

Abstract
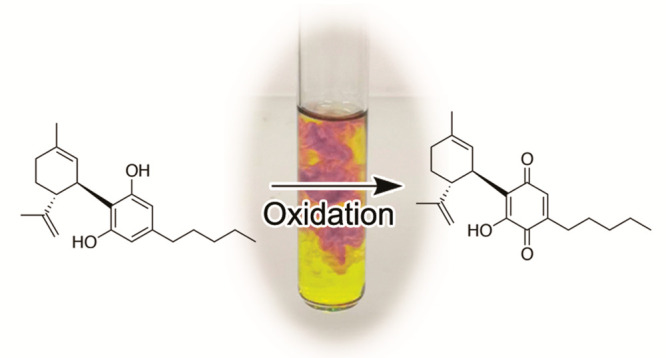
Spurred by a growing interest in cannabidiolquinone (CBDQ, HU-313, 2) as a degradation marker and alledged hepatotoxic metabolite of cannabidiol (CBD, 1), we performed a systematic study on the oxidation of CBD (1) to CBDQ (2) under a variety of experimental conditions (base-catalyzed aerobic oxidation, oxidation with metals, oxidation with hypervalent iodine reagents). The best results in terms of reproducibility and scalability were obtained with λ5-periodinanes (Dess-Martin periodinane, 1-hydroxy-1λ5,2-benziodoxole-1,3-dione (IBX), and SIBX, a stabilized, nonexplosive version of IBX). With these reagents, the oxidative dimerization that plagues the reaction under basic aerobic conditions was completely suppressed. A different reaction course was observed with the copper(II) chloride-hydroxylamine complex (Takehira reagent), which afforded a mixture of the hydroxyiminodienone 11 and the halogenated resorcinol 12. The λ5-periodinane oxidation was general for phytocannabinoids, turning cannabigerol (CBG, 18), cannabichromene (CBC, 10), and cannabinol (CBN, 19) into their corresponding hydroxyquinones (20, 21, and 22, respectively). All cannabinoquinoids modulated to a various extent peroxisome proliferator-activated receptor gamma (PPAR-γ) activity, outperforming their parent resorcinols in terms of potency, but the iminoquinone 11, the quinone dimers 3 and 23, and the haloresorcinol 12 were inactive, suggesting a specific role for the monomeric hydroxyquinone moiety in the interaction with PPAR-γ.
Color development has played a significat role in the early studies on Cannabis (Cannabis sativa L.) and cannabinoids. Thus, the first phytocannabinoids were purified from Cannabis red oil, a deep-red high-vacuum distillation fraction of Cannabis extracts.1,2 A red-purple color was also observed when fiber hemp or hashish was treated with methanolic KOH.3 Under these conditions, the development of a color is specific for Cannabis and Cannabis-derived products (marijuana, hashish),4 and the reaction has long been proved as an expeditious method for their identification in a forensic context (Beam test).4
The nature of the pigment from Cannabis red oil is still unclear,
but color formation in the Beam test is the result of the aerobic
oxidation of cannabidiol (CBD, 1) to the hydroxyquinone 2 (cannabidiolquinone, CBDQ, HU-331),5 a compound that has attracted considerable interest because of its
selective anticancer activity6,7 and catalytic inhibitory
properties on topoisomerase IIα.8 While development of 2 as a drug was abandoned, possibly
because of unfavorable stability properties (vide infra) and cellular
toxicity,9 distinct lines of research rekindled
interest in this compound. Thus, microsomial formation of 2 from CBD(1) has been associated with P450 covalent
inhibition and perturbation of hepatic xenobiotics metabolism,10 and a similar process could also underlie the
liver toxicity reported for high dosages of CBD.11 Furthermore, 2 is formed during long-term
storage of CBD under aerobic conditions,12 and its availability is therefore important for quality control
of this active pharmaceutical ingredient (API).
Despite the convergence of interest for CBDQ (2) from various areas of cannabinoid research, its only reported synthesis is the one inspired by the Beam test, that is, the aerobic oxidation of CBD in a cooled biphasic petroleum ether/5% ethanolic KOH system.5,6 Under these conditions, yields are erratic, scale-dependent, and modest (ca. 20% at best),5,6 while significant amounts of the dimeric quinone 3 are also formed by oxidative dimerization of CBDQ.5 Both reaction products, especially 3, are unstable and rapidly turn into a complex mixture of polar compounds.5 In our hands, the oxidation reaction was poorly reproducible and could not be scaled up over a few hundred milligrams of starting material, even when air or 80% oxygen was bubbled into the biphasic reaction system. A more reproducible behavior was observed with KH or LiH in tetrahydrofuran (THF) or toluene under heterogeneous conditions, but scale-up was still problematic. While Beam-type oxidation strategies were eventually abandoned, their mechanism is worth mentioning. Thus, the reaction is presumably triggered by formation of a phenolate anion, next oxidized to an electrophilic radical (4) that adds to dioxygen to form a hydroperoxy radical. The latter is reduced to the corresponding anion (5) by a second phenolate ion, and, after tautomerization to 6, the hydroperoxy anion is trapped by the para-carbonyl group. This generates the bridged keto-peroxyhemiacetal 7, whose α-deprotonation triggers cleavage of the peroxidic bond, eventually affording the hydroxylated quinone 2 via the hydrate 8 (Figure 1).
Figure 1.

Possible mechanism of the base-mediated aerobic formation of cannabidiolquinone (CBDQ (2)) from cannabidiol (CBD (1)) in ethanolic KOH. (R = 3-p-mentha-1,8-dienyl).
This process is reminiscent of the transformation
of vitamin K
hydroquinone into its epoxyquinone form,13 and the mechanism outlined in Figure 1 could explain the sensitivity of the reaction to radical
traps like butylated hydroxytoluene (BHT) as well as the unreactivity
of monoalkylated phytocannabinoids, like Δ9-tetrahydrocannabinol
(Δ9-THC, 9) and cannabichromene (CBC, 10), where the prototropic equilibrium required for the formation
of the peroxyhemiacetal is not possible (cf. the formation of 6 from 5 in Figure 1).
The reaction profile of the Beam
test was basically replicated,
without any substantial improvement of yield, by metal oxidants [FeCl3, K3[Fe(CN)6], MnO2, Cr6+-based reagents, CuCl, CuCl2, Ag2O,
NH4Ce(NO3)5] under both catalytic
and stoichiometric conditions, as well as by peroxides (tert-butyl hydroperoxide (TBHP), basic H2O2), with
significant amounts of the dimer 3 being always formed
under basic conditions or during the long reaction times required
to achieve a significant conversion. A surprising and notable exception
was the behavior of the Takehira complex (CuCl2-hydroxylamine),14 which afforded a mixture of the hydroxyiminodienone 11 and the chlororesorcinol 12. The regioselectivity
of the formation of 11 was deduced from the diagnostic 3J heteronuclear multiple bond correlation
(HMBC) cross-peaks of H-1″ with the hydroxyiminocarbonyl carbon.
The Takehira complex was originally developed for the oxidation of methylpolyphenols to their corresponding hydroxyquinones,14 a reaction of relevance for the industrial synthesis of vitamin E,14 and was later modified by replacement of hydroxylamine with other nitrogen bases.15 In control experiments, copper(II) chloride alone gave CBDQ (2) and the dimer 3 as the only reaction products, while the quinone 2 did not react with hydroxylamine, suggesting a role for hydroxylamine in the chemoselective halogenation reaction, possibly via the generation of an N-chlorinated species, and of copper(II) in the activation of the quinonecarbonyl carbon toward nucleophilic attack by hydroxylamine.
Hypervalent iodine derivatives have become increasingly popular for a wide range of oxidative reactions,16 and bis(trifluoroacetoxy)iodobenzene (BTIB) was reported to oxidize the mono-O-alkylated cannabinoid Δ9-THC (9), otherwise unreactive in Beam-type oxidations,6 to its corresponding hydroxyquinone.6 This λ3-iodane was also able to oxidize CBD to CBDQ, but λ5 iodanes like 2-iodoxybenzoic acid (1-hydroxy-1λ5,2-benziodoxole-1,3-dione, IBX, 13)17 and the Dess-Martin periodinane (DMP)18 gave much better and more reproducible yields, with a stabilized and not explosive version of IBX (SIBX)19 emerging as the reagent of choice. The superior behavior of SIBX compared to IBX might be related to the acidity of the stabilizing matrix (isophthalic and benzoic acids), which could help the hydrolytic cleavage of iodic esters formed in the reaction.19
The oxidation is presumably initiated by the sigmatropic rearrangement of the iodine–oxygen bond in the mixed λ5 iodane ester 14 formed by interaction of IBX and the C-1′ phenolic hydroxy group (Figure 2). The resulting C-2′ λ3-quinol 15, after oxidation to the corresponding λ5-iodane 16, is transformed by [3.3]-sigmatropic rearrangement of the carbon–oxygen bond into the C-4′ λ5-iodinane 17, with β-elimination eventually generating the hydroxyquinone 2 and a reduced λ-iodinane. Remarkably, dimerization was completely suppressed under iodinane oxidation, and yields in the range of 50–60% could be obtained at multigram reaction scale. CBDQ, an orange powder,20 is unstable in solution, rapidly degrading in both protic (methanol) and aprotic (acetone, CHCl3) solvents, with generation of the more polar dimer 3 next to a host of uncharacterized more polar products. On the other hand, it could be stored for at least 10 months as a powder at −18 °C in a sealed flask, or for additional time as a frozen benzene or dimethyl sulfoxide (DMSO) solution at 4 °C.21
Figure 2.

Possible mechanism for the SIBX-mediated formation of cannabidiolquinone (CBDQ (2)) from cannabidiol (CBD (1)) (R = 3-p-mentha-1,8-dienyl, R′ = n-pentyl).
The oxidation with SIBX is general
for phytocannabinoids, and,
apart from cannabigerol (18), it could also be applied
to monoetherified compounds [cannabichromene (CBC, 10), cannabinol (19)] that are unreactive under Beam-test
conditions, to afford their corresponding hydroxyquinones 20–22.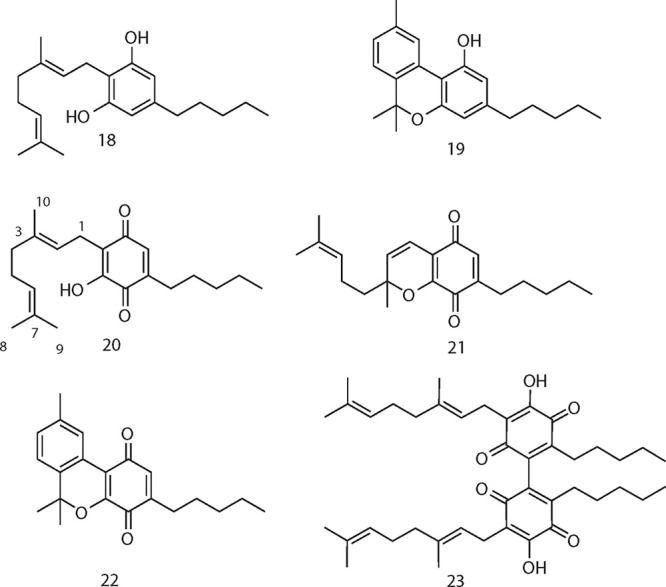
CBDQ (2) has been reported to be non-narcotic6 and lacks significant affinity for CB1 and CB2 receptors.9 Nevertheless, it showed powerful modulating activity on peroxisome proliferator-activated receptor gamma (PPAR-γ),9,22 and various degrees of PPAR-γ activating activity were also shown by the other cannabinoquinoids (Table 1). However, dimerization was detrimental for activity, and dimeric quinones were devoid of significant activity in PPARγ-activity assays.23 Dimeric quinones are axially chiral, and, since enantiomeric cannabinoids can show markedly different profiles of bioactivity,2 the one from CBG (CBGQ (23)) was resolved by chromatography on a chiral-phase column packed with amylose-tris(5-chloro-2-methylphenylcarbamate). However, both the (aR) and the (aS) enantiomers turned out to be inactive.23 Similarly, the hydroxyiminodienone 11 and the chlorinated resorcinol 12 were also devoid of activity.23
Table 1. PPAR-γ Modulation Activitya.
| compound | EC50 |
|---|---|
| 1 | >25 μM |
| 10 | >25 μM |
| 18 | 15.7 μM |
| 19 | >25 μM |
| 2 | 10.5 μM |
| 21 | 14.7 μM |
| 20 | 4.9 μM |
| 22 | 23.1 μM |
PPAR-γ modulation activity of the phytocannabinoids 1, 10, 18, and 19 and their corresponding cannabinoquinones (2, 21, 20, and 22). Rosiglitazone (1 μM) was used as positive control for PPAR-γ activation (50-fold induction over basal activity).
In conclusion, we have developed a reproducible and scalable synthesis of cannabinoquinoids, including CBDQ (2), significantly enhancing access to this compound24 of relevance not only for its bioactivity profile but also for the analytics of CBD, the study of its binding to P450 apoproteins, and its effects on liver function.
Experimental Section
General Experimental Procedures
IR spectra were recorded on an Avatar 370 FT-IR Techno-Nicolet apparatus. 1H (400 and 500 MHz) and 13C (100 and 125 MHz) NMR spectra were measured on Varian INOVA NMR spectrometers. Chemical shifts were referenced to the residual solvent signal (methanol-d4: δH = 3.34, δC = 49.0 or CDCl3: δH = 7.21, δC = 77.0). Homonuclear 1H connectivities were determined by the correlated spectroscopy (COSY) experiment. One-bond heteronuclear 1H–13C connectivities were determined with the heteronuclear single quantum coherence (HSQC) spectroscopy experiment. Two- and three-bond 1H–13C connectivities were determined by gradient two-dimensional (2D) heteronuclear multiple bond correlation (HMBC) experiments optimized for a 2,3J = 9 Hz. Low- and high-resolution electrospray ionization mass spectrometry (ESI-MS) data were determined on an LTQ OrbitrapXL (Thermo Scientific) mass spectrometer.
Reactions were monitored by thin-layer chromatography (TLC) on Merck 60 F254 (0.25 mm) plates, visualized by staining with 5% H2SO4 in EtOH and heating. Organic phases were dried with Na2SO4 before evaporation. Chemical reagents and solvents were purchased form Sigma-Aldrich and were used without further purification unless stated otherwise. Petroleum ether with boiling point of 40–60 °C was used. Silica gel 60 (70–230 mesh) was used for gravity column chromatography (GCC).
SIBX Oxidation of Phytocannabinoids. Reaction with CBD (1) as Example
To a cooled (ice bath) solution of CBD (5 g, 15,6 mmol) in ethyl acetate (EtOAc, 75 mL), SIBX (21.1 g, 31.5 mmol, 2 molar equiv) was addd in six portions of ca. 5 g each. The cooling bath was removed, and the suspension was stirred at room temperature for 18 h and then filtered over a pad of diatomaceous earth. The filtration cake was washed with EtOAc (50 mL), and the pooled filtrates were washed with saturated Na2S2O3 (4 × 75 mL) and next with brine. After the drying and evaporation, the residue was purified by GCC on silica gel (75 g, petroleum ether–EtOAc 9:1 as eluant) to obtain a brown oil that solidified upon storing in the refrigerator. Washing with cold petroleum ether removed some of the colored impurities and afforded an orange powder (3.17 g, 61%). The same protocol was used for the oxidation and the purification of the other phytocannabinoids investigated (CBC, 10; CBG, 18; CBN, 19). The scale was 100–200 mg, and the yields were 59, (CBCQ, 21), 37 (CBGQ, 20), and 58%, (CBNQ, 22).
Cannabigeroquinone (CBGQ, 20)
Red powder, IR νmax (KBr disc): 3272, 2955, 2923, 2856, 1644, 1637, 1350, 1316, 1191, 1175, 580 cm–1; 1H NMR (CDCl3, 400 MHz) δ 6.94 (1H, s, OH), 6.45 (1H, bs, H-2′), 5.13 (1H, t, J = 7.4 Hz, H-2), 5.04 (1H, t, J = 6.7 Hz, H-7), 3.13 (2H, d, J = 7.4 Hz, H-1), 2.41 (2H, t, J = 7.6, H-1″), 1.99–1.90 (4H, m, H-4, H-5), 1.73 (3H, s, H-8), 1.64 (3H, s, H-9), 1.57 (3H, s, H-10), 1.50 (2H, m, H-2′′), 1.33 (4H, m, H-3′′, H-4′′), 0.89 (3H, t, J = 6.8 Hz, H-5″); 13C NMR (CDCl3, 100 MHz) δ 187.7, 184.2, 150.9, 145.1, 137.3, 134.4, 131.5, 124.3, 120.2, 119.7, 39.8, 31.5, 28.3, 27.4, 26.7, 25.8, 22.5, 22.0, 17.8, 16.3, 14.0; ESI-MS: m/z 331 [M + H]+; high-resolution (HR) ESI-MS m/z 331.2262 [M + H]+, calcd. for C21H31O3, 331.2268.
Cannabichromenquinone (CBCQ, 21)
Red oil, IR νmax (KBr disc): 2957, 2926, 2852, 1648, 1580, 1324, 1078, 969, 891 cm–1; 1H NMR (CDCl3, 400 MHz) 6.47 (1H, d, J = 9.9 Hz, H-1), 6.40 (1H, bs, H-2′), 5.56 (1H, d, J = 9.9 Hz, H-2), 5.07 (1H, t, J = 6.9 Hz, H-6), 2.39 (2H, t, J = 7.6 Hz, H-1″), 2.08 (1H, m, H-5a), 1.88 (1H, m, H-5b), 1.66 (2H, overlapped, H-4), 1.64 (3H, s, H-8), 1.55 (3H, s, H-9), 1.49 (2H, m, H-2′′), 1.46 (3H, s, H-10), 1.32 (4H, m, H-3′′, H-4′′), 0.89 (3H, t, J = 6.7 Hz, H-5″); 13C NMR (CDCl3, 100 MHz) δ 184.6, 181.9, 150.8, 147.7, 132.2, 131.4, 128.8, 123.4, 115.4, 115.0, 83.0, 41.5, 31.4, 28.7, 27.4, 27.3, 25.6, 22.6, 22.4, 17.7, 13.9; ESI-MS m/z 329 [M + H]+; HR ESI-MS m/z 329.2107 [M + H]+, calcd for C21H29O3, 329.2111.
Cannabinolquinone (CBNQ, 22)
Red oil, IR νmax (KBr disc): 2955, 2924, 2855, 1649, 1382, 1145, 1110, 811 cm–1; 1H NMR (CDCl3, 400 MHz) δ 8.30 (1H, s, H-2), 7.09 (1H, d, J = 7.9 Hz, H-6), 7.02 (1H, d, J = 7.9 Hz, H-5), 6.63 (1H, t, J = 1.4 Hz, H-2′), 2.40 (2H, t, J = 7.7 Hz, H-1″), 2.36 (3H, s, H-7), 1.69 (6H, s, H-9, H-10), 1.56 (2H, m, H-2″), 1.32 (4H, m, H-3′′, H-4′′), 0.90 (3H, t, J = 6.8 Hz, H-5″); 13C NMR (CDCl3, 100 MHz) δ 180.2, 175.3, 163.3, 144.7, 138.1, 133.8, 131.8, 128.9, 125.7, 122.3, 111.0, 82.7, 53.6, 31.6, 29.8, 29.0, 28.3, 27.4, 22.4, 21.4, 13.9; ESI-MS m/z 325 [M + H]+; HR ESI-MS m/z 325.1791 [M + H]+, calcd. for C21H25O3, 325.1798.
Dimeric Cannabigeroquinone (23) and Chiral-Phase Chromatography
Red powder, IR νmax (KBr disc): 3280, 2955, 1350, 1188, cm–1; 1H NMR (CDCl3, 400 MHz) δ 6.98 (1H, s, OH), 5.17 (1H, t, J = 7.4 Hz, H-2), 5.06 (1H, t, J = 6.7 Hz, H-7), 3.16 (2H, d, J = 7.4 Hz, H-1), 2.32 (2H, t, J = 7.6, H-1″), 2.05–1.90 (4H, m, H-4, H-5), 1.73 (3H, s, H-8), 1.64 (3H, s, H-9), 1.57 (3H, s, H-10), 1.49 (2H, m, H-2′′), 1.32 (4H, m, H-3′′, H-4′′), 0.89 (3H, t, J = 6.8 Hz, H-5″). ESI-MS: m/z 645 [M + H]+; HR ESI-MS m/z 645.4159 [M + H]+, calcd. for C41H57O6, 645.4155.
A sample of compound 23 (2.0 mg) was separated on a chiral-phase Lux 5 μ Amylose-2 250 × 4.60 mm column, Phenomenex, eluent n-hexane/isopropyl alcohol 9:1 (0.2% trifluoroacetic acid (TFA)) with a flow of 0.7 mL/min, and two peaks were obtained with Rt = 8 min (0.9 mg) and Rt = 13 min (0.7 mg).
Oxidation of Cannabidiol (CBD, 1) with the Takehira Reagent
To a stirred solution of CBD (1, 200 mg, 0,64 mmol) in toluene–tert-butanol (3:1, 20 mL), copper(II) chloride (43 mg, 0.32 mmol, 0.5 molar equiv) and hydroxylamine hydrochloride (22 mg, 0.32 mmol, 0,.5 molar equiv) were added. The solution turned from yellow to brown and was stirred for 2 h at rt, worked up by dilution with 2N H2SO4 and extraction with EtOAc. The organic phase was washed with brine, dried with Na2SO4, filtered, and evaporated. The residue was purified by GCC (5 g silica gel, petroleum ether–EtOAc gradient, from to petroleum ether to 95:5 petroleum ether–EtOAc as eluent) to give 12 (135 mg, 34%) and 11 (20%).
Hydroxyiminocannabiquinone (11)
Brownish oil, IR νmax (KBr disc): 2960, 2924, 2856, 1617, 1420, 1420, 1260, 1092, 1016, 797 cm–1; 1H NMR (methanol-d4, 400 MHz) δ 6.25 (1H, s, H-2′), 5.12 (1H, s, H-2), 4.50 (1H, s, H-9a), 4.49 (1H, s, H-9b), 3.82 (1H, m, H-3), 2.92 (1H, td, J = 11.7, 3.2 Hz, H-4), 2.70 (2H, t, J = 7.5 Hz, H-1″), 2.18 (1H, m, H-6a), 1.99 (1H, m, H-6b), 1.73 (2H, overlapped, H-5), 1.65 (3H, s, H-7), 1.63 (3H, s, H-10), 1.60 (2H, overlapped, H-2′′), 1.34 (2H, overlapped, H-3′′), 1.33 (2H, overlapped, H-4′′), 0.91 (3H, t, J = 6.9 Hz, H-5″); 13C NMR (methanol-d4, 100 MHz) δ 176.3 (C-1′), 168.1 (C-5′), 150.2 (C-8), 148.7 (C-3′), 148.1 (C-4′), 133.6 (C-1), 125.6 (C-2), 120.6 (C-2′), 119.1 (C-6′), 110.8 (C-9), 45.6 (C-4), 36.2 (C-3), 32.8 (C-3″), 31.6 (C-6), 31.5 (C-1′′), 30.8 (C-5), 30.5 (C-2′′), 23.6 (C-7), 23.5 (C-4′′), 19.1 (C-10), 14.3 (C-5′′); ESI-MS m/z 344 [M + H]+; HR ESI-MS m/z 344.2210 [M + H]+ calcd for C21H30NO3, 344.2220.
2-Chlorocannabidiol (12)
Yellow oil, IR νmax (KBr disc): 3500, 3421, 2962, 2924, 2859, 1623, 1421, 1258, 1193, 1054, 888, 817, 698 cm–1; 1H NMR (methanol-d4, 400 MHz): δ 6.21 (1H, s, H-2′), 5.25 (1H, s, H-2), 4.46 (1H, s, H-9a), 4.44 (1H, s, H-9b), 3.99 (1H, m, H-3), 2.95 (1H, m, H-4), 2.56 (2H, t, J = 7.4 Hz, H-1″), 2.20 (1H, m, H-5a), 2.01 (1H, d, J = 17.1 Hz, H-5b), 1.76 (2H, m, H-6), 1.68 (3H, s, H-7), 1.65 (3H, s, H-10), 1.56 (2H, m, H-2″), 1.35 (4H, m, H-3′′-4′′), 0.91 (3H, t, J = 6.8 Hz, H-5″); 13C NMR (methanol-d4, 100 MHz): δ 156.1, 152.7, 150.2, 139.3, 134.2, 126.6, 118.2, 112.8, 110.7, 109.6, 46.2, 38.3, 34.7, 32.7, 31.7, 30.7, 30.5, 23.7, 23.5, 19.3, 14.4. ESI-MS m/z 349, 351 [M + H]+ ratio 3:1; HR ESI-MS m/z [M + H]+349.1919 (calcd for C21H3035ClO2, 349.1929).
PPAR-γ Activity Evaluation
Human embryonic kidney epithelial cells 293T cells were obtained from the American Type Culture Collection (CRL-3216) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal calf serum (FCS) and antibiotics. To analyze the PPAR-γ transcriptional activity, HEK-293T cells were cultured in 24-well plates (2 × 104 cells/well) and transiently cotransfected with GAL4-PPAR-γ (50 ng) and GAL4-luc (firefly luciferase, 50 ng) vectors using Roti-Fect (Carl Roth). Twenty hours after transfection the cells were stimulated with increasing concentrations of the compounds for 6 h, and luciferase activities were quantified using Dual-Luciferase Assay (Promega). Rosiglitazone (1 μM, Cayman Chemical), was used as a positive control for PPAR-γ activation (50-fold induction over basal activity). Test compounds and controls stocks were prepared in DMSO, and the final concentration of the solvent was always less than 0.5% v/v. The plasmid GAL4-PPAR-γ was obtained from Prof. C. Sinal (Dalhousie University). Half-maximal effective concentration (EC50) was estimated using Prism software (GraphPad). All transfection experiments were performed at least three times.
Acknowledgments
We are grateful to Dr. G. Grassi (CREA, Rovigo) for generously supplying the Carmagnola strain of Cannabis used for the isolation of cannabidiol. We thank MIUR for financial support to the groups in Novara and Naples (PRIN2017, Project No. 2017WN73PL, Bioactivity-directed exploration of the phytocannabinoid chemical space).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.9b01284.
1H and 13C NMR spectra for CBDQ (2) and the other cannabinoquinoids mentioned in this study (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Wood T. B.; Spivey W. T. N.; Easterfield T. H. J. Chem. Soc., Trans. 1896, 69, 539–546. 10.1039/CT8966900539. [DOI] [Google Scholar]
- Isolation and Structure Elucidation of Cannabidiol:; Adams R.; Pease D. C.; Clark J. H.; Baker B. R. J. Am. Chem. Soc. 1940, 62, 2197–2200. 10.1021/ja01865a081. [DOI] [Google Scholar]; Adams R.; Hunt M.; Clark J. H. J. Am. Chem. Soc. 1940, 62, 196–200. 10.1021/ja01858a058. [DOI] [Google Scholar]
- Beam W. 4th Report, Wellcome Trop. Research Lab., Rep. Sudan Gov. 1911, 25. [Google Scholar]
- Grlic L.A Comparative Study on Some Chemical and Biological Characteristics of Various Samples of Cannabis Resin. United Nations Office of Drugs and Crime, 1962. (https://www.unodc.org/unodc/en/data-and-analysis/bulletin/bulletin_1962-01-01_3_page005.html). [Google Scholar]
- Mechoulam R.; Ben-Zvi Z.; Gaoni Y. Tetrahedron 1968, 24, 5615–5624. 10.1016/0040-4020(68)88159-1. [DOI] [PubMed] [Google Scholar]
- Kogan N. M.; Rabinowitz R.; Levi P.; Gibson D.; Sandor P.; Schlesinger M.; Mechoulam R. J. Med. Chem. 2004, 47, 3800–3806. 10.1021/jm040042o. [DOI] [PubMed] [Google Scholar]
- Peters M.; Kogan N. M. Expert Opin. Invest. Drugs 2007, 16, 1405–1413. 10.1517/13543784.16.9.1405. [DOI] [PubMed] [Google Scholar]
- Regal K. M.; Mercer S. L.; Deweese J. E. Chem. Res. Toxicol. 2014, 27, 2044–2051. 10.1021/tx500245m. [DOI] [PubMed] [Google Scholar]
- del Río C.; Navarrete C.; Collado J. A.; Bellido M. L.; Gómez-Cañas M.; Pazos M. R.; Fernández-Ruiz J.; Pollastro F.; Appendino G.; Calzado M. A.; Cantarero I.; Muñoz E. Sci. Rep. 2016, 6, 21703. 10.1038/srep21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornheim L. M.; Grillo M. P. Chem. Res. Toxicol. 1998, 11, 1209–1216. 10.1021/tx9800598. [DOI] [PubMed] [Google Scholar]
- Ewing L. A.; Skinner C. M.; Quick C. M.; Kennon-McGill S.; McGill M. R.; Walker L. A.; ElSohly M. A.; Gurley B. J.; Koturbash I. Molecules 2019, 24, 1694. 10.3390/molecules24091694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appendino G.; Allegrini P.. Manuscript in preparation.
- Dowd P.; Hershline R.; Ham S.; Naganathan S. Science 1995, 269, 1684–1691. 10.1126/science.7569894. [DOI] [PubMed] [Google Scholar]
- Takehira K.; Shimizu M.; Watanabe Y.; Orita H.; Hayakawa T. J. Chem. Soc., Chem. Commun. 1989, 1705–1706. 10.1039/C39890001705. [DOI] [Google Scholar]
- Takehira K.; Shimizu M.; Watanabe Y.; Orita H.; Hayakawa T. Tetrahedron Lett. 1989, 30, 6691–6692. 10.1016/S0040-4039(00)70652-6. [DOI] [Google Scholar]
- Nicolaou K. C.; Montagnon T.; Baran P. S.; Zhong Y.-L. J. Am. Chem. Soc. 2002, 124, 2245–2258. 10.1021/ja012127+. [DOI] [PubMed] [Google Scholar]
- Frigerio M.; Santagostino M.; Sputore S. J. Org. Chem. 1999, 64, 4537–4538. 10.1021/jo9824596. [DOI] [Google Scholar]
- Dess D. B.; Martin J. C. J. Org. Chem. 1983, 48, 4155–4156. 10.1021/jo00170a070. [DOI] [Google Scholar]
- Ozanne A.; Pouysegu L.; Depernet D.; Francois B.; Quideau S. Org. Lett. 2003, 5, 2903–2906. We previously disclosed the use of SIBX for the telescoped synthesis of aminoderivatives of cannabinoquinoids in the proprietary literature: 10.1021/ol0349965. [DOI] [PubMed] [Google Scholar]; Appendino G.; Cabello de Alba M. L. B.; Munoz E.. WO158381, 2015.
- The product resulting from the Beam-type oxidation was reported as a brown powder.6 We confirmed this observation, presumably related to the formation of highly colored impurities under the basic conditions of the Beam oxidation that could not be removed by hexane washing or recrystallization.
- Appendino G.; Pollastro F.; Verotta L.; Ballero M.; Romano A.; Wyrembek P.; Szczuraszek K.; Mozrzymas J. W.; Taglialatela-Scafati O. J. Nat. Prod. 2009, 72, 962–965. 10.1021/np8007717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja A. G.; Carrillo-Salinas F.; Pagani A.; Gómez-Cañas M.; Negri R.; Navarrete C.; Mecha M.; Mestre L.; Fiebich B. L.; Cantarero I.; Calzado M. A.; Bellido M. L.; Fernandez-Ruiz J.; Appendino G.; Guaza C.; Muñoz E. J. Neuroimmune Pharmacol. 2012, 7, 1002–1016. 10.1007/s11481-012-9399-3. [DOI] [PubMed] [Google Scholar]
- Compounds were considered inactive if unable to induce 5-fold PPARC-induction at 50 M concentration.
- Hanuš L. O.; Tchilibon S.; Ponde D. E.; Breuer A.; Fride E.; Mechoulam R. Org. Biomol. Chem. 2005, 3, 1116–1123. 10.1039/b416943c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.