

ABSTRACT
In order to improve the safety of novel therapeutic drugs, better understanding of the mechanisms of action is important. Ado-trastuzumab emtansine (also known as T-DM1) is an antibody-drug conjugate (ADC) approved for the treatment of HER2-positive breast cancer. While the treatment with T-DM1 results in significant efficacy in the selected patient population, nonetheless, there are concerns with side effects such as thrombocytopenia and hepatotoxicity. While current understanding of the mechanism of T-DM1-mediated side effects is still incomplete, there have been several reports of HER2-dependent and/or -independent mechanisms that could be associated with the T-DM1-induced adverse events. This review highlights the importance of HER2-independent mechanism of T-DM1 to induce hepatotoxicity, which offers a new insight into a role for CKAP5 in the overall maytansinoid-based ADC (DM1 and DM4)-mediated cytotoxicity. This discovery provides a molecular basis for T-DM1-induced off-target toxicity and opens a new avenue for developing the next generation of ADCs.
Keywords: antibody-drug conjugate, HER2-positive metastatic breast cancer, payload, ado-trastuzumabemtansine/T-DM1, off-target toxicity
Statement of Significance: This review summarizes the results showing that ado-trastuzumab emtansine (T-DM1) binds to CKAP5 on the cell surface of hepatocytes via its payload component (DM1). This discovery provides a molecular basis for T-DM1-induced off-target toxicity and opens a new avenue for developing the next generation of ADCs.
ANTIBODY-DRUG CONJUGATE
The development of novel therapeutic drugs that incorporate molecular targets and previously un- and under-explored mechanisms of action has the potential to advance the field of cancer drug, but it also poses multiple product development hurdles [1]. Among others, unintended toxicities and unknown drug safety profiles could be introduced by such novel therapeutics [1]. Antibody-drug conjugate (ADC) is a relatively new class of therapeutic, which is generated by linking highly specific monoclonal antibodies (e.g., trastuzumab) to highly toxic chemical agents (e.g., DM1 or DM4) via different linker molecules [2,3]. This approach could enable specific targeting of antigen-expressing target cells with highly toxic chemical agents following ADC internalization and the release of cytotoxic agents inside the cells [2]. There are many antigens, chemical agents and linker types that have been explored for the design of ADCs, and the literature summary is available elsewhere [2,4]. Currently, there are about more than 100 ongoing clinical studies with ADCs for the treatment of human cancers, and 9 ADCs have been approved by the FDA for the treatment of different cancers (Table 1) [5–11]. Despite their preclinical promises, ~20 ADCs have been discontinued from clinical trials because of unforeseen or unacceptable toxicities [12]. The toxicities induced by ADC molecules may be driven by any of the components of ADCs [12].
Table 1.
FDA-approved ADCs
| Drug name | Approval year | Indication(s) |
|---|---|---|
| GO (trade name: Mylotarg) https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761060lbl.pdf |
2017; 2000 | Newly diagnosed and relapsed CD33-positive acute myeloid leukemia (AML) |
| Brentuximab vedotin (trade name: Adcetris) https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125388s099lbl.pdf |
2011 | Hodgkin lymphoma (HL) and systemic anaplastic large cell lymphoma (ALCL) |
| Ado-trastuzumab emtansine (trade name: Kadcyla) https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125427s105lbl.pdf |
2013 | HER2-positive, metastatic breast cancer and the adjuvant treatment of patients with HER2-positive early breast cancer |
| Inotuzumab ozogamicin (trade name: Besponsa) https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf |
2017 | Relapsed or refractory CD22-positive B-cell precursor acute lymphoblastic leukemia |
| Polatuzumab vedotin-piiq (trade name: Polivy) https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761121s000lbl.pdf |
2019 | Relapsed or refractory diffuse large B-cell lymphoma (DLBCL) |
| Enfortumab vedotin (trade name: Padcev) https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761137s000lbl.pdf |
2019 | Locally advanced or metastatic urothelial cancer |
| Trastuzumab deruxtecan (trade name: Enhertu) https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761139s000lbl.pdf |
2019 | Unresectable or metastatic HER2-positive breast cancer |
| Sacituzumab govitecan (trade name: Trodelvy) https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761115s000lbl.pdf |
2020 | Metastatic triple-negative breast cancer (mTNBC) |
| Belantamab mafodotin-blmf (trade name: Blenrep) https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761158s000lbl.pdf |
2020 | Relapsed or refractory multiple myeloma |
While a full understanding of the safety profile for any ADCs would require extensive nonclinical and clinical testing followed by careful post-market surveillance, any prior knowledge of potential safety concerns originating from different ADC components (such as antibody, linker and payload) may prove advantageous in clinical development strategy [1,13]. Some of the ADCs utilize components which have already been extensively tested in clinical studies and approved by regulatory authorities (e.g. approved monoclonal antibody trastuzumab used for the development of ADC ado-trastuzumab emtansine) [2,14]. Currently, the majority of ADCs are being developed for oncology indications. International Conference on Harmonization (ICH) S9 (2009) provides guidance on the nonclinical safety assessment of anticancer pharmaceuticals, including protein conjugates [1,15].
ADC-mediated targeting is expected to be relatively selective toward antigen-expressing tissues. Given that the hybrid nature of ADCs includes a small molecule component, it is conceivable that some of common ADC-associated toxicities may be due to the small molecule component and not due to the antibody or linker components [2,12]. The analysis performed on approved ADC therapeutics indicated that many of the dose-limiting toxicities, including bone marrow suppression, hepatotoxicity and neuropathy, are related to the small molecule component [1,13]. Better understanding of these mechanisms of antigen-dependent and/or -independent cytotoxicities induced by ADCs is warranted for future ADC development strategies.
T-DM1 AND T-DM1-INDUCED TOXICITIES
Ado-trastuzumab emtansine (also known as T-DM1) is an ADC approved by FDA for the treatment of trastuzumab-resistant human epidermal growth factor receptor 2 (HER2)-positive metastatic breast cancer and for use as an adjuvant treatment option for patients with HER2-positive early breast cancer who have residual invasive disease [16–18]. T-DM1 consists of a maytansinoid toxin DM1 linked to a humanized monoclonal antibody trastuzumab via a thioether-based chemical linker [19]. Upon binding to HER2, internalization and subsequent lysosomal proteolytic degradation of T-DM1 results in the release of Lys-MCC-DM1 metabolite in the cell where it targets microtubules and induces cell death [19]. Thrombocytopenia, elevated liver transaminases and peripheral neuropathy are the most common and frequent toxicities seen in T-DM1 therapy [5,20]. Interesting to note, some of these toxicities were previously noted for other approved ADC-based therapies [1,13].
MECHANISMS OF T-DM1-INDUCED OFF-TARGET TOXICITIES
The mechanisms which contribute to T-DM1-induced off-target toxicities are still elusive, and a better understanding has the potential to benefit the whole ADC therapeutic class. Although the preclinical studies alone are not sufficient for predicting the full spectrum of potential toxicities, these studies may provide very useful information to help understand the mechanisms underlying significant adverse events. For example, Uppal et al. demonstrated that T-DM1 inhibited megakaryocyte differentiation via the HER2-independent, FcγRIIa-dependent pathway by showing that the FcγRIIa receptor-blocking antibody, anti-CD32, blocked the T-DM1–cell surface binding and subsequent internalization of T-DM1 [21]. Based on the data described in that article, the authors concluded that FcγRIIa, at least partially, contributes to the mechanisms of T-DM1 binding and internalization to mediate the impairment of megakaryocytes’ differentiation. However, because the data did not support the co-localization of internalized T-DM1 with a lysosomal marker LAMP1 in the megakaryocytes, it is still not clear from this study as to how the ADC/FcγRIIa complex is internalized and how the payload of T-DM1, Lys-MCC-DM1, is released to the target microtubules in the megakaryocytes [21]. Thon et al. showed that T-DM1 internalization was dependent on the trastuzumab antibody and not through the HER2 or Fc receptor since the megakaryocytes and platelets did not express HER2 on the cell surface and the non-specific humanized antibody (5B6)-DM1 was not internalized in the mouse megakaryocytes [22]. This study also showed that T-DM1 disrupted the microtubule organization in both megakaryocytes and platelet cells [22]. Although there is still controversy in T-DM1-induced thrombocytopenia from the two independent groups, their studies have offered a possibility that HER2-independent T-DM1 internalization by megakaryocytes may result in thrombocytopenia.
Hepatotoxicity is one of the black box warnings for T-DM1 treatment since it may cause severe increases in liver enzymes, serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) [23]. In order to explore the molecular mechanisms underlying the T-DM1-induced hepatotoxicity, we have developed in vitro and in vivo models, such as immortalized human liver cells, which express lower HER2 levels compared to the T-DM1-sensitive and HER2-positive breast cancer cell lines, such as SKBR-3 and BT-474 [24]. Our data suggested that T-DM1 might cause hepatotoxicity via the HER2-dependent pathway [24]. We also found that the hepatocellular injury mediated by T-DM1 was further enhanced by a pro-inflammatory cytokine, TNF-α, that triggered a mitochondrial-dependent apoptosis due to the rupture of the outer mitochondrial membrane caused by T-DM1 [24]. Gemtuzumab ozogamicine (GO) is yet another example where hepatotoxicity is believed to be attributed to a specific antigen-dependent target of an ADC [25]. GO is an ADC consisting of an anti-CD33 antibody linked to a cytotoxic antibiotic calicheamicin [25–27]. GO was approved by the FDA in 2000 for the treatment of acute myeloid leukemia (AML) and was withdrawn from the market in 2010 because the post-approval study failed to show an improved survival when compared with chemotherapy alone in patients, as well as safety concerns, including significant hepatotoxicity [25,26]. Maniecki et al. demonstrated that the CD33 receptor was widely distributed in the liver tissue and on hepatocytes [28]. It is believed that the specific targeting of GO to CD33-expressing hepatocytes is related to hepatotoxicity [28].
CKAP5, A NOVEL TARGET OF T-DM1-INDUCED TOXICITY
In addition to HER2-dependent hepatotoxicity induced by T-DM1, we also noticed that DM1-conjugated to a non-targeting IgG can also cause a dose-dependent cell growth inhibition in vitro [23]. We hypothesized that a HER2-independent mechanism might also be involved in the T-DM1-induced hepatotoxicity. To search for the novel target molecule(s) involved in the T-DM1-induced hepatotoxicity, T-DM1 was used as a bait and was incubated with human and mouse hepatocytes in cell-culture dishes to allow for the immunoprecipitation analysis of T-DM1-bound cell surface molecules. This screening revealed a 230 kDa protein that specifically bound to T-DM1 but not to trastuzumab or control IgG [29]. This 230 kDa protein was identified by mass spectrometry as cytoskeleton-associated protein 5 (CKAP5, also known as ch-TOG or XMAP215), which is a member of the XMAP215/Dis 1 family that plays a critical role in the regulation of microtubule polymerization by binding to tubulin [29–31]. Regarding the tissue-distribution of CKAP5, based on The Human Protein Atlas (https://www.proteinatlas.org/), CKAP5 is ubiquitously expressed in a variety of human tissues. We further confirmed that CKAP5 binding to T-DM1 was mediated via the DM1 portion of T-DM1 and demonstrated that the interaction between DM1 and CKAP5 occurred on the cell surface, independent of trastuzumab, the antibody portion of T-DM1 [29]. Our data showed that upon binding to CKAP5 on the cell surface, T-DM1 began to damage cell plasma membrane followed by calcium influx into hepatocytes, resulting in the disruption of microtubule polymerization and apoptosis [29]. In our study, brentuximab vedotin consisting of MMAE failed to bind to CKAP5 and it did not cause the cell membrane damage and apoptosis of hepatocytes [29].
Our study identifies a novel mechanism of ADC (T-DM1)-mediated and target (HER2)-independent cytotoxicity in normal cells/tissues where the levels of HER2 expression are low, thus, highlights that the internalization of T-DM1 followed by payload release inside of the cells may not be necessary for T-DM1 to induce liver cell injury and hepatotoxicity. In addition, our study indicates that this novel mechanism may also be relevant for other DM1- and DM4-conjugated ADCs as the non-specific ADC molecules (α-CD22-DM1 and α-gD-DM4) were found to bind to CKAP5 and induce microtubules’ disorganization in hepatocytes [29]. These data further suggest that CKAP5 can be considered as a cellular target for off-target toxicity mediated by maytansinoid (DM1 or DM4)-conjugated ADCs and warrants further preclinical and clinical studies. Figure 1 summaries the mechanisms by which T-DM1 induces hepatotoxicity via both HER2- and CKAP5-dependent pathways.
Figure 1.
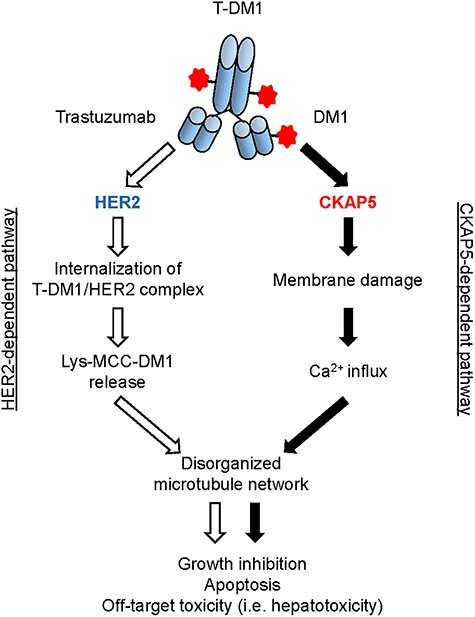
Proposed working model: T-DM1-mdiated cytotoxicity via both HER2-independent and -dependent mechanisms. CKAP5-dependent pathway: upon specific binding to CKAP5 via payload (DM1) of T-DM1 on cell surface of hepatocytes, T-DM1 begins to damage cell plasma membrane followed by calcium influx to the inside of hepatocytes. The increased calcium concentration in the cytosol of hepatocytes caused disorganized microtubule network, which resulted in cell growth inhibition, apoptosis and hepatotoxicity. T-MD1 payload (DM1)-mediated plasma membrane damage and calcium influx via interaction with CKAP5 on the cell surface may be particularly important for T-DM1-induced cytotoxicity of normal cells/tissues, where HER2 expression is relatively low or missing to induce “off-target” toxicity. HER2-dependent pathway: Upon T-DM1-mediated interaction with HER2 expressed on the surface of tumor cells, T-DM1/HER2 complex is internalized, followed by lysosomal degradation and payload release to target microtubules, resulting in apoptosis and cell death. HER2-dependent pathway plays a major role for the mechanisms of T-DM1 action in HER2-positive tumor cells.
FUTURE DIRECTIONS
Currently, maytansinoid-conjugated ADCs are a dominant proportion of all ADCs under clinical investigation [4,5]. Side effects observed in clinical studies with maytansinoid-conjugated ADCs appear to be related to the payload, DM1 and DM4 [12]. For example, gastrointestinal effects, thrombocytopenia and neutropenia are associated with DM1-conjugated ADCs, while ocular toxicity is the most commonly associated with DM4-conjugated ADCs [12]. Hepatotoxicity, however, is observed with both, DM1- and DM4-conjugated ADCs [32]. A meta-analysis of ADC (660 publications) between 2000 and January 2014 showed that there was a notable difference in the several key toxicities between DM1- and DM4-conjugated ADCs [33]. It is interesting to note that we also observed a difference in binding activity toward to CKAP5 between DM1- and DM4-conjugated ADCs [29], suggesting that it may be relevant for the differential toxicity profiles detected in the respective ADCs. Our study advances the understanding of the mechanisms of ADC-induced off-target toxicity. Thus, masking payloads or toxin in ADCs prior to the internalization step might be a useful approach to reduce target-independent but payloads/toxin-dependent side effects. The design of such ADCs, especially maytansinoid-conjugated ADCs, should benefit considerably from the better understanding of the payload-mediated and antigen-independent cytotoxicity in the normal cells.
While full understanding of the mechanisms of T-DM1-associated off-target toxicity remains elusive, the preclinical studies performed by our lab as well as by other laboratory clearly illustrate that ADC induces off-target toxicity/side effects via both antigen-dependent and antigen-independent mechanisms [21,22,24,29]. While the expression patterns may be available for the different targets/antigens of ADCs prior to ADC product development and thus such target-dependent toxicities could be potentially predicted, the understanding of the mechanisms contributing to the antigen-independent or payload-dependent toxicities is still rudimentary and more research efforts are needed in that field. The availability of multiple ADC therapeutics in advanced clinical testing and a better understanding of molecular mechanisms of “off-target” toxicity induced by ADCs can provide a good beginning of the framework in the future research efforts in dealing with ADC-mediated toxicity.
ACKNOWLEDGMENTS
We thank Drs. Tao Xie and Nozomi Sakakibara for critical internal review of the manuscript. This work is supported by US Food and Drug Administration (FDA) intramural research funding.
Contributor Information
Yukinori Endo, Division of Biotechnology Review and Research 1, Office of Biotechnology Products, Office of Pharmaceutical Quality, Center for Drug Evaluation and Research, U.S. Food and Drug Administration (FDA), Silver Spring, MD 20993, USA.
Nishant Mohan, Division of Biotechnology Review and Research 1, Office of Biotechnology Products, Office of Pharmaceutical Quality, Center for Drug Evaluation and Research, U.S. Food and Drug Administration (FDA), Silver Spring, MD 20993, USA.
Milos Dokmanovic, Division of Biotechnology Review and Research 1, Office of Biotechnology Products, Office of Pharmaceutical Quality, Center for Drug Evaluation and Research, U.S. Food and Drug Administration (FDA), Silver Spring, MD 20993, USA.
Wen Jin Wu, Division of Biotechnology Review and Research 1, Office of Biotechnology Products, Office of Pharmaceutical Quality, Center for Drug Evaluation and Research, U.S. Food and Drug Administration (FDA), Silver Spring, MD 20993, USA.
CONFLICT OF INTEREST STATEMENT
All authors declared no competing financial and/or non-financial interests in relation to the work described.
DISCLAIMER
This article reflects the views of the authors and should not be construed to represent FDA’s views or policies.
References
- 1. Lansita, JA, Burke, JM, Apgar, JFet al. An introduction to the regulatory and nonclinical aspects of the nonclinical development of antibody drug conjugates. Pharm Res 2015; 32: 3584–92. [DOI] [PubMed] [Google Scholar]
- 2. Dokmanovic, M, ElZarrad, MK, Hirsch, DSet al. Antibody-drug conjugates as therapeutic agents in oncology: overview and perspectives. In: Frontiers in Anti-Cancer Drug Discovery, Bentham Science Publishers, Vol.2. 2013, 139–89. [Google Scholar]
- 3. Ducry, L, Stump, B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem 2010; 21: 5–13. [DOI] [PubMed] [Google Scholar]
- 4. Beck, A, Goetsh, L, Dumontet, Cet al. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov 2017; 16: 315–37. [DOI] [PubMed] [Google Scholar]
- 5. Wolska-Washer, A, Robak, T. Safety and tolerability of antibody-drug conjugates in cancer. Drug Saf 2019; 42: 295–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chau, CH, Steeg, PS, Figg, WD. Antibody-drug conjugates for cancer. Lancet 2019; 394: 793–804. [DOI] [PubMed] [Google Scholar]
- 7. Deeks, ED. Polatuzumab vedotin: first global approval. Drugs 2019; 79: 1467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hanna, KS. Enfortumab vedotin to treat urothelial carcinoma. Drugs Today (Barc) 2020; 56: 329–35. [DOI] [PubMed] [Google Scholar]
- 9. Keam, SJ. Trastuzumab deruxtecan: first approval. Drugs 2020; 80: 501–8. [DOI] [PubMed] [Google Scholar]
- 10. Syed, YY. Sacituzumab govitecan: first approval. Drugs 2020; 80: 1019–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Markham, A. Belantamab mafodotin: first approval. Drugs 2020; 80: 1607–13. [DOI] [PubMed] [Google Scholar]
- 12. Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016; 8: 659–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saber, H, Leighton, JK. An FDA oncology analysis of antibody-drug conjugates. Regul Toxicol Pharmacol 2015; 71: 444–52. [DOI] [PubMed] [Google Scholar]
- 14. Lambert, JM, Chari, RV. Ado-trastuzumab emtansine (T-DM1): an antibody-drug conjugate (ADC) for HER2-positive breast cancer. J Med Chem 2014; 57: 6949–64. [DOI] [PubMed] [Google Scholar]
- 15.Food and Drug Administration, HHS. International Conference on Harmonisation; Guidance on S9 Nonclincal Evaluation for Anticancer Pharmaceuticals; availability. Notice. Fed Regist. 2010;75:10487–8. PMID: 20383918. [PubMed]
- 16. Giordano, SH, Temin, S, Kirshner, JJet al. Systemic therapy for patients with advanced human epidermal growth factor receptor 2-positive breast cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2014; 32: 2078–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Von Minckwitz, G, Huang, SH, Mano, MSet al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N Engl J Med 2019; 380: 617–28. [DOI] [PubMed] [Google Scholar]
- 18. Wedam, S, Fashoyin-Aje, L, Gao, Xet al. FDA approval summary: ado-trastuzumab emtansine for the adjuvant treatment of HER2-positive early breast cancer. Clin Cancer Res 2020; 26: 4180–5. [DOI] [PubMed] [Google Scholar]
- 19. Lewis Phillips, GD, Li, G, Dugger, DLet al. Targeting HER2- positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res 2008; 68: 9280–90. [DOI] [PubMed] [Google Scholar]
- 20. Boyraz, B, Sendur, MA, Aksoy, Set al. Tratuzumab emtansine (T-DM1) for HER2-potivite breast cancer. Curr Med Res Opin 2013; 29: 405–14. [DOI] [PubMed] [Google Scholar]
- 21. Uppal, H, Doudement, E, Mahapatra, Ket al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res 2015; 21: 123–33. [DOI] [PubMed] [Google Scholar]
- 22. Thon, JN, Devine, MT, Jurak Begonja, Aet al. High-content live-cell imaging assay used to establish mechanism of trastuzumab emtansine (T-DM1)–mediated inhibition of platelet production. Blood 2012; 120: 1975–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Diéras, V, Harbeck, N, Budd, GTet al. Trastuzumab emtansine in human epidermal growth factor receptor 2-positive metastatic breast cancer: an integrated safety analysis. J Clin Oncol 2014; 32: 2750–7. [DOI] [PubMed] [Google Scholar]
- 24. Yan, H, Endo, Y, Shen, Yet al. Ado-trastuzumab emtansine targets hepatocytes via human epidermal growth factor receptor 2 to induce hepatotoxicity. Mol Cancer Ther 2016; 13: 480–90. [DOI] [PubMed] [Google Scholar]
- 25. Voutsadakis, IA. Gemtuzumab ozogamicin (CMA-676, Mylotarg) for the treatment of CD33+ acute myeloid leukemia. Anti-Cancer Drugs 2002; 13: 685–92. [DOI] [PubMed] [Google Scholar]
- 26. Tanimoto, T, Tsubokura, M, Mori, Jet al. Differences in drug approval processes of 3 regulatory agencies: a case study of gemtuzumab ozogamicin. Investig New Drugs 2013; 31: 473–8. [DOI] [PubMed] [Google Scholar]
- 27. Rowe, JM, Löwenberg, B. Gemtuzumab ozogamicin in acute myeloid leukemia: a remarkable saga about an active drug. Blood 2013; 121: 4838–41. [DOI] [PubMed] [Google Scholar]
- 28. Maniecki, MB, Hasle, H, Bendix, Ket al. Is hepatotoxicity in patients treated with gemtuzumab ozogamicine due to specific targeting of hepatocytes? Leuk Res 2011; 35: e84–6. [DOI] [PubMed] [Google Scholar]
- 29. Endo, Y, Takeda, K, Mohan, Net al. Payload of T-DM1 binds to cell surface cytoskeleton-associated protein 5 to mediate cytotoxicity of hepatocytes. Oncotarget 2018; 9: 37200–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Al-Bassam, J, Larsen, NA, Hyman, AAet al. Crystal structure of a TOG domain: conserved feature of XMAP215/Dis-1-family TOG domains and implications of tubulin binding. Structure 2007; 15: 355–62. [DOI] [PubMed] [Google Scholar]
- 31. Brouhard, GJ, Stear, JH, Noetzel, TLet al. XMAP215 is a processive microtubule polymerase. Cell 2008; 132: 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Taplin, S, Vashisht, K, Walles, Met al. Hepatotoxicity with antibody maytansinoid conjugates: a review of preclinical and clinical findings. J Appl Toxicol 2018; 38: 600–15. [DOI] [PubMed] [Google Scholar]
- 33. Masters, JC, Nickens, DJ, Xuan, Det al. Clinical toxicity of antibody drug conjugates: a meta-analysis of payloads. Investig New Drugs 2018; 36: 121–35. [DOI] [PubMed] [Google Scholar]