

Abstract
Cell-to-cell heterogeneity in metabolism plays an unknown role in physiology and pharmacology. To functionally characterize cellular variability in metabolism, we treated cells with inhibitors of oxidative phosphorylation (OXPHOS) and monitored their responses with live-cell reporters for ATP, ADP/ATP, or activity of the energy-sensing kinase AMPK. Across multiple OXPHOS inhibitors and cell types, we identified a subpopulation of cells resistant to activation of AMPK and reduction of ADP/ATP ratio. This resistant state persists transiently for at least several hours and can be inherited during cell divisions. OXPHOS inhibition suppresses the mTORC1 and ERK growth signaling pathways in sensitive cells, but not in resistant cells. Resistance is linked to a multi-factorial combination of increased glucose uptake, reduced protein biosynthesis, and G0/G1 cell cycle status. Our results reveal dynamic fluctuations in cellular energetic balance and provide a basis for measuring and predicting the distribution of cellular responses to OXPHOS inhibition.
Graphical Abstract
eTOC blurb
Kosaisawe et al. develop a live-cell imaging method to interrogate cellular usage of oxidative phosphorylation (OXPHOS) for ATP generation. The single-cell resolution enabled by this approach reveals a distinct form of heterogeneity in cellular metabolism, in which certain cells rely on OXPHOS to maintain energy charge, whereas other cells do not. These states of sensitivity and resistance to OXPHOS inhibition interchange over time, modulate AMPK, ERK and mTOR activation, and are set by each cell’s balance of glycolytic capacity and ATP turnover.
Introduction
Metabolic functions vary across different cell types and tissues (Hensley et al., 2016; Konagaya et al., 2017; Tasdogan et al., 2020), but much less is known about cell-to-cell metabolic heterogeneity. Single-cell resolution is important because cellular heterogeneity limits drug efficacy (Altschuler and Wu, 2010), and targeting of metabolic functions for therapeutic purposes will require identification of resistant subpopulations of cells. Current approaches measure the expression profiles of metabolic enzymes (Hartmann et al., 2020; Xiao et al., 2019), rather than directly assessing metabolites or their flux. Other work has used biosensors to reveal variability in clonal cell lines responding to inhibitors of metabolism (Hung et al., 2017) and in cellular metabolic disposition based on responses to glucose withdrawal (Depaoli et al., 2018). However, while it is typical for single-cell approaches to reveal heterogeneity, it is not clear how the observed variation originates within an isogenic population of cells, and what impact it has on pathways downstream of the immediate response.
To approach these questions, we focused on inhibitors of oxidative phosphorylation (OXPHOS), which are important both as tools to probe cellular metabolism and as treatments for diabetes and cancer. Naturally occurring OXPHOS inhibitors, including oligomycin, antimycin, and rotenone are produced defensively in microorganisms and plants. Biguanides derived from the plant compound galegine, including metformin and phenformin, are weaker inhibitors of OXPHOS. Metformin is used widely to treat type II diabetes and also has anti-tumorigenic, anti-fibrotic, and pro-longevity effects (Anisimov, 2010; Dos Santos et al., 2018). These effects have spurred the search for additional OXPHOS inhibitors, leading to the development of compounds including IACS-010759 and Gboxin, which are being evaluated as cancer therapies (Molina et al., 2018; Shi et al., 2019). Many cancers upregulate OXPHOS and the tricarboxylic acid (TCA) cycle and rely on them for ATP production, biosynthesis (Vander Heiden and DeBerardinis, 2017) or resistance to chemotherapy (Vashisht Gopal et al., 2019).
By inhibiting ATP generation, OXPHOS inhibitors induce metabolic stress, which can range in severity from mild activation of stress pathways to a lethal energetic crisis. Understanding the cellular response to OXPHOS inhibition will improve on existing treatments for diabetes, cancer, and other conditions (Ashton et al., 2018; Stoker et al., 2019) and illuminate the fundamental cell biology of metabolic stress responses (Wu et al., 2016). Potent OXPHOS inhibitors (e.g. oligomycin) block oxygen consumption within seconds (Ruas et al., 2018), preventing TCA cycle turning and increasing flux through glycolysis to compensate for ATP loss (Fan et al., 2013); this adaptation can occur quickly enough that ATP levels remain nearly constant (Hao et al., 2010). Cellular responses to OXPHOS inhibition involve signaling between the mitochondria and stress response proteins in the cytoplasm and nucleus (Quirós et al., 2017). Extracellular nutrients modulate the response to OXPHOS inhibition by determining the metabolic pathways available to compensate for the loss of oxidative ATP production and NADH oxidation (Gui et al., 2016). However, essentially all the known elements of the OXPHOS inhibitor response have been established as bulk properties of cell populations, potentially overlooking distinct subpopulations of individual cells that vary widely, as tissues do, in their usage of OXPHOS.
OXPHOS usage can be evaluated by the acute change in oxygen consumption upon treatment with oligomycin (Buttgereit and Brand, 1995). This perturbation-based approach is useful because measuring metabolic fluxes through tracing of labeled metabolites (Jang et al., 2018) or model-based reconstruction (Orth et al., 2010) require comprehensive measurements (Fendt et al., 2013; Hackett et al., 2016) that are infeasible in single cells. In contrast, the response to OXPHOS perturbation can provide a simple, functional measurement of a cell’s usage of OXPHOS and glycolysis (Mookerjee et al., 2017) that can be compared across different cell types and tumors (Simões et al., 2015). At the single-cell level, measurements of oxygen consumption are possible (Dussmann et al., 2017), but other live-cell reporters may provide a more accessible quantification of OXPHOS activity. One such possibility is AMP-activated protein kinase (AMPK), a primary contributor to the OXPHOS inhibition response. AMPK directly binds ATP, ADP, and AMP, and in response to decreased cellular energy charge (AMP and ADP relative to ATP)(Hardie and Hawley, 2001; Oakhill et al., 2011), it phosphorylates an array of substrates to enhance catabolism and suppress anabolism (Gowans et al., 2013; Hardie, 2014; Xiao et al., 2011). While AMPK is not required for all the effects of OXPHOS inhibition (Griss et al., 2015), its activity and phosphorylation status, or phosphorylation of its effectors such as acetyl-CoA carboxylase (ACC), are useful indicators of cellular energetic status. Recently, fluorescent protein-based FRET reporters have enabled tracking of AMPK activity in living cells, revealing the localization and kinetics of its activity in response to different forms of metabolic perturbation, including OXPHOS inhibition (Hung et al., 2017; Konagaya et al., 2017; Tsou et al., 2011).
Here, we used live-cell measurements of AMPK activity in response to OXPHOS inhibition to quantify differences in OXPHOS usage between single cells. We found that prominent cell-to-cell differences in AMPK response are common across OXPHOS inhibitors, and we confirmed that these changes correspond to perturbation of energy charge, using reporters for intracellular ATP concentration (Imamura et al., 2009) or ADP/ATP ratio (Tantama et al., 2013) and various supporting assays to make unambiguous measurements of metabolic changes. We find that individual cells interconvert between sensitive and resistant states on the scale of hours, and we demonstrate that each cell’s response to OXPHOS inhibition is a function of the rate of insulin-stimulated glucose uptake relative to protein synthesis rate and cell cycle status. These findings establish that OXPHOS usage varies in a functionally important way between cells, and over time within the same cell.
Results
Variable AMPK responses to OXPHOS inhibition are common
In principle, strong activation of AMPK by OXPHOS inhibition indicates dependence on OXPHOS for ATP production (Gowans et al., 2013; Hao et al., 2010). Conversely, cells with adequate capacity to generate ATP through other routes such as glycolysis would not experience a loss in energy charge and activation of AMPK upon OXPHOS inhibitor treatment. In MCF10A non-tumor epithelial cells, OXPHOS inhibitors targeting complexes I, III, and V of the electron transport chain induced a similar pattern: a sharp but variable initial increase in AMPK activity, followed by alternating cycles of inactivity and activity with an approximate period of 3 hours (Fig. 1A). Metformin induced only a small initial peak, consistent with its weak inhibitory activity, but it induced subsequent oscillations similar to the other inhibitors (Fig. S1A). In contrast to OXPHOS inhibitors, the direct AMPK activator AICAR produced a gradual rise to a new steady state of AMPK activity, without oscillations (Fig. S1B).
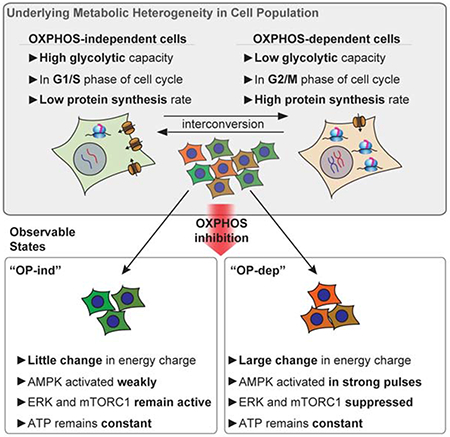
Figure 1 – Variable AMPK responses to OXPHOS inhibition are common.
A: AMPKAR2PHOS responses for cells grown in 17 mM glucose (see STAR Methods, imaging media table, for all formulations). Subplots represent single cells selected to depict the full range of responses, with population average and interquartile range in the bottom subplot. Gray shaded area shows the 2-hour time window used for analysis of peak height. N=2; see STAR Methods for definitions of replicates and cell numbers analyzed.
B: Single-cell measurements of AMPKAR2PHOS and pACC IF in MCF10A cells treated with 2.5 μg/ml oligomycin. AMPKAR2PHOS was measured in live cells 15-18 minutes after treatment; pACC was measured following fixation and linked to AMPKAR2PHOS for the same cell. R2 and p-value are shown for a fitted linear function (dashed line). N=2.
C: Schematic of AMPKAR2 pulse parameterization. Peak activity was defined as the local maximum value within 2 hours after perturbation; baseline was defined as the average of AMPKAR2 activity for one hour before treatment. Amplitude (AMPKARΔ) was calculated by subtraction of baseline from peak.
D: Histogram of AMPKAR2Δ values after treatment with 2.5 μg/ml oligomycin. Green and orange lines are fitted Gaussian distributions. The dashed line is defined by the intersection between distributions and used as the cutoff for determining the percentage of OP-ind or OP-dep cells. N=2.
E: Comparison of AMPK responses across cell lines. Top panels – representative AMPKAR2PHOS measurements for cells grown in 17 mM glucose without insulin and EGF, treated with 1.8 μg/ml oligomycin. Each subplot represents a single cell measurement, with population average and interquartile range in the bottom subplot. Middle panels - histograms of AMPKAR2Δ in response to oligomycin (OM) 1.8 μg/ml treatment. Dashed lines are defined by the intersection of fitted bimodal distributions using pooled data for treated and untreated cells within each cell line. Bottom panels - sample images of AMPKAR2 responses. N=3.
To quantify OXPHOS inhibitor responses, we developed metrics for single-cell AMPK activity. We first confirmed that the average AMPKAR2 FRET ratio correlates linearly with its phosphorylation status across the full range of metabolic conditions tested (Fig. S1C,D). AMPKAR2 phosphorylation ranged from ~30% in cells cultured with full growth medium to ~75% in cells deprived of glucose or treated with 2-deoxyglucose, ruling out saturation of the reporter and confirming that AMPK retains some activity even under full nutrient conditions (Gowans et al., 2013). We show all subsequent AMPKAR measurements as AMPKAR2PHOS, the calibrated fraction of AMPKAR2 phosphorylated. On a cell-by-cell basis, AMPKAR2PHOS correlated linearly with immunofluorescence (IF) for ACC phosphorylated at Ser-79 (pACC; R2=0.63; Fig. 1B and S1E). Both pACC and AMPKAR2PHOS were bimodal, and >80% of cells were either double-positive or double-negative. Because we expect the initial change in AMPK activity to correlate with reliance on OXPHOS for ATP production at the time of treatment, we evaluated the baseline-to-peak amplitude of AMPKAR2PHOS for each cell within 2 hours following oligomycin treatment (Fig. 1C), which we term AMPKAR2Δ. AMPKAR2Δ was not correlated with basal AMPKAR2PHOS (Fig. S1F) and was distributed bimodally (Fig. 1D). Similar distributions were found with rotenone, antimycin, and IACS-010759 (Fig. S1G). We termed cells with a low AMPKAR2Δ “OXPHOS-independent” (OP-ind) because they were able to withstand OXPHOS inhibition with little evidence of energy stress. Cells with a strong AMPK response were termed “OXPHOS-dependent” (OP-dep). In contrast to OXPHOS inhibitors, maximal doses of AICAR stimulated a uniform increase in AMPK activity across the entire population (Fig. S1H), showing that heterogeneous responses are a property of OXPHOS inhibition, rather than AMPK activation.
To establish whether heterogeneous AMPK responses are shared by other cell types, we stably expressed AMPKAR2 and measured distributions of AMPKAR2Δ in other cell lines, including 184A1 (mammary epithelial), MCF7 (breast cancer), U87 (glioblastoma), and A549 (non-small cell lung cancer, LKB1-deficient)(Fig. 1E). To compare equivalent conditions for all cells, oligomycin challenge was performed in the absence of insulin, which resulted in a lower fraction of OP-ind MCF10A cells (12%). Under the same conditions, AMPKAR2Δ in 184A1 cells was bimodally distributed, with a higher percentage of OP-ind cells (72%), suggesting that they have on average a higher capacity to maintain their ATP production independently of OXPHOS. In contrast, nearly 100% of MCF7 cells showed a strong AMPKAR2Δ, indicating a greater dependence on OXPHOS to maintain ATP homeostasis. U87 cells showed a broad distribution of responses, with both OP-ind and OP-dep populations. A549 cells, which are deficient for the AMPK activator LKB1, showed only weak AMPKAR2Δ, as expected. These data indicate that heterogeneity in the initial OXPHOS inhibitor response is common among human cell lines.
Heterogeneous AMPK responses propagate to downstream signaling activity
AMPK inhibits the activities of the RAS/ERK pathway (Shen et al., 2013) and mTORC1 (Gwinn et al., 2008; Inoki et al., 2003) (Fig. 2A). In MCF10A cells, we investigated whether heterogeneous activation of AMPK induces corresponding changes in these connected pathways. ERK activity was monitored simultaneously with AMPK using a translocation-based reporter, ERKTR (Regot et al., 2014). Upon oligomycin treatment, ERKTR detected an average decrease in ERK activity (Fig. 2B), consistent with inhibition of this pathway by active AMPK. On a cell-by-cell basis, the reduction of ERK activity correlated with the magnitude of AMPK activation for each cell (Fig. 2C,i), whereas no correlation was found in the absence of oligomycin. By IF, OP-dep cells showed a lower intensity of phospho-ERK staining (Fig. 2C,ii). Furthermore, when time courses of AMPKAR2 and ERKTR signals were tracked over time in individual cells, a significant anti-correlation was observed where pulses of AMPK activity were matched by depressions in ERK activity (Fig. 2D,i), with a lag time of 6 minutes or less (Fig. 2E,i,ii).
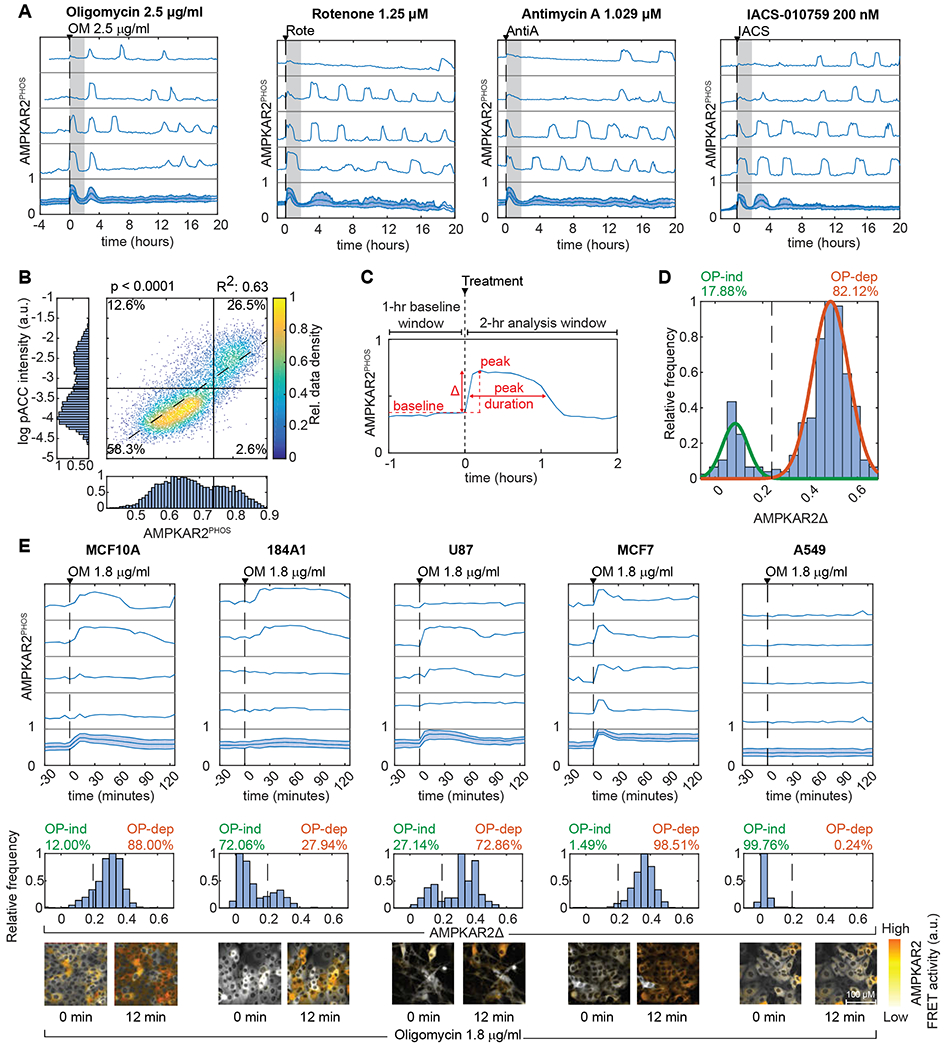
Figure 2 – Heterogeneous AMPK responses propagate to downstream signaling activity.
A: Known connections between AMPK, mTORC1 and ERK, and corresponding reporters for live-cell analysis. ERKTR and TFEB-TR indicate the activities of ERK and mTORC1, respectively, by their cytoplasmic to nuclear ratio.
B: Population average responses of AMPKAR2PHOS (blue) compared to ERKTR (orange) and TFEB-TR (green) after oligomycin (OM, upper panels) or vehicle (lower panels) treatment. Shaded areas indicate interquartile ranges. N=2.
C: Correlation of AMPKAR2Δ with signaling markers in single cells. Each dot indicates a single cell in which AMPKAR2Δ was measured in tandem with (i) ERKTR (live-cell), (ii) phosphorylated ERK (IF), (iii) TFEB-TR (live-cell), or (iv) phosphorylated 4E-BP1. For live-cell measurements, values represent amplitude of response. For IF measurements, values represent integrated staining intensity for cells fixed immediately following measurement of AMPKAR2Δ. R2 and p values are shown for linear regression against pooled data for both untreated and oligomycin-treated cells. N=2.
D: Dynamic relationship of AMPK activity with (i) ERK and (ii) mTORC1 reporters. Representative single-cell profiles of AMKPAR2PHOS were measured in the same cell as ERKTR (orange) or TFEB-TR (green).
E: Cross-correlation analysis for AMPK activity with ERK and mTORC1 reporters. (i) and (iii) show distributions of the lag time at which maximum anti-correlation is found between AMPKAR2 and ERKTR or between AMPKAR2 and TFEB-TR, respectively, (ii) and (iv) show the distribution of Pearson’s cross-correlation coefficients at the lag time with maximum correlation (0 minutes for ERKTR, −12 minutes for TFEB-TR). N=2.
To detect mTORC1 activity in live cells, we used the nuclear-to-cytosolic translocation of a fluorescent protein fusion to transcription factor EB (TFEB-TR), which is stimulated by mTORC1-mediated phosphorylation (Fig. 2A) (Li et al., 2018; Settembre et al., 2012). As in the case of ERKTR, TFEB-TR cytosolic-to-nuclear ratio was decreased following oligomycin treatment (Fig. 2B) and correlated to AMPKAR2PHOS at the single cell level (Fig. 2C,iii). IF for phospho-4E-BP1, an mTORC1 substrate, was also reduced in OP-dep cells (Fig. 2C,iv). Cycles of TFEB-TR translocation coincided with AMPK pulses, following a ~12 minute lag (Figs. 2D,ii and 2E,iii–iv). These results are consistent with dynamic regulation of mTORC1 by AMPK, although they do not rule out the possibility that OXPHOS inhibition suppresses mTORC1 independently of AMPK (Kalender et al., 2010). Together these data establish that heterogeneity in OXPHOS inhibitor responses has a functional impact on the AMPK signaling network.
AMPK responses to OXPHOS inhibition report the dynamics of ATP metabolism
Cell-to-cell variation in AMPKAR2Δ may reflect differences in cellular energy charge (Hardie, 2014), but this variation could also arise from variable drug uptake or other forms of AMPK regulation (Lin and Hardie, 2017; Zhang et al., 2017). To independently measure changes in energy charge, we used the ADP/ATP reporter PercevalHR (Berg et al., 2009; Tantama et al., 2013), which reports intracellular ADP/ATP ratio as a spectral shift in mVenus excitation, a ratio we refer to as PercevalEX (Fig. 3A). Similar to AMPKAR2PHOS, the immediate response of PercevalEX following OXPHOS inhibition was heterogeneous (Fig. 3B,i). However, unlike AMPK activity, PercevalEX lacked two distinct modes. Under continuous exposure to oligomycin, we observed pulses of PercevalEX 1-2 hours in duration, interspaced by 2-4 hours, similar to AMPKAR2PHOS in timing but more variable in amplitude. Staining of pACC was correlated with PercevalEX, with agreement of pACC staining and PercevalEX responses in ~80% of cells (Fig. 3B,ii and S2A). However, the distinction between high- and low-PercevalEX cells was not sharp, and cells at intermediate PercevalEX values were distributed between high- and low-pACC subpopulations, consistent with findings that factors other than energy charge can influence AMPK activity (Hawley et al., 2005; Zhang et al., 2017). Based on these data, differences in energy charge are a plausible cause for AMPK variation but are not strictly identical to AMPK activity within individual cells.
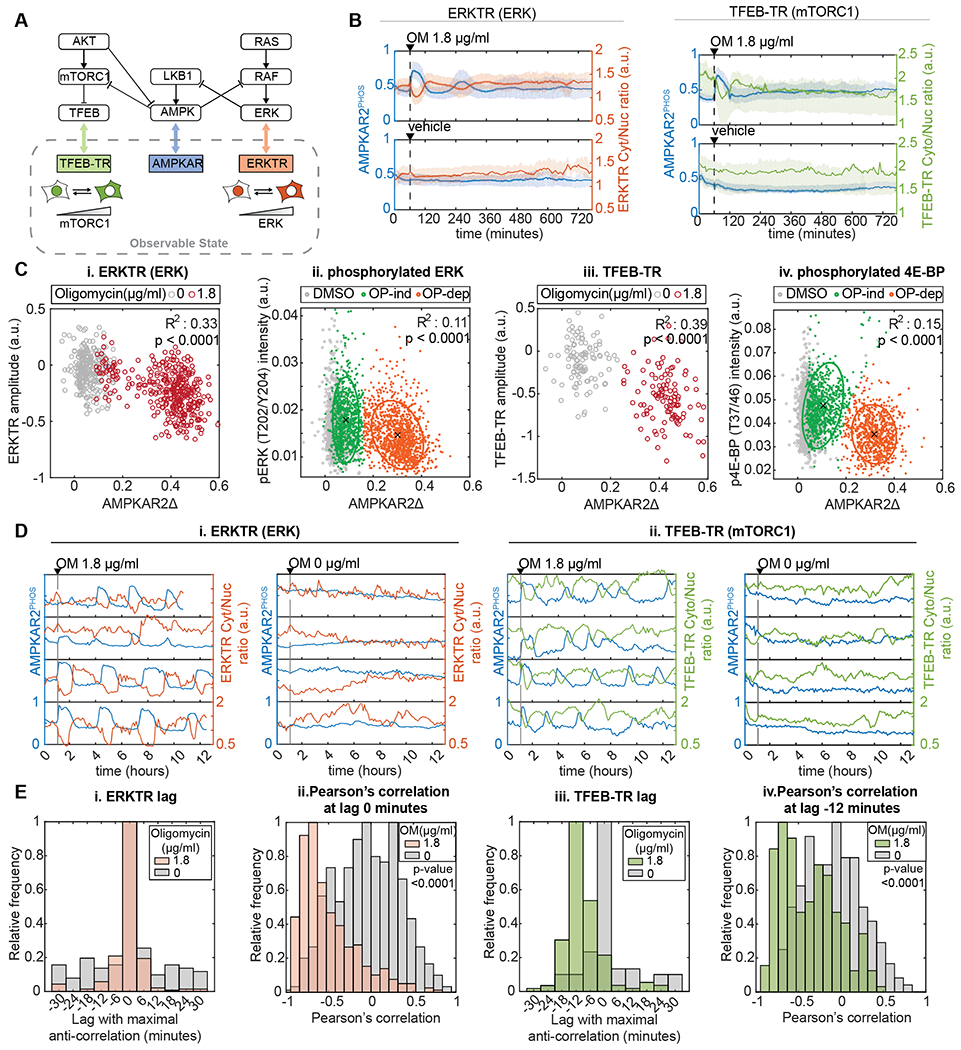
Figure 3 – AMPK responses to OXPHOS inhibition report the dynamics of ATP metabolism.
A: Schematic of ATP metabolism and reporters used. AMPKAR2 indicates AMPK kinase activity, PercevalHR reports intracellular ADP/ATP ratio, and ATeam1.03 reports intracellular ATP concentration.
B: Responses of ADP/ATP and ATP reporters to OXPHOS inhibition, (i) and (iii) show representative single-cell recordings of PercevalHR (i) or ATeam1.03 FRET activity (iii) after treatment with 2.5 μg/ml oligomycin. Each subplot represents a single cell measurement, with the population average and interquartile range shown at bottom, (ii) and (iv) show scatter plots of single-cell measurements of PercevalEX (ii) or ATeam1.03 FRET activity (iv) with phospho-ACC staining intensity in MCF10A cells treated with oligomycin (OM) 2.5 μg/ml. Numbers indicate the percentage of cells in each quadrant. R2 values are shown for linear fits to the data. N=2.
C, D, and E: AMPK, ADP/ATP, and ATP responses to OXPHOS inhibition when glucose is varied. Line plots (left) show the responses for each reporter after cells were cultured in media containing the indicated glucose concentration and then treated with 2.5 μg/ml oligomycin (OM). Light lines indicate individual cells, and heavy lines the population mean. Histograms (right) show the distribution of response amplitudes in each condition, calculated as in Figure 1. In (D) and (E), the light gray histograms indicate the distributions of reporter measurements for cells treated with vehicle at the same glucose concentration. N=2.
We next used the FRET-based ATP sensor ATeam 1.03 (Imamura et al., 2009) to track intracellular ATP concentrations under the same conditions. Following oligomycin treatment, we were unable to detect any change in ATP level, nor any pulsatile characteristics as observed for AMPKAR2PHOS or PercevalEX(Fig. 3B,iii). To confirm that the lack of ATeam response is not a result of out-of-range ATP concentration, we treated these cells with oligomycin in the absence of glucose, which resulted in an immediate and sharp decline in ATeam signal (Fig. S2C), followed within 4 hours by visible cell death. We confirmed this result using bulk ATP assays, which detected no OXPHOS inhibitor-induced change in ATP at 17 mM glucose but a >90% decrease upon inhibitor treatment in the absence of glucose (Fig. S2D). When ATeam cells were co-stained with pACC, we observed that the rare low-ATeam cells (~10%) were predominantly pACC-positive, as expected for cells with low ATP (Fig. 3B,iv and S2B). These results indicate that ATeam accurately reports ATP levels within MCF10A, and that cytoplasmic ATP remains stable during OXPHOS inhibition, as previously observed (Gowans et al., 2013; Hao et al., 2010).
The differences between AMPK activity, ADP/ATP ratio, and ATP concentration prompted us to investigate their relationship. To approach this question, we quantified AMPKAR2PHOS, PercevalEX, or ATeam responses following oligomycin treatment under varying concentrations of glucose (Fig. 3C–E). AMPKAR2PHOS responses remained bimodal across all conditions, with the frequency of OP-ind cells decreasing from >20% of cells at 17 mM glucose (standard MCF10A culture conditions) to 8-9% at 4.25 mM glucose (an intermediate physiological concentration) and falling to <1% at lower glucose concentrations (Fig. 3C). In contrast, PercevalEX was distributed unimodally in each condition, with a mean that increased gradually as glucose concentration was lowered (Fig. 3D). ATeam showed no response until glucose was reduced below 1 mM, at which point it showed a rapid decrease in all cells (Fig. 3E). Together, these data suggest a model consistent with previous observations, in which the absolute cytosolic concentration of ATP is maintained at a nearly constant level, provided that glycolysis can operate at a sufficiently high rate. Rapid equilibration of ATP with ADP and AMP prevents a large drop in absolute ATP levels but allows a significant shift in ADP/ATP and AMP/ATP ratios, which are detected by AMPK (Hardie et al., 2012). The gradually shifting broad distribution of PercevalEX indicates that these ratios vary from cell to cell and are sufficient to induce AMPK activity in some cells (OP-dep) but not others (OP-ind). The bimodality observed in AMPK activity but not ADP/ATP ratio is consistent with ultrasensitive activation of AMPK (Hardie et al., 1999). Thus, the differing relationships between pACC, AMPKAR2, PercevalEX, and ATeam can be understood as the outcome of the ATP/AMPK system as it operates in single cells that vary in their capacity to maintain ATP production when OXPHOS is inhibited.
The results above suggest that energy charge is the primary factor determining AMPK activity under OXPHOS inhibition. Bulk measurements of metabolites are consistent with this interpretation: oligomycin treatment induced strong and persistent suppression of TCA cycle intermediates (Fig. S2E,F), while inference of ATP production (Mookerjee et al., 2017) confirmed a nearly complete switch from OXPHOS to glycolysis during oligomycin treatment (Fig. S2G). Finally, comparison of OXPHOS inhibitor responses under different conditions argued that the lack of AMPK activity in OP-ind cells is not an artifact of incomplete OXPHOS inhibition (Fig. S3A–G). We conclude that OP-ind cells represent a subpopulation with metabolic characteristics inherently distinct from OP-dep cells.
Resistance to OXPHOS inhibition is a heritable but transient state
Our results raise the question of whether OXPHOS inhibitor resistance is a fixed or transient cellular property, which we approached using sister cell analysis (Spencer et al., 2009; Strasen et al., 2018). We tracked the history of cells prior to OXPHOS inhibitor treatment and compared AMPKAR2Δ for sister cell pairs as a function of time since their last mitosis (Fig. 4A). If resistance is a permanent characteristic, sister cells would remain similar in their response indefinitely (Fig. 4A,i), whereas if resistance is transient, they will be similar initially but diverge over time (Fig. 4A,ii). Both OP-ind and OP-dep cells were well represented at all times after the last division (Fig. 4B,C), and sister cell pairs within 2 hours of their shared mitosis were significantly more likely to have a similar AMPKAR2Δ response than random pairs of cells (Fig. 4D,E). However, this similarity in AMPKAR2Δ between daughters decayed gradually and approached the level of unrelated cells with a half-life of ~29 hours (Fig. 4E). These results indicate that OP-ind or OP-dep states are a heritable property that can persist for at least several hours, and that transitions between these states occur intermittently within the cell population.
Figure 4 – OXPHOS inhibitor resistance is a heritable but transient cellular state.
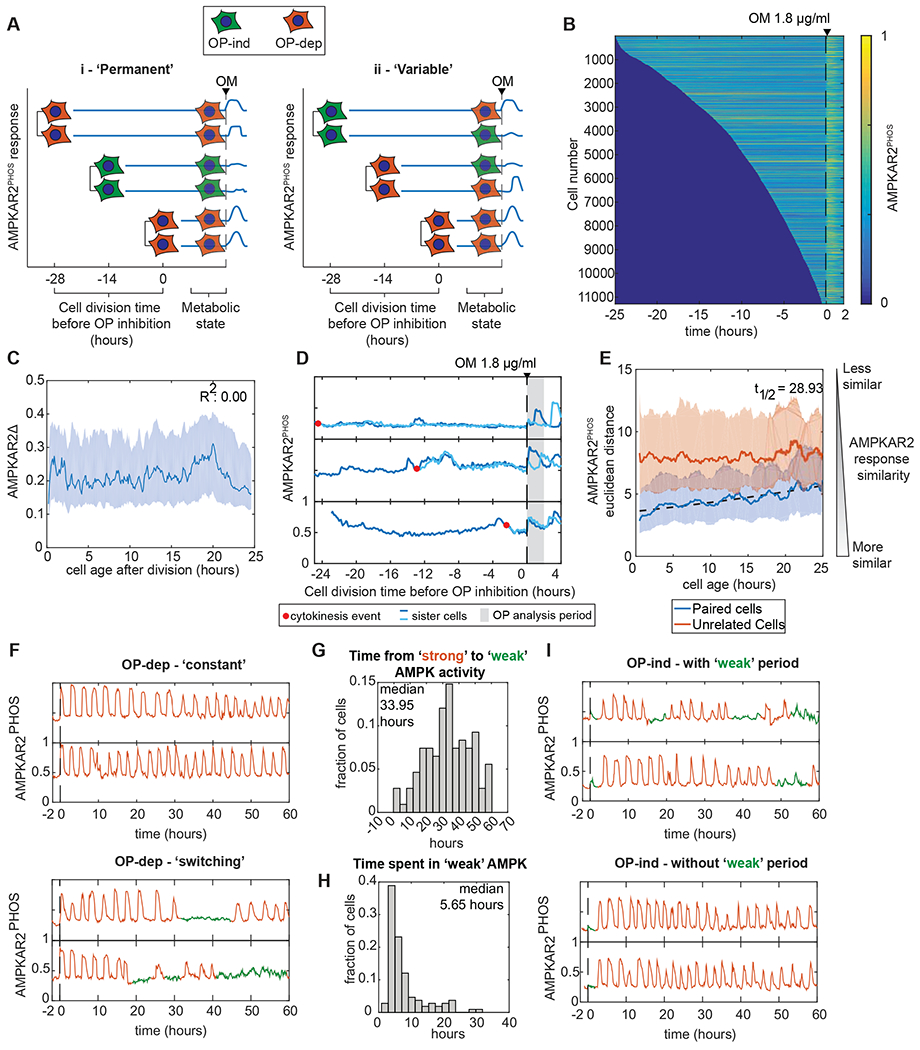
A: Schematic of sister cell analysis to distinguish between permanent and time-varying cell states determining the response to OXPHOS inhibition.
B: Heatmaps of AMPKAR2PHOS in individual cells. MCF10A-AMPKAR2 cells were imaged for 24 hours before treatment with 1.8 μg/ml oligomycin. Each horizontal line represents a single cell’s AMPKAR2PHOS profile, beginning with its most recent cell division and ending 2 hours after oligomycin (OM) treatment. Cells were sorted by the time of their last division. Analysis contains >11,000 individual cells. N=3.
C: Line plot of the relationship between a cell’s age at the time of oligomycin treatment and its recorded AMPKAR2Δ response.
D: Sample traces of AMPK activity from sister cell pairs.
E: Comparison of AMPKAR2PHOS responses in sister cells. Dissimilarity between the sisters of each division, or between randomly chosen pairs of cells was calculated (see Methods). Solid lines represent effect size, and the shaded areas represent interquartile range after bootstrapping. Dashed line represents a fitted exponential function for the decay of sister cell similarity over time.
F: Transitions in OXPHOS dependence within single cells. Top panels show two example cells in which strong AMPK activity (continued pulsing) persists for the remainder of the experiment. Bottom panels show two example cells that transition from strong activity (orange) to a state with weak AMPK activity (green). N=2.
G: Distribution of transition times from strong to weak AMPK activity. For all cells showing a transition as shown in the bottom panels of (F), the time between oligomycin treatment and the first transition is shown as a histogram.
H: Distribution of durations of weak AMPK activity states. For all cells showing both entry into and exit from an OP-ind state during the experiment (green phases in (F)), the interval between entry and exit is shown as a histogram.
I. Examples of cells transitioning from a weak to a strong AMPK state. Top panels show example cells that transition to strong and then return to weak activity. Bottom panels show instances of cells that transition to strong activity for the remainder of the experiment.
To observe transitions between sensitive and resistant states more directly, we performed long-term imaging of cells under continuous oligomycin treatment. Cells classified as OP-dep entered immediately into a regular oscillatory pattern of AMPK activity (as seen in Fig. 1A), with a period of approximately 3 hours (Fig. 4F, top). However, we noted that cells occasionally exited this oscillatory state and entered a phase with weaker, irregular AMPK activity (Fig. 4F, bottom and S4A). For cells that showed such a transition, the median time to transition ranged from 0 to 60 hours, with a median of 34 hours. Phases of weak AMPK activity persisted for 1-20 hours, with a median of 5.6 hours (Fig. 4H), before cells returned to the oscillatory phase. Similarly, cells that initially showed an OP-ind response typically underwent a transition into oscillatory activity within 6 hours after oligomycin treatment (Fig. 4I). These results are consistent with the sister-cell analysis and indicate that cells transition intermittently between an OP-dep-like phase with oscillatory AMPK activity, and an OP-ind-like phase with weak AMPK activity. Furthermore, comparison of the duration and frequencies of these phases to the overall frequency of OP-ind cells suggests that the primary source of initial OP-ind cells are the intermittent phases of weak AMPK activity, rather than the 3-hour oscillatory nature of the AMPK response (Fig. S4B).
Glucose uptake and protein synthesis modulate OXPHOS inhibitor resistance
We next addressed the molecular differences that underlie OP-dep and OP-ind responses, beginning with the capacity to produce ATP by glycolysis. Treatment with insulin, which stimulates glucose uptake and glycolysis through PI3K/AKT signaling, increased the frequency of OP-ind responses in MCF10A, MCF7, 184A1, and U87MG cells (Fig. 5A). Furthermore, inhibition of AKT, hexokinase, or mTORC1/2 shifted cells toward OP-dep responses (Fig. 5B). In contrast, inhibition of fatty acid oxidation or lipolysis failed to significantly alter OP-ind responses (Fig. S5A). While these results implicate regulation of glucose uptake through insulin/PI3K/AKT signaling, this pathway can potentially affect AMPK activation through multiple routes (Suzuki et al., 2013). To test whether OP-ind cells can result solely from increased glucose uptake capacity, we overexpressed the glucose transporter GLUT1 along with a co-translated red fluorescent protein (RFP) to quantify GLUT1 overexpression on a cell-by-cell basis. We removed insulin to eliminate the AKT-induced component of glucose uptake. Higher expression GLUT1-RFP correlated with weaker AMPK responses to OXPHOS inhibitor (Fig. 5C), with 60% of GLUT1-RFP-expressing cells showing OP-ind behavior, while almost 100% of cells not expressing GLUT1-RFP were OP-dep. When insulin was added, OP-ind responses shifted to include cells at lower GLUT1-RFP expression levels, consistent with the induction of endogenous (unlabeled) glucose transporters (Fig. 5D,E). Accordingly, the predictability of OXPHOS inhibitor response as a function of exogenous GLUT1 expression decayed (Fig. 5C–E, bottom panels). These data demonstrate that a cell’s OXPHOS inhibitor resistance status can be determined by its capacity to take up glucose, and that this status can be predicted based on glucose transporter expression when other factors are minimized.
Figure 5 – Glucose uptake capacity is sufficient to drive OXPHOS inhibitor resistance.

A: Increase in OP-ind responses stimulated by insulin. Histograms show AMPKAR2Δ responses to 1.8 μg/ml oligomycin. N=2.
B: Change in OP-dep responses in response to inhibitor treatment for MCF10A cells under 17 mM glucose. Inhibitors were added 30 minutes prior to oligomycin. Horizontal black lines indicate the fraction of OP-dep cells under control treatment (DMSO); points falling outside the gray region are considered significant by t-test. Points represent the mean, and error bars standard error of the mean; N=2.
C, D and E: Effect of increased glucose uptake on OXPHOS inhibitor responses. MCF10A-AMPKAR2 cells stably overexpressing GLUT1-IRES-NLS-mCherry were cultured with 0 (C), 100 (D), or 10000 ng/ml (E) insulin and exposed to oligomycin (OM). Each row in the heatmaps (upper panels) represents an individual cell; rows are sorted by relative mCherry intensity (corresponding to the level of GLUT1 overexpression), which is indicated by the color bar to the left. GLUT1 expression levels are normalized to the minimum and maximum expression levels in the population. Lower panels show scatter plots of mCherry intensity and AMPKAR2Δ following oligomycin treatment. N=2.
F, G: Increase in OP-ind responses following glucose starvation. (F) shows average AMPKAR2PHOS recordings for MCF10A cells grown in the absence of glucose for 24 hours and then treated with glucose at the specified concentrations, followed by 1.8 μg/ml oligomycin at 30 minutes or 1 minute after glucose addition. (G) shows histograms of AMPKAR2Δ values after oligomycin treatment for the conditions shown in (F). N=2.
Most cell lines are cultured in media containing high levels of glucose, and therefore OP-ind responses might result from glucose-induced changes in glycolytic gene expression or from increased storage metabolites (such as glycogen). To test these possibilities, we cultured cells in the absence of glucose for 24 hours, and then added glucose for a short time window (30 minutes or 1 minute) prior to treating with oligomycin (Fig. 5F). Because short exposure to glucose is unlikely to increase storage pools or expression of glycolytic enzymes, we expected that OP-ind cells relying on these mechanisms would become OP-dep during the starvation period. However, OP-ind cells were instead detected at a much higher frequency following glucose withdrawal (>90% at 17 mM glucose, Fig. 5G) than when cultured in glucose continuously (~25% at 17 mM glucose, see Fig. 3C). This result indicates that OP-ind cells are not simply the result of prolonged high glucose conditions. Rather, it may be that during the starvation period, ATP consumption rates decline more than glycolytic capacity, resulting in anabolically inactive cells that have relatively low demand but remain poised to utilize glucose when it is resupplied (Fig. S5B). Consistent with this interpretation, protein synthesis rate as measured by O-propargyl puromycin (OPP) incorporation was significantly lower following 24 hour glucose starvation (Fig. S5C). At the same time, the frequency of OP-ind cells remained dependent on the concentration of glucose that was resupplied for 1 minute (Fig. 5G). Thus, these results support that glycolytic capacity is needed for OP-ind responses and implicate ATP turnover as a potential additional factor that influences the OXPHOS inhibitor response. We also found that glutamine, which is provided in excess in some cell culture media, is not required for OP-ind responses but can increase their frequency (Fig. S5D).
Because our results and previous studies (Buttgereit and Brand, 1995) suggest that ATP usage (i.e., turnover) by protein synthesis may impact energy charge upon OXPHOS inhibition, we tested the effect of translation inhibition by cycloheximide (CHX) using both OPP incorporation and a live-cell translation reporter, TOP-H2B-YFP-DD (Han et al., 2014)(Fig. 6A–C). Pre-treatment with CHX decreased the fraction of OP-dep cells from 60% to <10% (Fig. 6D). Furthermore, even 15 minutes after an AMPK response was initiated, protein synthesis inhibitors immediately reduced AMPK activity, shortening the pulse length from 60 minutes to 20 minutes (Figure 6E, F). This observation suggests that reducing ATP consumption by translation lowers the impact of OXPHOS inhibitors on energy charge and AMPK activation.
Figure 6 – Reducing protein synthesis rate promotes OXPHOS inhibitor resistance.
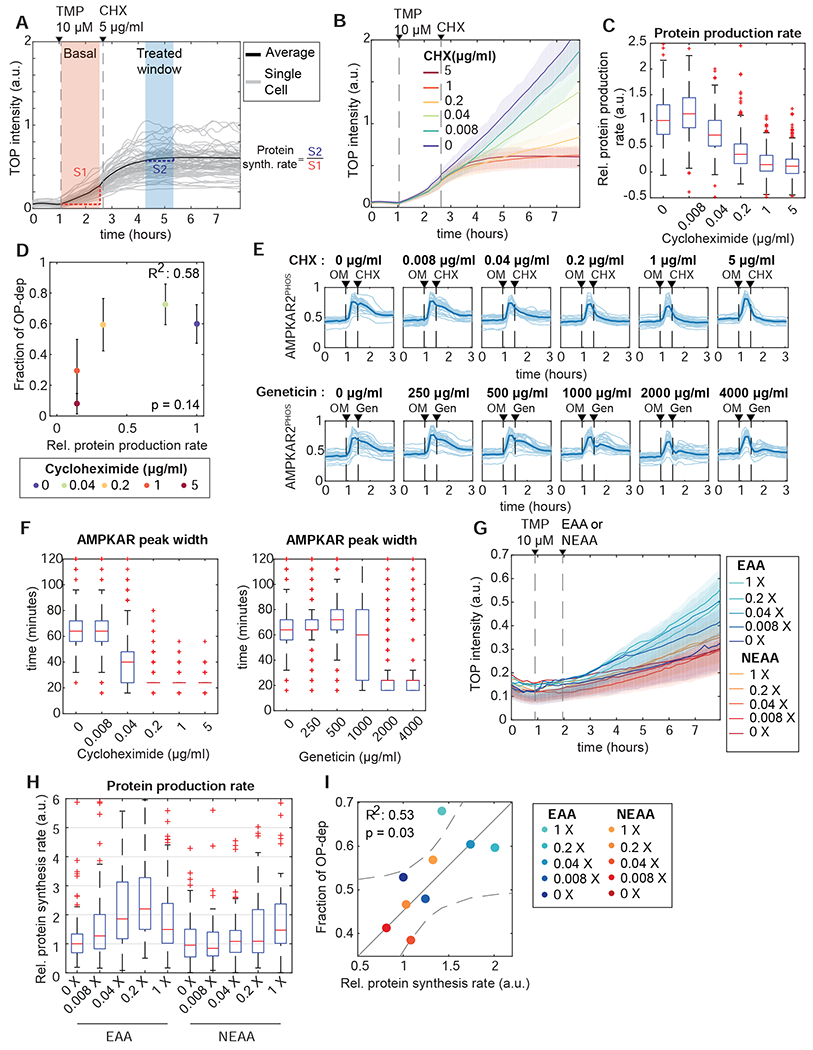
A: Measurement of protein synthesis rates in live cells. MCF10A cells stably expressing TOP-H2B-YFP-DD (TOP) were treated with the degron inhibitor trimethoprim (TMP). Protein production rate was calculated as the slope of YFP intensity change during the 60 minutes after TMP treatment (orange shaded area). The effect of CHX inhibition on protein production was quantified from the slope for a 60 minute period beginning 90 minutes after treatment (blue shaded area); relative protein production rate is calculated as the ratio of slopes in the blue and orange regions (S1 and S2 respectively).
B, C: Quantification of reduced protein synthesis rates. (B) shows mean TOP-H2B-YFP-DD intensity for a concentration series of CHX treatments. Shaded areas show interquartile ranges. (C) shows calculated single-cell relative protein production rates for each concentration of cycloheximide. Each box represents the distribution of >400 cells. N=2.
D: Relationship of average protein production rate to the fraction of OP-dep cells. Protein rate was measured as in (A-C). The corresponding fraction of OP-dep responses was determined by culturing cells in the same CHX concentrations, followed by oligomycin treatment. Points represent the mean, and error bars standard error of the mean; N=2.
E, F: Termination of AMPK activity pulses by protein synthesis inhibition. (E) shows single-cell traces (light lines) and means (dark lines) for cells treated with oligomycin (OM), followed by the protein synthesis inhibitors CHX or geneticin (Gen) at the concentrations indicated. (F) shows quantification of single cell AMPKAR2PHOS pulse widths after CHX (left panel) or geneticin (right panel) treatment. Pulse widths were calculated as the time at which AMPKARPHOS decreased to 50% of the maximum value for each cell following treatment with CHX or geneticin. N=2.
G-I: Modulation of protein synthesis and AMPK responses by amino acid availability. (G) shows representative mean TOP-H2B-YFP-DD intensity for MCF10A cells cultured in essential or non-essential amino acid at the indicated concentrations (X represents fold-change relative to the concentration in MEM). Shaded areas show interquartile ranges. (H) shows quantification of relative protein synthesis rates from the experiment shown in (G). Each box represents the distribution of >200 cells. (I) shows the mean protein synthesis rates from (G) plotted against the corresponding fraction of OP-dep cells, measured after oligomycin treatment in the same amino acid concentrations. Solid line represents a fitted linear model, and dashed lines the 95% confidence bounds. N=2.
Several observations supported the interpretation that inhibitors of translation limit AMPK activity due to a decrease in catabolic activity, rather than activation of feedback or stress responses. First, while extended incubation with CHX increased phosphorylation of S6 and 4E-BP1 as previously reported (Santos et al., 2019), this increase was moderate relative to the pre-existing mTORC1 activity level (Fig. S6D,E). Furthermore, culture of cells in varying concentrations of essential amino acids (EAA) or non-essential amino acids (NEAA) modulated the rate of protein synthesis as measured by TOP-H2B-YFP-DD (Fig. 6G,H). Under these conditions, the fraction of OP-dep cells measured following oligomycin treatment correlated with the protein synthesis rate (R2 =0.53, Fig. 6I). These results support that OP-ind cells depend on a low rate of protein synthesis, which makes it possible for glycolysis to maintain cellular energy charge above the threshold to trigger AMPK.
OXPHOS inhibitor response states are inherently multivariate
We investigated the cell division cycle as a potential source of variability, combining AMPKAR2 measurements with DNA content, a live-cell S/G2 reporter (mCherry-Geminin1-330), and staining for phosphorylated Rb protein (pRb). Plotting these measurements enabled cell cycle phases to be clearly distinguished (Fig. 7A). We then overlaid the oligomycin-induced AMPKAR2Δ measurement for each cell, recorded immediately prior to fixation (Fig. 7B). We noted that both high and low AMPKAR2Δ measurements were found in all stages of the cell cycle (Fig. 7C,D). However, comparisons of the cell cycle phase distributions of OP-dep and OP-ind cells revealed enrichment for different phases – OP-ind cells were 2-fold more likely to be in G1 phase relative to OP-dep cells, whereas OP-dep cells were more often found in S and G2 phases (Fig. 7E). We noted that OPP incorporation increased with progression of the cell cycle (Fig. S7A), providing a potential explanation for the higher number of OP-dep cells in S and G2. Live-cell recordings in cells expressing both AMPKAR2 and mCherry-Geminin1-330 and treated with oligomycin identified OP-ind phases immediately following cell division (Fig. 7F, top panel), consistent with their statistical enrichment in G0/G1. However, other cells showed continuous OP-dep responses in G0/G1 (Fig. 7F, bottom panel), indicating that OP-ind responses are probabilistically related to cell cycle position, but not strictly determined by it.
Figure 7 – OXPHOS inhibitor response states are inherently multivariate.
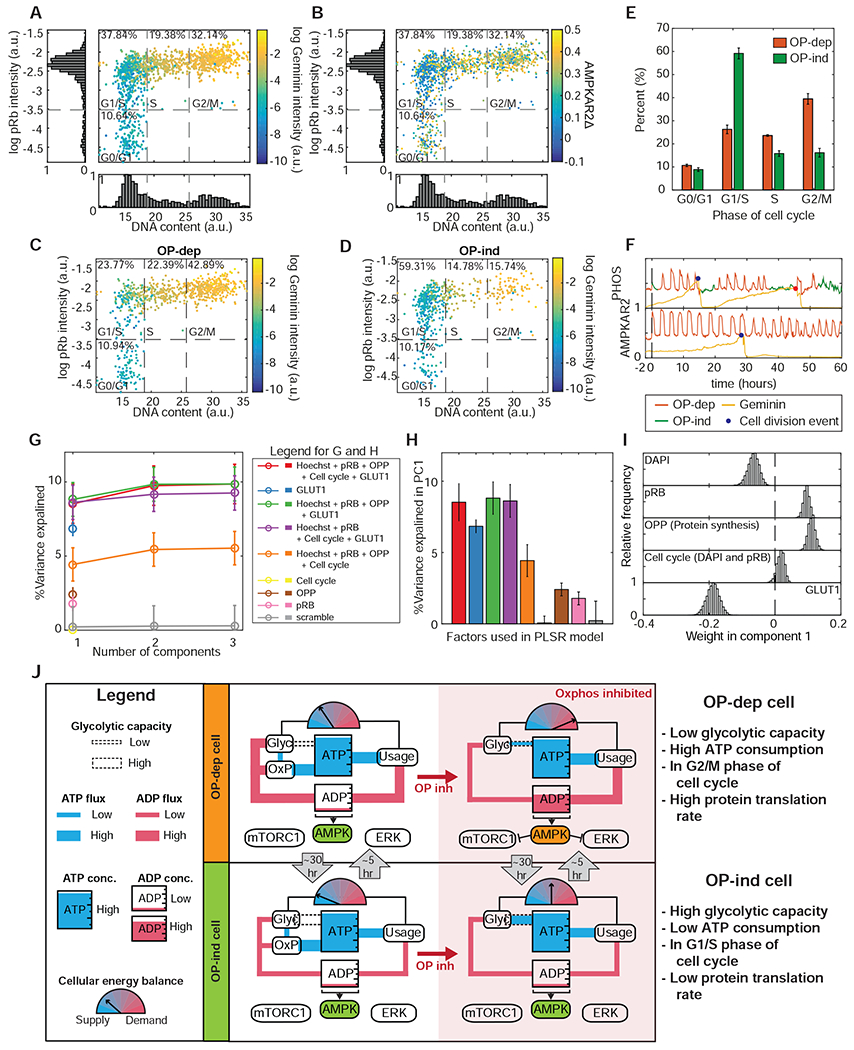
A: Scatter plot of single cell measurements of DNA content (Hoechst 33342 intensity) and phospho-Rb IF, colored by mCherry-Geminin1-330 intensity. Dotted lines divide the phases of the cell division cycle, with the percentage of cells in each phase indicated. N=3.
B: Scatter plot of cell cycle parameters as in (A), colored by AMPKAR2Δ values recorded during the 30 minutes immediately prior to fixation and staining. N=3.
C and D: Scatter plots of cell cycle parameters as in (A), divided between OP-dep cells (C) and OP-ind cells (D). N=3.
E: Bar graph comparing the distribution of cell cycle phases for OP-ind and OP-dep cells. Error bars represent standard errors of means; N=3.
F: Single cell traces of AMPK activity and mCherry-Geminin1-330 intensity for (top) a cell in which OP-ind occurs early in G0/G1, and (bottom) a cell in which no OP-ind phase occurs during G0/G1. Markers indicate the time of mitosis.
G: Predictivity of PLSR models of AMPKAR2Δ (percentage of total variance explained) including the indicated combinations of measurements. For the “scrambled” model, pairings between input and output measurements for each cell were randomly reassigned. Cell cycle position was estimated by using the Wanderlust algorithm for non-linear mapping. See STAR Methods and Figure S7D for details.
H: Bar chart showing percent of total variance explained by the first PLSR component for models using combinations of cellular processes measurement as indicated.
I: Contribution of measured processes to PLSR models for AMPKAR2PHOS response to oligomycin treatment. Distributions of parameter coefficients were generated by bootstrapping with replacement 10,000 times.
J: Simplified diagrams indicate the state of ATP metabolism in OP-ind and OP-dep cells. Blue “pipes” indicate flux of ATP, and red pipes the flux of ADP. Meter icons indicate the balance of ATP production capacity relative to ATP consumption. The dotted ‘pipe’ indicates reserve glycolytic capacity to supply ATP. OP-dep cells (top panel) have low glycolytic capacity with high ATP demand; upon OXPHOS inhibition, these cells maintain constant ATP concentration at the expense of an increase in ADP that triggers activation of AMPK and inhibition mTORC1 and ERK pathways. OP-ind cells (bottom panel) have high reserve glycolytic capacity and relatively low ATP demand. Upon OXPHOS inhibition, thesecells can maintain constant ATP and a low concentration of ADP via glycolysis, and AMPK consequently remains inactive.
To understand how multiple factors interact to determine OXPHOS inhibitor responses within individual cells, we performed a multivariate analysis using partial least squares regression (PLSR). We collected a multiplexed dataset for AMPKAR2Δ, OPP incorporation, cell cycle markers (Hoechst-33342 and pRb) and our GLUT1-RFP expression system as a surrogate measurement for glycolytic rate. We used Wanderlust (Bendall et al., 2014) to represent cell cycle position as a single continuous variable cycle based on DNA content and pRb measurements (Fig. S7D). PLSR models were generated to predict each cell’s AMPK response based on its individual combination of the other factors. Initially, we excluded cells with the highest GLUT1-RFP expression (Fig. S7E) to prevent them from dominating the model predictions. In the best fitting models, the first principal component (PC) captured approximately 8% of variability in AMPK response between single cells, which was increased to 10% by the second PC; additional PCs did not further improve predictivity (Fig. 7G and S7F). This relatively low predictive power is not unusual for single-cell models of signaling responses (Gillies et al., 2017) and indicates that unmeasured factors or stochastic variation contribute substantially to the variable AMPK response. Nonetheless, the variability captured by the model represents a strong signal relative to control models (Fig. 7G, gray line) that can be used to understand the interactions between measured variables. In the first PC, GLUT1-RFP was the highest weighted input and contributed negatively to AMPK response, while pRB and OPP incorporation had smaller positive contributions (Fig. 7I). Individually, GLUT1, OPP, and pRB predicted 6.9%, 2.4%, and 1.8% of AMPK variability, while the inferred cell cycle position was less predictive than pRb (Fig. 7H). As expected, when all levels of GLUT1-RFP expression were considered, overall predictivity rose to 35% (Fig. S7E,F) but was dominated by the contribution of GLUT1-RFP (Fig. S7G). Overall, this analysis demonstrates that each cell’s OXPHOS inhibitor response is a multivariate process determined by a combination of glycolytic rate, protein synthesis, and RB phosphorylation, as well as other unmeasured parameters.
Discussion
Cellular heterogeneity can influence the therapeutic efficacy of drugs (Altschuler and Wu, 2010). We identify a distinct form of heterogeneity that results from transient differences in each cell’s balance between the capacity to generate ATP through glycolysis and its ATP consumption or turnover rate. While protein synthesis, glucose uptake, and the cell cycle are well-known to affect cellular energy balance, our analysis reveals that these processes interact at the cellular level to create transient states of resistance to an acute metabolic challenge like OXPHOS inhibitor treatment. This variation impacts metabolic stress signaling through AMPK, mTOR, and ERK, which are among the intended targets for OXPHOS inhibition in both cancer and diabetes (Howell et al., 2017; Kim et al., 2012).
Our data indicate that the response to OXPHOS inhibition depends on multiple factors and is difficult to predict for any individual cell. Each cell carries on its own mixture of ATP-consuming processes, and at the same time has a certain maximal capacity for ATP production through glycolysis. The net balance of these processes determines the extent to which ATP production can continue when OXPHOS is inhibited (Fig. 7J). Therefore, while protein synthesis and AKT signaling are significant drivers of the OXPHOS response, for a specific cell they may not be the most consequential, depending on which other ATP-consuming or generating processes are active. Furthermore, our data suggest that as the prevalence of these processes changes over time in a given cell, so does its OXPHOS inhibition response. Only when components such as GLUT1 are overexpressed can a cell’s response be predicted reliably based on a single factor. This complexity makes the OXPHOS inhibitor response useful, as it can interrogate the net ATP production/turnover balance in a single measurement.
How does cellular variation in ATP turnover and production arise? While OP-dep and OP-ind responses correlate to some extent with cell cycle phases, this bias cannot explain most variation, as both types of response can be found at any point in the cell cycle. We suggest that, because protein synthesis increases throughout the cell cycle (Elliott and McLaughlin, 1978), ATP turnover increases and OP-dep states become increasingly likely during S and G2, explaining their observed enrichment in these phases. Can the remaining variability then be attributed to different rates of glucose uptake? This is possible, as we have observed that AMPK and AKT activity can fluctuate during normal growth conditions (Hung et al., 2017), which would be expected to result in different glucose uptake rates over time. Still another possibility is that, as in yeast (Cai and Tu, 2012; Tu et al., 2007) mammalian cells have an intrinsic cycle that controls flux through glycolysis and protein synthesis. Our results reveal the existence of at least two different but interrelated cellular rhythms: a regular oscillation between AMPK activity and inactivity with a period of ~3 hours during OXPHOS inhibition, and a longer-term shift between a state in which cells are competent for AMPK oscillations and a state in which AMPK activity remains dormant. The longer cycle, on the order of 20-30 hours, could be linked to the mammalian cell division cycle (Ahn et al., 2017) or circadian rhythms (Bass and Takahashi, 2010). However, it is less clear whether the shorter cycles have any relationship to the yeast metabolic cycle, which has a period of 2-3 hours but is closely linked to the cell cycle.
Our data reiterate the remarkable adaptability of ATP homeostasis that has previously been reported (Gowans et al., 2013; Hao et al., 2010). The stability of ATP concentration under severe perturbation of ATP production by OXPHOS inhibitor implies that a large fraction of ATP production can be shifted to glycolysis within seconds, despite its low yield of 2 ATP per glucose molecule relative to the ~30 produced by OXPHOS. Because ATP homeostasis is maintained even in cells without a detectable AMPK response (OP-ind), AMPK is likely not required for this initial adaptation. Rather, our data imply that during OXPHOS inhibitor treatment, flux through glycolysis is redirected from the production of biosynthetic intermediates, which are uncoupled from ATP production (Lunt and Vander Heiden, 2011), to prioritize the production of ATP. In OP-ind cells, the ATP consumption load is low enough that this shift can occur without a large perturbation of ADP/ATP ratio, precluding activation of AMPK. In OP-dep cells, this shift is sufficiently rapid to preserve ATP levels, but generates a large enough rise in ADP/ATP ratio to cross the ultrasensitive threshold for AMPK activation (Hardie et al., 1999). AMPK likely plays a longer-term role in metabolic adaptation, consistent with the finding that it is required for mammalian development (Viollet and Foretz, 2016) but not for cellular viability (O’Neill et al., 2011).
Recently developed pharmacological AMPK activators (Cokorinos et al., 2017; Myers et al., 2017) and OXPHOS inhibitors (Molina et al., 2018; Shi et al., 2019) are expected to be useful in the treatment of cancer, diabetes, and inflammatory conditions. Understanding the factors that underlie the heterogeneous OXPHOS inhibitor response will be important in optimizing this growing class of pharmacological compounds. The ability to predict and control the fraction of cells that respond to OXPHOS inhibitors may allow these drugs to be tailored toward different goals. Potent induction of energy stress in the largest number of cells possible may be desirable in the case of anti-cancer therapy, but heterogeneous activation may be preferable when trying to restore physiological energy balance in diabetes or metabolic syndrome. Measuring metabolic heterogeneity may also be important for predicting responses to other drugs, such as cytotoxic chemotherapies. Our study opens a new window into how cellular heterogeneity in drug responses can arise from underlying metabolic differences between cells.
Limitations of Study
This study relies on immortalized cell lines, which carry genetic abnormalities, and which are cultured in medium that does not correspond to physiological conditions. Thus, this study does not bear on whether the heterogeneity we observe occurs within the human body. While we identify factors that alter or predict heterogeneous metabolic behavior, our study does not identify the underlying process that creates alternating periods of OXPHOS inhibitor sensitivity or specify the nature of this process. Changes in gene expression profile could underlie shifts in sensitivity, but it is equally possible that post-translational modifications of proteins, or changes in metabolic pathway flux generate the observed variation; further work will be needed to distinguish these possibilities. Finally, our conclusions rely heavily on live-cell reporters of cellular metabolites or kinase activity, and though we provide validation of reporter data using alternate methods, we cannot exclude the possibility that the reporters show some cross-reactivity to additional factors in the cell.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact John Albeck (jgalbeck@ucdavis.edu).
Materials Availability
Plasmids generated in this study are forthcoming to Addgene. All cell lines and plasmids will be made available upon request from the Lead Contact.
Data and Code Availability
All data processing was performed in MATLAB using previously described methods (Gillies et al., 2020; Pargett and Albeck, 2018; Pargett et al., 2017). MATLAB scripts will be provided upon request by the Lead Contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and media
Routine cell culture for human mammary epithelial cells, MCF10A clone 5E (Janes et al., 2010) and 184A cells were performed as previously described (Debnath et al., 2003). MCF10A and 184A1 were grown in ‘DMEM/F12 growth medium’ (see Media table). Primary stocks from the original clonal derivation (MCF10A-5E) or the ATCC (184A1) were used in all experiments. MCF7, U87, and A549 cell lines were obtained from ATCC and cultured in ‘DMEM growth medium’ (see Media composition). All cells were routinely split when they are ~80% confluent.
In live microscopy experiments, we used a custom formulation, termed ‘imaging base-DMEM/F12’, which consists of DMEM/F12 lacking glucose, glutamine, riboflavin, folic acid, and phenol red (Life Technologies or UC Davis Veterinary Medicine Biological Media Service) which allows adjustment of available nutrients and avoids fluorescence background. All experiments involving MCF10A or 184A1 cell line were performed in ‘Imaging medium 1’ (see Media composition). ‘Imaging medium 1 – noAA’ was used in experiments that involved amino acid perturbation. For experiments with MCF7, U87 or A549 cell lines, ‘Imaging medium 2’ was used. For all experiments, ‘Imaging medium 1’, ‘Imaging medium 1 – noAA’ and ‘Imaging medium 2’ were supplied with glucose 17 mM and 25 mM, respectively, unless indicated otherwise.
Before imaging, cells were washed twice with their respective media and then cultured in imaging experiment media at least 2 hours prior to imaging, unless indicated otherwise. The cell to media ratio was maintained at 150-200 cells/μl for all experiments. For experiments involving titration of insulin or EGF concentrations, cells were placed in EGF- or insulin-deficient media for 4 – 6 hours prior to imaging.
Media composition
DMEM/F12 growth media
| Component | Vendor | Catalog number | Final Concentration |
|---|---|---|---|
| DMEM/F2 | Gibco | 11320-033 | - |
| Horse Serum | Invitrogen | 16050-122 | 5% |
| EGF | Peprotech | AF-100-15 | 20 ng/ml |
| Hydrocortisone | Sigma | H0888 | 0.5 mg/ml |
| Cholera toxin | Sigma | C8052 | 100 ng/ml |
| Insulin | Sigma | I9278 | 10 ug/ml |
DMEM growth medium
| Component | Vendor | Catalog number | Final Concentration |
|---|---|---|---|
| DMEM | Gibco | 11965-092 | - |
| Fetal bovine serum | Gemini bio products | 100-106 | 10% |
Imaging medium 1
| Component | Vendor | Catalog number | Final Concentration |
|---|---|---|---|
| Imaging base-DMEM/F12 | Gibco | Custom; equivalent to Gibco 11320-033 lacking glucose, glutamine, pyruvate, riboflavin, folic acid, and phenol red | - |
| D-glucose | Fisher | D16 | 17 mM |
| BSA | Invitrogen | 16050-122 | 0.1% w/v |
| Hydrocortisone | Sigma | H0888 | 0.5 mg/ml |
| Cholera toxin | Sigma | C8052 | 100 ng/ml |
| Penicillin-Streptomycin | Gibco | 15140122 | 100 U/ml |
| EGF | Peprotech | AF-100-15 | 20 ng/ml |
| Insulin | Sigma | I9278 | 10 ug/ml |
Imaging medium 2
| Component | Vendor | Catalog number | Final Concentration |
|---|---|---|---|
| Imaging base-DMEM/F12 | Gibco | Custom; equivalent to Gibco 11320-033 lacking glucose, glutamine, pyruvate, riboflavin, folic acid, and phenol red | - |
| D-glucose | Fisher | D16 | 25 mM |
| BSA | Invitrogen | 16050-122 | 0.1% w/v |
| Penicillin-Streptomycin | Gibco | 15140122 | 100 U/ml |
Imaging medium 1 - noAA
| Component | Vendor | Catalog number | Final Concentration |
|---|---|---|---|
| Imaging base-DMEM/F12-noAA | Gibco | Custom; equivalent to Gibco 11320-033 lacking glucose, glutamine, amino acids, pyruvate, riboflavin, folic acid, and phenol red | - |
| D-glucose | Fisher | D16 | 17 mM |
| BSA | Invitrogen | 16050-122 | 0.1% w/v |
| Hydrocortisone | Sigma | H0888 | 0.5 mg/ml |
| Cholera toxin | Sigma | C8052 | 100 ng/ml |
| Penicillin-Streptomycin | Gibco | 15140122 | 100 U/ml |
| EGF | Peprotech | AF-100-15 | 20 ng/ml |
| Insulin | Sigma | I9278 | 10 ug/ml |
Seahorse Assay Medium
| Component | Vendor | Catalog number | Final Concentration |
|---|---|---|---|
| Seahorse XF base medium | Agilent | 103334-100 | - |
| Sodium pyruvate | Gibco | 11360070 | 1 mM |
| L-Glutamine | Gibco | 35050079 | 2 mM |
METHOD DETAILS
Reporter construction
The reporters AMPKAR2 (Hung et al., 2017) and ERKTR-mCherry (Sparta et al., 2015) were previously described. PercevalHR (Tantama et al., 2013), ATeam1.03 (Imamura et al., 2009), and GLUT1 were obtained from Addgene. TOP-H2B-YFP-DD (Han et al., 2014) was generously provided by Tobias Meyer. PercevalHR was modified with a nuclear export sequence at the C-terminus to compartmentalize the sensor in the cytosol. AMPKAR2, PercevalHR, and Ateam1.03 sensors were cloned into a vector compatible with piggyBAC transposase-mediated delivery (Yusa et al., 2011) to minimize recombination between CFP and YFP. GLUT1-IRES-NLS-mCherry was constructed by cloning the GLUT1 coding sequence (Takanaga et al., 2008) into the retroviral vector pBabe-neo (BamHI/Xhol); a nuclear localization signal (NLS) was added to mCherry by PCR and was cloned into retroviral vector pBabe-neo (BamHI/EcoRI). IRES-NLS-mCherry was then inserted at the 3’ end of GLUT1 (Xhol/Sall). pLJM1-TFEB-TR. TFEBTR-mCardinal was constructed by inserting the coding sequence for TFEB residues 1-237 into pLJM1 upstream of and in-frame with the coding sequence of mCardinal. Correct insertions for all plasmids were confirmed by sequencing.
Reporter Delivery
Cell lines stably expressing biosensors were generated by retroviral transduction or transfection with the PiggyBac transposase system (Yusa et al., 2011). PiggyBac plasmids were delivered by electroporation (Amaxa II system, Lonza). After transfection or transduction, cells were selected with puromycin (1–2 μg/ml) or geneticin (300 μg/ml); single-cell clones were made by limiting dilution or flow cytometry sorting. For each reporter, we isolated multiple stable clones with homogenous expression; data reported in this study reflect representative behaviors that were consistent across clones for each reporter line. Main reporter cell lines were confirmed to be mycoplasma-negative by PCR; results were validated by third-party testing of selected lines (ATCC).
Live-cell fluorescence microscopy
Time-lapse wide-field microscopy was performed as described previously (Hung et al., 2017; Pargett et al., 2017). Briefly, 25,000 cells were plated one day prior to imaging in glass-bottom 96-well plates (Cellvis P96-1.5H-N, Mountain View, CA) pretreated with type I collagen (Gibco A10483-01) to promote cell adherence. For experiments with drug addition, cells were placed in imaging medium until the addition of the drug. For drugs dissolved in DMSO, the final DMSO concentration was <0.1%. Cells were maintained in 95% air and 5% CO2 at 37°C in an environmental chamber. Images were collected with a Nikon (Tokyo, Japan) 20/0.75 NA Plan Apo objective on a Nikon Eclipse Ti inverted microscope, equipped with a Lumencor SOLA or Lumencor SPECTRA X light engine. Fluorescence filters used in the experiment are: DAPI (custom ET395/25x - ET460/50m - T425lpxr, Chroma), CFP (49001, Chroma), Sapphire (custom ET420/10x - ET525/50m - T425lpxr, Chroma), GFP (49002, Chroma), YFP (49003, Chroma), Cherry (41043, Chroma) and Cy5 (49006, Chroma). For AMPKAR2 and Ateam1.03 biosensors, CFP and YFP filters were used to acquire images, while for the PercevalHR biosensor Sapphire and GFP filters were used. Images were acquired using AndorZyla 5.5 scMOS camera every 6 – 7 minutes with 2x2 binning. Exposure times for each channel were 25-50 ms for DAPI; 150 – 250 ms for CFP; 150 – 250 ms for YFP; 500 – 750 ms for Sapphire; 500 – 750 ms for GFP; 300 – 500 ms for Cherry and 300 – 500 ms for Cy5.
Immunofluorescence microscopy
At indicated times during live-cell imaging experiment, 8% paraformaldehyde was added directly into imaging media to make 2% paraformaldehyde final concentration. Paraformaldehyde fixation was performed for 15 minutes, followed by permeabilization with 100% methanol. Cells were then washed in PBS-T (0.1% Tween-20 in PBS) twice and blocked with Odyssey Blocking Buffer (Li-Cor, Lincoln, NE) for 1 hour at room temperature. Reporter fluorophores were bleached as described in the CyCIF protocol (Lin et al., 2015). Samples were then incubated with primary antibody at the indicated concentrations (see Antibody Table), diluted in blocking buffer, overnight at 4°C. Secondary staining was performed with Alexa 647-conjugated anti-rabbit (Life Technologies, A-21245, diluted at 1:1000 in blocking buffer), followed by DNA staining with Hoechst-33342 (Life Technologies, H3570, diluted at 1:1000 in PBS). Plates were imaged as described for live-cell microscopy, using DAPI and Cy5 filter sets. After imaging, the Afterwards, intensity of fixed-cell images in each condition were matched back to the corresponding time-lapse movies.
Single cell protein synthesis estimation by O-propargyl-puromycin (OPP)
To estimate global nascent protein synthesis rate, we pulse-labeled cells with the puromycin analog O-propargyl-puromycin (OPP; Click Chemistry Tools #1407) at 10 μM final concentration for 30 minutes before the end of live-cell imaging. After fixation, permeabilization, and fluorophore bleaching as described earlier, cells were then incubated with click chemistry reaction buffer (10 μM Azide dye + 4mM CuSO4 + 50 mM Ascorbic acid in 100 mM Tris Buffer pH 8.5) containing Alexa 647 Azide dye (Click Chemistry Tools #1299) for 1 hour. Then, samples were washed with PBS three times and imaged as described earlier.
Phos-Tag electrophoresis and western blot
All samples for western blot experiments were collected from cells cultured in 6 well-plates at 80% confluency. Samples were lysed with ice-cold RIPA buffer. For Phos-Tag™ gel electrophoresis, we used SuperSep™ Phos-tag™ Precast Gels (Wako; 195-17991). Samples were loaded at 3 ug/lane, as measured by BCA protein assay (Thermo Scientific 23225).
The electrophoresis running buffer was Tris-Glycine-SDS solution (25 mM Tris, 192 mM Glycine, 0.1% SDS, pH 8.3), supplied with 1.25 mM sodium bisulfite immediately before electrophoresis. Electrophoresis was performed at 100V, constant voltage for 3 hours at 4°C. After electrophoresis was completed, gels were washed in methanol-free transfer buffer (25 mM Tris, 192 mM Glycine, pH 8.3, 10 mM EDTA) 3 times, 10 minutes each in order to remove divalent cations that would immobilize phosphorylated proteins in the gel. Then gel was equilibrated in transfer buffer (25 mM Tris, 192 mM Glycine, pH 8.3, 10 mM EDTA, 20% v/v Methanol) twice, 10 minutes each. Separated proteins were transferred to PVDF membrane using wet blot transfer method at 18V, overnight at 4°C.
Following protein transfer, membranes were stained with 3% w/v Ponceau S to validate transfer efficiency, then thoroughly de-stained with Milli-Q water and 0.1%PBST (10 mM Tris–HCl (pH 7.5), 100 mM NaCl, and 0.1% v/v Tween-20). Non-specific antibody binding was blocked by incubating membranes in Odyssey blocking buffer (Licor; 927-40000) for 1 hour at room temperature. Primary antibodies (Rabbit Anti-GFP, CST 2956) were diluted to 1:1000 in blocking buffer and incubated with the membrane overnight at 4°C to detect the AMPKAR2 reporter, Following extensive washing in 0.1%PBST (3 times, 10 minutes each), membranes were incubated with diluted IRDye 800CW (Licor; 926-32211) secondary antibodies for 1 hour, at room temperature. After washing in 0.1%BST (3 times, 10 minutes each) immunoreactive bands were recorded with an Odyssey CLx imaging system.
Luminescence ATP determination
ATP concentration for bulk cell populations was determined using an ATP determination kit (Thermo Fisher, A22066), using protocol provided by the manufacturer with minor modification as follows. Cells were plated in 96-well plate at 25000 cell/well 1 day before the experiment and treated as previously described for live-cell microscopy. Samples were collected at indicated time points by incubation with Trichloracetic acid (TCA), final concentration of 2.5% v/v, at 4° C for 30 minutes. After cell lysis, samples were diluted five-fold to minimize TCA concentration (now 0.5% v/v). 10 μl of diluted sample was added to 90 μl reaction solution (see product manual), in 96-well plate assay plate (Corning 3603) followed by incubation for 15 minutes at room temperature. Luminescence was monitored by microplate reader (Molecular Device, SpectraMax M5) at 560 nM, room temperature.
Measurement of mitochondrial stress responses and ATP flux from glycolysis/oxidative phosphorylation
XF24 cell culture plates and sensor cartridges (100867–100) were purchased from Seahorse Bioscience (North Billerica, MA). Cells were plated in XF24 cell culture plates at a density determined by optimization experiments (35000 cells/well) and incubated at 37 °C with 5% CO2 overnight in growth medium; even distribution of cells was verified visually. For the mitochondrial stress test, the growth medium was completely removed 24 hours after plating, and cells were washed twice with 1,000 ml of pre-warmed imaging medium 1. 500 ml of imaging medium 1 was added to each well and cells were incubated in a 37 °C incubator without CO2for 1 hr to allow cell equilibration with Imaging medium 1 (see Media Table). Oxygen consumption rates were measured with the XF24 analyzer under this basal condition followed by sequential addition of different oligomycin concentration, as indicated in Supplementary Figure 4A. For ATP fluxes from glycolysis and oxidative phosphorylation estimation, the data collected in the previous study (Hung et al., 2017) using Seashorse Assay Medium (see Media Table) was applied to formula previously described by Mookerjee et al.(Mookerjee et al., 2017).
GC-TOF analysis of metabolites
For GCMS analysis, cells were plated in 10 cm plates at 107 cells per plate. After incubation overnight, the growth medium was replaced with 10 ml of ‘Imaging medium 1’ supplied with 17 mM glucose. After 4 hours of incubation, cells were treated with oligomycin 1.8 μg/ml. Samples were later prepared for gas chromatography study as described in (Fiehn, 2016). Briefly, samples were quenched by immediately replacing the media with 1 ml of pre-chilled, degassed 3:3:2 v/v acetonitrile:isopropanol:water (Fisher) at 0,30,60,150 and 270 minutes following oligomycin, representing the average first peak, trough, and second peak of the AMPKAR2PHOS response to OXPHOS inhibitors. After quenching, samples were flash-frozen in liquid nitrogen and stored in −80°C freezer.
Prior to GC-TOF analysis, all samples were thawed at room temperature and centrifuged at 14,000 rcf. Supernatants were removed, and samples evaporated to dryness using a CentrVap. To remove membrane lipids and triglycerides, dried samples were resuspended with 1:1 v/v acetonitrile:water, decanted and evaporated to dryness using a CentrVap. Internal standards, C8–C30 fatty acid methyl esters (FAMEs), were added to samples and derivatized with methoxyamine hydrochloride in pyridine followed by MSTFA (Sigma-Aldrich 69479) for trimethylsilylation of acidic protons. Derivatized samples were subsequently submitted for analysis by GC-TOFMS.
Primary metabolite data was collected using a Leco Pegasus IV time of flight (TOF) MS (Leco Corporation) coupled to an Agilent 6890 GC (Agilent Technologies) equipped with a 30 m long 0.25 mm id Rtx5Sil-MS column (30 m × 0.25 mm; 0.25 μm phase) and a Gerstel MPS2 automatic liner exchange system (Gerstel GMBH & Co. KG). The chromatographic gradient used a constant flow of 1 ml/min, and an oven temperature ramping from 50°C for to 330°C over 22 minutes. Mass spectrometry data were collected using 1525 V detector voltage at m/z 85–500 with 17 spectra/sec, electron ionization at −70 eV and an ion source temperature of 250°C. QC injections, blanks, and pooled human plasma were used for quality assurance throughout the run. Data were processed by BinBase (Fiehn et al., 2005) for deconvolution, peak picking, filtering, and metabolite identifications.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image processing
After background subtraction and flat field correction, image data were processed to segment and average pixels within each identified cell’s nucleus and cytoplasm, using a custom procedure written for MATLAB (Pargett et al., 2017), with modifications in the cytosolic identification protocol as described below. Image data were stored in ND2 files generated by NIS Elements and accessed using the Bio-Formats MATLAB toolbox. Individual cells were tracked overtime using uTrack 2.0 (Jaqaman et al., 2008). Cytoplasmic masks were created by watershed method (Vincent and Soille, 1991) using cytosolic YFP (for cell lines expressing AMPKAR2 or ATeam1.03) or GFP (for cell lines expressing PercevalHR) to identify the cytosolic boundary. The cytosolic area is further restricted to the area within 5 pixels from the nuclear border. The resulting single-cell time series traces were filtered for quality by a minimum length of trace and maximum number of contiguous missing or corrupt data points.
FRET reporter calibration and measurement
To quantify FRET biosensors (AMPKAR2 and ATeam1.03), we calculated FRET efficiency exactly as shown previously (Gillies et al., 2020), using a spectral model of light propagated through the microscopy system, including the live cell specimen. Since AMPKAR2 reporter is a substrate for AMPK kinase activity, it is possible to estimate the fraction of sensor that is phosphorylated using Phos-Tag™ electrophoresis, followed by immunoblot against GFP (see Phos-Tag electrophoresis and western blot). This measurement allows us to convert FRET ratios to the fraction of AMPKAR2 sensor that is phosphorylated, AMPKAR2PHOS. Western blot images were manually segmented to quantify protein bands using ImageJ and quantified as average band intensity. AMPKAR2 phosphorylation fraction was calculated by computing the ratio of the phosphorylated band over the summation of phosphorylated and unphosphorylated bands. Conditions shown in Supplementary Figure 1C–D (4 replicates per treatment) were selected because they exhibit sustained AMPKAR2 activity over a range of intensities. The average fraction of reporter phosphorylated was quantified in each condition and compared with the average FRET efficiency as calculated from live-cell experiments with corresponding treatments and time points. Linear fitting was performed, providing a calibrated measurement of the fraction of AMPKAR2 phosphorylated, based on live-cell measurements (eq 1).
| (1) |
Perceval reporter measurement
Unlike FRET reporters, PercevalHR has only one fluorophore, cp173 mVenus, that binds to ATP and ADP differentially, resulting in a shift of excitation spectra with peaks at 470 nM (ATP-bound) and 405 nM (ADP-bound)(Tantama et al., 2013). To measure the proportions of these forms, we imaged cells expressing PercevalHR reporter with Sapphire and GFP filters (see Live-cell fluorescence microscopy). To account for variation in microscope light source set up from experiment to experiment, we scaled image measurements by the relative excitation intensity and exposure time delivered in each channel. The ratio of intensity when excited by Sapphire and GFP filters, which we term PercevalEX, reflects the ratio of ADP to ATP.
Cell age and sister cell analysis
For sister cell analysis, we expressed an NLS-mCherry nuclear marker in MCF10A-AMPKAR2 cell line to improve nuclei tracking accuracy across cytokinesis. Cell division events were first automatically identified by uTrack2.0 (Jaqaman et al., 2008) and later manually verified. In total, we were able to record more than 5,500 cell division events (11,000 related cells) within 25 hours. This dataset gave us estimates on both each cell’s age and their lineage at the time they were challenged with oligomycin.
The similarity of AMPKAR2PHOS response between cell sister cells was calculated by computing the Euclidean distance of AMPKAR2PHOS responses within the 2-hour window after oligomycin treatment. To determine whether AMPKAR2PHOS response between sister cells was more similar than that of unrelated cells, we generated 1000 random pairs of cells that divided at the same time and computed the average AMPKAR2PHOS Euclidean distance. We were able to estimate the Euclidean distance of AMPKAR2PHOS between unrelated cells with 95% confidence interval. The age-dependent increase in the AMPKAR2PHOS Euclidean distance was fitted by an exponential function to estimate the half-life.
Analysis and statistics of kinetics in reporter signals
A custom MATLAB algorithm was designed to identify peaks (Gillies et al., 2017) in the time-lapse signals of AMPKAR2, PercevalHR, and Ateam1.03 activity. The AMPK, PercevalHR, and Ateam1.03 were first smoothened using Butterworth low pass filter with a 3-timepoint cutoff period to remove spurious noise. Peaks and associated valleys in the index were identified by setting two local cutoff values, based on maximum and minimum values of the data within a sliding time window (typically 120 minutes for AMPKAR2 and PercevalHR, 30 minutes for Ateaml .03). A peak was detected if both cutoff values were crossed by a rise and subsequent fall in the index. Typically, more than 300 individual cell recordings were scored for each condition and plotted as a histogram.
For long-term AMPK activity analysis, we identified the ‘strong’ phases of low AMPK activity (OP-ind) as follows. First, we applied the moving standard deviation with a 3-hour sliding window to AMPKAR traces from cells treated with oligomycin. We chose a 3-hour sliding window, because the peak-to - peak period of AMPK activity is around 2 hours, making 3 hours the Nyquist interval. Next, we used the 95th percentile of the calculated moving standard deviation values from all cells treated with vehicle to set a cut-off point for ‘strong’ AMPK activity (Figure S4A, upper panel). Any sections of AMPK activity trace that had a moving standard deviation period below the cut-off for longer than 3 hours were designated as ‘’ AMPK response phases
GC-TOF data analysis
Peak heights of each metabolite were used for further statistical analysis. First, data were normalized by using the sum of the knowns, or mTIC normalization, to scale each sample. Peak heights were then submitted using R to DeviumWeb (v0.3.2). The data were normalized further by log transformation and Pareto scaling. ANOVA analysis was performed with Tukey post hoc testing with an alpha of 0.05. The reported trends in metabolite abundance following oligomycin treatment were robust to the normalization scheme and could also be observed in raw peak values.
Distributions and statistical tests
Statistical tests, including ANOVA, t-tests, and Pearson’s correlations, were performed using standard functions in MATLAB. R2 values were calculated as the square of the Pearson correlation coefficient. Where indicated in the figure legends, a linear regression model was generated, using the MATLAB command fitlm. Box and whisker plots show the median value (red line), interquartile range (box), range (whiskers), and outliers (plus symbols); for all other cases, definition of centers and dispersion measures are listed in the figure legends. Distributions of single-cell measurements were plotted and inspected visually to confirm that they met the assumptions of the statistical tests used. To test for bimodality, data were fitted to a bimodal Gaussian mixture distribution and a panel of unimodal distributions, including (including normal, log-normal, generalized extreme value, and Weibull). The best-fitted distribution was selected using corrected Akaike’s Information Criterion, to account for additional parameter terms (Cavanaugh, 1997). Data were considered bimodally distributed if and only if the bimodal Gaussian mixture distribution was ranked as the best-fitted distribution.
Pearson’s cross-correlation of time series
The time series to be compared were normalized by subtracting by their corresponding averages. To quantify lag between reporters for each time series, the maximal cross-correlation value was computed using the MATLAB xcorr function. We assumed that each pair of reporters, namely AMPKAR2 and TFEB-TR or AMPKAR2 and ERKTR, had a characteristic lag time, estimated as the mode of calculated lags across all sampled cells. The lag times identified from this process were used to align two time-series data. Pearson’s correlation coefficient was computed from these aligned time series for each cell. Significance of the differences between the Pearson’s coefficient distributions for different treatments was calculated by t-test.
Partial least squares regression modeling
To evaluate the relative contributions of glycolytic activity, protein synthesis rate and cell cycle stage to AMPK responses to OXPHOS inhibition, we performed live-cell experiments using MCF10A cells expressing AMPKAR2 and GLUT1-NLS mCherry, in which allows glycolytic capacity can be estimated by measuring mCherry intensity. After 4 hours of live imaging, cells were pulse-labeled with OPP for 30 minutes and later treated with oligomycin 1.8 μg/ml for 15 minutes prior to fixation. Samples were bleached and stained with phospho-Rb and Hoechst 33342 as described earlier. Intensity of mCherry, OPP, phospho-Rb and Hoechst were used as proxies of glycolytic activity, protein synthesis rate, G1/S transition, and DNA content, respectively. Since the relationship between DNA content and phospho-Rb is not linear, as shown in Figure 6A, we created a linearized pseudotime variable for cell cycle progression using Wanderlust (Bendall et al., 2014). AMPKAR2Δ in response to oligomycin treatment was used as the output variable for PLSR models, while mCherry, OPP, DAPI, phospho-Rb and cell cycle pseudotime values were used as input variables. Data were first centered by subtracting the mean from each parameter and input parameters were scaled to unit variance. The MATLAB implementation of the SIMPLS partial least squares regression algorithm was employed to fit paired data sets to a linear model.
Replicates
Numbers of independent replicates are indicated in each figure legend as “N”; we define ‘independent replicate’ as a complete, separate performance of a time-lapse imaging experiment with similar culture and treatment conditions, beginning from the plating of cells from bulk culture on an imaging plate and occurring on different days from other replicates. For all independent replicates, a minimum of 200 cells were imaged and tracked in each condition. Unless noted otherwise, where single-cell recordings are shown, the displayed cells were chosen by random number generation in MATLAB with a threshold for minimum tracking time to eliminate cells in which recording was terminated prematurely due to failure of the tracking algorithm. The chosen tracks were manually verified to be representative of successfully tracked cells and consistent with the overall range of cell behaviors. Cellular measurements determined by manual inspection to have poor tracking or quantification accuracy were discarded.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-Rb (Ser807/811) (D20B12) | Cell Signaling Technology | RRID:AB_11178658 Cat#8516 |
| Phospho-Acetyl-CoA Carboxylase (Ser79) (D7D11) | Cell Signaling Technology | RRID:AB_2687505 Cat#11818 |
| Phospho-S6 Ribosomal Protein (Ser240/244) (D68F8) | Cell Signaling Technology | RRID:AB_10694233 Cat#5364 |
| Phospho-4E-BP1 (Thr37/46) (236B4) | Cell Signaling Technology | RRID:AB_560835 Cat#2855 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) | Cell Signaling Technology | RRID:AB_2315112 Cat#4370 |
| GFP (4B10) | Cell Signaling Technology | RRID:AB_1196614 Cat#2955 |
| Goat anti-Rabbit IgG Alexa 647 | Life Technologies | RRID:AB_2535813 A-21245 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Oligomycin A | Sigma | Cat#75351 |
| Insulin | Sigma | Cat#I9278 |
| EGF | Peprotech | Cat#100-15 |
| Hydrocortisone | Sigma | Cat#H0888 |
| Cholera Toxin | Sigma | Cat#C8052 |
| DMEM/F12 | Invitrogen | Cat#11330-032 |
| Pen/Strep | Invitrogen | Cat#15070-063 |
| Antimycin A | Sigma | Cat#A8674 |
| Rotenone | Sigma | Cat#45656 |
| FCCP | Sigma | Cat#C2920 |
| Glucose | Fisher | Cat#D16 |
| MK2206 | Selleckchem | Cat#S1078 |
| Torin1 | Selleckchem | Cat#S2827 |
| 2DG | Sigma | Cat#D8375 |
| 3PO | Selleckchem | Cat#S7639 |
| BU99006 | Santa Cruz Biotechnology | Cat#SC-300307 |
| CAS-648926-15-2 | EMD Millipore | Cat#361515 |
| Etomoxir | Sigma | Cat#E1905 |
| 6AN | Cayman Chemical | Cat#10009315 |
| Cycloheximide | EMD Millipore | Cat#239763 |
| Collagen I, Rat tail | Gibco | Cat#A10483-01 |
| Bafilomycin A | Cayman Chemical | Cat#11038 |
| SBI0206965 | Sigma | Cat#SML1540 |
| PF0175157 | Sigma | Cat#PZ0299 |
| Paraformaldehyde 8% | Electron Microscopy Sciences | Cat#157-8-100 |
| Hoechst 33342 | Life Technologies | Cat#H3570 |
| Cell Culture Media | ||
| DMEM/F-12 1:1 | Life Technologies | Cat#11320 |
| Horse Serum | Invitrogen | 16050-122 |
| DMEM | Life Technologies | 11965175 |
| FBS | Life Technologies | 11965175 |
| Experimental Models: Cell lines | ||
| MCF10A | Janes et al., 2010 | RRID:CVCL_0598 |
| 184A1 | ATCC | RRID:CVCL_3040 |
| MCF7 | ATCC | RRID:CVCL_0031 |
| U87 | ATCC | RRID:CVCL_0022 |
| A549 | ATCC | RRID:CVCL_0023 |
| MCF10A-AMPKAR2 | (Hung et al, 2017) | |
| MCF10A-AMPKAR2-ERKTR | This report | |
| MCF10A-AMPKAR2-TFEBTR | This report | |
| MCF10A-PercevalHR | This report | |
| MCF10A-Ateam | This report | |
| MCF10A-AMPKAR2-GLUT1_NLSmCherry | This report | |
| MCF10A-AMPKAR2-Geminin::mCherry | This report | |
| MCF10A-AMPKAR2-NLS::mCherry | This report | |
| MCF10A-TOP | This report | |
| 184A1-AMPKAR2 | This report | |
| MCF7-AMPKAR2 | This report | |
| U87-AMPKAR2 | This report | |
| A549-AMPKAR2 | This report | |
| Recombinant DNA | ||
| pPBJ-AMPKAR2-puro | (Hung et al, 2017) | |
| pLJM-ERKTR::mCherry | (Sparta et al., 2015) | |
| GW1-PercevalHR | (Tantama et al., 2013) | Addgene#49082 |
| pPBJ-PercevaIHR-puro | This report | |
| ATeam1.03-nD/nA/pcDNA3 | (Imamura et al., 2009) | Addgene#51958 |
| pPBJ-Ateam1.03-puro | This report | |
| TOP-H2B-YFP-DD | (Han et al., 2014) | Addgene#96891 |
| pLJM-geminin::mCherry | (Hung et al., 2017) | |
| pLJM-TFEBTR::mCardinal | This report | |
| pBabe-NLS::mCherry | This report | |
| pBabe-GLUT1-NLS::mCherry | This report | |
| Software and Algorithms | ||
| NIS-Elements AR ver. 4.20 | Nikon | RRID:SCR_014329 |
| Bio-Formats ver. 5.1.1 (May 2015) | OME | RRID:SCR_000450 |
| uTrack 2.0 | (Jaqaman et al., 2008) | http://www.utsouthwestern.edu/labs/danuser/software/ |
| MATLAB | Math works | SCR_001622 |
| Other | ||
| Glass Bottom Plates, #1.5 cover glass | In Vitro Scientific | Cat#P24-1.5H-N, P96-1.5H-N |
Highlights.
Live-cell imaging during OXPHOS inhibition probes energetic balance in single cells
Transient energetic resistance occurs in multiple cell types and persists for hours
Resistant cells lack AMPK responses and fail to suppress ERK and mTOR signals
The balance of glycolysis and ATP turnover determines OXPHOS inhibitor response
Acknowledgements
Funding for this work was provided by the American Cancer Society (IRG-95-125-13), the National Institute of General Medical Sciences (1R01GM115650), and the American Association for Cancer Research/Stand Up To Cancer (SU2C-AACR-IRG-01-16). Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. Flow-cytometry services were supported by the UC Davis Comprehensive Cancer Center Support Grant (CCSG) awarded by the National Cancer Institute (NCI P30CA093373). Metabolomics were performed in collaboration with West Coast Metabolomics Center at UC Davis. TOP-H2B-YFP-DD was kindly provided by Tobias Meyer. Seahorse assays were performed with the assistance of Gino Cortopassi, UC Davis School of Veterinary Medicine. We thank Marek Kochanczyk, Ben Tu, Ralph DeBerardinis, Joshua Rabinowitz, Sabrina Spencer, Isaac Harris, and Jon Coloff for helpful discussions.
Declaration of Interests
John Albeck has received research grants from Kirin Corporation. The other authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn E, Kumar P, Mukha D, Tzur A, and Shlomi T (2017). Temporal fluxomics reveals oscillations in TCA cycle flux throughout the mammalian cell cycle. Mol Syst Biol 13, 953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschuler SJ, and Wu LF (2010). Cellular heterogeneity: do differences make a difference? Cell 141, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimov VN (2010). Metformin for aging and cancer prevention. Aging (Albany NY) 2, 760–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton TM, McKenna WG, Kunz-Schughart LA, and Higgins GS (2018). Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin Cancer Res 24, 2482–2490. [DOI] [PubMed] [Google Scholar]
- Bass J, and Takahashi JS (2010). Circadian integration of metabolism and energetics. Science 330, 1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Davis KL, Amir el AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, and Pe’er D (2014). Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157, 714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg J, Hung YP, and Yellen G (2009). A genetically encoded fluorescent reporter of ATP:ADP ratio. Nat Methods 6, 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttgereit F, and Brand MD (1995). A hierarchy of ATP-consuming processes in mammalian cells. Biochem J 312 (Pt 1), 163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, and Tu BP (2012). Driving the cell cycle through metabolism. Annu Rev Cell Dev Biol 28, 59–87. [DOI] [PubMed] [Google Scholar]
- Cavanaugh JE (1997). Unifying the derivations for the Akaike and corrected Akaike information criteria. Statistics & Probability Letters 33, 201–208. [Google Scholar]
- Cokorinos EC, Delmore J, Reyes AR, Albuquerque B, Kjobsted R, Jorgensen NO, Tran JL, Jatkar A, Cialdea K, Esquejo RM, et al. (2017). Activation of Skeletal Muscle AMPK Promotes Glucose Disposal and Glucose Lowering in Non-human Primates and Mice. Cell Metab 25, 1147–1159 e1110. [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, and Brugge JS (2003). Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268. [DOI] [PubMed] [Google Scholar]
- Depaoli MR, Karsten F, Madreiter-Sokolowski CT, Klee C, Gottschalk B, Bischof H, Eroglu E, Waldeck-Weiermair M, Simmen T, Graier WF, et al. (2018). Real-Time Imaging of Mitochondrial ATP Dynamics Reveals the Metabolic Setting of Single Cells. Cell Rep 25, 501–512 e503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos CC, Gopal B, and Verma S (2018). Metformin: An Old Dog with a New Trick? Cell Metab 28, 334–336. [DOI] [PubMed] [Google Scholar]
- Dussmann H, Perez-Alvarez S, Anilkumar U, Papkovsky DB, and Prehn JH (2017). Single-cell time-lapse imaging of intracellular O2 in response to metabolic inhibition and mitochondrial cytochrome-c release. Cell Death Dis 8, e2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott SG, and McLaughlin CS (1978). Rate of macromolecular synthesis through the cell cycle of the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 75, 4384–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, and Rabinowitz JD (2013). Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol 9, 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt S-M, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, Vokes NI, Guarente L, Vander Heiden MG, and Stephanopoulos G (2013). Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat Commun 4, 2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn O (2016). Metabolomics by Gas Chromatography-Mass Spectrometry: Combined Targeted and Untargeted Profiling. Curr Protoc Mol Biol 114, 30.34.31–30.34.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn O, Wohlgemuth G, and Sholz M (2005). Setup and annotation of metabolomic experiments by integrating biological and mass spectrometric metadata. Dils LNBI 3615, 224–239. [Google Scholar]
- Gillies TE, Pargett M, Minguet M, Davies AE, and Albeck JG (2017). Linear Integration of ERK Activity Predominates over Persistence Detection in Fra-1 Regulation. Cell Syst 5, 549–563 e545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies TE, Pargett M, Silva JM, Teragawa CK, McCormick F, and Albeck JG (2020). Oncogenic mutant RAS signaling activity is rescaled by the ERK/MAPK pathway. Mol Syst Biol 16, e9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowans GJ, Hawley SA, Ross FA, and Hardie DG (2013). AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab 18, 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griss T, Vincent EE, Egnatchik R, Chen J, Ma EH, Faubert B, Viollet B, DeBerardinis RJ, and Jones RG (2015). Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol 13, e1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, Davidson SM, Freinkman E, Thomas CJ, and Vander Heiden MG (2016). Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab 24, 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, and Shaw RJ (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett SR, Zanotelli VRT, Xu W, Goya J, Park JO, Perlman DH, Gibney PA, Botstein D, Storey JD, and Rabinowitz JD (2016). Systems-level analysis of mechanisms regulating yeast metabolic flux. Science 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Jaimovich A, Dey G, Ruggero D, Meyuhas O, Sonenberg N, and Meyer T (2014). Parallel measurement of dynamic changes in translation rates in single cells. Nat Methods 11, 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Chang CP, Tsao CC, and Xu J (2010). Oligomycin-induced bioenergetic adaptation in cancer cells with heterogeneous bioenergetic organization. J Biol Chem 285, 12647–12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2014). AMPK—Sensing Energy while Talking to Other Signaling Pathways. Cell Metab 20, 939–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, and Hawley SA (2001). AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays 23, 1112–1119. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, and Hawley SA (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Salt IP, Hawley SA, and Davies SP (1999). AMP-activated protein kinase: an ultrasensitive system for monitoring cellular energy charge. Biochem J 338 (Pt 3), 717–722. [PMC free article] [PubMed] [Google Scholar]
- Hartmann FJ, Mrdjen D, McCaffrey E, Glass DR, Greenwald NF, Bharadwaj A, Khair Z, Verberk SGS, Baranski A, Baskar R, et al. (2020). Single-cell metabolic profiling of human cytotoxic T cells. Nat Biotechnol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, and Hardie DG (2005). Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2, 9–19. [DOI] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al. (2016). Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, Hoxhaj G, Saghatelian A, Shaw RJ, and Manning BD (2017). Metformin Inhibits Hepatic mTORCl Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab 25, 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung YP, Teragawa C, Kosaisawe N, Gillies TE, Pargett M, Minguet M, Distor K, Rocha-Gregg BL, Coloff JL, Keibler MA, et al. (2017). Akt regulation of glycolysis mediates bioenergetic stability in epithelial cells. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura H, Nhat KPH, Togawa H, Saito K, lino R, Kato-Yamada Y, Nagai T, and Noji H (2009). Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci U S A 106, 15651–15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, and Guan KL (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. [DOI] [PubMed] [Google Scholar]
- Janes KA, Wang C-C, Holmberg KJ, Cabral K, and Brugge JS (2010). Identifying single-cell molecular programs by stochastic profiling. Nat Methods 7, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Chen L, and Rabinowitz JD (2018). Metabolomics and Isotope Tracing. Cell 173, 822–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, and Danuser G (2008). Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods 5, 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viol let B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, et al. (2010). Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab 11, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Kim MJ, Kim EJ, Yang Y, Lee MS, and Lim JS (2012). Berberine-induced AMPK activation inhibits the metastatic potential of melanoma cells via reduction of ERK activity and COX-2 protein expression. Biochem Pharmacol 83, 385–394. [DOI] [PubMed] [Google Scholar]
- Konagaya Y, Terai K, Hirao Y, Takakura K, Imajo M, Kamioka Y, Sasaoka N, Kakizuka A, Sumiyama K, Asano T, et al. (2017). A Highly Sensitive FRET Biosensor for AMPK Exhibits Heterogeneous AMPK Responses among Cells and Organs. Cell Rep 21, 2628–2638. [DOI] [PubMed] [Google Scholar]
- Li L, Friedrichsen HJ, Andrews S, Picaud S, Volpon L, Ngeow K, Berridge G, Fischer R, Borden KLB, Filippakopoulos P, et al. (2018). A TFEB nuclear export signal integrates amino acid supply and glucose availability. Nat Commun 9, 2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J-R, Fallahi-Sichani M, and Sorger PK (2015). Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat Commun 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S-C, and Hardie DG (2017). AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. [DOI] [PubMed] [Google Scholar]
- Lunt SY, and Vander Heiden MG (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27, 441–464. [DOI] [PubMed] [Google Scholar]
- Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip A-NA, et al. (2018). An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. [DOI] [PubMed] [Google Scholar]
- Mookerjee SA, Gerencser AA, Nicholls DG, and Brand MD (2017). Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem 292, 7189–7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers RW, Guan HP, Ehrhart J, Petrov A, Prahalada S, Tozzo E, Yang X, Kurtz MM, Trujillo M, Gonzalez Trotter D, et al. (2017). Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 357, 507–511. [DOI] [PubMed] [Google Scholar]
- O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jørgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, et al. (2011). AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A 108, 16092–16097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, and Kemp BE (2011). AMPK is a direct adenylate charge-regulated protein kinase. Science 332, 1433–1435. [DOI] [PubMed] [Google Scholar]
- Orth JD, Thiele I, and Palsson BO (2010). What is flux balance analysis? Nat Biotechnol 28, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pargett M, and Albeck JG (2018). Live-Cell Imaging and Analysis with Multiple Genetically Encoded Reporters. Curr Protoc Cell Biol 78, 4.36.31–34.36.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pargett M, Gillies TE, Teragawa CK, Sparta B, and Albeck JG (2017). Single-Cell Imaging of ERK Signaling Using Fluorescent Biosensors. Methods Mol Biol 1636, 35–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirós PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, Gygi SP, and Auwerx J (2017). Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 216, 2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regot S, Hughey JJ, Bajar BT, Carrasco S, and Covert MW (2014). High-sensitivity measurements of multiple kinase activities in live single cells. Cell 157, 1724–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruas JS, Siqueira-Santos ES, Rodrigues-Silva E, and Castilho RF (2018). High glycolytic activity of tumor cells leads to underestimation of electron transport system capacity when mitochondrial ATP synthase is inhibited. Sci Rep 8, 17383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos DA, Shi L, Tu BP, and Weissman JS (2019). Cycloheximide can distort measurements of mRNA levels and translation efficiency. Nucleic Acids Res 47, 4974–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. (2012). A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31, 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen CH, Yuan P, Perez-Lorenzo R, Zhang Y, Lee SX, Ou Y, Asara JM, Cantley LC, and Zheng B (2013). Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation. Mol Cell 52, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lim SK, Liang Q, Iyer SV, Wang HY, Wang Z, Xie X, Sun D, Chen YJ, Tabar V, et al. (2019). Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 567, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões RV, Serganova IS, Kruchevsky N, Leftin A, Shestov AA, Thaler HT, Sukenick G, Locasale JW, Blasberg RG, Koutcher JA, et al. (2015). Metabolic plasticity of metastatic breast cancer cells: adaptation to changes in the microenvironment. Neoplasia 17, 671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparta B, Pargett M, Minguet M, Distor K, Bell G, and Albeck JG (2015). Receptor Level Mechanisms Are Required for Epidermal Growth Factor (EGF)-stimulated Extracellular Signal-regulated Kinase (ERK) Activity Pulses. J Biol Chem 290, 24784–24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SL, Gaudet S, Albeck JG, Burke JM, and Sorger PK (2009). Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoker ML, Newport E, Hulit JC, West AP, and Morten KJ (2019). Impact of pharmacological agents on mitochondrial function: a growing opportunity? Biochem Soc Trans 47, 1757–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasen J, Sarma U, Jentsch M, Bohn S, Sheng C, Horbelt D, Knaus P, Legewie S, and Loewer A (2018). Cell-specific responses to the cytokine TGFβ are determined by variability in protein levels. Mol Syst Biol 14, e7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Bridges D, Nakada D, Skiniotis G, Morrison SJ, Lin JD, Saltiel AR, and Inoki K (2013). Inhibition of AMPK catabolic action by GSK3. Mol Cell 50, 407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takanaga H, Chaudhuri B, and Frommer WB (2008). GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochimica et Biophysica Acta (BBA) - Biomembranes 1778, 1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantama M, Martinez-Francois JR, Mongeon R, and Yellen G (2013). Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun 4, 2550.24096541 [Google Scholar]
- Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen B, Solmonson A, Murphy MM, Gu Z, Gu W, Martin M, et al. (2020). Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577, 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou P, Zheng B, Hsu CH, Sasaki AT, and Cantley LC (2011). A fluorescent reporter of AMPK activity and cellular energy stress. Cell Metab 13, 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu BP, Mohler RE, Liu JC, Dombek KM, Young ET, Synovec RE, and McKnight SL (2007). Cyclic changes in metabolic state during the life of a yeast cell. Proc Natl Acad Sci U S A 104, 16886–16891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, and DeBerardinis RJ (2017). Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashisht Gopal YN, Gammon S, Prasad R, Knighton B, Pisaneschi F, Roszik J, Feng N, Johnson S, Pramanik S, Sudderth J, et al. (2019). A Novel Mitochondrial Inhibitor Blocks MAPK Pathway and Overcomes MAPK Inhibitor Resistance in Melanoma. Clin Cancer Res 25, 6429–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent L, and Soille P (1991). Watersheds in digital spaces: an efficient algorithm based on immersion simulations. IEEE Transactions on Pattern Analysis and Machine Intelligence 13, 583–598. [Google Scholar]
- Viollet B, and Foretz M (2016). Animal Models to Study AMPK. Exp Suppl 107, 441–469. [DOI] [PubMed] [Google Scholar]
- Wu L, Zhou B, Oshiro-Rapley N, Li M, Paulo JA, Webster CM, Mou F, Kacergis MC, Talkowski ME, Carr CE, et al. (2016). An Ancient, Unified Mechanism for Metformin Growth Inhibition in C. elegans and Cancer. Cell 167, 1705–1718.e1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, et al. (2011). Structure of mammalian AMPK and its regulation by ADP. Nature 472, 230–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Dai Z, and Locasale JW (2019). Metabolic landscape of the tumor microenvironment at single cell resolution. Nat Commun 10, 3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K, Zhou L, Li MA, Bradley A, and Craig NL (2011). A hyperactive piggyBac transposase for mammalian applications. Proceedings of the National Academy of Sciences 108, 1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C-S, Hawley SA, Zong Y, Li M, Wang Z, Gray A, Ma T, Cui J, Feng J-W, Zhu M, et al. (2017). Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data processing was performed in MATLAB using previously described methods (Gillies et al., 2020; Pargett and Albeck, 2018; Pargett et al., 2017). MATLAB scripts will be provided upon request by the Lead Contact.