

Abstract
Recent cancer sequencing efforts have uncovered asymmetry in DNA damage induced mutagenesis between the transcribed and non-transcribed strands of genes. Here, we investigate the major type of damage induced by ultraviolet (UV) radiation, the cyclobutane pyrimidine dimers (CPDs), which are formed primarily in TT dinucleotides. We reveal that a transcriptional asymmetry already exists at the level of TT dinucleotide frequency and therefore also in CPD damage formation. This asymmetry is conserved in vertebrates and invertebrates and is completely reversed between introns and exons. We show the asymmetry in introns is linked to the transcription process itself, and is also found in enhancer elements. In contrast, the asymmetry in exons is not correlated to transcription, and is associated with codon usage preferences. Reanalysis of nucleotide excision repair, normalizing repair to the underlying TT frequencies, we show repair of CPDs is more efficient in exons compared to introns, contributing to the maintenance and integrity of coding regions. Our results highlight the importance of considering the primary sequence of the DNA in determining DNA damage sensitivity and mutagenic potential.
INTRODUCTION
The genome is constantly subjected to damaging agents that target specific nucleotides. For example, certain base oxidations occur primarily on guanines, whereas base deamination occurs frequently at cytosines (1). The altered structure of damaged nucleotides can result in mis-pairing during replication and the introduction of mutations. Thus, specific base damages result in specific mutational signatures (2–5). Organisms from all kingdoms of life possess mechanisms to repair and tolerate base damages and minimize detrimental mutagenesis. The efficiency of DNA repair processes is not uniform throughout the genome, and is affected both by the chromatin compaction and transcriptional status of a region. As a result, mutagenesis is not uniform (4,6–8).
Open and transcribed regions of the genome, are generally associated with lower mutation rates (9–11). Lower mutagenesis has been attributed to the higher accessibility of the functional regions to repair enzymes (4,7,8,12). Within transcribed regions selective pressure also contributes to the lower mutation rates. Mutagenesis patterns differ between the transcribed and non-transcribed strands. Comparative genomic approaches show that C→T, A→G and G→T are all more prevalent in the coding strand (13,14). As a result, there is a reported asymmetry in the frequency of nucleotides between the two strands, and specifically depletion of T and G in the transcribed strand compared to the coding, or non-transcribed, strand. The asymmetric mutagenesis in genes could be driven either by preferential repair of transcribed strands, or by transcription-associated damage on the exposed non-transcribed strands (4,15).
One of the most studied DNA damages are damages induced by ultraviolet (UV) irradiation in sunlight. The most abundant type of damage induced by UV is the cyclobutyl pyrimidine dimer (CPD). CPD dimers form primarily in TpT (TT) pairs and to a lesser extent in TpC, CpT and CpC (16,17). These bulky damages pose an additional threat to genome function since they block both RNA and DNA polymerases. As a result, organisms of all kingdoms have evolved multiple mechanisms to deal with these damages, including multiple alternative DNA repair pathways, and specialized DNA polymerases that are able to bypass these lesions during replication in a relatively error-free manner (1,18,19). Despite these, UV radiation still induces a cell stress response, mutagenesis and cell death, and is a leading cancer-risk factor.
Recent advances in high resolution genomics have produced several single nucleotide resolution maps of CPDs in human cells (20–23). Together, they show that nucleosome and transcription factor binding could alter damage formation frequencies, primarily due to bending of the DNA to favorable angles for dimer formation (24,25). However, in general, CPD frequencies are relatively uniform and dictated primarily by the underlying genomic sequences. The higher the frequency of TT, the higher the frequency of damage. The immediate consequence of these damages is inhibition of gene expression (26,27). The CPDs block elongating RNA Pol II and induce its subsequent degradation, resulting in a transcriptional shutdown. Longer genes, which harbor more TTs, are more susceptible to damage and therefore to compromised expression (28).
The major mechanism for removal of UV dimers in mammalian cells is nucleotide excision repair (NER). In NER, damages can be recognized either directly (in global genome repair) or by the stalled RNA polymerase (in transcription-coupled repair). Genome-wide mapping of NER has shown that repair is highly influenced by chromatin accessibility, and that transcription-coupled repair is exclusive to the transcribed strand, explaining the non-uniform distribution of UV mutagenesis (29–32).
The high dependence of damage formation on the underlying sequence has motivated us to investigate the frequency of damage-forming dinucleotides in genes, and led us to uncover asymmetric dinucleotide distributions that are conserved in vertebrates and invertebrates. Our findings suggest dinucleotide and polynucleotide frequencies are under additional constraints beyond the ones single-nucleotide sequences are subjected to. Our results highlight the importance of considering the primary DNA sequence as a driver of damage formation and a factor influencing genome stability.
METHODS
Genomic coordinates
The annotation file for 28,712 protein coding genes was retrieved from UCSC table browser, RefSeq, assembly hg38. In case of multiple variants, the longest one was kept. Overlapping genes and genes that have nearby genes in a distance of at most 6 kb upstream were removed using bedtools overlap and closest commands (version v2.26.0) (33). Exon and introns annotation files were retrieved from the UCSC table browser by uploading the list of the non-overlapping transcripts. In order to avoid splicing junction biases, 100 bases from each side of the intron and 10 bases from each side of the exon were removed. Consequently, the filtering analysis resulted in 9449 transcripts, 90,839 exons and 73,771 introns.
Coordinates of non-stranded permissive enhancers were retrieved from Zenodo (https://zenodo.org/record/556775#.XrfJJagzaUl (34)), the left and right CAGE tags (columns 2 and 3 of the BED12 file) were considered as the corresponding eTSSs. Enhancers that have nearby enhancers or genes within distance of 2 Kb were excluded using bedtools closest commands leaving 12,849 enhancer elements (33). To plot average profiles, intervals of 750 bases were taken for each eTSS in the direction of transcription using bedtools slop and intersect commands. Due to the 5′→3′ directionality of transcription, the transcribed strand for the upstream eTSS (the left eTSS) is the plus strand, whereas the transcribed strand for the downstream eTSS (the right eTSS) is the minus strand.
Calculation of dimer frequencies
Dinucleotides frequencies of all genomic elements on the transcribed and non-transcribed strands were calculated using a custom python script. Since the direction of transcription is known, given the coding sequence (the non-transcribed strand) the sequence of its complementary strand (the transcribed strand) can be deduced based on DNA base pairing considerations. For example, TpT on the transcribed strand will be described as ApA on the coding sequence. In case of overlaps (e.g. TpTpT) the dinucleotide was counted twice, with the exception of the polyT analysis in Figure 2. Average plots of TT frequencies over human genes were generated using bedtools. The bedtools intersect command was used for getting TT coordinates in those regions, and the bedtools slop command for defining the upstream and downstream regions relative to the TSSs. Only non-overlapping genes of length > 10 Kb were taken for this analysis (6401 in total). TT coordinates in the entire human genome were extracted with FUZZNUC (version 6.6.0.0) from the EMBOSS package with the complement parameter (35). Strand-specific profiles were created using the R (version 3.5.1) Bioconductor genomation package (version 1.14.0) (36).
Figure 2.

Polynucleotide frequencies in genes are not explained solely by the frequencies of the single nucleotides composing them. (A) Comparison of the frequency of 10 possible dinucleotides (calculated for both strands, observed, green) compared to the frequency that would be expected by random pairing of the single nucleotides that compose them (expected, mint). (B) Comparison of the observed (burgundy) asymmetry score to the asymmetry score expected (pink) by random pairing of the single nucleotides that compose them. (**P < 0.0001, * P < 0.01 based on a Wilcoxon signed-rank test with Bonferroni correction). (C) Average profiles for polyT sequence frequencies surrounding the TSS of genes for Ts found in the context of T1 and up to T5. (D) The frequency of the Ts found in each of the contexts Tn out of all the Ts on each strand is calculated for T1 to T5. (** P < 0.0001 comparing transcribed and non-transcribed strands based on a paired t-test with Bonferroni correction). In all plots, boxes represent range between 75th and 25th percentile, the line represents the median and the diamond the mean. Outliers were discarded for the presentation.
Analysis of Damage-seq data
Damage-seq data of CPD from the NHF1 cell line was download from GEO (accession number GSE98025). Reads were processed following the steps mentioned in Hu et al. (21,37) and mapped to the human genome (hg38) with bowtie. Replicates were merged and only reads containing dipyrimidines (TT, CT, TC and CC) were kept (92%). Damage coverage of different genomic elements was calculated with bedtools coverage command.
Analysis of XR-seq data
Genome-wide maps of NER for CPDs in three cell lines: wild-type NHF1 skin fibroblasts, XP-C mutants and CS-B mutants were obtained from GEO (accession number GSE67941). For each of these cell lines, the sequencing reads were extracted, processed and mapped to the human genome, following the steps mentioned in Hu et al. (30). In Damage-seq analysis, 4nt sequences (−1 to 3 nt upstream from the fragment end) were used for analysis. In order to avoid biases in the repair to damage normalization, in XR-seq the read length was also reduced to 4 nt based on the identified dipyrimidine sites, taking the −1 and +1 flanking nucleotides.
Transcriptional strand asymmetry scores
Dinucleotide frequencies on the transcribed and non-transcribed strands were calculated as described above. The expected dinucleotide frequencies were calculated as the product of the frequencies of the individual nucleotides that compose the observed dinucleotide. For example, the expected frequency of TpT was calculated as f(T)2. The dinucleotide asymmetry score between the strands was calculated as [f(Dinuc.transcribed) – f(Dinuc.non-transcribed)]/ [f(Dinuc.transcribed) + f(Dinuc.non-transcribed)].
Identification of poly(T) tracks
To plot average profiles of poly T tracts loci corresponding to the motif XpolyTX, where X = [A, C, G] were identified using the FUZZNUC. Poly T tables for the boxplots analysis in Figure 2 were created using custom python scripts.
Codon usage analyses
Coding sequences (CDSs) of the non-overlapping genes (described above) were retrieved from the UCSC table browser, RefSeq, assembly hg38. In order to quantify the enrichment or reduction of each dinucleotide within codons, the observed and expected frequencies were compared, where:
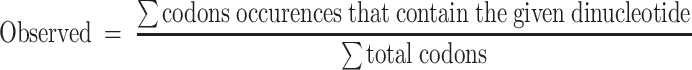 |
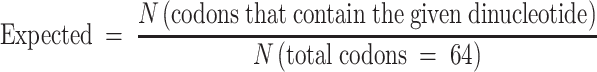 |
To check whether a specific codon pair is under- or over- represented, we used the following formulas for calculating the ratio of observed versus expected representation.
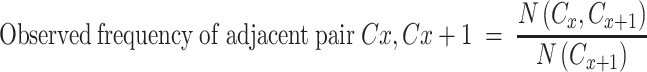 |
 |
where N(Cx, Cx + 1) represents all the occurrences of Cx as a first codon and Cx + 1 the second codon, and N(Cx + 1) represents all occurrences of Cx + 1 appear to be the second codon, where Cx is any given codon. The expected frequency of Cx represents its relative frequency regardless of its order. Stop codons were included.
RNA-seq analysis
For the comparative analysis between male germ cell expression levels and strand asymmetry, we obtained testis expression profiles containing the median TPM from 322 individuals from the genotype-tissue expression (GTEx) Portal (https://gtexportal.org/home/datasets/). In addition, male primordial germ cell (PGC) and H9 ESC RNA-seq data (Supplementary Figure S6) were obtained from Guo et al. and Dileep et al., respectively ((38,39), GEO accession number GSE79552, GSE130541). Exon and intron coordinates of protein coding genes were extracted from GENCODE 25 annotation file using custom python script. Exons and introns of a given gene were merged forming a continuous sequence, in case of multiple transcripts only the longest one was kept. The protein coding genes were grouped in expression levels quartiles based on their expression values (transcripts per million, TPM or fragments per Kb per million, FPKM) and the strand asymmetry score was calculated for the exons and introns of every group of genes as described above.
Genome 3D organization
A/B compartment scores at 50 Kb resolution of Hi-C data from H9 ESCs were obtained from Dileep et al. ((39), GEO accession number GSE130541). Sex chromosomes (X and Y), and chromosome 4 and 10 were removed by the authors. Exons and introns that overlap the 50 Kb intervals were obtained using the bedtools intersect command. Elements were classified as A or B compartment based on the given A/B compartment score of their overlapping 50 Kb intervals. Elements that overlapped both compartments were discarded.
Analysis of dinucleotide transcriptional asymmetry in multiple organisms
Genomic data of 222 vertebrate, 96 Invertebrate and 67 plant species was downloaded from Ensembl ((40), releases 98, 45 and 47, respectively). Exon and intron coordinates were extracted from the gff annotation file. GC content, and dinucleotide frequencies in the transcribed and non-transcribed strands of each organism were calculated as described above.
Analysis of RNA Polymerase II and ATAC-seq data
BigWig files of ChIP-seq data of elongating RNA polymerase II (RNAPII) and ATAC-seq data in un-irradiated human VH10 cells (Fibroblast cells immortalized with hTERT) were obtained from GEO accession numbers GSE83763 and GSE125181 (41,42), respectively. Introns and exons coordinates of hg19 were obtained as described above where first and last intron/exon were removed in order to avoid biases. Average profiles over introns and exons were generated as describe above. RNAPII and ATAC-seq coverage was calculated with bedtools coverage command.
Distribution of mutations across enhancers
Whole genome simple somatic mutation (SSM) data of 25 cancers (Supplementary Table S1) was obtained from the ICGC portion of the PCAWG consensus call sets for SNV/Indel (43). The same cancer types of different populations were merged. C→T transitions that are formed at sites of UV pyrimidine dimers were retrieved by mutationalPatterns package (44). G → A transitions on the plus strand were considered as C→T transitions on the minus strand. C→T transitions coverage across enhancers regions was calculated with bedtools coverage command for the transcribed and non-transcribed strand separately.
Statistical analysis
We used Spearman's correlation to compute the correlation between damage levels and TT frequencies. Statistical significance was computed by either Wilcoxon test or t-test depending on the data distribution. The P-values of multiple tests were adjusted by Bonferroni correction.
Codes and sample data for all analyses are provided in GitHub repository https://github.com/AdarLab/Damage_asymmetry.git.
RESULTS
Transcriptional asymmetry in T-tracks results in asymmetric damage formation
Genome-wide mapping of UV induced CPDs has shown that CPDs form primarily in TpT pairs (20–23). In Damage-seq, sites of damage are precisely mapped by isolating damaged DNA and identifying the sites where a DNA polymerase is blocked. CPDs, previously mapped by Damage-seq, correlate with the overall TT frequency, such that the higher the frequency of TT in a gene, the higher the frequency of damage (Figure 1A). Thus, the major determinant of damage formation is the underlying nucleotide composition. While overall damage distribution in the genome is relatively uniform, analysis of damage formation over genes uncovered non-random patterns of damage formation. CPDs are depleted at the transcription start site (TSS) and are enriched at the transcription end sites (Figure 1B). Analysis of TT distributions at these same regions show the pattern of CPD damage formation is explained by the underlying TT frequencies. TTs are depleted at the TSS (45) and enriched at the TES (Figure 1C). Most notably, there is a clear asymmetry in the frequency of TTs, and therefore CPDs, between the transcribed and non-transcribed strands of genes that extends throughout the gene body. While this asymmetry in the gene body is small, based on Wilcoxon signed-rank test with Bonferroni correction it is significant (P < 0.0001, Figure 1D and E).
Figure 1.

Asymmetric CPD damage formation is explained by asymmetric TT dinucleotide distributions. (A) CPD Damage-seq read density positively correlates with TT frequencies in genes, shown is Spearman correlation coefficient. (B) Average CPD Damage-seq read density profiles plotted separately for the transcribed (TS, light blue) and non-transcribed (NTS, purple) at the beginning and end of genes. TSS: transcription start site and TES: transcription end site. (C) Same as (B) except plotted is the average TT frequency. (D) CPD Damage-seq read frequencies over the transcribed and non-transcribed strands of gene bodies. Each data point represents normalized read count for an individual gene. (**P < 0.0001, based on Wilcoxon signed-rank test with Bonferroni correction). Boxes represent range between 75th and 25th percentile, the line represents the median and the diamond the mean. Outliers were discarded for the presentation (E) Same as (D) except plotted are TT frequencies. (F) CPD Damage-seq read density profiles at enhancers, starting from each of the bidirectional eTSSs as defined by the FANTOM consortium. Data plotted separately for the plus (yellow) and minus (green) strands upstream (left) and downstream (right) the respective eTSSs. (G) Same as (F) except plotted is the average TT frequency. (H) Same as D except CPD Damage-seq read frequencies are plotted over the transcribed and non-transcribed strands of the first 500b from the eTSSs in the direction of transcription. (I) Same as (H) except plotted are TT frequencies.
Enhancer elements in the genome are bi-directionally transcribed (46). We therefore conducted a similar analysis of damage and TT frequencies at enhancer elements identified by the Fantom consortium (34). For each enhancer, the sequence downstream of the left and right enhancer TSS (eTSS) was analyzed. Our results show that, similar to genes, CPD damage formation and TT frequencies are asymmetric at enhancers. This asymmetry is flipped between the left and right sides of enhancer centers. Given the directionality of transcription (from 5’ to 3’ of the nascent RNA), this shows that there are lower damage and TT frequencies on the actively transcribed strand in both directions (Figure 1F-I). These findings suggest that like genes, transcribed enhancers are subject to asymmetric mutagenesis. Indeed, as was previously reported for genes (3,47) analysis of melanoma mutations from whole genome sequencing of cancers (43) shows lower UV-linked C→T mutation frequencies on the transcribed, compared to non-transcribed strands of enhancers. This lower frequency of C→T mutations is specific for melanomas compared to other cancers, since other cancer types show no significant asymmetry (Supplementary Figure S1).
The transcriptional asymmetry in TT dinucleotide frequency is consistent with the previously reported enrichment of T nucleotides on non-transcribed strands (13,14). Therefore, the asymmetry of the dinucleotide frequencies could be explained by random adjacency of the two nucleotides composing them. We expanded our analysis to all the possible dinucleotides (showing only one of the complementary pairs, Figure 2). The overall frequency of dinucleotides in genes (counting both strands) cannot be explained solely by frequencies of the mononucleotides composing them (Figure 2A). While several dinucleotide frequencies, including TT are higher than expected in genes, the frequencies of others such as AT, TA and CG are depleted. We analyzed the asymmetry between the strands for all possible asymmetric pairs (excluding TA, AT, GC and CG pairs, that are inherently symmetric), and compared these distributions to those expected at random based on the mononucleotide frequencies (Figure 2B). For this we calculated a transcriptional asymmetry score as [f(Dinuc.transcribed) – f(Dinuc.non-transcribed)] / [f(Dinuc.transcribed) + f(Dinuc.non-transcribed)]. All dinucleotide pairs showed a degree of asymmetry between the strands. However, this asymmetry is not solely explained by the mononucleotide frequencies. For example, at random we would expect CT and TC frequencies to be identical, and similarly asymmetric. In fact, the frequency and degree of asymmetry of CT and TC are different (Figure 2A and B). Analysis of a set of random genomic regions (matched in number and lengths, but excluding transcribed regions) shows these enriched and depleted dinucleotide frequencies are also observed in non-transcribed regions; however, the asymmetry is strictly linked to transcription (Supplementary Figure S2).
Our analysis showed that of all dinucleotides, TT is the most asymmetric. The similarity between the observed and expected asymmetry levels indicates the majority of this asymmetry is explained by the asymmetric frequencies of the single T. It was previously reported that CPDs preferentially form in polyT-tracts (30,48). We therefore analyzed the asymmetry of specific poly-T sequences (up to 10) between strands. We observed higher asymmetries with higher T numbers (Figure 2C and Supplementary Figure S3A). To determine whether there was a specific preference for polyT over single T in each strand, we calculated the fraction of all the Ts in each strand that are within a single, or each of the poly-T contexts (Figure 2D and Supplementary Figure S3B). Indeed, it appears that the fraction of Ts in a single T context in the transcribed (52.87%) versus the non-transcribed (49.83%) strand is higher, whereas the fraction of polyT is lower. Thus, there is selective depletion of poly-T tracts in the transcribed strand.
Together, these results explain the asymmetric CPD damage formation in genes. Since CPDs block RNA polymerase transcription, depletion of these damages in the transcribed strands would be beneficial to any sun-exposed tissue and organism.
An opposite TT asymmetry in exons and introns
Previous analyses of asymmetric mutagenesis and nucleotide compositions focused on intronic sequences, excluding coding sequences that are under selective pressure (13,14). However, damages in transcribed strands are detrimental to transcription both in exons and introns. Therefore, we analyzed TT frequencies in the transcribed and non-transcribed strands for exons and introns separately. The asymmetry in TT, and therefore CPD formation, between the strands was flipped (Figure 3A–D and Supplementary Figure S4). While in introns, TTs were depleted in the transcribed strand relative to the non-transcribed strand, in exons TTs were more prevalent in the transcribed strand compared to the non-transcribed strand. This opposite asymmetry is maintained throughout the exon lengths and is not dictated by specialized sequences in the ends. Still, consistent with previous reports that exons were G/C rich, the overall TT frequency in transcribed strands of exons was lower than in introns, resulting in overall lower CPD damage frequencies in exons.
Figure 3.

Opposite asymmetries in exons and introns. (A) Average CPD Damage-seq read density downstream of intron (left) and exon (right) starts. The transcribed (TS, light blue) and non-transcribed (NTS, purple) are plotted separately. (B) Similar to A, except TT frequencies are plotted. (C) Damage-seq read frequencies over gene bodies calculated separately for the transcribed and non-transcribed strands of introns and exons (**P < 0.0001, based on Wilcoxon signed-rank test with Bonferroni correction). (D) Similar to A, except plotted are TT frequencies. (E) The ratio of the frequency of codons containing the indicated dinucleotides compared to the expected frequency in the absence of codon bias. (F) The ratio of observed to expected frequencies for codons starting with each of the four possible nucleotides appearing after codons ending with the indicated nucleotide. (G) Asymmetry scores of TT dinucleotide frequencies in introns (left) and exons (right) for genes that were stratified to four quartiles based on their expression levels in testis. In boxplots, boxes represent range between 75th and 25th percentile, the horizontal line represents the median and the diamond the mean. (H) Scatterplots depicting the average frequency (per Kb) of TT on the transcribed strand versus the non-transcribed strand in exons (orange) or introns (blue) for 222 vertebrate species (left) and 96 invertebrate species (right).
We tested whether the transcriptional asymmetry in TT distribution in exons can be explained by codon usage. Indeed, codons that contain AA pairs are more frequent than expected at random, whereas codons that contain TT are less frequent, consistent with higher TT frequencies on the transcribed strand in exons (Figure 3E). TT/AA sequences in coding sequences could also be a result of T/A appearing in adjacent codons. To measure the frequency of dinucleotide sequences created by adjacent codons, we calculated the frequency of codons starting with a specific nucleotide appearing after a codon ending with a specific nucleotide (Figure 3F), and compared it to the frequency expected by equal distribution of codons. This analysis identified a depletion of codons starting with G following codons ending with C (Supplementary Figure S4C), and depletion of codons starting with A following codons ending with T (Figure 3F). The depletion of CG and TA sequences (also observed in Figure 3E) was previously reported (49,50) and attributed to selection against CGs that are sites of DNA methylation, and TA sequences that result in nuclease-sensitive UA sequences in RNA. In contrast, the frequencies of adjacent codons that would result in AA formation (codons ending in A and starting with A) was similar to what would be expected by equal distribution of codons. The frequency of adjacent codons forming TT sequences (codons starting with T following a codon ending with T) was lower than expected by equal distribution of codons, consistent with the observed lower TT frequency on the non-transcribed (coding) strand. Together, both codon composition and order explain the opposite asymmetry in TT formation in exons compared to introns.
The transcriptional asymmetry in nucleotide composition in introns, which are not under the selective pressures of coding regions, is likely driven by asymmetric mutagenesis in transcribed regions in the germlines. This asymmetric mutagenesis can be a result of preferential repair of the transcribed strand, or higher damage of the exposed non-transcribed strand. Since it was reported that germline mutations are primarily paternal (51), we obtained testis RNA-seq data from GTEx dataset (52) in order to test the relationship between expression levels and TT asymmetry. The TT asymmetry in introns negatively correlates with expression levels in male germ cells. In contrast, there is no correlation between the asymmetry in exons and the expression levels in testis (Figure 3G). This observation is also supported by male PGC and embryonic stem cell RNA-seq data (Supplementary Figure S4D and E) obtained from Gou et al. and Dileep et al. (38,39). Analysis of the three dimensional organization of the genome in embryonic stem cells shows stronger asymmetry in introns in compartment A (active chromatin) compared to compartment B (inactive chromatin), consistent with the higher expression of genes in active chromatin (Supplementary Figure S4F). Thus, we conclude the asymmetry in introns, but not exons, is likely driven by the transcriptional process itself.
Opposite asymmetry in exons and introns is conserved through vertebrate and invertebrate evolution
To see if this asymmetry was conserved in other organisms, we downloaded the intron and exons sequences from all vertebrate, invertebrates, and plant genomes available in the Ensebml database (40). Fungal and bacterial genomes were not analyzed due to the low frequency of introns. For each organism, the average TT frequency was calculated for the transcribed and non-transcribed strands across all exons or introns. Indeed, there is a reversed asymmetry in TT frequency in the transcribed and non-transcribed strands between exons and introns in all vertebrates and invertebrates (Figure 3H). Of all the possible dinucleotide asymmetries, the asymmetry in introns, and the opposite asymmetry in exons is most pronounced for TT pairs (compare to Supplementary Figure S5). With five exceptions, the depletion of TT in the transcribed strands of introns is also observed in the majority of plant species (Supplementary Figure S4G). The exceptions were Cyanidioschyzon merolae, Oryza brachyantha, Chondrus crispus and Ostreococcus lucimarinus, which have very low intron frequencies, and the unicellular algae Chlamydomonas reinhardtii that has exceptionally high (61%) GC content. These were omitted in subsequent analyses. In contrast to vertebrates and invertebrates, in plants, the asymmetry in exons is lost. Using Arabidopsis thaliana as a model plant genome, we saw that the loss of asymmetry in exons can be attributed to the difference in codon usage (Supplementary Figure S4G–I), where both AA and TT containing codons are more prevalent than expected and codon order does not influence AA/TT formation. Thus in plants, the transcription-driven asymmetry in introns persists, but strand asymmetry in exons is lost due to altered codon usage. There is no correlation between G/C content and the asymmetry level in introns. In exons however, some correlation is observed, consistent with effects of G/C content on codon usage preferences (Supplementary Figure S6).
Higher efficiency of NER in exons
In light of this non-uniform distribution of TTs and therefore CPDs, we re-analyzed previous CPD DNA repair data generated by XR-seq (30) from three human cell lines: a Cockayne syndrome group B patient (CS-B) fibroblast cell line proficient in global genome repair only, a Xeroderma Pigmentosum group C (XP-C) patient fibroblast cell line proficient in transcription-coupled repair only (Figure 4) and a normal fibroblast cell line NHF1 proficient in both repair pathways (Supplementary Figure S7). In each cell type, the average XR-seq profiles were plotted separately for the transcribed and non-transcribed strands (Figure 4A and Supplementary Figure S7A). In XP-C cells, there is a clear enrichment of repair on the transcribed strand. Transcription-coupled repair is significantly elevated after the gene start, but there is a small dip in the signal immediately downstream of the TSS. Normalizing the XR-seq data by either the underlying TT, or initial CPD frequencies, resulted in a disappearance of this dip, and a smooth peak of DNA repair (Figure 4B and C; Supplementary Figure S7B and C). Similar to transcribed genes, normalized NER at enhancers displayed a strong transcription-coupled repair phenotype in XP-C cells (Figure 4D–F and Supplementary Figure S7D–F). In CS-B cells that do not have transcription-coupled repair but only global genome repair there is no enrichment of repair on the transcribed strand of genes. On the contrary, in the original XR-seq plots, there appears to be a slightly higher signal over the non-transcribed strand, starting from the gene start and proceeding into the gene body. Upon normalizing repair to the underlying TT or CPD frequencies, this preference for non-transcribed strand repair is lost. This is consistent with the higher observed repair being the result of the higher level of damage on the non-transcribed strand. However, near the TSS, there is in general higher efficiency of repair, likely due to the more open chromatin structure. Within this region—even after normalization of the data, there is still higher repair on the non-transcribed strand. While the strand difference is diminished, a similar pattern is observed at eTSS sites in enhancers. This could be due to stalled RNA polymerases that cannot elicit transcription-coupled repair, and inhibit access of global genome repair factors to the damage.
Figure 4.

Altered repair profiles after normalizing to underlying damage or dinucleotide composition. (A) Average profile of normalized XR-seq read counts over the first 10 Kb of gene intervals. Data plotted separately for the transcribed strand (TS, burgundy) and the non-transcribed strand (NTS, blue) (B) Average profile of XR-seq after normalization to the underlying CPD damage frequency determined by Damage seq. TS, black, NTS, green. (C) Same as (B) except data is normalized to the underlying TT frequency. TS, orange, NTS, purple. (D andE) Average profile of XR-seq after normalization to the underlying TT frequency at enhancers. Plotted separately are the plus (pink) and minus (blue) strands in the 750 bp transcribed intervals starting from the left and right eTSSs, respectively. (F) XR-seq normalized to TT frequencies over the transcribed and non-transcribed strands of 500 bp transcribed regions starting from the eTSSs. (**P < 0.0001 based on Wilcoxon signed-rank test with Bonferroni correction). Boxes represent range between 75th and 25th percentile, the line represents the median and the diamond the mean. Outliers were discarded for the presentation. In all plots, top panel refers to data from XP-C cells, proficient only in transcription-coupled repair, bottom panel refers to CS-B cells proficient only in global genome repair.
Given the difference in damage formation in exons and introns, we reanalyzed NER efficiencies in these regions, normalizing repair measurements by XR-seq to the underlying damage or TT frequencies (Figure 5A and B; Supplementary Figure S7G and H). Global genome repair (in CS-B cells) appear higher in exons compared to introns (Figure 5A and B). The repair, specifically in exons, shows a slight but significant preference for the non-transcribed strand (Supplementary Figure S7K). This may be due to stalled RNA polymerases occluding damage sites, as was observed at gene starts. Transcription-coupled repair (in XP-C cells) also shows slightly higher repair in exons; however, the difference is not statistically significant (Figure 5C and D). Since transcription coupled repair is initiated by actively elongating RNA Pol II, and global genome repair is highly influenced by chromatin accessibility, we analyzed elongating RNA Pol II occupancy and ATAC-seq accessibility profiles from VH10 skin fibroblasts over introns and exons (data obtained from (41,42), Figure 5E–J). Like transcription-coupled repair, RNA pol II showed higher, but not statistically significant, different levels on exons. ATAC-seq indicates exons are more accessible, explaining the higher global genome repair frequencies. Thus, actively transcribed exons are subject to more efficient repair.
Figure 5.

Differential NER in exons and introns. (A) Average profile of XR-seq after normalization to the underlying TT frequency. Plotted separately are the transcribed strand (TS, pink) and non-transcribed strand (NTS, purple) over the first 1.874 Kb of intron intervals in XP-C (top) and CS-B (bottom) cells. (B) Same as A, except plotted over 120 bases of exons. (C andD) XR-seq normalized to TT frequencies over the transcribed and non-transcribed strands, respectively, of exons and introns in XP-C (top) and CS-B (bottom) cells. (E) Average profile of ChIP-seq read densities of elongating RNA polymerase II (RNAPII) over the first 1.874 Kb of introns. Data plotted for both strands. (F) Same as (E), except plotted over the first 120 bases of exons. (G) Distribution of ChIP-seq reads of elongating RNA polymerase II (RNAPII) over introns and exons. (H) ATAC-seq read densities measuring chromatin accessibility over the first 1.874 Kb of introns. Data plotted for both strands. (I) Same as (H), except plotted over the first 120 bases of exons. (J) Distribution of ATAC-seq reads over introns and exons. In all plots, **P < 0.0001 based on Wilcoxon signed-rank test with Bonferroni correction. boxes represent range between 75th and 25th percentile, the line represents the median and the diamond the mean. Outliers were discarded for the presentation.
DISCUSSION
Recent analyses of UV induced DNA damages have shown that while NER efficiencies are very heterogenic, damage frequencies in the genome are relatively uniform regardless of chromatin context or gene expression levels (21,29). Certain nuclear factors can affect damage formation. These include the rotational setting of nucleosomes (24,25), and binding of transcription factors (21–23). However, the major determinant of damage distribution in the genome remains the underlying sequence composition. Here we show that this sequence composition is not uniform and therefore can influence both damage distribution and its transcriptional consequences.
Our results clearly show that the distribution of dinucleotides in transcribed DNA is not random, but also not dictated solely by the frequencies of the single nucleotides composing them. This observation extends beyond dinucleotides also to polynucleotide sequences. Asymmetry of mononucleotide repeats was also recently reported by Georgakopoulos-Soares (53), who showed insertion and deletion events are asymmetric in transcribed regions. Additional elements in the human genome harbor non-random distributions of dinucleotides. For instance, nucleosome positions are associated with depletion of AA and TT pairs, and enrichment of GG, CC and GC pairs (54). CG dinucleotides, which are the target of DNA methylation, are generally depleted, while TG dinucleotides are overrepresented most likely because of CG → TG transitions of methylated cytosines as was previously reported both in evolution and cancer genomic studies (2,55,56). Here we show that dinucleotides also present different degrees of transcriptional asymmetries.
Of all possible asymmetric dinucleotides, TT/AA are most asymmetric. Furthermore, this asymmetry is completely reversed between introns and exons. What are the driving forces of these asymmetries? Exons are under strong selective pressure, and the TT asymmetry is consistent both with codon usage preferences and codon order. In introns, there are other forces at play. The most likely explanation is the action of asymmetric mutagenic events. However, we do not think the asymmetry is driven by UV mutagenesis, or is driven by a selective pressure to remove TTs from transcribed strands. This is supported by several aspects of the data: first, the asymmetry in TT is very similar to the asymmetry expected by a random pairing of two Ts (Figure 2B and D), indicating there is no strong selection specifically against TT. Second, the asymmetry is observed in all vertebrates and invertebrates including organisms that are not exposed to sunlight (Figure 3). And last, UV primarily causes somatic mutations in sun-exposed organs, whereas germ cells of most organisms are protected from light. Beyond UV-dimers, there are other damaging agents that target dinucleotides, and recent cancer mutagenesis efforts have uncovered new dinucleotide mutational signatures (57).
While in exons the asymmetry is flipped, the absolute frequency of TT sequences in both the transcribed and non-transcribed strands is still lower than that of introns and the genome average. TT and TA sequences in coding strands produce RNA harboring UU and UA sequences, which are targets of nucleases, and could therefore be selected against to preserve RNA stability (58).
From the UV-damage viewpoint, the lower TT frequency on transcribed strands is fortuitous as it would result in a lower level of polymerase-blocking damages. Does UV mutagenesis play a role in this phenomenon? Unlikely, since in multicellular organisms, these selection processes must apply to the germline mutations and most germ cells are protected from UV. Could there be selection against UV-damage forming dinucleotides? That does not seem to be the case.
Regardless, the outcome is that there are differences in the frequencies of CPD-forming TTs in the two strands. Genomic studies of NER, and specifically transcription-coupled repair, should consider these frequencies in the analyses of differential repair between regions and between strands. Doing so, we show that repair, primarily global genome repair, is more efficient in exons compared to introns. This could be explained by the higher accessibility of exons. A related study showed mismatch repair is more efficient in exons (59). This higher efficiency of repair of coding sequences acts to preserve their integrity.
We show that like genes, transcribed enhancer elements are also characterized by asymmetric transcription-coupled repair, asymmetric distribution of nucleotides, and asymmetric damage formation. The recent availability of cancer whole genome sequencing data allow us to analyze mutational patterns at enhancers. Indeed, we show that as in genes, UV-related melanoma mutations exhibit a mutational strand-asymmetry at enhancers.
CONCLUSION
Genome sequence is constantly subject to two, mostly contradicting, forces: natural selection to preserve function, and mutagenic processes that drive diversity but carry possible deleterious consequences. Transcriptional asymmetries in nucleotide composition are a result of asymmetric mutagenesis, which can be the result of preferential DNA repair of the transcribed strand, or higher damage-sensitivity of the exposed non-transcribed strand. Using UV damages as an example, here we show that this also makes the primary sequence of the genome asymmetrically sensitive to the next round of damage as the cycle of damage and mutagenesis continues.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: S.A. and E.E.H. designed the study. E.E.H. and M.M. performed the bioinformatic analyses. S.A. and E.E.H. wrote the manuscript.
Contributor Information
Elisheva E Heilbrun, Department of Microbiology and Molecular Genetics, Institute for Medical Research Israel Canada, Faculty of Medicine, Hebrew University of Jerusalem, Ein Kerem, Jerusalem 91120, Israel.
May Merav, Department of Microbiology and Molecular Genetics, Institute for Medical Research Israel Canada, Faculty of Medicine, Hebrew University of Jerusalem, Ein Kerem, Jerusalem 91120, Israel.
Sheera Adar, Department of Microbiology and Molecular Genetics, Institute for Medical Research Israel Canada, Faculty of Medicine, Hebrew University of Jerusalem, Ein Kerem, Jerusalem 91120, Israel.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Israel Science Foundation [1710/17]; Israel Cancer Association [20191630]; Jacob and Lena Joels Memorial Fund Senior Lectureship (to S.A.); Malvina and Solomon Pollack Scholarship Fund (to E.E.H.); Israel Council for Higher Education Scholarship (to E.E.H.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Chatterjee N., Walker G.C.. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017; 58:235–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L.et al.. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haradhvala N.J., Polak P., Stojanov P., Covington K.R., Shinbrot E., Hess J.M., Rheinbay E., Kim J., Maruvka Y.E., Braunstein L.Z.et al.. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell. 2016; 164:538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tubbs A., Nussenzweig A.. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017; 168:644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Helleday T., Eshtad S., Nik-Zainal S.. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014; 15:585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gospodinov A., Herceg Z.. Shaping chromatin for repair. Mutat. Res. 2013; 752:45–60. [DOI] [PubMed] [Google Scholar]
- 7. Mao P., Wyrick J.J.. Organization of DNA damage, excision repair, and mutagenesis in chromatin: A genomic perspective. DNA Repair (Amst.). 2019; 81:102645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gonzalez-Perez A., Sabarinathan R., Lopez-Bigas N.. Local determinants of the mutational landscape of the human genome. Cell. 2019; 177:101–114. [DOI] [PubMed] [Google Scholar]
- 9. Polak P., Karlic R., Koren A., Thurman R., Sandstrom R., Lawrence M., Reynolds A., Rynes E., Vlahovicek K., Stamatoyannopoulos J.A.et al.. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015; 518:360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schuster-Bockler B., Lehner B.. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012; 488:504–507. [DOI] [PubMed] [Google Scholar]
- 11. Hodgkinson A., Eyre-Walker A.. Variation in the mutation rate across mammalian genomes. Nat. Rev. Genet. 2011; 12:756–766. [DOI] [PubMed] [Google Scholar]
- 12. Hu J.C., Choi J.H., Gaddameedhi S., Kemp M.G., Reardon J.T., Sancar A.. Nucleotide excision repair in human cells fate of the excised oligonucleotide carrying dna damage in vivo. J. Biol. Chem. 2013; 288:20918–20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Green P., Ewing B., Miller W., Thomas P.J., Program N.C.S., Green E.D. Transcription-associated mutational asymmetry in mammalian evolution. Nat. Genet. 2003; 33:514–517. [DOI] [PubMed] [Google Scholar]
- 14. Mugal C.F., von Grunberg H.H., Peifer M.. Transcription-induced mutational strand bias and its effect on substitution rates in human genes. Mol. Biol. Evol. 2009; 26:131–142. [DOI] [PubMed] [Google Scholar]
- 15. Hanawalt P.C., Spivak G.. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008; 9:958–970. [DOI] [PubMed] [Google Scholar]
- 16. Douki T., Cadet J.. Individual determination of the yield of the main UV-induced dimeric pyrimidine photoproducts in DNA suggests a high mutagenicity of CC photolesions. Biochemistry. 2001; 40:2495–2501. [DOI] [PubMed] [Google Scholar]
- 17. Mitchell D.L., Jen J., Cleaver J.E.. Sequence specificity of cyclobutane pyrimidine dimers in DNA treated with solar (ultraviolet B) radiation. Nucleic Acids Res. 1992; 20:225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sancar A. Mechanisms of DNA repair by photolyase and excision nuclease (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2016; 55:8502–8527. [DOI] [PubMed] [Google Scholar]
- 19. Yang W., Gao Y.. Translesion and repair DNA polymerases: diverse structure and mechanism. Annu. Rev. Biochem. 2018; 87:239–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elliott K., Bostrom M., Filges S., Lindberg M., Van den Eynden J., Stahlberg A., Clausen A.R., Larsson E.. Elevated pyrimidine dimer formation at distinct genomic bases underlies promoter mutation hotspots in UV-exposed cancers. PLos Genet. 2018; 14:e1007849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu J., Adebali O., Adar S., Sancar A.. Dynamic maps of UV damage formation and repair for the human genome. Proc. Natl Acad. Sci. U.S.A. 2017; 114:6758–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mao P., Brown A.J., Esaki S., Lockwood S., Poon G.M.K., Smerdon M.J., Roberts S.A., Wyrick J.J. ETS transcription factors induce a unique UV damage signature that drives recurrent mutagenesis in melanoma. Nat. Commun. 2018; 9:2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Premi S., Han L., Mehta S., Knight J., Zhao D., Palmatier M.A., Kornacker K., Brash D.E.. Genomic sites hypersensitive to ultraviolet radiation. Proc. Natl Acad. Sci. U.S.A. 2019; 116:24196–24205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mao P., Smerdon M.J., Roberts S.A., Wyrick J.J.. Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc. Natl Acad. Sci. U.S.A. 2016; 113:9057–9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pich O., Muinos F., Sabarinathan R., Reyes-Salazar I., Gonzalez-Perez A., Lopez-Bigas N.. Somatic and germline mutation periodicity follow the orientation of the DNA minor groove around nucleosomes. Cell. 2018; 175:1074–1087. [DOI] [PubMed] [Google Scholar]
- 26. Geijer M.E., Marteijn J.A.. What happens at the lesion does not stay at the lesion: transcription-coupled nucleotide excision repair and the effects of DNA damage on transcription in cis and trans. DNA Repair (Amst.). 2018; 71:56–68. [DOI] [PubMed] [Google Scholar]
- 27. Heilbrun E.E., Merav M., Parnas A., Adar S.. The hardwired transcriptional respnose to DNA damage. Curr. Opin. Syst. Biol. 2020; 19:1–7. [Google Scholar]
- 28. Andrade-Lima L.C., Veloso A., Paulsen M.T., Menck C.F., Ljungman M.. DNA repair and recovery of RNA synthesis following exposure to ultraviolet light are delayed in long genes. Nucleic Acids Res. 2015; 43:2744–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adar S., Hu J., Lieb J.D., Sancar A.. Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proc. Natl Acad. Sci. U.S.A. 2016; 113:E2124–E2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu J., Adar S., Selby C.P., Lieb J.D., Sancar A.. Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 2015; 29:948–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lim B., Mun J., Kim Y.S., Kim S.Y.. Variability in chromatin architecture and associated DNA repair at genomic positions containing somatic mutations. Cancer Res. 2017; 77:2822–2833. [DOI] [PubMed] [Google Scholar]
- 32. van der Weegen Y., Golan-Berman H., Mevissen T.E.T., Apelt K., Gonzalez-Prieto R., Goedhart J., Heilbrun E.E., Vertegaal A.C.O., van den Heuvel D., Walter J.C.et al.. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat. Commun. 2020; 11:2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rennie S., Dalby M., van Duin L., Andersson R.. Transcriptional decomposition reveals active chromatin architectures and cell specific regulatory interactions. Nat. Commun. 2018; 9:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rice P., Longden I., Bleasby A.. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000; 16:276–277. [DOI] [PubMed] [Google Scholar]
- 36. Akalin A., Franke V., Vlahovicek K., Mason C.E., Schubeler D. Genomation: a toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics. 2015; 31:1127–1129. [DOI] [PubMed] [Google Scholar]
- 37. Hu J., Lieb J.D., Sancar A., Adar S.. Cisplatin DNA damage and repair maps of the human genome at single-nucleotide resolution. Proc. NatlAcad. Sci. U.S.A. 2016; 113:11507–11512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guo H., Hu B., Yan L., Yong J., Wu Y., Gao Y., Guo F., Hou Y., Fan X., Dong J.et al.. DNA methylation and chromatin accessibility profiling of mouse and human fetal germ cells. Cell Res. 2017; 27:165–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dileep V., Wilson K.A., Marchal C., Lyu X., Zhao P.A., Li B., Poulet A., Bartlett D.A., Rivera-Mulia J.C., Qin Z.S.et al.. Rapid irreversible transcriptional reprogramming in human stem cells accompanied by discordance between replication timing and chromatin compartment. Stem Cell Reports. 2019; 13:193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yates A.D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R.et al.. Ensembl 2020. Nucleic Acids Res. 2020; 48:D682–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lavigne M.D., Konstantopoulos D., Ntakou-Zamplara K.Z., Liakos A., Fousteri M.. Global unleashing of transcription elongation waves in response to genotoxic stress restricts somatic mutation rate. Nat. Commun. 2017; 8:2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liakos A., Konstantopoulos D., Lavigne M.D., Fousteri M.. Continuous transcription initiation guarantees robust repair of all transcribed genes and regulatory regions. Nat. Commun. 2020; 11:916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E.et al.. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019; 47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Blokzijl F., Janssen R., van Boxtel R., Cuppen E.. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018; 10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Field Y., Kaplan N., Fondufe-Mittendorf Y., Moore I.K., Sharon E., Lubling Y., Widom J., Segal E.. Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comput. Biol. 2008; 4:e1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Core L.J., Martins A.L., Danko C.G., Waters C.T., Siepel A., Lis J.T.. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet. 2014; 46:1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tomkova M., Tomek J., Kriaucionis S., Schuster-Bockler B.. Mutational signature distribution varies with DNA replication timing and strand asymmetry. Genome Biol. 2018; 19:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bryan D.S., Ransom M., Adane B., York K., Hesselberth J.R.. High resolution mapping of modified DNA nucleobases using excision repair enzymes. Genome Res. 2014; 24:1534–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Beutler E., Gelbart T., Han J.H., Koziol J.A., Beutler B.. Evolution of the genome and the genetic code: selection at the dinucleotide level by methylation and polyribonucleotide cleavage. Proc. Natl Acad. Sci. U.S.A. 1989; 86:192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Halder B., Malakar A.K., Chakraborty S.. Dissimilar substitution rates between two strands of DNA influence codon usage pattern in some human genes. Gene. 2018; 645:179–187. [DOI] [PubMed] [Google Scholar]
- 51. Campbell C.D., Eichler E.E.. Properties and rates of germline mutations in humans. Trends Genet. 2013; 29:575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Consortium G.T. Human genomics. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Georgakopoulos-Soares I., Koh G., Momen S.E., Jiricny J., Hemberg M., Nik-Zainal S.. Transcription-coupled repair and mismatch repair contribute towards preserving genome integrity at mononucleotide repeat tracts. Nat. Commun. 2020; 11:1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Valouev A., Johnson S.M., Boyd S.D., Smith C.L., Fire A.Z., Sidow A.. Determinants of nucleosome organization in primary human cells. Nature. 2011; 474:516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Duncan B.K., Miller J.H.. Mutagenic deamination of cytosine residues in DNA. Nature. 1980; 287:560–561. [DOI] [PubMed] [Google Scholar]
- 56. Ehrlich M., Zhang X.Y., Inamdar N.M.. Spontaneous deamination of cytosine and 5-methylcytosine residues in DNA and replacement of 5-methylcytosine residues with cytosine residues. Mutat. Res. 1990; 238:277–286. [DOI] [PubMed] [Google Scholar]
- 57. Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Tian Ng A.W., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N.et al.. The repertoire of mutational signatures in human cancer. Nature. 2020; 578:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Al-Saif M., Khabar K.S.. UU/UA dinucleotide frequency reduction in coding regions results in increased mRNA stability and protein expression. Mol. Ther. 2012; 20:954–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Frigola J., Sabarinathan R., Mularoni L., Muinos F., Gonzalez-Perez A., Lopez-Bigas N.. Reduced mutation rate in exons due to differential mismatch repair. Nat. Genet. 2017; 49:1684–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.