, 1.8 Hz, 2H), 6.99 (d, J = 2.3 Hz, 1H), 6.79 (d, J = 16.4 Hz, 1H), 4.47 (q, J = 7.3 Hz, 2H), 1.52 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.79, 159.45, 147.56, 146.70, 145.88, 143.10, 137.54, 133.65, 132.67, 132.59, 131.84, 130.91, 129.31, 128.90, 128.83, 128.45, 126.92, 126.07, 125.58, 124.89, 124.04, 121.88, 120.60, 118.56, 117.70, 117.56, 47.83, 15.72. tR = 2.48 min (generic method). ESI-MS for C33H24ClN3O2: calculated 529.2, found m/z 530.1, 532.1 [M + H]+, 528.2, 530.2 [M – H]−.
6-Chloro-3-(5-(4-(2-ethyl-2H-indazol-5-yl)phenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (107c)
Compound 107c was synthesized via general procedure B using 107b (141 mg, 0.27 mmol) with hydrazine hydrate (26 μL, 0.54 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–15% EtOH/DCM) to afford 107c (123 mg, yield 83%). 1H NMR (400 MHz, DMSO-d6) δ 8.41 (d, J = 0.9 Hz, 1H), 7.92–7.88 (m, 1H), 7.67 (d, J = 9.0 Hz, 1H), 7.59–7.48 (m, 7H), 7.42 (d, J = 8.8 Hz, 1H), 7.37–7.33 (m, 1H), 7.25 (dq, J = 4.9, 1.9 Hz, 1H), 7.16 (dd, J = 9.3, 2.7 Hz, 3H), 6.90 (d, J = 2.4 Hz, 1H), 4.62 (ddd, J = 10.9, 9.4, 3.1 Hz, 1H), 4.47 (q, J = 7.3 Hz, 2H), 3.27 (dd, J = 16.5, 11.1 Hz, 1H), 2.62 (dd, J = 16.4, 9.5 Hz, 1H), 1.52 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 160.67, 148.34, 147.41, 145.61, 141.99, 139.64, 137.15, 135.26, 132.81, 130.34, 129.28, 128.75, 128.31, 128.21, 127.04, 126.81, 126.55, 125.72, 125.64, 125.23, 123.65, 121.93, 120.96, 117.77, 117.44, 62.61, 47.80, 44.49, 15.76. tR = 2.35 min (generic method). ESI-MS for C33H26ClN5O: calculated 543.2, found m/z 544.2, 546.2 [M + H]+, 542.3, 544.3 [M – H]−.
(E)-6-Chloro-4-phenyl-3-(3-(4-(1-propyl-1H-pyrazol-4-yl)phenyl)acryloyl)quinolin-2(1H)-one (108b)
Compound 108b was synthesized via general procedure A using 60a (324 mg, 1.10 mmol) and 4-(1-propyl-1H-pyrazol-4-yl)benzaldehyde 84 (236 mg, 1.10 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–8% EtOH/DCM) to afford 108b (387 mg, yield 72%). 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 8.26 (d, J = 0.8 Hz, 1H), 7.94 (d, J = 0.8 Hz, 1H), 7.68–7.55 (m, 5H), 7.50–7.38 (m, 5H), 7.33 (dd, J = 7.7, 1.8 Hz, 2H), 6.98 (d, J = 2.3 Hz, 1H), 6.73 (d, J = 16.4 Hz, 1H), 4.06 (t, J = 7.0 Hz, 2H), 1.96–1.87 (m, 2H), 0.84 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 193.72, 159.43, 146.62, 146.14, 137.50, 136.32, 135.30, 133.65, 132.61, 131.69, 130.87, 129.40, 128.88, 128.79, 128.42, 127.62, 126.26, 126.05, 125.56, 125.07, 120.85, 120.59, 117.67, 53.07, 23.07, 10.90. tR = 2.46 min (generic method). ESI-MS for C30H24ClN3O2: calculated 493.2, found m/z 494.0, 496.0 [M + H]+, 492.0, 494.0 [M – H]−.
6-Chloro-4-phenyl-3-(5-(4-(1-propyl-1H-pyrazol-4-yl)phenyl)-4,5-dihydro-1H-pyrazol-3-yl)quinolin-2(1H)-one (108c)
Compound 108c was synthesized via general procedure B using 108b (358 mg, 0.72 mmol) with hydrazine hydrate (70 μL, 1.44 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–10% EtOH/DCM) to afford 108c (326 mg, yield 89%). 1H NMR (400 MHz, DMSO-d6) δ 12.18 (s, 1H), 8.13 (d, J = 0.8 Hz, 1H), 7.83 (d, J = 0.8 Hz, 1H), 7.57 (dd, J = 8.8, 2.4 Hz, 1H), 7.51 (tdd, J = 6.6, 3.8, 1.8 Hz, 3H), 7.42 (dd, J = 8.5, 6.2 Hz, 3H), 7.34 (dt, J = 6.8, 1.7 Hz, 1H), 7.26–7.22 (m, 1H), 7.09 (d, J = 3.2 Hz, 1H), 7.08–7.03 (m, 2H), 6.90 (d, J = 2.3 Hz, 1H), 4.61–4.49 (m, 1H), 4.06 (t, J = 7.0 Hz, 2H), 3.22 (dd, J = 16.4, 11.0 Hz, 1H), 2.56 (dd, J = 16.4, 9.6 Hz, 1H), 1.96–1.87 (m, 2H), 0.85 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 160.53, 148.31, 145.48, 140.99, 136.97, 135.81, 135.21, 131.39, 130.34, 129.22, 128.71, 128.27, 128.19, 126.92, 126.85, 126.79, 125.75, 125.62, 124.76, 121.34, 120.92, 117.33, 62.66, 52.98, 44.43, 23.16, 10.94. tR = 2.34 min (generic method). ESI-MS for C30H26ClN5O: calculated 507.2, found m/z 508.0, 510.1 [M + H]+, 506.1, 508.1 [M – H]−.
(E)-3-(3-(4-Fluorophenyl)acryloyl)-4-phenylquinolin-2(1H)-one (109b)
Compound 109b was synthesized via general procedure A using 61a (265 mg, 1.00 mmol) and 4-fluorobenzaldehyde 62 (107 μL, 1.00 mmol). Title compound 109b was obtained after precipitation and filtration from the reaction crude (345 mg, yield 93%). 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 7.73 (d, J = 7.2 Hz, 2H), 7.65–7.00 (m, 12H), 6.73 (d, J = 16.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 194.31, 159.77, 147.76, 144.37, 134.36, 131.36, 130.92, 130.92, 128.92, 128.46, 128.23, 127.52, 126.86, 126.86, 124.54, 122.05, 119.21, 116.00, 115.92, 115.90, 115.88, 115.79. tR = 2.25 min (generic method). ESI-MS for C24H16FNO2: calculated 369.1, found m/z 370.5 [M + H]+; 368.4 [M – H]−.
3-(5-(4-Fluorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (109c)
Compound 109c was synthesized via general procedure B using 109b (300 mg, 0.81 mmol) with hydrazine hydrate (79 μL, 1.62 mmol). Title compound 109c was obtained after precipitation from DCM (292 mg, yield 94%). 1H NMR (400 MHz, DMSO-d6) δ 12.11 (s, 1H), 7.54–7.44 (m, 4H), 7.40 (dd, J = 8.3, 1.2 Hz, 1H), 7.32 (dt, J = 7.4, 1.7 Hz, 1H), 7.21 (dt, J = 6.8, 1.9 Hz, 1H), 7.14–7.02 (m, 6H), 6.99 (dd, J = 8.1, 1.4 Hz, 1H), 4.58 (td, J = 10.9, 3.1 Hz, 1H), 3.23 (dd, J = 16.4, 11.0 Hz, 1H), 2.56–2.52 (m, 1H). tR = 2.13 min (generic method). ESI-MS for C24H18FN3O: calculated 383.1, found m/z 384.5 [M + H]+, 382.5 [M – H]−.
(E)-3-(3-(4-Bromophenyl)acryloyl)-4-phenylquinolin-2(1H)-one (110b)
Compound 110b was synthesized via general procedure A using 61a (265 mg, 1.00 mmol) and 4-bromobenzaldehyde 64 (185 mg, 1.00 mmol). Title compound 110b was obtained after precipitation and filtration from the reaction crude (396 mg, yield 92%). 1H NMR (400 MHz, DMSO-d6) δ 7.67–7.50 (m, 5H), 7.45–7.36 (m, 5H), 7.29 (dd, J = 7.7, 1.8 Hz, 2H), 7.15–7.02 (m, 2H), 6.79 (d, J = 16.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 210.70, 133.70, 131.89, 131.23, 130.49, 129.00, 128.43, 128.25, 126.84, 124.03, 121.73, 119.29, 116.57, 99.54. tR = 2.43 min (generic method). ESI-MS for C24H16BrNO2: calculated 429.0, found m/z 430.4, 432.4 [M + H]+; 428.4, 430.4 [M – H]−.
3-(5-(4-Bromophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (110c)
Compound 110c was synthesized via general procedure B using 110b (300 mg, 0.70 mmol) with hydrazine hydrate (68 μL, 1.40 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0.2–3% EtOH/DCM) to afford 110c (213 mg, yield 69%). 1H NMR (400 MHz, DMSO-d6) δ 12.04 (s, 1H), 7.58–7.36 (m, 7H), 7.31 (dt, J = 7.3, 1.6 Hz, 1H), 7.20 (dt, J = 7.3, 2.0 Hz, 1H), 7.13–6.95 (m, 5H), 4.56 (ddd, J = 11.0, 9.3, 3.3 Hz, 1H), 3.30–3.14 (m, 1H), 2.57–2.51 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 160.71, 149.63, 145.96, 143.13, 138.24, 135.77, 131.05, 130.51, 129.28, 128.73, 128.69, 128.10, 128.06, 127.90, 126.94, 125.39, 121.92, 119.82, 119.57, 115.31, 62.01, 44.62. tR = 2.31 min (generic method). ESI-MS for C24H18BrN3O: calculated 443.1, found m/z 444.4, 446.5 [M + H]+.
(E)-3-(3-(3-Bromophenyl)acryloyl)-4-phenylquinolin-2(1H)-one (111b)
Compound 111b was synthesized via general procedure A using 61a (265 mg, 1.00 mmol) and 3-bromobenzaldehyde 85 (117 μL, 1.00 mmol). Title compound 111b was obtained after precipitation and filtration from the reaction crude (380 mg, yield 88%). 1H NMR (400 MHz, DMSO-d6) δ 12.18 (s, 1H), 7.93 (s, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.59 (ddt, J = 7.2, 3.9, 1.6 Hz, 2H), 7.51–7.37 (m, 5H), 7.37–7.27 (m, 3H), 7.16 (td, J = 7.6, 7.1, 1.2 Hz, 1H), 7.08 (dd, J = 8.2, 1.5 Hz, 1H), 6.84 (d, J = 16.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 194.29, 159.58, 148.05, 143.93, 138.80, 136.83, 134.24, 133.07, 131.37, 131.26, 131.06, 130.87, 128.92, 128.85, 128.52, 128.26, 127.14, 126.93, 122.24, 122.19, 119.23, 115.66. tR = 2.40 min (generic method). ESI-MS for C24H16BrNO2: calculated 429.0, found m/z 430.4, 432.4 [M + H]+; 428.3, 430.3 [M – H]−.
3-(5-(3-Bromophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (111c)
Compound 111c was synthesized via general procedure B using 111b (300 mg, 0.70 mmol) with hydrazine hydrate (68 μL, 1.40 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 1–5% EtOH/DCM) to afford 111c (256 mg, yield 84%). 1H NMR (400 MHz, DMSO-d6) δ 12.06 (s, 1H), 7.56–7.36 (m, 7H), 7.36–7.29 (m, 1H), 7.26–7.18 (m, 2H), 7.18–7.06 (m, 3H), 7.01 (dd, J = 8.2, 1.4 Hz, 1H), 4.59 (ddd, J = 11.1, 9.3, 3.4 Hz, 1H), 3.24 (dd, J = 16.6, 11.2 Hz, 1H), 2.56 (dd, J = 16.6, 9.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 160.67, 149.59, 146.52, 145.91, 138.21, 135.73, 130.47, 130.39, 129.66, 129.25, 129.10, 128.64, 128.02, 128.00, 127.87, 126.89, 125.61, 125.30, 121.87, 121.52, 119.47, 115.27, 61.94, 44.57. tR = 2.30 min (generic method). ESI-MS for C24H18BrN3O: calculated 443.0, found m/z 444.5, 446.4 [M + H]+; 442.6, 444.4 [M – H]−.
(E)-3-(3-(4-Chlorophenyl)acryloyl)-4-phenylquinolin-2(1H)-one (112b)
Compound 112b was synthesized via general procedure A using 61a (169 mg, 0.64 mmol) and 4-chlorobenzaldehyde 63 (90 mg, 0.64 mmol). Title compound 112b was obtained after precipitation and filtration from the reaction crude (246 mg, quantitative yield). 1H NMR (400 MHz, DMSO-d6) δ 12.18 (s, 1H), 7.71 (d, J = 8.3 Hz, 2H), 7.59 (td, J = 7.6, 7.0, 1.5 Hz, 1H), 7.52–7.37 (m, 7H), 7.34–7.29 (m, 2H), 7.17 (t, J = 7.6 Hz, 1H), 7.12–7.05 (m, 1H), 6.79 (d, J = 16.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 194.66, 160.05, 148.50, 144.66, 139.25, 135.67, 134.74, 133.75, 131.78, 131.55, 130.77, 129.39, 129.00, 128.73, 128.65, 127.42, 122.69, 119.69, 116.13. tR = 2.37 min (generic method). ESI-MS for C24H16ClNO2: calculated 385.1, found m/z 386.4, 388.4 [M + H]+; 384.4, 386.4 [M – H]−.
3-(5-(4-Chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (112c)
Compound 112c was synthesized via general procedure B using 112b (225 mg, 0.58 mmol) with hydrazine hydrate (56 μL, 1.16 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 1–10% EtOH/DCM) to afford 112c (185 mg, yield 79%). 1H NMR (400 MHz, DMSO-d6) δ 12.05 (s, 1H), 7.56–7.43 (m, 4H), 7.40 (dd, J = 8.3, 1.2 Hz, 1H), 7.35–7.26 (m, 3H), 7.21 (dt, J = 7.3, 2.0 Hz, 1H), 7.13–7.06 (m, 4H), 7.00 (dd, J = 8.2, 1.4 Hz, 1H), 4.58 (ddd, J = 10.9, 9.2, 3.2 Hz, 1H), 3.25 (dd, J = 16.5, 11.1 Hz, 1H), 2.58–2.52 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 161.18, 150.09, 146.43, 143.16, 138.71, 136.24, 131.79, 130.97, 129.76, 129.16, 128.82, 128.60, 128.56, 128.53, 128.36, 127.41, 125.88, 122.39, 120.04, 115.78, 62.43, 45.12. tR = 2.26 min (generic method). ESI-MS for C24H18ClN3O: calculated 399.1, found m/z 400.4, 402.4 [M + H]+; 398.4, 400.5 [M – H]−.
(E)-3-(3-(4-Methoxyphenyl)acryloyl)-4-phenylquinolin-2(1H)-one (113b)
Compound 113b was synthesized via general procedure A using 61a (265 mg, 1.00 mmol) and p-anisaldehyde 65 (122 μL, 1.00 mmol). Title compound 113b was obtained after precipitation and filtration from the reaction crude (304 mg, yield 80%). 1H NMR (400 MHz, DMSO-d6) δ 12.16 (s, 1H), 7.66–7.51 (m, 3H), 7.47–7.34 (m, 5H), 7.32–7.27 (m, 2H), 7.14 (t, J = 7.7 Hz, 1H), 7.06 (dd, J = 8.2, 1.4 Hz, 1H), 6.93 (d, J = 8.8 Hz, 2H), 6.60 (d, J = 16.3 Hz, 1H), 3.78 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 194.07, 161.37, 159.72, 147.51, 145.75, 138.92, 134.40, 131.59, 130.85, 130.48, 128.91, 128.41, 128.19, 126.83, 125.41, 122.04, 119.23, 115.76, 114.36, 55.33. tR = 2.19 min (generic method). ESI-MS for C25H19NO3: calculated 381.1, found m/z 382.5 [M + H]+, 380.5 [M – H]+.
3-(5-(4-Methoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)-4-phenylquinolin-2(1H)-one (113c)
Compound 113c was synthesized via general procedure B using 113b (300 mg, 0.79 mmol) with hydrazine hydrate (77 μL, 1.58 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0–5% EtOH/DCM) to afford 113c (212 mg, yield 69%). 1H NMR (400 MHz, DMSO-d6) δ 12.02 (s, 1H), 7.54–7.43 (m, 4H), 7.39 (dd, J = 8.3, 1.2 Hz, 1H), 7.31 (dq, J = 7.6, 1.4 Hz, 1H), 7.21 (ddd, J = 5.3, 4.0, 2.0 Hz, 1H), 7.09 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.00 (dt, J = 8.2, 2.2 Hz, 3H), 6.82–6.76 (m, 2H), 4.51 (t, J = 10.3 Hz, 1H), 3.72 (s, 3H), 3.16 (dd, J = 16.4, 10.9 Hz, 1H), 2.57–2.51 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 160.73, 158.22, 149.48, 146.05, 138.21, 135.81, 135.45, 130.43, 129.25, 128.74, 128.08, 128.05, 127.86, 127.59, 126.91, 125.62, 121.88, 119.59, 115.28, 113.57, 62.36, 55.02, 44.63. tR = 2.08 min (generic method). ESI-MS for C25H21N3O2: calculated 395.1, found m/z 396.5 [M + H]+; 394.6 [M – H]−.
(E)-4-Phenyl-3-(3-(4-(trifluoromethyl)phenyl)acryloyl)quinolin-2(1H)-one (114b)
Compound 114b was synthesized via general procedure A using 61a (301 mg, 1.10 mmol) and 4-trifluoromethylbenzaldehyde 68 (150 μL, 1.10 mmol). Title compound 114b was obtained after precipitation and filtration from the reaction crude (414 mg, yield 87%). 1H NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 7.81 (dd, J = 62.7, 7.9 Hz, 4H), 7.67–7.24 (m, 8H), 7.25–7.05 (m, 2H), 6.91 (d, J = 16.2 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 194.74, 160.08, 148.71, 143.97, 139.35, 138.85, 134.71, 131.63, 130.31, 129.64, 129.40, 129.04, 128.76, 127.45, 126.14, 122.71, 119.67, 116.20. tR = 2.41 min (generic method). ESI-MS for C25H16F3NO2: calculated 419.1, found m/z 420.5 [M + H]+; 418.5 [M – H]−.
4-Phenyl-3-(5-(4-(trifluoromethyl)phenyl)-4,5-dihydro-1H-pyrazol-3-yl)quinolin-2(1H)-one (114c)
Compound 114c was synthesized via general procedure B using 114b (256 mg, 0.60 mmol) with hydrazine hydrate (58 μL, 1.20 mmol). Purification was performed by direct phase flash chromatography (SiO2 gold 24 g; 0.8–6% EtOH/DCM) to afford 114c (214 mg, yield 80%). 1H NMR (400 MHz, DMSO-d6) δ 12.05 (s, 1H), 7.60 (d, J = 8.1 Hz, 2H), 7.55–7.37 (m, 5H), 7.32 (dd, J = 11.5, 7.6 Hz, 3H), 7.24–7.17 (m, 2H), 7.10 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.00 (dd, J = 8.2, 1.4 Hz, 1H), 4.69 (ddd, J = 11.6, 9.1, 2.8 Hz, 1H), 3.33 (m, 1H), 2.63–2.52 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 161.17, 150.18, 148.95, 146.44, 138.72, 136.22, 131.00, 129.76, 129.22, 129.13, 128.78, 128.57, 128.51, 128.37, 128.02, 127.72, 127.42, 126.14, 125.77, 125.62, 125.57, 125.53, 122.40, 120.02, 115.79, 62.59, 45.15. tR = 2.30 min (generic method). ESI-MS for C25H18F3N3O: calculated 433.1, found m/z 434.4 [M + H]+; 432.5 [M – H]−.
tert-Butyl (3-(3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-3-oxopropyl)carbamate (119)
In round-bottom flask, the commercially available 3-((tert-butoxycarbonyl)amino)propanoic acid 118 (182 mg, 0.96 mmol), HOBt (156 mg, 1.16 mmol), and EDCI (222 mg, 1.16 mmol) were stirred in DCM (15 mL) at rt for 1 h. Then a solution of the 86c (400 mg, 0.96 mmol) and Et3N (296 μL, 2.12 mmol) in DCM (5 mL) was added. The mixture was stirred at rt overnight. The solvent was removed under reduced pressure, the residue redissolved with EtOAc and then washed with H2O, 1 M NaHCO3, and finally 10% citric acid. The organic layer was dried over Na2SO4 and evaporated to dryness. Purification was performed by direct phase flash chromatography (0–30% EtOAc/DCM). Yield 72 mg, 25%. 1H NMR (400 MHz, DMSO-d6) δ 12.40 (s, 1H), 7.65 (dd, J = 4.0, 8.0 Hz, 1H), 7.59–7.50 (m, 4H), 7.46 (d, J = 8.0 Hz, 1H), 7.43–7.42 (m, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.04 (t, J = 8.0 Hz, 2H), 6.93 (d, J = 2.0 Hz, 1H), 6.80 (dd, J = 4.0, 8.0 Hz, 2H), 6.66 (t, J = 8.0 Hz, 1H), 5.32 (dd, J = 4.0, 12.0 Hz, 1H), 3.73 (dd, J = 12.0, 16.0 Hz, 1H), 2.99 (dd, J = 8.0, 12.0 Hz, 2H), 2.76 (dd, J = 4.0, 20.0 Hz, 1H) 2.41 (q, J = 8.0 Hz, 2H), 1.38 (s, 9H).
Methyl 3-(Methylsulfonamido)propanoate (123)
In a dried round-bottom flask, commercially available methyl 3-aminopropanoate 121 (745 mg, 5.34 mmol) and triethylamine (3.7 mL, 26.68 mmol) were stirred in anhydrous DCM (5.6 mL) prior to the addition of methanesulfonyl chloride 122 (1.65 mL, 10.67 mmol). After stirring at rt for 2 days, the reaction was quenched with NaHCO3 sat. solution and extracted three times with CHCl3. The organic layer was dried over Na2SO4 and evaporated to dryness. The title compound was obtained after purification over direct phase flash column chromatography (0–40% EtOAc/petroleum ether). Yield 791 mg, 82%. 1H NMR (400 MHz, CDCl3-d) δ 4.94 (s, 1H), 3.72 (s, 3H), m (3.42–3.37, 2H), 2.97 (s, 3H), 2.64 (t, J = 4 Hz, 2H). 13C NMR (101 MHz, CDCl3-d) δ 172.63, 52.18, 40.60, 38.89, 34.58.
3-(Methylsulfonamido)propanoic Acid (124)
In round-bottom flask, methyl 3-(methylsulfonamido)propanoate 123 (791 mg, 4.36 mmol) was stirred in MeOH/THF (1:1 v/v) prior to the addition of 2 M LiOH (2.5 mL). The reaction was stirred at rt overnight. The reaction mixture was acidified with 1 M HClaq (pH 2) and extracted three times with ethyl acetate. The organic layer was dried over Na2SO4, filtered and evaporated to dryness to obtain the title compound. Yield 600 mg, 82%. 1H NMR (400 MHz, DMSO-d6) δ 12.29 (s, 1H), 7.02 (t, J = 4.0 Hz, 1H), 7.15 (dd, J = 7.0, 12.6, 2H), 2.43 (t, J = 7.2 Hz, 2H).
Biology. ELISA Assay
Competitive ELISA screening assay using biotinylated BRC4 peptide to disrupt the BRC4–RAD51 interaction was performed by modifying the method described by Rajendra et al.33 BRC4-biotinylated peptide (N-term biotin-KEPTLLGFHTASGKKVKIAKESLDKVKNLFDEKEQ from Life Technologies) was used to coat 384-well plates (Nunc). After washing with PBS containing 0.05% Tween-20 (PBST) and blocking with the solution BSA 1% /PBST, overnight hybridization with human RAD51 protein (NP_002866 Creative Biomart, NY) was performed. Test compounds were added in dose response from 0.01 to 100 μM in triplicate with constant DMSO 1%. Antibody raised against RAD51 (Millipore) and HRP-secondary antibody staining to develop the 3,3′,5,5′-tetramethylbenzidine signal (Sigma) quenched with 1 M HCl was used as the assay readout. Colorimetric measure was read on a Victor5 (PerkinElmer) plate reader. BRC4 and Rad51 were included in the assay as positive control. Results were analyzed by using GraphPad software.
Protocol for the Expression and Purification of His-hRAD51
hRAD51 was expressed in E. coli Rosetta2(DE3)pLysS cells. A saturated overnight culture of Rosetta2(DE3)pLysS/pET15b-His-hRAD51 was diluted (1:1000) into a fresh TB-5052 autoinduction medium containing ampicillin (100 μg/mL). The flasks were shaken at 200 rpm at 20 °C for 72 h. The pellet was subsequently resuspended in an appropriate volume of buffer A (20 mM Tris-HCl (pH 8.00), 500 mM NaCl, 10 mM imidazole, 5 mM DTT, 10% (v/v) glycerol) supplemented with protease inhibitor cocktail (SIGMAFAST protease inhibitor cocktail tablets, EDTA-50 free). The cell suspension was lysed on ice trough sonication (24 rounds of 30 in.; amplitude 85%; Tip MS72; Bandelin Sonoplus HD2070 sonicator). The disrupted cell suspension was centrifuged for 1 h at 13 000 rpm. The supernatant fraction was filtered with a 0.45 μm (MiniSart syringe filter 0.45 μm) membrane to remove residual particulates before chromatography. The supernatant was applied onto a His-Trap column (His-TrapTM FF 5 mL, GE Healthcare) equilibrated with buffer A. A wash step was performed using 10% of buffer B (20 mM Tris-HCl (pH 8.00), 500 mM NaCl, 500 mM imidazole, 10% (v/v) glycerol). The protein was then eluted with a linear gradient from 10% to 100% of buffer B over 10 column volumes. Fractions (0.5 mL) were collected and analyzed by SDS–PAGE. Collected fractions corresponding to the recombinant protein were dialyzed overnight at 4 °C against buffer C (50 mM Tris-HCl (pH 8.00), 200 mM KCl, 0.25 mM EDTA, 2 mM DTT, 10% (v/v) glycerol). Dialyzed protein was loaded onto an anion exchange column (ResQ, GE Healthcare) equilibrated in buffer C. The elution was performed with a linear gradient of buffer B (50 mM Tris-HCl (pH 8.00), 1 M KCl, 0.25 mM EDTA, 2 mM DTT, 10% (v/v) glycerol). Fractions (0.5 mL) were collected and analyzed by SDS–PAGE. Fractions containing His-hRAD51 were pooled and dialyzed against the storage buffer (20 mM Hepes (pH 8.00), 250 mM KCl, 0.1 mM EDTA, 2 mM DTT, 10% (v/v) glycerol). The protein yield was determined from the optical absorption at 280 nm (extinction coefficient 14 900 M–1 cm–1) of the final sample.
Microscale Thermophoresis
The recombinant protein hRAD51 was labeled with the Monolith His-Tag labeling kit RED-tris-NTA 2nd Generation kit (NanoTemper Technologies). MST measurements were simultaneously performed on 16 capillaries containing a constant concentration (25 nM) of labeled RED-tris-NTA 2nd Generation His-hRAD51 protein and 16 different concentrations of 35d in order to determine a concentration-dependent MST binding curve. The highest 35d concentration tested was 40 μM. Measurements were carried out in MST buffer (20 mM Hepes (pH 8.00), 250 mM KCl, 0.1 mM EDTA, 5% (v/v) glycerol, 5% DMSO).
Cell Culture and Treatments
BxPC-3 and Capan-1 cells were grown in RPMI 1640 supplemented with 10% FBS, 100 U/mL penicillin/streptomycin, 2 mM glutamine. All media and supplements were from Sigma-Aldrich. Non-neoplastic, immortalized cells from human kidney (HK-2, ATCC CRL2190) were grown in DMEM:F12 medium containing 40 ng/mL dexamethasone and supplemented as described above.
All cultures were routinely tested for Mycoplasma contamination. Treatments (olaparib and BRCA2-RAD51 disruptors) were administered in culture medium supplemented with 0.6% DMSO. The same amount of DMSO was added to the control, untreated cultures.
Homologous Recombination Assay
Homologous recombination (HR) was assessed by using a commercially available assay (Norgen). This assay is based on cell transfection with two plasmids that, upon cell entry, recombine. The efficiency of HR can be assessed by real-time PCR, using primer mixtures included in the assay kit. Different primer mixtures allow one to discriminate between the original plasmid backbones and their recombination product.
BxPC3 cells (2 × 105 per well) were seeded in a 24-well plate and allowed to adhere overnight. Co-transfection with the two plasmids was performed in Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. During transfection (5 h), cells were exposed to different doses of RAD51-BRCA2 disruptors, dissolved in RPMI in the presence of 0.6% DMSO. After washing with PBS, cells were harvested, and DNA was isolated using Illustra Tissue and Cell Genomic Prep Mini Spin kit (GE Healthcare). Sample concentration was measured using an ONDA Nano Genius photometer. The efficiency of HR was assessed by real-time PCR, using 25 ng of template, the primer mixtures included in the assay kit and following the protocol indicated by the manufacturer. Data analysis was based on the ΔΔCt method: [recombination product/backbone plasmids]treated versus [recombination product/backbone plasmids]control.
Immunofluorescence
Immunofluorescence was used for studying RAD51 nuclear translocation and for evaluating DNA damage through the detection of γH2AX nuclear foci. To visualize RAD51 in cell nuclei, BxPC-3 cells were seeded on glass coverslips placed in a 6-well culture plate (2 × 105 cells/well) and allowed to adhere overnight. Cultures were then preincubated with 20 μM 35d for 1 h and subsequently exposed to 50 μM cisplatin for an additional 1.5 h. Medium was removed, and cells were maintained in the presence of 20 μM 35d for 4 h. After this time, cultures growing on coverslips were fixed in PBS containing 1% formalin for 20 min, permeabilized in 70% ethanol, air-dried, and washed twice with PBS. Samples were incubated in 10% bovine serum albumin (BSA) in PBS for 30 min at 37 °C and subsequently exposed to an anti-RAD51 mouse monoclonal antibody (Santa Cruz Biotechnology, 1:1000 in 5% BSA/PBS) overnight at 4 °C. After washing, coverslips were incubated with an anti-mouse FITC-conjugated secondary antibody (1:1000 in 1% BSA/PBS) for 1 h at 37 °C, washed, air-dried, and mounted with a solution of DAPI (2 μg/mL) and DABCO.
To evaluate DNA damage through γH2AX nuclear foci, BXPC3 and Capan1 cells were seeded on glass coverslips in 6-well tissue culture plate (2 × 105 cells/well) and allowed to adhere overnight. After 48 h treatment with olaparib (10 μM) or 35d (20 μM) given alone or in combination, cultures growing on coverslips were fixed and treated as described above. For this experiment, the used antibodies were a rabbit polyclonal anti-γH2AX (Abcam, 1:1000 in 5% BSA/PBS) and a secondary anti-rabbit rhodamine-labeled (Novus Biologicals, 1:1000 in 1% BSA/PBS). For both experiments, images were acquired using a Nikon fluorescent microscope equipped with filters for FITC, TRITC, and DAPI. The percentage of cells bearing nuclear foci was estimated by two independent observers, by analyzing 100–250 cells for each treatment sample.
Cell Viability Assay
Cell viability was assessed with the CellTiter-Glo luminescent cell viability assay from Promega. For this experiment, 1 × 104 cells in 200 μL of culture medium were seeded into each well of a 96-multiwell white body plate and allowed to adhere overnight. After 72 h incubation in the presence of olaparib (10 μM) and the RAD51-BRCA2 disruptors alone or in combination, the plate was allowed to equilibrate at room temperature for 30 min and the CellTiter-Glo reactive was directly added to each well. The plate was kept on a shaker for 10 min to induce cell lysis, and its luminescence was measured with a Fluoroskan Ascent FL reader (Labsystems).
Cytotoxicity Assay
Cell death was assessed by applying the CellTox Green cytotoxicity assay (Promega). Briefly, BxPC-3 (1 × 104/well) were plated in 96-well plates and treated for 72 h with olaparib (10 μM) and the BRCA2-RAD51 disruptors administered alone or in combination. At the end of treatment, the CellTox dye was added to cell cultures and the green fluorescence signal, which is produced by the binding interaction with dead cell DNA, was measured following the manufacturer’s instructions.
Assessment of Cell Death with Vital Dyes
BxPC-3 cells were grown on coverslips placed in a 6-multiwell plate (5 × 105 cells/well). After a 72 h treatment with olaparib (10 μM) or 35d (20 μM) given alone or in combination, wells were rapidly washed with PBS and filled with 500 μL of a PBS solution containing DAPI (4.6 μg/mL) and PI (50 μg/mL). After a 10 min incubation at room temperature under light-shielded condition, they were washed with PBS, fixed (10 min) with 10% neutral buffered formalin solution, and washed again to eliminate the fixative. Coverslips were then applied on glass slides using two drops of mounting media (DABCO). Images were acquired on a Nikon fluorescent microscope equipped with filters for DAPI and PI.
Micronuclei Visualization
For micronuclei visualization, cells were grown on coverslips placed in a 6-multiwell plate (5 × 105 cells/well). After a 72 h treatment with olaparib (10 μM) or 35d (20 μM) given alone or in combination, wells were rapidly washed with PBS and then fixed for 10 min with cold methanol. Coverslips were then air-dried and mounted on glass slides using a solution of DAPI (5 μg/mL)/DABCO. Images were acquired on a Nikon fluorescent microscope equipped with filters for DAPI. A cell was considered to contain micronuclei if the following criteria were met: (i) one or more round fluorescent bodies were present in the cytoplasm which did not touch the main nucleus; (ii) they were <1/3 of the main nucleus diameter; (iii) they were nonrefractile, to exclude foreign bodies. The percentage of cells bearing micronuclei was estimated by two independent observers, by analyzing 100–250 cells for each treatment sample.
Acknowledgments
This work was supported by the Associazione Italiana per la Ricerca sul Cancro AIRC (Progetto IG 2018, id 21386), the Italian Institute of Technology (IIT), and the University of Bologna. We thank Prof. Maurizio Recanatini for fruitful discussions.
Glossary
Abbreviations Used
- MeCN
acetonitrile
- Boc2O
di-tert-butyl dicarbonate
- CHX
cyclohexane
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DMSO
dimethylsulfoxide
- EDC
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- EtOH
ethanol
- ESI
electrospray ionization
- EtOAc
ethyl acetate
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- HOBt
hydroxybenzotriazole
- MS
mass spectrometry
- μWave
microwave
- MeI
iodomethane
- DMF
N,N-dimethylformamide
- MeOH
methanol
- NMR
nuclear magnetic resonance
- rt
room temperature
- THF
tetrahydrofuran
- TLC
thin layer chromatography
- Et3N
triethylamine
- DAPI
4′,6-diamidino-2-phenylindole
- PI
propidium iodide
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01526.
Molecular formula strings and some data (CSV)
Computational methods; separation and ELISA assay results of enantiomers 4d-I and 4d-II; experimental procedure and characterization data of aldehydes 67, 70, 74–84; 1H and 13C NMR spectra of final compounds 4d–10d, 14d–15d, 18d–57d; HPLC–MS analysis of selected final compounds 4d–10d, 14d–15d, 18–25d, 35d, 49d, 57d (PDF)
Author Present Address
○ F.F.: Molecular Horizon srl, Via Montelino 30, 06084 Bettona (PG), Italy.
Author Contributions
∇ G.B., D.M., and M.M. contributed equally to this work.
The authors declare the following competing financial interest(s): One patent application protecting the class of compounds disclosed in this article has been filed by the following authors: Greta Bagnolini, Domenico Milano, Marcella Manerba, Jose Antonio Ortega, Francesca De Franco, Roberto Pellicciari, Saverio Minucci, Giuseppina Di Stefano, Marinella Roberti, and Andrea Cavalli.
Supplementary Material
References
- Bridges C. B. The origin and variations in sexual and sex limited characters. Am. Nat. 1922, 56, 51–63. 10.1086/279847. [DOI] [Google Scholar]
- Dobzhansky T. Genetics of natural populations. Xiii. Recombination and variability in populations of Drosophila pseudoobscura. Genetics 1946, 31, 269–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi J. C. Synthetic lethality and semi-lethality among functionally related mutants of Drosophila melanfgaster. Genetics 1968, 59, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser C. A.; Schekman R. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell 1990, 61, 723–733. 10.1016/0092-8674(90)90483-U. [DOI] [PubMed] [Google Scholar]
- Hennessy K. M.; Lee A.; Chen E.; Botstein D. A group of interacting yeast DNA replication genes. Genes Dev. 1991, 5, 958–969. 10.1101/gad.5.6.958. [DOI] [PubMed] [Google Scholar]
- Kaelin W. G. Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- Gupta A.; Ahmad A.; Dar A. I.; Khan R. Synthetic Lethality: From research to precision cancer nanomedicine. Curr. Cancer Drug Targets 2018, 18, 337–346. 10.2174/1568009617666170630141931. [DOI] [PubMed] [Google Scholar]
- Chan D. A.; Giaccia A. J. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat. Rev. Drug Discovery 2011, 10, 351–364. 10.1038/nrd3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beijersbergen R.; Wessels L.; Bernards R. Synthetic lethality in cancer therapeutics. Annu. Rev. Cancer Biol. 2017, 1, 141–161. 10.1146/annurev-cancerbio-042016-073434. [DOI] [Google Scholar]
- Chen E. Targetin epigenetics using synthetic lethality in precision medicine. Cell. Mol. Life Sci. 2018, 75, 3381–3392. 10.1007/s00018-018-2866-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth A.; Lord C. J. Synthetic lethal therapies for cancer: what’s next after PARP inhibitors?. Nat. Rev. Clin. Oncol. 2018, 15, 564–576. 10.1038/s41571-018-0055-6. [DOI] [PubMed] [Google Scholar]
- Parameswaran S.; Kundapur D.; Vizeacoumar F. S.; Freywald A.; Uppalapati M.; Vizeacoumar F. J. A road map to personalizing targeted cancer therapies using synthetic lethality. Trends Cancer 2019, 5, 11–29. 10.1016/j.trecan.2018.11.001. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee S.; Nandi S. Synthetic lethality in DNA repair network: a novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 2017, 69, 929–937. 10.1002/iub.1696. [DOI] [PubMed] [Google Scholar]
- Minchom A.; Aversa C.; Lopez J. Dancing with the DNA damage response: next-generation anti-cancer therapeutic strategies. Ther. Adv. Med. Oncol. 2018, 10, 1–18. 10.1177/1758835918786658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilie P. G.; Tang C.; Mills G. B.; Yap T. A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. 10.1038/s41571-018-0114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Deeks E. D. Olaparib: first global approval. Drugs 2015, 75, 231–240. 10.1007/s40265-015-0345-6. [DOI] [PubMed] [Google Scholar]
- www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm592357.htm (accessed Sep 15, 2019).
- Golan T.; Hammel P.; Reni M.; Van Cutsem E.; Macarulla T.; Hall M. J.; Park J.; Hochhauser D.; Arnold D.; Oh D.; Reinacher-Schick A.; Tortora G.; Algul H.; O’Reilly E. M.; McGuinness D.; Cui K. Y.; Schlienger K.; Locker G. Y.; Kindler H. L. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 2019, 381, 317–327. 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A. A.; Masson J. Y.; McIlwraith M. J.; Stasiak A. Z.; Stasiak A.; Venkitaraman A. R.; West S. C. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol. Cell 2001, 7, 273–282. 10.1016/S1097-2765(01)00175-7. [DOI] [PubMed] [Google Scholar]
- Klein H. L. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair 2008, 7, 686–693. 10.1016/j.dnarep.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning W.; Sturzbecher H. W. Homologous recombination and cell cycle checkpoints: Rad51 in tumour progression and therapy resistance. Toxicology 2003, 193, 91–109. 10.1016/S0300-483X(03)00291-9. [DOI] [PubMed] [Google Scholar]
- Yanagisawa T.; Urade M.; Yamamoto Y.; Furuyama J. Increased expression of human DNA repair genes, XRCC1, XRCC3 and RAD51, in radioresistant human KB carcinoma cell line N10. Oral Oncol. 1998, 34, 524–528. 10.1016/S1368-8375(98)00045-1. [DOI] [PubMed] [Google Scholar]
- Pellegrini L.; Yu D. S.; Lo T.; Anand S.; Lee M.; Blundell T. M.; Venkitaraman A. R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 2002, 420, 287–293. 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- Bork P.; Blomberg N.; Nilges M. Internal repeats in the BRCA2 protein sequence. Nat. Genet. 1996, 13, 22–23. 10.1038/ng0596-22. [DOI] [PubMed] [Google Scholar]
- Wong A. K.; Pero R.; Ormonde P. A.; Tavtigian S. V.; Bartel P. L. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J. Biol. Chem. 1997, 272, 31941–31944. 10.1074/jbc.272.51.31941. [DOI] [PubMed] [Google Scholar]
- Bignell G.; Micklem G.; Stratton M. R.; Ashworth A.; Wooster R. The BRC repeats are conserved in mammalian BRCA2 proteins. Hum. Mol. Genet. 1997, 6, 53–58. 10.1093/hmg/6.1.53. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Zhou L.; Wu G.; Konig H.; Lin X.; Li G.; Qiu X.; Chen C.; Hu C.; Goldblatt E.; Bhatia R.; Chamberlin A. R.; Chen P.; Lee W. A novel small molecule RAD51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia. EMBO Mol. Med. 2013, 5, 353–365. 10.1002/emmm.201201760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. H.; Chen P. L.; Zhu J.. Composition and Methods for the Disruption of BRCA2-RAD51 Interaction. PCT Int. Appl. WO 2006/044933, 2006
- Falchi F.; Giacomini E.; Masini T.; Boutard N.; Di Ianni L.; Manerba M.; Farabegoli F.; Rossini L.; Robertson J.; Minucci S.; Pallavicini I.; Di Stefano G.; Roberti M.; Pellicciari R.; Cavalli A. Synthetic lethality triggered by combining olaparib with BRCA2-Rad51 disruptors. ACS Chem. Biol. 2017, 12, 2491–2497. 10.1021/acschembio.7b00707. [DOI] [PubMed] [Google Scholar]
- Roberti M.; Schipani F.; Bagnolini G.; Milano D.; Giacomini E.; Falchi F.; Balboni A.; Manerba M.; Farabegoli F.; De Franco F.; Robertson J.; Minucci S.; Pallavicini I.; Di Stefano G.; Girotto S.; Pellicciari R.; Cavalli A. Rad51/BRCA2 disruptors inhibit homologous recombination and synergize with olaparib in pancreatic cancer cells. Eur. J. Med. Chem. 2019, 165, 80–92. 10.1016/j.ejmech.2019.01.008. [DOI] [PubMed] [Google Scholar]
- Scott D. E.; Bayly A. R.; Abell C.; Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discovery 2016, 15, 533–550. 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- Rajendra E.; Venkitaraman A. R. Two modules in the BRC repeats of BRCA2 mediate structural and functional interactions with the RAD51 recombinase. Nucleic Acids Res. 2010, 38, 82–96. 10.1093/nar/gkp873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomme J.; Takizawa Y.; Martinez S. F.; Renodon-Corniere A.; Fleury F.; Weigel P.; Yamamoto K.; Kurumizaka H.; Takahashi M. Inhibition of filament formation of human Rad51 protein by a small peptide derived from the BRC-motif of the BRCA2 protein. Genes Cells 2008, 13, 471–481. 10.1111/j.1365-2443.2008.01180.x. [DOI] [PubMed] [Google Scholar]
- Nomme J.; Renodon-Corniere A.; Asanomi Y.; Sakaguchi K.; Stasiak A. Z.; Stasiak A.; Norden B.; Tran V.; Takahashi M. Design of potent inhibitors of human RAD51 recombinase based on BRC motifs of BRCA2 protein: modeling and experimental validation of a chimera peptide. J. Med. Chem. 2010, 53, 5782–5791. 10.1021/jm1002974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acker T. M.; Khatri A.; Vance K. M.; Slabber C.; Bacsa J.; Snyder J. P.; Traynelis S. F.; liotta D. C. Structure-activity relationships and pharmacophore model of a noncompetitive pyrazoline containing class of GluN2C/GluN2D selective antagonists. J. Med. Chem. 2013, 56, 6434–6456. 10.1021/jm400652r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran M.; Uvarani C.; Chandraprakash K.; Umamaheswari M.; Mohan P. S. An Expedient Approach for the Synthesis of Bioactive Pyrazole, Isoxazole and Benzodiazepine-Substituted Quinolin-2(1H)-one Derivatives. J. Heterocyclic Chem. 2015, 52, 1082–1092. 10.1002/jhet.2192. [DOI] [Google Scholar]
- Abbott D. W.; Freeman M. L.; Holt J. T. Double-strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. J. Natl. Cancer. Inst. 1998, 90, 978–985. 10.1093/jnci/90.13.978. [DOI] [PubMed] [Google Scholar]
- Cummings B. S.; Schnellmann R. G. Measurement of cell death in mammalian cells. Curr. Protoc. Pharmacol. 2004, 25, 12.8.1–12.8.22. 10.1002/0471141755.ph1208s25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.