

Abstract
People worldwide are living longer, and it is estimated that by 2050, the proportion of the world’s population over 60 years of age will nearly double. Natural lung aging is associated with molecular and physiological changes that cause alterations in lung function, diminished pulmonary remodeling and regenerative capacity, and increased susceptibility to acute and chronic lung diseases. As the aging population rapidly grows, it is essential to examine how alterations in cellular function and cell-to-cell interactions of pulmonary resident cells and systemic immune cells contribute to a higher risk of increased susceptibility to infection and development of chronic diseases, such as chronic obstructive pulmonary disease and interstitial pulmonary fibrosis. This review provides an overview of physiological, structural, and cellular changes in the aging lung and immune system that facilitate the development and progression of disease.
Keywords: lung aging, inflammation, chronic obstructive pulmonary disease, COPD, interstitial pulmonary fibrosis, IPF, acute respiratory distress syndrome, ARDS, pneumonia
INTRODUCTION
Longitudinal studies have illustrated that pulmonary function impairment is a predictor for morbidity and mortality and may promote the development of multiple disease processes (1, 2). As the aging population rapidly grows, it is essential to examine how physiological and cellular changes in the aging lung bring about the development and progression of lung diseases.
Age-associated changes in intrinsic mechanisms that aid in cell regeneration and repair, such as depletion of adult stem cell reservoirs, mitochondrial dysfunction, increased oxidative stress, and telomere shortening, contribute to an inability of lung cells to maintain baseline homeostasis. Senescent cells display a genetic and morphologic phenotype that is distinct from growth-competent cells and can have a detrimental impact on neighboring cells, structural components, and composition of the extracellular matrix (ECM) (3). In healthy aging, common cellular stresses can yield primary senescent cells that diminish tissue-repair capacity due to cell cycle arrest in progenitor cells and increase production of proinflammatory and matrix-degrading molecules (3, 4). Exposure to extrinsic time-related effects, such as chronic exposure to atmospheric pollution, dusts, particulates, and gases, induce low-level insults to cellular function and accelerate aging of the lung (3). DNA damage in response to compounds, such as those present in cigarette smoke, as well as telomere shortening due to increased demand for injured airway epithelium repair, can result in disease-related senescence and subsequent generation of a secondary subset of senescent cells (5). In contrast to normal aging, disease-induced senescence may be restricted to the lung and, owing to prolonged or intense exposure to disease-related senescence triggers, will result in an increased rate of senescent cell accumulation that further amplifies disease progression. While cellular senescence in resident cells in response to intrinsic and extrinsic factors may lead to both age- and disease-related changes in the lung, it is important to note that senescence in circulating immune cells can also impact lung aging and disease progression.
As lung aging is highly complex and can result from accumulated changes between injury and repair to lung cellular systems, this review provides an overview of physiological, structural, and cellular changes that occur during aging. Moreover, this review examines how genetic background and lifestyle modulate age-related changes in the lung, thereby contributing to the development and progression of airway diseases such as chronic obstructive pulmonary disease (COPD) and interstitial pulmonary fibrosis (IPF) as well as increased susceptibility to infectious stimuli and environmental exposures.
STRUCTURAL AND PHYSIOLOGIC CHANGES IN THE AGING LUNG
There is considerable change in lung structure and function with advancing age (summarized in Supplemental Table 1). Noninvasive studies in healthy subjects have illustrated age-dependent differences in acinar microstructure (6). The alveoli, with their capillary network for gas exchange, and the airways that allow the movement of air in and out of the alveoli are the principal functional respiratory components of the lungs. Although the number of alveoli, alveolar ducts, and capillary segments remain constant at adulthood, there is a marked increase in alveolar size and alveolar-capillary surface area with aging (6). Alterations in alveolar depth and acinar airway lumen that occur during compensatory remodeling are associated with advanced age (6). Elastic recoil of the lungs, due to a reduction in the surface tension forces caused by increased individual diameters of alveoli, is reduced with aging and gives rise to an increase in end-expiratory lung volume (7, 8). Larger alveolus, which results from changes in the coiling structure of elastin and other fibers around alveolar ducts, leads to an evenly distributed increase in the size of alveoli without destruction to the alveolar wall (9–12). A decline in elastic recoil combined with a reduction in the number of elastic attachments of supporting alveoli and increased collagen results in smaller airways closing at higher lung volumes (9, 10). As a result, some airways may be narrowed or closed during normal tidal breathing in older individuals, thereby increasing the functional residual capacity and reducing the expiratory airflow from the lungs (12, 13). As age-associated declines in elastic recoil are not uniform throughout the lung, nonuniform distribution of ventilation, due to regions of the lung being more or less compliant, can occur.
It is important to note that marked changes in chest wall function and respiratory muscle strength can also impact lung aging and the ability to clear mucus or foreign particulates from the lungs. The shape of the thorax, due to age-associated changes in thoracic posture and general stiffening of the ribs, can impact lung function (14–16). A reduction in elastic recoil of the lungs and changes in chest wall deformation give rise to increased functional residual capacity with age (7, 14, 17). A reduction in respiratory muscle and diaphragm strength also impacts lung function as a person ages (14–18). Narrowing of the intervertebral disk space can contribute to curvature of the spine and decreased space between the ribs, resulting in a smaller chest cavity. Despite an age-associated reduction in elastic recoil, due to decreased respiratory strength and chest wall compliance, total lung capacity does not significantly change with age (7, 12, 14, 19).
It is well appreciated that, even in healthy persons, lung function declines starting at the age of 35. Specifically, there is a decline in forced expiratory volume (FEV1) by approximately 30 mL/year and forced vital capacity (FVC) by approximately 20 mL/year, which results in an age-associated decline in the FEV1/FVC ratio (7, 12–14, 20, 21). As a person ages, there is a decrease in resting arterial oxygen tension, which gives rise to diminished gas exchange in the lungs (7, 12, 22). Despite changes to the alveolar surface area, the blood supply to the alveoli is not equally distributed with age. Age-associated changes in pulmonary circulation increase systolic pressure of pulmonary arteries and a reduction in lung-diffusing capacity of carbon dioxide (CO2) (22, 23). Reduced alveolar-capillary density as well as changes to pulmonary vasculature result in a gradual decrease in pulmonary capillary blood volume with aging (24–28). Consequently, standard deviation of alveolar ventilation and perfusion distribution across the lungs is increased and results in lower resting arterial oxygen in older persons (24, 25). A decline in expiratory flow, increased closing volume, and declining exercise performance result in a change in ventilation responses to exercise as well as increased CO2 production and O2 consumption at all levels of exertion (7, 12, 29–32). Due to the heterogeneity of the aging population and highly variable rates of lung aging, the interpretation of healthy or normal lung function in older persons and disease diagnosis can be problematic. Height, gender, weight, and ethnicity can be partly responsible for differences in FEV1/FVC, and the analysis of pulmonary function and the development of reference standards to examine lung function in older persons still remains to be fully elucidated.
CELLULAR CHANGES IN THE AGED LUNG
Normal lung aging is associated with multiple structural and functional changes in the respiratory tract (6, 10). Many of these changes, as highlighted in Figure 1, give rise to decreased lung function, altered pulmonary remodeling, diminished regeneration, and enhanced susceptibility to pulmonary disease (7, 10). This section focuses on several of the main cell types involved in tissue repair and remodeling of the conducting and respiratory airways and the impact of aging on cellular function.
Figure 1.
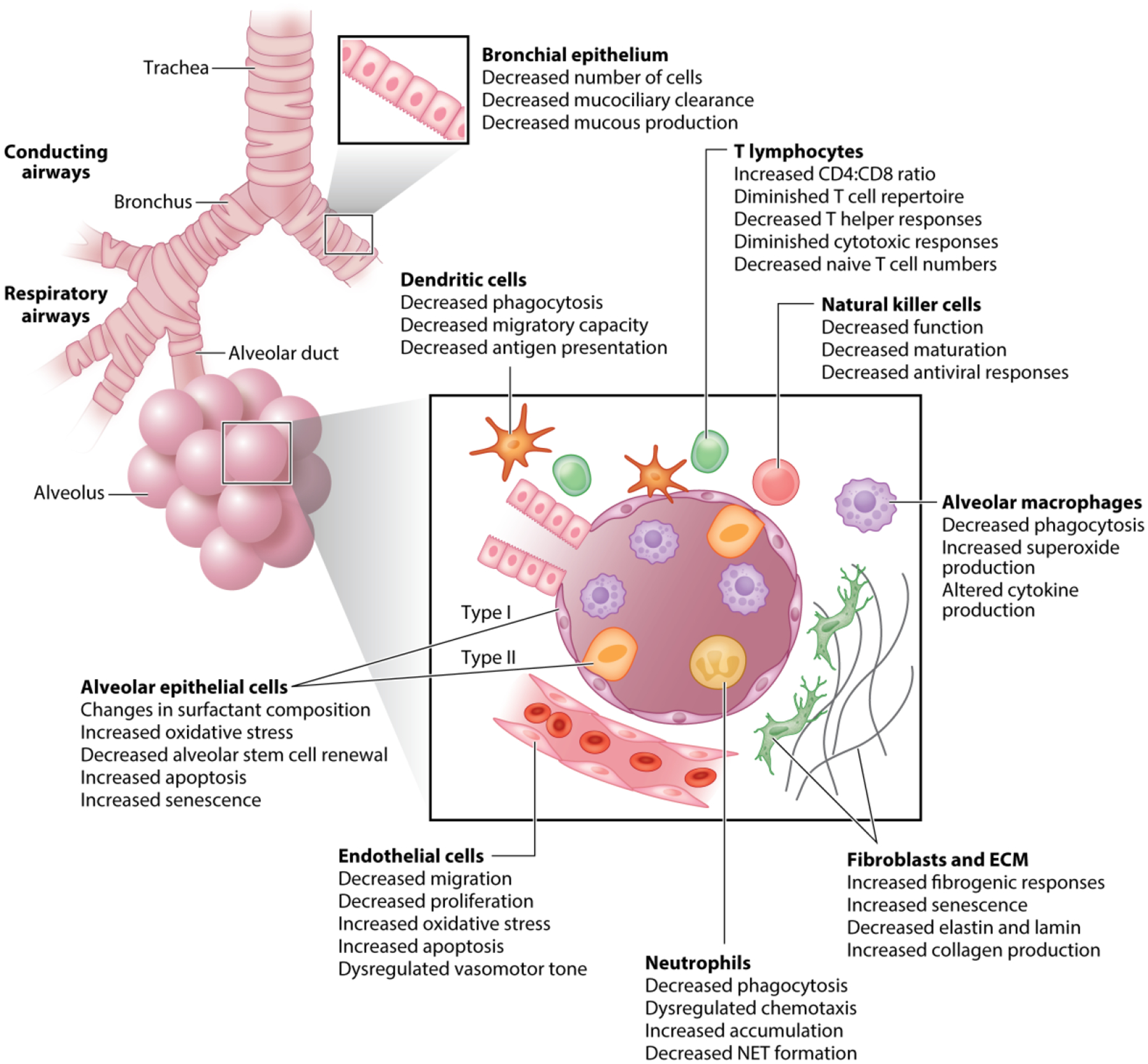
Cellular changes in the aged lung. The epithelial surface of the respiratory airways is a large, highly vascularized area where efficient gas exchange and host defense rely on the integrity of the epithelium; age-associated changes in type I and II epithelial, fibroblast, endothelial, and airway smooth muscle cell composition and function can contribute to the development and progression of lung disorders in the elderly. Poor prognosis and recovery in pulmonary inflammatory diseases has been attributed to immunosenescence or age-related changes in innate and adaptive immune responses in the lung. Abbreviations: ECM, extracellular matrix; NET, neutrophil extracellular trap.
Conducting Airways
The cartilaginous conducting airways of the lungs, from the trachea to terminal bronchioles, extend deep into the lung parenchyma and are surrounded by an abundance of submucosal glands that secrete an abundance of fluids, antimicrobial proteins, and mucins in the airways. The trachea, bronchi, and bronchioles are mainly lined by a ciliated pseudostratified epithelium. Respiratory epithelial cells create multiple barriers that act as a selective gate between the outside environment and the underlying tissue. Epithelial cells are polarized by the formation of specialized cell–cell junctions, known as apical junctional complexes, and are formed by tight and adherent junctions created by homotypic and heterotypic binding among various claudins expressed in pulmonary cells (33–35). Although ciliated cells are the predominant surface cells, other cell types that are influenced by injury and infection, such as serous, club, neuroendocrine, and goblet cells, are found in relatively low numbers in normal airways (36, 37). Basal cells, which are located beneath the surface epithelium, play a critical role in regeneration of injured airway epithelium and serve as progenitors of ciliated and secretory cells (38, 39). Myoepithelial, serous, goblet, basal, and ciliated cells line submucosal glands and secrete fluids and other host-defense proteins onto the airway surface at baseline and in response to environmental stimuli (36, 37, 40).
Cell-associated and secreted mucins create a barrier for the luminal surface of the conducting airways and prevent the access of microbes and particles to underlying epithelial cells (41–44). Polymeric glycoconjugates bind to and transport pathogens from the conducting airways (44). Mucin glycoproteins are large, heavily glycosylated proteins that share an abundance of repeated threonine-rich domains that serve as linkage sites for large carbohydrate structures (42–45). Membrane-associated mucins have transmembrane and cytosolic domains that allow them to tether to the epithelial cell plasma membrane, where they can function in cellular adhesion, pathogen binding, and signal transduction (46, 47). In addition, membrane-associated mucins, such as MUC4, MUC13, MUC16, and MUC21, can create a direct host defense barrier that can be shed by pathogen or host-associated proteases and transferred through the mucociliary escalator, which sweeps microbes and debris upwards for removal (40, 45). Secreted airway mucins, such as MUC5B, MUC5AC, and MUC2, form a mucous gel that disrupts bacterial aggregation and can directly prevent adherence of microbial pathogens to cell surfaces (40, 45). After synthesis and processing, secreted airway mucins are stored as intracellular granules within secretory cells, such as goblet cells, until stimulated for release (37). To generate linear and multimeric gel-like lattice structures of polymeric proteins that can be moved up the airway by the beating of cilia, mucins undergo extensive post-translational folding and carboxy and amino-terminal domain modifications (48). In sum, mucins maintain airway homeostasis and their synthesis and secretion aid in the removal of pathogens and cellular debris (33, 49).
Respiratory epithelial cells recognize microbial pathogens through pathogen recognition receptors (PRRs) and initiate signaling to recruit and guide immune cells (50). Signaling by PRRs, such as Toll-like receptors (TLRs) and nod-like receptors (NLRs), is critical for activating mucosal immune responses (51–53). Recognition of a pathogen-associated molecular pattern (PAMP) or danger-associated molecular pattern (DAMP) containing molecules results in the initiation of signaling cascades that alter epithelial gene expression, signal transduction, and cytokine production (54). Extrinsic epithelial responses to PAMPs or DAMPs are influenced by the type of pathogen and the inflammatory environment in the lung and can result in protective and/or pathological effects on airway barrier function (50, 54–56). Expression of cytokines and chemokines by epithelial cells influences the recruitment and activation of immune cells, thereby modulating inflammatory responses in the lung (50, 54–56). Epithelial cell–intrinsic responses to PRRs can activate signaling pathways that modulate the expression and production of mucins and antimicrobial peptides (57–59).
The vast majority of ciliated luminal epithelial cells function to transport the particles and pathogens entrapped within the mucus gel from the lungs. Axenomes, which use energy from ATP hydrolysis to drive rhythmic movements, are formed by the assembly of an organization of microtubules with dynein arm motor complexes (60). The biogenesis of motile cilia is directly linked to the differentiation from basal cells and other airway progenitors and can be directly influenced by toxicants and inflammatory processes (60). To move mucus toward the larynx, ciliary beat frequency and directionality along the airway epithelium in response to mechanical stress or neurochemical and inflammatory signals must be carefully coordinated (60). Impairment in ciliary function can detrimentally impact mucociliary clearance and lead to recurrent infections that are associated with multiple pulmonary diseases.
As the respiratory epithelium plays a critical role in the recognition of PAMPs and DAMPs in the airways, a balance between initiation of pathways that regulate cellular responses, recruitment of immune cells, maintenance of barrier function, and clearance of pathogens must be maintained. Experimental studies have illustrated that there is an age-associated increase in bronchoepithelial cell apoptosis, resulting in decreased cell numbers present in aged lung (61). Changes in cellular composition negatively affect the epithelium and can lead to increased epithelial thinning. In response to chronic inflammation, upregulation of surface receptors on epithelial cells, such as the platelet-activating factor receptor, may give rise to increased bacterial adhesion and accumulation in the aged lung and correlate with susceptibility to pneumonia (62). Abnormalities in microtubular composition and function in cilia can further negatively impact defense mechanisms in the aged lung (63). In healthy aging, a decrease in mucociliary clearance can be conducive to microbial invasion in the lower airways and alveoli (63–65). Decreased mucous production and composition can lead to diminished pathogen removal and increased susceptibility to infection and disease progression in the aged lung. Age-associated changes in the molecular composition of epithelial lining fluid, specifically a decrease in antioxidants, can also impact lung function (66). In sum, the conducting airways play a pivotal role in maintaining lung homeostasis and age-associated changes in composition and function can be partly responsible for increased susceptibility of older persons to lung disorders.
Respiratory Airways.
The epithelial surface of the respiratory airways is a large, highly vascularized area where efficient gas exchange and host defense is dependent on the integrity of the airway epithelial barrier. The coordinated functions of multiple cells types in the respiratory airways are essential to maintain and preserve tissue integrity, barrier function, and cellular communication. Age-associated changes in the composition and function of these diverse cell types can contribute to increased susceptibility of older persons to the development and progression of lung disorders.
Alveolar epithelial cells.
The epithelial surface of the respiratory airways is a large, highly vascularized area where efficient gas exchange and host defense rely on the integrity of the epithelium. The maintenance and repair of the distal gas exchange region of the lung are mediated by millions of alveoli organized into hundreds of clusters. In contrast to the diversity of cell types in the conducting airways, only two cell types line the alveolar epithelium. Squamous type I alveolar epithelial cells (AEC1s) are flat and cover approximately 90–95% of the alveolar surface in the adult lung (67). AEC1s interact closely with endothelial cells of the pulmonary capillaries and are the primary site for gas exchange (67). Cuboidal type II epithelial cells (AEC2s) are characterized by an abundance of lipid-rich secretory molecules, known as lamellar bodies (68). Long-lived AEC2s play a critical role in the synthesis of surfactant lipids and proteins that reduce the surface tension and prevent collapse of the lungs during the ventilatory cycle (69). The type and abundance of mucus secreted by AEC2s can differ along the proximal-peripheral airway of the lungs (45, 47). AEC2s serve as the main progenitor cells during repair of the alveoli. In response to injury and destruction of AEC1s, AEC2s function as progenitor cells in the alveoli and proliferate and differentiate into AEC1s (68, 69). Due to the importance of AECs, to meet the ongoing synthetic and metabolic requirements, there needs to be a delicate balance between energy expenditure for activities necessary to maintain cellular homeostasis and those necessary for responding to injury or infection. Increased cellular apoptosis, reactive oxygen species (ROS) production, DNA damage, diminished autophagy, and dysregulated unfolded protein responses can result in injury of AEC2s and can worsen the prognosis in acute lung injury (70–73).
AEC2s, and to a lesser extent AEC1s, are important effector cells in inflammatory responses in the lung. In response to infectious stimuli, AEC2s, through the release of antimicrobial molecules, cytokines, and chemokines, can recruit monocytes and macrophages as well as shape the magnitude and duration of the innate immune response (68, 69). Surfactant production, which is crucial for lowering the alveolar surface tension, also plays a key role in pulmonary inflammation. Surfactant forms a single, stable lipid layer at the air–liquid interface of alveoli. Surfactant proteins A (SP-A), B (SP-B), C (SP-C), and D (SP-D) are important to the structural and bioactive properties, metabolism, and microbial opsonization of lung surfactant (74–77). Surfactant proteins and lipids are synthesized, stored, and secreted into the alveoli by AEC2s. Tubulin myelin, which is a highly structured lipid-protein complex that has distinct roles in alveolar homeostasis, requires SP-A and SP-B for oligomerization (78). Tubular myelin serves as an extracellular reservoir of surfactant lipids that move throughout the air–liquid interface to reduce surface tension. Many innate antiviral and antibacterial defense proteins, such as lysosome, SP-B, SP-C, and SP-D, reside within tubular myelin scaffolding (78–82).
There is an age-related decrease in the ratio of proliferating to apoptotic AECs (83). Senescence in AEC2s can lead to a critical decline in alveolar epithelial stem cell renewal. Previous work in mice lacking telomerase function in AEC2s illustrated an elevation in apoptosis, increased inflammation, and enhanced susceptibility to acute lung injury (84). A marked accumulation of dysfunctional mitochondria and impaired autophagy contribute to increased oxidative stress and impaired structure and content of lamellar bodies in AEC2s (83). An age-associated impairment in AEC2 function, due to increased cellular senescence, contributes to the highly proinflammatory and oxidative environment within the lung (66). Persistent low-grade inflammation in the lower respiratory tract can augment proteolytic and oxidant-mediated injury to the lung matrix, resulting in alveolar loss and subsequent impairment of gas exchange across the alveolar membrane. Changes in SP-A and SP-D may be attributed to the more oxidative lung environment that occurs with advanced aging (66).
Fibroblasts and the extracellular matrix.
Fibroblasts, especially those in close proximity to the airway epithelium, play a key role in the regulation of local responses. Fibroblasts are located throughout the interstitium of the lungs, between epithelial and endothelial layers, and are involved in tissue repair. Fibroblasts, through the secretion of glycoproteins, matrix metalloproteases (MMPs), and collagens, are instrumental in the structural organization and remodeling of the ECM. Fibroblasts remain inactive until stimulated to proliferate or differentiate into myofibroblasts for the purpose of cellular replacement and repair in response to injury or cell death.
The ECM is a highly dynamic complex of fibrous proteins, glycoproteins, and proteoglycans that provide structural integrity to the lung. The composition of the ECM within the lung parenchyma has a direct impact on lung function and the development and progression of most senescence-related lung diseases (85, 86). Specifically, the growth factors, cytokines, and ECM-remodeling enzymes present within the ECM provide signaling cascades that define the differentiation, proliferation, survival, and recruitment of cells (85, 86). The ECM in the lung is restricted to two basic compartments: thin basement membranes located under all epithelial and endothelial layers of the lung and interstitial spaces that form within the parenchyma of the lung (87). Normal function of the lung is influenced by the dimensionality, molecular composition, and intrinsic stiffness of the ECM. Tensile strength of the lung is attributed to fibrillar proteins, such as collagens and fibronectins, while elastic recoil is accredited to elastin molecules (87). Changes in ECM stiffness may be attributed to an alteration in the composition of matrix molecules in the ECM, changes in protein cross-linking, and modified remodeling (87).
Age-associated changes in the expression, posttranslational modifications, and remodeling of fibrillar collagens are instrumental in the progression of ECM stiffness. Normal lung aging is characterized by increased collagen, which promotes age-related changes in elasticity and airspace enlargement (6). In response to injury in the aged lung, there is augmented profibrotic matrix production that is associated with increased recruitment of circulating pluripotent mesenchymal progenitor cells, called fibrocytes (88). In senescent cells, upregulated expression of MMPs, which degrade proteins such as collagen and elastin, can have a significant impact on the composition and function of the ECM (89, 90). A decrease in elastin content leads to the uncoupling of the elastin-lamin receptor, which alters signal transduction in parenchymal cells and results in increased collagen deposition (91, 92). Research using acellularized lungs, where all cells have been removed and only the ECM remains, has illustrated that ECM from aged lung contains less structural protein diversity (93). Repopulation of the ECM by inoculation of human lung epithelial cells and fibroblasts into the aged acellularized lungs resulted in decreased laminin, elastin, and fibronectin expression and was associated with elevated collagen (93). In summary, the ECM is important for providing support and anchorage for cells as well as regulating intercellular communication. Changes in fibroblast function and ECM composition can increase susceptibility of the aged lung to injury and disease progression.
Endothelial cells.
Endothelial cells are one of the most important cellular components of blood vessels. Cell–cell junctions are attachment sites between endothelial cells that play a pivotal role in maintaining tissue integrity, barrier function, and cellular communication (94). Endothelial cells, through the coordinated opening and closure of cell–cell junctions, control the infiltration of leukocytes, neutrophil sequestration, and plasma proteins into the walls of blood vessels (94). Endothelial cell junctions can function as signaling structures that communicate cell position, regulate vascular homeostasis, and aid in limiting cellular growth and apoptosis. Similar to epithelial cells, endothelial cells are also polarized by the formation of tight and adherens junctions. Endothelial cell adhesion at adherens junctions is mediated by vascular endothelial cadherin, which is linked to intracellular proteins, such as β-catenin, plakoglobin, and p120 (95, 96). At tight junctions, adhesion of endothelial cells is associated with intracellular proteins, such as occluden and members of the junctional adhesion molecule family (95, 96). Junctions are essential for maintaining vessel wall integrity and alteration of the molecular organization, and intracellular signaling of junctional proteins can result in complex effects on vascular homeostasis.
Endothelial cells play a pivotal role in the regulation and modulation of immune responses. In addition to forming a physical barrier, endothelial cells can synthesize and secrete mediators, such as chemokines and lipid mediators, which activate and recruit leukocytes (95, 96). Endothelial cells also modulate the expression of surface adhesion molecules and factors, such as intercellular adhesion molecule 1 (ICAM-1), plasminogen activator inhibitor 1 (PAI-1), vascular adhesion protein 1 (VCAM-1), and platelet endothelial adhesion molecule 1 (PECAM-1), to regulate leukocyte transmigration and vascular permeability during inflammatory responses (95).
In the aged lung, endothelial cell senescence and a decline in the concentration in vascular endothelial growth factors can directly impacts angiogenesis by limiting vascular growth. Age-associated changes in endothelial vasodilation have been attributed to decreased endothelial nitric oxide (NO) synthase activity and NO production (97). Augmented ROS levels in neighboring senescent cells as well as an increase in proatherogenic and prothrombogenic factors ICAM-1 and PAI-1 promote negative regulation of NO signaling (97). Increased susceptibility of surface membrane components, such as phospholipids, fatty acid chains, and lipoproteins, to oxidative stress promotes the generation of neo-epitopes (97). Recognition of these neo-epitopes by the innate immune system can initiate a series of responses that activate numerous pathological processes that accelerate luminal narrowing, such as increased apoptosis, dysregulated vasomotor tone, and dysregulated endothelial cell activation and migration (97). Taken together, diminished and/or dysregulated endothelial repair and regeneration in aged lung promotes increased susceptibility of older persons to disease processes such as pulmonary sepsis and pulmonary hypertension.
Airway smooth muscle.
Airway smooth muscle (ASM) plays a critical role in hyperresponsiveness and airway remodeling that occur with lung disease (98). ASM is a structural component of the bronchial airways that regulates bronchomotor tone in asthma and modulates airway inflammation. ASM cells can express adhesion molecules, such as ICAM-1 and VCAM-1, as well as integrins, which play a role in regulating the strength of smooth muscle contraction (99). By the expression and secretion of proinflammatory cytokines and mediators, ASM cells play a role in the induction of local inflammatory and remodeling responses (99).
Experimental aging models have illustrated decreased expression of γ-smooth muscle actin and desmin in aged lung, which may indicate that contractions of larger intrapulmonary airways decline with aging (100). Interestingly, α-smooth muscle actin and vimentin expression in the distal airway may be increased in aged lung, suggesting that mechanical forces are strengthened in the peripheral airways (100).
In summary, the epithelial surface of the respiratory airways is a large, highly vascularized area where efficient gas exchange and host defense rely on the integrity of the epithelium. Age-associated changes in the composition and function of AEC1s, AEC2s, fibroblasts and endothelial and ASM cells can be factors in the increased susceptibility of older persons to the development and progression of lung disorders.
IMMUNITY AND INFLAMMATION IN THE AGED LUNG
The lung has evolved multiple defense systems to preserve homeostasis and respond to foreign stimuli. As illustrated in Figure 1, a multitude of immune cell types are instrumental in the initiation and resolution of innate and adaptive immune responses in the lung. Immunosenescence, or aging of the immune system, results from an imbalance between inflammatory and anti-inflammatory mechanisms. Chronic antigen stimulation throughout a lifetime as well as oxidative stress and the production of oxygen free radicals augment production of proinflammatory cytokines. Poor prognosis and recovery in pulmonary inflammatory diseases have been attributed to age-related changes in innate and adaptive immune responses in the lung. In this section, we focus on several key immune cell types in the lung and the impact of aging on their function.
Innate Immunity.
In addition to innate immune sensing that occurs in the conducting and respiratory airways, resident and circulating immune cells aid in the recognition and clearance of foreign particles present in the pulmonary microenvironment. Innate immune responses are multifaceted, and the magnitude and specificity of a response is highly dependent on the regulation and function of multiple cell types. Age-associated changes in immune cell function can promote increased susceptibility of older persons to the development and progression of lung disorders.
Alveolar macrophages.
Alveolar macrophages (AMs) are long-lived resident innate immune cells of the airways and key effectors of recognition, initiation, and resolution of the host defense against microbes. Under steady-state conditions, AMs compose greater than 90% of the resident immune cell population of the lung. In normal lung, resting AMs are maintained in a quiescent state, but maintain the ability to rapidly respond to foreign microorganisms and particulate material. AMs play an intricate role in lung homeostasis by maintaining alveolar concentrations of surfactant lipids and proteins. In the absence of parenchymal secreted growth factors, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), impaired AM differentiation can result in decreased surfactant protein and lipid catabolism and subsequent accumulation of lipoprotein material within the alveoli (75). AM recognition of PAMP and DAMP molecules initiates a series of coordinated innate immune responses designed to provide defense against the foreign entity. Upon stimulation, AMs produce inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin (IL)-6, IL-12, and type I interferons, chemokines, lipid mediators, and antimicrobial peptides. The inflammatory environment within the lung heavily influences the recruitment, differentiation, and function of circulating monocytes. AMs are the primary phagocytes of the innate immune system and aid in clearing the air spaces of infectious, allergic, or toxic particles. Effective bacterial elimination by AMs proceeds in two separate phases: an initial period of macrophage viability and intracellular bacterial killing followed by a later induction of apoptosis and clearance of bacteria (101).
AMs also play an important role in tissue damage control. They do this by clearing away extracellular debris and apoptotic cells from the airways and producing anti-inflammatory cytokines, such as IL-10, as well as lipoxins and resolvins, that work in autocrine and paracrine fashion to resolve inflammation (102, 103). A reduction in macrophage phagocytic function and excessive apoptosis can give rise to severe lung pathologies and may play a role in chronic inflammatory lung disorders (104, 105). Additionally, if inflammatory AMs do not undergo apoptosis and are not removed from the lung, chronic inflammation or fibrosis can develop.
There is an age-associated decline in phagocytic capacity in alveolar and pulmonary macrophages, which results in impaired and/or delayed pathogen clearance from the lung (106–108). In addition, age-related alterations in pathogen recognition, decreased PRR expression, and diminished responses to secondary signaling may underlie increased susceptibility of older persons to infectious stimuli (109). An age-associated reduction or delay in cytokine, chemokine, or interferon production in response to stimulation can negatively impact innate immune responses in the lung (110). Similarly, chronic exposure to low levels of proinflammatory cytokines, such as TNF-α, can alter the responsiveness of circulating monocytes and macrophages to PAMPs. Altered migratory potential leads to increased recruitment of less mature myeloid cells with hyperinflammatory responses into the lung. Disrupted production of anti-inflammatory cytokines by AMs can augment inflammation and impair tissue restoration. Experimental studies have illustrated an age-associated increase in ROS levels that corresponds with decreased antioxidative responses in AM (111, 112). AM function in the elderly is greatly influenced by ROS, and the production of oxygen intermediates by dysregulated mitochondria can promote increased superoxide production (111). Changes in environmental and tissue-derived signals in the aged lung can directly and indirectly impact the functional response of AM, thereby disrupting cellular communication and augmenting inflammatory responses. Taken together, the diminished/dysregulated responses of AM to pathogenic stimuli and impaired ability to promote resolution contribute to excessive inflammation and tissue damage in the aged lung.
Dendritic cells.
Dendritic cells (DCs) are an immune cell subset that plays a role in shaping innate immune responses in lung tissue. Sentinel airway mucosal DCs (AMDCs) constantly sample antigens present on the luminal surface of the airway. While AMDCs lack the ability to present antigen through the secretion of chemokines, they can recruit immature DCs to the lung. DCs bind, internalize, and process antigens. In response to PRR stimulation, DCs rapidly produce high levels of proinflammatory cytokines, such as IL-12, TNF-α and IL-6, and upregulate costimulatory molecule expression. Interaction of DCs with epithelial cells can further shape the magnitude of the immune response to environmental stimuli. Upon activation, mature DCs migrate to the lung draining lymph nodes, where they present antigens to naïve T cells. In conjunction with activation markers, such as CD80 and CD86, and expression of major histocompatibility complex I/II molecules, DCs can aid in T cell activation and differentiation. Taken together, DCs regulate maintain self-tolerance as well as modulate inflammation and immunopathology in the lung.
Despite comparable cell numbers, differentiation, and morphology in the young and aged, DCs from older persons exhibit impaired phagocytosis of antigen (113, 114). In addition, aged DCs maintain the ability to upregulate chemokines and chemokine receptors but exhibit decreased migratory capacity in response to stimuli (114). In experimental models, DC migration into the aged lung during influenza infection is reduced (115). Altered PRR expression and/or changes in downstream signaling cascades may underlie dysregulated DC responses in older persons (114, 116). There is an age-associated reduction in the upregulation of costimulatory molecules critical for T cell priming and dysregulated cytokine production by DCs (115, 117). Diminished CD8 T cell activation may be due to decreased cross-presenting capacity of aged DCs (114, 118). Defects in DC function, in combination with intrinsic changes in T cells, contribute to blunted adaptive immune responses in the elderly lung.
Neutrophils.
Neutrophils are among the first responders to infectious stimuli. Neutrophils migrate into infected tissues soon after a pathogen is detected and secrete a variety of enzymes, toxic molecules, and radicals that play a role in pathogen clearance. In excess, they can induce tissue injury in the host. Neutrophils can eliminate microbes through phagocytosis, generation of ROS, secretion of microbial molecules and enzymes, and the release of neutrophil extracellular traps (NETs). NETs are networks of extracellular fibers containing neutrophil-derived chromatin, DNA, and histones that bind and immobilize pathogens. Neutrophils can work together with macrophages to contain and clear infections (119). Apoptosis of neutrophils and subsequent clearance by macrophages helps to regulate inflammation (119). Increased cell surface expression of apoptotic markers, such as phophatidylserine, promotes macrophage-mediated phagocytosis of apoptotic neutrophils as well as increased production of anti-inflammatory cytokines, such as TGF-β (119).
Although the number of circulating neutrophils is preserved in older persons, a decrease in CD16 expression results in augmented superoxide generation and impaired neutrophil-mediated phagocytosis (120, 121). An age-associated alteration in pathogen-mediated destruction by NETs further contributes to impaired bacterial clearance and may underlie increase susceptibility of older persons to infection (122). In healthy older adults, there is an increased proportion of neutrophils and a lower percentage of macrophages present in bronchoalveolar lavage (BAL) fluids (121). Experimental models of infection or injury have demonstrated that neutrophil chemotaxis is dysregulated (123). Specifically, there is an age-associated delay in neutrophil recruitment or, in some instances, increased neutrophil accumulation due to a failure to disperse at the site of infection (123). Changes in cytokine signaling pathways may also increase the susceptibility of neutrophils undergoing apoptosis (124). Increased and prolonged neutrophil recruitment in the aged lung can have devastating effects on lung parenchyma (125). Increasing numbers of recruited neutrophils can promote the development of edema, enhanced disease severity, prolonged inflammation, and pulmonary tissue damage (125, 126).
Innate lymphoid cells.
Innate lymphoid cells (ILCs) represent a heterogeneous cell population that can regulate immunity, inflammation, and help restore airway epithelial cell integrity. ILCs are classified based on their functional characteristics, cytokine profile, and the transcription factor needed for development. ILC1 cells, including natural killer (NK) cells, can produce IFN-γ and TNF-α and advance the immune response against intracellular bacteria and parasites. ILC2 cells, produce Th2-related cytokines, such as IL-5, IL-9, and IL-13, and help parasite specific and allergic inflammatory responses. ILC3 cells are able to produce IL-17 and can be subdivided into natural cytotoxicity-triggering receptor positive or negative cells. Taken together, ILCs play an important role in maintaining tissue homeostasis, restoring airway epithelial integrity, and coordinating innate immune responses to infectious stimuli. Although little is currently known regarding the impact of aging on ILCs, there is a potential for age-associated changes in ILC function and frequency.
Natural killer cells.
NK cells are cytotoxic lymphocytes that, through their ability to recognize and eliminate infected and/or damaged cells from the lung, play an important role in immunity against foreign pathogens. NK cells also function in the resolution of inflammation as well as the elimination of senescent or stressed cells. Cytokine production at the site of infection recruits NK cells. NK cells provide rapid responses to infected cells and can secrete perforin and granzyme-containing granules to induce apoptosis or osmotic cell lysis in target cells. In addition, NK cells can produce cytokines, such as IFN-γ, which activate macrophage phagocytic functions. NK cells play an important role in mediating antibody-dependent cellular cytotoxicity of opsonized target cells. Cross talk between NK cells and DCs as well as NK cells and CD8+ T cells is essential for the development of efficient and potent innate and adaptive immune responses.
Experimental studies have illustrated that altered function and phenotype of NK cells with advanced aged underlies increased susceptibility of older persons to infectious stimuli (127). An age-associated increase in NK cell senescence has been shown to impair antiviral immune responses in response to influenza (3, 128). Low levels of inflammation present in normal aged lung as well as changes in the nonhematopoietic environment may underlie reduced recruitment and maturation of NK cells (129).
In summary, the lung has evolved multiple innate immune defense systems to preserve homeostasis and respond to foreign stimuli. Age-associated changes in regulation and function of innate cell types, such as AMs, DCs, and NK cells, can promote increased susceptibility of older persons to the development and progression of lung disorders.
Adaptive Immunity
The adaptive immune response is highly specific and can provide long lasting cell-mediated immunity against pathogens invading the lung microenvironment. Upon recognition of foreign endogenous and exogenous “non-self” antigens, lymphocytes can directly eliminate specific pathogens and pathogen infected cells as well as mediate the immune response. Antibody and cell-mediated immune responses play an essential role in specific pathogen elimination and diminished adaptive immunity can increase the likelihood of lung disorders in the elderly.
CD4+ and CD8+ lymphocytes.
Adaptive immune responses by T lymphocytes are critical for cell-mediated immunity and pathogen-specific defense. Progenitor T cells migrate from the bone marrow to the thymus, where they undergo a programmed process of T cell development. Naïve T cells migrate to secondary lymphoid tissues where they survey antigens presented by DCs. In response to a specific antigen, naïve T cells become activated and subsequently proliferate and differentiate into effector T cells. Effector T cells migrate to sites of infection or inflammation and, upon clearance or resolution, will either undergo activation-induced cell death or will become memory T cells that provide long-term immune protection against repeat pathogens. CD4+ T cells are categorized, depending on the type of cytokine produced, into three subsets: Th1 (intracellular microorganisms, IFN-γ), Th2 (parasites, IL-5, and IL-13), and Th17 (extracellular microorganisms, IL-17). Effector CD8+ T cells can secrete cytotoxic perforin and granzyme B–containing molecules and aid in the clearance of foreign pathogens. Adaptive T cell–mediated immune responses allow the host to quickly and effectively respond to repeat stimuli, such as viruses and bacteria. T cell antigen specificity is localized to T cell receptors and, upon antigen presentation, undergoes selective expansion of T cell clones. While lower in number than other immune cell types, T cells have been identified in the lung, with the number and subset varying between the mucociliary epithelium, pulmonary interstitium, and bronchoalveolar space (130).
Multiple age-associated changes in T cell numbers and function may underlie increased susceptibility of older persons to environmental stimuli. The numbers of CD3+, CD4+, and CD8+ T cell populations decrease with advancing age. There is an age-associated reduction in naïve T cell numbers that corresponds with increased memory T cell numbers (131). Specifically, the ratio of CD4+ to CD8+ lymphocytes in the BAL fluid has been shown to increase with age, suggesting a decrease in the number of naïve T cells available that can be converted to memory cells against a novel antigen (131, 132). Further, memory T cell responses, T cell receptor repertoire diversity, T helper cell differentiation, and T helper cell activity diminish with age (131, 132). Decreased CD4+ and CD8+ T cell responses in the elderly can impair immunity to influenza vaccination and cytotoxicity against the influenza virus, respectively (132, 133). In summary, age-associated changes in T cell–adaptive immune responses can lead to increased susceptibility to infectious stimuli and prolonged and more severe disease severity.
B lymphocytes.
Through a tightly regulated process, lymphoid progenitor cells mature through pro- and pre-B cell stages into immature B cells with unique B cell receptors. Rearrangement of heavy and light chain immunoglobulins (Ig) generates a highly diverse antibody repertoire. Depending on the extent of B cell receptor engagement, B cells can mature into marginal zone or follicular B cells. Marginal zone B cells can respond to blood-borne pathogens and differentiate into Ig-secreting plasma cells. Long-lived plasma cells are present within the airway submucosa and produce polymeric IgA and IgM that are secreted into the lumen (134). Memory B cells are present within the peribronchial regions of the lung and in response to infectious stimuli can rapidly produce antibodies (134). Circulating B cells migrate into pulmonary lymph nodes (134). Of note, in response to chronic pulmonary inflammation, persistent exposure to antigen, and/or tissue injury, activated B cells can give rise to lymphoid follicle formation in the lung.
Multiple studies have illustrated an age-associated decline in B cell development, specifically a decreased transition from the presence of naïve to mature B cells (135). Specifically, experimental murine studies have demonstrated a decline in the number pro-B, pre-B, and immature B cell subsets present in older animals (135). Changes in immature B cell migration and an increase in age-associated B cells can decrease repertoire diversity (135). While the functional ability to produce antibody remains intact, age-associated changes in antibody specificity and antigen affinity result in a diminished ability to mount robust antibody responses in older persons (135).
In summary, chronic antigen stimulation throughout a lifetime as well as oxidative stress and the production of oxygen free radicals impair adaptive immune responses in the lung. Poor prognosis and recovery in pulmonary diseases have been attributed to age-related changes in innate and adaptive immune responses in the lung. Specifically, augmented and prolonged innate inflammation and diminished adaptive regulation can increase the likelihood of lung disorders in the elderly.
THE IMPACT OF AGING ON LUNG DISEASE
Due to changes in lung function and anatomy as well as lung antioxidant defenses, the aged lung is more susceptible to environmental exposure–induced injury (19, 86). The human lung is constantly exposed to oxidative stress–inducing particulates, such as cigarette smoke, airborne aerosols, diesel emission particulates, and other exogenous toxins. Multiple antioxidant defenses are present in the lung to help defend itself from the deleterious effects of oxidative stress (136). The BAL contains antioxidants, such as superoxide dismutase (SOD), glutathione peroxidases, catalase, metal binding proteins, vitamins, and surfactant, to minimize oxidative injury to the respiratory epithelium (66, 136). An age-associated reduction in BAL antioxidant levels and composition may underlie increased susceptibility of older persons to exposure with environmental toxins, such as ozone, cigarette smoke, and particulate matter (66). In this section, we discuss how physiological and cellular changes in the aging lung are instrumental in the development and progression of lung diseases, such as COPD, IPF, acute respiratory distress syndrome (ARDS), and pneumonia.
Chronic Obstructive Pulmonary Disease
COPD is a heterogeneous, chronic inflammatory lung disease that manifests clinically later in life and can lead to significant morbidity and premature death. COPD is a common disorder in the elderly, with cigarette smoking being the greatest risk factor for COPD development in genetically susceptible individuals (137, 138). COPD accounts for one-fifth of all hospitalizations in persons 75 years or older and is associated with increased prevalence of comorbidities, such as congestive heart failure, vascular disease, diabetes, and cancer, that can complicate the course of the disease (64, 137). Age-associated changes in the structure, function, and control of the respiratory system can greatly influence COPD susceptibility in older persons (6–10). Interestingly, many of the anatomical and physiological changes seen in COPD, such as airspace dilation resulting from loss of supporting tissue without alveolar wall destruction, have also been described in the similarly aged lungs of nonsmokers, further illustrating that the process of aging is a contributing factor for disease progression (Figure 2) (7, 10, 16). Specifically, shortening of telomeres, increased cellular senescence, and increased DNA damage are present in both aged and COPD lungs. However, when compared to aging lungs, there is a significant increase in collagen, fibronectin, and laminin, with more disorganized collagen fibers present in COPD lungs (139). Exposure to chronic bronchoconstricting inflammatory stimuli promotes increased bronchoconstriction and long-term mechanical adaptations through ECM remodeling of airways and the parenchyma (138).
Figure 2.

Comparison of aged and chronic obstructive pulmonary disease (COPD) lung. Many of the anatomical and physiological changes seen in COPD, such as airspace dilation resulting from loss of supporting tissue without alveolar wall destruction, have also been described in the similarly aged lungs of nonsmokers, further illustrating that the process of aging is a contributing factor for disease progression. However, when compared to aging lungs, there is a significant increase in collagen, fibronectin, and laminin, with more disorganized collagen fibers present in COPD lungs. Chronic bronchitis, with increased airway wall thickening, inflammation, and heightened mucus production as well as emphysema, with alveolar wall destruction, hyperinflation and impaired gas exchange, are two common phenotypic manifestations of COPD.
Age-associated immunosenescence and cellular senescence in response to exposure to cigarette smoke can greatly impact COPD pathogenesis (3, 19, 86). There is increasing evidence that many cell types, such as AECs, fibroblasts, and endothelial cells, become senescent in COPD lungs. Specifically, when compared to non-COPD controls, fibroblasts isolated from COPD patients exhibit a senescent phenotype with an abnormal repair capacity (139). A reduced number of circulating progenitor cells and changes in the regenerative capacity of airway basal cells and AEC2s result in decreased cellular repair and renewal in the COPD lung (69, 84, 139). Elastin degradation and increased circulation of elastin fragments can induce inflammatory responses that lead to lung tissue destruction (139). Increased production of proinflammatory cytokines IL-6 and TNF-α is associated with heightened COPD obstruction, and dysregulated synthesis of additional inflammatory markers can increase the risk of developing COPD-related complications (85, 86, 139). Upregulation of compensatory innate immune responsiveness, chemotaxis, and innate cell retention can be conducive to creating a proinflammatory state in COPD airways in the elderly (85, 86, 139). Specifically, macrophages, which can produce multiple proinflammatory cytokines and secrete MMPs, are the most abundant cell type in the BAL fluid of COPD patients, and increased numbers correlate with disease severity (85, 86, 104). Impaired phagocytosis and clearance of cellular debris by airway macrophages in COPD result in persistent inflammation (85, 104). Neutrophils, which can produce multiple proteases and ROS, are also present in the sputum and BAL of COPD patients (86). Increased and prolonged neutrophil recruitment in the COPD lung can have devastating effects on lung parenchyma and can enhance disease severity, prolong inflammation, and cause further tissue damage (107, 125, 126). An age-dependent increase in cytotoxic CD8+ and memory CD4+ T cells in the airways of COPD patients may further disease progression (86, 130, 132). Heightened expression of tissue-specific chemokine receptors, such as CXCR3, on epithelial and endothelial cells in the lungs of smokers with COPD has been shown to correlate with increased disease severity (85).
COPD is also associated with abnormalities in ciliary function and decreased mucociliary clearance, which can lead to increased incidence of recurrent infections, bronchiectasis, and airway obstruction (19, 63, 64, 86, 140). Chronic bronchitis, with increased airway wall thickening, inflammation, and heightened mucus production, as well as emphysema, with alveolar wall destruction, hyperinflation, and impaired gas exchange, are two common phenotypic manifestations of COPD (Figure 2). Acute exacerbations of COPD (AECOPD) are common in patients with severe and very severe COPD and are characterized by increased dyspnea, purulence, and augmented sputum volume (86, 137). Bacterial and viral infections are the most frequent causes of AECOPD, but air pollution and environmental temperature changes may also be partly responsible. Acute exacerbations place COPD patients at risk of pulmonary failure and are associated with worse quality of life, accelerated decline in lung function, and increased mortality.
Aging and Interstitial Lung Disease
Aging is associated with increased susceptibility to interstitial lung diseases, primarily IPF. This chronic and progressive disease of unknown etiology results in impaired gas exchange and respiratory failure. IPF carries a poor prognosis, with a survival of less than five years post diagnosis. IPF is a process that is characterized by epithelial cell injury, fibroblast expansion, augmented ECM remodeling, and lung architecture destruction (87, 141). The histopathological appearance of IPF is usually interstitial pneumonia, which is characterized by the heterogeneous appearance of architectural distortions in combination with areas of honeycomb changes or scars and small areas of active fibrosis by fibroblasts/myofibroblasts (141, 142). Glycolytic reprogramming has been shown to play an important role in the pathogenesis of lung fibrosis, with higher glycolytic activity detected in fibrotic areas (143). Specifically, aged fibroblasts have augmented glucose utilization and increased resistance to apoptosis, which may underlie increased susceptibility of the aged lung to fibrogenesis (144).
As illustrated in Figure 3, age-related morphological and physiological changes in the lung, dysregulated innate and adaptive immune responses, abnormal AEC and fibroblast activation, and increased oxidative stress can increase susceptibility to disrepair (83, 87–91). Further, age-associated changes in genomic instability, telomere shortening, epigenetic alterations, cellular senescence, impaired autophagy, and mitochondrial dysfunction may promote IPF development and progression (3, 12, 62, 84, 88, 89, 116, 145). Gene polymorphisms and transcriptional changes impair epithelial integrity and spatial orientation and underlie the inability of AECs to respond to repeat injuries and mechanical stretch associated with ventilation (72, 145). Age-associated alterations in the unfolded protein response promote excessive or unresolved endoplasmic reticulum stress and mitochondrial dysfunction, which enhance apoptotic responses of AECs (72). Increased telomere shortening in aging diminishes AEC1 regeneration by AEC2s (83, 84). Aberrant epithelial cells secrete an excessive variety of mediators that induce fibroblast proliferation, basement membrane disruption, ECM production, and airway remodeling (33, 68, 69, 145). Environmental cues generated by increased collagen deposition and ECM stiffness as well as aberrant expression of multiple genes, miRNAs, and polymorphisms promote an excessive fibroproliferative phenotype in fibroblasts (87, 90, 125, 145). Fibroblasts differentiate into myofibroblasts, which through the production of cytokines can enhance the inflammatory response in the lung (87, 145). IPF myofibroblasts can produce mediators that induce epithelial apoptosis and subsequently destabilize the epithelium and increase alveolar damage.
Figure 3.

Cellular and molecular mechanisms that contribute to interstitial pulmonary fibrosis (IPF). In panel a, IPF is characterized by repetitive epithelial cell injury, increased alveolar epithelial cell senescence, production by profibrotic mediators contributing to elevated matrix deposition by myofibroblasts, changes in microbiome composition, and abnormalities in host immune defense. Therapeutic interventions are shown in green. In panels b (low power) and c (high power), histological features (hematoxylin and eosin staining) of usual interstitial pneumonia (UIP) are illustrated. Fibroblastic foci, a prominent, but nonspecific feature of IPF, are illustrated by an asterisk. Abbreviations: AEC, alveolar epithelial cell; CCL2, chemokine (C-C motif) ligand 2; CTGF, connective tissue growth factor; CXCL12, C-X-C motif chemokine ligand 12; FGF-2, fibroblast growth factor 2; FGFR1, fibroblast growth factor receptor 1; LOXL2, lysyl oxidase-like 2; LPA, lysophosphatidic acid; LPA1, lysophosphatidic acid receptor type 1; LPC, lysophosphatidylcholine; MCP-1, monocyte chemoattractant protein 1; MMP, matrix metalloproteinase; PDGF, platelet-derived growth factor; PDGFR: PDGF receptor; SDF-1, stromal cell–derived factor 1; SNP, single nucleotide polymorphism; TGF-β, transforming growth factor β; TGFβR1/2, TGFβ receptor type 1 or 2; TIMP, tissue inhibitor of metalloproteinases; TRPV4, transient receptor potential cation channel subfamily V member 4; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor. Adapted with permission from Reference 141. Copyright 2018, Massachusetts Medical Society.
Acute exacerbations in IPF patients are characterized by worsened hypoxemic respiratory failure (141). Exacerbations can be triggered by bacterial and viral infections, aspiration, or drug toxicity, but in many instances they remain idiopathic (141). Acute exacerbations place IPF patients at risk of pulmonary failure and are associated with worse quality of life, accelerated decline in lung function, and increased mortality.
Age-Associated Lung Function Decline with Asthma
Asthma is characterized by bronchoconstriction, airway mucus plugging, and airway edema. Structural changes, such as subepithelial collagen deposition, goblet cell and mucous gland hyperplasia, as well as ASM hypertrophy and hyperplasia, illustrate that remodeling of airways occurs in asthma patients (98, 99). Despite an age-associated decline in IgE production, a relationship still exists between heightened IgE levels and the development of asthma in the elderly (140). While mortality from asthma has declined in the majority of age groups, the number of deaths in elderly has remained constant (140). While it is appreciated that there is a decline in lung function in healthy older persons, increased airway remodeling and inflammation that occur with asthma can contribute to an accelerated decline in lung function. Age-associated impairments in innate and adaptive immune responses in the lung may contribute to increased susceptibility of older persons developing an asthma exacerbation (3, 19, 107, 109, 110). Taken together, although allergies and asthma can affect all age groups, asthma in the elderly in regards to inflammation and clinical manifestation is different from asthma in younger patients.
Sepsis and Acute Respiratory Distress Syndrome in the Elderly
ARDS, the most severe form of acute lung injury, is a severe respiratory illness, with older patients having a higher risk of ARDS development (146). Many causes underlie the development of ARDS, but sepsis and reperfusion syndrome are most prevalent (146, 147). ARDS is often a manifestation of multiple organ dysfunctions and, due to its heterogeneous presentations, ARDS can differ between individual patient disease groups (146). Sepsis is an inflammatory response to infection, with severe sepsis and septic shock being its more severe forms. Due to reduced immunity, coexisting comorbidities, and age-associated functional limitations, the incidence of severe sepsis and septic shock increases in older individuals (104, 146). Mortality rates associated with severe sepsis also increase with age, with a 50–60% mortality rate in elderly patients (146). Dysregulated cytokine production can predispose the elderly to systemic infection by microbial pathogens and can lead to poor outcomes in septic patients (148). Age-associated alterations in oxidative stress and ROS production, specifically in senescent pulmonary vasculature as well as epithelial and immune cells, can promote worse clinical outcomes in elderly patients with sepsis (53, 149). Further, elderly patients exhibit reduced tolerance to systemic inflammation, with increased mortality when compared with younger septic individuals (146–148). Aberrant responses of pulmonary endothelium to inflammatory mediators may increase susceptibility of the aged lung to infection. Patients with severe sepsis and septic shock often require mechanical ventilation, which in the elderly is independently associated with increased mortality (146, 147).
Pneumonia
Sepsis and septic shock can result from an infection anywhere in the body; however, the most common source of sepsis in elderly patients is the respiratory tract. Community-acquired pneumonia (CAP) is the most common type of pneumonia and remains a leading cause of morbidity and mortality worldwide. Bacteria, especially Streptococcus pneumoniae, are the most common pathogens that commonly cause CAP in the elderly (62). Many age-associated risk factors contribute to the increased susceptibility of older persons to bacterial CAP (Supplemental Table 2). In healthy young individuals, despite frequent colonization of the upper airways by pathogenic bacteria, multiple innate mechanisms help to protect the lower airways, and CAP is relatively uncommon. Viral pneumonia can directly or indirectly, through predisposing the lung to a secondary infection, significantly impact morbidity and mortality in older persons. Age-associated alterations in immune surveillance, specifically decreased innate and adaptive immune responses, and dysfunctional immune response regulation can be responsible for increased lung pathology in response to infectious stimuli (3, 62, 106, 110, 114, 135). Upregulation of epithelial cell surface receptors in response to chronic inflammation increases bacterial adhesion and accumulation in the aged lung and correlates with susceptibility to pneumonia (62, 97). Also, depressed clearance mechanisms, such as cough, oral and mucociliary clearance, and swallowing disorders can also increase susceptibility (40, 63, 64, 140). The presence of lung diseases that alter lung structure, such as IPF or COPD, and systemic diseases, such as congestive heart failure and diabetes, can greatly impact the development and progression of pneumonia.
CONCLUSIONS AND FUTURE DIRECTIONS
Lung aging is highly complex and results from accumulated changes between injury and repair to lung cellular systems. Age-associated changes in intrinsic mechanisms that aid in cell regeneration and repair, such as depletion of adult stem cell reservoirs, mitochondrial dysfunction, increased oxidative stress, and telomere shortening, contribute to an inability of lung cells to maintain baseline homeostasis. Normal lung aging is associated with multiple structural and functional changes in the respiratory tract, with many of these changes contributing to decreased lung function, altered pulmonary remodeling, diminished regeneration, and enhanced susceptibility to pulmonary disease. The lung has evolved multiple innate and adaptive defense systems to preserve homeostasis and respond to foreign stimuli. Age-associated changes in composition and function of AEC1s, AEC2s, fibroblasts, and endothelial and ASM cells underlie increased susceptibility of older persons to the development and progression of lung disorders. Immunosenescence contributes to the production of oxygen free radicals and augments production of proinflammatory cytokines. Sustained inflammation of the lower respiratory tract may predispose older adults to increased vulnerability to toxic environmental exposure and accelerated lung function decline. Poor prognosis and recovery in pulmonary inflammatory diseases have been attributed to age-related changes in innate and adaptive immune responses in the lung. Genetic background and lifestyle promote age-related changes in the lung and contribute to the development and progression of airway diseases and increased susceptibility to infectious stimuli and toxins.
Over the last several decades, many critical insights into the cellular and molecular mechanisms that underlie lung aging and increased susceptibility to disease have been generated. More detailed mechanistic studies are needed to validate many of the genetic determinants that have been associated with disease progression and how the process of aging gives rise to these genetic signatures. Additional research is needed to understand the interaction between various immune and lung cell types and how aging impacts these populations. Understanding how and when the process of aging in the lung begins will also need to be clarified. Specifically, how biologic hallmarks of aging are associated with varying lung phenotypes and disease progression.
Supplementary Material
SUMMARY POINTS.
Age-associated changes in intrinsic mechanisms that aid in cell regeneration and repair, such as depletion of adult stem cell reservoirs, mitochondrial dysfunction, increased oxidative stress, and telomere shortening, contribute to an inability of lung cells to maintain baseline homeostasis.
Normal lung aging is associated with multiple structural and functional changes in the respiratory tract, with many of these changes giving rise to decreased lung function, altered pulmonary remodeling, diminished regeneration, and enhanced susceptibility to pulmonary disease (7, 10).
Impairment of pulmonary function is a predictor for morbidity and mortality and may promote the development of multiple disease processes (1, 2).
The conducting airways play a pivotal role in maintaining lung homeostasis and age-associated changes in composition and function can be partly responsible for increased susceptibility of older persons to lung disorders.
The epithelial surface of the respiratory airways is a large, highly vascularized area where efficient gas exchange and host defense rely on the integrity of the epithelium; age-associated changes in the composition and function of AEC1s, AEC2s, fibroblasts and endothelial and ASM cells can contribute to the development and progression of lung disorders in the elderly.
Poor prognosis and recovery in pulmonary inflammatory diseases have been attributed to immunosenescence or age-related changes in innate and adaptive immune responses in the lung.
An age-associated reduction in BAL antioxidant levels and composition may underlie increased susceptibility of older persons to exposure with environmental toxins, such as ozone, cigarette smoke, and particulate matter (66).
FUTURE ISSUES.
What defines normal aging of the lung, and can combined molecular, cellular, imaging, and functional studies over a lifetime help to provide a better phenotype?
When does aging in the lung begin, and what are the dominant cell types and molecular signatures that drive this process?
What are the genetic and/or molecular processes that underlie the development of divergent lung phenotypes in the elderly, such as COPD or IPF?
Can an age-associated impact on innate lymphoid cell phenotypes and function contribute to lung disease progression?
As a person ages, how do different cell types of the conducting and respiratory airways adjust to metabolic changes in the host and still perform their essential duties?
When does immunosenescence begin in the lung, what molecular/genetic alterations occur with aging contribute to its development, and can new therapeutic interventions decrease the deleterious effects this process?
ACKNOWLEDGMENTS
The authors acknowledge the following funding sources: grants R01AG052530 (H. W. S-D), R01AG056699 (H. W. S-D), and K08HL138285 (S.J.C) from the US National Institutes of Health and an American Lung Association Biomedical Research Grant (S.J.C).
KEY TERMS
- Senescence
diminished tissue-repair capacity due to cell cycle arrest in progenitor cells and increase production of proinflammatory and matrix-degrading molecules
- Immunosenescence
age-associated decline in immune system capacity to respond to infections and develop long-term immune memory
- FEV
forced expiratory volume measures how much air a person can exhale during a forced breath
- FVC
forced vital capacity is the amount of air exhaled during the FEV test
- COPD
chronic obstructive pulmonary disease is a chronic inflammatory lung disease that causes fixed airflow obstruction
- IPF
idiopathic pulmonary fibrosis is a lung disease that results in scarring of the lung interstitium
- ARDS
acute respiratory distress syndrome occurs when fluid leaks into the lungs, making it difficult or impossible to breathe
- Pneumonia
an infection that causes the air sacs in one or both lungs to fill up with fluid or purulent material
- AEC1
squamous type I alveolar epithelial cells interact closely with endothelial cells of the pulmonary capillaries
- AEC2
cuboidal type II alveolar epithelial cells function as progenitor cells in the alveoli and proliferate and differentiate into AEC1s
- ECM
extracellular matrix is a highly dynamic complex of fibrous proteins, glycoproteins, and proteoglycans that provide structural integrity to the lung
- Fibroblast
maintains the integrity of the alveolar structure by repairing injured areas and ECM production
- Myofibroblast
α-smooth muscle actin–expressing fibroblast that produces type I collagen and fibrogenic/inflammatory cytokines in fibrotic lesions
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Mannino DM, Buist AS, Petty TL, Enright PL, Redd SC. 2003. Lung function and mortality in the United States: data from the First National Health and Nutrition Examination Survey follow up study. Thorax 58:388–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beaty TH, Cohen BH, Newill CA, Menkes HA, Diamond EL, Chen CJ. 1982. Impaired pulmonary function as a risk factor for mortality. Am. J. Epidemiol 116:102–13 [DOI] [PubMed] [Google Scholar]
- 3.Childs BG, Durik M, Baker DJ, van Deursen JM. 2015. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat. Med 21:1424–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu D, Hornsby PJ. 2007. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 67:3117–26 [DOI] [PubMed] [Google Scholar]
- 5.Walters MS, De BP, Salit J, Buro-Auriemma LJ, Wilson T, et al. 2014. Smoking accelerates aging of the small airway epithelium. Respir. Res 15:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quirk JD, Sukstanskii AL, Woods JC, Lutey BA, Conradi MS, et al. 2016. Experimental evidence of age-related adaptive changes in human acinar airways. J. Appl. Physiol 120:159–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verbeken EK, Cauberghs M, Mertens I, Clement J, Lauweryns JM, Van de Woestijne KP. 1992. The senile lung. Comparison with normal and emphysematous lungs. 2. Functional aspects. Chest 101:800–9 [DOI] [PubMed] [Google Scholar]
- 8.Babb TG, Rodarte JR. 2000. Mechanism of reduced maximal expiratory flow with aging. J. Appl. Physiol 89:505–11 [DOI] [PubMed] [Google Scholar]
- 9.Turner JM, Mead J, Wohl ME. 1968. Elasticity of human lungs in relation to age. J. Appl. Physiol 25:664–71 [DOI] [PubMed] [Google Scholar]
- 10.Verbeken EK, Cauberghs M, Mertens I, Clement J, Lauweryns JM, Van de Woestijne KP. 1992. The senile lung. Comparison with normal and emphysematous lungs. 1. Structural aspects. Chest 101:793–99 [DOI] [PubMed] [Google Scholar]
- 11.Forrest JB. 1970. The effect of changes in lung volume on the size and shape of alveoli. J. Physiol 210:533–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janssens JP, Pache JC, Nicod LP. 1999. Physiological changes in respiratory function associated with ageing. Eur. Respir. J 13:197–205 [DOI] [PubMed] [Google Scholar]
- 13.Kerstjens HA, Rijcken B, Schouten JP, Postma DS. 1997. Decline of FEV1 by age and smoking status: facts, figures, and fallacies. Thorax 52:820–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mittmann C, Edelman NH, Norris AH, Shock NW. 1965. Relationship between chest wall and pulmonary compliance and age. J. Appl. Physiol 20:1211–16 [Google Scholar]
- 15.Enright PL, Kronmal RA, Higgins MW, Schenker MB, Haponik EF. 1994. Prevalence and correlates of respiratory symptoms and disease in the elderly. Chest 106:827–34 [DOI] [PubMed] [Google Scholar]
- 16.Enright PL, Kronmal RA, Manolio TA, Schenker MB, Hyatt RE. 1994. Respiratory muscle strength in the elderly: correlates and reference values. Am. J. Respir. Crit. Care Med 149:430–38 [DOI] [PubMed] [Google Scholar]
- 17.Polkey MI, Harris ML, Hughes PD, Hamnegard CH, Lyons D, et al. 1997. The contractile properties of the elderly human diaphragm. Am. J. Respir. Crit. Care Med 155:1560–64 [DOI] [PubMed] [Google Scholar]
- 18.Tolep K, Higgins N, Muza S, Criner G, Kelsen SG. 1995. Comparison of diaphragm strength between healthy adult elderly and young men. Am. J. Respir. Crit. Care Med 152:677–82 [DOI] [PubMed] [Google Scholar]
- 19.Sharma G, Goodwin J. 2006. Effect of aging on respiratory system physiology and immunology. Clin. Interv. Aging 1:253–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crapo RO, Morris AH, Clayton PD, Nixon CR. 1982. Lung volumes in healthy nonsmoking adults. Bull. Eur. Physiopathol. Respir 18:419–25 [PubMed] [Google Scholar]
- 21.Xu X, Laird N, Dockery DW, Schouten JP, Rijcken B, Weiss ST. 1995. Age, period, and cohort effects on pulmonary function in a 24-year longitudinal study. Am. J. Epidemiol 141:554–66 [DOI] [PubMed] [Google Scholar]
- 22.Stam H, Hrachovina V, Stijnen T, Versprille A. 1994. Diffusing capacity dependent on lung volume and age in normal subjects. J. Appl. Physiol 76:2356–63 [DOI] [PubMed] [Google Scholar]
- 23.Hardie JA, Vollmer WM, Buist AS, Ellingsen I, Morkve O. 2004. Reference values for arterial blood gases in the elderly. Chest 125:2053–60 [DOI] [PubMed] [Google Scholar]
- 24.Kronenberg RS, Drage CW. 1973. Attenuation of the ventilatory and heart rate responses to hypoxia and hypercapnia with aging in normal men. J. Clin. Investig 52:1812–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peterson DD, Pack AI, Silage DA, Fishman AP. 1981. Effects of aging on ventilatory and occlusion pressure responses to hypoxia and hypercapnia. Am. Rev. Respir. Dis 124:387–91 [DOI] [PubMed] [Google Scholar]
- 26.Cardus J, Burgos F, Diaz O, Roca J, Barbera JA, et al. 1997. Increase in pulmonary ventilation-perfusion inequality with age in healthy individuals. Am. J. Respir. Crit. Care Med 156:648–53 [DOI] [PubMed] [Google Scholar]
- 27.Mackay EH, Banks J, Sykes B, Lee G. 1978. Structural basis for the changing physical properties of human pulmonary vessels with age. Thorax 33:335–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emirgil C, Sobol BJ, Campodonico S, Herbert WH, Mechkati R. 1967. Pulmonary circulation in the aged. J. Appl. Physiol 23:631–40 [DOI] [PubMed] [Google Scholar]
- 29.McClaran SR, Babcock MA, Pegelow DF, Reddan WG, Dempsey JA. 1995. Longitudinal effects of aging on lung function at rest and exercise in healthy active fit elderly adults. J. Appl. Physiol 78:1957–68 [DOI] [PubMed] [Google Scholar]
- 30.O’Kroy JA, Lawler JM, Stone J, Babb TG. 2000. Airflow limitation and control of end-expiratory lung volume during exercise. Respir. Physiol 119:57–68 [DOI] [PubMed] [Google Scholar]
- 31.McConnell AK, Davies CT. 1992. A comparison of the ventilatory responses to exercise of elderly and younger humans. J. Gerontol 47:B137–41 [DOI] [PubMed] [Google Scholar]
- 32.McConnell AK, Semple ES, Davies CT. 1993. Ventilatory responses to exercise and carbon dioxide in elderly and younger humans. Eur. J. Appl. Physiol. Occup. Physiol 66:332–37 [DOI] [PubMed] [Google Scholar]
- 33.Whitsett JA, Alenghat T. 2015. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol 16:27–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koval M 2013. Differential pathways of claudin oligomerization and integration into tight junctions. Tissue Barriers 1:e24518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koval M 2013. Claudin heterogeneity and control of lung tight junctions. Annu. Rev. Physiol 75:551–67 [DOI] [PubMed] [Google Scholar]
- 36.Crapo JD, Barry BE, Gehr P, Bachofen M, Weibel ER. 1982. Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis 125:740–45 [DOI] [PubMed] [Google Scholar]
- 37.Rogers DF. 1994. Airway goblet cells: responsive and adaptable front-line defenders. Eur. Respir. J 7:1690–706 [PubMed] [Google Scholar]
- 38.Kotton DN, Morrisey EE. 2014. Lung regeneration: mechanisms, applications and emerging stem cell populations. Nat. Med 20:822–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Y, Riccio P, Schotsaert M, Mori M, Lu J, et al. 2018. Spatial-temporal lineage restrictions of embryonic p63+ progenitors establish distinct stem cell pools in adult airways. Dev. Cell 44:752–61.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitsett JA. 2018. Airway epithelial differentiation and mucociliary clearance. Ann. Am. Thorac. Soc 15:S143–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rose MC, Nickola TJ, Voynow JA. 2001. Airway mucus obstruction: mucin glycoproteins, MUC gene regulation and goblet cell hyperplasia. Am. J. Respir. Cell Mol. Biol 25:533–37 [DOI] [PubMed] [Google Scholar]
- 42.Rose MC. 1992. Mucins: structure, function, and role in pulmonary diseases. Am. J. Physiol 263:L413–29 [DOI] [PubMed] [Google Scholar]
- 43.Rose MC, Brown CF, Jacoby JZ 3rd, Lynn WS, Kaufman B. 1987. Biochemical properties of tracheobronchial mucins from cystic fibrosis and non-cystic fibrosis individuals. Pediatr. Res 22:545–51 [DOI] [PubMed] [Google Scholar]
- 44.Gum JR Jr. 1992. Mucin genes and the proteins they encode: structure, diversity, and regulation. Am. J. Respir. Cell Mol. Biol 7:557–64 [DOI] [PubMed] [Google Scholar]
- 45.Voynow JA, Rubin BK. 2009. Mucins, mucus, and sputum. Chest 135:505–12 [DOI] [PubMed] [Google Scholar]
- 46.Evans CM, Koo JS. 2009. Airway mucus: the good, the bad, the sticky. Pharmacol. Ther 121:332–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams OW, Sharafkhaneh A, Kim V, Dickey BF, Evans CM. 2006. Airway mucus: from production to secretion. Am. J. Respir. Cell Mol. Biol 34:527–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Button B, Cai LH, Ehre C, Kesimer M, Hill DB, et al. 2012. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 337:937–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rajavelu P, Chen G, Xu Y, Kitzmiller JA, Korfhagen TR, Whitsett JA. 2015. Airway epithelial SPDEF integrates goblet cell differentiation and pulmonary Th2 inflammation. J. Clin. Investig 125:2021–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lambrecht BN, Hammad H. 2012. The airway epithelium in asthma. Nat. Med 18:684–92 [DOI] [PubMed] [Google Scholar]
- 51.Greene CM, McElvaney NG. 2005. Toll-like receptor expression and function in airway epithelial cells. Arch. Immunol. Ther. Exp 53:418–27 [PubMed] [Google Scholar]
- 52.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. 2009. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat. Med 15:410–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cleaver JO, You D, Michaud DR, Pruneda FA, Juarez MM, et al. 2014. Lung epithelial cells are essential effectors of inducible resistance to pneumonia. Mucosal Immunol. 7:78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryu JH, Kim CH, Yoon JH. 2010. Innate immune responses of the airway epithelium. Mol. Cells 30:173–83 [DOI] [PubMed] [Google Scholar]
- 55.Parker D, Martin FJ, Soong G, Harfenist BS, Aguilar JL, et al. 2011. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. MBio 2:e00016–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parker D, Prince A. 2011. Type I interferon response to extracellular bacteria in the airway epithelium. Trends Immunol. 32:582–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu L, Lee PK, Lee WM, Zhao Y, Yu D, Chen Y. 2009. Rhinovirus-induced major airway mucin production involves a novel TLR3-EGFR-dependent pathway. Am. J. Respir. Cell Mol. Biol 40:610–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen R, Lim JH, Jono H, Gu XX, Kim YS, et al. 2004. Nontypeable Haemophilus influenzae lipoprotein P6 induces MUC5AC mucin transcription via TLR2-TAK1-dependent p38 MAPK-AP1 and IKKβ-IκBα-NF-κB signaling pathways. Biochem. Biophys. Res. Commun 324:1087–94 [DOI] [PubMed] [Google Scholar]
- 59.Bals R 2000. Epithelial antimicrobial peptides in host defense against infection. Respir. Res 1:141–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choksi SP, Lauter G, Swoboda P, Roy S. 2014. Switching on cilia: transcriptional networks regulating ciliogenesis. Development 141:1427–41 [DOI] [PubMed] [Google Scholar]
- 61.Wansleeben C, Bowie E, Hotten DF, Yu YR, Hogan BL. 2014. Age-related changes in the cellular composition and epithelial organization of the mouse trachea. PLOS ONE 9:e93496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shivshankar P, Boyd AR, Le Saux CJ, Yeh IT, Orihuela CJ. 2011. Cellular senescence increases expression of bacterial ligands in the lungs and is positively correlated with increased susceptibility to pneumococcal pneumonia. Aging Cell 10:798–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ho JC, Chan KN, Hu WH, Lam WK, Zheng L, et al. 2001. The effect of aging on nasal mucociliary clearance, beat frequency, and ultrastructure of respiratory cilia. Am. J. Respir. Crit. Care Med 163:983–88 [DOI] [PubMed] [Google Scholar]
- 64.Proenca de Oliveira-Maul J, Barbosa de Carvalho H, Goto DM, Maia RM, Fló C, et al. 2013. Aging, diabetes, and hypertension are associated with decreased nasal mucociliary clearance. Chest 143:1091–97 [DOI] [PubMed] [Google Scholar]
- 65.Svartengren M, Falk R, Philipson K. 2005. Long-term clearance from small airways decreases with age. Eur. Respir. J 26:609–15 [DOI] [PubMed] [Google Scholar]
- 66.Moliva JI, Rajaram MV, Sidiki S, Sasindran SJ, Guirado E, et al. 2014. Molecular composition of the alveolar lining fluid in the aging lung. Age 36:9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Williams MC. 2003. Alveolar type I cells: molecular phenotype and development. Annu. Rev. Physiol 65:669–95 [DOI] [PubMed] [Google Scholar]
- 68.Castranova V, Rabovsky J, Tucker JH, Miles PR. 1988. The alveolar type II epithelial cell: a multifunctional pneumocyte. Toxicol. Appl. Pharmacol 93:472–83 [DOI] [PubMed] [Google Scholar]
- 69.Fehrenbach H 2001. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir. Res 2:33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Choi AM, Ryter SW, Levine B. 2013. Autophagy in human health and disease. N. Engl. J. Med 368:651–62 [DOI] [PubMed] [Google Scholar]
- 71.Nakahira K, Choi AM. 2013. Autophagy: a potential therapeutic target in lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol 305:L93–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Osorio F, Lambrecht B, Janssens S. 2013. The UPR and lung disease. Semin. Immunopathol 35:293–306 [DOI] [PubMed] [Google Scholar]
- 73.Ryter SW, Cloonan SM, Choi AM. 2013. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol. Cells 36:7–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weaver TE, Whitsett JA. 1991. Function and regulation of expression of pulmonary surfactant-associated proteins. Biochem. J 273(Part 2):249–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Whitsett JA, Weaver TE. 2002. Hydrophobic surfactant proteins in lung function and disease. N. Engl. J. Med 347:2141–48 [DOI] [PubMed] [Google Scholar]
- 76.Zhang L, Ikegami M, Dey CR, Korfhagen TR, Whitsett JA. 2002. Reversibility of pulmonary abnormalities by conditional replacement of surfactant protein D (SP-D) in vivo. J. Biol. Chem 277:38709–13 [DOI] [PubMed] [Google Scholar]
- 77.Trapnell BC, Whitsett JA. 2002. GM-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu. Rev. Physiol 64:775–802 [DOI] [PubMed] [Google Scholar]
- 78.McCormack FX, Whitsett JA. 2002. The pulmonary collectins, SP-A and SP-D, orchestrate innate immunity in the lung. J. Clin. Investig 109:707–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Glasser SW, Maxfield MD, Ruetschilling TL, Akinbi HT, Baatz JE, et al. 2013. Persistence of LPS-induced lung inflammation in surfactant protein-C-deficient mice. Am. J. Respir. Cell Mol. Biol 49:845–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang L, Johansson J, Ridsdale R, Willander H, Fitzen M, et al. 2010. Surfactant protein B propeptide contains a saposin-like protein domain with antimicrobial activity at low pH. J. Immunol 184:975–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ryan MA, Akinbi HT, Serrano AG, Perez-Gil J, Wu H, et al. 2006. Antimicrobial activity of native and synthetic surfactant protein B peptides. J. Immunol 176:416–25 [DOI] [PubMed] [Google Scholar]
- 82.Augusto LA, Li J, Synguelakis M, Johansson J, Chaby R. 2002. Structural basis for interactions between lung surfactant protein C and bacterial lipopolysaccharide. J. Biol. Chem 277:23484–92 [DOI] [PubMed] [Google Scholar]
- 83.Walski M, Pokorski M, Antosiewicz J, Rekawek A, Frontczak-Baniewicz M, et al. 2009. Pulmonary surfactant: ultrastructural features and putative mechanisms of aging. J. Physiol. Pharmacol 60(Suppl. 5):121–25 [PubMed] [Google Scholar]
- 84.Alder JK, Barkauskas CE, Limjunyawong N, Stanley SE, Kembou F, et al. 2015. Telomere dysfunction causes alveolar stem cell failure. PNAS 112:5099–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grumelli S, Corry DB, Song LZ, Song L, Green L, et al. 2004. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLOS Med. 1:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sharma G, Hanania NA, Shim YM. 2009. The aging immune system and its relationship to the development of chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc 6:573–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.White ES. 2015. Lung extracellular matrix and fibroblast function. Ann. Am. Thorac. Soc 12(Suppl. 1):S30–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sueblinvong V, Neveu WA, Neujahr DC, Mills ST, Rojas M, et al. 2014. Aging promotes pro-fibrotic matrix production and increases fibrocyte recruitment during acute lung injury. Adv. Biosci. Biotechnol 5:19–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Campisi J 2005. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120:513–22 [DOI] [PubMed] [Google Scholar]
- 90.Parrinello S, Coppe JP, Krtolica A, Campisi J. 2005. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci 118:485–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.D’Errico A, Scarani P, Colosimo E, Spina M, Grigioni WF, Mancini AM. 1989. Changes in the alveolar connective tissue of the ageing lung. An immunohistochemical study. Virchows Arch. A Pathol. Anat. Histopathol 415:137–44 [DOI] [PubMed] [Google Scholar]
- 92.Fulop T Jr., Douziech N, Jacob MP, Hauck M, Wallach J, Robert L. 2001. Age-related alterations in the signal transduction pathways of the elastin-laminin receptor. Pathol. Biol 49:339–48 [DOI] [PubMed] [Google Scholar]
- 93.Godin LM, Sandri BJ, Wagner DE, Meyer CM, Price AP, et al. 2016. Decreased laminin expression by human lung epithelial cells and fibroblasts cultured in acellular lung scaffolds from aged mice. PLOS ONE 11:e0150966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shasby DM. 2007. Cell-cell adhesion in lung endothelium. Am. J. Physiol. Lung Cell. Mol. Physiol 292:L593–607 [DOI] [PubMed] [Google Scholar]
- 95.Langer HF, Chavakis T. 2009. Leukocyte-endothelial interactions in inflammation. J. Cell Mol. Med 13:1211–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Petri B, Phillipson M, Kubes P. 2008. The physiology of leukocyte recruitment: an in vivo perspective. J. Immunol 180:6439–46 [DOI] [PubMed] [Google Scholar]
- 97.Jane-Wit D, Chun HJ. 2012. Mechanisms of dysfunction in senescent pulmonary endothelium. J. Gerontol. A Biol. Sci. Med. Sci 67:236–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Prakash YS. 2016. Emerging concepts in smooth muscle contributions to airway structure and function: implications for health and disease. Am. J. Physiol. Lung Cell. Mol. Physiol 311:L1113–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Damera G, Tliba O, Panettieri RA Jr. 2009. Airway smooth muscle as an immunomodulatory cell. Pulm. Pharmacol. Ther 22:353–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yamamoto Y, Tanaka A, Kanamaru A, Tanaka S, Tsubone H, et al. 2003. Morphology of aging lung in F344/N rat: alveolar size, connective tissue, and smooth muscle cell markers. Anat. Rec. A Discov. Mol. Cell. Evol. Biol 272:538–47 [DOI] [PubMed] [Google Scholar]
- 101.Marriott HM, Bingle CD, Read RC, Braley KE, Kroemer G, et al. 2005. Dynamic changes in Mcl-1 expression regulate macrophage viability or commitment to apoptosis during bacterial clearance. J. Clin. Investig 115:359–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O’Garra A. 1991. IL-10 inhibits cytokine production by activated macrophages. J. Immunol 147:3815–22 [PubMed] [Google Scholar]
- 103.Mosser DM, Zhang X. 2008. Interleukin-10: new perspectives on an old cytokine. Immunol. Rev 226:205–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. 2003. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol. Cell Biol 81:289–96 [DOI] [PubMed] [Google Scholar]
- 105.Morimoto K, Janssen WJ, Terada M. 2012. Defective efferocytosis by alveolar macrophages in IPF patients. Respir. Med 106:1800–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hearps AC, Martin GE, Angelovich TA, Cheng WJ, Maisa A, et al. 2012. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 11:867–75 [DOI] [PubMed] [Google Scholar]
- 107.Li Z, Jiao Y, Fan EK, Scott MJ, Li Y, et al. 2017. Aging-impaired filamentous actin polymerization signaling reduces alveolar macrophage phagocytosis of bacteria. J. Immunol 199:3176–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wong CK, Smith CA, Sakamoto K, Kaminski N, Koff JL, Goldstein DR. 2017. Aging impairs alveolar macrophage phagocytosis and increases influenza-induced mortality in mice. J. Immunol 199:1060–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shaw AC, Panda A, Joshi SR, Qian F, Allore HG, Montgomery RR. 2011. Dysregulation of human Toll-like receptor function in aging. Ageing Res. Rev 10:346–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Metcalf TU, Cubas RA, Ghneim K, Cartwright MJ, Grevenynghe JV, et al. 2015. Global analyses revealed age-related alterations in innate immune responses after stimulation of pathogen recognition receptors. Aging Cell 14:421–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Plataki M, Cho SJ, Harris RM, Huang HR, Yun HS, et al. 2019. Mitochondrial dysfunction in aged macrophages and lung during primary Streptococcus pneumoniae infection is improved with pirfenidone. Sci. Rep 9:971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Suzuki M, Betsuyaku T, Ito Y, Nagai K, Nasuhara Y, et al. 2008. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol 39:673–82 [DOI] [PubMed] [Google Scholar]
- 113.Agrawal A, Gupta S. 2011. Impact of aging on dendritic cell functions in humans. Ageing Res. Rev 10:336–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Agrawal A, Agrawal S, Cao JN, Su H, Osann K, Gupta S. 2007. Altered innate immune functioning of dendritic cells in elderly humans: a role of phosphoinositide 3-kinase-signaling pathway. J. Immunol 178:6912–22 [DOI] [PubMed] [Google Scholar]
- 115.Toapanta FR, Ross TM. 2009. Impaired immune responses in the lungs of aged mice following influenza infection. Respir. Res 10:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Panda A, Qian F, Mohanty S, van Duin D, Newman FK, et al. 2010. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J. Immunol 184:2518–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Plowden J, Renshaw-Hoelscher M, Gangappa S, Engleman C, Katz JM, Sambhara S. 2004. Impaired antigen-induced CD8+ T cell clonal expansion in aging is due to defects in antigen presenting cell function. Cell Immunol. 229:86–92 [DOI] [PubMed] [Google Scholar]
- 118.Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, et al. 2015. Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J. Immunol 195:2624–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Silva MT, Correia-Neves M. 2012. Neutrophils and macrophages: the main partners of phagocyte cell systems. Front. Immunol 3:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen MM, Palmer JL, Plackett TP, Deburghgraeve CR, Kovacs EJ. 2014. Age-related differences in the neutrophil response to pulmonary pseudomonas infection. Exp. Gerontol 54:42–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Corberand J, Ngyen F, Laharrague P, Fontanilles AM, Gleyzes B, et al. 1981. Polymorphonuclear functions and aging in humans. J. Am. Geriatr. Soc 29:391–97 [DOI] [PubMed] [Google Scholar]
- 122.Brinkmann V, Zychlinsky A. 2007. Beneficial suicide: why neutrophils die to make NETs. Nat. Rev. Microbiol 5:577–82 [DOI] [PubMed] [Google Scholar]
- 123.Nomellini V, Brubaker AL, Mahbub S, Palmer JL, Gomez CR, Kovacs EJ. 2012. Dysregulation of neutrophil CXCR2 and pulmonary endothelial icam-1 promotes age-related pulmonary inflammation. Aging Dis. 3:234–47 [PMC free article] [PubMed] [Google Scholar]
- 124.Fulop T Jr., Fouquet C, Allaire P, Perrin N, Lacombe G, et al. 1997. Changes in apoptosis of human polymorphonuclear granulocytes with aging. Mech. Ageing Dev 96:15–34 [DOI] [PubMed] [Google Scholar]
- 125.Starr ME, Ueda J, Yamamoto S, Evers BM, Saito H. 2011. The effects of aging on pulmonary oxidative damage, protein nitration, and extracellular superoxide dismutase down-regulation during systemic inflammation. Free Radic. Biol. Med 50:371–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zemans RL, Colgan SP, Downey GP. 2009. Transepithelial migration of neutrophils: mechanisms and implications for acute lung injury. Am. J. Respir. Cell Mol. Biol 40:519–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nogusa S, Ritz BW, Kassim SH, Jennings SR, Gardner EM. 2008. Characterization of age-related changes in natural killer cells during primary influenza infection in mice. Mech. Ageing Dev 129:223–30 [DOI] [PubMed] [Google Scholar]
- 128.Beli E, Clinthorne JF, Duriancik DM, Hwang I, Kim S, Gardner EM. 2011. Natural killer cell function is altered during the primary response of aged mice to influenza infection. Mech. Ageing Dev 132:503–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shehata HM, Hoebe K, Chougnet CA. 2015. The aged nonhematopoietic environment impairs natural killer cell maturation and function. Aging Cell 14:191–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pabst R, Tschernig T. 1995. Lymphocytes in the lung: an often neglected cell. Numbers, characterization and compartmentalization. Anat. Embryol 92:293–99 [DOI] [PubMed] [Google Scholar]
- 131.Haynes L, Swain SL. 2006. Why aging T cells fail: implications for vaccination. Immunity 24:663–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kovaiou RD, Grubeck-Loebenstein B. 2006. Age-associated changes within CD4+ T cells. Immunol. Lett 107:8–14 [DOI] [PubMed] [Google Scholar]
- 133.Zhou X, McElhaney JE. 2011. Age-related changes in memory and effector T cells responding to influenza A/H3N2 and pandemic A/H1N1 strains in humans. Vaccine 29:2169–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Allie SR, Bradley JE, Mudunuru U, Schultz MD, Graf BA, et al. 2019. The establishment of resident memory B cells in the lung requires local antigen encounter. Nat. Immunol 20:97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Holodick NE, Rothstein TL. 2015. B cells in the aging immune system: time to consider B-1 cells. Ann. N. Y. Acad. Sci 1362:176–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Comhair SA, Erzurum SC. 2002. Antioxidant responses to oxidant-mediated lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol 283:L246–55 [DOI] [PubMed] [Google Scholar]
- 137.van Durme Y, Verhamme KMC, Stijnen T, van Rooij FJA, Van Pottelberge GR, et al. 2009. Prevalence, incidence, and lifetime risk for the development of COPD in the elderly: the Rotterdam study. Chest 135:368–77 [DOI] [PubMed] [Google Scholar]
- 138.Wu X, Yuan B, Lopez E, Bai C, Wang X. 2014. Gene polymorphisms and chronic obstructive pulmonary disease. J. Cell Mol. Med 18:15–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Brandsma CA, de Vries M, Costa R, Woldhuis RR, Konigshoff M, Timens W. 2017. Lung ageing and COPD: Is there a role for ageing in abnormal tissue repair? Eur. Respir. Rev 26:170073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Incalzi RA, Maini CL, Fuso L, Giordano A, Carbonin PU, Galli G. 1989. Effects of aging on mucociliary clearance. Compr. Gerontol. A 3(Suppl.):65–68 [PubMed] [Google Scholar]
- 141.Lederer DJ, Martinez FJ. 2018. Idiopathic pulmonary fibrosis. N. Engl. J. Med 378:1811–23 [DOI] [PubMed] [Google Scholar]
- 142.Katzenstein AL, Mukhopadhyay S, Myers JL. 2008. Diagnosis of usual interstitial pneumonia and distinction from other fibrosing interstitial lung diseases. Hum. Pathol 39:1275–94 [DOI] [PubMed] [Google Scholar]
- 143.Xie N, Tan Z, Banerjee S, Cui H, Ge J, et al. 2015. Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am. J. Respir. Crit. Care Med 192:1462–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cho SJ, Moon JS, Lee CM, Choi AM, Stout-Delgado HW. 2017. Glucose transporter 1-dependent glycolysis is increased during aging-related lung fibrosis, and phloretin inhibits lung fibrosis. Am. J. Respir. Cell Mol. Biol 56:521–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Selman M, Pardo A. 2014. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am. J. Respir. Crit. Care Med 189:1161–72 [DOI] [PubMed] [Google Scholar]
- 146.Ely EW, Wheeler AP, Thompson BT, Ancukiewicz M, Steinberg KP, Bernard GR. 2002. Recovery rate and prognosis in older persons who develop acute lung injury and the acute respiratory distress syndrome. Ann. Intern. Med 136:25–36 [PubMed] [Google Scholar]
- 147.Siner JM, Pisani MA. 2007. Mechanical ventilation and acute respiratory distress syndrome in older patients. Clin. Chest Med 28:783–91 [DOI] [PubMed] [Google Scholar]
- 148.Bruunsgaard H, Skinhøj P, Qvist J, Pedersen BK. 1999. Elderly humans show prolonged in vivo inflammatory activity during pneumococcal infections. J. Infect. Dis 180:551–54 [DOI] [PubMed] [Google Scholar]
- 149.Al-Shaer MH, Choueiri NE, Correia ML, Sinkey CA, Barenz TA, Haynes WG. 2006. Effects of aging and atherosclerosis on endothelial and vascular smooth muscle function in humans. Int. J. Cardiol 109:201–6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.