

Abstract
Thiazoles, their benzofused systems, and thiazolidinone derivatives are widely recognized as nuclei of great value for obtaining molecules with various biological activities, including analgesic, anti-inflammatory, anti-HIV, antidiabetic, antitumor, and antimicrobial. In particular, in the past decade, many compounds bearing these heterocycles have been studied for their promising antibacterial properties due to their action on different microbial targets. Here we assess the recent development of this class of compounds to address mechanisms underlying antibiotic resistance at both bacterial-cell and community levels (biofilms). We also explore the SAR and the prospective clinical application of thiazole and its benzofused derivatives, which act as inhibitors of mechanisms underlying antibiotic resistance in the treatment of severe drug-resistant infections. In addition, we examined all bacterial targets involved in their antimicrobial activity reporting, when described, their spontaneous frequencies of resistance.
1. Introduction
Antibiotic resistance (AMR) is among the most relevant health problems of this century. At least 700 000 people die of no longer treatable infections every year in the world.1
AMR crisis involves any class of antibiotic including the second- and third-line antibiotics once considered the last resort drugs to tackle common infections. We are losing our ability to keep under control many common infections; consequently, one of the pillars on which modern medicine is based threatens to collapse.
The phenomenon of AMR at biochemical and physiological levels may manifest in any single bacterial cell (planktonic growth) or in a sessile complex microbial community (biofilm).2 Bacteria might become resistant to antibiotics at the cellular level, and bacteria can become resistant to antibiotics by inactivating or altering the molecular structure of an antibiotic, modifying the antibiotic target, and decreasing the intracellular drug concentration by expressing efflux pumps. These biochemical mechanisms are present in any single bacterial cell embedded in the biofilm structure too, however, a set of adaptive and intrinsic mechanisms due to growth as a community contribute to making biofilms up to a thousand times more resistant than single bacterial cells.3
Within a global strategy for fighting antibiotic resistance, in 2005, the World Health Organization (WHO) published for the first time a list called Critical Important Antimicrobials (CIA). This list includes antibiotics and antimicrobials widely used in the world, divided in three categories of clinical importance (critically important, divided in highest priority and high priority; highly important; important) with the scope of preserving the clinical use and the rational employment of these molecules. A recent recommendation of the Interagency Coordination Group on Antimicrobial Resistance (IACG) of the United Nations is to stop the use of antibiotics categorized as the highest priority critically (i.e., third and higher generation cephalosporins, macrolide, quinolones and fluoroquinolones, glycopeptides, macrolides and ketolides, polymyxins) in nonclinical settings (farms, food, and feed production).4
There are indeed many scientists who fear that the world is going toward a “post-antibiotic era” where common infections, which were easily cured, could become chronic or fatal. As a consequence, about 10 million deaths caused by infectious diseases treatment failure could be reached in 2050.5
In this scenario, the development of new small molecules able to counteract the commonest mechanisms underlying antibiotic resistance including enzymatic inactivation of antibiotics, alterations in cell penetrability, efflux pumps activity or biofilm formation is urgently required.6,7
Many heterocyclic compounds have been synthesized in the past decade in the attempt to obtain new antimicrobials, which would be able to treat infections caused by resistant bacterial strains. Herein we focused on synthetic molecules bearing the thiazole, benzothiazole, and thiazolidinone scaffolds, which are widely recognized as nuclei of great value for obtaining molecules endowed with various biological activities,8 including analgesic,9 anti-inflammatory,9 antidiabetic,10 antitumor,11 and antimicrobial.12
Sulfur containing compounds such as thiazole, compared to other five-membered heterocycles such as oxazole and imidazole, possess unique features due to the low lying C–S σ* orbitals that, conferring small regions of low electron density on sulfur (σ-holes), may play a role in drug–target interactions.13 The presence of sulfur exerts also significant effect on bond angles and the topology of substituents, it being associated with longer lengths and smaller bond angles. Moreover, thiazole ring systemshows cLogP and cLogD values near to 0.5 and pKa and pKBHX (the latter related to the H-bond interaction properties) of 2.53 and 1.37, respectively. Although the benzo-fusion of thiazoles causes a significant reduction of pKa values, this modification has a limited effect on pKBHX values (Table 1).14
Table 1. Chemical-Physical Properties of Thiazole, Imidazole, and Oxazole Scaffolds.

In particular, we assess the recent development (2008–2019) of this class of compounds in the treatment of antibiotic resistance mechanisms at both bacterial cell and community levels. We also review the SAR and the potential clinical implementation of thiazole and benzothiazole with antibacterial properties in the therapies for severe drug-resistant infections.
2. DNA Gyrase and Topoisomerase IV Inhibitors
To obtain more potent therapeutic strategies, many targets were investigated, among them, type II topoisomerase enzymes, which include bacterial DNA gyrase and topoisomerase IV. These enzymes were discovered in 1976 in the Gram-negative pathogen Escherichia coli by Gellert and co-workers and are at the center of many studies because of their commercial success with the development of the fluoroquinolones.15 Gyrase maintains negative supercoiling of the bacterial chromosome, while topoisomerase IV is responsible for untangling daughter chromosomes. Type II topoisomerases are crucial enzymes for maintaining DNA integrity during the replication playing key roles in DNA replication, transcription, and repair, recombination, and transposition.16 One of the most important advantage to consider the bacterial type II topoisomerases as targets for the design of novel therapeutic classes of antibiotics is that they are highly conserved enzymes in bacterial strains and therefore their inhibition could lead to a broad spectrum activity.17
Fluoroquinolones represent an important class of dual inhibitors of bacterial type II topoisomerases. Despite their clinical success in the past, their use is now limited by growing antibiotic resistance, which strongly compromised their effectiveness. Both for DNA gyrase and topoisomerase IV, fluoroquinolones, interacting with the enzyme-bound DNA complex, determine conformational modifications preventing the assembly of DNA strands that impede the normal enzyme activity. This causes the inhibition of DNA synthesis and the consequent death of the bacterial cell.
Several searches have been carried out in order to obtain new drugs able to inhibit both enzymes, DNA gyrase and topoisomerase IV, reducing the probability that bacteria develop antibiotic resistance mechanisms.18 A special effort must be made in identifying selective inhibitors toward the bacterial form which are inactive against the eukaryotic type II topoisomerases. This desirable selectivity can be achieved because there are distinct structural differences from the homodimeric mammalian enzyme counterparts.
Structurally, DNA gyrase is composed of two heterodimeric subunits GyrA and two GyrB in complex, whereas topoisomerase IV consists of two ParC and two ParE subunits.
With the aim of obtaining specific inhibitors of GyrB, which is considered an interesting target to overcome the cross-resistance to quinolones, Ronkin and collaborators performed a high-throughput screen. This screen identified compound 1 (Table 2), which showed an inhibitory activity against GyrB in E. coli with a Ki value of 2.9 μM.19,20 The X-ray crystal structure of this derivative inside the GyrB active site of Staphylococcus aureus allowed the identification of structural features responsible for the interactions with the enzyme, guiding the synthesis of the analogues (1a–d and 2a–y). It was observed that (i) the pyrazole moiety was involved in a hydrogen bond with Asp81 and a highly conserved structural water molecule. (ii) the presence in position 2 of the thiazole ring of a 3-pyridyl moiety (2e), assuring an additional hydrogen bond with Arg136, improved the activity by 5–10-fold compared to 1a and 1d (Figure 1), (iii) the substituent at the 4 position of the thiazole ring affected the activity of the compound, substituents larger than methyl should be tolerated and lipophilic groups gave better results, (iv) the position 5 of the pyrazole ring can be substituted with carboethoxy, amide, or carbamate groups. In particular, the ethyl amide compounds are twice as potent as the corresponding ethyl esters. Another key feature is the presence of a carbamate group, which was able to form an additional hydrogen bond with Asn46, as observed in derivatives 2t–y (Table 2), which were the most potent in term of enzymatic inhibition showing Ki values in the range 0.040–3.4 μM. Despite their interesting activity against GyrB, carbamates 2t–y did not show antibacterial activity against wild-type Escherichia coli strains as well as S. aureus ATCC 29213 and Streptococcus pneumoniae ATCC 10015; this is probably due to the efflux pumps antimicrobial resistance mechanism.
Table 2. Chemical Structures and E. coli GyrB Inhibitory Activity of Compounds 1, 1a–d, and 2a–y.
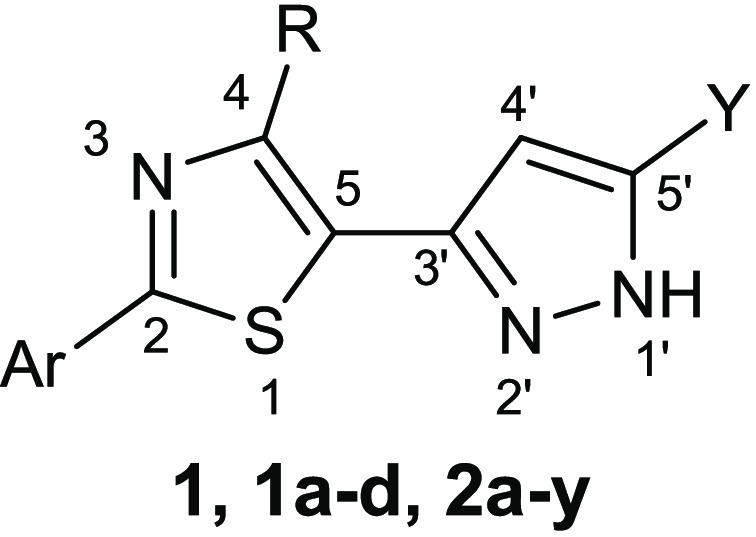

Figure 1.
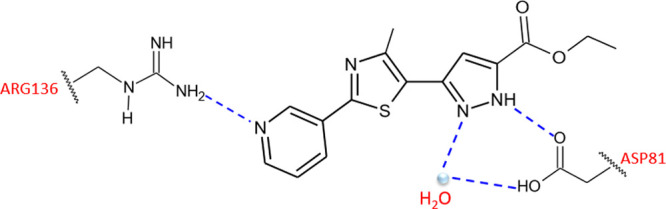
Schematic representation of the binding mode in GyrB active site of S. aureus of compound 2e. Hydrogen bonds are represented as blue dashed line.
In an attempt to discover innovative inhibitors of both GyrB and ParE, Stokes and collaborators synthesized two benzothiazole ethyl urea compounds 3a,b (Figure 2), which potently inhibited in vitro the ATPase activity of E. coli DNA gyrase and topoisomerase IV, eliciting IC50s in the range from 0.0033 to 0.046 μg/mL.21 Derivatives 3a,b were extremely more potent against topoisomerase enzymes than the reference drug novobiocin. Moreover, differently from compounds 1 and 2, they showed a significant antibacterial activity against a broad range of Gram-positive/negative pathogens with MIC values of 0.008, 0.03, and 0.06 μg/mL against S. pneumoniae ATCC 49619, Staphylococcus epidermidis ATCC 1228, and Streptococcus pyogenes ATCC 51339, respectively.
Figure 2.

Chemical structures of compounds 3a,b, 4, 5, and chlorobiocin (6).
Compounds 3a,b were also tested for evaluating their spontaneous frequencies of resistance (FoRs). It is known that antibacterial compounds acting on a single bacterial target show FoRs in the range 10–6 to 10–9,22 conversely, the FoR values for the two benzothiazole ethyl urea derivatives 3a,b against S. aureus ATCC 29213 were <2.3 × 10–10 and <2.3 × 10–11, respectively, at concentrations 8-fold the MIC. This behavior seems to be due to the capability of the compounds 3a,b of simultaneously inhibiting two intracellular targets, i.e., GyrB and ParE. Additionally, these compounds proved to be not toxic when assayed against the HepG2 human liver cell line.
It was observed that the presence of a cyclic amine between the carboxylate and the C-5 aryl group enhanced the antibacterial activity and the solubility compared to the derivatives without this group.23 Further improvement in the PK profile was obtained with the inclusion of an α substituent to the carboxylic acid, which showed enhanced solubility, excellent oral bioavailability, and lower clearance. Remarkably, compounds 3a,b had powerful inhibitory activity against S. aureus topoisomerase IV (0.012 and 0.008 μg/mL, respectively), proving their selectivity toward the bacterial isoform without affecting human topoisomerase II.
Most of the known bacterial topoisomerase inhibitors have been identified with the aid of computational techniques such as virtual screenings and docking studies in the active sites.24
Brvar and collaborators employed the crystal structure of the N-terminal portion of the B subunit of DNA gyrase, the G24 protein (PDB 1KZN), cocrystallized with the natural inhibitor, chlorobiocin (6) (Figure 2) to generate a structure-based pharmacophore model.25 Twelve features involved in the interaction between the inhibitor and the enzyme were identified: (i) the presence of two hydrogen bond donors, (ii) two hydrogen bond acceptors, (iii) an aromatic ring, and (iv) seven hydrophobic portions. This pharmacophore was successfully validated by assessing its ability to recognize the bioactive conformation of 6, and then was used to perform a large scale virtual screening of 5 million compounds, which allowed the identification of 400 new hit compounds belonging to different classes.
Further in silico studies, including a docking calculation for the potential interactions of the G24 active-site with the binding site for ATP, resulted in the identification of new strong antigyrase agents with 4,5′-bithiazole-2,2′-diamine structure. Among these agents, compound 4 (Figure 2) had the most promising DNA gyrase inhibitory activity displaying an IC50 of 5.5 μM.
Subsequently, a commercially available library of 240 4′-methyl-N2-phenyl-[4,5′-bithiazole]-2,2′-diamines was docked into the G24 ATP-binding site, using the software GOLD. This study focused on compounds capable to bind the previously selected residues Asp73, Thr165 through W1001 water molecule and Arg136. Eight compounds were selected to be examined for their inhibitory activity on DNA gyrase inhibitory activity, and the bithiazole 5 (Figure 2) emerged for its potency, showing an IC50 of 1.1 μM. The binding mode of this derivative was confirmed by X-ray crystallography (Figure 3), and its structure has been reported in the RCSB Protein Data Bank (PDB 4DUH).25 Despite their structural diversity, compound 5 and the coumarin inhibitor chlorobiocin (6) showed a very similar binding pattern. As observed for 6, the bithiazole 5 formed two hydrogen bonds with (1) Asp73, through the 2′-propionylamido group, which was oriented in the active site as the 5-methylpyrrole ring of 6, and (2) the conserved water molecules that were coordinated with the residues Asp73, Gly77, and Thr165. The propionyl moiety participated also to the hydrophobic interactions with the binding pocket made by Val43, Val71, Val120, and Val 167. This is the same binding pocket, occupied by the methylpyrrole group of 6. Additionally, the second thiazole ring was oriented in a manner analogous to the methyl groups of the sugar part of 6 and formed hydrophobic interactions with the residues Ile78, Ile90, and Val120. The 2-aminothiazole moiety seems essential in the DNA gyrase inhibition as it contains an acceptor–donor interaction pattern, which is a fundamental requirement for this activity. Similarly, the presence of a hydrogen bond acceptor on the para position of the phenyl ring seems to play a key role in the enzymatic activity.
Figure 3.
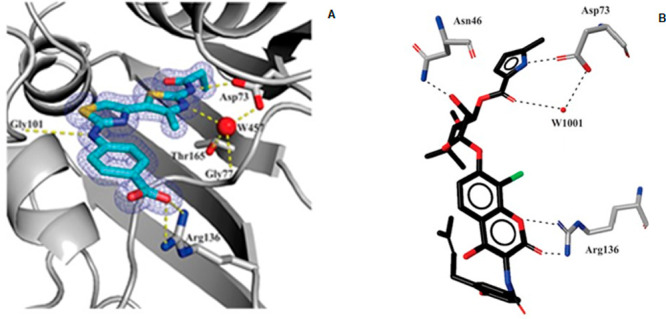
(A) Bithiazole 5 in G24 active site. Hydrogen bonds are designed as dotted lines; electronic density, contoured at 2σ, is reported as meshed net. (B) Chlorobiocin (6) binding mode into the G24 ATP-binding.25Reproduced with permission from ref (25). Copyright 2012 American Chemical Society.
An important class of gyrase inhibitors is constituted by marine alkaloids, which are widely recognized as molecules of great value in terms of biological properties and many derivatives in the past decade have been successfully developed for different therapeutic purposes.26−28
However, the marine alkaloids oroidin analogues have attracted great attention for their significant antibacterial and antibiofilm activities.29−32
Performing a virtual screening of a library of oroidin analogues employing the E. coli GyrB crystal structure (PDB 4DUH), Tomǎsič and co-workers identified a 4,5,6,7-tetrahydrobenzo[1,2-d]thiazole derivative 7a (Table 3) able to inhibit E. coli DNA gyrase with an IC50 of 12 μM.33 To better understand how to optimize the structure of the compound 7a in order to obtain more potent inhibitors, its binding mode in the ATP-binding site of E. coli GyrB (PDB 4DUH) was investigated. The 4,5-dibromo-1H-pyrrole-2-carboxamide moiety is involved in the interaction with the hydrophobic pocket and is able to make hydrogen bonds with the Asp73 side chain as well as with the conserved water molecule, miming ATP interactions with the same residue, whereas the position of the 2-amino group on the thiazole ring allows substitution with functional groups, such as oxalyl, malonyl, and succinyl (Figure 4), for interaction with Arg76 and/or Arg136.
Table 3. Inhibitory Activity of E. coli and S. aureus DNA Gyrase and Topoisomerase IV by the 4,5,6,7-Tetrahydrobenzo[1,2-d]thiazoles Containing the 4,5-Dibromo-1H-pyrrole (7a–h), 4,5-Dichloro-1H-pyrrole Moiety (7i–n), or 1H-Indole Moiety (7o–s).

| DNA
gyrase IC50 (Kda) [μM] or RA [%]b |
topoisomerase
IV IC50 (Kda) [μM] or RA [%]b |
E.
coli MIC [μg/mL] |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| compd | * | R | X | n | E. coli | S. aureus | E. coli | S. aureus | wt | tolC | impA |
| negc | 100% | 100% | 100% | 100% | |||||||
| NBd | 0.17 μM | 0.040 μM | 11 μM | 27 μM | |||||||
| 7a | S | 12 μM | 90% | 101% | 102% | >256 | 64 | 64 | |||
| 7b | R | 47 μM | 118% | 96% | 92% | >256 | 64 | 64 | |||
| 7c | S | OC2H5 | Br | 0 | 0.10 μM | 80 μM | 74% | 180 μM | >256 | 128 | >256 |
| 7d | S | OCH3 | Br | 1 | 0.096 μM | 110 μM | 86 μM | 74% | >256 | 16 | 32 |
| 7e | S | OCH3 | Br | 2 | 0.093 μM | 113% | 97% | 99% | >256 | 32 | 64 |
| 7f | S | OH | Br | 0 | 0.058 μM | 120 μM | 200 μM | 78 μM | >256 | 256 | >256 |
| 7g | S | OH | Br | 1 | 0.069 μM | 86 μM | 74 μM | 76 μM | >256 | 256 | >256 |
| 7h | S | OH | Br | 2 | 0.049 μM | 270 μM | 90% | 110 μM | >256 | 256 | >256 |
| 7i | OC2H5 | Cl | 0 | 0.40 μM | 320 μM | 300 μM | 290 μM | >256 | 256 | >256 | |
| 7j | OCH3 | Cl | 1 | 0.40 μM | 63% | 97% | 101% | >256 | 32 | 32 | |
| 7k | OCH3 | Cl | 2 | 0.89 μM | 86% | 100% | 93% | >256 | >256 | >256 | |
| 7l | OH | Cl | 0 | 0.59 μM | 300 μM | 82% | 170 μM | >256 | >256 | >256 | |
| 7m | OH | Cl | 1 | 0.13 μM | 87% | 98% | 102% | >256 | >256 | >256 | |
| 7n | OH | Cl | 2 | 0.30 μM | 10 μM | 72% | 97% | >256 | >256 | >256 | |
| 7o | OC2H5 | 0 | 10 μM | 76% | 101% | 99% | nt | nt | nt | ||
| 7p | OCH3 | 2 | 9.1 μM | 87% | 101% | 104% | nt | nt | nt | ||
| 7q | OH | 0 | 7.7 μM | 79% | 47% | 99% | >256 | >256 | 128 | ||
| 7r | OH | 1 | 7.6 μM | 117% | 96% | 103% | >256 | >256 | >256 | ||
| 7s | OH | 2 | 8.1 μM | 95% | 99% | 79% | >256 | >256 | >256 | ||
K determined by surface plasmon resonance.
Residual activity of the enzyme at 100 μM concentration of the tested compound. nt: not tested.
Figure 4.
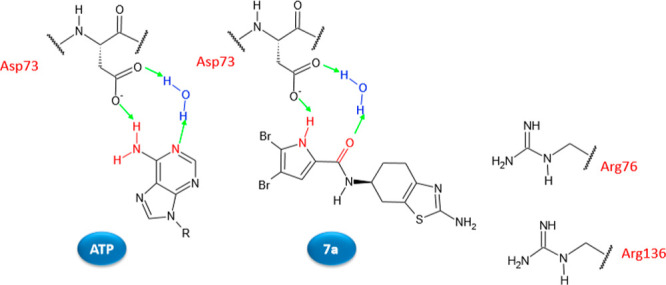
Schematic representation of the binding mode in the E. coli GyrB ATP-binding site of ATP and compound 7a. Hydrogen bonds are represented as green arrows.
Many analogues were prepared to get more insights into the SAR of the 4,5,6,7-tetrahydrobenzo[1,2-d]thiazole class, as well as to improve the affinity toward the enzyme. These studies also evaluated the effect of the replacement of the 4,5-dibromo-1H-pyrrole with the 4,5-dichloro-1H-pyrrole or 1H-indole moieties. In addition, experiments with the (R)-isomer (7b) of compound 7a assessed the influence of chirality on enzymatic inhibition.
All the synthesized compounds were tested against E. coli DNA gyrase. Then the compounds with IC50s lower than 50 μM were evaluated for their inhibitory activities against S. aureus DNA gyrase as well as E. coli and S. aureus topoisomerases IV (Table 3).
The substituents on the pyrrole ring play a pivotal role in the activity of these compounds. This might be explained by the hydrophobic interactions of the bromine atoms with the pocket of the ATP-binding site. The introduction of an oxalyl, malonyl, or succinyl group, as a hydrogen bond acceptor, led to derivatives (7c,d,e), as suggested by computational studies. These compounds enhanced the inhibition of E. coli DNA gyrase, with IC50s of 0.10, 0.096, and 0.093 μM, respectively, probably due to their capacity to interact with Arg136. The stereochemistry seems also to have an important role in determining activity (S-isomers, in fact, showed stronger inhibitory activity than the (R)-counterparts.
In the indole series (compounds 7o–s), the activity was maintained in the low micromolar range. Conversely, the substitution of the bromine with chlorine atoms on the pyrrole ring (compounds 7i–n) led to a decrease inpotency toward DNA gyrase as well as topoisomerase IV.
Unfortunately, despite the activity showed by compounds 7 in the enzymatic assay, no derivative of the series showed antibacterial activity at the concentration of 50 μM against the Gram-negative E. coli ATCC 25922, P. aeruginosa ATCC 27853, and the Gram-positive E. faecalis ATCC 29212 and S. aureus ATCC 25923 bacterial strains. Only compounds 7d against E. faecalis and 7e and 7j against S. aureus displayed percentages of growth inhibition higher than 50%.
To investigate if the lack of antibacterial effects against the Gram-negative pathogens of this class was ascribable to permeability problems or to efflux pumps, the 4,5,6,7-tetrahydrobenzo[1,2-d]thiazoles 7 were tested against permeabilized (impA) and efflux pump knockout (ΔtolC) strains of E. coli. However, among the tested compounds, only five derivatives, 7a, 7b, 7d, 7e, and 7j, showed better antibacterial effects toward impA and ΔtolCE. coli strains than the wild-type.
Even if DNA gyrase is a fundamental enzyme for the bacterial viability, often it was observed that its inhibition in a biochemical assay does not correspond to an antibacterial effect at the cellular level.
Structural modifications of the most active E. coli DNA gyrase inhibitor 7f, which showed an IC50 value of 58 nM, led to two series of nanomolar inhibitors 8a–e (Table 4) and 9a–g (Table 5).34,35 In particular, the replacement of the 4,5,6,7-tetrahydrobenzo[1,2-d]thiazole scaffold with a planar benzothiazole-2,6-diamine moiety and the move of the 4,5-dibromopyrrole-2-carbonyl group from N-6 to N-2 of the central core proved to be advantageous for the enzymatic activity. The most potent inhibitor was the derivative 8d, which displayed an IC50 of 38 nM (Table 4). However, even if the DNA gyrase inhibitory activity increased, no improvement in the antibacterial properties was observed, probably because efflux pumps inactivated these compounds. The compounds, in fact, proved to be significantly more potent toward the efflux deficient strain, ΔtolCE. coli than the wild type.
Table 4. Inhibition of E. coli and S. aureus DNA Gyrase and Topoisomerase IV by the Benzothiazole Compounds with the Pyrrole-2-carboxamido Moiety at Position 6 on the Benzothiazole Core (8a–e).

| DNA
gyrase IC50 [μM] |
topoisomerase
IV IC50 [μM] |
|||||
|---|---|---|---|---|---|---|
| compd | n | R | E. coli | S. aureus | E. coli | S. aureus |
| novobiocin | 0.17 | 0.040 | 11 | 27 | ||
| 8a | 0 | CH2CH3 | 0.081 ± 0.05 | >100 | >100 | >100 |
| 8b | 1 | CH3 | 0.24 ± 0.02 | >100 | >100 | >100 |
| 8c | 2 | CH3 | 0.87 ± 0.21 | >100 | >100 | >100 |
| 8d | 0 | H | 0.038 ± 0.001 | >100 | 5.4 ± 1.0 | 2.5 ± 0.2 |
| 8e | 2 | H | 0.057 ± 0.018 | >100 | 4.5 ± 0.7 | 2.2 ± 0.3 |
Table 5. Inhibition of E. coli and S. aureus DNA Gyrase and Topoisomerase IV by the Benzothiazole Compounds with the Pyrrole-2-carboxamido Moiety at Position 2 on the Benzothiazole Core (9a–g).


For the compounds of the series 9, the highest activity was reached against the E. coli DNA gyrase by the derivative 9d, which showed an IC50 value against this enzyme of 33 nM (Table 5).
Taking into account the interesting antibacterial properties described for benzothiazole and thiazolidinone scaffolds, Haroun and collaborators synthesized a new series of compounds 10a–l (Table 6), bearing both bioactive nuclei. All compounds were tested for their activity against the Gram-negative E. coli (ATCC 35210), Enterobacter cloacae, Pseudomonas aeruginosa (ATCC 27853), Salmonella typhimurium (ATCC 13311) pathogens, as well as against the Gram-positive Listeria monocytogenes (NCTC 7973), Bacillus cereus (clinical isolate), Micrococcus flavus (ATCC 10240), and S. aureus (ATCC 6538). All of these novel derivatives showed good antibacterial potency, proving to be more active than reference drugs, such as ampicillin and streptomycin, with MIC values in the range of 0.0018–0.28 μmol/mL and MBC values in the range of 0.0036–0.37 μmol/mL. An initial hypothesis on the mode of action of these compounds came from an evaluation of docking studies conducted on topoisomerase II DNA gyrase (1KZN) and E. coli Mur B (2Q85) (Figure 5A,B), showing that these compounds were capable of binding to the active sites of both enzymes, with a specific affinity toward MurB. In particular, the best binding score and binding energy values toward both enzymes were found for compound 10a, which interacted with the binding site of topoisomerase II DNA gyrase through two hydrogen bonds, between the nitrogen atom of the central chain and the Thr165 and between the keto group of thiazolidinone ring and Asn46. Moreover, the phenyl ring is involved in hydrophobic interactions with Val71, Val167, Ala47, and Val43. The greater affinity toward MurB observed for derivative 10a was due to the ability of this compound to form four hydrogen bonds with the active site, two with Gly123, and the other two with Arg124 and Tyr190. In addition, the benzothiazole scaffold formed hydrophobic interactions with Val129, Leu218, and Ala124. MurB is a UDP-N-acetylenolpyruvoylglucosamine reductase, which leads the catalysis the second committed reaction in the biosynthesis of peptidoglycan, and therefore it is fundamental for bacterial growth. However, assays on the enzyme were not conducted to confirm the computationally obtained data, therefore the target of this series remains unknown. Additionally, it should be useful to investigate the potential of compounds 10a–l as pan assay interference compounds (PAINS), because many subclasses of aminothiazoles were classified in this category and, therefore, could give false positive results.36
Table 6. Chemical Structures and Antibacterial Activity of Compounds 10a–l.

| MIC
values (MBC values) μmol/mL ×
102 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | R | B. cereus | M. flavus | S. aureus | L. monocytogenes | E. coli | E. cloacae | P. aeruginosa | S. typhimurium |
| 10a | H | 1.49 (2.97) | 2.97 (5.94) | 1.19 (1.49) | 1.49 (2.97) | 0.23 (2.97) | 1.49 (2.97) | 4.45 (5.94) | 1.49 (2.97) |
| 10b | 4-OH | 4.90 (9.80) | 9.80 (19.6) | 4.90 (9.80) | 4.90 (9.8) | 9.80 (19.6) | 4.90 (9.80) | 4.90 (9.80) | 4.90 (9.80) |
| 10c | 4-OCH3 | 4.80 (19.2) | 9.60 (19.2) | 4.80 (9.60) | 9.60 (19.2) | 14.4 (19.2) | 7.20 (9.60) | 9.60 (19.2) | 4.80 (9.60) |
| 10d | 4-OH; 3-OCH3 | 4.60 (9.20) | 13.9 (18.6) | 9.20 (37.3) | 18.6 (37.3) | 13.9 (18.6) | 6.90 (9.20) | 9.20 (37.3) | 3.45 (4.60) |
| 10e | 2-Cl | 4.80 (9.60) | 9.60 (19.2) | 4.80 (9.60) | 9.60 (19.2) | 9.60 (19.2) | 4.80 (9.60) | 9.60 (19.2) | 4.80 (9.60) |
| 10f | 3-Cl | 4.60 (9.20) | 4.60 (9.20) | 4.60 (9.20) | 4.60 (9.20) | 0.18 (0.36) | 4.60 (9.20) | 4.60 (9.20) | 4.60 (9.20) |
| 10g | 4-Cl | 4.80 (19.2) | 9.60 (19.2) | 4.80 (9.60) | 9.60 (19.2) | 9.60 (19.2) | 4.80 (9.60) | 9.60 (19.2) | 2.40 (4.80) |
| 10h | 2-NO2 | 9.10 (18.2) | 9.10 (18.2) | 9.10 (18.2) | 4.50 (9.10) | 9.10 (18.2) | 4.50 (9.10) | 4.50 (9.10) | 9.10 (18.2) |
| 10i | 3-NO2 | 14.0 (18.4) | 18.4 (36.8) | 14.0 (18.4) | 18.4 (36.8) | 23.3 (36.8) | 23.3 (36.8) | 18.4 (36.8) | 4.60 (9.20) |
| 10j | 4-NO2 | 0.58 (1.16) | 9.20 (18.4) | 0.58 (1.16) | 9.20 (18.4) | 9.20 (18.4) | 2.30 (9.20) | 9.20 (18.4) | 0.58 (1.16) |
| 10k | 4-OH; 3,5-OCH3 | 8.80 (17.6) | 8.80 (17.6) | 4.40 (8.80) | 2.20 (8.80) | 8.80 (17.6) | 4.40 (8.80) | 2.20 (8.80) | 2.20 (8.80) |
| 10l | 4-OH; 3-OCH3; 5-I | 7.20 (28.8) | 7.20 (14.4) | 3.60 (7.20) | 28.8 (36.0) | 7.20 (14.4) | 3.60 (14.4) | 14.4 (28.8) | 7.20 (14.4) |
Figure 5.
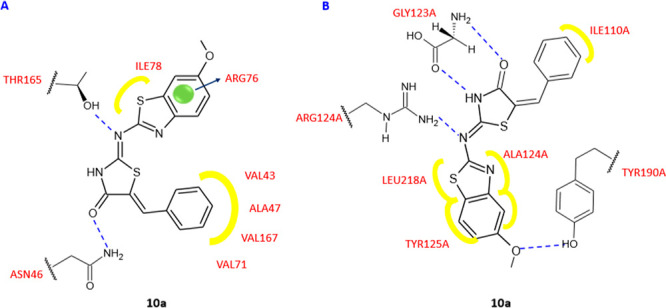
Binding mode of compound 10a in the topoisomerase II DNA gyrase active site (1KZN) (A) and binding mode of compound 10a in the E. coli Mur B active site (2Q85) (B). Yellow, van der Waals interactions; blue dashed line, hydrogen bond; blue arrow, π interaction.
Azoalkyl ether imidazo[2,1-b]benzothiazoles 11a–j (Table 7) were described as antibacterial drugs with a wide spectrum of activity against both Gram-positive and Gram-negative pathogens, showing in some cases higher potency than the reference drugs chloromycin (12) and norfloxacin (13).37 In particular, compound 11d with a propyl linker was up to 8-fold more effective against methicillin-resistant S. aureus (MRSA) and S. typhi eliciting a MIC values of 2 and 1 μg/mL, respectively. To evaluate the capability of derivative 11d to stimulate bacterial resistance, a time-kill kinetic experiment was performed on MRSA. The results highlighted an instant bactericidal effect against MRSA without effects on the onset of antibiotic resistance. The possible mechanism of action was investigated through (i) a docking study aiming to evaluate the interaction of derivative 11d with the binding site of S. aureus DNA gyrase (PDB 2XCS) and (ii) a UV–vis spectroscopic analysis to clarify the binding mode with MRSA DNA. The computational study disclosed the possibility of compound 11d to interact with the complex of gyrase–DNA by π–π stacking between the two carbonyl groups of the imidazo[2,1-b] benzothiazole backbone and base pairs DG-11 and DG-10 of DNA. The nitrogen atom of the azole cycle was able to form a hydrogen bond with Met1121, whereas the nitro group bond to residue Asp1083 (Figure 6). Additionally, UV–vis spectroscopy showed that compound 11d is capable to enter into MRSA–DNA, preventing the replication process of DNA.
Table 7. Chemical Structures and Antibacterial Activity of Compounds 11a–j, Chloromycin (12), and Norfloxacin (13).

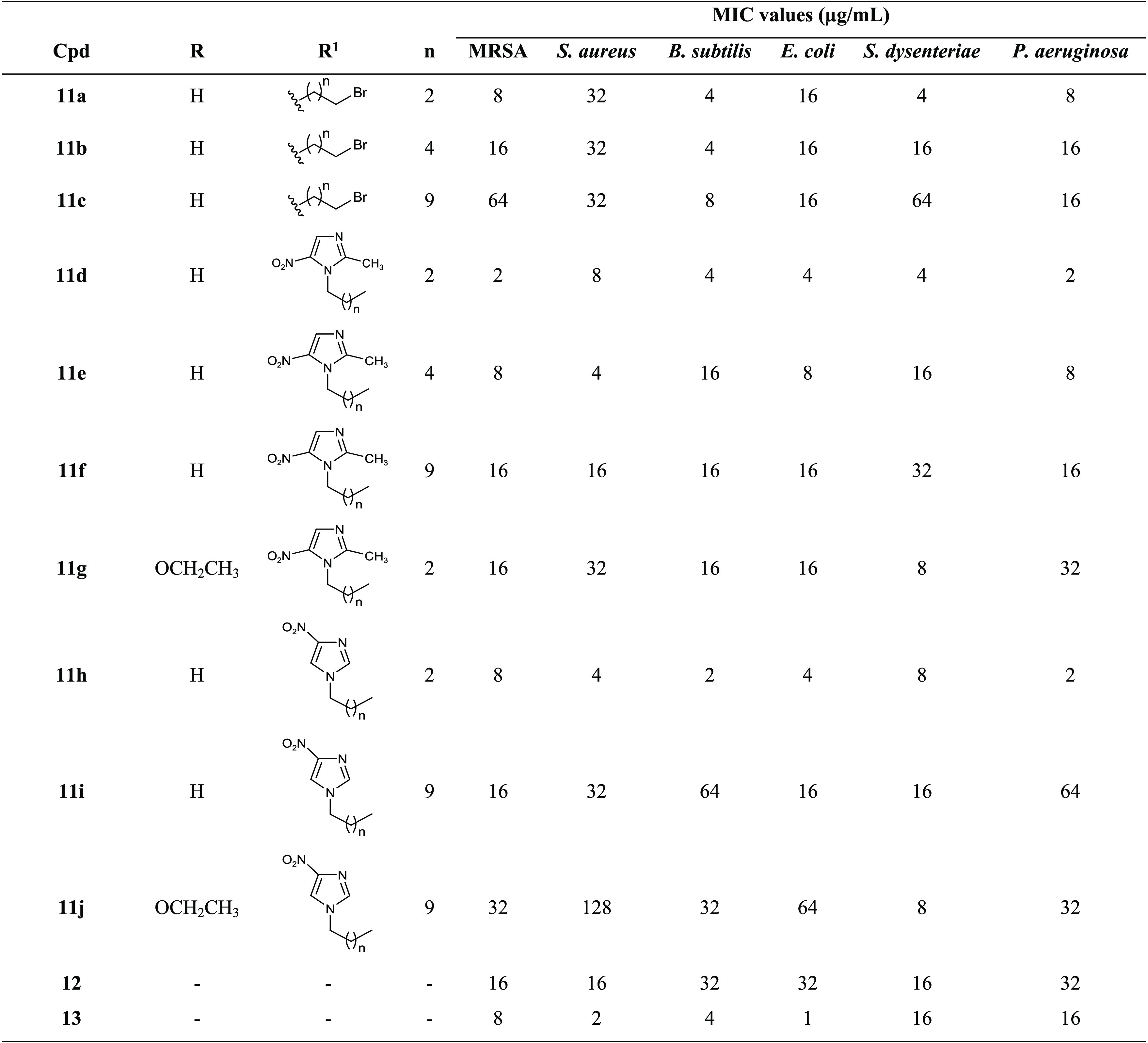
Figure 6.
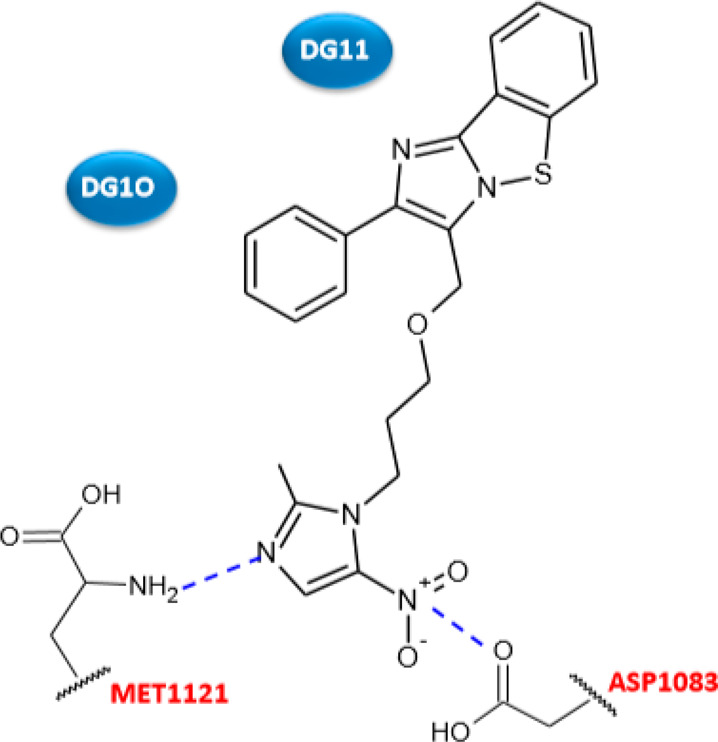
Schematic representation of the binding of compound 11d into the active site of S. aureus DNA gyrase. Hydrogen bonds in blue dashed lines.
Schiff bases constitute a class of compounds endowed with a broad spectrum of biological activity, probably ascribable to the presence of the electrophilic carbon atom and the nucleophilic nitrogen atom of the imine group, which may provide precious binding sites for the interaction with the biological target. Schiff bases also demonstrate to be sufficiently stable in in vitro and in vivo assays.38 On the basis of this knowledge, a series of Schiff bases of thiazolyl-triazole 14a–g (Table 8) were reported as antibacterial compounds and potential DNA gyrase inhibitors.39 All of these compounds were evaluated against the Gram-positive S. aureus ATCC 49444 and Listeria monocytogenes ATCC 19115 and the Gram-negative P. aeruginosa ATCC 27853 and S. typhimurium ATCC 14028. L. monocytogenes was the most susceptible strain, with MIC values of 1.9–3.9 μg/mL, which are lower than ciprofloxacin (15). Among the Gram-negative, P. aeruginosa was potently inhibited by derivatives 14a–g (MIC = 1.9 μg/mL). The ratio between MBC and MIC disclosed a bactericidal effect for these molecules. Docking studies on the DNA gyrase from L. monocytogenes suggested a mechanism of action in which this enzyme is involved. In particular, all the Schiff bases seem to prevent the entry of O-(5′-phospho-DNA)-tyrosine intermediate in the binding site.
Table 8. Chemical Structures and Antibacterial Activities of Compounds 14a–g and Ciprofloxacin (15).

| MIC
(μg/mL) |
|||||
|---|---|---|---|---|---|
| compd | Ar | S. aureus ATCC 49444 | L. monocytogenes ATCC 19115 | P. aeruginosa ATCC 27853 | S. typhimurium ATCC 14028 |
| 14a | 4-Br-phenyl | 31.2 | 1.9 | 1.9 | 62.5 |
| 14b | 4-F-phenyl | 31.2 | 1.9 | 1.9 | 62.5 |
| 14c | 4-NO2-phenyl | 62.5 | 1.9 | 1.9 | 62.5 |
| 14d | 2-OCH3-phenyl | 31.2 | 1.9 | 1.9 | 62.5 |
| 14e | 3-OCH3-phenyl | 31.2 | 1.9 | 1.9 | 31.2 |
| 14f | 2-thienyl | 15.6 | 1.9 | 1.9 | 62.5 |
| 14g | 4-(CH3)2N-phenyl | 31.2 | 1.9 | 1.9 | 62.5 |
| 15 | 1.9 | 3.9 | 3.9 | 0.97 | |
Eakin et al. identified a series of pyrrolamides with potent DNA gyrase inhibitory activity, employing a fragment-based lead generation approach (FBLG) with NMR screening to discover compounds with low molecular weight able to bind the ATP-binding pocket of E. coli GyrB.40 Starting from almost 1000 compounds with molecular masses ranging from 100 to 370 Da, the pyrrole 16 (Figure 7) showed a DNA gyrase IC50 of 3 μM and was chosen as the lead compound for the design of more potent inhibitors. The structural optimization of pyrrolamide 16 had mainly the purpose of improving the antibacterial activity of this class of compounds. In fact, despite its potent inhibitory activity against the enzyme, the compound 16 was ineffective in vitro against the tested bacterial strains (E. coli ARC523, E. coli ARC524, Haemophilus influenzae KW20, S. aureus ARC516, S. pneumoniae ARC548, Enterococcus faecium ARC521). Structural modifications of pyrrolamide 16, suggested by computational and X-ray crystallography studies, created a powerful GyrB inhibitor, incorporating the thiazole nucleus, 17 (Figure 7), endowed with a significant antibacterial activity against S. pneumoniae using a mouse model of lung infection. The crystallographic structure of derivative 16 inside the ATP-binding domain of S. aureus GyrB revealed a common interaction pattern with the natural substrate, in particular, the pyrrole moiety occupied the same pocket of the ATP adenine, creating a hydrogen bond with Asp81, whereas, the carbonyl group was involved in a hydrogen bond with a conserved water molecule.
Figure 7.

Graphic representation of the binding mode of compounds 16 and 17. Hydrogen bonds are represented with dashed blue lines, while hydrophobic interactions are in yellow.
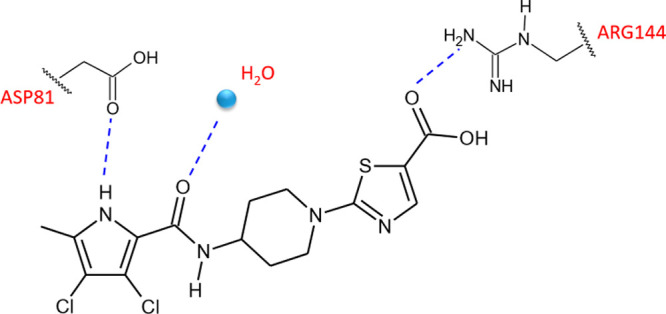
The introduction in compound 17 of the two chlorine atoms in the pyrrole moiety proved to be particularly advantageous for the affinity toward the enzyme. These atoms indeed increased the hydrophobic interactions with the adenine pocket and because of their electron withdrawing effect and improved the ability of the pyrrole NH group in establishing a hydrogen bond with the Asp81. Moreover, the presence of a carboxyl group on the thiazole ring enhanced the affinity toward the enzyme, forming two additional hydrogen bonds with the Arg144 of the active site. Pyrrolamide 17 showed MBC values against S. aureus, H. influenzae, and S. pneumoniae of 8, 4, and 0.5 μg/mL, respectively, eliciting no cytotoxicity toward mammalian cells.
The ability of 17 to induce spontaneous resistance was evaluated at the concentration of 8 μg/mL in S. aureus ARC516 and a frequency of 2.5 × 10–9 was observed. Importantly, in vivo efficacy of compound 17 assessed in an S. pneumoniae lung infection model in mice (1 × 105 CFU/lung) has been found at the oral dose of 160 mg/kg.
The fluorine atom on the 3-piperidine position characterizes 18 (Figure 8), which was obtained as mixture of four possible distereoisomers, including the (3S,4R)-distereoisomer, which showed enhanced inhibitory activity against DNA gyrase and antibacterial potency. The presence of fluorine atom, which occupied a region close to the hydrophobic pocket of the enzyme, determined an improvement in the activity by 4–8-fold. This might be due to a modification in the piperidine conformation, which allowed stronger interactions of the pyrrole carboxamide and the thiazole carboxylate with the Asp81 and Arg84, respectively. Additionally, the carboxyl group of the thiazole ring is involved in a salt bridge with Arg144 (Figure 9). This new enantiomerically pure thiazole analogue showed improved in vivo potency against S. pneumoniae in a mouse model of pneumonia.41 The compound was orally administered 18 h postinfection with 105 CFU/lung of S. pneumonia ARC548, and the in vivo activity was evaluated through viable counts in dilutions of lung homogenates. A dose-dependent reduction in viable bacterial counts in the lung was observed, eliciting the highest response at the dose of 80 mg/kg.
Figure 8.

Chemical structures of compound 18 and AZD5099(19).
Figure 9.
Representative model of the binding mode of compound 18 in complex with S. aureus GyrB (3TTZ) illustrated by ball and stick design.
Further SAR studies, carried out with purpose of optimizing the structure of these compounds, resulted in the identification of the antibacterial agent AZD5099 (19) (Figure 8), which entered phase I clinical trials as a novel treatment of Gram-positive and Gram-negative infections.42 The replacement of the fluorine atom at the 3-position of the piperidine scaffold with a methoxyl group was particularly advantageous in terms of enzyme inhibitory activity and antibacterial properties. Additionally, the carboxylic group in the thiazole nucleus was crucial for the activity because it is involved in a salt bridge interaction with the residue Arg144. AZD5099 (19) proved to be efficacious in different in vivo models, in particular, for cure of nosocomial lung and skin infective diseases caused by relevant Gram-positive such as S. aureus. Studies on the pharmacokinetic and bioavailability highlighted the suitability of AZD5099 (19) for both parenteral and oral administration.
3. Modulators of Bacterial Cell Wall Synthesis and Permeability
3.1. MurB Inhibitors
The MurB enzyme is an NADPH dependent UDP-N-acetylenolpyruvylglucosamine reductase that plays a key role in the second step of bacterial peptidoglycan biosynthesis.
In particular, the Mur pathway enzymes MurA and MurB catalyze the synthesis of UDP-N-acetylmuramic acid (UDP-MurNAc) from UDP-N-acetylglucosamine (UDP-GlcNAc). Subsequently, MurC, MurD, MurE, and MurF add amino acids to UDP-MurNAc and generate the UDP-MurNAc-pentapeptide, which is incorporated into the nascent bacterial peptidoglycan.43 MurB emerged as an interesting target for novel antibacterial drugs for many reasons: (i) its inhibition leads to a bactericidal effect because it is fundamental for the bacterial growth, (ii) there is no analogue in the eukaryotic cell therefore the selectivity should be easily obtainable, and (iii) being present in both Gram-positive and Gram-negative pathogens its inhibition should result in a broad spectrum antibacterial activity (Figure 10).44
Figure 10.
Representative examples of small molecules with MurB inhibitory activity.
Among the thiazole compounds, 4-thiazolidinones, which are the tetrahydro derivative of thiazole bearing a carbonyl group, are widely described in recent years as they present different biological activities.45−50
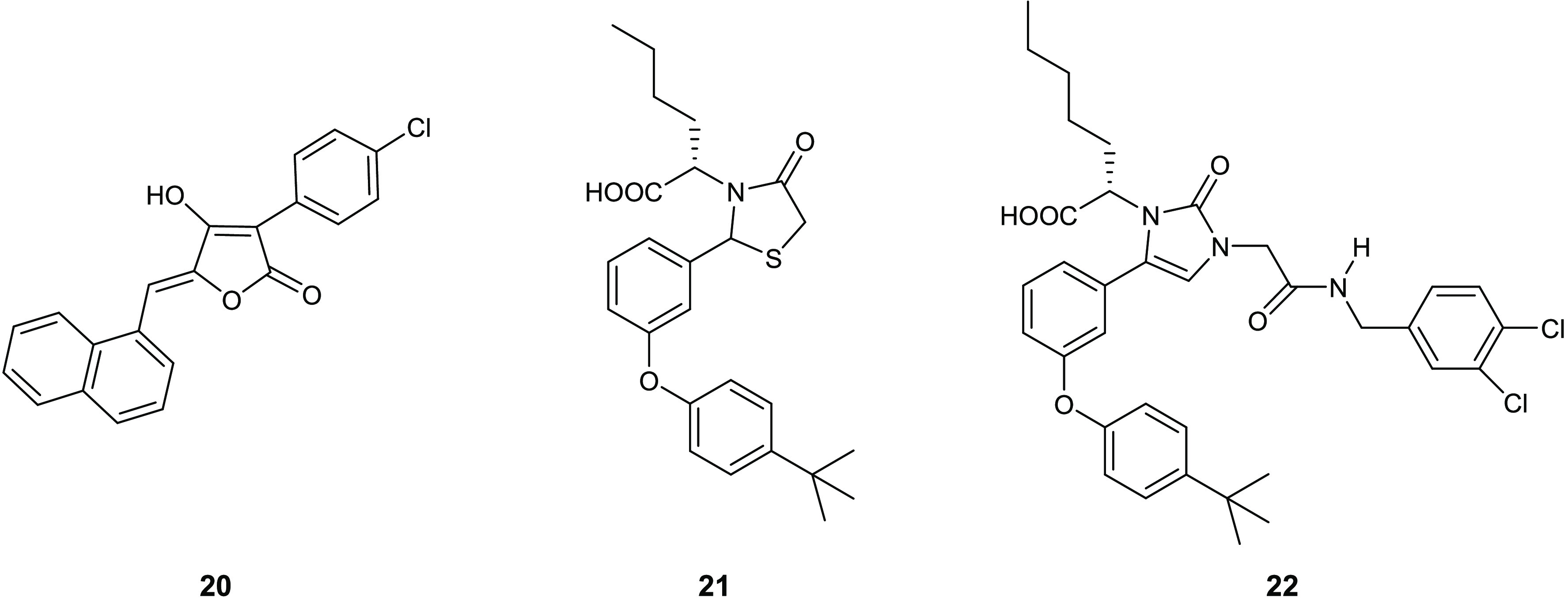
The ability of the 4-thiazolidinone scaffold in inhibiting this enzyme was first described in 2000 by Andres and co-workers, who identified three derivatives 20–22 endowed with potent E. coli MurB inhibitory activity at low micromolar level (Figure 10).51 Among them, the 4-thiazolidinonederivative 21 results the most potent compoundwith an IC50 value of 7.7 μM, which is 2-fold lower than those obtained by derivatives 20 and 22 (IC50 values of 14 and 15 μM, respectively). It was hypothesized, as supported by in silico studies reported by the authors, that 4-thiazolidinone scaffold may act as MurB inhibitor by mimicking the diphosphate group.
3.2. Penicillin Binding Protein Inhibitors
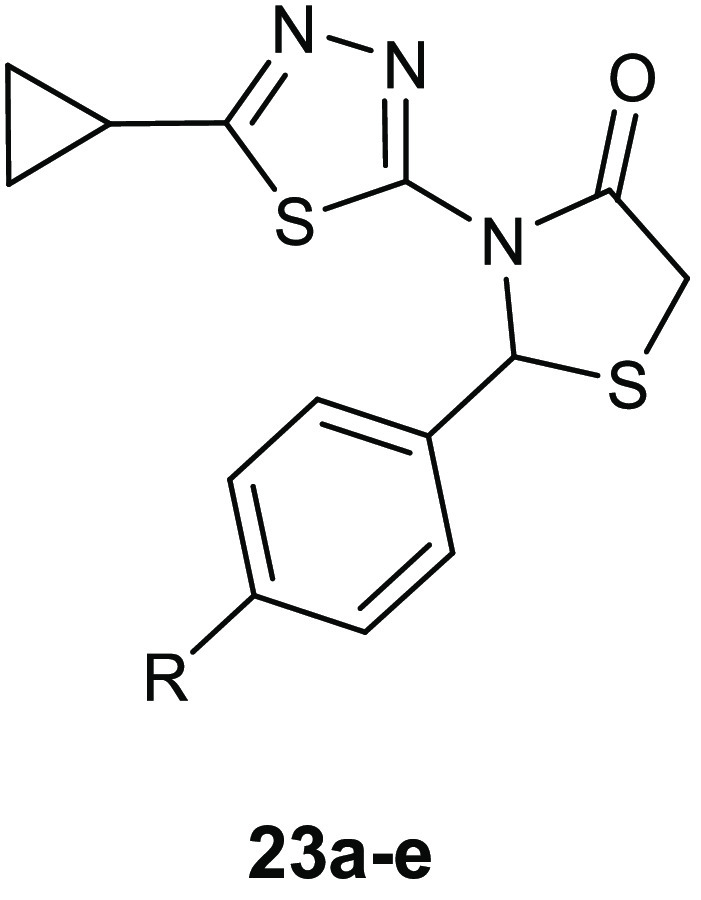
A series of thiazolidinone derivatives 23a–e (Table 9) was synthesized and examined for inhibitory activity toward transpeptidases and β-lactamase.52
Table 9. Chemical Structures and Antibacterial Activity of Compounds 23a–e.
| MIC
values (μg/mL) |
|||||
|---|---|---|---|---|---|
| compd | R | S. aureus ATCC 25923 | B. subtilis ATCC 6633 | E. coli ATCC 25922 | P. aeruginosa ATCC 27853 |
| 23a | Cl | 31.25 | 31.25 | 125 | 125 |
| 23b | CN | 62.50 | 62.50 | 250 | 250 |
| 23c | OCH3 | 125 | 125 | 250 | 250 |
| 23d | N(CH3)2 | 250 | 250 | 500 | 500 |
| 23e | NO2 | 15.60 | 31.25 | 62.50 | 125 |
When assayed against the Gram-positive bacteria S. aureus ATCC 25923 and Bacillus subtilis ATCC 6633, the Gram-negative bacteria E. coli ATCC 25922, and P. aeruginosa ATCC 27853 compounds 23a–e showed MIC values within the range of 15.60–500 μg/mL, exhibiting a strong selectivity toward Gram-positive pathogens. The most active derivative 23e elicited MIC values of 15.60, 31.25, and 62.50 μg/mL against S. aureus, B. subtilis, and E. coli, respectively. Studies on the mechanism of action were carried out by employing the method of Heipieper for evaluating the leakage of UV260 and UV280 (mostly nucleic acid material and protein) absorbing material. In these studies, 23e was able to alter the membrane permeability causing cell death. In addition, docking studies into the active site of penicillin binding protein (PBP) (PDB 1FXV) and β-lactamase (PDB 4FH2) enzymes highlighted a higher affinity for the PTB transpeptidase, supporting the hypothesis of a mechanism of inhibition of the synthesis of peptidoglycan by inhibiting the enzyme PTB transpeptidase. The compound showed a binding pattern similar to that of ampicillin, forming a hydrogen bond with the key residue of the binding site Ser 1B. The determination of the leakage of UV260- and UV280-absorbing material, which mainly consists of nucleic acid material and protein, confirmed that the compound acts by altering membrane permeability.
The authors utilized for their docking studies the conformation reported in Table 9 instead of the preferred topology involving the C=O oxygen atom proximal to S stabilized by an O to S interaction.13,14 Therefore, the authors have jeopardized the reliability of the obtained results.
Further biochemical studies on the enzyme should be carried out to confirm this hypothetical mechanism of action and for validating the docking results.
The thiazole scaffold is also present in a novel injectable siderophore cephalosporin bearing a catechol moiety, known as cefiderocol (24) (Figure 11), which demonstrated powerful antibacterial activity against Gram-negative pathogens. This activity is caused by the inhibition of penicillin binding proteins and thus interfering with the bacterial cell wall synthesis.53,54 Cefiderocol (24), although structurally very similar to the third- and fourth-generation cephalosporins ceftazidime (25) and cefepime (26) (Figure 11), elicited higher stability toward different β-lactamases such as AmpC and extended-spectrum β-lactamases.
Figure 11.

Chemical structures of cefiderocol (24), ceftazidime (25), and cefepime (26).
The key role of the aminothiazole ring, common to many broad-spectrum cephalosporins, in enhancing the activity against Gram-negative bacteria is well described. Thus, high resolution crystal structures of the P. aeruginosa PBP3 with many compounds including marketed β-lactams, as well as in silico studies, highlighted that ceftazidime (25) affinity for B3-PBPs was due to the interaction of its aminothiazole ring with the binding pocket.55 The amino acid residues of the aminothiazole binding pocket are conserved in many B3-PBPs. Therefore, the presence of aminothiazole, determining an increase in the affinity toward these proteins, usually is responsible of the enhancement of activity against Gram-negative bacteria.56 In fact, none of the many cephalosporin derivatives synthesized using different heterocyclic systems instead of aminothiazole ring showed comparable potency.57 Additionally, the carboxypropyl-oxyimino group is responsible for the increased stability of the compound toward β-lactamases hydrolysis. Of note, the presence of a catechol group on the side chain improved significantly the periplasmic concentrations of cefiderocol (24), with respect to the other cephalosporins, because of its ability to chelate iron, which makes possible the use of iron transportation systems present in the outer membrane (OM) of Gram-negative pathogens (“Trojan horse strategy”).
Cefiderocol (24) proved to be efficacious against serious Gram-negative infections, showing a strong activity against infections caused by carbapenem-resistant and MDR Gram-negative bacteria, including MDR Enterobacteriales, P. aeruginosa, Acinetobacter baumannii, and Stenotrophomonas maltophilia.58 This compound is being investigated in phase III clinical studies. Previous clinical studies highlighted the efficacy and safety of intravenous cefiderocol (24) in patients with complicated urinary tract infections (cUTIs) and in the treatment of many Gram-negative infections, such as healthcare-associated pneumonia, hospital-acquired pneumonia, and ventilator-associated pneumonia.59
3.3. Inhibitors of Heptose Synthesis
Lipopolysaccharide (LPS) is a highly acylated saccharolipid, which constitutes the main component of the outer leaflet of the OM of Gram-negative pathogens. LPS has a crucial role in maintaining the barrier function by avoiding the passive diffusion of hydrophobic solutes such as antibiotics and detergents into the cell.60
LPS is formed by lipid A, a core oligosaccharide and a repeating polysaccharide O-antigen. The core oligosaccharide is constituted by an inner core of 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo) and heptose residues and an outer core of hexoses derivatives.
Inhibition of heptose synthesis leads to vulnerable bacteria with incomplete LPS, which are more sensitive to host defenses and to many antibiotics because of their increased membrane or cell wall permeability. The identification of small molecules interfering with this process is currently recognized as a valuable goal for the development of novel antivirulence agents against Gram-negative pathogens because heptose is not fundamental for Gram-negative survival, but the inhibition of its synthesis causes an attenuation of virulence.
The bacterial synthesis of heptose in E. coli involves many enzymes including the transketolase TktA, the ketose–aldose isomerase GmhA, the kinase HldE, the phosphatase GmhB, and the epimerase HldD.61 Desroy and collaborators focused their attention on the first step of heptose synthesis, looking for molecules that can inhibit the kinase activity of E. coli HldE.62
HldE in E. coli is a bifunctional cytoplasmic ATP-dependent kinase, which uses its two functional domains (carbohydrate kinase and adenylyltransferase) to catalyze the transformation of d-glycero-d-manno-heptose-7-phosphate in d-glycero-β-d-manno-heptose-1,7-bisphosphate and of d-glycero-β-d-manno-heptose-1-phosphate in ADP-D-glycero-β-d-manno-heptose (Scheme 1).63
Scheme 1. Synthesis of ADP-l-glycero-β-d-manno-heptose from Sedoheptulose-7-phosphate and Its Incorporation into LPS.

HldE can be considered a promising target for obtaining antimicrobial agents against Gram-negative pathogens. Because this enzyme is well conserved among bacteria and there are not homologues in humans, the synthesis of selective inhibitors with low toxicity should be feasible. However, heptose synthesis inhibitors are ineffective toward pathogens which have a LPS without heptose such as Acinetobacter, Chlamydia, and Moraxella.64
Through a high-throughput screen of 40 000 compounds performed on HldE-kinase activity of E. coli, two new inhibitors 27a,b (Figure 12) were identified. Compounds 27a,b showed IC50 values of 51 and 69 μM, respectively, behaving as competitive reversible inhibitors toward ATP in the reaction catalyzed by HldE kinase. SAR studies of these compounds led to the identification of more potent derivatives bearing the benzothiazole scaffold. The effect on the HldE kinase activity of various substituents on the phenyl ring was evaluated, and it was observed that (i) the presence of an amino group in ortho or para position was particularly advantageous for the activity, while, in contrast, it caused a loss of potency in meta position, (ii) a nitro group in ortho or in para position was detrimental for the activity, whereas it proved to be favorable in meta, (iii) trifluoromethyl and methoxy groups afforded an improvement in the activity in meta position but caused significant reduction in activity in ortho and para and, finally, (iv) a bromine atom was advantageous for the activity in any position. The replacement of the phenyl ring with heteroaromatic rings or with an ethyl group led to inactive compounds, highlighting the key role of the phenyl moiety for the activity on HldE kinase. In contrast, the oxazole core could be substituted with other heterocycles including thiazole, furan, or pyrazole without any effect on the activity. These results underlined also the importance of the carboxylic acid on the amide side chain because its replacement with other groups, such as amide, alcohol, acylsulfonamide, or tetrazole, led to inactive derivatives. On the basis of these findings and of the observation that the meta substitution of the phenyl ring was also relevant for the activity, a derivative 28 (Figure 12), bearing a pyrazole ring in that position, was synthesized and evaluated for its activity against HldE kinase. Among the derivatives of this series, compound 28 elicited the highest activity with an IC50 against the enzyme of 0.11 μM.
Figure 12.

Chemical structures of compounds 27a,b and 28.
The inhibitory activities against HldE-kinase and ribokinase (RK) of E. coli and HldA of Neisseria meningitidis (the equivalent of HldE-kinase in E. coli), which share a very similar ATP binding site, were also compared and the results showed a significant selectivity toward HldE/HldA enzymes of Gram-negative bacteria. These compounds are able to interfere with heptose synthesis, altering the barrier function of the LPS but showing no effects on E. coli viability, displayed the typical profile of an antivirulence agent. Such compounds, acting on virulence factors without interfering with the bacterial life cycle, impose low selective pressure for the development of bacterial antibiotic resistance mechanisms.
Other benzothiazole derivatives acting as membrane perturbing agents able to depolarize the cytoplasmatic membrane of S. aureus and E. coli are compounds 29a,b and 30a,b (Figure 13).65 When assayed for their antibacterial activity against S. aureus (ATCC 25323), E. coli (ATCC 35218), P. aeruginosa (ATCC 27893), K. pneumonia (ATCC 31488), E. faecalis (clinical isolate), and S. typhi (MTCC 3216), the benzothiazole 29a, bearing a chlorine atom and a methoxy group on the phenyl ring, demonstrated the highest potency with MIC values in the range of 3.91–31.2 μg/mL. The effects of this series in perturbing the permeability of S. aureus and E. coli membrane was evaluated by using the cationic membrane potential-sensitive cyanine dye, which allows a determination of the alteration of membrane potential and the formation of pores, caused by the compounds, by an increase in fluorescence intensity. Compounds 29a,b were able to damage the membrane structure in S. aureus and E. coli causing the dispersion of the bacterial cell contents. It was observed that, after interaction with the membrane, compounds 29a,b and 30a,b entered into the cytoplasm and bound to bacterial DNA enhancing the antibacterial effect due to the membrane permeabilization.
Figure 13.

Chemical structures of compounds 29a,b and 30a,b.
4. Tryptophanyl-tRNA Synthetase Inhibitors
The aminoacyl-tRNA synthetases are enzymes with important roles in RNA translation because they catalyze the aminoacylation reaction by covalently linking an amino acid to its corresponding tRNA.66 These enzymes are essential in bacterial growth and survival because they are involved in many metabolic and signaling pathways essential for cell viability.67
Compounds that are able to selectively inhibit the bacterial aminoacyl-tRNA synthetases without interfering with their mammalian analogues can be useful candidates for the development of new classes of antimicrobial agents.
Recently, Stana and co-workers reported a series of thiazolin-4-one derivatives 31 (Figure 14) endowed with moderate to good antibacterial activity against the Gram-negative bacterial strain E. coli ATCC 25922 and the Gram-positive bacterial strain S. aureus ATCC 49444.68 All compounds were more effective against Gram-positive pathogens, and compounds 31a and 31b showed MIC and MBC values against S. aureus ATCC 49444 of 0.97 and 1.95 μg/mL, respectively. Because of the structural similarity between compounds 31a and 31b with the known tryptophanyl-tRNA synthetase (TrpRS) inhibitor indolmycin (32), the authors hypothesized a mode of action involving TrpRS inhibition.
Figure 14.

Chemical structures of compounds 31a,b and of the known TrpRS inhibitor indolmycin (32).
The affinity of the new compounds toward the TrpRS from S. aureus (1I6K_P67592) and E. coli (5 V0I) was evaluated in silico through molecular docking studies. Results revealed a good affinity of the thiazolin-4-ones due to the formation of polar contacts between exocyclic secondary amine group and the carbonyl group from the thiazolin-4-one ring with amino acid residues of the binding site of the enzyme. Most of the synthesized compounds showed, in silico, better affinity toward TrpRS than the reference compound indolmycin (32). The best binding affinity was observed for the compounds incorporating voluminous moieties, such as the α-naphthylamino group, at position 2 of the thiazolin-4-one ring.
Molecular docking studies suggested the key role of the thiazolin-4-one scaffold in polar interactions with the TrpRS binding site. Most compounds showed a common binding pattern involving the carbonyl group of the thiazolin-4-one in the case of S. aureus TrpRS. Additionally, the nitrogen atom of the thiazolinone ring hypothesized to be responsible of a polar interaction with the Gly9 residue of the binding site of the most active derivative 31b against E. coli.
5. Sortase A (SrtA) Inhibitors
SrtA is a cysteine transpeptidase which anchors the surface proteins, MSRAMMs, to the peptidoglycan of the bacterial cell wall in Gram-positive pathogens. After the recognition of the LPXTG motif in MSRAMMs, SrtA catalyzes the following reactions: (i) thioesterification in which the enzyme cleaves the LPXTG between Thr and Gly, leading to the synthesis of thioester acyl-enzyme intermediate and (ii) the transpeptidation, which allows the formation of a bond between the C-terminal Thr residue of the protein to pentaglycine cross-bridges. The pivotal role of this enzyme in bacterial adhesion and bacterial pathogenesis is well documented and represents therefore an excellent potential target for antivirulence drug development because it is involved in the adhesion of bacteria to host tissues and in biofilm formation, but it is not essential for bacterial viability.69 Additionally, SrtA is a membrane enzyme, therefore much more easily accessible than intracellular targets, and there are no analogues of this enzyme in humans so SrtA inhibitors should have low toxicity and high selectivity.
A new series of 2-phenylthiazole derivatives 33a–h (Table 10) were synthesized as SrtA inhibitors.70 The antimicrobial activity of the new compounds was tested against five Gram-positive and two Gram-negative bacterial strains. All of the compounds showed good activity against S. aureus, in which the lowest MIC value (16 μg/mL) was observed for derivative 33f. The highest potency was reached by the chlorophenyl derivative 33h toward Staphylococcus saprophyticus ATCC 15305 (MIC = 2 μg/mL).
Table 10. Chemical Structures, Antibacterial Activities, and Antibiofilm Activities of Compounds 33a–h.

| MIC (mg/mL) |
BIC (mg/mL) | ||||
|---|---|---|---|---|---|
| compd | Ar | R | E. faecalis | S. saprophyticus | E. faecalis |
| 33a | Ph | CH3 | 0.25 | >1.0 | 0.004 |
| 33b | Ph | H | 0.125 | 0.032 | 0.002 |
| 33c | 4-NO2-Ph | H | 0.062 | 0.032 | 0.002 |
| 33d | 4-OCH3-Ph | H | 0.062 | 0.125 | 0.004 |
| 33e | 4-CN-Ph | H | 0.062 | 0.016 | 0.002 |
| 33f | 1-naphthalene | H | 0.016 | 0.004 | |
| 33g | 3-CONH2,4-OH-Ph | H | 0.25 | 0.008 | |
| 33h | 4-Cl-Ph | H | 0.032 | 0.002 | 0.016 |
Because the inhibition of SrtA is correlated to the inhibition of biofilm formation in Gram-positive pathogens, the derivatives were also evaluated for their antibiofilm properties against all the tested strains.
These compounds proved to be more potent against E. faecalis, showing BIC50, which is defined as the lowest concentration of compound that showed 50% inhibition on the biofilm formation, in the range 2–16 μg/mL. Aiming at corroborating the mechanism of action involving SrtA, molecular docking studies evaluated the active sites of SrtA from both bacterial species. Results highlighted higher affinity toward E. faecalis SrtA and a common binding pattern, which involved the thiazole nucleus in polar contacts through the nitrogen atoms with the hydroxyl group of Thr122. The presence of polar substituents on the phenyl ring at position 4 of the thiazole nucleus enhanced the affinity toward the transpeptidase through the formation of additional hydrogen bonds with the residues Arg224 and Asn221. Despite the fact that the in silico results were in line with the results of biofilm analyses, highlighting more favorable binding in the case of E. faecalis respect to S. aureus, further in vitro tests are warranted in order to confirm this mechanism of action.
6. Antibiofilm Compounds
Biofilm is studied as one of the critical bacterial virulence factor responsible for serious chronic infections which proved to be resistant to the majority of antibiotic therapies.71
Despite, many investigations focused on the development of novel antivirulence compounds with an antibiofilm mechanism of action,72,73 no antibiofilm agents have entered in the clinical practice. This is mainly caused by the lack of in vivo experiments for validating the efficacy of the new agents in preventing biofilm-associated infection.
At present, more than 80% of chronic infectious diseases are biofilm-mediated, therefore new strategies able to combat the formation of biofilm or to eliminate preformed biofilm are urgently needed. The identification of new agents that can inhibit bacterial biofilm but are not affecting microbial growth could lead the development of new antivirulence strategies with reduced selective pressure for the onset of drug resistance.
6.1. 4-Thiazolidinone Derivatives
Coagulase-negative S. epidermidis is currently considered the most common source of infection related to implanted medical devices. The ability of S. epidermidis to create biofilms on the facets of medical devices, significantly more resistant to standard antibiotics with respect to the planktonic form, often determine serious chronic infection. Starting from the knowledge on the interesting antibiofilm and antibacterial activity (BIC50 = 15.52 μg/mL, MIC = 3.88 μg/mL) of 3-(5-((6-(ethoxycarbonyl)-5-(benzo[1,3]dioxol-5-yl)-3-oxo-7-phenyl-thiazolo[3,2-a]pyrimidin-2(5H)-ylidene)methyl)furan-2-yl)benzoic acid 34 (Figure 15) against S. epidermidis,74 Pan and collaborators designed novel thiazolidiones 35 (Table 11) in order to synthesize more effective S. epidermidis biofilm inhibitors.75,76 The strongest compound, 35e, resulted in 4-fold more activity in dispersing S. epidermidis preformed biofilm (BIC50 = 3.08 μg/mL) and in inhibiting bacterial growth of the planktonic form (MIC = 1.54 μg/mL) than the parent compound 34.
Figure 15.

Chemical structures of compound 34.
Table 11. Chemical Structures, Antibacterial Activities (MIC50, μg/mL), and Antibiofilm Activities (BIC50, μg/mL) of Derivatives 35a–e.

| compd | R1 | R2 | MIC50S. epidermidis | BIC50S. epidermidis |
|---|---|---|---|---|
| 35a | 4-OCH3-Ph-CH2– | 3-COOH | 27.73 | 55.46 |
| 35b | 4-OCH3-Ph– | 3-COOH | 3.29 | 3.29 |
| 35c | 4-CH3-Ph-CH2– | 3-COOH | 3.27 | 6.54 |
| 35d | 2-CH3-Ph-CH2– | 3-COOH | 6.54 | 13.08 |
| 35e | 2-CH3-Ph– | 4-COOH | 1.54 | 3.08 |
The marine alkaloid oroidin showed a BIC50 against P. aeruginosa biofilms of 190 μM. To create more potent antibiofilm compounds, a series of 4-thiazolidinone derivatives 36 (Table 12) of the marine bromopyrrole was developed and evaluated for both antibacterial and antibiofilm activity against S. epidermidis ATCC 12228, S. aureus ATCC29213, and E. faecalis ATCC 29212. All of these compounds displayed encouraging effects against the tested strains.77
Table 12. Chemical Structures and Antibacterial Activities (BIC50, μg/mL) of Derivatives 36a–i and 37a,b.

| compd | R1 | BIC50S. aureus |
|---|---|---|
| 36a | Ph | 3.125 |
| 36b | 4-OCH3-Ph | 0.78 |
| 36c | 4-NO2-Ph– | 0.78 |
| 36d | 2-OH,4-OCH3-Ph– | 1.56 |
| 36e | 3-OH,4-OCH3-Ph– | 3.125 |
| 36f | 2,5-OH-Ph– | 1.56 |
| 36g | 4-F-Ph– | 1.56 |
| 36h | 4-Cl-Ph– | 1.56 |
| 36i | cinnamyl | 6.125 |
| 37a | 4-OCH3-Ph | 6.25 |
| 37b | 4-NO2-Ph– | 6.25 |
Importantly, compounds 36b and 36c were 3-fold more potent than the reference drug vancomycin, demonstrating antibiofilm effects against S. aureus ATCC29213 at a concentration of 0.78 μg/mL.
Biological data revealed the importance of the 1,3-thiazolidin-4-one group for the antibiofilm activity, in fact, its replacement with a six-membered ring such as the 1,3-thiazinan-4-ones led to a significant decrease in the activity, as it was demonstrated by the BIC values of compounds 37a,b; the corresponding six-membered analogues of the most active compounds 36b,c.
The condensation of the 4-thiazolidinone ring with a pyrimidine nucleus led to compounds 38a–d (Table 13) that showed antibacterial activity against staphylococcal strains, with MIC values in the range of 0.95–3.82 μg/mL. In addition, all of these compounds exhibited promising antibiofilm activity at 30 μg/mL. The assay of inhibitory activity on YycG histidine kinase suggested that the antibacterial activity of the most promising compound 38a is based on inhibiting this enzyme (IC50 of 7.73 μg/mL), which is an indispensable enzyme in the signal transduction pathway for the cell-wall metabolism.78
Table 13. Chemical Structures and Antibacterial Activities (MIC, μg/mL) of Derivatives 38a–d.

| compd | R1 | R2 | S. epidermidis ATCC35984 | S.epidermidis ATCC12282 | S.aureus ATCC25923 |
|---|---|---|---|---|---|
| 38a | 4-Cl | 3-COOH | 0.95 | 1.91 | 3.82 |
| 38b | 4-F | 3-COOH | 3.72 | 1.86 | 3.72 |
| 38c | 4-Cl | 4-COOH | 3.82 | 1.91 | 3.82 |
| 38d | 4-F | 4-COOH | 3.72 | 3.72 | 3.72 |
6.2. Thiazoles
Because the antibacterial activity of thiazoles and different types of Schiff bases is widely documented,79,80 a new series of 4-(o-methoxyphenyl)-2-aminothiazoles 39 (Figure 16) was prepared by a microwave-assisted synthesis and evaluated for its antibacterial and antibiofilm effect against P. aeruginosa, Bacillus subtilis, and E. coli.81 The best antibacterial activity was observed for compounds 39a and 39b against the planktonic form of B. subtilis, with MIC values ranging from 25 to 50 μg/mL.
Figure 16.
Chemical structures of compounds 39a,b, sulfathiazole (40), ceftriaxone (41), aztreonam (42), and compounds 43a,b.
These derivatives were also studied with scanning electron microscope (SEM) and confocal laser scanning microscope (CLSM) analysis in order to evaluate their biofilm inhibition in P. aeruginosa, and a significant reduction in the formation of biofilm was found at subinhibitory concentrations. Obtained results suggested a quorum sensing (QS) mediated inhibition of biofilm formation, it being regulated by an intercell communication system aided by released chemical signals.
The QS system is recognized as an appealing target for the development of efficacious antivirulence agents. Because QS is not directly implicated in vital and growth processes of bacteria, its modulation imposes only a minor pressure for the development of mechanisms underlying antibiotic resistance. A therapeutic strategy that fights QS signaling, rather than the bacteria life cycle, may find application in different fields including medicine, agriculture, and food technology.
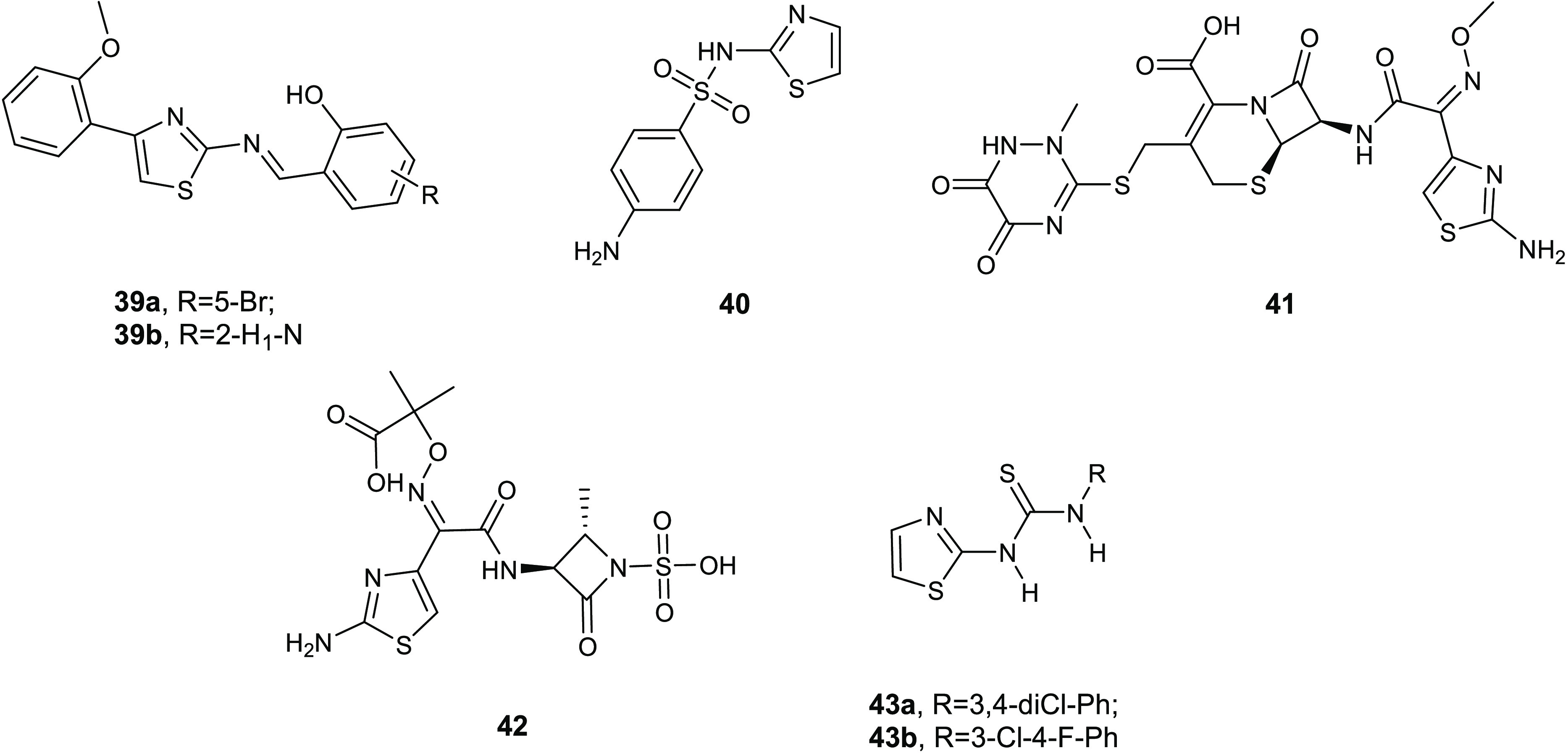
2-Amino-1,3-thiazole scaffold is known as a qualified pharmacophore for the development of antibacterial agents and other biologically active molecules.82 Numerous molecules bearing the 2-aminothiazole structural motif, including sulfathiazole (40), ceftriaxone (41), and aztreonam (42), have reached the clinical or preclinical phase. On the basis of the interesting antibacterial property described for this nucleus, Stefanska and co-workers synthesized a library of novel thiourea derivatives of type 40 (Figure 16), which were examined in vitro against several microorganisms, including relevant Gram-positive and Gram-negative pathogens.83
Compounds 43a and 43b showed the most promising activity against the staphylococcal planktonic forms eliciting MIC values toward MRSA strains and S. epidermidis in the range of 4–16 μg/mL.
The antimicrobial activity is affected by the type and the position of the substituent on phenyl ring. In particular, the 3-chloro-4-fluorophenyl substituted 43b showed the strongest activity against both standard and hospital Gram-positive pathogen strains.
These thiourea conjugates 43 were further tested, at concentrations 1–16 μg/mL, to evaluate their ability to inhibit the formation of the formation of biofilm in eight methicillin-resistant (MRSE) and two standard (ATCC 12228, ATCC 35984) strains of S. epidermidis. In addition to the antimicrobial activity against the free-swimming forms, compounds 43a and 43b were the most effective antibiofilm agents, with IC50s ranging from 0.35 to 7.32 μg/mL.
Marine natural products (MNP) or marine-derived molecules, such as marine sponge-derived compounds, have been under the spotlight because of their exclusive biodiversity and different structural features as compared to terrestrial products.84 Eight of these compounds are used in a number of therapeutic areas, while other compounds are under development in different phases of the clinical pipeline. Only a minority are original MNPs, while most of these compounds are derivatives synthesized through molecular lead optimization.85
Bis-indolyl alkaloids, which have two indole units bound to a spacer through their position 3, represent a class of deep-sea sponge metabolites with powerful biological effects, mainly antitumor86−88 and antimicrobial properties.89−94
Recent studies showed the antibiofilm activity of the new series of bis-indolyl alkaloid nortopsentin analogues 44 in which the imidazole core of the natural product is substituted by the thiazole ring.95 Compounds 44a–l (Table 14) did not affect the growth of the planktonic form of the Gram-positive S. aureus ATCC 25923, S. aureus ATCC 6538, and the Gram-negative P. aeuruginosa ATCC 15442 bacteria, showing MICs above 100 μg/mL.
Table 14. Chemical Structures and Inhibition of Biofilm Formation (IC50, μg/mL) of Thiazole Derivatives 44a–l.

| compd | R | R1 | R2 | S. aureus ATCC 25923 (μg/mL) | S. aureus ATCC 6538 (μg/mL) | P. aeuriginosa ATCC 15442 (μg/mL) |
|---|---|---|---|---|---|---|
| 44a | H | CH2CH2NHBoc | H | 3.9 ± 0.2 | 5.2 ± 0.3 | ns |
| 44b | H | CH2CH2NH2 | H | 4.7 ± 0.3 | 9.7 ± 0.9 | 22.7 ± 2.1 |
| 44c | Br | CH2CH2NH2 | H | 4.4 ± 0.1 | 3.3 ± 0.08 | 7.8 ± 0.09 |
| 44d | F | CH2CH2NH2 | H | 1.5 ± 0.1 | 6.3 ± 0.4 | 4.5 ± 0.4 |
| 44e | F | CH2CH2NH2 | CH3 | 0.5 ± 0.02 | 5.2 ± 0.08 | 3.9 ± 0.07 |
| 44f | OCH3 | CH2CH2OCH3 | CH3 | 1.2 ± 0.03 | 11.5 ± 0.7 | ns |
| 44g | Br | CH2CH2OCH3 | H | 0.79 ± 0.009 | 9.4 ± 0.3 | 4.4 ± 0.08 |
| 44h | Br | CH2CH2OCH3 | CH3 | 0.95 ± 0.01 | 11.2 ± 1.1 | 19.1 ± 0.1 |
| 44i | Br | CH2CH2OCH3 | CH2CH2OCH3 | 2.9 ± 0.02 | 18.8 ± 1.5 | ns |
| 44j | Br | CH3 | CH2CH2OCH3 | 2.5 ± 0.02 | ns | ns |
| 44k | F | CH2CH2OCH3 | CH3 | 0.2 ± 0.006 | 21.0 ± 1.7 | ns |
| 44l | H | Boc | CH2CH2OCH3 | 1.8 ± 0.1 | 6.9 ± 0.1 | ns |
Almost all derivatives were strong inhibitors of the formation of staphylococcal biofilm (Table 14). Thiazole derivatives 44e, 44g, 44h, and 44k were the most active against S. aureus ATCC 25923, showing IC50s of 0.5, 0.79, 0.95, and 0.2 μg/mL, respectively.
The lack of activity of the compounds 44 against the planktonic form was desirable in order to obtain antivirulence agents, which cause only a low selective pressure for the development of antibiotic-resistant strains. All of the new compounds were also evaluated at the concentration of 100 μg/mL to examine their capability to disperse preformed biofilm. However, none of these derivatives was able to disrupt biofilm architecture. Compounds 44 interfered with the first step of the formation of biofilm without effecting microbial growth nor preformed-biofilm, showing a significant selectivity against the Gram-positive pathogens respect to the Gram-negative. A mechanism of action involving the inhibition of the transpeptidase SrtA was hypothesized.96,97 Compounds 44a and 44l, which showed the best biofilm formation inhibitory effects as well as the highest selectivity against Gram-positive pathogens, were chosen to validate this hypothesis. Unfortunately, only compound 44a was able to moderately inhibit SrtA at the concentration of 100 μM, with a percentage of inhibition around 48%. Therefore, the antiadhesion activity observed for these derivatives was not correlated to the inhibition of the transpeptidase.
6.3. Benzothiazoles
The synthesis of hybrid molecules bearing two or more biologically active scaffolds in the same structure, is currently considered a promising approach in order to obtain new therapeutic strategies to treat antibiotic-resistance. The main advantage of this approach consists in the simultaneous presence of two pharmacophores, which can lead to a synergism of the biological activities, thus obtaining molecules able to act toward more than one target.98,99
Gondru and co-workers adopted the hybridization approach for the design of the new molecules 45a–g (Table 15), which bear simultaneously in one molecular framework the four pharmacophores pyrazole, thiazole, coumarin, and benzothiazole.100
Table 15. Chemical Structures and Antibacterial Activity of Compounds 45a–g.


Compounds 45a–g were evaluated for their antibacterial activity in vitro against Gram-positive (S. aureus MTCC 96, B. subtilis MTCC 121, S. aureus MLS16 MTCC 2940, Micrococcus luteus MTCC 2470) and Gram-negative pathogens (Klebsiella planticola MTCC 530, E. coli MTCC 739, P. aeruginosa MTCC 2453), and almost all of these derivatives showed antimicrobial activity at concentrations in the low micromolar range. The strongest derivative 45a displayed MIC values against all the tested strains in a range of 1.9–7.8 μg/mL. Biofilm inhibition assay revealed promising antibiofilm activity of compound 45e against S. aureus MTCC 96, with a BIC50 of 7.8 μg/mL. Additionally, derivative 45f elicited biofilm inhibitory activity against S. aureus MTCC 96, S. aureus MLS16 MTCC 2940, K. planticola MTCC 530, and E. coli MTCC 739, with BIC50 values in the range of 8.3–32.6 μg/mL.
The toxicity profile of these compounds was investigated against RAW 264.7 macrophages at concentrations of 2.5× and 10× MIC and, unfortunately, derivatives 45a–g were toxic at the lowest tested concentration. It was hypothesized that the high toxicity of this series was related to the presence of the benzothiazole ring, which undergoes ring-opening generating hydroxylamines with mutagenic and carcinogenic properties. However, this hypothesis was not evaluated in further experimental studies.
GroEL is a homo-oligomeric complex composed of 14 58 kDa subunits arranged in two seven-membered rings stacked back to back, which plays an important role in refolding polypeptides through a process very different from the other molecular chaperones. The folding reaction mediated by GroEL is an ATP-dependent reaction, and it requires a co-chaperone, named GroES.101 Because the GroEL/ES system has a key function for bacterial viability, the identification of small molecule able to block such function should be an innovative antibacterial strategy.
With the aim to identify new antimicrobial agents able to perturb protein folding pathways, Kunkle and co-workers performed a high-throughput screen for novel inhibitors of the GroEL/ES folding cycle.102 They identified 235 inhibitors of the E. coli GroEL/ES chaperonin system and selected a subset of 22 compounds to be further evaluated for their antibacterial activity against the so-called “ESKAPE” pathogens (Klebsiella pneumonia, Acinetobacter baumannii, P. aeruginosa, and Enterobacter cloacae).103 The results allowed the identification of the benzothiazole 46a (Table 16) as a hit compound for the synthesis of a novel class of antibacterial agents because it showed comparable bactericidal effects to vancomycin against S. aureus without toxic effects on human liver (THLE-3) or kidney (HEK 293) cell lines. Optimization of 46a has allowed the observation that (i) the presence of a benzothiazole group and the R2-hydroxyl are essential for robust inhibition, and (ii) the halogenation at the R1 position with a chlorine atom and at R3/R4 positions with a bromine atom further increases the GroEL/ES inhibitory activity. The strongest compounds of the series (46a–d) are reported in Table 16. Their effectiveness in the inhibition of GroEL/ES-mediated folding cycle was evaluated by using E. coli GroEL/ES as the surrogate chaperonin system for refolding of the enzymes malate dehydrogenase (MDH) and rhodanese (Rho). The results highlighted a strong correlation between the activities toward the two refolding assays, showing in the case of derivatives 46a–c IC50 values in the 0.70–4.7 μg/mL range against both systems. Even if studies on the selectivity of these compounds revealed a high affinity also toward the human heat shock protein HSP60/10, the selectivity indices of antibacterial activity and cytotoxicity against human kidney or liver cells were >50-fold.
Table 16. Chemical Structures of Compounds 46a–d and Their EC50 Values for Inhibitors Tested in Assays for the Formation of Biofilm and Penetration/Bactericidal Activity.

|
S. aureus proliferation
and biofilm assay EC50 [μg/mL] |
||||||
|---|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | planktonic growth | preventing biofilm formation | killing bacteria in biofilms |
| vancomycin | 0.97 | 0.78 | >145 | |||
| 46a | Cl | Br | Br | 0.20 | 0.41 | 1.37 |
| 46b | Cl | Br | H | 0.22 | 0.49 | 2.75 |
| 46c | H | Br | Br | 0.23 | 0.29 | 1.23 |
| 46d | H | H | H | 0.07 | 0.34 | 0.76 |
Compounds 46a–d showed a potent antibacterial activity against the planktonic form of S. aureus with EC50s in the range from 0.07 to 0.23 μg/mL. Most importantly, the GroEL inhibitor 46a was particularly effective toward MRSA strain without generating antibiotic resistance when tested following the procedure described by Kim and collaborators to evaluate the ability of compounds to induce drug resistance.104
Compounds 46a–d also prevented S. aureus biofilm formation eliciting BIC50 values of only 0.29–0.49 μg/mL. Being the derivatives equipotent against the planktonic form of the same strain, it is possible that the antibiofilm effect of these compounds is strongly associated with the antibacterial property. Therefore, the authors investigated the ability of this series to kill the bacterial cells inside the biofilm, evaluating their activity on preformed biofilm. In these experiments, compounds 46a–d showed EC50 values from 5 to 15-fold higher than the MICs, but they had however higher activity than the reference drug vancomycin.
In an attempt to obtain new compounds able to overcome the antibiotic-resistance of S. aureus, a dibenzyl (benzo[d]thiazol-2-yl(hydroxy)methyl) phosphonate 47 (Figure 17) was prepared and tested for its antibacterial and antibiofilm properties against strains of S. aureus resistant to penicillin, ampicillin, and methicillin.105 Phosphonates are valuable organophosphorus scaffolds characterized by the presence of a stable C–P bond, which is usually resistant to biochemical and photochemical destruction. The electron withdrawing effects of the thiazole ring may contribute to enhance its stability. The biofilm inhibitory activity of compound 47 was evaluated both as inhibition of biofilm formation and as disruption of preformed biofilm. This phosphonate derivative exhibited an interesting activity against the planktonic form and against the biofilms of S. aureus ATCC12600 as well as 12 different drug-resistant strains. It indeed determined the maximum of the activity at the concentration of 160 μg/mL with the total disappearance of bacterial aggregates. Studies on the mechanism of action revealed the lysis of Protein-A (FnBPA), which is an important surface protein belonging to the MSCRAMMs that binds fibronectin and fibrinogen and thus is a key determinant in biofilm formation and colonization.106
Figure 17.

Chemical structures of compounds 47, 48, and 49.
P. aeruginosa is among the most important biofilm-forming strains of Gram-negative pathogens and commonly causes serious chronic infections, especially in the respiratory system of patients suffering from cystic fibrosis. In P. aeruginosa biofilms, the matrix is essentially constituted by three different extracellular polymeric substance (EPS) molecules: alginate, Pel, and Psl, which are different for their chemical structure as well as for their biosynthetic mechanisms.107
The roles of Pel and Psl in antibiotic resistance, biofilm formation, and immune evasion are well-known. Their overproduction is correlated to a considerable increase in bacterial virulence.108 Pel also has important functions in cross-linking eDNA and consequentially in forming the biofilm structure. Deletion of pelB results in a severe biofilm decrease, whereas Psl forms fiber-like structures which are fundamental in interactions at cell surface, matrix development, and architecture of the biofilm. The Pel and Psl involvement in antimicrobial resistance has been widely investigated: Pel proved to be crucial for the resistance to aminoglycosides,109 while Psl is responsible for the tolerance to polymyxins, aminoglycosides, and fluoroquinolone antibiotics by sequestering them and also by reducing the recognition by the immune system.110
With the aim of obtaining novel inhibitors of P. aeruginosa biofilm formation targeting Pel and Psl and consequently the EPS secretion, Bernardes and collaborators carried out a high-throughput screen (HTS) for repressors of the gene expression of EPS, pelB::lux.111
Among the pel repressors identified, the benzothiazole derivatives 48 and 49 (Figure 17) showed significant antibiofilm activity against PAO1. These compounds were further evaluated for their antivirulence effect against PAO1 in the nematode Caenorhabditis elegans slow killing assay. The results of these studies elucidated the capability of these compounds to reduce the virulence of the wild-type PAO1, highlighting the roles of pel and psl EPS biosynthesis genes as promising tools for the development of antivirulence drugs.
7. Thiazoles as Pilicides
Targeting the adhesion of bacteria to host tissue is an emerging antivirulence strategy to counteract the resistance toward antibiotics because this process is necessary for pathogenesis, but it is not fundamental for the microbial growth and viability.112
The bacterial cell envelope can be extremely different among bacterial species presenting lipids, proteins, and exopolysaccharides, as well as fimbrial and nonfimbrial structures.
In Gram-negative pathogens, filamentous protein extensions, known as pili, are important virulence factors involved in nonspecific initial adhesion to abiotic surface and tissue colonization.113
Among the Gram-negative bacteria, uropathogenic E. coli (UPEC) is the primary cause of urinary tract infections (UTIs), which are among the most frequent diseases in community and hospital environments.114 In E. coli, pilus assembly is mediated by the chaperone/usher pathway (CUP pili), which is granted as a valuable target for the study of antivirulence agents able to prevent E. coli adhesion and biofilm formation.
A series of dihydrothiazole derivatives 50 (Figure 18) is described as pilicides able to bind the PapD–PapH chaperone–subunit complex, as revealed by X-ray crystallographic studies.115 Many of the C-2 aryl and heteroaryl-substituted derivatives of this class efficiently inhibited E. coli UTI89 biofilm formation, showing IC50s in the micromolar range, in particular the benzyl substituted displayed the highest potency in the biofilm inhibition evaluation assay with an IC50 of 7 μM. The introduction of a phenyl group at C-2 position significantly improved the inhibitory activity of pilus formation due to the establishment of additional hydrophobic interactions with the chaperone shallow pocket formed by the residues Pro30, Leu32, Ile93, and Pro95. The presence of a C-2 phenyl substituent caused a conformational change in the side chain of Leu32 that generated a surface pocket capable of accommodating it.
Figure 18.
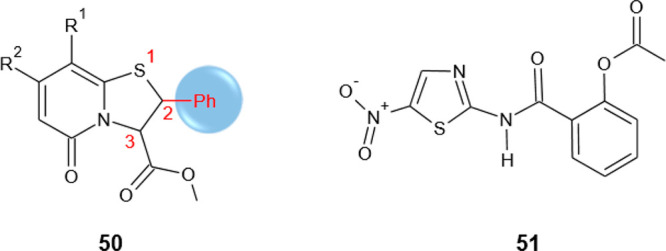
Chemical structures of compounds 50 and nitaxozanide (51).
Recent studies described the pilicides activity of the nitrothiazolyl-salicylamide nitazoxanide (51) (NTZ) (Figure 18), which is known for its therapeutic properties toward intestinal diseases including giardiasis and cryptosporidiosis.116 NTZ inhibits pili biogenesis by affecting CU pathways. In particular, it was observed that the mode of action is associated with a specific interference with proper maturation of the usher protein in the bacterial outer membrane (OM).
8. Compounds with Synergistic Effect in Association with Conventional Antibiotics
Resistance-nodulation division (RND)-type efflux pumps, causing the cellular extrusion of a number of antibiotics, such as β-lactams and β-lactamase inhibitors, fluoroquinolones, tetracyclines, and oxizolidines, play a key role in the MDR phenotype in Gram-negative pathogens.
Structurally RND pumps are formed by an integral membrane pump protein (AcrB), an OM channel (TolC) and a protein adapter (AcrA) (Figure 19).117
Figure 19.
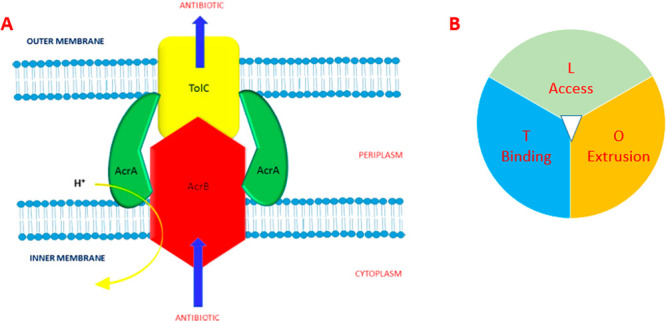
(A) RND efflux pump structure: integral membrane pump protein (AcrB), an outer membrane (OM) channel protein (TolC), and a periplasmic membrane fusion protein (AcrA).117 (B) AcrB structure.
Compounds that are able to inhibit efflux pumps are strongly required for restoring or increasing the antimicrobial activity of currently used antibiotics.
The design of new efflux pumps inhibitors (EPI) to overcome the MDR in Gram-negative pathogens have to take into account the structural features of both bacterial OM and RND. Whereas the penetration of the OM is favored for zwitterionic or hydrophilic molecules, contrarily, the binding with RND pumps needs a hydrophobic structure.
Despite many efforts in this field and the development of many EPIs, none of these compounds has reached the clinic to date.
A set of 2-substituted benzothiazoles 52a–n (Table 17) was able to rescue the antibacterial effect of ciprofloxacin (15) (CIP) in the AcrAB-TolC overexpressor E. coli AG102 mutant.118
Table 17. Chemical Structures and Observed MIC Values against the AcrAB-TolC Efflux Pump Overexpressor E. coli AG102 Strain of 2-Substituted Benzothiazoles 52a–n.

| compd | R | X | Y | MIC (μg/mL)a | E. coli AG102 combination with CIP | MIC (μg/mL)b |
|---|---|---|---|---|---|---|
| ciprofloxacin (15, CIP) | 0.125 | |||||
| 52a | H | 256 | CIP + 24a | 0.03 | ||
| 52b | OCH2(CH2)C2H5 | 64 | CIP + 24b | 0.5 | ||
| 52c | C2H5 | 128 | CIP + 24c | 0.016 | ||
| 52d | OCH3 | CH2 | 256 | CIP + 24d | 0.008 | |
| 52e | F | CH2 | 128 | CIP + 24e | 0.03 | |
| 52f | CH3 | CH2 | 512 | CIP + 24f | 0.004 | |
| 52g | H | CH2 | 256 | CIP + 24g | 0.125 | |
| 52h | F | CH2 | CH2 | 256 | CIP + 24h | 0.03 |
| 52i | F | CH2 | 128 | CIP + 24i | 0.03 | |
| 52j | Br | CH2 | 128 | CIP + 24j | 0.03 | |
| 52k | NO2 | CH2 | 128 | CIP + 24k | 0.06 | |
| 52l | C2H5 | CH2 | 64 | CIP + 24l | 0.06 | |
| 52m | H | CH2 | 256 | CIP + 24m | 0.016 | |
| 52n | H | C2H4 | 512 | CIP + 24n | 0.004 |
Observed MIC values of compounds tested alone.
Observed MIC values of CIP tested in combination with each compound.
Derivatives 52f and 52n showed a 10-fold reduction in the MIC value of CIP against E. coli AG102 strain, while 52d reduced it by about 8-fold (Table 17). These derivatives did not show intrinsic antibacterial activity but displayed a significant synergistic effect in combination with CIP against E. coli AG102. Compounds 52f and 52n associated with CIP indeed exhibited the highest antibacterial activity eliciting MIC values of 0.004 μg/mL.
Results of docking studies performed on 52, which have low molecular weights (in the range 330–402), in the crystal structure of the binding monomer T of AcrB (PDB 2DRD) were in agreement with the data obtained from the cellular assay. In fact, the major affinity toward the binding site of the monomer AcrB was shown by the derivatives 52d, 52f, and 52n, which displayed stronger binding interactions energies in comparison with CIP. All results suggested a mechanism of inhibition of AcrB by binding to the phenylalanine-rich region in the distal pocket. In particular, derivatives 52d and 52f docking poses showed a hydrogen bond with Asn274 and a π–π interaction with Phe178 and Tyr772, respectively. However, compound 52n showed a π–π interaction with Tyr772 and two π–cation interactions with Lys292 (Figure 20). The three compounds bound to the same region in the deep distal binding pocket of the T monomer, inhibiting or blocking CIP binding site. Studies of the binding energies of 52d, 52f, and 52n showed, for derivatives 52f and 52n, stronger binding energies than CIP, while for 52d, a lower energy was found. Therefore, two different mechanisms were hypothesized: a competitive inhibition of the CIP binding site for compounds 52f and 52n and an uncompetitive inhibition for 52d.
Figure 20.

Schematic representation of docking poses of compounds 52d, 52f, and 52n in the AcrB binding monomer crystal structure (PDB 2DRD). Hydrogen bonds in green, a π–π interactions in blue, and π–cation interactions in red.
AcrB is a protein of 1049 amino acids organized in three monomers or protomers characterized by different conformations: (i) loose (L), which is the access protomer, (ii) tight (T), which is the binding protomer, and (iii) open (O), which works as extrusion protomer. Crystallographic studies of AcrB with its substrates minocycline (53) and doxorubicin (54) highlighted that only one protomer is involved in the binding with the ligand and it depends by the molecular mass of the substrate. Compounds with low molecular mass, including minocycline (53) and doxorubicin (54), were bound in the T protomer in the phenylalanine-rich region. Conversely, compounds with high molecular mass, such as rifampicin (55) and erythromycin (56), docked the L monomer. The two different binding pockets are known as distal and proximal, respectively (Figure 21).
Figure 21.

Chemical structures of minocycline (53), doxorubicin (54), rifampicin (55), and erythromycin (56).
Most recently, another class of thiazole derivatives endowed with interesting synergistic effect with conventional antibiotics was discovered within a study aimed to obtain new sulfonamide derivatives as dihydropteroate synthase (DPHS) inhibitors.119
The antibacterial activity of sulfonamides is due to the modulation of the folate pathway by inhibiting DPHS, which drives the formation of dihydropteroate (DHPt) from p-aminobenzoic acid (PABA) and 6-hydroxymethyl-7,8-dihydropterin-pyrophosphate (DHPPP).
Compounds 57a–c and 58a,b (Figure 22) were active against all the tested strains, i.e., B. cerus, S. aureus, E. coli, and P. aeruginosa and showed a peculiar selectivity toward the Gram-negative E. coli, with MIC values in the range of 3.1–12.5 μg/mL. In particular, when compared with the reference drugs, the thiazole derivative 58a (MIC, 3.1 μg/mL) was equipotent to sulfamethoxazole (59) (Figure 22) and two times more potent than chloramphenicol (60) (MIC, 6.2 μg/mL) (Figure 22) against E. coli and P. aeruginosa. Further studies evaluated the synergistic effect of the new compounds in terms of fractional inhibitory concentration (FIC) through a combination study with sulfamethoxazole (59) and chloramphenicol (60) against all four bacterial strains. FIC values of 0.24 and 0.25 were observed in all tested strains. The compounds used in association with sulfamethoxazole (59) and chloramphenicol (60) demonstrated their ability to diminish the MIC of the antibiotic of 8-fold against E. coli. Remarkably, the combined MIC value (0.1953 μg/mL) of compound 58b with chloramphenicol (60) was 32-fold lower than the original MIC of 6.2 μg/mL. Such results highlighted the value of these compounds as efficacious adjuvants in an antibacterial combination approach.
Figure 22.

Chemical structures of compounds 57a–c, 58a,b, sulfamethoxazole (59), and chloramphenicol (60).
9. Antibacterial Activity of Thiazole Derivatives with Unknown Mechanism of Action
9.1. Thiazoles
Very recently, new thiazole-based compounds, 61(120) (Figure 23) and 62a–g (Table 18),121 were synthesized and tested for their antibacterial activity. The derivative 61 showed MIC values ranging from 78.1 to 396 μg/mL against the Gram-positive S. aureus and Bacillus subtilis and against the Gram-negative E. coli and Proteus vulgaris pathogens, resulting in being more efficacious toward S. aureus. The presence of p-tolyl and ethoxycarbonyl groups at positions 1 and 3, respectively, of the pyrazole ring was advantageous for the antibacterial activity of this class of compounds.
Figure 23.

Chemical structure of compound 61.
Table 18. Chemical Structures and Antibacterial Activities of Compounds 62a,b.

| MIC
values (μg/mL) |
|||||
|---|---|---|---|---|---|
| compd | R | S. aureus | B. subtilis | P. aeruginosa | K. pneumoniae |
| 62a | NO2 | 25 | 25 | 6.25 | 12.5 |
| 62b | F | 50 | 50 | 25 | 25 |
| chloramphenicol (60) | 6.25 | 6.25 | 6.25 | 12.5 | |
For the thiazole series 62, the antibacterial activity against S. aureus, B. subtilis, P. aeruginosa, and K. pneumoniae was positively influenced by the presence of electron withdrawing groups on the phenyl ring, as it has been confirmed by MIC values in the range of 6.25–50 μg/mL for the compounds 62a,b (bearing a nitro group or fluorine atom, respectively). SAR studies also highlighted the key role of the thiazole ring for the antibacterial activity. The in vitro antimicrobial studies confirmed the compound 62a exhibit selective activity toward P. aeruginosa and K. pneumonia, proving to be equipotent to the standard drug chloramphenicol (60) (Table 18).
9.2. Benzothiazoles
To unravel the SAR of the 2-mercaptobenzothiazole scaffold for the development of antibacterial agents, Franchini et al. studied if the replacement of the hydrogen at the 6-position of the heterocyclic moiety with groups able to form electronic and electrostatic interactions that could be advantageous for the biological properties.
With this purpose, a series of 2-mercaptobenzothiazole derivatives 63 (Table 19) was synthesized and tested for their antibacterial activity against Gram-positive (S. aureus, Bacillus cereus, B. subtilis, E. faecalis) and Gram-negative (E. coli, A. baumannii, K. pneumoniae, P. aeruginosa) pathogens. Among the new compounds, derivatives 63a,b were the most active; in particular, they showed the highest antibacterial activity against S. aureus ATCC 29213 bacteria strain, eliciting MIC values of 3.12 and 12.5 μg/mL. Additionally, compounds 63a,b proved to be active also against the resistant, overexpressing NorA efflux pump, S. aureus bacterial strains, demonstrating that they manage to overcome this mechanism of antibiotic resistance.122
Table 19. Chemical Structures of Compounds 63a,b, 64a–e, and 65a–c.

| compd | R | R1 | MIC (μg/mL) | compd | R | R1 | MIC (μg/mL) |
|---|---|---|---|---|---|---|---|
| 63a | CF3 | 3.12a | 64d | NH3+Cl– | H | 8b | |
| 63b | NO2 | 12.5a | 64e | NH3+Cl– | NH3+Cl– | 16b | |
| 64a | H | NO2 | 16b | 65a | 2,4-diOH-Ph | 20c | |
| 64b | H | NH2 | 8b | 65b | 2,4-diOCH3-Ph | 22c | |
| 64c | H | NH3+Cl– | 16b | 65c | 3,4,5- triOH-Ph | 18c |
The in vitro minimal inhibitory concentrations (MICs) were determined against S. aureus ATCC 29213 bacterial strain.
The in vitro minimal inhibitory concentrations (MICs) were determined against E. faecalis bacteria strain.
The in vitro minimal inhibitory concentrations (MICs) were determined against methicillin-resistant S. aureus (MRSA090) bacterial strain.
Other thiazole derivatives described for their antibacterial activity were the 2-benzothiazolyl benzo[b]thieno-2-carboxamides 64a–e (Table 19), which showed interesting activity against E. faecalis with MIC values in the range 8–16 μg/mL,123 and benzo[d]thiazole-hydrazones analogues 65a–c (Table 19),124 in which the electron donating groups, such as OH or OCH3, play a pivotal role in the antibacterial activity, as it has been confirmed by good MIC values in the range 18–22 μg/mL, obtained for the compounds 65a–c against methicillin-resistant S. aureus (MRSA090) bacterial strain.
Also, benzothiazole complexes are reported as potent antibacterial agents. Recently, the antimicrobial properties of two cationic Au(I) complexes derived from aryl-benzothiazoles: [(PPh3)Au(pbt)](OTf) 66 and [(PPh3)Au(qbt)](OTf) 67 (pbt = 2-(pyridyl)benzothiazole, qbt = (quinolyl)benzothiazole, and OTf = trifluoromethanesulfonate anion) were described (Figure 24).125 Complexes 66 and 67 showed strong antibacterial effects against the Gram negative bacteria A. baumannii and P. aeruginosa in a skin and soft tissue infection (SSTI) model.
Figure 24.

Chemical structures of compounds 66 and 67.
Through this model, the gradual penetration of bacteria deeper into the skin was evaluated using a two-layer agar system in which there is a layer of dispersed bacterial cells on the top and a nutrient-rich bottom layer. The gradient determines the slow migration of the bacteria from the top layer to the bottom layer, imitating the infectious process of the skin.
Complexes 66 and 67 were able to migrate and inhibit the infections from A. baumannii and P. aeruginosa more potently with respect to the neutral starting material PPh3AuCl. Results highlighted that the bactericidal effect was mainly due to the interaction between the cationic gold complexes and the bacterial cell membrane. Additionally, they are able to generate the reactive species Ph3PAu+ inside of the bacterial cell, which binds many biomolecules crucial for bacterial viability. Unfortunately, no information is available on their effects on the onset of antibiotic resistance.
A similar approach was used to obtain a class of imidazo[2,1-b]benzothiazolyl triazolium analogues, 68a–r and 69a–f (Table 20), which were tested in vitro for evaluating their antibacterial activity against the Gram-positive pathogens MRSA, S. aureus, B. subtilis, and M. luteus and the Gram-negative E. coli, Shigella dysenteriae, P. aeruginosa, S. typhi, and B. proteus. Some of them acted as potent antimicrobial agents eliciting MIC values in the low micromolar range.126 Of note, the unsubtituted derivatives 68a–i showed higher potency compared to the compounds bearing the ethoxy group at R1 position (68j–r). The series 69 proved to be less effective than 68, suggesting a detrimental effect of the alkyl chain for the antibacterial activity. It was observed that the biological activity in this series was influenced by the chain length, in fact, the pentyl derivatives 69a and 69b showed the highest potency, exhibiting in the case of compound 69b a MIC value of 8 μg/mL against S. aureus and B. proteus and of 2 μg/mL against M. luteus and S. dysenteriae. The presence of longer chains led to a decrease in the growth inhibition. Among the more potent series, 68 compounds substituted with two halogen atoms were more effective than the monosubstituted, in particular, the dichloro derivatives 68g, 68h, and 68q proved to me more potent than chloromycin (12), eliciting MIC values against P. aeruginosa of 8, 16, and 4 μg/mL, respectively. Interaction studies of calf thymus DNA with the most active compound of the series 68q elucidated a possible mechanism of action involving the intercalation into the DNA and the consequent block of replication.
Table 20. Chemical Structures of Compounds 68a–r and 69a–f.

| compd | n | R1 | R2 | R3 | R4 | compd | n | R1 | R2 | R3 | R4 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 68a | H | Cl | H | H | 68m | OEt | F | H | H | ||
| 68b | H | H | Cl | H | 68n | OEt | H | F | H | ||
| 68c | H | H | H | Cl | 68o | OEt | H | H | F | ||
| 68d | H | F | H | H | 68p | OEt | Cl | H | Cl | ||
| 68e | H | H | F | H | 68q | OEt | H | Cl | Cl | ||
| 68f | H | H | H | F | 68r | OEt | H | H | NO2 | ||
| 68g | H | Cl | H | Cl | 69a | 4 | H | ||||
| 68h | H | H | Cl | Cl | 69b | 5 | H | ||||
| 68i | H | H | H | NO2 | 69c | 6 | H | ||||
| 68j | OEt | Cl | H | H | 69d | 4 | OEt | ||||
| 68k | OEt | H | Cl | H | 69e | 5 | OEt | ||||
| 68l | OEt | H | H | Cl | 69f | 6 | OEt |
9.3. 4-Thiazolidinone Derivatives
A series of 4-thiazolidinone derivatives of the type 70 (Figure 25), bearing thiazole, thiazolidinone, and adamantane scaffolds, was synthesized and evaluated for the antibacterial activity against a panel of Gram-positive and Gram-negative bacteria including B. cereus (clinical isolate), M. flavus (ATCC 10240), L. monocytogenes (NCTC 7973), S. aureus (ATCC 6538), E. coli (ATCC 35210), P. aeruginosa (ATCC 27853), S. typhimurium (ATCC 13311), and Proteus mirabilis (human isolate).127 All of these compounds elicited a significant antibacterial activity against all the tested strains showing MIC values ranging from 5.01 to 20.8 μg/mL. Determination of MBC, which was almost 2-fold higher than the corresponding MIC, elucidated a bacteriostatic mechanism.
Figure 25.

Chemical structures of compounds 70.
The introduction of arylidene moieties in the thiazolidinone ring improved significantly the antimicrobial activity, and various substitutions on this nucleus were well tolerated. However, despite the excellent antibacterial activity described for derivatives 70 (Figure 25), two critical issues must be taken into account and need further studies: the knowledge of the mechanism of action and the possibility that they were PAINS compounds because of their potential to be Michael acceptor.
Other 4-thiazolidinone derivatives reported for their antibacterial activity were derivatives 71a–l (Table 21), which showed significant properties against the Gram-negative P. aeruginosa and E. coli, with MIC values ranging from 1.56 to 12.5 μg/mL, and against the Gram-positive S. aureus and B. subtilis, with MIC values between 1.56 and 6.25 μg/mL.128 The highest activity was obtained for derivatives with electron withdrawing group, including a bromine atom or nitro group on the aromatic ring.
Table 21. Chemical Structures and Antibacterial Activities of Compounds 71a–l.

| MIC
values (μg/mL) |
||||||
|---|---|---|---|---|---|---|
| compd | Ar | Ar1 | E. coli MTCC 40 | P. aeruginosa MTCC 2453 | S. aureus MTCC 121 | B. subtilis MTCC 96 |
| 71a | Ph | Ph | 6.25 | 6.25 | 6.25 | 3.12 |
| 71b | Ph | 3-NO2-Ph | 6.25 | 3.12 | 3.12 | 1.56 |
| 71c | Ph | 4-Cl-Ph | 3.12 | 12.5 | 3.12 | 6.25 |
| 71d | Ph | 3-Br-Ph | 12.5 | 6.25 | 3.12 | 6.25 |
| 71e | Ph | 3-OCH3-Ph | 6.25 | 6.25 | 6.25 | 1.56 |
| 71f | 3-Br-Ph | Ph | 6.25 | 3.12 | 3.12 | 3.12 |
| 71g | 3-Br-Ph | 3-NO2-Ph | 3.12 | 1.56 | 1.56 | 1.56 |
| 71h | 3-Br-Ph | 4-Cl-Ph | 12.5 | 6.25 | 6.25 | 6.25 |
| 71i | 3-Br-Ph | 3-Br-Ph | 6.25 | 6.25 | 3.12 | 3.12 |
| 71j | 3-Br-Ph | 4-OCH3-Ph | 1.56 | 12.5 | 3.12 | 1.56 |
| 71k | 3-F-Ph | Ph | 6.25 | 12.5 | 6.25 | 3.12 |
| 71l | 3-F-Ph | 3-NO2-Ph | 12.5 | 3.12 | 6.25 | 6.25 |
Compounds 72a–c (Table 22), containing the 4-thiazolidinone scaffold, displayed promising antimicrobial activity against the planktonic form as well as the biofilm of K. planticola, which is a main responsible cause of nosocomial infections of urinary tract.129 These derivatives elicited MIC and MBC values of 3.9 and 15.6 μg/mL, respectively. Additionally, they were able to inhibit K. planticola biofilm formation with BIC50 ranging from 20.28 to 20.79 μg/mL. The exact target of these molecules is not known, and consequently, the role of the thiazolidinone scaffold for the biological activity is not clear. However, modification on this nucleus influenced the antimicrobial properties. In particular, the presence of electron withdrawing substituents on the thiazolidinone scaffold was advantageous for the antibacterial activity, whereas the substitution of the phenyl ring with different heterocycles caused the loss of this activity. These observations suggest the involvement of the thiazolidinone ring in driving the biological activity.
Table 22. Chemical Structures and Antibacterial Activities of Compounds 72a–c.
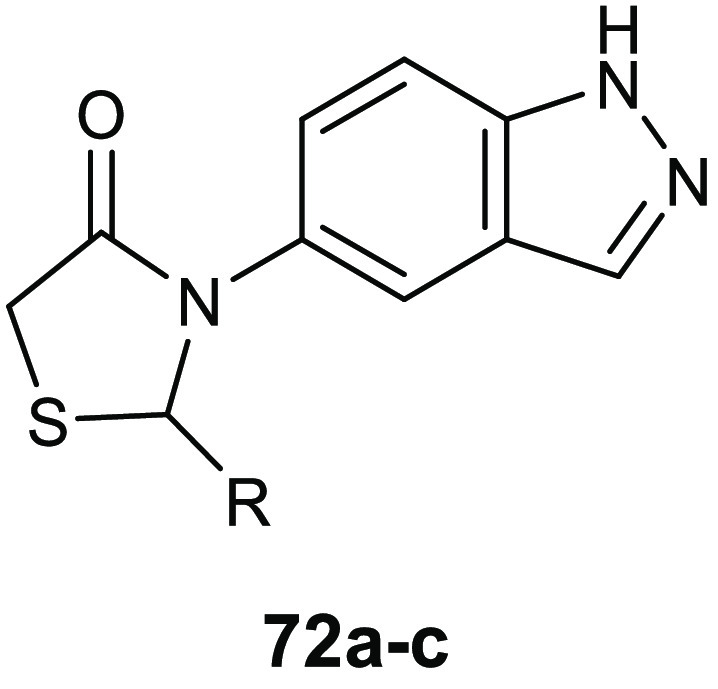
|
K. planticola MTCC 530 |
||||
|---|---|---|---|---|
| compd | R | MIC (μg/mL) | MBC (μg/mL) | BIC (μg/mL) |
| 72a | 4-CF3-Ph | 3.9 | 15.6 | 20.28 |
| 72b | 3-CF3-Ph | 3.9 | 15.6 | 20.72 |
| 72c | 4-OCF3-Ph | 3.9 | 15.6 | 20.79 |
Pitta and collaborators combined the three bioactive scaffolds thiazole, adamantane, and 4-thiazolidinone in order to obtain a new series of derivatives 73 (Table 23) with a remarkable antibacterial activity against a wide spectrum of Gram-positive and Gram-negative bacteria.130
Table 23. Chemical Structures and Antibacterial Activity of Compounds 73a–j.
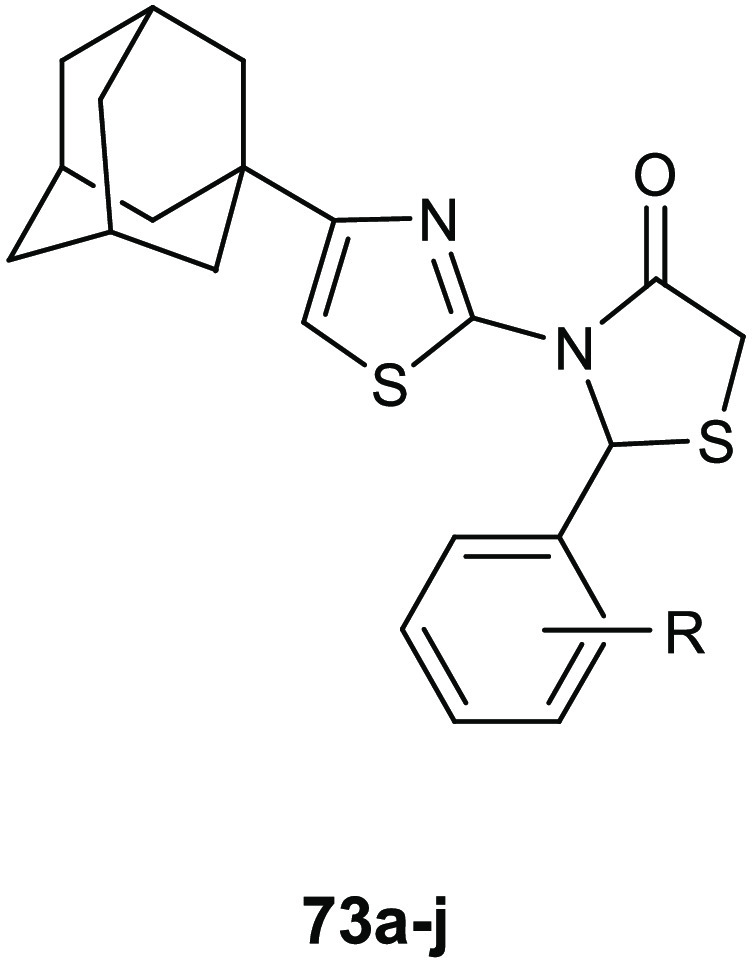
| MIC
values (μg/mL) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | R | B. cereus | M. flavus | S. aureus | L. monocytogenes | E. coli | E. coclae | P. aeruginosa | S. typhimurium |
| 73a | 2-Cl | 13.2 | 19.4 | 26.9 | 7.0 | 26.9 | 13.2 | 26.9 | 26.9 |
| 73b | 3-Cl | 7.0 | 26.9 | 13.2 | 26.9 | 26.9 | 26.9 | 29.6 | 13.2 |
| 73c | 4-Cl | 7.0 | 13.2 | 7.0 | 26.9 | 26.9 | 26.9 | 7.0 | 7.0 |
| 73d | 2,3-diCl | 14.2 | 14.2 | 4.18 | 29.0 | 29.0 | 14.2 | 7.1 | 7.1 |
| 73e | 2,6-diCl | 200 | 100 | 200 | 200 | 100 | 200 | 200 | 100 |
| 73f | 3-F | 124 | 12.7 | 124 | 6.33 | 6.33 | 12.7 | 3.7 | 3.7 |
| 73g | 4-Br | 47 | 100 | 200 | 200 | 100 | 9.5 | 200 | 100 |
| 73h | 4-NO2 | 7.2 | 27 | 13.5 | 27 | 37.5 | 13.5 | 13.5 | 13.5 |
| 73i | 4-OCH3 | 6.9 | 26.6 | 13.0 | 26.6 | 13.0 | 13 | 13.0 | 13.0 |
| 73j | 2,5-diOCH3 | 100 | 100 | 200 | 200 | 100 | 100 | 200 | 100 |
These compounds were more potent than the references drugs ampicillin and streptomycin, showing MIC values ranging from 3.7 to 200 μg/mL against the Gram-positive Bacillus cereus (clinical isolate), Micrococcus flavus (ATCC 10240), S. aureus (ATCC 6538), and Listeria monocytogenes (NCTC 7973), and the Gram-negative E. coli (ATCC 35210), Enterobacter cloacae (human isolate), P. aeruginosa (ATCC 27853), and Salmonella typhimurium (ATCC 13311).
The highest antibacterial activity was found for compound 73d, which elicited MIC values in the range of 4.18–29 μg/mL against all the tested strains. Notably, the position of the chlorine atom on the phenyl ring did not influence the biological activity of this class of compounds, whereas the introduction of a second chlorine was advantageous for the activity in the case of 2,3-disubstituted compounds but was detrimental in the case of the 2,6-disubstituted derivative. The presence of a fluorine atom at position 4 in the phenyl ring led to an improvement in the antimicrobial activity against all the bacterial strains except for B. cereus and S. aureus.
To investigate the potential mechanism of action involving the enzyme MurB, the most active compounds 73d and 73e were docked in the S. aureus MurB active site (PDB 1HSK).
Compound 73d (R-isomer) formed hydrogen bond contacts with the residues Gly249 and Arg242, and it was also involved in a Cl−π interaction with Phe274 of the active site. The phenyl ring of the 2,3-dichlorophenyl moiety is proposed to be involved in hydrophobic interactions with the amino acids Val239 and Gly273 and in π–π interaction with Phe274. Although in silico studies suggested a good affinity between derivatives 73 and MurB, no in vitro experiments on the enzyme have been carried out to validate this hypothesis.
A new series of 3-(2H-1,2,4-triazol-5-yl)-1,3-thiazolidin-4-one derivatives 74a–e (Table 24) was created and tested in vitro for the antibacterial activity and in silico for the MurB affinity.131 The highest activity was observed for derivative 74a, which showed MIC values of 8 μg/mL against S. aureus and 16 μg/mL against E. faecalis, B. subtilis, and E. coli. This was attributed to the good lipophilic features of this compound (log P value of 3.84), which makes possible the crossing of the lipid coat of bacteria. SAR studies highlighted that the presence of a methoxy group at the R position and fluorine, nitro, methoxy, and methyl at the R1 are advantageous for the antibacterial activity of these compounds.
Table 24. Chemical Structures and Antibacterial Activity of Compounds 74a–ea.

| MIC
values (μg/mL) |
|||||
|---|---|---|---|---|---|
| compd | R | S. aureus | E. faecalis | B. subtilis | E. coli |
| 74a | Cl | 8 | 16 | 16 | 16 |
| 74b | F | 128 | ND | ND | 64 |
| 74c | NO2 | 16 | ND | ND | 32 |
| 74d | OCH3 | 128 | ND | ND | 6 |
| 74e | CH3 | 64 | ND | ND | 32 |
ND = not determined.
Studies of molecular modeling with 74a revealed a good affinity toward the enzyme MurB with a binding energy of −12.18234 kcal/mol, but also in this case, the computational results are not supported by enzymatic assays.
A series of 4-thiazolidinone derivatives 75a–e (Table 25) was described for their potent antimicrobial activity against S. aureus, B. subtilis, and E. coli, showing comparable potency to norfloxacin used as reference drug.132 The highest activities were elicited by compounds 75b and 75d, against the Gram-negative E. coli pathogen, with a MIC value of 3.07 μg/mL, and against S. aureus, with a MIC value of 6.51 μg/mL, respectively. Against Gram-positive B. subtilis pathogen, the derivative 75a showed the best MIC value of 6.24 mg/mL. The new compounds showed a bacteriostatic activity because their MBC values were 3-fold higher than the MICs. The introduction of electron withdrawing groups increased the antibacterial activity of this class of compounds against S. aureus and B. subtilis, while electron donating groups were advantageous for the activity against E. coli.
Table 25. Chemical Structures and Antibacterial Activities of the 4-Thiazolidinones 75a–e.

| MIC (μg/mL) |
||||
|---|---|---|---|---|
| compd | R | S. aureus | B. subtilis | E. coli |
| 75a | 2-Cl | 12.5 | 6.24 | 6.24 |
| 75b | 4-Br | 6.51 | 47.8 | 12.16 |
| 75c | 3-NO2 | 25.23 | 12.41 | 25.23 |
| 75d | 4-N(CH3CH2)2 | 12.37 | 12.37 | 3.07 |
| 75e | 4-CH3 | 12.56 | 12.56 | 12.56 |
Recently, a sulfurated analogue of thiazolidinone compounds, the 1,3-thiazolidin-2-thione 76 (Figure 26), was found to act as an antibacterial agent equipotent to chloramphenicol (60) against S aureus, with a MIC value of 3.12 μg/mL, and with a significant activity also against Bacillus thuringiensis, with a MIC value of 6.25 μg/mL.133 Despite this interesting activity, thiazole derivative 76 did not demonstrate a significant increase in activity with respect to the pyridine analogue previously reported. Conversely, the authors observed a decrease in the activity against the Gram-negative pathogens was. Therefore, in this case, the presence of the thiazole scaffold seems not relevant for the antibacterial activity.
Figure 26.

Chemical structure of 76.
10. Conclusions and Future Directions
There is a need of a prompt intervention to face the global emergency of antibiotic resistance.
In addition to the rational use of the conventional antibiotics, which requires coherent strategy in human, animal, plant, and environmental health (One Health strategy), in order to preserve their efficacy in treating infectious diseases, other actions, such as development of new antimicrobial agents, are needed.
In the past decade, many thiazoles and benzothiazoles were described for their interesting antibacterial activity, in many cases also against MDR strains. One of the most significant feature of this class of compounds consists in their ability to interfere with diverse bacterial targets, including DNA gyrase, topoisomerase IV, biofilm formation, cell wall permeability, and tryptophanyl-tRNA synthetase. Consequently, they proved to be active against a broad spectrum of relevant Gram-positive and Gram-negative pathogens, with MIC values in the low micromolar range. Regarding the mechanisms of action, the most investigated is the inhibition of type II topoisomerases. In particular, the benzothiazoles 3a,b potently inhibited in vitro E. coli DNA gyrase and topoisomerase IV, with IC50 in the range 0.0033–0.046 μg/mL, eliciting MIC values between 0.008 to 0.06 μg/mL against S. pneumoniae, S. epidermidis, and S. pyogenes. In addition, because DNA gyrase is a crucial enzyme for bacterial viability, its inhibition could cause the development of antibiotic resistance strains, as was observed for the fluoroquinolone antibiotic class. Therefore, particularly relevant has been the study performed for evaluating their ability to induce resistance in terms of FoR, which established that these benzothiazole ethyl urea derivatives had a low capability to determine drug resistance, then they could be considered interesting lead compounds worthy of being developed.
Topoisomerases can be considered valuable targets for drug discovery as they are conserved across numerous bacterial species, so a broad spectrum of activity should be feasible. Moreover, in the past decade, new insights have been obtained in the molecular mechanisms of DNA gyrase and topoisomerase IV inhibition, the individuation of the structural features required for the interaction with the binding site and the factors responsible for the resistance development. This knowledge allowed identifying clinical drug candidates such as AZD5099 (19), which proved to be efficacious in different in vivo models for the treatment of nosocomial lung and skin infections caused by relevant Gram-positive pathogens.
Importantly, AZD5099 (19) showed FoR lower than the detection limit of 9.6 × 10–10 and impressive selectivity against the bacterial topoisomerase II compared to the human enzyme.
The modulation of the bacterial cell wall synthesis and permeability still remains a good target for the development of innovative antimicrobials, and many thiazoles are able to inhibit enzymes involved in these processes. However, assays to investigate the FoR values should be carried out because targets responsible for the integrity of the cell wall are needed for the bacterial viability, consequently the evolution of antibiotic resistance is unavoidable. Particularly relevant the data observed for the cephalosporin Cefiderocol, whose effectiveness and safety has been demonstrated for the treatment of cUTI and acute uncomplicated pyelonephritis in a clinical phase II trial.
An important aspect to take into consideration during the design of new antibacterial compounds bearing the thiazole scaffold is the classification of 14 subclasses of 2-aminothiazoles (2-ATs) as PAINS.134 PAINS compounds are promiscuous molecules that give multiple positive results, often not reliable, to different biological assays, and for this reason they should be removed from screening libraries and biological assay.135,136 There are different reasons for their promiscuous behavior, including their photoreactivity, the presence of impurities generated by bromomethyl ketones often used as chemical precursors in their preparation, and their chemical feature as thiol-reactive species.137 On the other side, it must be considered that the biological and therapeutic value of 2-ATs is widely recognized because many marketed drugs, including antibiotics like cefepime, cefetamet, cefoselis, cefotaxime, cefotiam, cefpodoxime, cefpirome ceftazidime, ceftibuten, and ceftriaxone are bearing this fragment in their structure.
Also, for the thiazolidinone derivatives, already known for their interesting pharmacological profile, it is important to consider, in the design of new antibacterial agents, their potential as PAINS. Classes such as 5-ene-4-thiazolidinones, indeed, often showed good activities within a range of different assays and against numerous proteins. Therefore, studies to exclude that the results are not related to the nature of Michael acceptors are needed.
It is important to consider that the possibility of being Michael acceptors is not necessarily associated with the behavior as PAINS compounds and it is often not confirmed experimentally under physiological conditions.138 Additionally, advantageous toxicological profile of several classes of 4-thiazolidinones strongly encourage their development and study as new drugs.139
The thiazole derivatives, which hit virulence factors, but do not interfere with the microbial growth, could be considered “evolution proof” and for sure worthy to be developed as antivirulence agents. Among them, the inhibitors of biofilm formation, compounds 44 and 46, that potently inhibited biofilm formation of relevant pathogens without affecting their viability, can be considered deserving for the design of novel antivirulence compounds.
Acknowledgments
Figure 3 is a reproduction of Figure 8 from ref (25), and for this reason we thank the authors.
Glossary
Abbreviations Used
- AMR
antibiotic-resistance
- 2-ATs
2-aminothiazoles
- AUC
area under the curve
- CLSM
confocal laser scanning microscope
- CIA
critical important antimicrobials
- CIP
ciprofloxacin
- CUP
chaperone/usher pathway
- DHPPP
6-hydroxymethyl-7,8-dihydropterin-pyrophosphate
- DHPt
dihydropteroate
- DPHS
dihydropteroate synthase
- EPI
efflux pumps inhibitors
- EPS
extracellular polymeric substances
- FBLG
fragment-based lead generation approach
- FIC
fractional inhibitory concentration
- FnBPA
fibronectin binding protein A
- FoRs
spontaneous frequencies of resistance
- HTS
high-throughput screening
- LPS
lipopolysaccharide
- MIC
minimum inhibitory concentrations
- MurB
uridine diphosphate-UDP-N-acetylenolpyruvylglucosamine reductase
- NTZ
nitazoxanide
- OM
outer membrane
- OTf
trifluoromethanesulfonate anion
- PABA
p-aminobenzoic acid
- pbt
2-(pyridyl)benzothiazole
- PAINS
pan assay interference compound
- PK
pharmacokinetic
- qbt
(quinolyl)benzothiazole
- QS
quorum sensing
- SEM
scanning electron microscope
- SrtA
sortase A
- TrpRS
tryptophanyl-tRNA synthetase
- UPEC
uropathogenic Escherichia coli
- UTIs
urinary tract infections
Biographies
Stella Cascioferro graduated in Pharmacy with honors in 1999, and she got her Ph.D. in Medicinal Chemistry in 2004 at the University of Palermo. In 2004, she joined the Physical and Theoretical Chemistry Laboratory at the University of Oxford as part of the group led by Professor Graham Richards. Her research interests include the design, synthesis, and biological evaluation of heterocyclic compounds as antitumoral and antinfective agents. Currently, she is researcher at the University of Palermo, Italy. She is the author of 58 scientific papers published in peer reviewed international journals of medicinal chemistry.
Barbara Parrino graduated in Medicinal Chemistry and Technology at the University of Palermo with full marks with honors in 2007. She got her Ph.D. in Pharmaceutical Sciences in March 2012, during which she was a Ph.D. visiting student at Semmelweiss University and at the University of Nottingham, and for which she received the “Doctor Europaeus” label. On September 2012, she was awarded the P. Ehrlich MedChem Euro-PhD Network Certificate. In 2015 she was recipient of the Medicinal Chemistry Division of the Italian Chemical Society Prize. From November 2012 to October 2017, she was a Postdoc Research fellow at the University of Palermo. Currently, she is researcher at the University of Palermo and author of 51 papers and one Italian patent.
Daniela Carbone holds a master’s degree in Pharmacy (full marks with honors) from the University of Palermo in 2015. From July to November 2018, she was a Ph.D. visiting student at VU University Medical Center of Amsterdam, and she earned her Ph.D. in Molecular and Biomolecular Sciences, with additional certification of Doctor Europaeus, in February 2019. Since April 2019, she has been a Research fellow at the Department of Biological, Chemical and Pharmaceutical Sciences and Technologies (University of Palermo, Italy). Her current research interests are focused on the medicinal chemistry field, particularly the design, synthesis, and biological evaluation of heterocyclic compounds, analogues of natural marine alkaloids, with antitumor and anti-infective properties.
Domenico Schillaci is a microbiologist and is the head of the Laboratory of Microbiology and Biological Assays of the Department of Biological, Chemical, and Pharmaceutical Science and Technology (University of Palermo, Italy). He is the head of, or collaborates with, research projects regarding the discovery of new anti-infective and antibiofilm agents, in particular against staphylococcal biofilms. He concentrates on both synthetic organic compounds and natural products as antimicrobial peptides. He is the author of 91 publications in peer reviewed journals. From 2001 to date, he has been teaching General Microbiology to the students of the School of Pharmacy of the University of Palermo. He has served as a reviewer for several journals in the field of applied microbiology.
Elisa Giovannetti is associate Professor of Pharmacology at the VU University Medical Center, Amsterdam, and Principal Investigator at the Cancer Pharmacology Lab (University of Pisa). She is member of the Steering Committee of the Pharmacology and Molecular Mechanisms (PAMM) group of the EORTC, and her studies are funded by grants from Italian Association for Research against Cancer (AIRC), Netherlands Organization for Scientific Research (NWO), Cancer Center Amsterdam (CCA) Foundation, and Dutch Cancer Society (KWF).
Girolamo Cirrincione is Emeritus Professor of Medicinal Chemistry at the University of Palermo. He is a member of the Drug Discovery Commettee of the Pharmacology and Molecular Mechanisms (PAMM) Group of the European Organization for Research and Treatment of Cancer, the Italian Chemical Society, where he has been President of the Medicinal Chemistry Division, and the International Society of Heterocyclic Chemistry, where he served as vice president for 2004–2005. He was Pro-rector for Research of the University of Palermo for 2015–2018.
Patrizia Diana is full Professor of Medicinal Chemistry at the University of Palermo. She is currently coordinator of the Ph.D. course of Molecular and Biomolecular Sciences and of the Medicinal and Biological Section of the Department of Science and Technology: Chemical, Biological and Pharmaceutical (STEBICEF) of the University of Palermo. She is a member of the Italian Chemical Society and International Society of Heterocyclic Chemistry and of the PAMM group of the ORTC.
The authors declare no competing financial interest.
References
- Prestinaci F.; Pezzotti P.; Pantosti A. Antimicrobial Resistance: A Global Multifaceted Phenomenon. Pathog. Global Health 2015, 109, 309–318. 10.1179/2047773215Y.0000000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D.; Misba L.; Khan A. U. Antibiotics versus Biofilm: An Emerging Battleground in Microbial Communities. Antimicrob. Resist. Infect. Control 2019, 8, 76. 10.1186/s13756-019-0533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebreyohannes G.; Nyerere A.; Bii C.; Sbhatu D. B. Challenges of Intervention, Treatment, and Antibiotic Resistance of Biofilm-Forming Microorganisms. Heliyon 2019, 5, e02192 10.1016/j.heliyon.2019.e02192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler C. I. R. Current Accounts of Antimicrobial Resistance: Stabilisation, Individualisation and Antibiotics as Infrastructure. Palgrave Commun. 2019, 5, 53. 10.1057/s41599-019-0263-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kraker M. E. A.; Stewardson A. J.; Harbarth S. Will 10 Million People Die a Year Due to Antimicrobial Resistance by 2050?. PLoS Medicine 2016, 13, e1002184 10.1371/journal.pmed.1002184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schillaci D.; Spanò V.; Parrino B.; Carbone A.; Montalbano A.; Barraja P.; Diana P.; Cirrincione G.; Cascioferro S. Pharmaceutical Approaches to Target Antibiotic Resistance Mechanisms. J. Med. Chem. 2017, 60, 8268–8297. 10.1021/acs.jmedchem.7b00215. [DOI] [PubMed] [Google Scholar]
- Cascioferro S. The Future of Antibiotic: From the Magic Bullet to the Smart Bullet. J. Microb. Biochem. Technol. 2014, 6, e118 10.4172/1948-5948.1000e118. [DOI] [Google Scholar]
- Sharma D.; Bansal K. K.; Sharma A.; Pathak M.; Sharma P. C. A Brief Literature and Review of Patents on Thiazole Related Derivatives. Curr. Bioact. Compd. 2019, 15, 304–315. 10.2174/1573407214666180827094725. [DOI] [Google Scholar]
- Muhammad Z. A.; Masaret G. S.; Amin M. M.; Abdallah M. A.; Farghaly T. A. Anti-Inflammatory, Analgesic and Anti-Ulcerogenic Activities of Novel Bis-Thiadiazoles, Bis-Thiazoles and Bis-Formazanes. Med. Chem. 2017, 13, 226–238. 10.2174/1573406412666160920091146. [DOI] [PubMed] [Google Scholar]
- Hosseinzadeh N.; Seraj S.; Bakhshi-Dezffoli M. E.; Hasani M.; Khoshneviszadeh M.; Fallah-Bonekohal S.; Abdollahi M.; Foroumadi A.; Shafiee A. Synthesis and Antidiabetic Evaluation of Benzenesulfonamide Derivatives. Iran J. Pharm. Res. 2013, 12, 325–330. [PMC free article] [PubMed] [Google Scholar]
- Morigi R.; Locatelli A.; Leoni A.; Rambaldi M. Recent Patents on Thiazole Derivatives Endowed with Antitumor Activity. Recent Pat. Anti-Cancer Drug Discovery 2015, 10, 280–297. 10.2174/1574892810666150708110432. [DOI] [PubMed] [Google Scholar]
- Pucci M. J.; Bronson J. J.; Barrett J. F.; DenBleyker K. L.; Discotto L. F.; Fung-Tomc J. C.; Ueda Y. Antimicrobial Evaluation of Nocathiacins, a Thiazole Peptide Class of Antibiotics. Antimicrob. Antimicrob. Agents Chemother. 2004, 48, 3697–3701. 10.1128/AAC.48.10.3697-3701.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beno B. R.; Yeung K.-S.; Bartberger M. D.; Pennington L. D.; Meanwell N. A. A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J. Med. Chem. 2015, 58 (11), 4383–4438. 10.1021/jm501853m. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. A Synopsis of the Properties and Applications of Heteroaromatic Rings in Medicinal Chemistry. Adv. Heterocycl. Chem. 2017, 123, 245–361. 10.1016/bs.aihch.2016.11.002. [DOI] [Google Scholar]
- Wolfson J. S.; Hooper D. C. The Fluoroquinolones: Structures, Mechanisms of Action and Resistance, and Spectra of Activity in Vitro. Antimicrob. Agents Chemother. 1985, 28, 581–586. 10.1128/AAC.28.4.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece R. J.; Maxwell A. DNA Gyrase: Structure and Function. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 335–375. 10.3109/10409239109114072. [DOI] [PubMed] [Google Scholar]
- Pietrusiński M.; Staczek P. Bacterial type II topoisomerases as targets for antibacterial drugs. Postepy Biochem. 2006, 52, 271–282. [PubMed] [Google Scholar]
- Bradbury B. J.; Pucci M. J. Recent Advances in Bacterial Topoisomerase Inhibitors. Curr. Opin. Pharmacol. 2008, 8, 574–581. 10.1016/j.coph.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Ronkin S. M.; Badia M.; Bellon S.; Grillot A.-L.; Gross C. H.; Grossman T. H.; Mani N.; Parsons J. D.; Stamos D.; Trudeau M.; Wei Y.; Charifson P. S. Discovery of Pyrazolthiazoles as Novel and Potent Inhibitors of Bacterial Gyrase. Bioorg. Med. Chem. Lett. 2010, 20, 2828–2831. 10.1016/j.bmcl.2010.03.052. [DOI] [PubMed] [Google Scholar]
- Bisacchi G. S.; Manchester J. I. A New-Class Antibacterial—Almost. Lessons in Drug Discovery and Development: A Critical Analysis of More than 50 Years of Effort toward ATPase Inhibitors of DNA Gyrase and Topoisomerase IV. ACS Infect. Dis. 2015, 1, 4–41. 10.1021/id500013t. [DOI] [PubMed] [Google Scholar]
- Stokes N. R.; Thomaides-Brears H. B.; Barker S.; Bennett J. M.; Berry J.; Collins I.; Czaplewski L. G.; Gamble V.; Lancett P.; Logan A.; Lunniss C. J.; Peasley H.; Pommier S.; Price D.; Smee C.; Haydon D. J. Biological Evaluation of Benzothiazole Ethyl Urea Inhibitors of Bacterial Type II Topoisomerases. Antimicrob. Agents Chemother. 2013, 57, 5977–5986. 10.1128/AAC.00719-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill A. J.; Chopra I. Preclinical Evaluation of Novel Antibacterial Agents by Microbiological and Molecular Techniques. Expert Opin. Invest. Drugs 2004, 13, 1045–1063. 10.1517/13543784.13.8.1045. [DOI] [PubMed] [Google Scholar]
- Axford L. C.; Agarwal P. K.; Anderson K. H.; Andrau L. N.; Atherall J.; Barker S.; Bennett J. M.; Blair M.; Collins I.; Czaplewski L. G.; Davies D. T.; Gannon C. T.; Kumar D.; Lancett P.; Logan A.; Lunniss C. J.; Mitchell D. R.; Offermann D. A.; Palmer J. T.; Palmer N.; Pitt G. R.; Pommier S.; Price D.; Narasinga Rao B.; Saxena R.; Shukla T.; Singh A. K.; Singh M.; Srivastava A.; Steele C.; Stokes N. R.; Thomaides-Brears H. B.; Tyndall E. M.; Watson D.; Haydon D. J. Design, Synthesis and Biological Evaluation of α-Substituted Isonipecotic Acid Benzothiazole Analogues as Potent Bacterial Type II Topoisomerase Inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 6598–6603. 10.1016/j.bmcl.2013.10.058. [DOI] [PubMed] [Google Scholar]
- Barančoková M.; Kikelj D.; Ilaš J. Recent Progress in the Discovery and Development of DNA Gyrase B Inhibitors. Future Med. Chem. 2018, 10, 1207–1227. 10.4155/fmc-2017-0257. [DOI] [PubMed] [Google Scholar]
- Brvar M.; Perdih A.; Renko M.; Anderluh G.; Turk D.; Solmajer T. Structure-Based Discovery of Substituted 4,5′-Bithiazoles as Novel DNA Gyrase Inhibitors. J. Med. Chem. 2012, 55, 6413–6426. 10.1021/jm300395d. [DOI] [PubMed] [Google Scholar]
- Liu J.; Li X.-W.; Guo Y.-W. Recent Advances in the Isolation, Synthesis and Biological Activity of Marine Guanidine Alkaloids. Mar. Drugs 2017, 15, 324. 10.3390/md15100324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netz N.; Opatz T. Marine Indole Alkaloids. Mar. Drugs 2015, 13, 4814–4914. 10.3390/md13084814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrino B.; Attanzio A.; Spanò V.; Cascioferro S.; Montalbano A.; Barraja P.; Tesoriere L.; Diana P.; Cirrincione G.; Carbone A. Synthesis, Antitumor Activity and CDK1 Inhibiton of New Thiazole Nortopsentin Analogues. Eur. J. Med. Chem. 2017, 138, 371–383. 10.1016/j.ejmech.2017.06.052. [DOI] [PubMed] [Google Scholar]
- Parrino B.; Schillaci D.; Carnevale I.; Giovannetti E.; Diana P.; Cirrincione G.; Cascioferro S. Synthetic Small Molecules as Anti-Biofilm Agents in the Struggle against Antibiotic Resistance. Eur. J. Med. Chem. 2019, 161, 154–178. 10.1016/j.ejmech.2018.10.036. [DOI] [PubMed] [Google Scholar]
- Ballard T. E.; Richards J. J.; Aquino A.; Reed C. S.; Melander C. Antibiofilm activiy of a diverse oroidin library, generated through reductive acylation. J. Org. Chem. 2009, 74, 1755–1758. 10.1021/jo802260t. [DOI] [PubMed] [Google Scholar]
- Richards J. J.; Ballard T. E.; Huigens R. W.; Melander C. Synthesis and Screening of an Oroidin Library against Pseudomonas Aeruginosa Biofilms. ChemBioChem 2008, 9, 1267–1279. 10.1002/cbic.200700774. [DOI] [PubMed] [Google Scholar]
- Hodnik Ž.; Łoś J. M.; Žula A.; Zidar N.; Jakopin Ž.; Łoś M.; Sollner Dolenc M.; Ilaš J.; Wȩgrzyn G.; Peterlin Mašič L.; Kikelj D. Inhibition of Biofilm Formation by Conformationally Constrained Indole-Based Analogues of the Marine Alkaloid Oroidin. Bioorg. Med. Chem. Lett. 2014, 24, 2530–2534. 10.1016/j.bmcl.2014.03.094. [DOI] [PubMed] [Google Scholar]
- Tomašič T.; Katsamakas S.; Hodnik Ž.; Ilaš J.; Brvar M.; Solmajer T.; Montalvão S.; Tammela P.; Banjanac M.; Ergović G.; Anderluh M.; Peterlin Mašič L.; Kikelj D. Discovery of 4,5,6,7-Tetrahydrobenzo[1,2-d]Thiazoles as Novel DNA Gyrase Inhibitors Targeting the ATP-Binding Site. J. Med. Chem. 2015, 58, 5501–5521. 10.1021/acs.jmedchem.5b00489. [DOI] [PubMed] [Google Scholar]
- Gjorgjieva M.; Tomašič T.; Barančokova M.; Katsamakas S.; Ilaš J.; Tammela P.; Peterlin Mašič L.; Kikelj D. Discovery of Benzothiazole Scaffold-Based DNA Gyrase B Inhibitors. J. Med. Chem. 2016, 59, 8941–8954. 10.1021/acs.jmedchem.6b00864. [DOI] [PubMed] [Google Scholar]
- Tomašič T.; Mirt M.; Barančoková M.; Ilaš J.; Zidar N.; Tammela P.; Kikelj D. Design, Synthesis and Biological Evaluation of 4,5-Dibromo-N-(Thiazol-2-yl)-1H-Pyrrole-2-Carboxamide Derivatives as Novel DNA Gyrase Inhibitors. Bioorg. Med. Chem. 2017, 25, 338–349. 10.1016/j.bmc.2016.10.038. [DOI] [PubMed] [Google Scholar]
- Haroun M.; Tratrat C.; Kositzi K.; Tsolaki E.; Petrou A.; Aldhubiab B.; Attimarad M.; Harsha S.; Geronikaki A.; Venugopala K. N.; Elsewedy H. S.; Sokovic M.; Glamoclija J.; Ciric A. New Benzothiazole-Based Thiazolidinones as Potent Antimicrobial Agents. Design, Synthesis and Biological Evaluation. Curr. Top. Med. Chem. 2018, 18, 75–87. 10.2174/1568026618666180206101814. [DOI] [PubMed] [Google Scholar]
- Maddili S. K.; Li Z.-Z.; Kannekanti V. K.; Bheemanaboina R. R. Y.; Tuniki B.; Tangadanchu V. K. R.; Zhou C.-H. Azoalkyl Ether Imidazo[2,1-b]Benzothiazoles as Potentially Antimicrobial Agents with Novel Structural Skeleton. Bioorg. Med. Chem. Lett. 2018, 28, 2426–2431. 10.1016/j.bmcl.2018.06.016. [DOI] [PubMed] [Google Scholar]
- Hameed A.; Al-Rashida M.; Uroos M.; Abid Ali S.; Khan K. M. Schiff bases in medicinal chemistry: a patent review (2010–2015). Expert Opin. Ther. Pat. 2017, 27, 63–79. 10.1080/13543776.2017.1252752. [DOI] [PubMed] [Google Scholar]
- Nastasǎ C.; Vodnar D. C.; Ionuţ I.; Stana A.; Benedec D.; Tamaian R.; Oniga O.; Tiperciuc B. Antibacterial Evaluation and Virtual Screening of New Thiazolyl-Triazole Schiff Bases as Potential DNA-Gyrase Inhibitors. Int. J. Mol. Sci. 2018, 19, 222 10.3390/ijms19010222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eakin A. E.; Green O.; Hales N.; Walkup G. K.; Bist S.; Singh A.; Mullen G.; Bryant J.; Embrey K.; Gao N.; Breeze A.; Timms D.; Andrews B.; Uria-Nickelsen M.; Demeritt J.; Loch J. T. 3rd; Hull K.; Blodgett A.; Illingworth R. N.; Prince B.; Boriack-Sjodin P. A.; Hauck S.; MacPherson L. J.; Ni H.; Sherer B. Pyrrolamide DNA Gyrase Inhibitors: Fragment-Based Nuclear Magnetic Resonance Screening to Identify Antibacterial Agents. Antimicrob. Agents Chemother. 2012, 56, 1240–1246. 10.1128/AAC.05485-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer B. A.; Hull K.; Green O.; Basarab G.; Hauck S.; Hill P.; Loch J. T.; Mullen G.; Bist S.; Bryant J.; Boriack-Sjodin A.; Read J.; DeGrace N.; Uria-Nickelsen M.; Illingworth R. N.; Eakin A. E. Pyrrolamide DNA Gyrase Inhibitors: Optimization of Antibacterial Activity and Efficacy. Bioorg. Med. Chem. Lett. 2011, 21, 7416–7420. 10.1016/j.bmcl.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Basarab G. S.; Hill P. J.; Garner C. E.; Hull K.; Green O.; Sherer B. A.; Dangel P. B.; Manchester J. I.; Bist S.; Hauck S.; Zhou F.; Uria-Nickelsen M.; Illingworth R. N.; Alm R.; Rooney M.; Eakin A. E. Optimization of Pyrrolamide Topoisomerase II Inhibitors Toward Identification of an Antibacterial Clinical Candidate (AZD5099). J. Med. Chem. 2014, 57, 6060–6082. 10.1021/jm500462x. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Severin A.; Chopra R.; Krishnamurthy G.; Singh G.; Hu W.; Keeney D.; Svenson K.; Petersen P. J.; Labthavikul P.; Shlaes D. M.; Rasmussen B. A.; Failli A. A.; Shumsky J. S.; Kutterer K. M.; Gilbert A.; Mansour T. S. 3,5-Dioxopyrazolidines, Novel Inhibitors of UDP-N- Acetylenolpyruvylglucosamine Reductase (MurB) with Activity against Gram-Positive Bacteria. Antimicrob. Agents Chemother. 2006, 50, 556–564. 10.1128/AAC.50.2.556-564.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson J. J.; DenBleyker K. L.; Falk P. J.; Mate R. A.; Ho H.-T.; Pucci M. J.; Snyder L. B. Discovery of the First Antibacterial Small Molecule Inhibitors of MurB. Bioorg. Med. Chem. Lett. 2003, 13, 873–875. 10.1016/S0960-894X(02)01076-4. [DOI] [PubMed] [Google Scholar]
- Liaras K.; Fesatidou M.; Geronikaki A. Thiazoles and Thiazolidinones as COX/LOX Inhibitors. Molecules 2018, 23, 685. 10.3390/molecules23030685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattan S. R.; Kekare P.; Patil A.; Nikalje A.; Kittur B. S. Studies on the Synthesis of Novel 2,4-Thiazolidinedione Derivatives with Antidiabetic Activity. Iran. J. Pharm. Sci. 2009, 5, 225–230. [Google Scholar]
- Suryawanshi R.; Jadhav S.; Makwana N.; Desai D.; Chaturbhuj D.; Sonawani A.; Idicula-Thomas S.; Murugesan V.; Katti S. B.; Tripathy S.; Paranjape R.; Kulkarni S. Evaluation of 4-Thiazolidinone Derivatives as Potential Reverse Transcriptase Inhibitors against HIV-1 Drug Resistant Strains. Bioorg. Chem. 2017, 71, 211–218. 10.1016/j.bioorg.2017.02.007. [DOI] [PubMed] [Google Scholar]
- Kesicki E. A.; Bailey M. A.; Ovechkina Y.; Early J. V.; Alling T.; Bowman J.; Zuniga E. S.; Dalai S.; Kumar N.; Masquelin T.; Hipskind P. A.; Odingo J. O.; Parish T. Synthesis and Evaluation of the 2-Aminothiazoles as Anti-Tubercular Agents. PLoS One 2016, 11, e0155209 10.1371/journal.pone.0155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques G. H.; Kunzler A.; Bareño V. D. O.; Drawanz B. B.; Mastelloto H. G.; Leite F. R. M.; Nascimento G. G.; Nascente P. S.; Siqueira G. M.; Cunico W. Antifungal Activity of 3-(Heteroaryl-2-yl-methyl)Thiazolidinone Derivatives. Med. Chem. 2014, 10, 355–360. 10.2174/15734064113099990030. [DOI] [PubMed] [Google Scholar]
- Gupta A.; Singh R.; Sonar P. K.; Saraf S. K. Novel 4-Thiazolidinone Derivatives as Anti-Infective Agents: Synthesis, Characterization, and Antimicrobial Evaluation. Biochem. Res. Int. 2016, 2016 (216), 8086762. 10.1155/2016/8086762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres C. J.; Bronson J. J.; D’Andrea S. V.; Deshpande M. S.; Falk P. J.; Grant-Young K. A.; Harte W. E.; Ho H.-T.; Misco P. F.; Robertson J. G.; Stock D.; Sun Y.; Walsh A. W. 4-Thiazolidinones: Novel Inhibitors of the Bacterial Enzyme MurB. Bioorg. Med. Chem. Lett. 2000, 10, 715–717. 10.1016/S0960-894X(00)00073-1. [DOI] [PubMed] [Google Scholar]
- Patel H.; Mishra L.; Noolvi M.; Karpoormath R.; Singh Cameotra S. Synthesis, in Vitro Evaluation, and Molecular Docking Studies of Azetidinones and Thiazolidinones of 2-Amino-5-Cyclopropyl-1,3,4-Thiadiazole as Antibacterial Agents. Arch. Pharm. 2014, 347, 668–684. 10.1002/ardp.201400140. [DOI] [PubMed] [Google Scholar]
- Ito A.; Nishikawa T.; Matsumoto S.; Yoshizawa H.; Sato T.; Nakamura R.; Tsuji M.; Yamano Y. Siderophore Cephalosporin Cefiderocol Utilizes Ferric Iron Transporter Systems for Antibacterial Activity against Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 7396–7401. 10.1128/AAC.01405-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhanel G. G.; Golden A. R.; Zelenitsky S.; Wiebe K.; Lawrence C. K.; Adam H. J.; Idowu T.; Domalaon R.; Schweizer F.; Zhanel M. A.; Lagacé-Wiens P. R. S.; Walkty A. J.; Noreddin A.; Lynch J. P. III; Karlowsky J. A. Cefiderocol: A Siderophore Cephalosporin with Activity Against Carbapenem-Resistant and Multidrug-Resistant Gram-Negative Bacilli. Drugs 2019, 79, 271–289. 10.1007/s40265-019-1055-2. [DOI] [PubMed] [Google Scholar]
- Han S.; Zaniewski R. P.; Marr E. S.; Lacey B. M.; Tomaras A. P.; Evdokimov A.; Miller J. R.; Shanmugasundaram V. Structural Basis for Effectiveness of Siderophore-Conjugated Monocarbams against Clinically Relevant Strains of Pseudomonas Aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 22002–22007. 10.1073/pnas.1013092107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvage E.; Terrak M. Glycosyltransferases and Transpeptidases/Penicillin-Binding Proteins: Valuable Targets for New Antibacterials. Antibiotics 2016, 5, 12. 10.3390/antibiotics5010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn G. L. Ceftizoxime and Other Third-Generation Cephalosporins: Structure-Activity Relationships. J. Antimicrob. Chemother. 1982, 10, 1–10. 10.1093/jac/10.suppl_C.1. [DOI] [PubMed] [Google Scholar]
- Kohira N.; West J.; Ito A.; Ito-Horiyama T.; Nakamura R.; Sato T.; Rittenhouse S.; Tsuji M.; Yamano Y. In Vitro Antimicrobial Activity of a Siderophore Cephalosporin, S-649266, against Enterobacteriaceae Clinical Isolates, Including Carbapenem-Resistant Strains. Antimicrob. Agents Chemother. 2016, 60, 729–734. 10.1128/AAC.01695-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portsmouth S.; van Veenhuyzen D.; Echols R.; Machida M.; Ferreira J. C. A.; Ariyasu M.; Tenke P.; Nagata T. D. Cefiderocol versus Imipenem-Cilastatin for the Treatment of Complicated Urinary Tract Infections Caused by Gram-Negative Uropathogens: A Phase 2, Randomised, Double-Blind, Non-Inferiority Trial. Lancet Infect. Dis. 2018, 18, 1319–1328. 10.1016/S1473-3099(18)30554-1. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Meredith T. C.; Kahne D. On the Essentiality of Lipopolysaccharide to Gram-Negative Bacteria. Curr. Opin. Microbiol. 2013, 16, 779–785. 10.1016/j.mib.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneidinger B.; Marolda C.; Graninger M.; Zamyatina A.; McArthur F.; Kosma P.; Valvano M. A.; Messner P. Biosynthesis Pathway of ADP-L-Glycero-Beta-D-Manno-Heptose in Escherichia Coli. J. Bacteriol. 2002, 184, 363–369. 10.1128/JB.184.2.363-369.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desroy N.; Moreau F.; Briet S.; Le Fralliec G.; Floquet S.; Durant L.; Vongsouthi V.; Gerusz V.; Denis A.; Escaich S. Towards Gram-Negative Antivirulence Drugs: New Inhibitors of HldE Kinase. Bioorg. Med. Chem. 2009, 17, 1276–1289. 10.1016/j.bmc.2008.12.021. [DOI] [PubMed] [Google Scholar]
- De Leon G. P.; Elowe N. H.; Koteva K. P.; Valvano M. A.; Wright G. D. An in Vitro Screen of Bacterial Lipopolysaccharide Biosynthetic Enzymes Identifies an Inhibitor of ADP-Heptose Biosynthesis. Chem. Biol. 2006, 13, 437–441. 10.1016/j.chembiol.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Caroff M.; Karibian D. Structure of Bacterial Lipopolysaccharides. Carbohydr. Res. 2003, 338, 2431–2447. 10.1016/j.carres.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Singh M.; Kumar Singh S.; Gangwar M.; Sellamuthu S.; Nath G.; Singh S. K. Design, Synthesis and Mode of Action of Some New 2-(4’-Aminophenyl) Benzothiazole Derivatives as Potent Antimicrobial Agents. Lett. Drug Design Discovery 2016, 13, 429–437. 10.2174/1570180812666150821003220. [DOI] [Google Scholar]
- Rajendran V.; Kalita P.; Shukla H.; Kumar A.; Tripathi T. Aminoacyl-TRNA Synthetases: Structure, Function, and Drug Discovery. Int. J. Biol. Macromol. 2018, 111, 400–414. 10.1016/j.ijbiomac.2017.12.157. [DOI] [PubMed] [Google Scholar]
- Giegé R.; Springer M. Aminoacyl-TRNA Synthetases in the Bacterial World. EcoSal Plus 2016, 7, ESP-0002-2016. 10.1128/ecosalplus.ESP-0002-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stana A.; Vodnar D. C.; Marc G.; Benedec D.; Tiperciuc B.; Tamaian R.; Oniga O. Antioxidant Activity and Antibacterial Evaluation of New Thiazolin-4-One Derivatives as Potential Tryptophanyl-TRNA Synthetase Inhibitors. J. Enzyme Inhib. Med. Chem. 2019, 34, 898–908. 10.1080/14756366.2019.1596086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascioferro S.; Totsika M.; Schillaci D. Sortase A: An Ideal Target for Anti-Virulence Drug Development. Microb. Pathog. 2014, 77C, 105–112. 10.1016/j.micpath.2014.10.007. [DOI] [PubMed] [Google Scholar]
- Oniga S. D.; Araniciu C.; Palage M. D.; Popa M.; Chifiriuc M.-C.; Marc G.; Pirnau A.; Stoica C. I.; Lagoudis I.; Dragoumis T.; Oniga O. New 2-Phenylthiazoles as Potential Sortase A Inhibitors: Synthesis, Biological Evaluation and Molecular Docking. Molecules 2017, 22, 1827. 10.3390/molecules22111827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrino B.; Diana P.; Cirrincione G.; Cascioferro S. Bacterial Biofilm Inhibition in the Development of Effective Anti-Virulence Strategy. Open Med. Chem. J. 2018, 12, 84–87. 10.2174/1874104501812010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.-H.; Lee J.-H. Antibiofilm Agents: A New Perspective for Antimicrobial Strategy. J. Microbiol. 2017, 55, 753–766. 10.1007/s12275-017-7274-x. [DOI] [PubMed] [Google Scholar]
- Cascioferro S.; Parrino B.; Petri G. L.; Cusimano M. G.; Schillaci D.; Di Sarno V.; Musella S.; Giovannetti E.; Cirrincione G.; Diana P. 2,6-Disubstituted Imidazo[2,1-b][1,3,4]Thiadiazole Derivatives as Potent Staphylococcal Biofilm Inhibitors. Eur. J. Med. Chem. 2019, 167, 200–210. 10.1016/j.ejmech.2019.02.007. [DOI] [PubMed] [Google Scholar]
- Qin Z.; Zhang J.; Xu B.; Chen L.; Wu Y.; Yang X.; Shen X.; Molin S.; Danchin A.; Jiang H.; Qu D. Structure-Based Discovery of Inhibitors of the YycG Histidine Kinase: New Chemical Leads to Combat Staphylococcus Epidermidis Infections. BMC Microbiol. 2006, 6, 96. 10.1186/1471-2180-6-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B.; Huang R.; Zheng L.; Chen C.; Han S.; Qu D.; Zhu M.; Wei P. Thiazolidione Derivatives as Novel Antibiofilm Agents: Design, Synthesis, Biological Evaluation, and Structure-Activity Relationships. Eur. J. Med. Chem. 2011, 46, 819–824. 10.1016/j.ejmech.2010.12.014. [DOI] [PubMed] [Google Scholar]
- Pan B.; Huang R.-Z.; Han S.-Q.; Qu D.; Zhu M.-L.; Wei P.; Ying H.-J. Design, Synthesis, and Antibiofilm Activity of 2-Arylimino-3-Aryl-Thiazolidine-4-Ones. Bioorg. Med. Chem. Lett. 2010, 20, 2461–2464. 10.1016/j.bmcl.2010.03.013. [DOI] [PubMed] [Google Scholar]
- Rane R. A.; Sahu N. U.; Shah C. P. Synthesis and Antibiofilm Activity of Marine Natural Product-Based 4-Thiazolidinones Derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 7131–7134. 10.1016/j.bmcl.2012.09.073. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Chen C.; Liu H.; Zheng L.; Tong Y.; Qu D.; Han S. Biological Evaluation of Halogenated Thiazolo[3,2-a]Pyrimidin-3-One Carboxylic Acid Derivatives Targeting the YycG Histidine Kinase. Eur. J. Med. Chem. 2014, 87, 500–507. 10.1016/j.ejmech.2014.09.096. [DOI] [PubMed] [Google Scholar]
- Siddiqui N.; Arya S. K.; Ahsan W.; Azad B. Diverse Biological Activities of Thiazoles: A Retrospect. Int.J. Drug Develop. Res. 2011, 3, 55–57. [Google Scholar]
- da Silva C. M.; da Silva D. L.; Modolo L. V.; Alves R. B.; de Resende M. A.; Martins C. V. B. de; de Fátima Â. Schiff Bases: A Short Review of Their Antimicrobial Activities. J. Advanc. Res. 2011, 2, 1–8. 10.1016/j.jare.2010.05.004. [DOI] [Google Scholar]
- More P. G.; Karale N. N.; Lawand A. S.; Narang N.; Patil R. H. Synthesis and Anti-Biofilm Activity of Thiazole Schiff Bases. Med. Chem. Res. 2014, 23, 790–799. 10.1007/s00044-013-0672-7. [DOI] [Google Scholar]
- Ayati A.; Emami S.; Asadipour A.; Shafiee A.; Foroumadi A. Recent Applications of 1,3-Thiazole Core Structure in the Identification of New Lead Compounds and Drug Discovery. Eur. J. Med. Chem. 2015, 97, 699–718. 10.1016/j.ejmech.2015.04.015. [DOI] [PubMed] [Google Scholar]
- Stefanska J.; Nowicka G.; Struga M.; Szulczyk D.; Koziol A. E.; Augustynowicz-Kopec E.; Napiorkowska A.; Bielenica A.; Filipowski W.; Filipowska A.; Drzewiecka A.; Giliberti G.; Madeddu S.; Boi S.; La Colla P.; Sanna G. Antimicrobial and Anti-Biofilm Activity of Thiourea Derivatives Incorporating a 2-Aminothiazole Scaffold. Chem. Pharm. Bull. 2015, 63, 225–236. 10.1248/cpb.c14-00837. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. Natural Products as Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins A.; Vieira H.; Gaspar H.; Santos S. Marketed Marine Natural Products in the Pharmaceutical and Cosmeceutical Industries: Tips for Success. Mar. Drugs 2014, 12, 1066–1101. 10.3390/md12021066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascioferro S.; Attanzio A.; Di Sarno V.; Musella S.; Tesoriere L.; Cirrincione G.; Diana P.; Parrino B. New 1,2,4-Oxadiazole Nortopsentin Derivatives with Cytotoxic Activity. Mar. Drugs 2019, 17, 35. 10.3390/md17010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanò V.; Attanzio A.; Cascioferro S.; Carbone A.; Montalbano A.; Barraja P.; Tesoriere L.; Cirrincione G.; Diana P.; Parrino B. Synthesis and Antitumor Activity of New Thiazole Nortopsentin Analogs. Mar. Drugs 2016, 14, 226. 10.3390/md14120226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrino B.; Carbone A.; Ciancimino C.; Spanò V.; Montalbano A.; Barraja P.; Cirrincione G.; Diana P.; Sissi C.; Palumbo M.; Pinato O.; Pennati M.; Beretta G.; Folini M.; Matyus P.; Balogh B.; Zaffaroni N. Water-Soluble Isoindolo[2,1-a]Quinoxalin-6-Imines: In Vitro Antiproliferative Activity and Molecular Mechanism(s) of Action. Eur. J. Med. Chem. 2015, 94, 149–162. 10.1016/j.ejmech.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Oh K.-B.; Mar W.; Kim S.; Kim J.-Y.; Lee T.-H.; Kim J.-G.; Shin D.; Sim C. J.; Shin J. Antimicrobial Activity and Cytotoxicity of Bis(Indole) Alkaloids from the Sponge Spongosorites Sp. Biol. Pharm. Bull. 2006, 29, 570–573. 10.1248/bpb.29.570. [DOI] [PubMed] [Google Scholar]
- Oh K.-B.; Mar W.; Kim S.; Kim J.-Y.; Oh M.-N.; Kim J.-G.; Shin D.; Sim C. J.; Shin J. Bis(Indole) Alkaloids as Sortase A Inhibitors from the Sponge Spongosorites Sp. Bioorg. Med. Chem. Lett. 2005, 15, 4927–4931. 10.1016/j.bmcl.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Gul W.; Hamann M. T. Indole Alkaloid Marine Natural Products: An Established Source of Cancer Drug Leads with Considerable Promise for the Control of Parasitic, Neurological and Other Diseases. Life Sci. 2005, 78, 442–453. 10.1016/j.lfs.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright A. E.; Pomponi S. A.; Cross S. S.; McCarthy P. A New Bis-(Indole) Alkaloid from a Deep-Water Marine Sponge of the Genus Spongosorites. J. Org. Chem. 1992, 57, 4772–4775. 10.1021/jo00043a045. [DOI] [Google Scholar]
- Bao B.; Sun Q.; Yao X.; Hong J.; Lee C.-O.; Sim C. J.; Im K. S.; Jung J. H. Cytotoxic Bisindole Alkaloids from a Marine Sponge Spongosorites Sp. J. Nat. Prod. 2005, 68, 711–715. 10.1021/np049577a. [DOI] [PubMed] [Google Scholar]
- Sun H. H.; Sakemi S.; Gunasekera S.; Kashman Y.; Lui M.; Burres N.; McCarthy P.. Bis-Indole Imidazole Compounds Which Are Useful Antitumor and Antimicrobial Agents. US 4970226A, 1990.
- Carbone A.; Parrino B.; Cusimano M. G.; Spanò V.; Montalbano A.; Barraja P.; Schillaci D.; Cirrincione G.; Diana P.; Cascioferro S. New Thiazole Nortopsentin Analogues Inhibit Bacterial Biofilm Formation. Mar. Drugs 2018, 16, 274. 10.3390/md16080274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian S. K.; Liu G.; Jensen E. R.; Lenoy E.; Schneewind O. Staphylococcus Aureus Sortase Mutants Defective in the Display of Surface Proteins and in the Pathogenesis of Animal Infections. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 5510–5515. 10.1073/pnas.080520697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascioferro S.; Raffa D.; Maggio B.; Raimondi M. V.; Schillaci D.; Daidone G. Sortase A Inhibitors: Recent Advances and Future Perspectives. J. Med. Chem. 2015, 58, 9108–9123. 10.1021/acs.jmedchem.5b00779. [DOI] [PubMed] [Google Scholar]
- Chen L.; Yang D.; Pan Z.; Lai L.; Liu J.; Fang B.; Shi S. Synthesis and Antimicrobial Activity of the Hybrid Molecules between Sulfonamides and Active Antimicrobial Pleuromutilin Derivative. Chem. Biol. Drug Des. 2015, 86, 239–245. 10.1111/cbdd.12486. [DOI] [PubMed] [Google Scholar]
- Klahn P.; Brönstrup M. Bifunctional Antimicrobial Conjugates and Hybrid Antimicrobials. Nat. Prod. Rep. 2017, 34, 832–885. 10.1039/C7NP00006E. [DOI] [PubMed] [Google Scholar]
- Gondru R.; Sirisha K.; Raj S.; Gunda S. K.; Kumar C. G.; Pasupuleti M.; Bavantula R. Design, Synthesis, In Vitro Evaluation and Docking Studies of Pyrazole-Thiazole Hybrids as Antimicrobial and Antibiofilm Agents. ChemistrySelect 2018, 3, 8270–8276. 10.1002/slct.201801391. [DOI] [Google Scholar]
- Horwich A. L.; Farr G. W.; Fenton W. A. GroEL-GroES-Mediated Protein Folding. Chem. Rev. 2006, 106, 1917–1930. 10.1021/cr040435v. [DOI] [PubMed] [Google Scholar]
- Johnson S. M.; Sharif O.; Mak P. A.; Wang H.-T.; Engels I. H.; Brinker A.; Schultz P. G.; Horwich A. L.; Chapman E. A Biochemical Screen for GroEL/GroES Inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 786–789. 10.1016/j.bmcl.2013.12.100. [DOI] [PubMed] [Google Scholar]
- Abdeen S.; Salim N.; Mammadova N.; Summers C. M.; Frankson R.; Ambrose A. J.; Anderson G. G.; Schultz P. G.; Horwich A. L.; Chapman E.; Johnson S. M. GroEL/ES Inhibitors as Potential Antibiotics. Bioorg. Med. Chem. Lett. 2016, 26, 3127–3134. 10.1016/j.bmcl.2016.04.089. [DOI] [PubMed] [Google Scholar]
- Kim S.; Lieberman T. D.; Kishony R. Alternating Antibiotic Treatments Constrain Evolutionary Paths to Multidrug Resistance. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 14494–14499. 10.1073/pnas.1409800111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeswanth S.; Chandra Sekhar K.; Chaudhary A.; Sarma P. V. G. K. Anti-Microbial and Anti-Biofilm Activity of a Novel Dibenzyl (Benzo[d] Thiazol-2-yl-(Hydroxy)-Methyl) Phosphonate by Inducing Protease Expression in Staphylococcus Aureus. Med. Chem. Res. 2018, 27, 785–795. 10.1007/s00044-017-2102-8. [DOI] [Google Scholar]
- Hymes J. P.; Klaenhammer T. R. Stuck in the Middle: Fibronectin-Binding Proteins in Gram-Positive Bacteria. Front. Microbiol. 2016, 7, 1504. 10.3389/fmicb.2016.01504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin M. J.; Nivens D. E.; Weadge J. T.; Howell P. L. Biosynthesis of the Pseudomonas Aeruginosa Extracellular Polysaccharides, Alginate, Pel, and Psl. Front. Microbiol. 2011, 2, 167. 10.3389/fmicb.2011.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin K. M.; Irie Y.; Tart C. S.; Urbano R.; Whitney J. C.; Ryder C.; Howell P. L.; Wozniak D. J.; Parsek M. R. The Pel and Psl Polysaccharides Provide Pseudomonas Aeruginosa Structural Redundancy within the Biofilm Matrix. Environ. Microbiol. 2012, 14, 1913–1928. 10.1111/j.1462-2920.2011.02657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin K. M.; Gordon V. D.; Murakami K.; Borlee B. R.; Wozniak D. J.; Wong G. C. L.; Parsek M. R. The Pel Polysaccharide Can Serve a Structural and Protective Role in the Biofilm Matrix of Pseudomonas Aeruginosa. PLoS Pathog. 2011, 7, e1001264 10.1371/journal.ppat.1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings N.; Ramirez Millan M.; Caldara M.; Rusconi R.; Tarasova Y.; Stocker R.; Ribbeck K. The Extracellular Matrix Component Psl Provides Fast-Acting Antibiotic Defense in Pseudomonas Aeruginosa Biofilms. PLoS Pathog. 2013, 9, e1003526 10.1371/journal.ppat.1003526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tilburg Bernardes E.; Charron-Mazenod L.; Reading D. J.; Reckseidler-Zenteno S. L.; Lewenza S. Exopolysaccharide-Repressing Small Molecules with Antibiofilm and Antivirulence Activity against Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2017, 61, e01997-16 10.1128/AAC.01997-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascioferro S.; Cusimano M. G.; Schillaci D. Antiadhesion Agents against Gram-Positive Pathogens. Future Microbiol. 2014, 9, 1209–1220. 10.2217/fmb.14.56. [DOI] [PubMed] [Google Scholar]
- Berne C.; Ellison C. K.; Ducret A.; Brun Y. V. Bacterial Adhesion at the Single-Cell Level. Nat. Rev. Microbiol. 2018, 16, 616–627. 10.1038/s41579-018-0057-5. [DOI] [PubMed] [Google Scholar]
- Terlizzi M. E.; Gribaudo G.; Maffei M. E. UroPathogenic Escherichia Coli (UPEC) Infections: Virulence Factors, Bladder Responses, Antibiotic, and Non-Antibiotic Antimicrobial Strategies. Front. Microbiol. 2017, 8, 1566. 10.3389/fmicb.2017.01566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorell E.; Pinkner J. S.; Phan G.; Edvinsson S.; Buelens F.; Remaut H.; Waksman G.; Hultgren S. J.; Almqvist F. Design and Synthesis of C-2 Substituted Thiazolo and Dihydrothiazolo Ring-Fused 2-Pyridones: Pilicides with Increased Antivirulence Activity. J. Med. Chem. 2010, 53, 5690–5695. 10.1021/jm100470t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahales P.; Hoffman P. S.; Thanassi D. G. Nitazoxanide Inhibits Pilus Biogenesis by Interfering with Folding of the Usher Protein in the Outer Membrane. Antimicrob. Agents Chemother. 2016, 60, 2028–2038. 10.1128/AAC.02221-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opperman T. J.; Nguyen S. T. Recent Advances toward a Molecular Mechanism of Efflux Pump Inhibition. Front. Microbiol. 2015, 6, 421. 10.3389/fmicb.2015.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz S.; Altinkanat-Gelmez G.; Bolelli K.; Guneser-Merdan D.; Ufuk Over-Hasdemir M.; Aki-Yalcin E.; Yalcin I. Binding Site Feature Description of 2-Substituted Benzothiazoles as Potential AcrAB-TolC Efflux Pump Inhibitors in E. Coli. SAR QSAR Environ. Res. 2015, 26, 853–871. 10.1080/1062936X.2015.1106581. [DOI] [PubMed] [Google Scholar]
- Naaz F.; Srivastava R.; Singh A.; Singh N.; Verma R.; Singh V. K.; Singh R. K. Molecular Modeling, Synthesis, Antibacterial and Cytotoxicity Evaluation of Sulfonamide Derivatives of Benzimidazole, Indazole, Benzothiazole and Thiazole. Bioorg. Med. Chem. 2018, 26, 3414–3428. 10.1016/j.bmc.2018.05.015. [DOI] [PubMed] [Google Scholar]
- Abu-Melha S.; Edrees M. M.; Salem H. H.; Kheder N. A.; Gomha S. M.; Abdelaziz M. R. Synthesis and Biological Evaluation of Some Novel Thiazole-Based Heterocycles as Potential Anticancer and Antimicrobial Agents. Molecules 2019, 24, 539. 10.3390/molecules24030539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy N. B.; Zyryanov G. V.; Reddy G. M.; Balakrishna A.; Padmaja A.; Padmavathi V.; Reddy C. S.; Garcia J. R.; Sravya G. Design and Synthesis of Some New Benzimidazole Containing Pyrazoles and Pyrazolyl Thiazoles as Potential Antimicrobial Agents. J. Heterocyclic Chem. 2019, 56, 589–596. 10.1002/jhet.3435. [DOI] [Google Scholar]
- Franchini C.; Muraglia M.; Corbo F.; Florio M. A.; Di Mola A.; Rosato A.; Matucci R.; Nesi M.; van Bambeke F.; Vitali C. Synthesis and Biological Evaluation of 2-Mercapto-1,3-Benzothiazole Derivatives with Potential Antimicrobial Activity. Arch. Pharm. 2009, 342, 605–613. 10.1002/ardp.200900092. [DOI] [PubMed] [Google Scholar]
- Cindrić M.; Perić M.; Kralj M.; Martin-Kleiner I.; David-Cordonnier M.-H.; Paljetak H. Č.; Matijašić M.; Verbanac D.; Karminski-Zamola G.; Hranjec M. Antibacterial and Antiproliferative Activity of Novel 2-Benzimidazolyl- and 2-Benzothiazolyl-Substituted Benzo[b]Thieno-2-Carboxamides. Mol. Diversity 2018, 22, 637–646. 10.1007/s11030-018-9822-7. [DOI] [PubMed] [Google Scholar]
- Zha G.-F.; Leng J.; Darshini N.; Shubhavathi T.; Vivek H. K.; Asiri A. M.; Marwani H. M.; Rakesh K. P.; Mallesha N.; Qin H.-L. Synthesis, SAR and Molecular Docking Studies of Benzo[d]Thiazole-Hydrazones as Potential Antibacterial and Antifungal Agents. Bioorg. Med. Chem. Lett. 2017, 27, 3148–3155. 10.1016/j.bmcl.2017.05.032. [DOI] [PubMed] [Google Scholar]
- Stenger-Smith J.; Chakraborty I.; Mascharak P. K. Cationic Au(I) Complexes with Aryl-Benzothiazoles and Their Antibacterial Activity. J. Inorg. Biochem. 2018, 185, 80–85. 10.1016/j.jinorgbio.2018.05.003. [DOI] [PubMed] [Google Scholar]
- Maddili S. K.; Katla R.; Kannekanti V. K.; Bejjanki N. K.; Tuniki B.; Zhou C.-H.; Gandham H. Molecular Interaction of Novel Benzothiazolyl Triazolium Analogues with Calf Thymus DNA and HSA-Their Biological Investigation as Potent Antimicrobial Agents. Eur. J. Med. Chem. 2018, 150, 228–247. 10.1016/j.ejmech.2018.02.056. [DOI] [PubMed] [Google Scholar]
- Omar K.; Geronikaki A.; Zoumpoulakis P.; Camoutsis C.; Soković M.; Cirić A.; Glamoclija J. Novel 4-Thiazolidinone Derivatives as Potential Antifungal and Antibacterial Drugs. Bioorg. Med. Chem. 2010, 18, 426–432. 10.1016/j.bmc.2009.10.041. [DOI] [PubMed] [Google Scholar]
- Deep A.; Jain S.; Sharma P. C.; Mittal S. K.; Phogat P.; Malhotra M. Synthesis, Characterization and Antimicrobial Evaluation of 2,5-Disubstituted-4-Thiazolidinone Derivatives. Arabian J. Chem. 2014, 7, 287–291. 10.1016/j.arabjc.2010.10.032. [DOI] [Google Scholar]
- Angapelly S.; Sri Ramya P. V.; SunithaRani R.; Kumar C. G.; Kamal A.; Arifuddin M. Ultrasound Assisted, VOSO4 Catalyzed Synthesis of 4-Thiazolidinones: Antimicrobial Evaluation of Indazole-4-Thiazolidinone Derivatives. Tetrahedron Lett. 2017, 58, 4632–4637. 10.1016/j.tetlet.2017.10.070. [DOI] [Google Scholar]
- Pitta E.; Tsolaki E.; Geronikaki A.; Petrović J.; Glamočlija J.; Soković M.; Crespan E.; Maga G.; Bhunia S. S.; Saxena A. K. 4-Thiazolidinone Derivatives as Potent Antimicrobial Agents: Microwave-Assisted Synthesis, Biological Evaluation and Docking Studies. MedChemComm 2015, 6, 319–326. 10.1039/C4MD00399C. [DOI] [Google Scholar]
- Ahmed S.; Zayed M. F.; El-Messery S. M.; Al-Agamy M. H.; Abdel-Rahman H. M. Design, Synthesis, Antimicrobial Evaluation and Molecular Modeling Study of 1,2,4-Triazole-Based 4-Thiazolidinones. Molecules 2016, 21, 568. 10.3390/molecules21050568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deep A.; Narasimhan B.; Lim S. M.; Ramasamy K.; Mishra R. K.; Mani V. 4-Thiazolidinone Derivatives: Synthesis, Antimicrobial, Anticancer Evaluation and QSAR Studies. RSC Adv. 2016, 6, 109485–109494. 10.1039/C6RA23006G. [DOI] [Google Scholar]
- El-Sayed E. H.; Fadda A. A. Synthesis and Antimicrobial Activity of Some Novel Bis Polyfunctional Pyridine, Pyran, and Thiazole Derivatives. J. Heterocyclic Chem. 2018, 55, 2251–2260. 10.1002/jhet.3276. [DOI] [Google Scholar]
- Devine S. M.; Mulcair M. D.; Debono C. O.; Leung E. W. W.; Nissink J. W. M.; Lim S. S.; Chandrashekaran I. R.; Vazirani M.; Mohanty B.; Simpson J. S.; Baell J. B.; Scammells P. J.; Norton R. S.; Scanlon M. J. Promiscuous 2-Aminothiazoles (PrATs): A Frequent Hitting Scaffold. J. Med. Chem. 2015, 58, 1205–1214. 10.1021/jm501402x. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Nissink J. W. M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. 10.1021/acschembio.7b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Dahlin J. L.; Nissink J. W. M.; Strasser J. M.; Francis S.; Higgins L.; Zhou H.; Zhang Z.; Walters M. A. PAINS in the Assay: Chemical Mechanisms of Assay Interference and Promiscuous Enzymatic Inhibition Observed during a Sulfhydryl-Scavenging HTS. J. Med. Chem. 2015, 58, 2091–2113. 10.1021/jm5019093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman H. J.; Davies K. J. A.; Ursini F. How Do Nutritional Antioxidants Really Work: Nucleophilic Tone and Para-Hormesis versus Free Radical Scavenging in Vivo. Free Radical Biol. Med. 2014, 66, 24–35. 10.1016/j.freeradbiomed.2013.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminskyy D.; Kryshchyshyn A.; Lesyk R. 5-Ene-4-Thiazolidinones - An Efficient Tool in Medicinal Chemistry. Eur. J. Med. Chem. 2017, 140, 542–594. 10.1016/j.ejmech.2017.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]