

Abstract
Motivation
Circular RNAs (circRNAs), a class of non-coding RNAs generated from non-canonical back-splicing events, have emerged to play key roles in many biological processes. Though numerous tools have been developed to detect circRNAs from rRNA-depleted RNA-seq data based on back-splicing junction-spanning reads, computational tools to identify critical genomic features regulating circRNA biogenesis are still lacking. In addition, rigorous statistical methods to perform differential expression (DE) analysis of circRNAs remain under-developed.
Results
We present circMeta, a unified computational framework for circRNA analyses. circMeta has three primary functional modules: (i) a pipeline for comprehensive genomic feature annotation related to circRNA biogenesis, including length of introns flanking circularized exons, repetitive elements such as Alu elements and SINEs, competition score for forming circulation and RNA editing in back-splicing flanking introns; (ii) a two-stage DE approach of circRNAs based on circular junction reads to quantitatively compare circRNA levels and (iii) a Bayesian hierarchical model for DE analysis of circRNAs based on the ratio of circular reads to linear reads in back-splicing sites to study spatial and temporal regulation of circRNA production. Both proposed DE methods without and with considering host genes outperform existing methods by obtaining better control of false discovery rate and comparable statistical power. Moreover, the identified DE circRNAs by the proposed two-stage DE approach display potential biological functions in Gene Ontology and circRNA-miRNA–mRNA networks that are not able to be detected using existing mRNA DE methods. Furthermore, top DE circRNAs have been further validated by RT-qPCR using divergent primers spanning back-splicing junctions.
Availability and implementation
The software circMeta is freely available at https://github.com/lichen-lab/circMeta.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Circular RNAs (circRNAs) are a class of single-stranded and covalently ‘head-to-tail’ joined RNA species, initially described in the human genome in the 1990s with scrambled exon order (Nigro et al., 1991), and recently emerged as a multi-functional family of RNAs in eukaryotes (Chen, 2016; Li et al., 2018; Wilusz, 2018). circRNAs are derived from a non-canonical form of alternative splicing when the pre-mRNA splicing machinery ‘back splices’ to ligate a downstream splice donor site to an upstream splice acceptor (SA) site (Wilusz, 2018). Recent studies using RNA-seq data revealed that thousands of circRNAs could be detected in eukaryotes (Szabo and Salzman, 2016; Wilusz, 2018). Many circRNAs display tissue-specific expression, implying their potential critical biological functions. For example, a recent analysis showed that circRNAs are highly abundant in the mammalian brain compared to other analyzed tissues. Importantly, many circRNAs are upregulated during neurodevelopment and become highly enriched in synapses (Rybak-Wolf et al., 2015). Loss of mammalian circRNA Cdr1as leads to miRNA network dysregulation and impaired brain functions (Piwecka et al., 2017). These observations together highlighted the critical roles that circRNA plays in mammalian central nervous systems. Besides the important roles of circRNAs in neurodegenerative diseases, several recent studies have revealed circRNAs are abundant and functional in cancer and could serve as potential biomarkers (Chen et al., 2019; Vo et al., 2019).
Due to the scramble exon ordering of circRNA, most circRNA detection algorithms such as findcirc (Memczak et al., 2013), CIRCexplorer (Zhang et al., 2014) and CIRI (Gao et al., 2015) use back-splicing junction-spanning reads from rRNA-depleted RNA sequencing data to predict the landscape of circRNAs. These algorithms utilize different read aligners, such as Tophat2 (Kim et al., 2013), Bowtie2 (Langdon, 2015) or BWA (Li and Durbin, 2009), to map back-splicing junction-spanning reads to the whole genome or transcriptome. As a consequence, large numbers of circRNAs identified are mainly from exonic regions. Though tools for predicting circRNAs are well-established, downstream methods for critical genomic feature annotation involved in circRNA biogenesis, as well as optimized differential expression (DE) methods for circRNAs in a unified framework are not readily available.
Several key genomic features have been recently suggested to play important roles in circRNA biogenesis. For instance, the flanking introns of circularized exons are substantially longer than those that are randomly chosen, which could introduce more cis-regulatory elements that promote circRNA formation (Zhang et al., 2014). Intronic repetitive elements such as Alu elements and short interspersed nuclear elements (SINEs), could form RNA duplexes through orientation-opposite complementary sequences termed inverted repeated Alu pairs (IRAlus) or inverted repeated SINE pairs (IRSINEs), which facilitate the effective back-splicing by bringing back-splicing sites into proximity through intron pairing. It has been shown that the efficiency to form circRNAs depends on the overall score of IRAlus or IRSINEs pairing within or across the flanking introns. In addition to the simple base complementarity, recent evidence indicates RNA adenosine-to-inosine (A-to-I) editing, often enriched in Alu elements, affects the thermodynamics of flanking intron pairing (Li et al., 2018). Knockdown of the RNA-specific adenosine deaminase elevates circRNA formation, possibly due to the downregulation of inosine and enhancement of RNA pairing (Rybak-Wolf et al., 2015). Although these genomic features in flanking introns provide critical insights for the formation of circRNAs, a systematic and unified framework to identify and characterize these features is lacking.
Different from annotated genes (e.g. ∼20K in human), the number and back-splicing sites of circRNAs vary a lot across different biological conditions (Nicolet et al., 2018; Rybak-Wolf et al., 2015), providing a sophisticated post-transcriptional gene regulatory network. Moreover, the distribution assumption for back-splicing junction reads of circRNAs may differ from linear reads in mRNAs, rendering standard DE methods for mRNAs less optimal. Therefore, it is important to develop a DE method specifically for circRNAs based on junction reads. In addition, it has been shown that expression level of circRNAs determined by back-splicing junction reads may vary relative to expression level of their host gene measured by linear splice junction reads (Westholm et al., 2014). The relative expression level of circRNAs and their parental linear mRNA could differ significantly between cell types, indicating they could be subjected to independent regulatory mechanisms (Salzman et al., 2013). In order to accurately cross-compare circRNA differential levels, several publications employed circular-to-linear ratio (CLR) to define expression levels of circRNAs among differential cell types (Nicolet et al., 2018; Rybak-Wolf et al., 2015; Zhou et al., 2017). However, rigorous statistical methods are under-developed to identify differential CLR where the expression of circRNAs and linear isoform could be highly correlated or diverged.
In this work, we develop a unified computational framework ‘circMeta’, to perform comprehensive analyses of predicted circRNA including three primary tasks (i) back-splicing flanking intron identification and genomic feature annotation for flanking introns including host genes, intron length, repetitive elements, circulation competition score and A-to-I editing (ii) a two-stage method for DE analysis of circRNAs based on junction reads to quantitatively compare circRNA levels (iii) a Bayesian hierarchical model for DE analysis of circRNAs based on CLR with considering host genes to study spatial and temporal regulation of circRNA production. Top DE circRNAs have been further validated by RT-qPCR using divergent primers spanning back-splicing junctions. Our proposed DE methods outperform existing methods based on simulation studies and real data applications, and the results provide important biological insights by an example of regulatory roles of circRNAs in different human brain regions.
2 Materials and methods
2.1 Datasets and methods for predicting circRNAs
We collect ribosomal RNA (rRNA) depleted RNA-seq datasets and matched miRNA-seq datasets in three brain regions including frontal cortex, cerebellum and diencephalon with two replicates each from ENCODE project (Thomas et al., 2007). These datasets will be used to demonstrate the workflow and effectiveness of circMeta. As findcirc, CIRCexplorer and CIRI are the most popular circRNA prediction methods that allow accurate prediction of circRNAs based on back-splicing junction reads from rRNA-depleted RNA-seq data, circMeta accommodates the identified circRNAs in the output format of the three methods to perform genomic feature annotation and DE analysis in a unified framework. In the following analyses, we use the common circRNAs identified by CIRI, findcirc and CIRCexplorer as input for circMeta, as circRNA prediction algorithms should ideally be combined to achieve reliable predictions (Hansen et al., 2016).
2.2 Genomic feature annotations of circRNAs
The current version of circMeta provides the following genomic features to annotate circRNAs, which will help better predict and quantify circRNAs.
2.2.1. Host or nearest gene
We first classify predicted circRNAs into exonic, intronic and intergenic circRNAs based on their origin. Exonic circRNAs contain at least one back-splicing exon. Recent studies indicate the dynamics between canonical and back-splicing depends on the availability of core spliceosome, suggesting the circRNA-linear mRNA balance control could contribute to the post-transcriptional gene regulation (Liang et al., 2017). The information of host or adjacent genes could potentially provide biological insights of circRNA-linear mRNA interplay. We annotate the host genes that harbor these intronic and exonic circRNAs, and define the adjacent genes to the circRNAs derived from intergenic sequences.
2.2.2. Back-splicing flanking intron
For exonic circRNAs, we obtain the flanking introns of the back-splicing sites since they are crucial in forming RNA pairing to facilitate efficient back-splicing.
2.2.3. Repetitive elements
As both computational analyses and experimental evidence confirm that flanking intron RNA pairing, mainly through IRAlus or IRSINEs, promotes circRNA formation (Zhang et al., 2014), circMeta calculates the number and orientations of Alu elements in circRNA flanking introns and offers a fast way to calculate IRAlus across or within individual flanking introns. We follow the definition of IRAlus-within as the number of pairing inverted repeated Alu elements within one flanking intron that may prevent the formation of circulation and IRAlus-across as the number of pairing inverted repeated Alu elements across two flanking introns that may promote the formation of circulation (Zhang et al., 2014). We then calculate the IRAlus-score that reflects the competition between IRAlus-within and IRAlus-across to serve as likelihood indication for RNA pairing across the flanking introns for circRNA biogenesis (Zhang et al., 2014).
2.2.4. RNA modification
Based on the fact that inosine can form a base pair with cytidine (C) thereby being converted to guanosine (G) in the cDNA sequence by reverse transcription, the A to G conversion at any given site can be regarded as a potential RNA editing site (Suzuki et al., 2015). RNA-seq sequence alignments could be obtained from intermediate steps in these circRNA prediction methods. The number of RNA-seq reads supporting the edited (G in the sense of transcription) and unedited (A in the sense of transcription) sequences could be further detected using the samtools and bcftools (Li et al., 2009). circMeta could obtain and filter A-to-I sites by reported RNA editing sites in RNA editing database such as RADAR (Ramaswami and Li, 2014) and DARNED (Kiran and Baranov, 2010).
2.2.5. Circular-to-linear ratio
circMeta first detects the linear reads in the back-splicing junctions and then calculates CLR as the ratio of circular junction read count to linear junction read count.
2.3 Differential expression analysis of circRNAs without considering host genes
Since circRNA is mostly defined based on circular junction reads spanning the back-splicing sites, many published works directly take the circular read counts as the expression level of circRNAs. Though the negative binomial distribution (NB) is the most widely used for modeling mRNA counts and for performing DE analysis, it remains unclear whether the direct extension of NB for modeling circular read counts and downstream NB-based DE analysis is optimal, given circular read counts might be less over-dispersed than mRNA read counts. Considering this, we propose a two-stage DE analysis. First, we perform deviance goodness of fit (GOF) test for both NB and Poisson distributions for each circRNA. The deviance is defined as , which is twice the difference between the log-likelihood of the saturated model and the log-likelihood of the fitted model. The deviance follows the χ2 distribution and we use the χ2 test to calculate the P-value for each fitting. A good fitting is indicated by P-value <0.05. Second, we adopt the DE method with the distribution assumption that obtains overall better GOF. To be specific, we let xijl be the observed circular read counts of ith circRNA, jth condition and lth replicate. xijl can be assumed to follow NB as or Poisson distribution as . In the above, sijl is the size factor such as sequencing depth, μij and denote the mean expression and dispersion of ith circRNA in jth condition, respectively. It should be noted that offers the flexibility to model the mean–variance relationship in the way of in NB. If NB achieves better GOF, circMeta uses edgeR (Robinson et al., 2010) as default. If Poisson distribution obtains better GOF instead, circMeta uses approximated z-test for testing the Poisson rates between ith circRNA in two conditions as
If the circular read count is low, a square root transform helps the z-test statistic approach normality, which is given by
In brief, circMeta has two stages for performing DE analysis of circRNAs based on circular reads: (i) GOF test for NB or Poisson distribution and (ii) NB-based test (edgeR) or Poisson-based test (z-test). As a result, circMeta outputs the circular read counts, fold change, P-value and FDR for all circRNAs.
2.4 Differential expression analysis of circRNAs with considering host genes
Similarly, we define xijl as the observed circular read counts, nijl is the total read counts including circular and linear reads covering back-splicing junctions of ith circRNA, jth condition and lth replicate. Thus, CLR could be calculated as . To achieve an easier statistical modeling, we alternatively use CLP (circular-to-linear proportion) as a representative of CLR. We define pijl as the underlying CLP and assume xijl follows a binomial distribution as . Since pijl is bounded between 0 and 1, we further assume that pijl follows a beta distribution as . The beta distribution is parameterized by mean μij and a dispersion parameter ρijl. The mean and variance of the beta distribution hold the relationship as . Actually, the above parameterizations are similar to the model for the proportion of methylated rate for CpG in single nucleotide resolution sequencing data (Feng et al., 2014).
Thus, we employ a beta-binomial model to model circRNA expression relative to that of the host gene among multiple biological conditions. Though either moment of moments (MOM) or maximum likelihood estimation (MLE) could obtain the estimates of ρij and μij, we choose MOM for its simplicity. However, the replicates of RNA-seq for each condition are limited, ranging from two to five for a typical study. It is therefore important to employ a Bayesian approach to estimate the parameters for each circRNA by borrowing information from others. Especially, the Bayesian approach will help stabilize the variance estimate and thus might improve the power in DE analysis. To stabilize the variance estimate , we adopt a shrinkage approach on dispersion parameter ρij. Specifically, we will build up another level of the beta-binomial model by imposing a distribution for ρij. To choose a reasonable distribution, we explore the empirical distribution of ρ estimated from all circRNAs detected in frontal cortex with two replicates RNA-seq and observe the distribution approximates a log-normal distribution (Fig. 2B). Thus, we assume and we have the final Bayesian hierarchical model as follows,
Fig. 2.
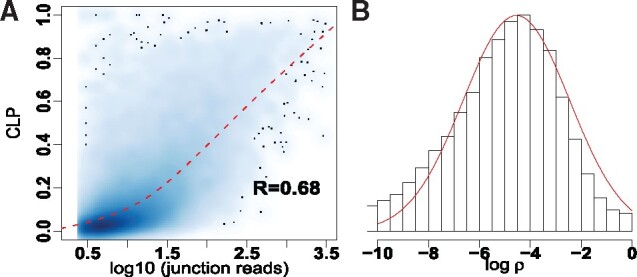
(A) Correlation of logarithmic circular read counts and circular-to-linear proportion (CLP) for common circRNAs identified by findcirc, CIRCexplorer and CIRI in frontal cortex. (B) Histogram of the logarithmic estimated dispersion ρ from common circRNAs identified by findcirc, CIRCexplorer and CIRI in frontal cortex. The solid line is the theoretical density curve for a normal distribution with parameters estimated from sample mean and variance of logarithm of ρ
To make the statistical inference, we first obtain using MOM and and based on in the log-normal distribution. To obtain a shrunk estimate of ρij, we adopt a similar penalized approach used by DSS (Feng et al., 2014) by maximizing the conditional likelihood of ρij as follows,
After all parameters are estimated, hypothesis tests can be performed at each circRNA in two groups with the null hypothesis . We first use Wald test for ith circRNA as,
The details of obtain can be found in Supplementary Material.
We also use likelihood ratio test (LRT) with one degree freedom as,
where l0 is the log-likelihood under H0 and l1 is the log-likelihood under Ha. After P-values are obtained, false discovery rate (FDR) can be obtained using Benjamini and Hochberg (1995).
3 Results
3.1 Genomic feature annotation of circRNAs
We use circMeta to obtain flanking intron length, Alu and SINE enrichment, IRAlus-score and IRSINEs-score from the top and bottom 1K predicted circRNAs ranked by circular read counts in three brain regions and plot these genomic features (Fig. 1). We find that top circRNAs usually have longer flanking introns, more abundant Alu and SINE (excluding Alu), higher IRAlus-score and IRSINEs-score than bottom circRNAs (P-value <0.05), which is consistent with previous observations (Zhang et al., 2014). As expected, we also find more than 90% identified A-to-I editing sites are within intronic Alu. Since circRNAs have been shown to display tissue-specific or developmental-specific expression (Nicolet et al., 2018; Rybak-Wolf et al., 2015; Zhou et al., 2017), it is useful to demonstrate the relationship between circRNAs and their host genes by CLP or CLR. We thus calculate CLP and fit a smoothing spline between natural logarithm of circular reads and CLP (Fig. 2A, Supplementary Fig. S1) and find a strong correlation between circular reads and CLP (R = 0.680, 0.705, 0.695) in three brain regions. The correlations indicate that, while many circRNAs and their host genes remain correlated, some circRNAs are independent of their host genes and could possess unique tissue-specific functions.
Fig. 1.
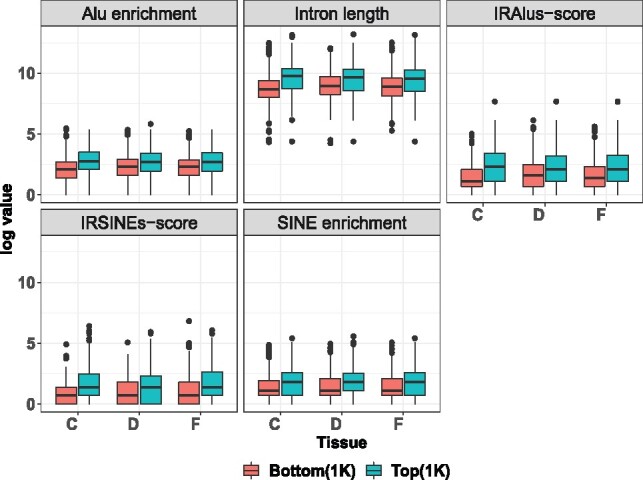
Genomic annotations of common circRNAs identified by findcirc, CIRCexplorer and CIRI in frontal cortex, cerebellum and diencephalon (F, frontal cortex; C, cerebellum; D, diencephalon). Distribution of natural logarithm of junction intron length, Alu enrichment, IRAlus-score, SINE enrichment and IRSINEs-score in top and bottom 1K of identified circRNAs. SINE excludes Alu elements
3.2 Differential expression analysis of circRNAs without considering host genes
We first perform simulation studies to demonstrate the workflow and performance of the two-stage DE analysis approach. In the first stage, we perform the GOF test for all predicted circRNAs in each brain region and find that Poisson distribution actually achieves overall better GOF than NB (Table 1, Supplementary Fig. S2). In the second stage, we thus choose and apply Poisson-based z-test statistic on simulated datasets generated based on predicted circRNAs in frontal cortex. In each simulation, we assume there are 10 000 circRNAs with randomly sampled estimated Poisson rate μi from all estimates. To evaluate the power, we let for all circRNAs except 5% randomly sampled circRNAs differentially expressed between two groups with a log-fold change of 2 in either or . We further allow two replicates for each condition and generate the circular read counts from Poisson distribution. Finally, the simulation is repeated 50 times and the average power (True Positive Rate), FDR (False Discovery Rate), TP (True Positive) and FP (False Positive) for Poisson-based z-test and NB-based edgeR, DESeq2 (Love et al., 2014) are reported respectively. The simulation results are presented in Table 2. We could observe that z-test achieves the highest power and estimated FDR closest to the nominal level 0.05 (0.033 versus 0.05), edgeR obtains the second highest power but significantly over-estimated FDR (0.169 versus 0.05) and DESeq2 is the most conservative test with the lowest power and no FP is reported. As expected, z-test is a more powerful DE test statistic if Poisson distribution fits the data better.
Table 1.
GOF test for circRNAs identified in three brain tissues (P-value <0.05)
| Brain tissue | Poisson (GOF) | NB (GOF) | #circRNA |
|---|---|---|---|
| F | 14 799 | 5633 | 28 674 |
| D | 11 076 | 3729 | 16 466 |
| C | 6262 | 1701 | 14 855 |
F, frontal cortex; C, cerebellum; D, diencephalon.
Table 2.
Simulations when DE is declared with nominal FDR 0.05
| TPR | FDR | TP | FP | |
|---|---|---|---|---|
| edgeR | 0.845 | 0.169 | 422.560 | 88.380 |
| DESeq2 | 0.560 | 0.000 | 280.100 | 0.000 |
| z-test | 0.884 | 0.033 | 441.760 | 15.140 |
We next apply three tests to detect DE circRNAs in the pairwise comparisons of three brain regions. Similar to the observations from the simulation study, we find z-test obtains most DE circRNAs (FDR < 0.05), followed by edgeR. DESeq2 is the most conservative test with the fewest number of DE circRNAs identified (Table 3). We further evaluate common DE circRNAs among three DE methods in each pairwise comparison. In the comparison between frontal cortex and cerebellum, all 351 DE circRNAs identified by edgeR are readily detected by z-test. Meanwhile, 129 out of 138 DE circRNAs identified by DESeq2 are detected by z-test. We also find the trend holds similarly between frontal cortex and diencephalon where 75 out of 99 DE circRNAs identified by edgeR and all DE circRNAs identified by DESeq2 are detected by z-test, respectively. Comparing cerebellum to diencephalon, all DE circRNAs identified by edgeR and DESeq2 are detected by z-test. These observations indicate that most, if not all, DE circRNAs identified by edgeR and DESeq2 can also be detected by z-test, whereas z-test can further detect high-confident DE circRNAs that are missed in existing methods without losing the stringency.
Table 3.
Real data DE results for pairwise comparisons between three brain tissues
| edgeR | DESeq2 | z-test | #circRNA | |
|---|---|---|---|---|
| F versus C | 351 | 138 | 1248 | 19 796 |
| F versus D | 99 | 40 | 366 | 20 001 |
| C versus D | 40 | 8 | 419 | 11 516 |
F, frontal cortex; C, cerebellum; D, diencephalon.
In order to first shed light on the biological roles of circRNAs, we perform Gene Ontology (GO) analyses using host genes harboring DE circRNAs between cerebellum and frontal cortex identified by z-test. Interestingly, while there are common GO terms, host genes harboring circRNAs expressed high in cerebellum are enriched in pathways such as cerebellum and hindbrain development (Fig. 3A). On the other hand, host genes harboring circRNAs expressed higher in frontal cortex are involved in biological pathways including neuron projection and cognition (Supplementary Fig. S3A). In addition, cerebellum–diencephalon comparison also renders numerous GO terms related to brain development and functions (Supplementary Figs S4A and S5A). These findings indicate that host genes harboring DE circRNAs identified by z-test are potentially related to key brain development and functions.
Fig. 3.
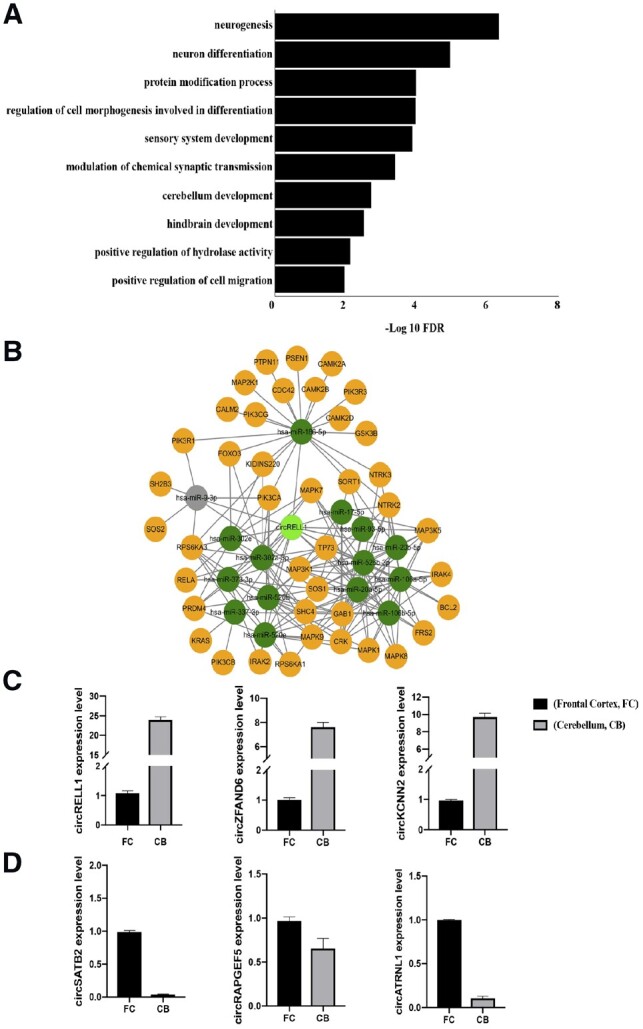
(A) Gene ontology (GO) analysis using host genes harboring circRNAs expressed higher in cerebellum than infrontal cortex. Bar charts showing selected biological processes ranked by false discovery rate (FDR) (FDR <0.05). (B) The putative miRNAs that could be sponged by circRELL1 are predicted by starBase database and indicated in dark green. The mRNAs regulated by miRNA changes are shown in yellow. miRNAs that are not expressed in corresponding tissues are marked in gray. The circRNA-miRNA–mRNA network is related to neurotrophin signaling pathway. (C) The expression of three cerebellum enriched circRNA predicted by circMeta, namely circRELL1, circZFAND6 and circKCNN2, is experimentally validated by RT-qPCR. (D) The expression of three frontal cortex enriched circRNA predicted by circMeta, namely circSATB2, circRAPGEF5 and circATRNL1, is experimentally validated by RT-qPCR
Since circRNAs have been suggested to serve as ‘miRNA sponge’ to interfere with miRNA-mediated gene regulation (Chen, 2016; Li et al., 2018; Wilusz, 2018), we use starBase database (Li et al., 2014; Yang et al., 2011) to predict potential miRNAs bound to top DE circRNAs identified between cerebellum and frontal cortex to explore the biological roles of DE circRNAs in gene regulation. As an example, circRELL1, one of the top circRNAs expressed higher in cerebellum, could potentially interfere with a number of miRNAs through base pair complementarity. In addition, the DIANA-miRPath analysis (Vlachos et al., 2015) reveals a circRNA-centered miRNA–mRNA network that could be regulated by circRELL1. This network is potentially related to many biological events, such as neurotrophin signaling pathway that has been shown in brain development (Huang and Reichardt, 2001) (Fig. 3B). Moreover, circSATB2, expressed higher in frontal cortex, could potentially regulate the ErbB signaling pathway (Supplementary Fig. S3B). Similarly, many biological events could also be regulated by top DE circRNAs identified between cerebellum and diencephalon (Supplementary Figs S4B and S5B). Importantly, most of the predicted miRNA bound to these circRNAs are expressed in their respective tissues determined by miRNA-seq (Supplementary Tables S1 and S2). These results together demonstrate DE circRNAs identified by circMeta could have independent and distinctive biological roles in brain development and functions from their linear mRNA counterpart.
To further validate the top DE circRNAs identified by circMeta, we obtain cerebellar and cortical RNA samples from fetal brains and perform RT-qPCR using divergent primers (Supplementary Table S3) amplifying back-splicing junctions of these top DE circRNAs (Rybak-Wolf et al., 2015; Zhang et al., 2014). Importantly, RT-qPCR validates top DE circRNAs identified by circMeta (FDR < 0.05). For example, three top DE circRNAs (circRELL1, circZFAND6, circKCNN2) identified by circMeta expressed higher in cerebellum than cortex, indeed show the same trend in RT-qPCR (Fig. 3C). Similarly, circSATB2 circRAPGEF5, circATRNL1 with a higher expression in frontal cortex also show the same trend in RT-qPCR (Fig. 3D). Taken together, these RT-qPCR validations further support the accuracy and power of circMeta optimized to identify DE circRNAs.
Based on the results from both the in silico simulation study and the real data analysis, we find that z-test outperforms the traditional mRNA DE methods for the nature of less over-dispersed circular read counts. The host genes harboring identified DE circRNAs by z-test are enriched in a circRNA-centered miRNA–mRNA network involved in brain development and functions. In contrast, DE circRNAs identified by either edgeR or DESeq2 do not show significantly enriched GO terms. Thus, the two-stage DE approach (z-test) is well optimized with comparable power and better FDR control than DE methods not designed specifically for circRNAs. This approach is suitable to quantitatively detect circRNA levels and study their downstream functions, such as miRNA sponge, decoy for RNA-binding protein or circRNA translation.
3.3 Differential expression analysis of circRNAs with considering host genes
We employ the real data-based simulation study to evaluate the performance of the proposed DE method with considering host genes. Based on circular reads and linear reads obtained from predicted circRNAs in frontal cortex, parameters such as μi, ρi and ni resembling real circular proportion, dispersion and total read coverage for ith circRNA could be estimated. For each simulation, we assume 10 000 circRNAs with simultaneously randomly sampled triples of μi, ρi and ni from all estimates. To evaluate the power, we allow 5% randomly sampled μij differential between two groups with a log-fold change of 2 in either or . We allow two replicates for each condition and generate the circular read counts xij from beta-binomial distribution. We repeat the simulation 50 times and report the average power, FDR, TP and FP for our proposed Wald test and LRT with shrunk variance estimate and their naive counterparts. We further include χ2 test and Fisher’s exact test as comparisons. In addition, we add CircTest (Cheng et al., 2016) that is based on an ANOVA-based LRT in beta-binomial distribution for DE circRNA analysis as another comparison.
In Table 4, Fisher’s exact test and χ2 test achieve the highest power by detecting almost all DE circRNAs. However, they suffer from a significant high FDR, making these tests undesirable. Although both Wald test with shrinkage and LRT with shrinkage obtain comparable power compared to their naive counterparts, they achieve a much better FDR control (0.069 versus 0.297 for Wald test; 0.104 versus 0.489 for LRT). Comparing Wald test with shrinkage, LRT with shrinkage obtains higher power (0.825 versus 0.744) but suffers from a little loss of FDR control (0.104 versus 0.069) on a FDR nominal level of 0.05. Furthermore, both Wald test with shrinkage and LRT with shrinkage achieve higher power and better FDR control than CircTest.
Table 4.
Simulations when DE is declared with nominal FDR 0.05
| TPR | FDR | TP | FP | |
|---|---|---|---|---|
| Wald | 0.741 | 0.297 | 370.580 | 156.440 |
| Wald (shrunk) | 0.744 | 0.069 | 371.980 | 27.420 |
| LRT | 0.890 | 0.489 | 445.220 | 426.400 |
| LRT (shrunk) | 0.825 | 0.104 | 412.480 | 48.160 |
| Fisher | 0.966 | 0.758 | 483.240 | 1511.720 |
| χ 2 | 0.965 | 0.754 | 482.440 | 1482.820 |
| CircTest | 0.628 | 0.167 | 313.780 | 63.180 |
We again apply all DE methods to identify DE circRNAs with considering host genes in the pairwise comparisons among three brain regions. The number of DE circRNAs (FDR < 0.05) is demonstrated in Table 5. We observe that the overall trends are consistent with the results of the simulation study. χ2 and Fisher’s exact test detect most DE circRNAs. Wald test with shrinkage detects more DE circRNAs than naive Wald test in all three comparisons. With or without shrinkage, LRT detects more DE circRNA than Wald test. Moreover, CircTest detects the fewest DE circRNAs out of all three comparisons. Especially, CircTest only detects six DE circRNAs between frontal cortex and diencephalon, and one DE circRNAs between cerebellum and diencephalon, respectively.
Table 5.
Real data DE results for pairwise comparisons between three brain tissues
| Wald | Wald (shrunk) | LRT | LRT (shrunk) | Fisher | χ 2 | CircTest | #circRNA | |
|---|---|---|---|---|---|---|---|---|
| F versus C | 2071 | 2734 | 2818 | 2826 | 5695 | 5329 | 1418 | 19 796 |
| F versus D | 880 | 1114 | 1238 | 1257 | 3547 | 3241 | 6 | 20 001 |
| C versus D | 71 | 87 | 184 | 135 | 1206 | 1087 | 1 | 11 516 |
F, frontal cortex; C, cerebellum; D, diencephalon.
We plot the distribution of Wald test statistic with and without shrinkage and corresponding FDR distribution in the comparison between frontal cortex versus cerebellum (Fig. 4A and B). We also plot the distribution of LRT statistic with or without shrinkage and corresponding FDR distribution (Fig. 4C and D). We find that Wald test statistic approximates to a normal distribution and LRT statistics approximates to a χ2 distribution. As expected, either Wald test or LRT with shrinkage achieves an overall lower FDR estimate than their naive counterparts without shrinkage. In addition, we do the same plots for frontal cortex versus diencephalon comparison and cerebellum versus diencephalon comparison (Supplementary Figs S6 and S7) and observe similar trends.
Fig. 4.
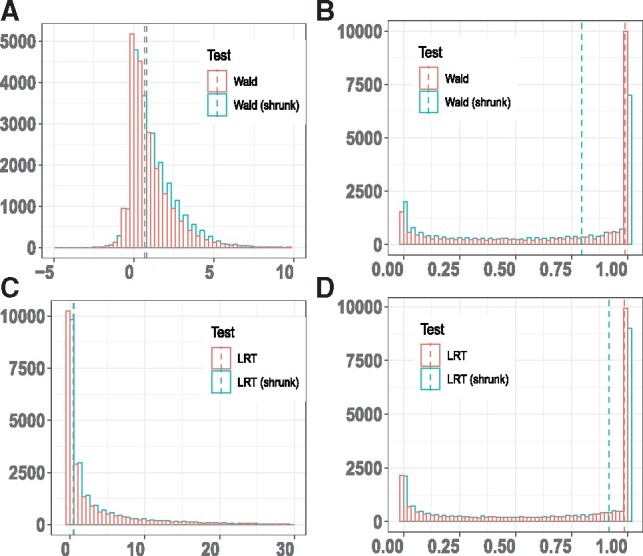
Differential expressed circRNA analysis between frontal cortex and cerebellum. (A, B) Comparison of statistics distribution and FDR distribution between Wald test versus Wald test (shrunk); (C, D) Comparison of statistics distribution and FDR distribution between LRT versus LRT (shrunk). The median of test statistics or FDR is indicated by a dashed vertical line
From both the simulation study and the real data analysis, we find that our proposed test statistics outperform other methods by achieving a much better FDR control. Between Wald test with shrinkage and LRT with shrinkage, we find that LRT with shrinkage obtains a higher power at the cost of inflated FDR. We include both tests in circMeta and users can choose either of them depending on the need.
4 Discussion
In this work, we develop circMeta, a unified and comprehensive computational workflow for state-of-the-art circRNA analyses. circMeta contains several key modules for circRNA analyses: identifying and categorizing circRNA based on back-splicing junction reads from rRNA-depleted RNA-seq data; annotating various functional genomic features including intron length, repetitive elements and A-to-I editing that play a critical role in the circRNA biogenesis; calculating the competition score to provide the likelihood for flanking intron pairing and circRNA formation; calculating linear read counts for back-splicing sites and corresponding CLR; and performing DE analysis with or without considering host genes.
Through real data analysis on three distinctive brain regions using three of the most widely used circRNA prediction methods, we consistently find that intron length, number and competition score for Alu or SINE, as well as CLR, are positively correlated with back-splicing junction reads, confirming their critical roles in circRNA biogenesis. These functional genomic features could be used to integrate with junction reads for better predicting, quantifying and validating circRNAs in vivo. The information could also serve as key targets to experimentally modulate circRNA expression, such as mutating or deleting critical Alu elements or inosine sites to affect endogenous circRNA formation.
As the number and back-splicing sites of circRNAs vary dramatically compared to mRNA, the current DE methods designed for mRNA expression might not be optimal to identify DE circRNAs. To address this, we first merge circRNAs identified in different conditions before carrying out DE analysis, and we develop DE methods for circRNAs with or without considering host genes, respectively. Without considering the host genes, we develop a data-driven two-stage DE approach for circRNAs based on circular junction reads: we first check the GOF of the junction reads under the assumption of Poisson distribution and NB respectively and adopt an appropriate DE method with the distribution assumption of overall better GOF. The two-stage DE approach is suitable to quantitatively detect circRNA levels and study their downstream functions, such as miRNA sponge. Since we find that circular junction reads in DE circRNAs in all three brain regions fit a Poisson distribution better than NB, we adopt a Poisson-based z-test for both simulation and real data analysis. In the simulation study, we find that Poisson-based z-test outperforms other methods by obtaining high power and better FDR control compared to standard DE methods for mRNAs such as edgeR and DESeq2 that are based on the assumption of NB. The results of the real data analysis are consistent with the simulation study: our approach identified the most DE circRNAs. Moreover, host genes bearing DE circRNAs identified by Poisson-based z-test are enriched in GO terms related to brain development and functions and interact with potential miRNAs. It should be noted that we do not exclude the possibility that NB may fit better in other datasets when NB-based test such as edgeR may be more powerful.
A great number of circRNAs have a low CLR (e.g. CLR < 1 or CLP < 0.5), indicating the parental linear mRNA are dominant, with a small amount of circRNAs being produced. In contrast, many circRNAs show a high value of CLR (e.g. CLR > 1 or CLP > 0.5) suggesting that the levels of circRNAs are even higher than their host genes. This observation indicates that circular junction reads do not fully represent the CLR, particularly when a high level of circRNAs is produced in a given loci. With this consideration of the host genes, we adopt a Bayesian hierarchical model to perform DE analysis based on CLR. By adopting a similar shrinkage strategy used in detecting differential methylation loci from single nucleotide resolution DNA methylation data, we find both Wald test and LRT with shrunk variance estimate achieve better FDR control than their naive counterparts without shrinkage in the simulation. Moreover, the proposed approach outperforms CircTest, that is the only published DE method for circRNAs based on CLR, by achieving higher power and better FDR control. Furthermore, Fisher’s exact test and χ2 test suffer significant loss of FDR control and thus are undesirable. The DE approach using CLR could provide important insights on spatial and temporal regulation of circRNA production. circMeta is the first to offer two separate, fully optimized algorithms to determine DE of circRNAs.
A recent study (Hansen et al., 2016) performed a systematic comparison of circRNA prediction tools, and concluded that combining any two algorithms would greatly decrease the false positive rate and in general strengthen the output quality. To reduce the false positives generated from individual circRNA prediction algorithm, we recommend using the common circRNAs predicted by multiple circRNA prediction methods as input to circMeta, as performed in this study.
Public research database consortia provide an unprecedented opportunity for researchers to explore the prevalence and functional roles of circRNAs in different diseases. For example, Accelerating Medicines Partnership—Alzheimer’s Disease (AMP—AD) is a multi-institutional, multidisciplinary project and publicly accessible database funded by National Institute of Aging that offers a wide cohort studies of both normal control subjects and severe terminal AD patients with corresponding rRNA-depleted RNA-seq datasets. Other consortia such as TCGA and ENCODE provide a rich collection of rRNA-depleted RNA-seq datasets across various cancer types. By utilizing the datasets from these consortia, researchers could apply circMeta to investigate the genomic features that foster RNA circulation formation and detect the cancer-specific DE circRNAs or AD-associated circRNAs between normal subjects and disease patients, which may offer additional insights into the onset and progression of AD or cancer.
In conclusion, we provide an integrative computational framework for analyzing circRNAs. The proposed framework could be easily extended to include more functional genomic features such as N6-methyladenosine (m6A) and more complex study designs in DE analysis. In our future work, we plan to extend our model from a two group comparison to multifactorial experimental designs, to include more genomic features, and to integrate genomic features with circular junction reads for better predicting and quantifying circRNAs.
Supplementary Material
Acknowledgement
We thank Dr Peng Jin from Emory University for providing human fetal cortex and cerebellum samples.
Funding
This work was supported by the National Institutes of Health [R01 MH117122 to L.C. and B.Y.]. B.Y. is supported by Emory Alzheimer’s Disease Research Center pilot grant.
Conflict of Interest: none declared.
Contributor Information
Li Chen, Department of Health Outcomes Research and Policy, Auburn University, Auburn, AL 36849, USA.
Feng Wang, Department of Human Genetics, Emory University School of Medicine, Atlanta, GA 30322, USA.
Emily C Bruggeman, Department of Human Genetics, Emory University School of Medicine, Atlanta, GA 30322, USA.
Chao Li, Department of Health Outcomes Research and Policy, Auburn University, Auburn, AL 36849, USA.
Bing Yao, Department of Human Genetics, Emory University School of Medicine, Atlanta, GA 30322, USA.
References
- Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Methodol., 57, 289–300. [Google Scholar]
- Chen L.L. (2016) The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol. Cell Biol., 17, 205–211. [DOI] [PubMed] [Google Scholar]
- Chen S. et al. (2019) Widespread and functional RNA circularization in localized prostate cancer. Cell, 176, 831–843. [DOI] [PubMed] [Google Scholar]
- Cheng J. et al. (2016) Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics, 32, 1094–1096. [DOI] [PubMed] [Google Scholar]
- Feng H. et al. (2014) A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res., 42, e69.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y. et al. (2015) CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol, 16, 4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen T.B. et al. (2016) Comparison of circular RNA prediction tools. Nucleic Acids Res., 44, e58.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E.J., Reichardt L.F. (2001) Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci., 24, 677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. et al. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol., 14, R36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiran A., Baranov P.V. (2010) DARNED: a database of RNA editing in humans. Bioinformatics, 26, 1772–1776. [DOI] [PubMed] [Google Scholar]
- Langdon W.B. (2015) Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min., 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.H. et al. (2014) starBase v2.0: decoding miRNA–ceRNA, miRNA–ncRNA and protein–RNA interaction networks from large-scale clip-seq data. Nucleic Acids Res., 42, D92–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. et al. (2018) The biogenesis, functions, and challenges of circular RNAs. Mol. Cell, 71, 428–442. [DOI] [PubMed] [Google Scholar]
- Liang D. et al. (2017) The output of protein-coding genes shifts to circular RNAs when the pre-mRNA processing machinery is limiting. Mol. Cell, 68, 940–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I. et al. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S. et al. (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature, 495, 333–338. [DOI] [PubMed] [Google Scholar]
- Nicolet B.P. et al. (2018) Circular RNA expression in human hematopoietic cells is widespread and cell-type specific. Nucleic Acids Res., 46, 8168–8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro J.M. et al. (1991) Scrambled exons. Cell, 64, 607–613. [DOI] [PubMed] [Google Scholar]
- Piwecka M. et al. (2017) Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science, 357, eaam8526.. [DOI] [PubMed] [Google Scholar]
- Ramaswami G., Li J.B. (2014) RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res., 42, D109–D103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D. et al. (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak-Wolf A. et al. (2015) Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell, 58, 870–885. [DOI] [PubMed] [Google Scholar]
- Salzman J. et al. (2013) Cell-type specific features of circular RNA expression. PLoS Genet., 9, e1003777.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T. et al. (2015) Transcriptome-wide identification of adenosine-to-inosine editing using the ice-seq method. Nat. Protoc., 10, 715–732. [DOI] [PubMed] [Google Scholar]
- Szabo L., Salzman J. (2016) Detecting circular RNAs: bioinformatic and experimental challenges. Nat. Rev. Genet., 17, 679–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. et al. (2007) The ENCODE project at UC Santa Cruz. Nucleic Acids Res., 35, D663.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos I.S. et al. (2015) DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res., 43, W460–W466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo J.N. et al. (2019) The landscape of circular RNA in cancer. Cell, 176, 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westholm J.O. et al. (2014) Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep., 9, 1966–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz J.E. (2018) A 360 degrees view of circular RNAs: from biogenesis to functions. Wiley Interdiscip. Rev. RNA, 9, e1478.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.H. et al. (2011) starBase: a database for exploring microRNA–mRNA interaction maps from Argonaute clip-seq and degradome-seq data. Nucleic Acids Res., 39, D202–D209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.O. et al. (2014) Complementary sequence-mediated exon circularization. Cell, 159, 134–147. [DOI] [PubMed] [Google Scholar]
- Zhou C. et al. (2017) Genome-wide maps of m6a circRNAs identify widespread and cell-type-specific methylation patterns that are distinct from mRNAs. Cell Rep., 20, 2262–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.