

Abstract
A high fat diet and obesity have been linked to the development of metabolic dysfunction and the promotion of multiple cancers. The causative cellular signals are multifactorial and not yet completely understood. In this report, we show that Inositol Polyphosphate-4-Phosphatase Type II B (INPP4B) signaling protects mice from diet-induced metabolic dysfunction. INPP4B suppresses AKT and PKC signaling in the liver thereby improving insulin sensitivity. INPP4B loss results in the proteolytic cleavage and activation of a key regulator in de novo lipogenesis and lipid storage, SREBP1. In mice fed with the high fat diet, SREBP1 increases expression and activity of PPARG and other lipogenic pathways, leading to obesity and non-alcoholic fatty liver disease (NAFLD). Inpp4b−/− male mice have reduced energy expenditure and respiratory exchange ratio leading to increased adiposity and insulin resistance. When treated with high fat diet, Inpp4b−/− males develop type II diabetes and inflammation of adipose tissue and prostate. In turn, inflammation drives the development of high-grade prostatic intraepithelial neoplasia (PIN). Thus, INPP4B plays a crucial role in maintenance of overall metabolic health and protects from prostate neoplasms associated with metabolic dysfunction.
Subject terms: Non-alcoholic fatty liver disease, Insulin signalling, Type 2 diabetes
Characterizing mice lacking Inositol Polyphosphate-4-Phosphatase Type II B (INPP4B), Zhang et al. discovered that SREBP1 signaling is activated in livers of Inpp4b−/− males leading to development of NAFLD and insulin resistance. When fed high fat diet, Inpp4b−/− males develop type II diabetes, inflammation of adipose tissue, and prostate neoplasia. These findings provide insights into INPP4B protective role from metabolic syndrome.
Introduction
In 2017, overconsumption and unhealthy diets contributed to 11 million deaths globally. Diet-associated deaths from cancers and diabetes were ranked second and third behind cardiovascular disease1. The National Center for Health Statistics reported in 2016 that over 80% of adult males aged 20 years and older in the United States were overweight or obese. Among this age group, 70% of obese men and 30% of normal weight men will develop metabolic dysfunction2. The consumption of a high-fat diet (HFD) is recognized as a leading cause of obesity, metabolic dysfunction, elevated chronic inflammation3, and cancer mortality4. Major indicators of metabolic dysfunction include elevated glucose levels, insulin resistance, and the expansion of mesenteric and omental adipose tissues5. Importantly for men, HFD and resulting obesity are tightly associated with lower urinary tract syndromes, including benign prostatic hyperplasia (BPH)6, accelerated progression of prostate cancer, and decreased prostate cancer patient survival rates7,8.
Increasing evidence suggests that non-alcoholic fatty liver disease (NAFLD) is the best clinical indicator of metabolic dysfunction in both obese and lean individuals9,10. Hepatic lipid storage occurs as a result of dysregulation in the insulin signaling pathway. Activation of the insulin receptor in liver, muscle, and fat cells leads to the activation of PI3K/AKT signaling pathways11. High levels of circulating diacylglycerol (DAG) lipids that are associated with HFDs activate PKCs, which, in turn, phosphorylate and inhibit the insulin receptor, lowering the insulin sensitivity12. In both mice and men, obesity elevates DAG levels and activates PKC signaling in multiple tissues, leading to the development of NAFLD and insulin resistance. Clinically, the levels of circulating DAG and PKCε activation are the strongest predictors of insulin resistance in obese men13.
Importantly, a HFD is not exclusively responsible for metabolic syndrome as it has been reported that some obese individuals maintain a healthy metabolic profile12,14,15. There are several allelic variants that may impact the effects of known determinants of metabolic health. Polymorphisms in peroxisome proliferator-activated receptor γ (PPARG), adiponectin (ADIPOQ), leptin receptor (LEPR), and insulin receptor substrate 2 (IRS2) have been linked to obesity and type 2 diabetes mellitus (T2D)16–19. However, the specific signaling pathways that are disrupted during the initial stages of metabolic dysfunction are still not known.
Inositol Polyphosphate-4-Phosphatase Type II B (INPP4B) dephosphorylates both membrane lipids and phosphoproteins20. We have shown that INPP4B suppresses the AKT and PKC pathways20–22, which are the major mediators of metabolic dysfunction, suggesting a role for INPP4B in the regulation of metabolic health. We tested this hypothesis using Inpp4b−/− male mice to compare their metabolic fitness to that of the wild-type (WT) males. We found that INPP4B has diet-dependent and independent actions in the mouse liver, adipose tissue, pancreas, and prostate. When fed a low-fat diet (LFD), 3-month-old Inpp4b−/− males increased their fat-to-lean body mass ratio, developed liver steatosis and hyperglycemia. This metabolic dysfunction likely resulted from reduced ambulatory activity, energy expenditure, and respiratory exchange ratios. When fed a HFD, in addition to liver steatosis, Inpp4b−/− males also developed type II diabetes (T2D), inflammation of the visceral white adipose tissue (WAT) and prostate, and prostatic intraepithelial neoplasia (PIN). Thus, the loss of INPP4B drives metabolic dysfunction by increasing hepatic lipogenesis, elevating systemic and local inflammation, and promoting the neoplastic transformation of the prostate in obese males.
Results
Accelerated weight gain in Inpp4b−/− male mice fed with a high-fat diet
Inpp4b expression was previously reported in multiple tissues23. Analysis of mouse tissues showed that the highest expression of Inpp4b is in the testis, mammary gland fat pad, epididymal white adipose tissue (eWAT), muscle, bladder, and the ventral and dorsolateral lobes of the prostate. Among distinct adipose tissues, highest level of Inpp4b expression was observed in mesenteric WAT (mWAT) and lowest in brown adipose tissue (BAT). Lower levels of Inpp4b mRNA were also detected in the pancreas and liver (Fig. 1a, b), indicating that Inpp4b is expressed in all major organs involved in insulin resistance. Inpp4b−/− males fed with a HFD gained significantly more weight than age-matched WT controls (Fig. 1c). At 3 months of age, obesity in Inpp4b−/− males was not accompanied by changes in body length, blood pressure, or heart rate (Supplementary Fig. 1a–c). A comparison of two representative fat depots, visceral WAT, the #4 mammary fat pad, and eWAT, revealed a significant weight increase in the HFD-fed Inpp4b−/− males compared to the WT males (Supplementary Fig. 1d, e). Total body composition analysis by QMR revealed that knockout males have an increased fat-to-lean mass ratio (Fig. 1d–f). While overall body weights of experimental animals did not change in the course of CLAMS analysis, total food and water intake as well as the home cage activities were significantly decreased in knockout mice (Fig. 1g–i). Consistently, energy expenditure and respiratory exchange ratios were significantly decreased in Inpp4b−/− males during light cycle (Fig. 1j, k and Supplementary Fig. 1f, g). Histological comparison of the inguinal, retroperitoneal, mesenteric and eWAT, as well as BAT, did not reveal visible differences between 4 experimental groups (Supplementary Fig. 2).
Fig. 1. Inpp4b−/− males accumulate more fat due to decreased activity, energy expenditure, and respiratory exchange ratio.

a Expression of Inpp4b in mouse tissues. b Expression of Inpp4b in epididymal, retroperitoneal, mesenteric, inguinal, and brown adipose tissue of FVB males. c Body weights of LFD WT (N = 14), LFD Inpp4b−/− (N = 12), HFD WT (N = 9), and HFD Inpp4b−/− (N = 10) mice were recorded every week beginning on post-partum day 2. LFD WT, LFD Inpp4b−/−, HFD WT, and HFD Inpp4b−/− groups are represented by the blue line with squares, red line with up-pointing triangles, green line with down-pointing triangles, and purple line with circles, respectively (**p = 0.0048, ***p = 0.001). d–f Percent fat (**p = 0.0012) (d), and percent lean (**p = 0.0014) (e) were determined by NMR and the ratios of fat-to-lean mass (**p = 0.00015) (f) were calculated for WT (N = 6) or Inpp4b−/− (N = 10) mice. g, h Hourly food (**p = 0.0027) and water intake (***p = 0.00023) were recorded for the WT (N = 6) or Inpp4b−/− (N = 8) mice. i Mouse ambulatory activity was detected with XYZ beam array over 92 h in WT (N = 6) or Inpp4b−/− (N = 10) mice. Total distances were acquired by MetaScreen and processed using ExpeData. j, k Animals were acclimated in cages for the first 42 h. Time course of energy expenditure (j) and respiratory exchange ratio (k) over subsequent 55 h in WT (N = 6) or Inpp4b−/− (N = 8) male mice using CLAMS analysis.
Metabolic and inflammatory changes in adipose tissue of Inpp4b−/− males
In eWAT, neither diet nor Inpp4b knockout altered the protein levels of HK2, an enzyme that mediates the commitment step of glycolysis (Fig. 2a and Supplementary Fig. 3a). Expression of fatty acid synthase (Fasn) mRNA was increased with HFD (Supplementary Fig. 3b) and the FASN protein levels were further increased in eWAT of HFD-fed Inpp4b−/− mice (Fig. 2a, b), suggesting a role for INPP4B in lipid accumulation. Expression of Inpp4b was not affected in eWAT tissue by the HFD (Supplementary Fig. 3c). Thus, the loss of INPP4B combined with a HFD-activated lipogenesis led to a significant adipose expansion.
Fig. 2. Comparison of the metabolic and inflammatory profiles of adipose tissues from WT and Inpp4b−/− males.

a Protein lysates from eWAT were analyzed for FAS, HK2, CD68, and tubulin by western blot. b Quantification of FAS protein expression in (a). The FAS values were normalized to tubulin and are shown as fold change (N = 3 per group). c–g RNA was extracted from mouse eWAT of LFD WT (N = 12), LFD Inpp4b−/− (N = 11), HFD WT (N = 8), and HFD Inpp4b−/− (N = 9) mice. The expression levels of Lep/Adipoq (c), Il6 (d), Tnf (e), Adgre1 (f), and Cd68 (g) were assayed by qRT-PCR using 18S as an internal control. h Quantification of CD68 protein in (a). Average expression in LFD WT mice was designated as 1 (N = 3 per group). i–l Concentrations of leptin (i), insulin (j), C-peptide (k), Resistin (l) in serum of LFD WT (N = 11), LFD Inpp4b−/− (N = 11), HFD WT (N = 9), and HFD Inpp4b−/− (N = 9) mice (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Exact p values are provided in Supplementary Data 2).
WAT secretes a broad variety of adipokines and cytokines that modulate inflammation and insulin signaling24–27. We tested whether INPP4B regulates the inflammatory status of adipose tissue in HFD-fed males. The eWAT of HFD-fed Inpp4b−/− mice had a significantly increased ratio of leptin-to-adiponectin expression (Fig. 2c) and elevated expression of the inflammatory cytokines Il6 and Tnf (Fig. 2d, e). The adipose tissue in HFD-fed knockout males showed an increased expression of the macrophage markers Adgre1 and CD68 (Fig. 2f, g). The highest protein levels of CD68 were observed in the eWAT of the HFD Inpp4b−/− group, suggesting that the INPP4B deficiency promotes the inflammatory response of adipose tissue in diet-induced obesity (Fig. 2a, h). As shown in Fig. 2d–h, both HFD and INPP4B loss contributed to the increases in inflammatory cytokines and infiltrating macrophage markers in the adipose tissue.
Using adipokine arrays, we compared the protein levels of 40 adipokines from the eWAT of WT and Inpp4b−/− mice fed with LFD or HFD (Supplementary Fig. 3d). The array analysis confirmed that compared to LFD WT males, adipose tissue leptin/adiponectin protein-level ratios were increased in LFD Inpp4b−/−, HFD WT, and HFD Inpp4b−/− groups. However, the differences between these 3 groups did not reach statistical significance (Supplementary Fig. 3d). Consistent with the increased presence of CD68+ cells, the eWAT from Inpp4b−/− mice expressed significantly higher levels of the proinflammatory chemokine, monocyte chemoattractant protein-1 (MCP-1), which promotes macrophage infiltration and is known to be increased in obese diabetic mice28,29. Adipose expansion and inflammation are downregulated by INPP4B in obese animals, suggesting a protective role for INPP4B from metabolic syndrome. Analysis of metabolic hormones in the serum showed significant increases in leptin, insulin, C-peptide, and resistin in HFD Inpp4b−/− males compared to WT on either diet and LFD Inpp4b−/− mice (Fig. 2i–l and Supplementary Fig. 3e).
Inpp4b-deficient mice develop hyperglycemia on low-fat diet and type 2 diabetes on high-fat diet
The expansion and increased inflammation of adipose tissues suggested that Inpp4b−/− animals are insulin resistant. In an independently conducted QTL analysis in mice, Mu et al. have shown an association between the D8Mit195 locus, mapped immediately next to the Inpp4b gene, and elevated plasma glucose levels (LRS = 10.4, LOD = 2.3)30. The increase in the Lep/Adipoq ratio observed in HFD Inpp4b−/− group is characteristic to obese diabetic mice and men31,32. Glucose tolerance tests revealed that WT HFD-fed mice had an elevated fasting blood glucose level compared to the lean WT males. Upon administration of a bolus of oral glucose to WT males, serum glucose levels rose higher in HFD group than in LFD but recovered to normal after 120 min. When fed with either LFD or HFD, Inpp4b−/− mice had elevated glucose levels that they were unable to fully clear after 120 min (Fig. 3a, b). Insulin tolerance testing revealed that Inpp4b−/− males were resistant to insulin treatment (Supplementary Fig. 4a). The rapid increase of glucose to nearly 600 mg/dl during the glucose tolerance test, insulin resistance, and the inability to clear blood glucose in obese Inpp4b−/− males were consistent with obesity-induced T2D33. Circulating, non-fasting glucose and insulin levels were also significantly elevated in HFD-fed mutant animals compared to every other group (Fig. 3c, d). Despite normal levels of circulating insulin in LFD Inpp4b−/− males, the rate of hepatic gluconeogenesis was increased, confirming that lean knockout males developed hyperglycemia (Supplementary Fig. 4b). Similar to our HFD Inpp4b−/− male mice, gene expression analysis in the livers of non-diabetic (N = 9) and T2D obese individuals (N = 9) (GSE15653) revealed that INPP4B expression was decreased in the obese, diabetic group (Fig. 3e)34. As expected, pancreatic islets were positive for insulin staining by IHC (Fig. 3f). Consistent with the increase in blood insulin levels, the expression of proinsulin convertase, Pcsk1, was increased in the pancreata of LFD-fed Inpp4b−/− mice and in both HFD-fed groups (Supplementary Fig. 4c). Both HFD and Inpp4b knockout decreased expression of the peptide hormone cholecystokinin (Cck), an appetite suppressant and stimulant of pancreatic enzyme secretion35 (Supplementary Fig. 4d). The markers of pancreatic β-cell viability and inflammation, Furin and Il6, and the unfolded protein response (UPR), Mafa, did not significantly change (Supplementary Fig. 4e–g) at 3 months of age. Thus, our results and results from other groups suggest that INPP4B is a metabolic modulator that protects from diet-induced T2D.
Fig. 3. INPP4B protects mice from hyperglycemia and diabetes.

a Oral glucose tolerance test of LFD WT (N = 7), LFD Inpp4b−/− (N = 7), HFD WT (N = 9), and HFD Inpp4b−/− (N = 6) mice following a 2 g/kg oral glucose challenge. # is used to indicate statistically significant difference between LFD groups and * is used to indicate statistically significant difference for HFD groups. LFD WT, LFD Inpp4b−/−, HFD WT, and HFD Inpp4b−/− groups are represented by blue line with squares, red line with up-pointing triangles, green line with down-pointing triangles, and purple line with circles, respectively. b, c Blood glucose was measured in the same group as (a) after 6 h of fasting (b) or non-fasting (c) LFD WT, LFD Inpp4b−/−, HFD WT and HFD Inpp4b−/− mice. d Blood insulin was measured by ELISA in the same groups as (a). e INPP4B expression in livers of subjects with or without type II diabetes (T2D) were exported from GSE15653. f Representative pancreata sections from LFD WT, LFD Inpp4b−/−, HFD WT, and HFD Inpp4b−/− mice were stained for insulin and counterstained with hematoxylin (*p < 0.05, ***p < 0.001, ****p < 0.0001. Exact p values are provided in Supplementary Data 2).
Loss of INPP4B leads to NAFLD
NAFLD is the best predictor of insulin resistance9. To determine whether insulin resistance in Inpp4b−/− mice is associated with NAFLD, we first compared the liver weights in WT and Inpp4b−/− males fed with LFD or HFD. Strikingly, the liver weights were significantly elevated in Inpp4b−/− knockouts independent of the diet (Fig. 4a). Morphometric quantification of H&E stained slides revealed severe steatosis in the livers of all Inpp4b−/− mice (Fig. 4b, c). Livers in knockout animals featured all major histological characteristics of NAFLD, such as accumulation of fat (Fig. 4d), microvesicular and macrovesicular steatosis, hepatocellular hypertrophy, pyknotic nuclei, and Mallory bodies (Fig. 4e–g)36. Liver triglyceride amounts were significantly increased in Inpp4b−/− mice compared to WT controls in both LFD and HFD groups (Supplementary Fig. 5a). In the livers of WT male mice, the HFD caused modest increases in the expression of Inpp4b and Cd68, a marker of infiltrating macrophages (Supplementary Fig. 5b, c). In the human liver, INPP4B was detected in the cytoplasm and cell membrane of normal hepatocytes and its protein levels decreased in primary and, especially, metastatic hepatocellular carcinoma37, which undergo significant metabolic reprogramming. Similar to the mouse liver, hepatic INPP4B expression in patients with NAFLD was significantly reduced compared to healthy controls (GSE126848)38 and there was a highly significant correlation between steatosis score and INPP4B mRNA levels (Fig. 4h, i). Thus, the loss of INPP4B in mice is similar to the decline in INPP4B expression observed in steatotic livers of NAFLD patients38.
Fig. 4. INPP4B protects mice from liver steatosis.

a Livers were dissected from LFD WT (N = 11), LFD Inpp4b−/− (N = 13), HFD WT (N = 9), and HFD Inpp4b−/− (N = 10) mice. Liver weights in 4 experimental groups (*p = 0.0422 for LFD groups and *p = 0.0374 for HFD groups). b Representative H&E staining of liver sections from designated experimental groups used for steatosis analysis. c Morphometric quantification of the unstained area on H&E stained liver sections (*p = 0.111, ***p = 0.0004). d Representative images of the Oil Red O/hematoxylin staining of frozen liver sections in designated groups. e Characteristic features of hepatocellular steatosis in livers of HFD and LFD Inpp4b−/− males. Examples of microvesicular (mic) and macrovesicular (mac) steatoses, Mallory bodies (closed arrowhead) and pyknotic nuclei (open arrowhead). f Number of individual features per 100 cells. g Total number of steatotic features per 100 cells in each treatment group (*p = 0.0118, **p = 0.0052). h INPP4B expression in Reads Per Kilobase of transcript, per Million mapped reads (RPKM) in normal liver tissue (N = 14), obese (N = 12 patients), and NAFLD (N = 15 patients) in GSE126848 dataset (exact p values are provided in the Source data). One-way ANOVA (Dunnett’s multiple comparison test) was used. i Correlation between INPP4B expression with the steatosis score in patients with NAFLD or NASH. Data were exported from GSE126848.
NAFLD in Inpp4b−/− males is caused by increased lipogenesis, WAT inflammation, and activation of AKT and PKC signaling
To determine the molecular pathways that protect WT mice on HFD from NAFLD, we performed RNA-seq analysis of liver samples from WT and Inpp4b−/− males on a HFD. Analysis of differentially expressed genes (DEG) with DAVID functional annotation module and Gene Set Enrichment Analysis (GSEA) revealed that Inpp4b deletion affected lipid and glucose metabolisms, drug detoxification, peroxisome biogenesis, and expression of the T2D-associated genes (Fig. 5a, b and Supplementary Data 2). Lipid metabolism and peroxisome biogenesis are known targets of the PPAR family of transcription factors37,39,40. PPARG is a transcriptional regulator of fatty acid uptake and storage, while PPARA activity stimulates fatty acid oxidation41. Expression of Pparg increased in both LFD- and HFD-fed Inpp4b−/− males compared to WT controls (Fig. 5c). Correspondingly, expression of PPARG target genes regulating lipid delivery, internalization, triglyceride synthesis, and fatty acid metabolism increased in Inpp4b−/− livers (Fig. 5d–g). Strikingly, a group of genes involved in sexually dimorphic drug and lipid metabolism, Cyp2b9, Cyp2b13, and Hao2 (GSE 89091)42, were strongly downregulated by INPP4B as their expression dramatically increased in the livers of knockout males (Fig. 5g–i). Ppara expression was modestly increased in LFD Inpp4b−/− group only (Supplementary Fig. 5d), potentially contributing to partial NAFLD resistance in lean knockout animals. Similar to Inpp4b−/− mice, liver samples collected in the course of Roux-en-Y gastric bypass surgery from 1008 obese patients (GSE24335) exhibited a highly significant negative correlation between INPP4B and PPARG expression43 (Fig. 5j). Thus, expression data in mice and men support our finding that INPP4B regulates PPARG levels in the liver.
Fig. 5. INPP4B regulates metabolic and PPAR pathways in mouse liver.

a RNAs were extracted from livers of HFD WT (N = 4) and HFD Inpp4b−/− (N = 4) mice and submitted for RNA sequencing. The changes in gene expression were compared between HFD WT and HFD Inpp4b−/− mice. b KEGG pathway functional enrichment analysis was done using DAVID functional annotation module or GSEA. The vertical axis represents the KEGG pathway terms significantly enriched by the loss of INPP4B in mouse liver. The horizontal axis shows the logarithmic scale of the p value. c–i RNA from LFD WT (N = 10), LFD Inpp4b−/− (N = 12), HFD WT (N = 8), and HFD Inpp4b−/− (N = 10) mice were analyzed for Pparg (c), Cd36 (d), Mogat1 (e), Vldlr (f), Cyp2b9 (g), Cyp2b13 (h), and Hao2 (i) by qRT-PCR using 18S as an internal control. j Correlation between INPP4B expression and PPARG expression in 289 human liver samples (GSE24335) (**p < 0.01, ***p < 0.001, ****p < 0.0001. Exact p values are provided in the Source data).
Disparate spatial localization of these proteins suggests the existence of an intermediate regulatory protein. SREBP1 is a transcriptional activator of many lipogenic enzymes, including Pparg44,45. Based on our RNA-seq analysis, the levels of Srebf1 mRNA did not significantly increase in Inpp4b−/− livers (Fig. 6a). However, activation of SREBP1 requires proteolytic cleavage and nuclear translocation46,47. Comparison of the precursor (125 kDa) and transcriptionally active (55 kDa) forms of SREBP1 showed a significant increase in both proteins’ levels due to Inpp4b knockout and HFD treatment (Fig. 6b, c). We and others have previously reported that INPP4B regulates Akt and PKC21,22,48 pathways. Consistently, phosphorylation of AKT, PKCβII, and PKCζ was increased in the livers of HFD-fed and knockout animals with the highest expression in HFD Inpp4b−/− group (Fig. 6d–f). Interestingly, phosphorylation of PKCɛ remained unchanged in all groups. Conversely, 16-h treatment of Inpp4b−/− males with the Akt inhibitor, AZD5363, and pan-PKC inhibitor, BIM-I, led to a significant decline in both the precursor and mature forms of SREBP1 (Fig. 6g, h). Importantly, the expression of primary SREBP1 target genes, Mogat1, Cyp2b13, and Cyp2b949, were significantly decreased in the livers of mice treated with the inhibitors BIM-I and AZD5363 confirming direct involvement of Akt and PKC in the regulation of SREBP1 signaling (Fig. 6i).
Fig. 6. INPP4B regulates lipogenesis and AKT and PKC signaling pathways in mouse liver.

a RNA from Fig. 4(c–i) were analyzed for Srebf1 expression by qRT-PCR using 18S as control. b Proteins were extracted from livers of LFD WT, LFD Inpp4b−/−, HFD WT, and HFD Inpp4b−/− mice and used to compare levels of unprocessed SREBP1, processed SREBP1, and tubulin by western blotting. c Quantification of processed SREBP1 levels were presented as an average of three independent samples. The protein levels were normalized to tubulin and were shown in fold change from LFD WT. d Protein lysates from (b) were analyzed for pAKT, total AKT, pPKCβII, pPKCζ, pPKCε, and tubulin using western blotting. e, f Quantification of pAKT and pPKCβII protein from (d) (N = 3). g–i The Inpp4b−/− animals were treated with vehicle DMSO (N = 4), 6 mg/kg of AZD5363 (N = 2) or BIM-I (N = 4) for 16 h. Livers were dissected and used for protein and RNA purification. One-way ANOVA (Dunnett’s multiple comparison test) was used. (g) Fifty µg of protein extracted from livers of Inpp4b−/− animals treated with vehicle, BIM-I, or AZD5363 were used to compare levels of unprocessed and processed forms of SREBP1 and tubulin. (h) Quantification of processed or unprocessed SREBP1 proteins in (g) (N = 4). i RNAs purified from livers of Inpp4b−/− animals treated with vehicle, BIM-I, or AZD5363 were used to compare expression of SREBP1 target genes Mogat1, Cyp2b13, and Cyp2b9 by qRT-PCR using 18S as control. j WT or Inpp4b−/− mice were fasted overnight. Protein lysates from fasted and fed mice were isolated and analyzed for pAKT, precursor and processed forms of SREBP1, and tubulin using western blotting. k, l Quantification of pAKT and processed SREBP1 protein levels from two independent experiments (N = 4 for each individual bar) (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Exact p values are provided in the Source data).
Fasting suppresses lipogenesis by reducing the processing, and therefore activity, of the SREBP1 transcription factor. Remarkably, we found that downregulation of Akt signaling and SREBP1 processing during fasting was abolished in Inpp4b−/− males (Fig. 6j–l). There is strong evidence that SREBP1 expression and proteolysis in the liver is induced by insulin, Akt, retinoids, PKC, and its own activated form46,47,50–55. Thus, the activation of insulin, retinol, Akt, and PKC signaling pathways in obese knockout males contributes to the proteolytic cleavage of SREBP1 in the livers of HFD Inpp4b−/−. The cleaved form of SREBP1 is responsible for activating the expression of Pparg and other lipogenic enzymes that lead to the development of NAFLD (Figs. 2a, 3, 4, and 5b–f). The inability to cease de novo lipogenesis (DNL) during fasting cycles likely stimulates hepatic accumulation of lipids in Inpp4b−/− males.
HFD causes neoplastic changes in prostates of Inpp4b−/− mice
We have previously reported that Inpp4b−/− mice on regular chow have the same weight as WT animals and that they do not develop tumors in the first 4 months of age, while some hyperplastic changes developed in 1-year-old animals56. The increased leptin/adiponectin ratios and adipose inflammation are risk factors for the development of prostate cancer in men57,58 and were observed in HFD Inpp4b−/− males. We tested whether metabolic dysfunction triggered by the HFD would lead to an earlier and more severe prostate phenotype in knockout male mice. Indeed, all Inpp4b−/− males (N = 11) developed high-grade PIN by 11 weeks of age, while none of the WT males (N = 8) on the same diet did. Anterior prostate (AP), dorsolateral prostate (DLP), and ventral prostate (VP) lobes exhibited various degrees of epithelial expansion and loss of polarity. The most extensive histological changes were observed in DLP glands, including epithelial and basal layer expansion, piling up of epithelial cells (Fig. 7a, b), loss of polarity (Supplementary Fig. 6a), a discontinuous fibromuscular layer around individual glands (Supplementary Fig. 6b), and nuclear atypia (Supplementary Fig. 6c).
Fig. 7. Loss of Inpp4b leads to the development of PIN and inflammation in obese males.

a H&E staining of prostates from LFD WT, LFD Inpp4b−/−, HFD WT, and HFD Inpp4b−/− males. b Representative tissue sections from LFD WT, LFD Inpp4b−/−, HFD WT, and HFD Inpp4b−/− prostates were stained for α-SMA and counterstained with hematoxylin. Anterior prostate (AP), dorsolateral prostate (DLP), and ventral prostate (VP) were dissected from LFD WT (N = 8), LFD Inpp4b−/− (N = 8), HFD WT (N = 9), and HFD Inpp4b−/− (N = 9) mice. c–j RNA was extracted and the expression levels of Pbsn (c), Apof (d), Nkx3.1 (e), Msmb (f), Igfbp3 (g), Il6 (h), Tnf (i), and Il1b (j) were compared by qRT-PCR using 18S as an internal control. k Protein concentrations of IL-6 in mouse DLPs were measured using IL-6 ELISA (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Exact p values are provided in the Source data).
Androgen receptor activity, cell signaling, and inflammation are among the factors most strongly contributing to neoplastic transformation of the prostate59. The protein and mRNA levels of AR and PTEN were not significantly altered by INPP4B loss on either diet (Supplementary Fig. 7a–d). While the HFD led to increased macrophage infiltration in the prostates of both WT and mutant males, there was no difference between the two genotypes in Cd68 expression (Supplementary Fig. 7e). Inflammatory infiltrate was observed in the prostate stromata of obese WT and knockout males (Supplementary Fig. 7f, marked with *). Staining with the Ki67 antibody indicated increased cellular proliferation in the prostates of HFD Inpp4b−/− males (Supplementary Fig. 8a, b). While AR protein levels did not change, its transcriptional activity was affected. Expression of AR-induced target genes, Pbsn, Apof, Nkx3.1, and Msmb, were significantly decreased in the DLP of LFD Inpp4b−/− males (Fig. 7c–f). HFD further decreased expression of AR target genes Nkx3.1 and Msmb (Fig. 7e, f). However, we observed no change in the ability of AR to repress transcription of its target gene Igfbp3 (Fig. 7g). Interestingly, HFD decreased the levels of Inpp4b expression in DLP by almost two-fold (Supplementary Fig. 8c). Levels of pAkt were increased in prostates of LFD Inpp4b−/− mice; no further increase was detected in HFD-fed groups (Supplementary Fig. 8d, e). Similarly, levels of PKCβII and pPKCζ were slightly increased in DLP and VP lobes of LFD Inpp4b−/− males and treatment with HFD did not further increase the signal (data not shown).
HFD leads to prostate inflammation in Inpp4b−/− males
Inflammation plays a key role in the etiology of prostate cancer60. We tested whether the development of prostate neoplasms in the HFD Inpp4b−/− males was accompanied by inflammation. In the dorsolateral lobe of the prostates of obese males, Il6 and Tnf mRNA levels were significantly increased in Inpp4b−/− mice when compared to WT (Fig. 7h, i). As expected, expression of the potent proinflammatory cytokine, Il1b, was undetectable in the prostates of LFD mice. Consumption of HFD induced the expression of Il1b in the prostates in both genotypes. In the DLP, the loss of INPP4B resulted in a significant increase in Il1b expression when compared to WT (Fig. 7j). Protein levels of the proinflammatory cytokine IL6 were significantly increased in the DLP of HFD-fed Inpp4b−/− mice (Fig. 7k). While there are anatomical differences between mice and humans, the mouse DLP is considered most similar to the human peripheral zone, the site of origin of most human prostate cancers61. It is also important to note that in the prostate, the dorsolateral region features high level of Inpp4b expression (Supplementary Fig. 8c), suggesting a physiological function for INPP4B protein in that lobe.
Discussion
Metabolic dysfunction is the cause of significant morbidity and mortality due to diabetes, cardiovascular abnormalities, and an increased risk of cancer incidence and mortality. Though epidemiological links between increased calorie consumption, cancer, and diabetes are well established, less is known about the specific genes that protect some obese individuals from metabolic dysfunction and neoplasia. In this report, we show that INPP4B is a key regulator of metabolic health in lean male mice and protects obese mice from diabetes and prostate neoplasia.
INPP4B has been shown to play a tumor suppressor role in prostate, breast, and other cancers22,48,62,63, however, Inpp4b−/− mice do not develop malignancies64. Our data demonstrated significant metabolic changes in Inpp4b−/− males at 3 months of age and show that HFD exacerbated these changes. Inpp4b−/− males had an elevated fat-to-lean body mass ratio despite a somewhat lower food intake. This was likely due to their reduced activity and associated decline in energy expenditure and respiratory exchange ratio (Fig. 1 and Supplementary Fig. 1). INPP4B-dependent regulation of insulin sensitivity and its downstream Akt and PKC pathways led to the increased circulating levels of insulin and accelerated weight gain in HFD Inpp4b−/− males. Hepatic Akt and PKC signaling pathways are required for insulin sensitivity12, and, in fact, we observed inefficient glucose clearance and insulin resistance in both LFD and HFD Inpp4b−/− animals (Fig. 3 and Supplementary Fig. 4a). Inpp4b−/− mice developed hyperglycemia on an LFD and T2D on a HFD. Insulin promotes hepatic lipogenesis largely through upregulation of SREBP1, a transcription factor that potently activates lipogenic pathways65. Taniguchi et al. reported that in mouse liver, Akt mediates glucose homeostasis and insulin tolerance while PKCζ stimulates expression of SREBP153,66. Akt and PKC signaling also induce proteolytic cleavage of the SREBP1 precursor protein into a mature transcription factor46,47 which regulates the expression and activity of PPARG67,68. Consistently, we found that INPP4B loss and a HFD cooperatively induced the levels of pAkt, pPKCβII, pPKCζ, and SREBP1 cleavage, leading to increased expression of Pparg and other lipogenic transcripts during fed state (Figs. 5 and 6). SREBP1 processing declines dramatically during fasting to inhibit lipogenesis in the liver. Our data suggest that fasting-induced inhibition of SREBP1 processing is regulated by INPP4B (Fig. 6j–l). As a direct result of these transcriptional and posttranslational changes in the expression and activation of crucial cellular signaling factors, we observed the development of hepatosteatosis, WAT expansion, and inflammation in Inpp4b−/− males which in turn were further exacerbated by a HFD. At 3 months of age, both Inpp4b knockout and HFD increased circulating levels of the metabolic hormones insulin, C-peptide, leptin, and resistin (Fig. 2). Insulin and C-peptide are the products of the proteolytic cleavage of proinsulin. Increased leptin concentrations correlated with the increases in adipose tissue mass caused by HFD treatment and INPP4B loss (Figs. 1c and 2i). Increased resistin level is also consistent with our phenotype as this hormone mediates insulin resistance and links obesity to T2D in mice and men69,70. It is important to note that the Inpp4b−/− mouse model closely resembles human NAFLD. The downregulation of hepatic INPP4B expression in patients with T2D and advanced hepatosteatosis, as well as a highly significant negative correlation between the expression of INPP4B and PPARG was observed in livers of obese patients (Figs. 4g–i and 5j).
Metabolic dysfunction and adipose inflammation in HFD Inpp4b−/− males affected prostate gland development, as is often observed in men with this condition. HFD Inpp4b−/− males were significantly more susceptible to obesity-induced neoplastic transformation of the prostate than the WT males (Fig. 7a, b). Circulating leptin, local inflammation, and activated immune cells in prostate stroma likely contribute to the inflammatory environment of the prostate71–73. At 3 months of age, only local inflammation was detected in the prostate (Fig. 7h–k). No significant changes between 4 experimental groups in serum levels of IL-6, TNFα, and IL-1β inflammatory cytokines were detected at 3 months of age. It was established that IL-6 is elevated in tissues of BPH and prostate carcinomas in men74 and intratumoral IL-1β levels significantly correlate with biochemical recurrence in prostate cancer patients75. Importantly, prostatic neoplasms were detected only in HFD Inpp4b−/− males, suggesting a modulatory role for INPP4B in obesity-induced prostatic neoplasms. We have shown that the prostatic glands displayed increased proliferation of the epithelial layer and occasional breach of prostate glands’ fibromuscular membranes (Fig. 7a, b and Supplementary Fig. 6b). Distribution of E-cadherin in normal prostate glands was restricted to the lateral and basal membranes of the secretory epithelium. In neoplastic glands of HFD Inpp4b−/−, epithelial cells lost their polarity and E-cadherin distributed evenly around the cellular membranes (Supplementary Fig. 6a). Similar to previously reported c-myc-induced neoplastic lesions in the mouse prostate76,77, neoplastic lesions in HFD Inpp4b−/− males display hypochromatic, enlarged atypical nuclei with prominent enlarged nucleoli when compared to the more compact and densely stained nuclei of normal prostate epithelium cells (Supplementary Fig. 6c).
To the best of our knowledge, we discovered a novel function of INPP4B. We showed that INPP4B is an essential regulator of metabolic health. INPP4B expression protects the liver from steatosis, mediates insulin sensitivity, and links obesity to neoplastic changes in the prostate epithelium. As shown in Fig. 8, Inpp4b-deficient animals developed hyperglycemia on normal chow and T2D on HFD due to decreased insulin sensitivity. The loss of INPP4B induced the activation of hepatic SREBP1, ultimately leading to the development of NAFLD. Only the loss of INPP4B in HFD-fed mice promoted prostate neoplasia, suggesting that INPP4B might provide a functional link between obesity and increased prostate cancer incidence. In both mice and men, a decline in hepatic INPP4B expression correlated with increasing severity of NAFLD. Thus, Inpp4b−/− males faithfully reproduce the complex metabolic syndrome in men and, accordingly, the INPP4B signaling cascade should be considered for therapeutic intervention in this wide-spread disease.
Fig. 8. Physiological functions of INPP4B in liver, WAT, and prostate.
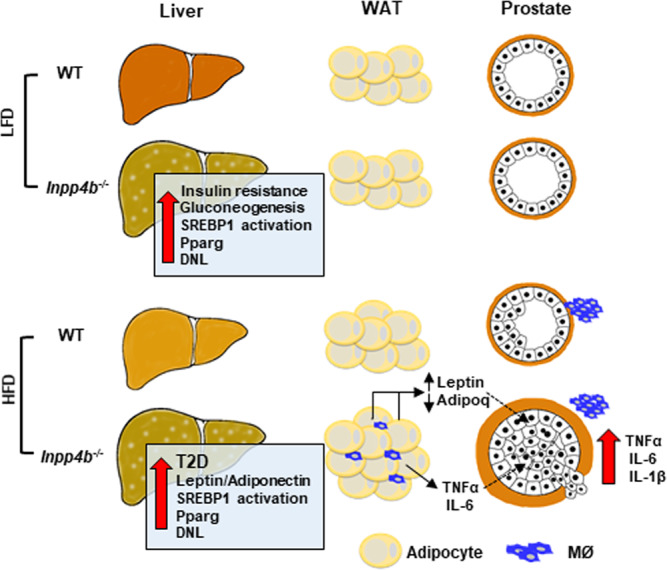
The loss of INPP4B activates SREBP1, stimulates hepatic expression of Pparg, causing liver steatosis, insulin resistance, de novo lipogenesis (DNL), and an increased rate of gluconeogenesis. When consuming a HFD, the Inpp4b−/− males developed type 2 diabetes, inflammation, and macrophage (MØ) infiltration of WAT, DNL, and an increased leptin/adiponectin ratio, all of which were shown to stimulate the development of PIN. Consumption of a HFD promoted macrophage infiltration in the prostates of both WT and Inpp4b−/− mice; however, increased production of proinflammatory cytokines was only evident in dorsolateral lobes of prostates of Inpp4b−/− mice.
Methods
Animal studies
Animals were housed at AAALAC certified facility at Florida International University. All protocols were approved by Florida International University Institutional Animal Care and Use Committee. Inpp4b−/− mice were provided by Dr. Vacher and bred into an FVB background56,78. WT and Inpp4b−/− males were fed with either LFD (LabDiet 5V75, St. Louis, MO) or HFD (TestDiet 58R3, St. Louis, MO). The HFD consisted of 59.4% fat, 25.7% carbohydrate, and 14.9% protein (total 22.8 kJ/g), whereas the regular chow contained 12.9% fat, 63.8% carbohydrate, and 23.2% protein (total 13.6 kJ/g). LFD groups were continuously maintained on LFD. For HFD cohorts, dams were fed the designated diet for 1 month prior to mating, during pregnancy, and 3 weeks lactation. Male pups were maintained on the HFD until euthanasia79,80. Mouse body weights were measured once a week. All LFD WT, LFD Inpp4b−/−, HFD WT, and HFD Inpp4b−/− mice were euthanized and dissected at 12 weeks and their tissues were collected for analysis. Six-month-old Inpp4b−/− mice were treated with DMSO control, or 6 mg/kg of Akt inhibitor AZD5363 (SelleckChem, Houston, TX) or PKC inhibitor BIM-I (AdipoGen, San Diego, CA) by intraperitoneal injection. After 16-h treatment, mice were euthanized, and livers were collected for gene and protein analyses.
Hemodynamic measurements
Eleven-week-old HFD, WT, and Inpp4b−/− mice were anesthetized with 2% isoflurane in 100% oxygen. Each mouse was placed on a heating platform with the temperature between 38 and 40 °C. A tail cuff connected to the pressurizing tubing was placed at the base of the mouse tail and attached to the CODA monitor (Kent Scientific, Torrington, CT). The heart rate, systolic, diastolic, and mean blood pressure where recorded using a non-invasive blood pressure system Coda Monitor Noninvasive Blood Pressure System with Coda Mouse Cuff Kit (Kent Scientific, Torrington, CT).
Indirect calorimetry and body composition analysis
Indirect calorimetry and body composition analysis were performed at Vanderbilt Mouse Metabolic Phenotyping Center. Eight WT and ten Inpp4b−/− males were individually placed in metabolic cages (identical to home cages with bedding) in a 12 h light/dark cycle, temperature/humidity-controlled dedicated room located in the Vanderbilt MMPC. Energy expenditure measures were obtained by indirect calorimetry (Promethion, Sable Systems, Las Vegas, NV). The calorimetry system consists of cages identical to home cages with bedding equipped with water bottles and food hoppers connected to load cells for food and water intake monitoring. All animals had ad libitum access to standard rodent chow and water. The air within the cages is sampled through microperforated stainless-steel sampling tubes that ensure uniform cage air sampling. Promethion utilizes a pull-mode, negative pressure system with an excurrent flow rate set at 2000 ml/min. Water vapor is continuously measured and its dilution effect on O2 and CO2 are mathematically compensated for in the analysis stream81. O2 consumption and CO2 production are measured for each mouse every 5 min for 30 s. Incurrent air reference values are determined every 4 cages. Respiratory quotient (RQ) is calculated as the ratio of CO2 production over O2 consumption. Energy expenditure is calculated using the Weir equation: EE (kcal/h) = 60 × (0.003941 × VO2(ml/min) + 0.001106 × VCO2(ml/min))82. Ambulatory activity was determined every second with XYZ beams. Data acquisition and instrument control were coordinated by MetaScreen v2.2.18 and the raw data were processed using ExpeData v1.7.30 (Sable Systems). Body composition in awake animals was determined at the Vanderbilt Mouse Metabolic Phenotyping Center by NMR using Bruker Minispec body composition analyzer (Bruker Optics, Billerica, MA).
Gene expression analysis
RNA was isolated from tissues using Tri Reagent (Molecular Research Center, Cincinnati, OH) and reverse transcribed using a Verso cDNA synthesis Kit (Thermo Fisher Scientific, Waltham, MA). The quantitative real-time PCR was performed using primers and probes from Universal Probe Library (Supplementary Data 1) (Roche, Basel, Switzerland) and a Roche 480 LightCycler (Roche). For gene expression analysis, samples were from LFD WT (N = 12), LFD Inpp4b−/− (N = 11), HFD WT (N = 9), and HFD Inpp4b−/− (N = 11) mice.
Glucose tolerance test (GTT) and pyruvate tolerance test (PTT)
The glucose tolerance tests were performed on 11-week-old males as previously described83. Briefly, after 6 h of fasting, mice (N ≥ 8) were weighed and given 2 g/kg of glucose (Sigma-Aldrich, St. Louis, MO) through a gavage needle (18 G, 5.08 cm; Cadence Science, Cranston, RI). A drop of blood was sampled from each mouse at 0, 15, 30, 60, 90, and 120 min after oral gavage and the glucose concentration was determined immediately using the ACCU-CHEK Nano SmartView glucometer (Roche Diagnostics, Indianapolis, IN). For pyruvate tolerance testing, mice were fasted overnight, followed by intraperitoneal injection of pyruvate (1.5 g/kg, Sigma-Aldrich, St. Louis, MO). The blood glucose levels were measured as above.
Insulin tolerance test (ITT)
Four-month-old LFD WT (N = 5) and LFD Inpp4b−/− (N = 9) male mice were fasted for 3 h in clean cages with standard light/dark cycle and free access to water. The mice were weighed and given 0.75 U/kg Humulin R insulin (Eli Lilly and Company, Indianapolis, IN) by intraperitoneal injection. A drop of blood was sampled from each mouse immediately before insulin injection and 30, 60, 90, and 120 min after. Glucose concentrations were determined using the ACCU-CHEK Nano SmartView glucometer (Roche Diagnostics, Indianapolis, IN).
ELISA and adipokine array
Blood insulin levels were determined using the Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem, Elk Grove Village, IL) according to the manufacturer’s instructions. Briefly, 5 µl of blood serum from LFD WT (N = 23), LFD Inpp4b−/− (N = 20), HFD WT (N = 9), and HFD Inpp4b−/− (N = 9) mice were used for the test. The insulin concentrations were measured using a CLARIOstar plate reader (BMG Labtech, Cary, NC). A Mouse IL6 ELISA kit (Sigma-Aldrich) was used to determine the IL6 level in LFD WT (N = 6), LFD Inpp4b−/− (N = 10), HFD WT (N = 5), and HFD Inpp4b−/− (N = 5) mouse DLPs. Adipokines and proinflammatory cytokines were detected in WAT protein extracts using the Proteome Profiler Mouse Adipokine Array Kit (R&D Systems, Minneapolis, MN). Samples from five mice in each group were pooled and used for analysis. The signal was captured using an ImageQuant LAS 500 imager (GE Healthcare, Chicago, IL).
Multiplex analysis of metabolic cytokines and hormones
The serum from LFD WT (N = 11), LFD Inpp4b−/− (N = 11), HFD WT (N = 9), HFD Inpp4b−/− (N = 9) male mice was analyzed for the following cytokines and chemokines: Amylin (active), C-Peptide 2, GIP (total), GLP-1(active), Ghrelin (active), Glucagon, Insulin, Leptin, PP, PYY, and Resistin. The analysis was done using the Mouse Metabolic 11-plex Arrays (Eve Technologies, Calgary, AB, Canada).
Morphometric quantification of steatosis
Liver steatosis was evaluated by measuring the surface of the unstained area of the H&E stained liver sections per field using 40X magnification. Five mice were selected from each group and 10 different areas from each mouse were counted using ImageJ software (National Institute of Mental Health, Bethesda, USA).
Oil red O staining
Fresh liver tissue was placed in OCT medium and snap frozen in liquid nitrogen. Liver samples were cut into 12 μm sections and attached to glass slides. Oil red O was dissolved in hot propylene glycol and filtered through Whatman #2 filter paper. Slides were fixed in 4% PFA, rinsed with distilled water and propylene glycol followed by staining with Oil red O and counterstained with hematoxylin. All images were acquired using an AxioCam MRc5 camera (Zeiss, Thornwood, NY).
Determination of triglycerides in mouse liver
The total triglycerides in liver were measured in LFD WT (N = 8), LFD Inpp4b−/− (N = 8), HFD WT (N = 6), and HFD Inpp4b−/− (N = 4) mice using a Triglyceride Colorimetric Assay Kit (Cayman Chemical, Ann Arbor, MI). The assay was performed according to the manufacturer’s instruction. The total amount of triglycerides in liver was calculated.
RNA sequencing and data analysis
RNA was isolated as described above from HFD WT (N = 4) and HFD Inpp4b−/− (N = 4), and submitted to Novogene for RNA sequencing. The libraries were generated using the NEBNext® Ultra™ RNA Library Prep Kit and used for Illumina 150-bp paired-end sequencing. Quality control assessment was done using Illumina RNA-seq pipeline to estimate genomic coverage, percent alignment, and nucleotide quality. Raw reads were mapped to the reference mouse genome (the most recent build GRCm38) using HISAT284 and STAR85 software. The reads for each gene aligned by HISAT2 were counted using HTSeq software86. Alignment by STAR was run with the option “—quantMode Gene Counts” that allowed counting of reads aligned to each gene. Raw counts from HTSeq and STAR were imported into Bioconductor/R package DESeq287, normalized, and tested for differential gene expression. This analysis was done separately for the files produced by each aligner. In each analysis we selected genes that were differentially expressed based on the criteria of false discovery rate (FDR) <10% and fold change >1.3 in either direction. Genes that showed differential expression after analysis of the files from both aligners have been selected for further analysis. Data has been uploaded to NCBI’s gene expression omnibus (GEO) database with accession number GSE134466.
Pathway analysis and Gene Ontology (GO) analysis
Biological processes and pathways affected by INPP4B knockout in mouse liver were performed using GSEA (http://www.broad.mit.edu/gsea/index.html) and DAVID (the Database for Annotation, Visualization and Integrated Discovery) analysis with DEGs obtained from the RNA-seq analysis. The gene sets for KEGG pathways (c2 KEGG curated) and GO analysis (c5 curated) were acquired from Molecular Signatures Database (http://software.broadinstitute.org/gsea/msigdb/index.jsp). The normalized enrichment score (NES) with FDR < 25% or p value < 0.05 was considered significant88.
Immunohistochemistry
Paraffin-embedded tissues were sectioned at 5 µM, and deparaffinized. For H&E, sections were then stained with Harris’ Alum Hematoxylin (#638A-85, Millipore, Burlington, MA) and Eosin-Y-Solution (#314–630, Thermo Fisher Scientific)56. For immunohistochemistry, antigen retrieval was done by heating slides at 99 °C for 15 min in 10 mM citrate buffer, pH = 6. Sections were blocked in 1% H2O2 in water and 5% BSA in PBS. Primary antibodies were diluted in PBS at 1:6000 for α-SMA (#ab5694, AbCam, Cambridge, MA), 1:60,000 for insulin (#ab181547, AbCam), 1:500 for E-cadherin (#3195, Cell Signaling, Danvers, MA), and 1:200 in Ki67 (#RB-1510-P1, Thermo Fisher Scientific). Antibodies were incubated overnight at 4 °C or for 1 h at room temperature. Sections were washed and incubated with biotinylated anti-rabbit secondary antibody for 18 min, followed by incubation with streptavidin-conjugated peroxidase for 30 min using the Vectastain ABC Kit (Vector Laboratories, Burlingame, CA). The staining was developed using the ImmPACT DAB Peroxidase Substrate Kit (Vector Labs, Burlingame, CA) and then the slides were counterstained with hematoxylin.
Western blotting
Mouse tissues were homogenized with a glass grinder in ice-cold RIPA buffer with protease and phosphatase inhibitors (GenDepot, Barker, TX). Cleared lysates were diluted to 4 µg/µl and 15–30 µg of protein was resolved on a 7.5–10% SDS-PAGE. For immunoblotting, the following primary antibodies and dilutions were used: rabbit antibodies total Akt (1:1000, #4691, Cell Signaling, Danvers, MA), pPKCζ T410 (1:1000, # 2060, Cell Signaling), pPKCβII S660 (1:1000, #9371, Cell Signaling), FAS (1:1000, #3180, Cell Signaling), HK2 (1:1000, #2867, Cell Signaling), PTEN (1:1000, #9188, Cell Signaling), primary mouse antibodies pAkt S473 (1:1000, #4051, Cell Signaling), β-tubulin (1:5000, #05–661, Millipore), SREBP1 (1:1000, NB #600-582, Novus Biologicals, Littleton, CO), CD68 (1:800, #ab 125212, AbCam, Cambridge, MA), and primary sheep antibody PKCε (1:2000, AF5134, R&D Systems, Minneapolis, MN). The following secondary antibodies were used: rabbit IgG (1:2000, W4011, Promega, Madison, WI), mouse IgG (1:2000, W4021, Promega), and sheep IgG (1:1000, HAF016, R&D systems). The signal was captured using an ImageQuant LAS 500 imager and analyzed by ImageQuant TL software (GE Healthcare, Chicago, IL).
Statistics and reproducibility
All data are presented as mean ± SEM, unless otherwise stated. Two-way ANOVA (Sidak multiple comparison test) for 4 groups and two-tailed Student’s t-test for 2 groups were performed by Prism 7.0. The p value < 0.05 and confidence level of 95% were considered statistically significant. Individual p values are reported in figure legends and/or in Source data (reported in Supplementary Data 2). For the box plots, the whiskers go down to the smallest value and up to the largest. The box extends from the 25th to 75th percentiles. Center line is plotted at the median.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This work was supported by NCI grant R15 CA179287-01A1 (I.U.A. and M.Z.), NIDDK 1R01DK110167 (A.I.A., I.U.A., and E.M.K.), and Canadian Institutes of Health Research grant #123343 (J.V.). The indirect calorimetry study was performed by the Vanderbilt Mouse Metabolic Phenotyping Center (DK059637 and 1S10RR028101-01).
Author contributions
I.U.A. designed and supervised the study; A.I.A. provided expertise for the mouse hepatosteatosis studies; M.M.I. conducted comparative histological analysis of the WT and the Inpp4b−/− mouse prostates; M.Z., Y.C., E.M.K., and J.L.V. performed experiments and prepared the figures; L.N. has conducted RNA-seq, bioinformatics, and data analysis; J.V. provided the Inpp4b−/− mouse model; F.K.K. provided expertise in transcriptomics in normal and steatotic human livers; I.U.A. and M.Z. wrote the manuscript with substantive contributions from all co-authors.
Data availability
Source data are available in Supplementary Data 2. RNA-sequencing data have been deposited in NCBI’s Sequence Read Archive (SRA) database with accession number GSE134466. The datasets that support the findings of this study are available in (https://www.ncbi.nlm.nih.gov/geo/) with the accession numbers and DOIs: GSE15653 (10.1210/jc.2009-0212), GSE126848 (10.1152/ajpgi.00358.2018), GSE89091 (10.1038/s41374-018-0088-6) and GSE24335 (10.1101/gr.112821.110). Source data for Figs. 1–7 and Supplementary Figs. 1, 3, 4, 5, 7, 8 are available in Supplementary Data 2.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-021-01940-6.
References
- 1.Collaborators GBDD. Health effects of dietary risks in 195 countries, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2019;393:1958–1972. doi: 10.1016/S0140-6736(19)30041-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wildman RP, et al. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: prevalence and correlates of 2 phenotypes among the US population (NHANES 1999-2004) Arch. Intern. Med. 2008;168:1617–1624. doi: 10.1001/archinte.168.15.1617. [DOI] [PubMed] [Google Scholar]
- 3.Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012;18:363–374. doi: 10.1038/nm.2627. [DOI] [PubMed] [Google Scholar]
- 4.Akinyemiju T, et al. Metabolic dysregulation and cancer mortality in a national cohort of blacks and whites. BMC Cancer. 2017;17:856. doi: 10.1186/s12885-017-3807-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quail DF, Dannenberg AJ. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 2018;15:139–154. doi: 10.1038/s41574-018-0126-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rohrmann S, Smit E, Giovannucci E, Platz EA. Association between markers of the metabolic syndrome and lower urinary tract symptoms in the Third National Health and Nutrition Examination Survey (NHANES III) Int. J. Obes. (Lond.) 2005;29:310–316. doi: 10.1038/sj.ijo.0802881. [DOI] [PubMed] [Google Scholar]
- 7.Cao Y, Ma J. Body mass index, prostate cancer-specific mortality, and biochemical recurrence: a systematic review and meta-analysis. Cancer Prev. Res (Phila.) 2011;4:486–501. doi: 10.1158/1940-6207.CAPR-10-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liss MA, et al. Higher baseline dietary fat and fatty acid intake is associated with increased risk of incident prostate cancer in the SABOR study. Prostate Cancer Prostatic Dis. 2019;22:244–251. doi: 10.1038/s41391-018-0105-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fabbrini E, et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc. Natl Acad. Sci. USA. 2009;106:15430–15435. doi: 10.1073/pnas.0904944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feldman A, et al. Clinical and metabolic characterization of lean Caucasian subjects with non-alcoholic fatty liver. Am. J. Gastroenterol. 2017;112:102–110. doi: 10.1038/ajg.2016.318. [DOI] [PubMed] [Google Scholar]
- 11.Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J. Clin. Invest. 2016;126:12–22. doi: 10.1172/JCI77812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samuel VT, Shulman GI. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 2018;27:22–41. doi: 10.1016/j.cmet.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumashiro N, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl Acad. Sci. USA. 2011;108:16381–16385. doi: 10.1073/pnas.1113359108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510:84–91. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vegiopoulos A, Rohm M, Herzig S. Adipose tissue: between the extremes. EMBO J. 2017;36:1999–2017. doi: 10.15252/embj.201696206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang F, Li S, Pan L, Jia C. Association of the G1057D polymorphism in insulin receptor substrate 2 gene with type 2 diabetes mellitus: a meta-analysis. J. Diabetes Complications. 2015;29:731–736. doi: 10.1016/j.jdiacomp.2015.04.017. [DOI] [PubMed] [Google Scholar]
- 17.Ristow M, Muller-Wieland D, Pfeiffer A, Krone W, Kahn CR. Obesity associated with a mutation in a genetic regulator of adipocyte differentiation. N. Engl. J. Med. 1998;339:953–959. doi: 10.1056/NEJM199810013391403. [DOI] [PubMed] [Google Scholar]
- 18.Vasseur F, Meyre D, Froguel P. Adiponectin, type 2 diabetes and the metabolic syndrome: lessons from human genetic studies. Expert Rev. Mol. Med. 2006;8:1–12. doi: 10.1017/S1462399406000147. [DOI] [PubMed] [Google Scholar]
- 19.Yang MM, et al. Variations in the obesity gene “LEPR” contribute to risk of type 2 diabetes mellitus: evidence from a meta-analysis. J. Diabetes Res. 2016;2016:5412084. doi: 10.1155/2016/5412084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez SM, et al. Determinants of the tumor suppressor INPP4B protein and lipid phosphatase activities. Biochem. Biophys. Res. Commun. 2013;440:277–282. doi: 10.1016/j.bbrc.2013.09.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodgson MC, et al. INPP4B suppresses prostate cancer cell invasion. Cell Commun. Signal. 2014;12:61. doi: 10.1186/s12964-014-0061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodgson MC, et al. Decreased expression and androgen regulation of the tumor suppressor gene INPP4B in prostate cancer. Cancer Res. 2011;71:572–582. doi: 10.1158/0008-5472.CAN-10-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferron M, Vacher J. Characterization of the murine Inpp4b gene and identification of a novel isoform. Gene. 2006;376:152–161. doi: 10.1016/j.gene.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 24.Stern JH, Rutkowski JM, Scherer PE. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 2016;23:770–784. doi: 10.1016/j.cmet.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fasshauer M, Bluher M. Adipokines in health and disease. Trends Pharm. Sci. 2015;36:461–470. doi: 10.1016/j.tips.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 26.Gupta RK. Adipocytes. Curr. Biol. 2014;24:R988–993. doi: 10.1016/j.cub.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 27.Fantuzzi G. Adipose tissue, adipokines, and inflammation. J. Allergy Clin. Immunol. 2005;115:911–919. doi: 10.1016/j.jaci.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 28.Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl Acad. Sci. USA. 2003;100:7265–7270. doi: 10.1073/pnas.1133870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amano SU, et al. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 2014;19:162–171. doi: 10.1016/j.cmet.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mu JL, et al. Quantitative trait loci analysis for the differences in susceptibility to atherosclerosis and diabetes between inbred mouse strains C57BL/6J and C57BLKS/J. J. Lipid Res. 1999;40:1328–1335. doi: 10.1016/S0022-2275(20)33495-7. [DOI] [PubMed] [Google Scholar]
- 31.Scherer PE. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes. 2006;55:1537–1545. doi: 10.2337/db06-0263. [DOI] [PubMed] [Google Scholar]
- 32.Kim JY, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Invest. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clee SM, Attie AD. The genetic landscape of type 2 diabetes in mice. Endocr. Rev. 2007;28:48–83. doi: 10.1210/er.2006-0035. [DOI] [PubMed] [Google Scholar]
- 34.Pihlajamaki J, et al. Thyroid hormone-related regulation of gene expression in human fatty liver. J. Clin. Endocrinol. Metab. 2009;94:3521–3529. doi: 10.1210/jc.2009-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rehfeld JF. Cholecystokinin-from local gut hormone to ubiquitous messenger. Front Endocrinol. (Lausanne) 2017;8:47. doi: 10.3389/fendo.2017.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang W, et al. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE. 2014;9:e115922. doi: 10.1371/journal.pone.0115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang W, et al. INPP4B inhibits cell proliferation, invasion and chemoresistance in human hepatocellular carcinoma. Onco Targets Ther. 2019;12:3491–3507. doi: 10.2147/OTT.S196832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suppli MP, et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am. J. Physiol. Gastrointest. Liver Physiol. 2019;316:G462–G472. doi: 10.1152/ajpgi.00358.2018. [DOI] [PubMed] [Google Scholar]
- 39.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat. Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 40.Vamecq J, Latruffe N. Medical significance of peroxisome proliferator-activated receptors. Lancet. 1999;354:141–148. doi: 10.1016/S0140-6736(98)10364-1. [DOI] [PubMed] [Google Scholar]
- 41.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev. Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 42.Chikada H, et al. Establishment and analysis of a mouse model that regulates sex-related differences in liver drug metabolism. Lab. Invest. 2018;98:1500–1511. doi: 10.1038/s41374-018-0088-6. [DOI] [PubMed] [Google Scholar]
- 43.Greenawalt DM, et al. A survey of the genetics of stomach, liver, and adipose gene expression from a morbidly obese cohort. Genome Res. 2011;21:1008–1016. doi: 10.1101/gr.112821.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JB, Spiegelman BM. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996;10:1096–1107. doi: 10.1101/gad.10.9.1096. [DOI] [PubMed] [Google Scholar]
- 45.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002;109:1125–1131. doi: 10.1172/JCI0215593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat. Rev. Endocrinol. 2017;13:710–730. doi: 10.1038/nrendo.2017.91. [DOI] [PubMed] [Google Scholar]
- 47.Sajan MP, et al. The critical role of atypical protein kinase C in activating hepatic SREBP-1c and NFkappaB in obesity. J. Lipid Res. 2009;50:1133–1145. doi: 10.1194/jlr.M800520-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gewinner C, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–125. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seo YK, et al. Genome-wide analysis of SREBP-1 binding in mouse liver chromatin reveals a preference for promoter proximal binding to a new motif. Proc. Natl Acad. Sci. USA. 2009;106:13765–13769. doi: 10.1073/pnas.0904246106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fleischmann M, Iynedjian PB. Regulation of sterol regulatory-element binding protein 1 gene expression in liver: role of insulin and protein kinase B/cAkt. Biochem. J. 2000;349:13–17. doi: 10.1042/bj3490013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hagiwara A, et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012;15:725–738. doi: 10.1016/j.cmet.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 52.Porstmann T, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taniguchi CM, et al. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKClambda/zeta. Cell Metab. 2006;3:343–353. doi: 10.1016/j.cmet.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 54.Yamamoto T, et al. Protein kinase Cbeta mediates hepatic induction of sterol-regulatory element binding protein-1c by insulin. J. Lipid Res. 2010;51:1859–1870. doi: 10.1194/jlr.M004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yecies JL, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011;14:21–32. doi: 10.1016/j.cmet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang M, et al. Inositol polyphosphate 4-phosphatase type II regulation of androgen receptor activity. Oncogene. 2019;38:1121–1135. doi: 10.1038/s41388-018-0498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gucalp A, et al. Periprostatic adipose inflammation is associated with high-grade prostate cancer. Prostate Cancer Prostatic Dis. 2017;20:418–423. doi: 10.1038/pcan.2017.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rhee H, Vela I, Chung E. Metabolic syndrome and prostate cancer: a review of complex interplay amongst various endocrine factors in the pathophysiology and progression of prostate cancer. Horm. Cancer. 2016;7:75–83. doi: 10.1007/s12672-015-0238-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nelson WG, De Marzo AM, Isaacs WB. Prostate cancer. N. Engl. J. Med. 2003;349:366–381. doi: 10.1056/NEJMra021562. [DOI] [PubMed] [Google Scholar]
- 60.De Marzo AM, et al. Inflammation in prostate carcinogenesis. Nat. Rev. Cancer. 2007;7:256–269. doi: 10.1038/nrc2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shappell SB, et al. Prostate pathology of genetically engineered mice: definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004;64:2270–2305. doi: 10.1158/0008-5472.CAN-03-0946. [DOI] [PubMed] [Google Scholar]
- 62.Fedele CG, et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc. Natl Acad. Sci. USA. 2010;107:22231–22236. doi: 10.1073/pnas.1015245107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Westbrook TF, et al. A genetic screen for candidate tumor suppressors identifies REST. Cell. 2005;121:837–848. doi: 10.1016/j.cell.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 64.Kofuji S, et al. INPP4B is a PtdIns(3,4,5)P3 phosphatase that can act as a tumor suppressor. Cancer Discov. 2015;5:730–739. doi: 10.1158/2159-8290.CD-14-1329. [DOI] [PubMed] [Google Scholar]
- 65.Shimomura I, et al. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl Acad. Sci. USA. 1999;96:13656–13661. doi: 10.1073/pnas.96.24.13656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shimano H, et al. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J. Clin. Invest. 1996;98:1575–1584. doi: 10.1172/JCI118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fajas L, et al. Regulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: implications for adipocyte differentiation and metabolism. Mol. Cell Biol. 1999;19:5495–5503. doi: 10.1128/MCB.19.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc. Natl Acad. Sci. USA. 1998;95:4333–4337. doi: 10.1073/pnas.95.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Steppan CM, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 70.Savage DB, et al. Resistin/Fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-gamma action in humans. Diabetes. 2001;50:2199–2202. doi: 10.2337/diabetes.50.10.2199. [DOI] [PubMed] [Google Scholar]
- 71.Dib LH, Ortega MT, Fleming SD, Chapes SK, Melgarejo T. Bone marrow leptin signaling mediates obesity-associated adipose tissue inflammation in male mice. Endocrinology. 2014;155:40–46. doi: 10.1210/en.2013-1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sfanos KS, De Marzo AM. Prostate cancer and inflammation: the evidence. Histopathology. 2012;60:199–215. doi: 10.1111/j.1365-2559.2011.04033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu P, et al. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006;124:615–629. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 74.Siegsmund MJ, Yamazaki H, Pastan I. Interleukin 6 receptor mRNA in prostate carcinomas and benign prostate hyperplasia. J. Urol. 1994;151:1396–1399. doi: 10.1016/S0022-5347(17)35267-9. [DOI] [PubMed] [Google Scholar]
- 75.Eiro N, et al. Analysis of the expression of interleukins, interferon beta, and nuclear factor-kappa B in prostate cancer and their relationship with biochemical recurrence. J. Immunother. 2014;37:366–373. doi: 10.1097/CJI.0000000000000045. [DOI] [PubMed] [Google Scholar]
- 76.Ellwood-Yen K, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–238. doi: 10.1016/S1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 77.Iwata T, et al. MYC overexpression induces prostatic intraepithelial neoplasia and loss of Nkx3.1 in mouse luminal epithelial cells. PLoS ONE. 2010;5:e9427. doi: 10.1371/journal.pone.0009427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ferron M, et al. Inositol polyphosphate 4-phosphatase B as a regulator of bone mass in mice and humans. Cell Metab. 2011;14:466–477. doi: 10.1016/j.cmet.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ceyhan Y, et al. Deletion of inositol polyphosphate 4-phosphatase type-II B affects spermatogenesis in mice. PLoS ONE. 2020;15:e0233163. doi: 10.1371/journal.pone.0233163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Benesh EC, Humphrey PA, Wang Q, Moley KH. Maternal high-fat diet induces hyperproliferation and alters Pten/Akt signaling in prostates of offspring. Sci. Rep. 2013;3:3466. doi: 10.1038/srep03466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lighton, J. R. B. Measuring Metabolic Rates (Oxford University Press, 2008).
- 82.Weir JB. New methods for calculating metabolic rate with special reference to protein metabolism. J. Physiol. 1949;109:1–9. doi: 10.1113/jphysiol.1949.sp004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J. Evaluating the glucose tolerance test in mice. Am. J. Physiol. Endocrinol. Metab. 2008;295:E1323–1332. doi: 10.1152/ajpendo.90617.2008. [DOI] [PubMed] [Google Scholar]
- 84.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dobin A, Gingeras TR. Mapping RNA-seq reads with STAR. Curr. Protoc. Bioinformatics. 2015;51:11–19. doi: 10.1002/0471250953.bi1114s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
Source data are available in Supplementary Data 2. RNA-sequencing data have been deposited in NCBI’s Sequence Read Archive (SRA) database with accession number GSE134466. The datasets that support the findings of this study are available in (https://www.ncbi.nlm.nih.gov/geo/) with the accession numbers and DOIs: GSE15653 (10.1210/jc.2009-0212), GSE126848 (10.1152/ajpgi.00358.2018), GSE89091 (10.1038/s41374-018-0088-6) and GSE24335 (10.1101/gr.112821.110). Source data for Figs. 1–7 and Supplementary Figs. 1, 3, 4, 5, 7, 8 are available in Supplementary Data 2.