

Abstract
Tau, a microtubule-associated protein that modifies the dynamic properties and organization of microtubules in neurons and affects axonal transport, shows remarkable heterogeneity, with multiple isoforms (45–65 kDa) generated by alternative splicing. A high-molecular-weight (HMW) isoform (110 kDa) that contains an additional large exon termed 4a was discovered more than 25 years ago. This isoform, called Big tau, is expressed mainly in the adult peripheral nervous system (PNS), but also in adult neurons of the central nervous system (CNS) that extend processes into the periphery. Surprisingly little has been learned about Big tau since its initial characterization, leaving a significant gap in knowledge about how the dramatic switch to Big tau affects the properties of neurons in the context of development, disease, or injury. Here we review what was learned about the structure and distribution of Big tau in those earlier studies, and add contemporary insights to resurrect interest in the mysteries of Big tau and thereby set a path for future studies.
Tau as a Microtubule-Associated Protein
Microtubules are a key component of the cellular cytoskeleton and, in neurons, they provide the framework for neuronal structure, while also acting as substrates for motor proteins that regulate intracellular trafficking. Intracellular transport also occurs in dendrites, but is particularly well studied in axons, where the nearly uniform polarity orientation of the microtubules allows for the bidirectional regulation of cargo movements in properly functional neurons, across all neuronal dimensions. The dynamic properties and organization of microtubules are mediated by post-translational modifications of their tubulin subunits as well as by interactions with an array of regulatory proteins, including classic fibrous microtubule-associated proteins, such as tau [1]. Tau was originally isolated on the basis of its ability to bind microtubules and promote their assembly through repeated microtubule-binding domains (R1–4). Recent studies confirm the binding of tau to microtubule protofilaments [2], but call into question the commonly touted role of tau as a microtubule stabilizer [3]. Tau has long been considered a microtubule stabilizer in the axon because of studies showing that it can stabilize microtubules when in the test tube or when overexpressed in cells [4–6]. More recent work suggests that tau, by outcompeting genuine microtubule stabilizers, enables axonal microtubules to have long labile domains [3]. Other studies have shown that tau is involved in mediating axonal transport, synaptic structure, and signaling pathways in neurons [1]. Tau is also a contributor to neurodegenerative diseases, including Alzheimer’s disease (AD), and other ‘tauopathies’, such as frontotemporal dementia, in which tau forms insoluble toxic aggregates and fibrils (e.g., neurofibrillary tangles) [7,8].
The Structure of Tau
In both health and disease, tau displays remarkable heterogeneity from a well-characterized 6-kb transcript corresponding to the MAPT gene. Genebank has a list of 203 tau sequences from mammals, birds, reptiles, amphibians, and bony fishi. As the result of alternative splicing, there are six developmentally expressed isoforms containing different numbers of repeats (three or four related to exon 10) of the microtubule-binding domain as well as the presence or absence of exons 2 and 3 (Figure 1A). Approximately 25 years ago, a HMW tau isoform of ~110 kDa (termed ‘Big tau’) was discovered in the adult PNS, optic nerve, and cell lines derived from neural crest [9,10]. The cloning and publication of Big tau sequence, distribution, and developmental regulation, described herein, are from work in mouse and rat as well as from cell lines derived from these species. Data on human Big tau isoforms and other species have been derived mostly from genomic analysis based on transcript alignments with the exception of Xenopus (discussed later). Big tau is encoded by an 8-kb transcript that contains an additional exon (termed 4a), the expression of which begins late during embryonic development and gradually increases postnatally [9,11]. Variants of Big tau transcripts exist with or without exon 6 (Figure 1B), as evidenced by a predominant isoform of 110 kDa and another of 90–95 kDa in some CNS regions, including the retina and optic nerve [12,13]. Big tau is sometimes referred to as HMW tau, but there is value in avoiding that term because it has also been used to describe aggregates of tau [14].
Figure 1. Structure of (A) Low-Molecular-Weight (LMW) Tau and (B) Big Tau.
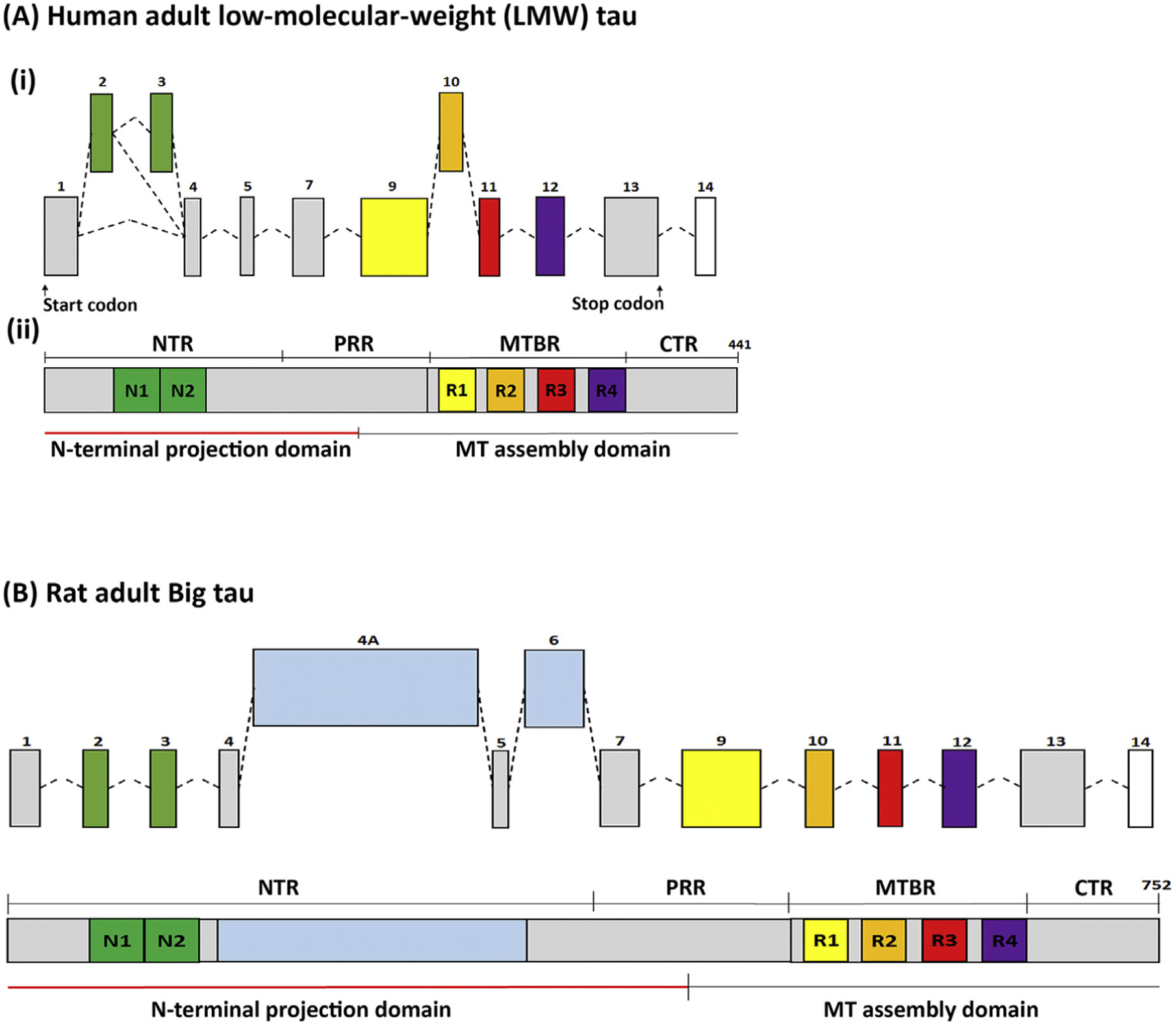
(A) (i) The transcriptional organization of LMW human tau with exons 2/3 (N1 and N2 at the N-terminal) and exon 10 (R2 at the microtubule binding domain) as alternative splicing exons, which are developmentally regulated. LMW tau lacks exons 6 and 8. These transcriptional events give rise to 6 LMW isoforms. (ii) The largest 4R isoforms, with both N1/N2 comprising 441 amino acids, show on Western blots with apparent MW of 48–67 kDa (the true MW being 37–46 kDa). The tau protein comprises the microtubule (MT)-assembly domain, which includes the C-terminal region and the four microtubule-binding repeats (R1–4) and the N-terminal projection domain, including the N-terminal N1 and N2 exons and part of the proline-rich region. (B) Big tau (structure shown for rat) is an isoform that includes exons 4a and alternatively spliced exon 6, expressing proteins of apparent MW of 110 kDa. Big tau has a similar microtubule-assembly domain to LMW tau, but has an N-terminal projection domain that is 510 aa, which is more than double the size relative to the projection domain of LMW tau. Abbreviations: CTR, C-terminal region; MTBR, MT-binding region; NTR, N-terminal region; PRR, proline-rich region.
Over two decades ago, polyclonal antibodies that recognize the entire 4a exon region specific to Big tau (Figure 2) were generated and used to determine the cellular localization of Big tau during development and in the adult CNS [15]. In addition to confirming expression in PNS neurons and the optic nerve, these studies showed that, in rat, Big tau is expressed in almost all CNS neurons that extend processes into the PNS, including spinal motor neurons, almost all cranial nerve motor nuclei (e.g., oculomotor, trochlear, trigeminal, abducens, facial, glossopharyngeal, vagus, and hypoglossal), and central processes of most sensory ganglia, but is absent from bipolar neurons of the olfactory, vestibular, and spiral ganglia. In subsequent studies, expression and selective distribution of Big tau in dorsal root ganglia (DRG) were shown during development and regeneration [16,17].
Figure 2. Distribution of Big Tau in the Adult Nervous System.
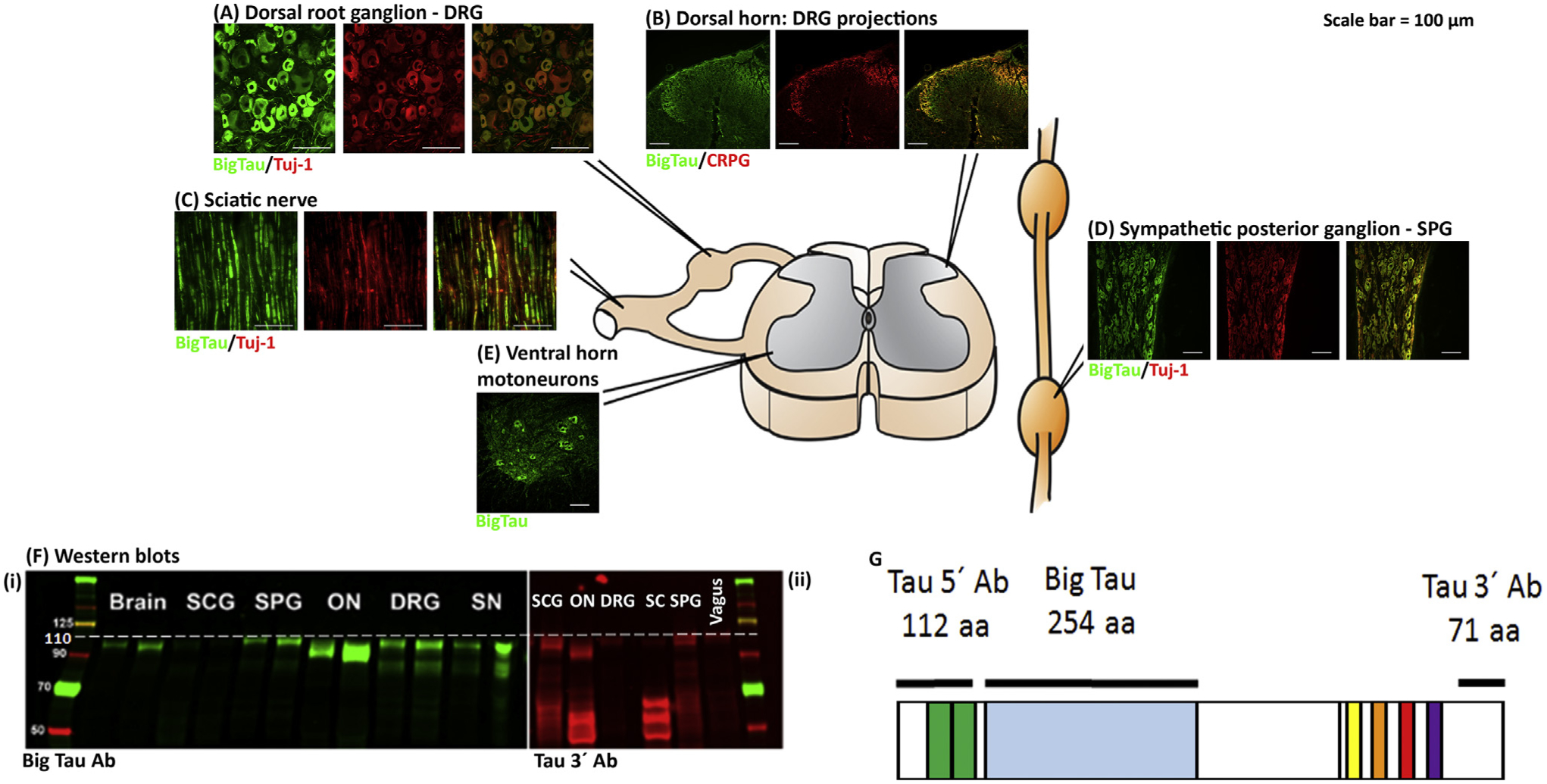
The images show the distribution of Big tau in the adult rat nervous system, consistent with published data [15,16], assessed using antibodies specific to Big tau. Staining of dorsal root ganglia (DRGs) (A) with the Big tau antibody compared with Tuj-1 (neuronal marker) shows expression of Big tau in small and medium-sized neurons, but not the large ones. (B) and (D) show the expression of Big tau in the spinal cord, with intense staining of motor neurons and their dendrites (D) as well as the dorsal horn (B), where the calcitonin gene-related peptide (CGRP)-positive central processes of DRG axons project and terminate. (C) Big tau expression in sciatic nerve colocalized with Tuj. Analysis of the sympathetic chain of ganglion cells (E) shows an example of the superior posterior ganglion (SPG), where all neurons appear to express Big tau. Not shown but discussed in the main text is the expression in brain, where all cranial nerves express Big tau with the exception of olfactory, vestibular, and spiral ganglia [15]. (F) Western blot results of different tissues from peripheral and central nervous systems (PNS and CNS) using an antibody specific to Big tau (i) as well as tau 3′ antibody (ii). (G) Specificity of Big tau antibodies [prepared against the entire 4a exon of 254 amino acids (aa) of rat tau], which confirms the expression of Big tau using Western blot (Fi) in SPG, optic nerve (ON), DRGs, and sciatic nerve (SN). The pattern of Big tau expression was verified with the tau 3′ antibodies (prepared against 71 amino acids at the C terminus), which recognize all tau isoforms (Fii). Images courtesy of Ying Jin and Theresa Connors.
The Challenges of Big Tau
Almost 25 years have elapsed with little progress since the original descriptive studies on Big tau. This leaves an astonishing gap in the progression of knowledge about an important neuronal protein that essentially doubles in size in specific neuronal systems, including sensory and motor neurons, with many questions remaining. Does the unique insert of Big tau alter its functions relative to the other isoforms of tau that lack the insert? Is the Big tau isoform, similar to other isoforms, modified by phosphorylation events that define a heterogeneous population of the protein with different developmental and distributional profiles? What is it about certain neurons and axonal projections (and not others) that benefit from such a dramatic switch in tau isoform profile? What can be learned about the properties of these neurons during: (i) nervous system development; (ii) normal physiological conditions of adult life; (iii) regeneration following injury; and (iv) degenerative disorders? Here, we review what was learned about Big tau in those earlier studies with respect to its structure and distribution, and add contemporary insights into potential mechanisms and function, to resurrect the mysteries of Big tau, and thereby establish a path forward for future work.
‘Conventional’ Low-Molecular-Weight Tau: A Short Summary
The structural and functional properties of tau have been discussed in recent reviews [1,3], as has the involvement of tau in neurodegenerative disorders, such as AD [8,18]. Human tau is encoded by a single gene located on chromosome 17 with multiple isoforms that are generated by alternative splicing to form six isoforms of 352–441 amino acid (aa) residues [19] and apparent MW of 48–67 kDa (the true MW being 37–46 kDa). Developmental studies of tau expression in human brain have shown major changes in the profile of the protein from two to three immature isoforms at early postnatal stages to five to six isoforms in the adult (Figure 1A), with similar changes also observed in rodents. Immature tau lacks exons 2 and 3, with exon 3 only expressed together with exon 2. The maturation process results in a switch from three to four repeats of the microtubule-binding domains (3R and 4R, respectively) located near the C terminus of tau [20]. There are also variants in the N-terminal sequence, which define part of the projection domain of tau, with immature tau lacking the expression of exons 2 and 3. The additional repeat domain in 4R tau confers a higher binding affinity to microtubules relative to 3R tau, and may have a role in promoting microtubule assembly by binding or bridging the interfaces of tubulin heterodimers [4]. Exons 2 and 3 and the N-terminal region may regulate the subcellular distribution of tau into axons and mediate the interaction of the projection region with other proteins (e.g., annexin, synapsin 1, and the dynactin complex) (Figure 3, Key Figure). These exons may also have a role in tau aggregation [21,22]. Accordingly, the subcellular distribution of tau is regulated during development, becoming enriched in axons as neurons establish their polarity via mechanisms that may include isoform specificity, local synthesis of tau in axons, preferential binding of tau to axonal microtubules, and/or preferential axonal transport of the protein [1,3,23,24]. Isoform expression of tau is regulated by splicing factors, for example, with exon 10 usage regulated by CDC2-like kinases CLK1, 2, 3, and 4 that phosphorylate proteins that regulate pre-mRNA splicing [25]. Tau has also been cloned and sequenced in other species, including mouse [26] and rat [27], where it shows a high degree of amino acid homology with human tau [28]. However, the occurrence of each of the six isoforms is variable; species that are phylogenetically related express a similar pattern of isoforms. Indeed, the unique expression pattern of tau isoforms in the human CNS may be related to its vulnerability to neurodegenerative disorders with tauopathies. [29].
In addition to splice variants, tau exhibits complex post-translational modifications, of which phosphorylation has been best studied, with hyperphosphorylation being associated with aberrant tau [8]. Other modifications include glycosylation, acetylation, nitration, methylation, prolyl-isomerization, ubiquitylation, sumoylation, and glycation. These modifications can affect the ability of tau to bind to microtubules, as well as its turnover, localization, and aggregation into pathological structures, such as paired helical fibrils and neurofibrillary tangles [30].
Big Tau: The Early Studies
An early indication of Big tau came during the 1980s, when rodent sympathetic neurons in culture were shown to express a HMW isoform in addition to the LMW variants [31,32]. Similar HMW isoforms were also described in PC12 cells [10,33] and N115 neuroblastoma cells [34,35]. Subsequent studies reported that, in mature rat DRGs, only one tau isoform was expressed, and this protein (110 kDa) was considerable larger in size than the predominant tau isoforms found in the brain [9]. The size of mRNA encoding Big tau in DRGs was also larger, 8 kb compared with 6 kb. Developmental studies showed that early postnatal DRGs in rat express both the 6-kb and 8-kb transcripts corresponding to LMW and Big tau, respectively. By postnatal day 7 (in rat) only Big tau is present and persists throughout adulthood in DRGs and their axons (sciatic nerve and the central process projecting to the dorsal horn of the spinal cord). Analysis of tau from the optic nerve showed a complex expression of tau isoforms that included the LMW, a middle MW of ~95 kDa and small amounts of Big tau [12]. Subsequent studies confirmed that these proteins are produced in retinal ganglion cells [13] and transported into the optic nerve (Figure 2).
Key Figure
Tau Functional Domains.
Figure 3.
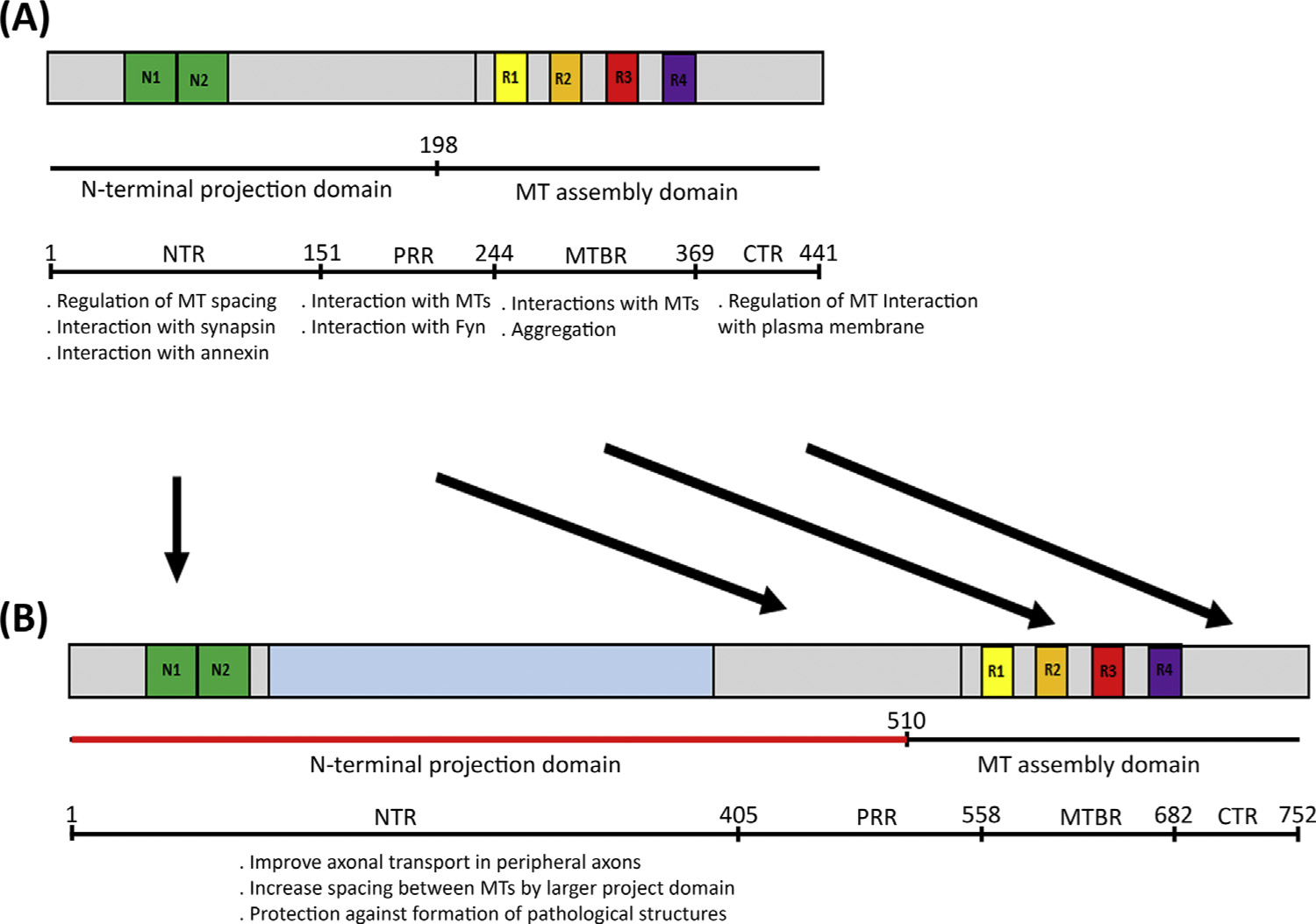
The figure shows a partial list of known functions ascribed to different domains of the tau molecule: N-terminal region (NTR), proline-rich region (PRR), microtubule-binding region (MTBR), and C-terminal region (CTR). These same domains are present in low-molecular-weight (LMW) tau (A) and Big tau (B). The knowledge of these various other domains underscores the unknown function of the large 4a insert of the Big tau molecule. Speculated in (B) are potential functional modifications in the molecule due to the unique presence in Big tau of the large 4a insert in the projection domain. Abbreviation: MT, microtubule.
Big Tau Cloning and Sequencing
The discovery of HMW isoforms at the protein level in the PNS and some cell lines during the early 1990s eventually led to the cloning of Big tau from rat DRGs [11]. The analysis of the sequencing data showed that the corresponding protein contains a sequence identical to the previously cloned LMW tau but with an additional 254-aa insert in the amino-terminal half in rat and 237 aa in mouse encoded by an exon termed 4a (Figure 1B). This sequence was identical to the HMW tau obtained from rat PC12 cells, but different from the sequence of a N115 neural crest cell line, which, in addition to the 4a exon, also contained exon 6, which is not found in DRGs [34]. The variability of exon 6 expression in Big tau present in DRGs, spinal cord, brain and neural cell lines is not entirely clear because Western blots show a similar band at 110 kDa with the exception of the optic nerve, where the Big tau isoform is at 95 kDa (Figure 2). These studies need to be revisited to establish the isoform profile of Big tau with respect to exon 6 as well as other exons, such as 2/3 [35].
Big tau is produced from an 8-kb mRNA generated by alternative splicing from the same gene that encodes LMW tau. Production of Big tau from the cloned sequence generates a protein with an apparent molecular mass of 110 kDa that aligns on SDS/PAGE with Big tau protein extracted from peripheral ganglia. RNA blots show that, in peripheral ganglia from adult rats, only the 8-kb mRNA band corresponding to Big tau is found, whereas, in ganglia from newborn rats, both 6-kb and 8-kb tau mRNA transcripts are found. Several reports have also noted small amounts of Big tau in the spinal cord and brain [12,36], which were initially detected only by PCR, but as discussed in subsequent sections, using antibodies specific to the 4a exon allowed researchers to define the specific cells and nuclei that express Big tau [11–13]. The expression of multiple tau isoforms in the optic nerve appears to be complex, but what is clear is the expression of a Big tau isoform containing the large 4a exon, and that the source of this protein is its expression by retinal ganglion cells [12,13].
Big Tau: Distribution in PNS and CNS
After Big tau was cloned in rat, the insert corresponding to exon 4a, which contains 254 aa, was used in its entirety to generate highly specific polyclonal antibodies against Big tau [15]. Polyclonal antibodies were also generated using 112 aa of the N-terminal (tau 5′) and 71 aa of the C-terminal (tau 3′) regions to detect all isoforms of tau [31] (Figure 2). The specificity of the Big tau antibody was demonstrated by Western blotting, which revealed only the 110-kDa protein (e.g., at high levels in DRGs and sciatic nerve and low levels in spinal cord and brain), while the general tau antibodies detected both Big tau and the LMW tau isoforms. The distribution of Big tau was probed by immunohistochemistry in the rat CNS and PNS (Figure 2) by comparing the staining profile of these two sets of antibodies. Big tau was found in the soma, dendrites, and axons of neurons that extended processes into the periphery [15]. In the developing spinal cord, Big tau was first expressed in the central projections of the DRG neurons and in motor neurons at embryonic day (E)18 and postnatal day 2, respectively, with the switch from LMW tau to Big tau complete by the second week postnatally. In the adult rat CNS, almost all neurons that extended processes into the PNS were shown to express Big tau, including all cranial nerve motor nuclei and central processes of most sensory ganglia; of these ganglia, only the bipolar neurons of the olfactory, vestibular, and spiral ganglia did not express Big tau [15]. Retinal ganglion cells are the only CNS neurons the processes of which remain entirely within the CNS that were shown to express Big tau. Big tau was not detected in other regions of the brain, including cerebral cortex and thalamus [15]. In the PNS, low levels of Big tau were observed in some E18 DRG neurons, with levels of Big tau increasing significantly by P0 and reaching high levels in adults in a subset of small and medium-sized neurons corresponding to DRGs that express calcitonin gene-related peptide (CGRP) and substance P [16]. Analysis of tau expression and transport within retinal ganglion cells revealed in optic axons a set of 50–60-kDa tau isoforms and a set of 90–95-kDa isoforms, which were shown to contain the domain of tau encoded by exon 4a, likely the form of Big tau without exon 6 [13]. Analysis of protein transport along optic axons showed that all detected tau isoforms, including Big tau, moved at a rate of 0.2–0.4 mm/d, while other microtubule-associated proteins (e.g., MAP1A) moved three- to five-fold more rapidly. By contrast, tubulins advanced at 0.1–0.2 mm/d, significantly more slowly than tau or other microtubule-associated proteins [12]. These studies established that tau is not cotransported with tubulin or microtubules in a simple 1:1 fashion, indicating that associations of tau with microtubules within axons are dynamic. For example, tau may be transported together with the fastest-moving microtubules [37], perhaps even being a factor in those rapid transit rates. Taken together, these results underscore the heterogeneity of tau expression in retinal ganglion cells and DRGs, with different complements of tau isoforms in specific subpopulations of neurons.
Big Tau: Function?
The functional reason for the switch from LMW tau to Big tau in specific neuronal populations is still unknown. Axons of these populations have in common that they often project to the periphery and are some of the longest in the body. Oblinger’s group reported that immature rat DRGs initially express both Big tau and LMW tau [9], with Big tau becoming prominent by postnatal day 7 and in the adult. They also examined the level of Big tau protein in adult DRGs after sciatic nerve crush at different time points and found that Big tau levels decreased. They speculated that Big tau has a role in stabilizing the mature axonal cytoskeleton compared with LMW tau, which is more conducive to axonal growth. By immunohistochemical staining, Fischer’s group examined the distribution of Big tau immunoreactivity in rat DRGs and sciatic nerves at different time points after peripheral nerve axotomy [16]. They found no changes in selective expression of Big tau during axonal regeneration and, hence, concluded that the distribution and expression of Big tau during axonal regeneration in the adult does not recapitulate the developmental pattern. Nevertheless, it is provocative to consider the possibility that most CNS neurons do not express Big tau so that their axons can remain comparatively dynamic relative to the axons of neurons that eventually express only Big tau, which are more vested in maintaining long-range connections as they project to the periphery.
Interestingly, Big tau is expressed in Xenopus, but the amino acid sequence corresponding to the exon 4a insert of the same size has only 22% sequence identity to that of its mammalian counterpart. Moreover, unlike mammalian Big tau with its two different isoforms, Xenopus Big tau has at least four isoforms with or without exons 4 and 6 [38]. By contrast, the microtubule-binding domains have 75–83% sequence identity between Xenopus and mammals, which may indicate that the Big tau insert is not a functional domain in the sense of interacting with other proteins or structures, but rather simply endows greater length to the projection domain. Nevertheless, this may have functional consequences, as indicated by experiments showing that increasing the expression of Big tau, but not the smallest tau isoform, in frog enhanced axonal outgrowth [38]. The low sequence identity of exon 4a, which defines Big tau, means that identification and analysis of Big tau in other species using the antibodies against the rat 4a domain is not possible.
Perhaps switching to Big tau in neurons with long or high caliber axons is associated with the amplified need for robust and efficient axonal transport (Figure 4). Analysis of tau interactions with microtubules suggest that the N-terminal domain projects away from microtubules (represented by the first 198 aa), acting as a spacer between neighboring microtubules or between microtubules and other cellular components. The modified structure of Big tau, which adds another 254 aa, more than doubles the projection domain from 198 aa to 510 aa (Figure 3) and may significantly affect these functions. Indeed, cross-sections of axonal microtubules reveal a larger spacing of ~35 nm when induced by Big tau compared with when induced by LMW tau at ~20 nm, suggesting that the larger spacing lowers the resistance of the axoplasm and, thus, reduces the energy required for transport along the microtubules [39]. Another interesting possibility relates to the phosphatase activating domain (PAD) of tau, which is present on the projection domain near the microtubule–motor interface on LMW tau. Through a multi-step pathway, the PAD can result in local phosphorylation of motor proteins that reduce their activity, and this is believed to be accentuated in pathologies due to the misfolding of tau that constitutively exposes the PAD [40]. In the case of Big tau, the PAD becomes located more distantly from the microtubule–motor interface, eliminating its ability to negatively regulate organelle transport in the axon (Figure 4). Another possibility is that the long relatively flexible projection domain of Big tau is more advantageous to the movement of large organelles, such as mitochondria, than the short project domain of LMW tau, which is stiffened by its phosphorylation [41]. Clues to the evolution of dramatic structural changes in tau structure and their functional implications may be derived from parallels to (but also differences from) MAP2, with the LMW isoform of MAP2C switching during development to the HMW isoforms of MAP2A/B (Box 1).
Figure 4. Putative Models of Tau Interactions with Microtubules (MT).
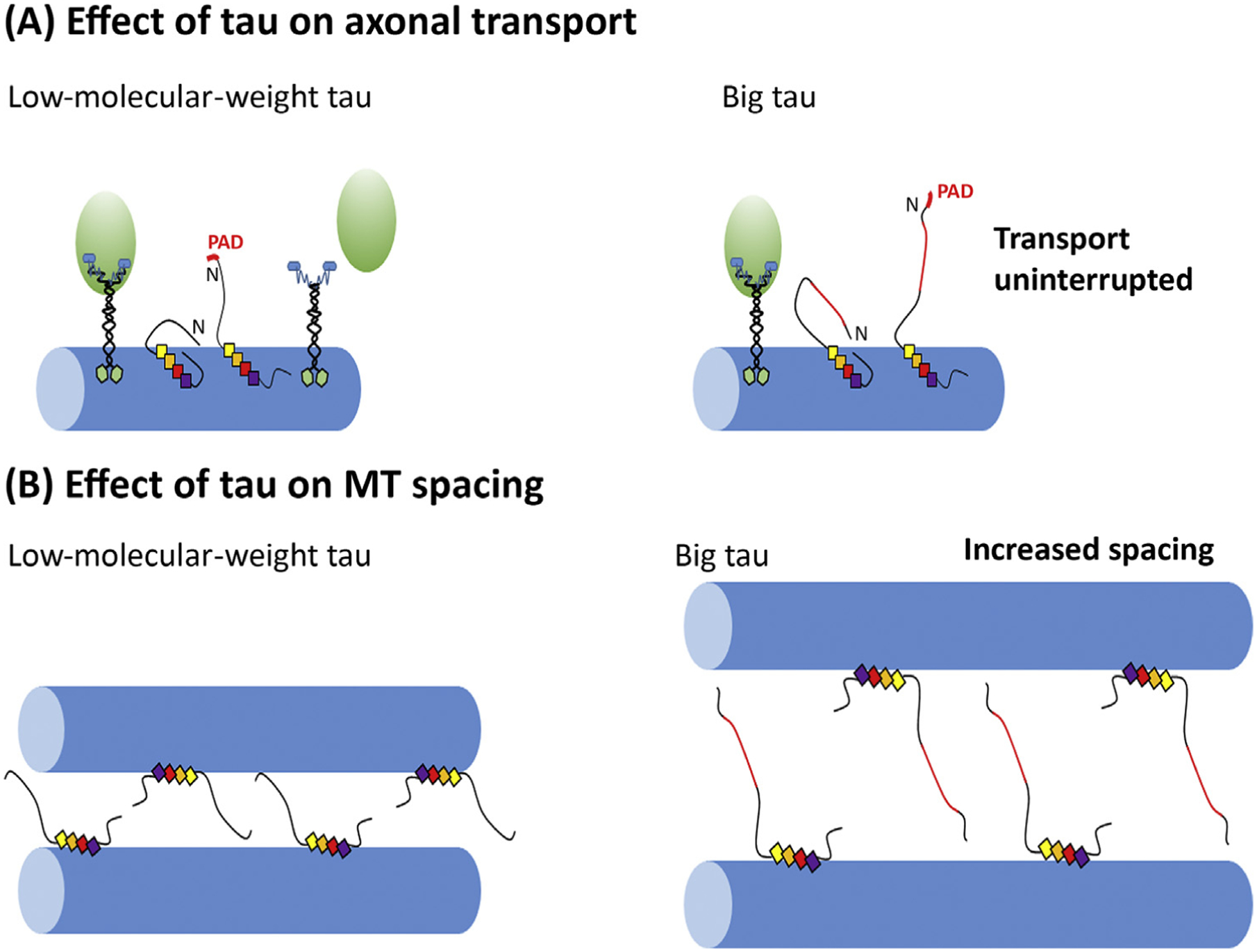
(A) A putative model of tau structural changes that affect axonal transport, in this case the interaction of kinesin with the hidden and exposed phosphatase-activating domain (PAD) of tau [40]. (B) A putative model of the effects of tau on microtubule spacing and potential differences in the distances in the case of Big tau compared with low-molecular-weight (LMW) tau.
Box 1. Parallels between Tau and MAP2.
Comparing the isoform structures of tau and MAP2 is potentially informative. Both are members of the same MAP family, together with MAP4, and share a relatively conserved C-terminal domain with microtubule-binding repeats as well as an N-terminal projection domain of varying size [54,55]. More important for present consideration is the similarity in the isoform structure between tau and MAP2, specifically the transition between the LMW and HMW isoforms. For tau, those are the six LMW isoforms (48–67 kDa) and Big tau (110 kDa), whereas, for MAP2, it is the LMW isoform called MAP2C (60 kDa) and the HMW isoforms called MAP2A/B (250 kDa). In both proteins, the increase in size is mediated by additional exon(s) to generate a larger projection domain. For tau, it is exon 4a (~250 aa) and for MAP2 it is exons 9–11 (~1300 aa). In the case of the 4a exon of tau, it is almost entirely devoid of putative phosphorylation sites. In the case of exon 9–11 of MAP2, the density of phosphorylation sites is dramatically lower than the rest of the molecule (~20 over 1300 aa versus 20 over the other 500).
Further analogy is that the LMW isoforms of tau are downregulated during development in the PNS and distinct regions of the CNS (as discussed in the main text) that express only Big tau in the adult; similarly, MAP2C (the juvenile isoform of MAP2 [56]) is downregulated after the early stages of neuronal development and eventually completely replaced by MAP2B and MAP2A [57]. However, as documented in not only mouse brain, but also other species, MAP2C continues to be expressed postnatally only in especially plastic neurons, such as the olfactory bulb [58], whereas LMW tau is the predominant form of tau in most of the CNS throughout life.
During development, tau gradually segregates into axons, while MAP2 is segregated into dendrites. The differential spatial segregation of tau mainly in axons and MAP2 in cell bodies and dendrites is part of the polarity process in neurons, but may also represent similar strategies for modulating microtubule structure and dynamics as well as intracellular transport in the two different compartments. Another difference in the adult distribution is that Big tau expression is limited to the PNS and specific areas of the CNS, while MAP2A/B has a more general distribution in all dendrites.
The similarity of the LMW isoforms of tau (e.g., the longest, 441-aa isoform) and MAP2 (e.g., MAP2C isoform at 551 aa) has been studied. Both proteins have intrinsically disordered structures and transient structural motifs related to their normal physiology and pathology of aggregation and neurodegeneration. The gap in knowledge is related to the similarities in the HMW isoforms of tau (Big tau, exon 4a) and MAP2 (MAP2A/B, exons 17–19). Are there structural similarities between these exons with respect to their primary sequence and/or active phosphorylation sites? Are there similarities in the evolution of the transition from LMW to HMW isoforms with respect to increased size and complexity of the nervous system?
Concluding Remarks and Future Perspectives
Curiously, most reviews on tau over the past decades have either ignored Big tau or have only mentioned it in passing, despite the widespread developmental transition from LMW tau to Big tau in PNS neurons and optic nerve, as well as most CNS neurons that extend processes into the PNS. This is surprising given that tau is one of the most-studied proteins in the nervous system, with hundreds of investigators around the world seeking to break new ground. Structural studies have shown that the native form of tau is of an unfolded protein with a flexible conformation, low level of secondary structure, and a potential to fold by interactions of domains that include charged amino acids. Indeed, biophysical analysis suggests possible folding of tau into a ‘paperclip’ conformation bringing into close proximity the C and N termini (Figures 4 and 5). This can be affected by the abundant phosphorylation sites of the tau protein, but could also theoretically be even more dramatically altered in the case of Big tau, which is almost devoid of potential phosphorylation sites (see later). Investigators using biophysical tools are hereby invited to model the conformational changes resulting from the inclusion of the Big tau insert, with another intriguing possibility being that the insert protects Big tau from pathologically misfolding under conditions that allow for misfolding of the shorter isoforms (Figure 5). Although the core of the misfolded tau protein in pathological states, such as AD, remains unchanged at the microtubule repeat region, the much-increased length of the projection domain of Big tau may nevertheless affect the conformational changes or the formation of the abnormal filament. Indeed, one review has argued (citing the authors’ unpublished data) that Big tau has a lower propensity to polymerize into fibrils compared with LMW variants of tau [42], and it also appears logical that a larger protein would be less able to be released from the neuron and propagate from neuron to neuron, as has been shown of LMW variants of tau [43]. Such a scenario could explain why tauopathies that prominently affect the brain have little or no comparable impact on the PNS, with only relatively rare reports of tau tangles in peripheral neurons [44,45]. If this is the case, could gene therapy be used to expand the size of tau to prevent its toxic misfolding offer a path toward therapy for tauopathy?
Figure 5. Aspects of the Pathology of Low-Molecular-Weight (LMW) Tau that May be Partially or Completely Ameliorated by the Presence of the 4a Insert in Big Tau.
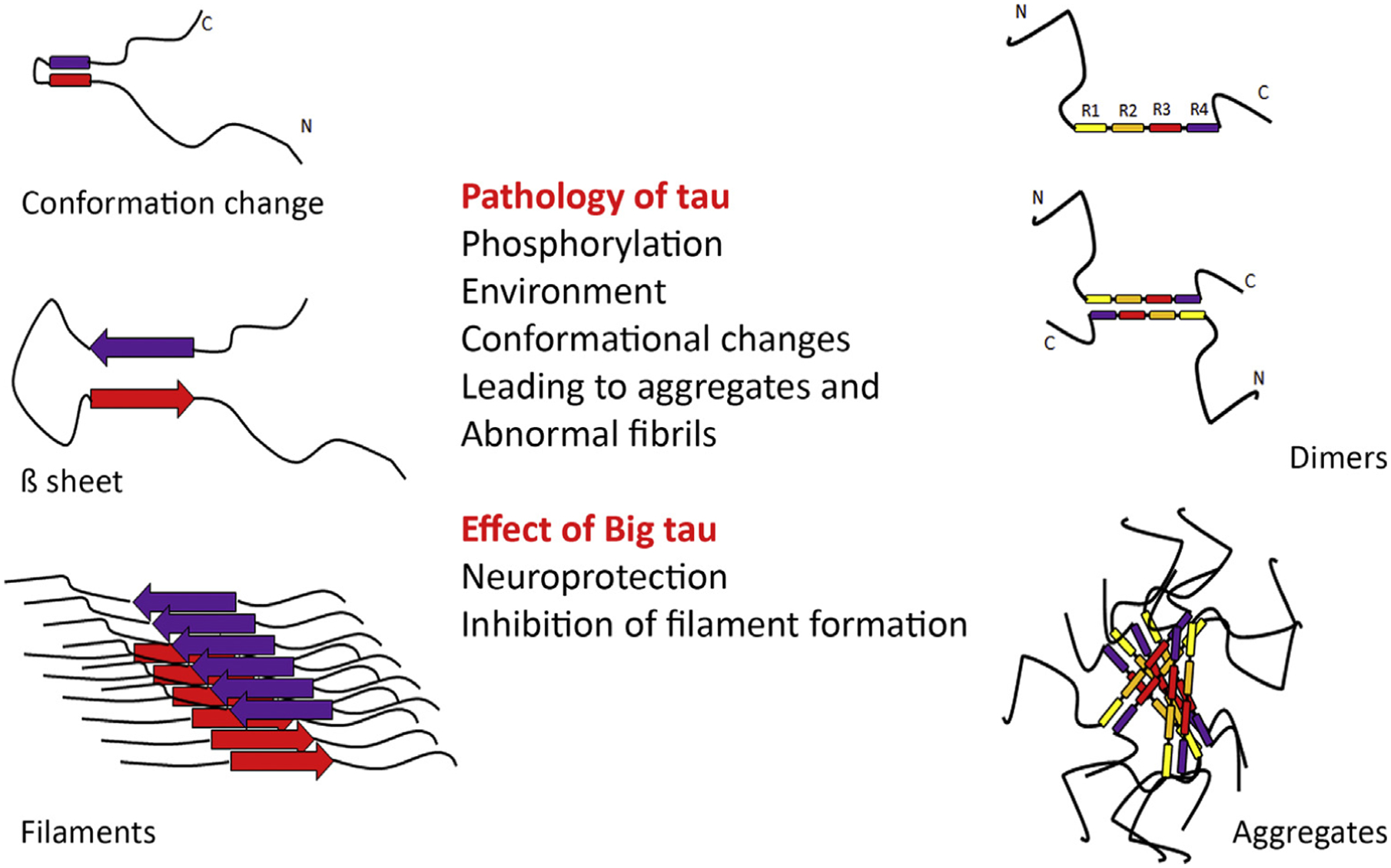
The figure depicts putative models of abnormal fibrils and aggregates generated by conformational changes and misfolding of tau. On the left is shown the conformational transition to β sheets and filaments, and on the right the transition to dimers and aggregates, which are known to be affected by the phosphorylation of tau and environmental factors. The schematic introduces the idea that Big tau may not misfold into pathological filaments or toxic aggregates because of the large insert that appears to be a ‘phospho-dead island’ in a phospho-rich molecule.
Alternatively (or additionally), might Big tau be even more generally neuroprotective to the axon? In the context of the corticospinal tracts, could neuroprotection by Big tau explain why peripheral nerves can acquire such great lengths without succumbing to degeneration in the face of cytotoxic conditions that dramatically impact the long axons that comprise these tracts? This, for example, might explain why hereditary spastic paraplegias that cause adult-onset corticospinal degeneration at all levels, from the lumbar to upper cervical spinal cord [46], have no apparent effect on similarly long peripheral nerves, such as the sciatic nerve, which express Big tau. There might also be important differences between Big tau and the LMW isoforms with regard to: (i) the capacity of tau to interact with other players, such as actin filaments, membrane, or microtubule plus-end tracking proteins (+TIPs); (ii) the competition of tau with other microtubule-associated proteins for interaction with the microtubule lattice; or (iii) as yet undiscovered properties of Big tau not shared by any of the other tau isoforms such as, for example, the capacity to form crosslinks with structures at greater distances from the microtubule (Figures 3 and 4).
Technologies have existed for decades that could have been utilized to make progress on these ideas with relatively straightforward experiments. A DNA construct could have been made to the protein sequence specific to Big tau and expressed in cells as a GFP-fusion to assay for potential interactions with other cellular structures. In addition, LMW tau and Big tau could have been compared in this manner, as could have pull-downs been performed for additional information on protein–protein interactions. In vitro and in vivo studies on protein misfolding, the role of tau in regulating organelle transport on microtubules, and the role of microtubule stability could have been performed. The capacity of neurofibrillary seeds to induce the aggregation of tau appears to be a relatively straightforward experiment to compare LMW tau and Big tau, and the issue could have been pursued to ascertain the minimal portion of the Big tau insert that elicits the same effect. With current gene-editing knowledge and technology, building on the work already done in Xenopus discussed earlier [38], cells or animals could be made in which LMW tau is replaced by Big tau, or vice versa, to ascertain the functional consequences in health, disease, and regeneration after injury. All of these ideas and more are readily available for exploration by the contemporary tau community.
According to a contemporary databaseii, unlike the rest of the tau protein, which is highly phosphorylated (>80 sites), the insert specific to Big tau has only two phosphorylated sites. This is especially interesting, given that the insert region specific to Big tau has >40 aa that could be phosphorylated, but apparently are not. In other databases (searched relatively extensively by the authors of this article), no compelling evidence could be found that the insert has much homology with known proteins or functional domains of proteins. It appears possible, on this basis, that the Big tau insert may be a phospho-dead, functionally inert zone that provides length to the projection domain of tau, and that nothing else is apparent from its sequence (Figures 3 and 4). This idea is supported by the Xenopus observations discussed earlier [38], and the fact that mutations and polymorphisms identified in exon 4a of humans do not appear to be pathogenic [47–51]. Collectively, these considerations suggest that the evolutionary origin of the insert was from an intron as opposed to an exon of a different protein [52]. Perhaps its sole evolutionary advantage is neuroprotection, as outlined earlier, with the evolutionary pressures never impacting the brain because CNS tauopathy generally manifests at ages other than the reproductive years. By contrast, early manifestations of PNS tau pathology might have been deleterious to reproductive success. Alternatively, proper brain function may require that Big tau is not expressed if, for example, its presence diminishes the plasticity of the brain needed for ongoing learning and memory. Indeed, the N-terminal region of tau, which is involved in important biological processes, has shown increased evolutionary complexity and phosphorylation sites to support the development of novel interactions [53]. While these changes have benefits, they may become a detriment at advanced ages due to increased propensity for misregulation and aggregation. Could Big tau counteract these vulnerabilities? These are all fascinating questions and possibilities that are well within the grasp of modern technology to address, and are framed collectively in the Outstanding Questions.
Outstanding Questions.
How does the large 4a exon insert in the projection domain of Big tau alter its functions and properties relative to the classic lower MW isoforms of tau?
What is it about adult peripheral neurons or central neurons that project their axons into the periphery that involve such a dramatic switch from LMW isoforms of tau to Big tau?
Does Big tau offer advantages relative to lower MW isoforms in functions of tau, such as modulation of microtubule dynamics, interactions with other proteins, and/or facilitation of axonal transport?
Does Big tau provide a protective mechanism to long axons with high metabolic stress, potentially avoiding toxic aggregation?
What is the evolutionary explanation for the existence of Big tau?
Does the almost complete lack of putative phosphorylation sites in the Big tau insert region provide clues as to its evolutionary origin and/or functional significance?
Will greater knowledge of Big tau reveal novel avenues toward protecting CNS neurons from tauopathy?
Highlights.
Tau is a highly studied protein, but most work on tau ignores the existence of Big tau, an isoform (discovered over 25 years ago) with a large additional exon (termed 4a) that doubles the size of the protein.
The switch from low-molecular-weight (LMW) isoforms of tau to Big tau occurs in most PNS neurons as they mature into adulthood, with the selective expression of Big tau in these neurons as well as certain neuronal populations of the CNS (those extending axons into the periphery) remaining a puzzle.
CNS neurons with axons projecting to the periphery also express Big tau as they mature into adulthood, including spinal motor neurons, retinal ganglion cells, and many cranial nerve neurons.
The region of Big tau corresponding to exon 4a has almost no homology to known proteins and almost no putative phosphorylation sites, suggesting that it arose evolutionarily from an intron of another protein.
Functional distinctions between Big tau and LMW isoforms may involve differences in their impact on axonal transport and microtubule spacing, as well as the lower propensity of Big tau to form toxic aggregates and fibrils.
Acknowledgments
The authors acknowledge the experimental work performed by Ying Jin and Theresa Connors that was used in Figure 2, and are grateful to expert graphic work and data search provided by Julien Bouyer in preparation of other figures, as well as Philip Yates for critical comments on the manuscript and Sandhya Kortagere for helpful discussions. Requests for Big tau antibodies should be addressed to I.F. Related work in the Baas laboratory is funded by the National Institutes of Health grant (R01 NS28785).
Footnotes
References
- 1.Tapia-Rojas C et al. (2019) It’s all about tau. Prog. Neurobiol 175, 54–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kellogg EH et al. (2018) Near-atomic model of microtubule-tau interactions. Science 360, 1242–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baas PW and Qiang L (2019) Tau: it’s not what you think. Trends Cell Biol 29, 452–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kadavath H et al. (2015) Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. U. S. A 112, 7501–7506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montejo de Garcini E et al. (1994) Overexpression of tau protein in COS-1 cells results in the stabilization of centrosome-independent microtubules and extension of cytoplasmic processes. Mol. Cell. Biochem 130, 187–196 [DOI] [PubMed] [Google Scholar]
- 6.Baas PW et al. (1994) Tau confers drug stability but not cold stability to microtubules in living cells. J. Cell Sci 107, 135–143 [DOI] [PubMed] [Google Scholar]
- 7.Arendt T et al. (2016) Tau and tauopathies. Brain Res. Bull 126, 238–292 [DOI] [PubMed] [Google Scholar]
- 8.Wang Y and Mandelkow E (2016) Tau in physiology and pathology. Nat. Rev. Neurosci 17, 5–21 [DOI] [PubMed] [Google Scholar]
- 9.Oblinger MM et al. (1991) Tau gene expression in rat sensory neurons during development and regeneration. J. Neurosci 11, 2453–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drubin D et al. (1988. May) Regulation of microtubule protein levels during cellular morphogenesis in nerve growth factor-treated PC12 cells. J. Cell Biol 106, 1583–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goedert M et al. (1992) Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc. Natl. Acad. Sci. U. S. A 89, 1983–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taleghany N and Oblinger MM (1992) Regional distribution and biochemical characteristics of high molecular weight tau in the nervous system. J. Neurosci. Res 33, 257–265 [DOI] [PubMed] [Google Scholar]
- 13.Mercken M et al. (1995) Three distinct axonal transport rates for tau, tubulin, and other microtubule-associated proteins: evidence for dynamic interactions of tau with microtubules in vivo. J. Neurosci 15, 8259–8267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeda S et al. (2016) Seed-competent high-molecular-weight tau species accumulates in the cerebrospinal fluid of Alzheimer’s disease mouse model and human patients. Ann. Neurol 80, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyne LJ et al. (1995) Distribution of Big tau in the central nervous system of the adult and developing rat. J. Comp. Neurol 358, 279–293 [DOI] [PubMed] [Google Scholar]
- 16.Nothias F et al. (1995) The expression and distribution of tau proteins and messenger RNA in rat dorsal root ganglion neurons during development and regeneration. Neuroscience 66, 707–719 [DOI] [PubMed] [Google Scholar]
- 17.Nunez J and Fischer I (1997) Microtubule-associated proteins (MAPs) in the peripheral nervous system during development and regeneration. J. Mol. Neurosci 8, 207–222 [DOI] [PubMed] [Google Scholar]
- 18.Chen XQ and Mobley WC (2019) Alzheimer disease pathogenesis: insights from molecular and cellular biology studies of oligomeric abeta and tau species. Front. Neurosci 13, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spillantini MG and Goedert M (1998) Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 21, 428–433 [DOI] [PubMed] [Google Scholar]
- 20.Himmler A et al. (1989) Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol. Cell. Biol 9, 1381–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo T et al. (2017) Roles of tau protein in health and disease. Acta Neuropathol. 133, 665–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong Q et al. (2012) Tau isoform composition influences rate and extent of filament formation. J. Biol. Chem 287, 20711–20719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janke C et al. (1996) Distribution of isoforms of the microtubule-associated protein tau in grey and white matter areas of human brain: a two-dimensional gelelectrophoretic analysis. FEBS Lett 379, 222–226 [DOI] [PubMed] [Google Scholar]
- 24.Zempel H et al. (2017) Axodendritic sorting and pathological missorting of Tau are isoform-specific and determined by axon initial segment architecture. J. Biol. Chem 292, 12192–12207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hartmann AM et al. (2001) Regulation of alternative splicing of human tau exon 10 by phosphorylation of splicing factors. Mol. Cell. Neurosci 18, 80–90 [DOI] [PubMed] [Google Scholar]
- 26.Lee G et al. (1988) The primary structure and heterogeneity of tau protein from mouse brain. Science 239, 285–288 [DOI] [PubMed] [Google Scholar]
- 27.Kosik KS et al. (1989) Developmentally regulated expression of specific tau sequences. Neuron 2, 1389–1397 [DOI] [PubMed] [Google Scholar]
- 28.Goedert M et al. (1988) Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. U. S. A 85, 4051–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janke C et al. (1999) Phylogenetic diversity of the expression of the microtubule-associated protein tau: implications for neurodegenerative disorders. Brain Res. Mol. Brain Res 68, 119–128 [DOI] [PubMed] [Google Scholar]
- 30.Bakota L et al. (2017) Systemic and network functions of the microtubule-associated protein tau: Implications for tau-based therapies. Mol. Cell. Neurosci 84, 132–141 [DOI] [PubMed] [Google Scholar]
- 31.Black MM et al. (1996) Tau is enriched on dynamic microtubules in the distal region of growing axons. J. Neurosci 16, 3601–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng I et al. (1985) Cultured neurons contain a variety of microtubule-associated proteins. Brain Res. 361, 200–211 [DOI] [PubMed] [Google Scholar]
- 33.Drubin DG et al. (1985) Nerve growth factor-induced neurite outgrowth in PC12 cells involves the coordinate induction of microtubule assembly and assembly-promoting factors. J. Cell Biol 101, 1799–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Couchie D et al. (1992) Primary structure of high molecular weight tau present in the peripheral nervous system. Proc. Natl. Acad. Sci. U. S. A 89, 4378–4381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gache Y et al. (1994) High molecular weight tau distribution and microtubule stability in neuroblastoma N115 cells. Exp. Brain Res 100, 267–275 [DOI] [PubMed] [Google Scholar]
- 36.Georgieff IS et al. (1991) High molecular weight tau: preferential localization in the peripheral nervous system. J. Cell Sci 100, 55–60 [DOI] [PubMed] [Google Scholar]
- 37.Konzack S et al. (2007) Swimming against the tide: mobility of the microtubule-associated protein tau in neurons. J. Neurosci 27, 9916–9927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y et al. (2015) Microtubule-associated protein tau promotes neuronal class II beta-tubulin microtubule formation and axon elongation in embryonic Xenopus laevis. Eur. J. Neurosci 41, 1263–1275 [DOI] [PubMed] [Google Scholar]
- 39.Frappier TF et al. (1994) tau Regulation of microtubule-microtubule spacing and bundling. J. Neurochem 63, 2288–2294 [DOI] [PubMed] [Google Scholar]
- 40.Kanaan NM et al. (2012) Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol. Aging 33, 826.e15–826.e30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hagestedt T et al. (1989) Tau protein becomes long and stiff upon phosphorylation: correlation between paracrystalline structure and degree of phosphorylation. J. Cell Biol 109, 1643–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avila J (2000) Tau aggregation into fibrillar polymers: taupathies. FEBS Lett. 476, 89–92 [DOI] [PubMed] [Google Scholar]
- 43.Dujardin S and Hyman BT (2019) Tau prion-like propagation: state of the art and current challenges. Adv. Exp. Med. Biol 1184, 305–325 [DOI] [PubMed] [Google Scholar]
- 44.Wakabayashi K et al. (1999) Neurofibrillary tangles in the peripheral sympathetic ganglia of non-Alzheimer elderly individuals. Clin. Neuropathol 18, 171–175 [PubMed] [Google Scholar]
- 45.Nishimura M et al. (1993) Neurofibrillary tangles in the neurons of spinal dorsal root ganglia of patients with progressive supranuclear palsy. Acta Neuropathol. 85, 453–457 [DOI] [PubMed] [Google Scholar]
- 46.Deluca GC et al. (2004) The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol. Appl. Neurobiol 30, 576–584 [DOI] [PubMed] [Google Scholar]
- 47.Lilius L et al. (1999) Tau gene polymorphisms and apolipoprotein E epsilon4 may interact to increase risk for Alzheimer’s disease. Neurosci. Lett 277, 29–32 [DOI] [PubMed] [Google Scholar]
- 48.Rademakers R et al. (2004) The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum. Mutat 24, 277–295 [DOI] [PubMed] [Google Scholar]
- 49.Poorkaj P et al. (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol 43, 815–825 [DOI] [PubMed] [Google Scholar]
- 50.Higgins JJ et al. (1999) Mutational analysis of the tau gene in progressive supranuclear palsy. Neurology 53, 1421–1424 [DOI] [PubMed] [Google Scholar]
- 51.Jin SC et al. (2012) Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer’s disease Ibero-American cohort. Alzheimers Res. Ther 4, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sorek R (2007) The birth of new exons: mechanisms and evolutionary consequences. RNA 13, 1603–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trushina NI et al. (2019) The evolution of Tau phosphorylation and interactions. Front. Aging Neurosci 11, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dehmelt L and Halpain S (2005) The MAP2/Tau family of microtubule-associated proteins. Genome Biol 6, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lewis SA et al. (1988) Microtubule-associated protein MAP2 shares a microtubule binding motif with tau protein. Science 242, 936–939 [DOI] [PubMed] [Google Scholar]
- 56.Garner CC et al. (1988) A 70-kilodalton microtubule-associated protein (MAP2c), related to MAP2. J. Neurochem 50, 609–615 [DOI] [PubMed] [Google Scholar]
- 57.Chung WJ et al. (1996) MAP2a, an alternatively spliced variant of microtubule-associated protein 2. J. Neurochem 66, 1273–1281 [DOI] [PubMed] [Google Scholar]
- 58.Crandall JE and Fischer I (1989) Developmental regulation of microtubule-associated protein 2 expression in regions of mouse brain. J. Neurochem 53, 1910–1917 [DOI] [PubMed] [Google Scholar]