

Abstract
Various factors, including the insect host, diet, and surrounding ecosystem can shape the structure of the bacterial communities of insects. We have employed next generation, high-throughput sequencing of the 16S rRNA to characterize the bacteriome of wild Zeugodacus (Bactrocera) cucurbitae (Coquillett) flies from three regions of Bangladesh. The tested populations developed distinct bacterial communities with differences in bacterial composition, suggesting that geography has an impact on the fly bacteriome. The dominant bacteria belonged to the families Enterobacteriaceae, Dysgomonadaceae and Orbaceae, with the genera Dysgonomonas, Orbus and Citrobacter showing the highest relative abundance across populations. Network analysis indicated variable interactions between operational taxonomic units (OTUs), with cases of mutual exclusion and copresence. Certain bacterial genera with high relative abundance were also characterized by a high degree of interactions. Interestingly, genera with a low relative abundance like Shimwellia, Gilliamella, and Chishuiella were among those that showed abundant interactions, suggesting that they are also important components of the bacterial community. Such knowledge could help us identify ideal wild populations for domestication in the context of the sterile insect technique or similar biotechnological methods. Further characterization of this bacterial diversity with transcriptomic and metabolic approaches, could also reveal their specific role in Z. cucurbitae physiology.
Keywords: melon fly, natural population, symbiome, 16S rRNA gene, next generation sequencing (NGS)
1. Introduction
Symbiosis is the process that occurs when two different organisms live together to form a mutually beneficial partnership. Most insects are symbiotically associated with bacteria [1]. However, the interactions between insects and microorganisms may also be commensal or pathogenic. Microbial symbionts play a significant role in the biology, including nutrition, immunity, reproduction, ecology, and evolution of many insect groups [2,3]. Especially, tephritid flies harbor different bacterial symbionts in their digestive system, which influence different developmental and fitness parameters [4]. This functional contribution of symbiotic microorganisms to insect physiology could find application in mass-rearing facilities, where the manipulation of insects often results in the deterioration of crucial biological parameters. Bacterial strains isolated from stable microbial communities of wild individuals can be provided to mass-reared insects as supplements, in an attempt to replicate the natural microbiome and improve fitness and mating success [5,6,7]. Studies dealing with tephritid microbiomics use either samples from laboratory colonies [8,9,10,11,12,13] or wild populations [10,14,15,16,17,18], which are generally characterized by higher microbial diversity compared to laboratory strains [10,19]. Therefore, it is important to identify and exploit this large portion of diversity, but also to determine how it is affected by the geographical isolation of wild populations.
Fruit flies of the family Tephritidae have a cosmopolitan distribution and are important pests in fruit production [20]. The genus Bactrocera is highly diverse and abundant, with many species still to be described [21]. At least 28 Bactrocera subgenera have been described and these are divided into four groups, namely Bactrocera, Melanodacus, Queenslandacus, and Zeugodacus [22].
Zeugodacus currently includes 192 species [23]. At least 50% of the species included in the Zeugodacus group, for which host plant records are available, are cucurbit feeders [23]. Amongst them, the melon fly, Zeugodacus (Bactrocera) cucurbitae (Coquillett), is of particular economic importance. Recently the systematic position of Zeugodacus was revised as Bactrocera, Dacus and the subgenera of the Zeugodacus group have different evolutionary histories [24,25]. In this study, we refer to the classification proposed by Virgilio et al. (2015) by using the new generic combination Zeugodacus (Zeugodacus) cucurbitae for the melon fly, although most existing literature refers to it under the former combination, Bactrocera (Zeugodacus) cucurbitae.
Even though Z. cucurbitae was initially described from the Hawaiian Islands, its presence was the result of accidental human-mediated introduction [26]. Nowadays, it is distributed across a range of climatic regions such as Central and East Asia (including Pakistan, India, Bangladesh, Nepal, China, Indonesia and the Philippines) and Oceania (including New Guinea and the Mariana Islands) [27]. Due to its vast adaptability, high reproduction potential and invasion capacity, the melon fly has been the subject of a worldwide pest management program [28].
Culture-dependent and culture-independent approaches have been employed to characterize the structure of the symbiotic community of Tephritidae species, including the melon fly [5,29,30,31]. Hadapad et al. (2015) investigated the composition and diversity of the microbial community in the midgut of melon fly, obtained from nine wild populations (India), and resulted in the dominant species inhabiting the midgut of the melon fly, which were from the genera Enterobacter (34.6%), Klebsiella (19.2%), Citrobacter (7.7%), Bacillus (15.4%) and Providencia (7.7%), and 3.8% each of Micrococcus, Staphylococcus, Leclercia and Exiguobacterium [30]. Another reported study on cultivable microbiota of Zeugodacus cucurbitae was performed by Gujjar et al. and shed light on a distinct pattern of gender specific gut bacterial colonization. The cultivable diversity from females of Z. cucurbitae comprised mainly of Morganella morganii and Bacillus pumilis while Z. cucurbitae males were predominantly colonized by aerobic endospore formers viz., Bacillus cereus, B. licheniformis and B. subtilis [32].
Beyond the insect host and its diet, the insect microbiota may be interconnected with the surrounding ecosystem. The present study aimed to investigate the structure of the bacterial symbiome of Z. cucurbitae flies in different locations distributed in Bangladesh, trying to reveal any association between the insect’s bacterial community profile and their environment. The Illumina MiSeq platform for high-throughput sequencing of 16S rRNA gene was utilized to analyze and characterize the whole fly bacteriome.
2. Materials and Methods
2.1. Z. Cucurbitae Collection and Storage
Male adult Z. cucurbitae flies were collected from three locations (Table 1) in Bangladesh during the period of May and June 2013, with the use of cue-lure traps, which contained highly attractive kairomone lures for male melon flies [33]. The collected melon flies were placed individually in plastic vials to prevent cross-contamination of bacteria between flies of the same location as well as from different locations. Collected flies were preserved in vials containing 800 μL of absolute ethanol to protect them from regurgitation within the tubes, and stored at −20 °C.
Table 1.
Sampling locations in Bangladesh and the number of samples that were collected from each location.
| Region | Location | Coordinates | Number of Insects | |
|---|---|---|---|---|
| Latitude | Longitude | Male | ||
| Dinajpur | Northwest | 25.819010 | 88.649265 | 20 |
| Jessore | Southwest | 23.177112 | 89.180159 | 20 |
| Rajshahi | Northwest | 24.489453 | 88.612312 | 20 |
2.2. DNA Extraction, 1st Step PCR Amplification and Purification
The selected samples were firstly surface sterilized with sterile water and thereupon transferred to new tubes containing lysis buffer for grinding with a homogenizer. Total DNA extraction was performed following a simplified CTAB protocol [34]. The quality of the DNA preparations and the concentration of double-stranded DNA were both analyzed by microvolume spectrophotometry.
Polymerase chain reaction (PCR) was performed with KAPA HiFi HotStart ReadyMix PCR Kit (KAPA BioSystems, USA) and the previously extracted DNA as a template. The variable V3–V4 region of the bacterial 16S rRNA gene sequences was amplified with the primer pair U341F-MiSeq 5′-CCT ACG GGR SGC AGC AG-3′ and 805R-MiSeq 5′-GA CTA CHV GGG TAT CTA ATC C-3′ [35]. Each 25 μL reaction contained 5μL of KAPA HiFi Fidelity Buffer (5X), 0.7 μL of dNTPs solution (10 mM each), 0.7 μL of each primer solution (10 μM), 0.3 μL of KAPA HiFi HotStart DNA Polymerase solution (1 U/μL), 1 μL from the template DNA solution and was finalized with 16.6 μL of sterile deionized water. The PCR amplifications were performed with a 3-min incubation at 95 °C followed by 30 cycles of 98 °C for 20 s, 60 °C for 15 s and 72 °C for 45 s, and a final 1-min extension at 72 °C. Negative and positive controls were always performed in parallel. All PCR products were separated in a 1.5 % (w/v) agarose gel in TAE buffer (40 mM Tris–acetate, 1 mM EDTA). The desired approximately 550 bp amplification product was visualized in Bio-Rad’s Gel Doc XR+ system. Positive PCR products were purified from unincorporated primers and nucleotides with a 20 % PEG, 2.5 M NaCl solution, centrifuged at 14,000× g for 20 min and the precipitate was washed twice with 125 μL of a 70 % v/v ethanol solution and centrifuged at 14,000× g for 10 min. The dried precipitates were suspended in 15 μL of sterile deionized water and the concentration was measured with a Quawell Q5000 micro-volume UV−Vis spectrophotometer.
2.3. Index PCR Amplification and Purification
The resulting PCR amplicons were diluted up to 10 ng/μL and then used as templates within the second-step PCR for further amplification, and to include the indexes (barcodes) as well as the Illumina adaptors. The combinatorial use of index primers resulted in unique samples that were pooled and sequenced on one Illumina MiSeq run. In more detail, amplification reaction was performed using the KAPA HiFi HotStart PCR Kit in a final volume of 50 μL. Each reaction contained 10 μL of KAPA HiFi Fidelity Buffer (5×), 1.5 μL of dNTPs solution (10 mM each), 5 μL of the forward indexing primer (10 μM), 5 μL of the reverse indexing primer (10 μΜ), 1 μL of KAPA HiFi HotStart DNA Polymerase (1 U/μL), 2 μL from the diluted PCR product (10 ng/μL) and 25.5 μL of sterile deionized water. The PCR amplifications were performed with a 3-min incubation at 95 °C followed by 8 cycles of 95 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s, and a final 5-min terminator reaction at 72 °C. The resulting amplicons from indexing PCR were purified using Macherey-Nagel’s NucleoMag® NGS Clean-up and Size Selection kit according to the manufacturer’s recommendations. Amplicons from different samples were quantified with a Quawell Q5000 micro-volume UV−Vis spectrophotometer and merged in equimolar ratios (8 nM).
2.4. NGS Sequencing and Bioinformatics Analysis
Sequencing was performed on the Illumina MiSeq platform, in 300 bp paired-end runs, by Macrogen Inc. (Seoul, Korea). Raw sequencing reads were demultiplexed, converted to FASTQ, and the Illumina adapters were trimmed using Illumina standard algorithms. Bioinformatic analysis of raw sequencing reads was conducted in usearch v. 11 [36]. Paired-end reads were assembled, trimmed by length and further corrected for error and quality using the -fastq_mergepairs option. The quality of the assembled sequences was further improved using -fastq_filter, followed by identifying unique read sequences and abundances with the -fastx_uniques option. Sequences were clustered into Operational Taxonomic Units (OTUs) using the -cluster_otus command [37]. Clustering was performed at 97, 94, 90 and 81% sequence similarity. Cross-talk errors were identified and filtered with –uncross option based on the UNCROSS2 algorithm [38]. Taxonomy was assigned with Qiime2 [39] using the BLAST+ algorithm against the SILVA 128 release database [40].
Alpha diversity analyses were performed with the “diversity” function of the R package “vegan”, and alpha diversity indices were plotted using “ggplot” function from package “ggplot2”. Statistical differences in bacterial composition and relative abundance between populations were detected using the non-parametric Kruskal−Wallis Rank Sum Test along with Wilcoxon Rank Sum Test [41]. Beta-diversity analysis was calculated using generalized UniFrac [42]. Visualization of the multidimensional distance matrix in a space of two dimensions was performed by the robust nonmetric version of Multi-Dimensional Scaling (NMDS) [43]. A permutational multivariate analysis of variance using distance matrices was performed with the function “adonis” of the package “vegan” in order to determine if the separation of groups was significant, as a whole and in pairs [44]. The 16S rRNA sequences reported in this study have been deposited in NCBI under Bioproject number PRJNA701541.
Interactions between microorganisms were investigated and visualized through co-occurrence networks. These interactions refer to microorganisms performing similar or complementary functions and/or sharing similar environmental conditions, but not necessarily having physical interactions [45,46] Network analysis of OTUs was performed using the CoNet plugin [47] in Cytoscape 3.8.2 (Institute for system biology, Seattle, WA, USA), and co-occurrence graphs were obtained using Gephi 0.9.2 (Gephi, WebAtlas, Paris, France). To build the network, the Pearson and Spearman correlation coefficients, Mutual Information, and the Bray–Curtis and Kullback–Leibler dissimilarity indices were combined. To compute the statistical significance of the copresence/mutual exclusion, edge-specific permutation and bootstrap score distributions were calculated with 1000 iterations. Edges with original scores outside the 0.95 range of their bootstrap distribution were discarded, and p-values were corrected using the Benjamini–Hochberg method. Nodes in each network visualization corresponded to microbial OTUs and edges to the microbial associations. The size of each node was proportional to the degree of interactions.
3. Results
3.1. 16S rDNA Sequence Reads
In the present study, the microbiota diversity across three wild Z. cucurbitae populations from Bangladesh, was unraveled utilizing a deep-sequencing approach on the Illumina MiSeq platform. In total 60 samples were analyzed, 20 from each population. After demultiplexing, quality filtering and chimera removal, the data set consisted of 1.27 million high-quality paired end sequences. Samples contained on average 21,273 sequences that were divided into 523 OTUs. In total, 47 OTUs contained more than 0.1% of total sequences and were kept for the rest of the analysis (Supplementary Table S1). Based on 97% sequence similarity, 28 OTUs were classified into three phyla, four classes and 15 genera, while 19 remained unassigned and were classified at lower similarities (Supplementary Table S2). Seven remained unclassified at 94 and two at 90% sequence similarity. These unassigned OTUs could potentially belong to new uncharacterized taxa. Based on 81% sequence similarity, all OTUs classified into three phyla, four classes and 20 genera.
3.2. The Environment Shapes the Insect Bacteriome
3.2.1. Bacterial Diversity between Wild Populations
Beta-diversity analysis revealed that bacterial communities of wild Z. cucurbitae were clustered according to the geographic origin of their population. The NMDS plot based on generalized UniFrac showed that even though groups overlapped, they differed significantly (PERMANOVA, p < 0.001, Figure 1). The same picture was also observed after pairwise comparison between the three territories (PERMANOVA, p < 0.002, Supplementary Figure S1).
Figure 1.

Nonmetric multidimensional scaling (NMDS) plot of bacterial communities for Z. cucurbitae samples collected from Dinajpur (red), Jessore (green) and Rajshahi (blue) (p < 0.001). The ‘d’ indicates dissimilarity scale of the grid (d = 0.2 mean that the distance between two grid lines represents approximately 20% dissimilarity between the samples).
3.2.2. Comparing Alpha-Diversity between Different Populations
Differences in bacterial richness and diversity were observed between the three wild populations (Figure 2). The population from Jessore exhibited the highest richness and the population from Rajshahi the highest diversity. In terms of richness, Jessore showed marginally significantly higher values than Dinajpur (pairwise Wilcoxon Rank Sum Test: adjusted p < 0.05) but not Rajshahi. Additionally, based on Simpson and Shannon indices, bacterial diversity was significantly higher in Jessore and Rajshahi compared to Dinajpur (pairwise Wilcoxon Rank Sum Test: adjusted p < 0.05). Pielou’s eveness index also showed a statistical difference even between Rajshahi and Jessore (pairwise Wilcoxon Rank Sum Test: adjusted p < 0.05).
Figure 2.

Species richness and diversity indices with significance differences in natural populations of Z. cucurbitae samples collected from the regions of Dinajpur, Jessore, and Rajshahi. Boxes represent interquartile range (IQR), the line within the boxes is the median, and the dots represent samples.
3.2.3. Bacterial Composition of Wild Z. cucurbitae Populations
Sequences from three bacterial phyla were present in all wild Z. cucurbitae populations. All three territories, Dinajpur, Jessore and Rajshahi, showed similar composition at phylum level dominated by Proteobacteria (52.0 ± 4.2, 76.0 ± 2.1 and 65.5 ± 2.3%, respectively), Bacteroidetes (38.3 ± 4.1, 21.4 ± 2.0 and 26.4 ± 1.8%, respectively) and Firmicutes (9.6 ± 3.2, 2.7 ± 0.5 and 8.1 ± 1.6%, respectively). Firmicutes and Bacteroidetes were each represented by a single class, Bacilli and Bacteroidia, respectively (Figure 3). Two classes were found in Proteobacteria. The most abundant were Gammaproteobacteria (51.8 ± 4.2 Dinajpur; 69.9 ± 2.2 Jessore and 62.7 ± 2.2% Rajshahi), followed by a smaller portion of Deltaproteobacteria (0.3 ± 0.1 in Dinajpur; 6.1 ± 2.5 in Jessore and 2.7 ± 1.3% in Rajshahi) (Figure 3). Sequences that belonged to the families, Desulfovibrionaceae, Dysgonomonadaceae, Enterobacteriaceae, Enterococcaceae, Halomonadaceae, Orbaceae, Streptococcaceae and Weeksellaceae were identified (Figure 4). Among them, the most prevalent were Enterobacteriaceae (40.6 ± 3.9), Dysgonomonadaceae (27.3 ± 2.6), Orbaceae (20.6 ± 2.5) and Enterococcaceae (6.7 ± 1.7). At the genus level, Dysgonomonas was the most frequent in Dinajpur and Rajshahi (37.1 ± 4.0 and 25.2 ± 1.8%), and Orbus in Jessore (20.5 ± 2.8%), although Dysgonomonas retained high relative abundance in those samples too (19.7 ± 2.1%) (Figure 3 and Figure 5). The majority of OTUs were present in all three populations with varying frequencies, except for three genera (Supplementary Table S1), among them, an uncultured genus from the family Orbaceae (OTU13) that was only identified in Jessore (3.0 ± 3.0%). Ambiguous taxa, on the other hand, which are present in all populations (average 12.4 ± 2.2%), contain sequences that belong to an unclassified genus of the Enterobacteriaceae family (OTU4). Pairwise comparisons between populations revealed significant differences in the relative abundance of 14 OTUs (pairwise Wilcoxon Rank Sum Test; adjusted p-value < 0.05) (Supplementary Figure S2). Among them, sequences identified as Enterobacter (OTU123) were significantly more abundant in the population from Dinajpur compared to the other two and Klebsiella were significantly higher in Dinajpur and Jessore compared to Rajshahi (OTU45) (pairwise Wilcoxon Rank Sum Test; adjusted p-value < 0.05).
Figure 3.

Relative abundance of wild Z. cucurbitae microbiota at the Class (D_2) and Genus (D_5) Level.
Figure 4.

Heat map of bacterial families identified in populations of Z. cucurbitae from Bangladesh.
Figure 5.

Heat map of bacterial genera observed in wild populations of Z. cucurbitae from Bangladesh.
3.2.4. Mutual Exclusion/Co-Occurrence Network Analysis
Potential interactions between bacterial partners of each region were investigated with co-occurrence and mutual exclusion network analysis. The networks for each region were visualized at the family (Figure 6) and genus level, and detailed information about their topology is included in Supplementary Figure S3, Supplementary Tables S3 and S4. The three areas were characterized by a different number of OTUs (nodes), interactions (edges) and clustering coefficients. Dinajpur and Jessore were characterized by almost the same number of nodes (43 and 45, respectively), which was higher than Rajshahi (36 nodes). Moreover, bacterial communities from Dinajpur showed more interactions (113) and lower clustering coefficient (0.205) compared to Jessore (77 edges and 0.349 coefficient) and Rajshahi (40 edges and 0.291 coefficient). Most of the interactions were classified as mutual exclusions (59–78%), while 22–41% were cases of copresence. Proteobacteria dominated in terms of interactions in all three areas. At the genus level, Pectobacterium (OTU16 and OTU20) showed the highest degree of interactions in Dinajpur, Orbaceae (OTU13) and Dysgonomonas (OTU9) in Jessore, and Klebsiella (OTU281) with Enterobacter (OTU123) in Rajshahi.
Figure 6.
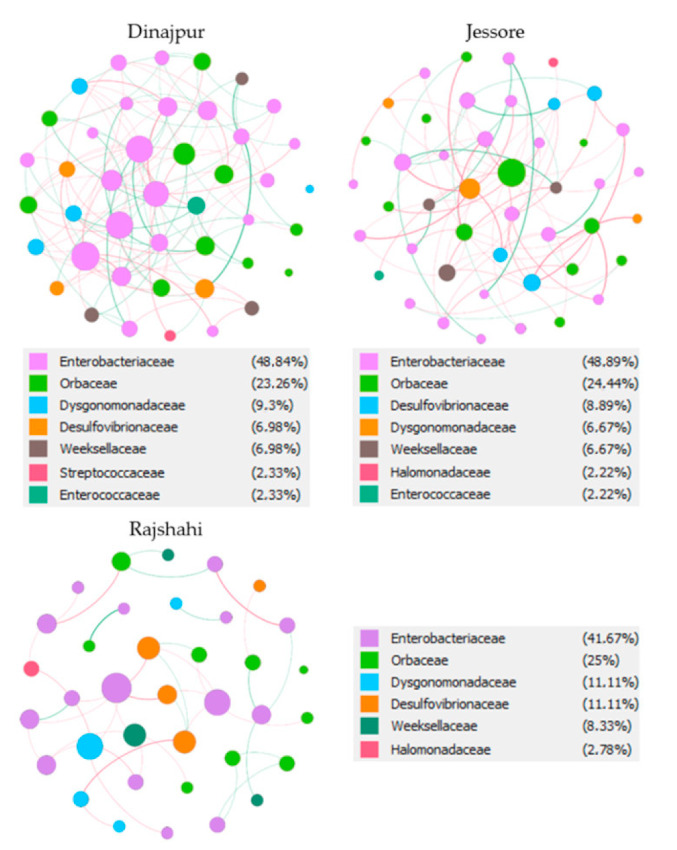
Mutual exclusion and co-occurrence networks at the family level for the OTUs that compose the bacterial communities of wild adult Z. cucurbitae flies from Bangladesh. The size of each node is proportional to the degree of interactions. Green edges represent cases of copresence and red edges mutual exclusion. The numbers in parentheses next to each OTU describe the percentage of nodes that belong to each family in the network.
4. Discussion
The aim of this study was to investigate the bacterial community structure of wild Z. cucurbitae flies that were collected in three districts of Bangladesh and assess the geographical impact on its composition. The analysis of the community was based on high throughput sequencing of the V3–V4 section of the bacterial 16S rDNA.
Taxonomic assignment of sequences revealed a large portion of uncharacterized bacterial diversity that resides within these flies. After OTU classification at 97%, 19 OTUs remained unassigned. Twelve of them were classified at 94% sequence similarity, five at 90% and two at 81%. Although the sequence similarity check is restricted at 460 bp and not the whole 16S rDNA, this result may still suggest the presence of new bacterial species, genera, and families. It is noteworthy that 40% (19 out of 47) of bacterial OTUs identified in the wild Z. cucurbitae could potentially belong to new families (2 out of 19), genera (5 out of 19) or species (12 out of 19), suggesting that there is a large portion of unexplored diversity in these populations. Characterizing this hidden bacterial diversity could help us identify important components of the melon fly bacteriome or taxa with interesting overall properties. This knowledge could prove useful, especially when we want to reconstruct the bacterial community of wild populations that have been introduced to the lab for mass-rearing purposes, in order to improve fitness and mating success. The process of colony adaptation typically results in the development of distinct bacterial communities between wild and laboratory populations, with laboratory populations usually experiencing a loss in diversity [10,19,48,49]. This could lead to the loss of important bacteria for the physiology of the mass-reared flies and a subsequent loss of fitness. Sex related differences in the microbiota have been documented in both wild and laboratory populations of the fly [10,13]. In this work however, only male subjects were analyzed, due to the fact that samples were collected using traps with male attractants.
Results suggest that the composition of the bacterial communities was significantly associated to the origin of the population. Changes in the structure of microbial communities among populations with different geographic distributions have been recorded in other Z. cucurbitae populations [16,50], but also in other fly species or insects [18,19,51,52,53]. Seasonal changes in climate conditions may shift the composition of the insect microbiome [54]. In our study, sampling took place within a short period (May−June) with fairly stable environmental conditions in all three areas. In order to better assess the effect of climate on the microbial profile, sampling could be performed within a longer period with seasonal weather variations. Other parameters affecting this differentiation could be related to host availability/adaptation or diet, since diet plays an important role in shaping the intestinal bacterial community of flies [49,55,56,57]. The diet of adult flies mainly includes plant exudates, honeydew and bird droppings scattered on leaves and fruits [58], while larvae develop in cucurbits [23]. This difference in dietary habits is reflected in the distinct microbial communities between developmental stages of the fly, especially between larval and adult stages [50,59]. However, regarding larvae, their microbiome did not seem significantly affected by different host plants [50]. In our case, since samples were collected with traps placed in orchards with various cucurbit crops, it was difficult to assess the specific adult diet or larval host, and therefore their impact on the identified bacterial communities.
More specifically, flies from Rajshahi and Jessore exhibited higher diversity and richness than Dinajpur. Proteobacteria, were the prevalent phylum in all samples, followed by a significant portion of Bacteroidetes. So far, Proteobacteria seem to be the prevalent phylum in Z. cucurbitae flies, whether they originate from laboratory or wild populations [10,13,16]. However, their relative abundance varies. Hadapad et al. identified higher relative abundance (87.7%) in wild Z. cucurbitae, from Karnataka, India, compared to the average relative abundance of our populations (64.5%) and even to the population with the highest percentage (76% in Jessore). Yong et al. [16] found on average 75.8% (59.82 to 97.69%) in three populations from Thailand, Peninsular Malaysia, and Sarawak.
Such differences could be explained by the impact of geography, host availability (different cucurbit hosts, e.g., pumpkin, bitter gourd, sweet gourd, bottle gourd, cucumber, etc.) or dietary habits. Other factors that might contribute to these differences are technical issues, such as the nature of samples (whole flies or specific tissue used, e.g., gut tissue), sample preservation and handling, or the number of reads per sample acquired during sequencing. For instance, it has been observed that preservation in ethanol has an impact on the resulting microbiome profiles, affecting the presence of both dominant and less represented OTUs [60].
Proteobacteria are generally associated with the gastrointestinal tract of flies, so their relative abundance is expected to increase when only the gut tissue is considered. This phylum was also dominant in mass-reared Z. cucurbitae from India (64.15%) [10] and Bangladesh (97%) [13]. Similarly, Bacteroidetes was the second most frequent phylum in all studies (21.4–38.3 our study; ~3–17 Hadapad et al.; 8.55–22.76 Yong et al.) and Firmicutes the third (2.7–9.6 our study; ~1.5–2.5 Hadapad et al.; 3.44–14.05 Yong et al.). Hadapad et al. also identified the presence of Cyanobacteria/Chloroplast but with very low relative abundance (<0.5%), while Yong et al. found traces of Actinobacteria (<0.01%). It seems that Z. cucurbitae flies contain a microbiome that consists of bacterial strains belonging to three phyla, Proteobacteria, Bacteroidetes and Firmicutes, with this specific order in terms of prevalence, but varying relative abundances. These phyla are also very common components of the microbiome of closely related Bactrocera species [11,14,18,61,62,63].
At family level, Z. cucurbitae populations exhibit differences. Our samples were characterized by the presence of Enterobacteriaceae, Dysgonomonadaceae, Orbaceae and Enterococcaceae. Yong et al. found 39 families across all samples, with six of them, namely Porphyromonadaceae, Enterococcaceae (3.5–14.1) Burkholderiaceae, Enterobacteriaceae, Pectobacteriaceae, and Orbaceae (7.4–14.7), being present in all three areas. Enterobacteriaceae was the prevalent family (43–67%). Dysgonomonadaceae was only found in very low frequencies, 0.2–1.4% [16]. The population from India mostly contained Enterobacteriaceae (dominant, 68.7%), Flavobacteriaceae, Brucellaceae, Pseudomonadaceae, Sphingobacteriaceae and Streptococcaceae. Enterococcaceae and Orbaceae were present but with relatively low frequencies, while Dysgonomonadaceae were not detected [10]. Enterobacteriaceae is usually among the dominant families in closely related Bactrocera or other fly species [11,14,18,19,31,49,61,62,63,64,65,66,67].
Bacteria of the genus Dysgonomonas showed the highest overall prevalence in the studied populations from Bangladesh. They were dominant in two populations and second only to Orbus in Jessore. Yong et al. identified the genus in all studied populations from Thailand, and Malaysia [16], but with low relative abundance (0–3.2%). In contrast, they were not found in wild Z. cucurbitae from India, but were highly abundant in mass reared adult flies [10]. A low presence of Dysgonomonas was also detected in wild populations of the relative species Bactrocera dorsalis from Hainan and Guizhou provinces in China [18]. A very low presence was detected in different developmental stages of B. dorsalis from Huizhou and Nansha from China [31]. Dysgonomonas sequences were also identified in all developmental stages of B. dorsalis from Wuhan [15]. Predominance of Dysgonomonas bacteria has been found in certain insects, such as wild Phormia regina flies (36.8%) [68] and bombardier beetles, Brachinus elongatulus (5.6–54.4%) [69]. Dysgonomonas are facultatively anaerobic cocci that produce acids by fermenting several carbohydrates [70]. Representatives of the genus that were isolated from the digestive tract of fungus-growing and wood-feeding species of termites exhibited such hydrolytic activity [71,72]. Genome sequencing and preliminary analysis of one of the isolated strains, revealed the presence of an array of hydrolytic enzymes related to decomposing lignocellulose, and carbohydrate degradation in general [73]. It is therefore possible that the bacteria are related to nutrition also in Z. cucurbitae flies, by participating in sugar fermentation.
The genus Orbus was overall the second most frequent in the studied populations. It was also among the dominant genera in wild populations from Thailand, and Malaysia, occurring in all populations with relative abundance values between 0–24.2% [16]. On the other hand, the bacteria were absent from Indian populations either when tested with high throughput sequencing [10] or culture dependent approaches [30,74]. Orbus bacteria were found in the closely related species from Chinese populations with 13.8–42.8% relative abundance [18], in B. dorsalis from Wuhan (0.04%) [15], but also in B. tryoni (Froggatt) (2%) and B. cacuminata (4.8%) from Australia [61]. The genus was also found in wild-collected Drosophila (29%) [49] and butterflies (9.8%) [75,76]. A Gram-negative, facultatively anaerobic, coccoid Orbus strain was isolated from the gut of the butterfly Sasakia charonda and exhibited similar carbohydrate hydrolytic activity to the Dysgonomonas bacteria mentioned previously [77]. The closely related Orbaceae species Gilliamella apicola, also provide their insect hosts, honeybees (Apis) and bumble bees (Bombus), with fatty acids by degrading plant carbohydrates [78]. An additional role to their host physiology is the detoxification of poisonous sugars [79].
As expected, many members of the dominant Enterobacteriaceae family were identified in our Z. cucurbitae samples. Among them Citrobacter were the most frequent, followed by Klebsiella, Providencia, Enterobacter and Pectobacterium. All of them were also identified in Thai and Malay populations with varying relative abundance [16]. In this case however, Klebsiella showed higher values (14–61.3%) compared to Citrobacter (0.4–35.2%), Providencia (0–9.4%), Pectobacterium (0.02–5.6%), and Enterobacter (0.07–0.5%). It is notable that only Providencia were identified in the samples from India [10], with this result probably reflecting differences in geographical origin, host availability and adaptation. However, four of them, were found in populations from India by culture dependent approaches, specifically Enterobacter (34.6%), Klebsiella (19.2%), Citrobacter (7.7%) and Providencia (7.7%) [30]. Isolates belonging to Enterobacter, Klebsiella, and Citrobacter were also found in another Indian population [74]. Klebsiella, Citrobacter, Pectobacterium and Enterobacter bacteria are mostly related to nitrogen fixation or exhibit pectinolytic activity. In this way, they provide their hosts with the nitrogen resources required to synthesize essential amino acids that they are otherwise unable to receive from their nitrogen-poor diet [64,65,80]. Some strains have also probiotic properties when provided as food supplements in mass-rearing facilities and could be used for improving important fitness parameters of hosts [5,6,8,81,82,83,84,85]. On the other hand, certain Providencia bacteria exhibit pathogenicity in insects [86,87]. However, their complete role in insects has not been fully deciphered, and thus a positive impact of these bacteria cannot be excluded.
Sequences belonging to Vagococcus and Desulfovibrio were also identified in our study. Vagococcus were overall more frequent, except for the population from Jessore. Yong et al. also identified both genera in all studied Z. cucurbitae populations, however, with different mean relative abundances (0–0.03% for Vagococcus and 0–28.1% for Desulfovibrio) [16]. Vagococcus were also detected in wild samples from India, while Desulfovibrio were absent [10]. Even though they are uncommon, these bacteria are present in other related species. Vagococcus sequences were present in B. dorsalis and B. minax from regions of China, with various frequencies, ranging from 0.001–11% [11,15,18,19,31,66]. On the other hand, Desulfovibrio are less common in B. dorsalis, with very low relative abundance (less than 0.1%) [31]. Similar to members of the Enterobacteriaceae, Desulfovibrio bacteria are possibly related to nutrition through nitrogen fixation for their hosts [64,80]. Bacteria of the genus Vagococcus have been isolated among others, from wasps [88], mosquitoes [89] and various species of flies [90,91]. Even though their importance in insect host physiology has not been fully explored, they have been shown to inhibit La Crosse virus in Aedes albopictus mosquitoes in vitro [92]. Some strains exhibit pathogenicity in the rainbow trout, Oncorhynchus mykiss [93], while others exhibit a probiotic effect, by stimulating the immune system of aquaculture fish or by protecting them from known pathogens, e.g., Vibrio anguillarum, a similar effect to that described in mosquitoes [94,95].
Network analysis indicated that the community from Dinajpur developed more interactions compared to the other two regions. Interestingly, certain OTUs with low relative abundance (even below 1%), like Shimwellia (OTU52), Gilliamella (OTU17), Chishuiella (OTU12), uncultured Orbaceae (OTU13) and Candidatus Schmidhempelia (OTU10) developed a high number of interactions, suggesting that even low represented OTUs may have an important role in the symbiotic community. For example, in the case of Dinajpur, even though Shimwellia (OTU52) and Escherichia-Shigella (OTU319) show only 0.1% relative abundance, they exhibit the highest number of positive interactions in the community.
5. Conclusions
Geographic distribution was clearly associated with the composition of bacterial communities in wild Z. cucurbitae flies from Bangladesh. All three studied districts/territories had developed distinct bacterial communities. However, the samples contained specific bacterial genera. The predominant were Dysgonomonas, Orbus, Citrobacter, Vagococcus, Klebsiella, Providencia, Desulfovibrio and Enterobacter. All genera were identified in previously studied populations from Thailand and Malaysia, but with differences in relative abundance. Most of these bacteria are related to nutrition in various insect species, except for Providencia that are generally pathogenic. Further characterization of this bacterial diversity with transcriptomic or metabolomic analyses, could shed light on their specific role in Z. cucurbitae natural populations. This metabolic potential, if it exists in Z. cucurbitae flies, could be exploited with the aim of improving its fitness during mass-rearing for SIT purposes.
Supplementary Materials
The following are available online at https://www.mdpi.com/2076-2607/9/3/659/s1. Figure S1: Pairwise non-metric multidimensional scaling (NMDS) plot of bacterial communities for Z. cucurbitae samples collected from Dinajpur (red), Jessore (green) and Rajshahi (blue). All p-values are below 0.05 indicating statistically significant differences. ‘d’ indicates dissimilarity scale of the grid (d = 0.2 mean that the distance between two grid lines represents approximately 20% dissimilarity between the samples); Figure S2: Alpha diversity indices and pairwise comparisons of OTUs that show statistically significant differences (p-value < 0.05) among the three tested areas; Figure S3: Mutual exclusion and co-occurrence networks among OTUs (genera) in the three tested areas; Table S1: Number of reads and the relative abundance of each OTU in the three tested populations; Table S2: Nineteen OTUs remained unassigned and were classified at lower similarities. These unassigned OTUs could potentially belong to new uncharacterized taxa. The column titled “Taxonomy assigned at” contains the % similarity threshold that each sequence was classified during taxonomy assignment; Table S3: Network statistics for each region; Table S4: Network statistics for the OTUs in the three tested areas. The column titled “degree” contains the total number of interactions. The column titled “posdegree” contains the number of positive interactions, and “negdegree” negative interactions.
Author Contributions
All authors contributed to the study conceptualization; Samples were collected and provided by M.K.; methodology, software, validation, formal analysis, and data curation, E.A., P.S., A.S., K.K., C.B., M.K. and G.T.; writing—original draft preparation, E.A. and P.S.; writing—review and editing, E.A., P.S., A.S., K.K., C.B., M.K. and G.T.; supervision, G.T.; All authors have read and agreed to the published version of the manuscript.
Funding
This research has been conducted with the support of the Erasmus+ Programme of the European Union. This work was also co-funded (a) by Greece and the European Union in the framework of the action titled “Innovation in Aquaculture” managed by the Special Management Service for Fisheries and Sea, and (b) by the International Atomic Energy research contract no. 22662 as part of the Coordinated Research Project ‘Improvement of Colony Management in Insect Mass-rearing for SIT Applications’.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Douglas A.E. Lessons from studying insect symbioses. Cell Host Microbe. 2011;10:359–367. doi: 10.1016/j.chom.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baumann P. Biology of bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu. Rev. Microbiol. 2005;59:155–189. doi: 10.1146/annurev.micro.59.030804.121041. [DOI] [PubMed] [Google Scholar]
- 3.Bourtzis K., Miller T.A., editors. Insect Symbiosis. 1st ed. Volume 3. CRC Press; Boca Raton, FL, USA: 2008. [Google Scholar]
- 4.Khan M., Pramanik K., Mahin A.-A., Miah A.B. Isolation and Identification of Mid-Gut Bacterial Community of Bactrocera dorsalis (Hendel) (Diptera: Tephritidae) Res. J. Microbiol. 2014;9:278–286. doi: 10.3923/jm.2014.278.286. [DOI] [Google Scholar]
- 5.Augustinos A.A., Kyritsis G.A., Papadopoulos N.T., Abd-Alla A.M.M., Cáceres C., Bourtzis K. Exploitation of the medfly gut microbiota for the enhancement of Sterile Insect Technique: Use of Enterobacter sp. in larval diet-based probiotic applications. PLoS ONE. 2015;10:e0136459. doi: 10.1371/journal.pone.0136459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao M., Zhang H., Cai P., Gu X., Wang D., Ji Q. Enhanced fitness of a Bactrocera cucurbitae genetic sexing strain based on the addition of gut-isolated probiotics (Enterobacter spec.) to the larval diet. Entomol. Exp. Et Appl. 2017;162:197–203. doi: 10.1111/eea.12529. [DOI] [Google Scholar]
- 7.Ras E., Beukeboom L.W., Cáceres C., Bourtzis K. Review of the role of gut microbiota in mass rearing of the olive fruit fly, Bactrocera oleae, and its parasitoids. Entomol. Exp. Appl. 2017;164:237–256. doi: 10.1111/eea.12609. [DOI] [Google Scholar]
- 8.Ben Ami E., Yuval B., Jurkevitch E. Manipulation of the microbiota of mass-reared Mediterranean fruit flies Ceratitis capitata (Diptera: Tephritidae) improves sterile male sexual performance. ISME J. 2010;4:28–37. doi: 10.1038/ismej.2009.82. [DOI] [PubMed] [Google Scholar]
- 9.Bai Z., Liu L., Noman M.S., Zeng L., Luo M., Li Z. The influence of antibiotics on gut bacteria diversity associated with laboratory-reared Bactrocera dorsalis. Bull. Entomol. Res. 2018;109:1–10. doi: 10.1017/S0007485318000834. [DOI] [PubMed] [Google Scholar]
- 10.Hadapad A.B., Shettigar S.K.G., Hire R.S. Bacterial communities in the gut of wild and mass-reared Zeugodacus cucurbitae and Bactrocera dorsalis revealed by metagenomic sequencing. BMC Microbiol. 2019;19:282. doi: 10.1186/s12866-019-1647-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang A., Yao Z., Zheng W., Zhang H. Bacterial Communities in the gut and reproductive organs of Bactrocera minax (Diptera: Tephritidae) based on 454 pyrosequencing. PLoS ONE. 2014;9:e106988. doi: 10.1371/journal.pone.0106988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stathopoulou P., Asimakis E.D., Khan M., Caceres C., Bourtzis K., Tsiamis G. Irradiation effect on the structure of bacterial communities associated with the Oriental fruit fly, Bactrocera dorsalis. Entomol. Exp. Et Appl. 2019;167:209–219. doi: 10.1111/eea.12770. [DOI] [Google Scholar]
- 13.Asimakis E.D., Khan M., Stathopoulou P., Caceres C., Bourtzis K., Tsiamis G. The effect of diet and radiation on the bacterial symbiome of the melon fly, Zeugodacus cucurbitae (Coquillett) BMC Biotechnol. 2019;19:88. doi: 10.1186/s12896-019-0578-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andongma A.A., Wan L., Dong Y.-C., Wang Y.-L., He J., Niu C.-Y. Assessment of the bacteria community structure across life stages of the Chinese citrus fly, Bactrocera minax (Diptera: Tephritidae) BMC Microbiol. 2019;19:285. doi: 10.1186/s12866-019-1646-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andongma A.A., Wan L., Dong Y.-C., Li P., Desneux N., White J.A., Niu C.-Y. Pyrosequencing reveals a shift in symbiotic bacteria populations across life stages of Bactrocera dorsalis. Sci. Rep. 2015;5:9470. doi: 10.1038/srep09470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yong H.-S., Song S.-L., Eamsobhana P., Pasartvit A., Lim P.-E. Differential abundance and core members of the bacterial community associated with wild male Zeugodacus cucurbitae fruit flies (Insecta: Tephritidae) from three geographical regions of Southeast Asia. Mol. Biol. Rep. 2019;46:3765–3776. doi: 10.1007/s11033-019-04818-3. [DOI] [PubMed] [Google Scholar]
- 17.Nikolouli K., Augustinos A.A., Stathopoulou P., Asimakis E., Mintzas A., Bourtzis K., Tsiamis G. Genetic structure and symbiotic profile of worldwide natural populations of the Mediterranean fruit fly, Ceratitis capitata. BMC Genet. 2020;21:128. doi: 10.1186/s12863-020-00946-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu S.-H., Chen Y., Li W., Tang G.-H., Yang Y., Jiang H.-B., Dou W., Wang J.-J. Diversity of bacterial communities in the intestinal tracts of two geographically distant populations of Bactrocera dorsalis (Diptera: Tephritidae) J. Econ. Entomol. 2018;111:2861–2868. doi: 10.1093/jee/toy231. [DOI] [PubMed] [Google Scholar]
- 19.Liu L.J., Martinez-Sañudo I., Mazzon L., Prabhakar C.S., Girolami V., Deng Y.L., Dai Y., Li Z.H. Bacterial communities associated with invasive populations of Bactrocera dorsalis (Diptera: Tephritidae) in China. Bull. Entomol. Res. 2016;106:718–728. doi: 10.1017/S0007485316000390. [DOI] [PubMed] [Google Scholar]
- 20.White I.M., Elson-Harris M.M. Fruit Flies of Economic Significance: Their Identification and Bionomics. CAB International; London, UK: 1992. [Google Scholar]
- 21.Vargas R.I., Piñero J.C., Leblanc L. An overview of pest species of Bactrocera fruit flies (Diptera: Tephritidae) and the integration of biopesticides with other biological approaches for their management with a focus on the Pacific region. Insects. 2015;6:297–318. doi: 10.3390/insects6020297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drew R.A.I. The tropical fruit flies (Diptera: Tephritidae: Dacinae) of the Australasian and Oceanian regions. Corrigendum to Memoirs of the Queensland Museum 26: 1-521. Mem. Qld. Mus. 1990;28:664. [Google Scholar]
- 23.De Meyer M., Delatte H., Mwatawala M., Quilici S., Vayssieres J.-F., Virgilio M. A review of the current knowledge on Zeugodacus cucurbitae (Coquillett) (Diptera, Tephritidae) in Africa, with a list of species included in Zeugodacus. ZooKeys. 2015;540:539–557. doi: 10.3897/zookeys.540.9672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krosch M.N., Schutze M.K., Armstrong K.F., Graham G.C., Yeates D.K., Clarke A.R. A molecular phylogeny for the Tribe Dacini (Diptera: Tephritidae): Systematic and biogeographic implications. Mol. Phylogenetics Evol. 2012;64:513–523. doi: 10.1016/j.ympev.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 25.Virgilio M., Jordaens K., Verwimp C., White I.M., De Meyer M. Higher phylogeny of frugivorous flies (Diptera, Tephritidae, Dacini): Localised partition conflicts and a novel generic classification. Mol. Phylogenetics Evol. 2015;85:171–179. doi: 10.1016/j.ympev.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Bess H.A., Van Den Bosch R., Haramoto F.H. Fruit fly parasites and their activities in Hawaii. Hawaii Entomol. Soc. 1961;17:367–378. [Google Scholar]
- 27.Dhillon M.K., Singh R., Naresh J.S., Sharma H.C. The melon fruit fly, Bactrocera cucurbitae: A review of its biology and management. J. Insect Sci. 2005;5:40. doi: 10.1093/jis/5.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koyama J., Kakinohana H., Miyatake T. Eradication of the melon fly, Bactrocera cucurbitae, in Japan: Importance of behavior, ecology, genetics, and evolution. Annu. Rev. Entomol. 2004;49:331–349. doi: 10.1146/annurev.ento.49.061802.123224. [DOI] [PubMed] [Google Scholar]
- 29.Aharon Y., Pasternak Z., Yosef M.B., Behar A., Lauzon C., Yuval B., Jurkevitch E. Phylogenetic, metabolic, and taxonomic diversities shape Mediterranean fruit fly microbiotas during ontogeny. Appl. Environ. Microbiol. 2013;79:303–313. doi: 10.1128/AEM.02761-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hadapad A.B., Prabhakar C.S., Chandekar S.C., Tripathi J., Hire R.S. Diversity of bacterial communities in the midgut of Bactrocera cucurbitae (Diptera: Tephritidae) Populations and Their Potential Use as Attractants. Pest. Manag. Sci. 2016;72:1222–1230. doi: 10.1002/ps.4102. [DOI] [PubMed] [Google Scholar]
- 31.Zhao X., Zhang X., Chen Z., Wang Z., Lu Y., Cheng D. The divergence in bacterial components associated with Bactrocera dorsalis across developmental stages. Front. Microbiol. 2018;9 doi: 10.3389/fmicb.2018.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gujjar N., Selvakumar G., Verghese A., Subramaniam S., More R. Diversity of the cultivable gut bacterial communities associated with the fruit flies Bactrocera dorsalis and Bactrocera cucurbitae (Diptera: Tephritidae) Phytoparasitica. 2017;45:453–460. doi: 10.1007/s12600-017-0604-z. [DOI] [Google Scholar]
- 33.Vargas R.I., Stark J.D., Kido M.H., Ketter H.M., Whitehand L.C. Methyl eugenol and cue-lure traps for suppression of male Oriental fruit flies and melon flies (Diptera: Tephritidae) in Hawaii: Effects of lure mixtures and weathering. J. Econ. Entomol. 2000;93:81–87. doi: 10.1603/0022-0493-93.1.81. [DOI] [PubMed] [Google Scholar]
- 34.Augustinos A.A., Santos-Garcia D., Dionyssopoulou E., Moreira M., Papapanagiotou A., Scarvelakis M., Doudoumis V., Ramos S., Aguiar A.F., Borges P.A.V., et al. Detection and characterization of Wolbachia infections in natural populations of aphids: Is the hidden diversity fully unraveled? PLoS ONE. 2011;6:e28695. doi: 10.1371/journal.pone.0028695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., Glöckner F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and Next-Generation Sequencing-based diversity studies. Nucleic Acids Res. 2013;41 doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edgar R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 37.Edgar R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 38.Edgar R.C. UNCROSS2: Identification of cross-talk in 16S rRNA OTU tables. bioRxiv. 2018:400762. doi: 10.1101/400762. [DOI] [Google Scholar]
- 39.Bolyen E., Rideout J.R., Dillon M.R., Bokulich N.A., Abnet C.C., Al-Ghalith G.A., Alexander H., Alm E.J., Arumugam M., Asnicar F., et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glöckner F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hollander M., Wolfe D.A., Chicken E. Nonparametric Statistical Methods. 3rd ed. John Wiley & Sons; Hoboken, NJ, USA: 2013. [Google Scholar]
- 42.Chen J., Bittinger K., Charlson E.S., Hoffmann C., Lewis J., Wu G.D., Collman R.G., Bushman F.D., Li H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics. 2012;28:2106–2113. doi: 10.1093/bioinformatics/bts342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minchin P.R. An evaluation of the relative robustness of techniques for ecological ordination. Vegetatio. 1987;69:89–107. doi: 10.1007/BF00038690. [DOI] [Google Scholar]
- 44.Anderson M.J. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 2001;26:32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x. [DOI] [Google Scholar]
- 45.Steele J.A., Countway P.D., Xia L., Vigil P.D., Beman J.M., Kim D.Y., Chow C.-E.T., Sachdeva R., Jones A.C., Schwalbach M.S., et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 2011;5:1414–1425. doi: 10.1038/ismej.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou J., Deng Y., Luo F., He Z., Yang Y. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. mBio. 2011;2 doi: 10.1128/mBio.00122-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faust K., Raes J. CoNet App: Inference of biological association networks using Cytoscape. F1000Res. 2016;5:1519. doi: 10.12688/f1000research.9050.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zouache K., Voronin D., Tran-Van V., Mavingui P. Composition of bacterial communities associated with natural and laboratory populations of Asobara tabida infected with Wolbachia. Appl. Environ. Microbiol. 2009;75:3755–3764. doi: 10.1128/AEM.02964-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chandler J.A., Lang J.M., Bhatnagar S., Eisen J.A., Kopp A. Bacterial communities of diverse Drosophila species: Ecological context of a host–microbe model system. PloS Genet. 2011;7:e1002272. doi: 10.1371/journal.pgen.1002272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Cock M., Virgilio M., Vandamme P., Bourtzis K., De Meyer M., Willems A. Comparative microbiomics of tephritid frugivorous pests (Diptera: Tephritidae) from the field: A tale of high variability across and within species. Front. Microbiol. 2020;11 doi: 10.3389/fmicb.2020.01890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yun J.-H., Roh S.W., Whon T.W., Jung M.-J., Kim M.-S., Park D.-S., Yoon C., Nam Y.-D., Kim Y.-J., Choi J.-H., et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 2014;80:5254–5264. doi: 10.1128/AEM.01226-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koskinioti P., Ras E., Augustinos A.A., Tsiamis G., Beukeboom L.W., Caceres C., Bourtzis K. The effects of geographic origin and antibiotic treatment on the gut symbiotic communities of Bactrocera oleae populations. Entomol. Exp. Et Appl. 2019;167:197–208. doi: 10.1111/eea.12764. [DOI] [Google Scholar]
- 53.Bel Mokhtar N., Maurady A., Britel M.R., El Bouhssini M., Batargias C., Stathopoulou P., Asimakis E., Tsiamis G. Detection of Wolbachia infections in natural and laboratory populations of the Moroccan hessian fly, Mayetiola destructor (Say) Insects. 2020;11:340. doi: 10.3390/insects11060340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferguson L.V., Dhakal P., Lebenzon J.E., Heinrichs D.E., Bucking C., Sinclair B.J. Seasonal shifts in the insect gut microbiome are concurrent with changes in cold tolerance and immunity. Funct. Ecol. 2018;32:2357–2368. doi: 10.1111/1365-2435.13153. [DOI] [Google Scholar]
- 55.Colman D.R., Toolson E.C., Takacs-Vesbach C.D. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 2012;21:5124–5137. doi: 10.1111/j.1365-294X.2012.05752.x. [DOI] [PubMed] [Google Scholar]
- 56.Engel P., Moran N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013;37:699–735. doi: 10.1111/1574-6976.12025. [DOI] [PubMed] [Google Scholar]
- 57.Lü J., Guo W., Chen S., Guo M., Qiu B., Yang C., Lian T., Pan H. Host plants influence the composition of the gut bacteria in Henosepilachna vigintioctopunctata. PLoS ONE. 2019;14:e0224213. doi: 10.1371/journal.pone.0224213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ben-Yosef M., Pasternak Z., Jurkevitch E., Yuval B. Symbiotic bacteria enable olive flies (Bactrocera oleae) to exploit intractable sources of nitrogen. J. Evol. Biol. 2014;27:2695–2705. doi: 10.1111/jeb.12527. [DOI] [PubMed] [Google Scholar]
- 59.Choudhary J.S., Naaz N., Prabhakar C.S., Das B., Singh A.K., Bhatt B.P. High taxonomic and functional diversity of bacterial communities associated with melon fly, Zeugodacus cucurbitae (Diptera: Tephritidae) Curr. Microbiol. 2021;78:611–623. doi: 10.1007/s00284-020-02327-2. [DOI] [PubMed] [Google Scholar]
- 60.De Cock M., Virgilio M., Vandamme P., Augustinos A., Bourtzis K., Willems A., De Meyer M. Impact of sample preservation and manipulation on insect gut microbiome profiling. A test case with fruit flies (Diptera, Tephritidae) Front. Microbiol. 2019;10 doi: 10.3389/fmicb.2019.02833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morrow J.L., Frommer M., Shearman D.C.A., Riegler M. The microbiome of field-caught and laboratory-adapted Australian tephritid fruit fly species with different host plant use and specialisation. Microb. Ecol. 2015;70:498–508. doi: 10.1007/s00248-015-0571-1. [DOI] [PubMed] [Google Scholar]
- 62.Yong H.-S., Song S.-L., Chua K.-O., Lim P.-E. Microbiota associated with Bactrocera carambolae and B dorsalis (Insecta: Tephritidae) revealed by Next-Generation Sequencing of 16S rRNA gene. Meta Gene. 2017;11:189–196. doi: 10.1016/j.mgene.2016.10.009. [DOI] [Google Scholar]
- 63.Yong H.-S., Song S.-L., Chua K.-O., Lim P.-E. High diversity of bacterial communities in developmental stages of Bactrocera carambolae (Insecta: Tephritidae) revealed by Illumina MiSeq sequencing of 16S rRNA gene. Curr. Microbiol. 2017;74:1076–1082. doi: 10.1007/s00284-017-1287-x. [DOI] [PubMed] [Google Scholar]
- 64.Behar A., Yuval B., Jurkevitch E. Enterobacteria-mediated nitrogen fixation in natural populations of the fruit fly Ceratitis capitata. Mol. Ecol. 2005;14:2637–2643. doi: 10.1111/j.1365-294X.2005.02615.x. [DOI] [PubMed] [Google Scholar]
- 65.Behar A., Jurkevitch E., Yuval B. Bringing back the fruit into fruit fly–bacteria interactions. Mol. Ecol. 2008;17:1375–1386. doi: 10.1111/j.1365-294X.2008.03674.x. [DOI] [PubMed] [Google Scholar]
- 66.Wang H., Jin L., Zhang H. Comparison of the diversity of the bacterial communities in the intestinal tract of adult Bactrocera dorsalis from three different populations. J. Appl. Microbiol. 2011;110:1390–1401. doi: 10.1111/j.1365-2672.2011.05001.x. [DOI] [PubMed] [Google Scholar]
- 67.Deutscher A.T., Burke C.M., Darling A.E., Riegler M., Reynolds O.L., Chapman T.A. Near full-length 16S rRNA gene Next-Generation Sequencing revealed Asaia as a common midgut bacterium of wild and domesticated Queensland fruit fly larvae. Microbiome. 2018;6:85. doi: 10.1186/s40168-018-0463-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deguenon J.M., Travanty N., Zhu J., Carr A., Denning S., Reiskind M.H., Watson D.W., Michael Roe R., Ponnusamy L. Exogenous and endogenous microbiomes of wild-caught Phormia regina (Diptera: Calliphoridae) flies from a suburban farm by 16S rRNA gene sequencing. Sci. Rep. 2019;9:20365. doi: 10.1038/s41598-019-56733-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McManus R., Ravenscraft A., Moore W. Bacterial associates of a gregarious riparian beetle with explosive defensive chemistry. Front. Microbiol. 2018;9 doi: 10.3389/fmicb.2018.02361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hofstad T., Olsen I., Eribe E.R., Falsen E., Collins M.D., Lawson P.A. Dysgonomonas gen. nov. to accommodate Dysgonomonas gadei sp. nov., an organism isolated from a human gall bladder, and Dysgonomonas capnocytophagoides (formerly CDC group DF-3) Int. J. Syst. Evol. Microbiol. 2000;50:2189–2195. doi: 10.1099/00207713-50-6-2189. [DOI] [PubMed] [Google Scholar]
- 71.Yang Y.-J., Zhang N., Ji S.-Q., Lan X., Zhang K., Shen Y.-L., Li F.-L., Ni J.-F. Dysgonomonas macrotermitis sp. nov., isolated from the hindgut of a fungus-growing termite. Int. J. Syst. Evol. Microbiol. 2014;64:2956–2961. doi: 10.1099/ijs.0.061739-0. [DOI] [PubMed] [Google Scholar]
- 72.Pramono A.K., Sakamoto M., Iino T., Hongoh Y., Ohkuma M. Dysgonomonas termitidis sp. nov., isolated from the gut of the subterranean termite Reticulitermes speratus. Int. J. Syst. Evol. Microbiol. 2015;65:681–685. doi: 10.1099/ijs.0.070391-0. [DOI] [PubMed] [Google Scholar]
- 73.Sun X., Yang Y., Zhang N., Shen Y., Ni J. Draft genome sequence of Dysgonomonas macrotermitis strain JCM 19375T, isolated from the gut of a termite. Genome Announc. 2015;3 doi: 10.1128/genomeA.00963-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mishra M., Sharma K., Subramanian S. Characterization of culturable gut bacterial isolates from wild population of melon fruit fly (Bactrocera cucurbitae) and assessing their attractancy potential for sustainable pest management. Phytoparasitica. 2018;46:583–594. doi: 10.1007/s12600-018-0694-2. [DOI] [Google Scholar]
- 75.Hammer T.J., McMillan W.O., Fierer N. Metamorphosis of a butterfly-associated bacterial community. PLoS ONE. 2014;9:e86995. doi: 10.1371/journal.pone.0086995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ravenscraft A., Berry M., Hammer T., Peay K., Boggs C. Structure and function of the bacterial and fungal gut microbiota of neotropical butterflies. Ecol. Monogr. 2019;89:e01346. doi: 10.1002/ecm.1346. [DOI] [Google Scholar]
- 77.Kim J.Y., Lee J., Shin N.-R., Yun J.-H., Whon T.W., Kim M.-S., Jung M.-J., Roh S.W., Hyun D.-W., Bae J.-W. Orbus sasakiae sp. nov., a bacterium isolated from the gut of the butterfly Sasakia charonda, and emended description of the Genus Orbus. Int. J. Syst. Evol. Microbiol. 2013;63:1766–1770. doi: 10.1099/ijs.0.041871-0. [DOI] [PubMed] [Google Scholar]
- 78.Kwong W.K., Moran N.A. Cultivation and characterization of the gut symbionts of honey bees and bumble bees: Description of Snodgrassella alvi gen. nov., sp. nov., a member of the Family Neisseriaceae of the Betaproteobacteria, and Gilliamella apicola gen. nov., sp. nov., a member of Orbaceae fam. nov., Orbales ord. nov., a sister taxon to the Order ‘Enterobacteriales’ of the Gammaproteobacteria. Int. J. Syst. Evol. Microbiol. 2013;63:2008–2018. doi: 10.1099/ijs.0.044875-0. [DOI] [PubMed] [Google Scholar]
- 79.Zheng H., Nishida A., Kwong W.K., Koch H., Engel P., Steele M.I., Moran N.A. Metabolism of toxic sugars by strains of the bee gut symbiont Gilliamella apicola. mBio. 2016:7. doi: 10.1128/mBio.01326-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ohkuma M., Noda S., Kudo T. Phylogenetic Diversity of nitrogen fixation genes in the symbiotic microbial community in the gut of diverse termites. Appl Env. Microbiol. 1999;65:4926–4934. doi: 10.1128/AEM.65.11.4926-4934.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gavriel S., Jurkevitch E., Gazit Y., Yuval B. Bacterially enriched diet improves sexual performance of sterile male Mediterranean fruit flies. J. Appl. Entomol. 2011;135:564–573. doi: 10.1111/j.1439-0418.2010.01605.x. [DOI] [Google Scholar]
- 82.Hamden H., Guerfali M.M., Fadhl S., Saidi M., Chevrier C. Fitness improvement of mass-reared sterile males of Ceratitis capitata (Vienna 8 Strain) (Diptera: Tephritidae) after gut enrichment with probiotics. J Econ Entomol. 2013;106:641–647. doi: 10.1603/EC12362. [DOI] [PubMed] [Google Scholar]
- 83.Rashid M.A., Andongma A.A., Dong Y.-C., Ren X.-M., Niu C.-Y. Effect of gut bacteria on fitness of the Chinese citrus fly, Bactrocera minax (Diptera: Tephritidae) Symbiosis. 2018;76:63–69. doi: 10.1007/s13199-018-0537-4. [DOI] [Google Scholar]
- 84.Cai Z., Yao Z., Li Y., Xi Z., Bourtzis K., Zhao Z., Bai S., Zhang H. Intestinal probiotics restore the ecological fitness decline of Bactrocera dorsalis by irradiation. Evol. Appl. 2018;11:1946–1963. doi: 10.1111/eva.12698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shuttleworth L.A., Khan M.A.M., Osborne T., Collins D., Srivastava M., Reynolds O.L. A walk on the wild side: Gut bacteria fed to mass-reared larvae of Queensland fruit fly [Bactrocera tryoni (Froggatt)] Influence Development. BMC Biotechnol. 2019;19:95. doi: 10.1186/s12896-019-0579-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Galac M.R., Lazzaro B.P. Comparative pathology of bacteria in the Genus Providencia to a natural host, Drosophila melanogaster. Microbes Infect. 2011;13:673–683. doi: 10.1016/j.micinf.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guerfali M.M., Djobbi W., Charaabi K., Hamden H., Fadhl S., Marzouki W., Dhaouedi F., Chevrier C. Evaluation of Providencia rettgeri pathogenicity against laboratory Mediterranean fruit fly strain (Ceratitis capitata) PLoS ONE. 2018;13:e0196343. doi: 10.1371/journal.pone.0196343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Killer J., Švec P., Sedláček I., Černohlávková J., Benada O., Hroncová Z., Havlík J., Vlková E., Rada V., Kopečný J., et al. Vagococcus entomophilus sp. nov., from the digestive tract of a wasp (Vespula vulgaris) Int. J. Syst. Evol. Microbiol. 2014;64:731–737. doi: 10.1099/ijs.0.054940-0. [DOI] [PubMed] [Google Scholar]
- 89.Chandel K., Parikh R.Y., Mendki M.J., Shouche Y.S., Veer V. Isolation and characterization of Vagococcus sp from midgut of Culex quinquefasciatus (Say) Mosquito. J. Vector. Borne Dis. 2015;52:52–57. [PubMed] [Google Scholar]
- 90.Gupta A.K., Nayduch D., Verma P., Shah B., Ghate H.V., Patole M.S., Shouche Y.S. Phylogenetic characterization of bacteria in the gut of house flies (Musca domestica L.) FEMS Microbiol. Ecol. 2012;79:581–593. doi: 10.1111/j.1574-6941.2011.01248.x. [DOI] [PubMed] [Google Scholar]
- 91.Malele I., Nyingilili H., Lyaruu E., Tauzin M., Bernard Ollivier B., Cayol J.-L., Fardeau M.-L., Geiger A. Bacterial diversity obtained by culturable approaches in the gut of Glossina pallidipes population from a non sleeping sickness focus in Tanzania: Preliminary Results. BMC Microbiol. 2018;18 doi: 10.1186/s12866-018-1288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Joyce J.D., Nogueira J.R., Bales A.A., Pittman K.E., Anderson J.R. Interactions between La Crosse Virus and bacteria isolated from the digestive tract of Aedes albopictus (Diptera: Culicidae) J. Med. Entomol. 2011;48:389–394. doi: 10.1603/ME09268. [DOI] [PubMed] [Google Scholar]
- 93.Ruiz-Zarzuela I., de Bias I., Gironés O., Ghittino C., MúAzquiz J.L. Isolation of Vagococcus salmoninarum in rainbow trout, Oncorhynchus mykiss (Walbaum), broodstocks: Characterization of the pathogen. Vet. Res. Commun. 2005;29:553–562. doi: 10.1007/s11259-005-2493-8. [DOI] [PubMed] [Google Scholar]
- 94.Sorroza L., Padilla D., Acosta F., Román L., Grasso V., Vega J., Real F. Characterization of the probiotic strain Vagococcus fluvialis in the protection of European sea bass (Dicentrarchus labrax) against vibriosis by Vibrio anguillarum. Vet. Microbiol. 2012;155:369–373. doi: 10.1016/j.vetmic.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 95.Román L., Real F., Sorroza L., Padilla D., Acosta B., Grasso V., Bravo J., Acosta F. The in vitro effect of probiotic Vagococcus fluvialis on the innate immune parameters of Sparus aurata and Dicentrarchus labrax. Fish Shellfish Immunol. 2012;33:1071–1075. doi: 10.1016/j.fsi.2012.06.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data sharing is not applicable to this article.