

Abstract
Hypertension, including transient events, is a major risk factor for developing late-onset dementia and Alzheimer’s disease (AD). Anti-hypertensive drugs facilitate restoration of normotension without amelioration of increased dementia risk suggesting that transient hypertensive insults cause irreversible damage. This study characterized the contribution of transient hypertension to sustained brain damage as a function of normal aging and AD. To model transient hypertension, we treated F344TgAD and non-transgenic littermate rats with L-NG-Nitroarginine methyl ester (L-NAME) for one month, ceased treatment and allowed for a month of normotensive recovery. We then examined the changes in the structure and function of the cerebrovasculature, integrity of white matter, and progression of AD pathology. As independent factors, both transient hypertension and AD compromised structural and functional integrity across the vascular bed, while combined effects of hypertension and AD yielded the largest deficits. Combined effects of transient hypertension and AD genotype resulted in loss of cortical myelin particularly in the cingulate cortex which is crucial for cognitive function. Increased cerebral amyloid angiopathy, a prominent pathology of AD, was detected after transient hypertension as were up- and down-regulation of proteins associated with cerebrovascular remodeling – osteopontin, ROCK1 and ROCK2, in F344TgAD rats even 30 days after restoration of normotension. In conclusion, transient hypertension caused permanent cerebrovasculature and brain parenchymal damage in both normal aging and AD. Our results corroborate human studies that have found close correlation between transient hypertension in midlife and white matter lesions later in life outlining vascular pathologies as pathological links to increased risk of dementia.
Keywords: Alzheimer’s disease, hypertension, F344TgAD rat, vascular reactivity, myelin, cerebral amyloid angiopathy
Graphical Abstract
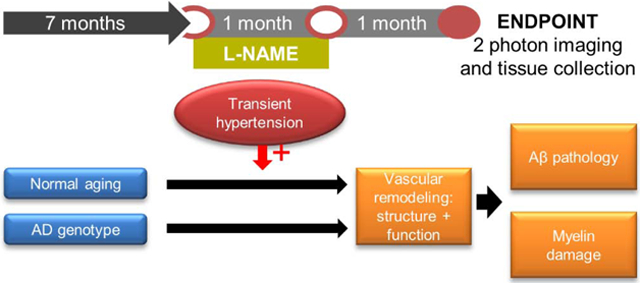
1. Introduction
Alzheimer’s disease (AD) is the most common type of dementia among the elderly population and includes a dominantly-inherited early-onset form and a much more prevalent late-onset form (Querfurth and LaFerla, 2010). The specific etiological causes of late-onset AD remain unclear although several lines of evidence suggest a key role for cerebrovascular dysfunction: diseases of the brain vasculature have a higher rate of comorbidity with AD compared to other neurological disorders (Toledo et al., 2013). Additionally, cerebrovascular pathologies accelerate cognitive decline in AD patients (Toledo et al., 2013). A spatial-temporal analysis of biomarkers revealed that markers of vascular deficits precede that of Aβ deposition suggesting that dysfunction in the cerebrovasculature may play a key etiological role in AD pathogenesis (Iturria-Medina et al., 2016).
Midlife hypertension is widely recognized as an independent and important risk factor of AD (Feldstein, 2012; Launer et al., 2000; Lennon et al., 2019). Most clinical cases of hypertension have a transient nature as the majority of patients receive treatments that regulate blood pressure (Joffres et al., 2013). Some studies have reported that these treatments lower the incidence of dementia, however, a significant number of studies have found either no reduction (in’t Veld et al., 2001; Khachaturian et al., 2006; Walker et al., 2017; Xu et al., 2017; Yasar et al., 2013) or an elevation in incidence suggesting that hypertensive events, though transient, can cause irreversible damage to the brain. The mechanistic links between hypertension and AD development is not well understood: High blood pressure leads to reduced lumen diameter and increased wall thickness in cerebral arteries as well as remodeling of overall vascular structure and loss of vascular function (Pires et al., 2013). A common manifestation of vascular impairment is white matter abnormalities; epidemiological evidence implicates hypertension as an important risk factor for developing white matter lesions (Basile et al., 2006; Liao et al., 1996). Corroboratively, region-specific white matter lesions correlate with both AD incidence and cognitive decline in AD (Brickman et al., 2015; Tabei et al., 2017). Taken together, we propose that the interaction between transient hypertension and AD pathogenesis involve a series of remodeling throughout the cerebrovascular network culminating in impairment of vascular function, loss of cerebral white matter, and progression of AD pathology.
Although several studies have successfully modeled persistent hypertension in animal models of AD by either surgically or pharmacologically constricting blood vessels (Cifuentes et al., 2017, 2015; Díaz-Ruiz et al., 2009; Kruyer et al., 2015; Shih et al., 2018), transient hypertensive events are rarely examined. Thus, we model transient hypertension by administration of N-nitro-L-arginine methyl ester (L-NAME) to F344TgAD (TgAD) and non-transgenic (NTg) littermate rats for a month, cessation of treatment followed by another month of recovery. L-NAME induces hypertension through inhibition of nitric oxide (NO) synthase, as its lowering of NO signaling within the vasculature induces hypertension, inflammation and oxidative stress leading to changes in the cerebrovascular wall structure (Delbosc et al., 2008; Dupuis et al., 2004; Husain and Hazelrigg, 2002). L-NAME-induced inhibition of endothelial nitric oxide synthase causes increased production of superoxide anions within the vasculature that further impairs NO signaling leading to impairment of endothelial-dependent arterial relaxation (Santhanam et al., 2015). Furthermore, attenuated NO signaling has been observed in hypertensive patients (Husain, 2002). The use of L-NAME thus allows us to recapitulate a spectrum of pathologies and facilitate a broad interrogation of downstream pathways that may contribute to long term vascular compromise. Henceforth, we modeled hypertension by L-NAME administration for one month to induce vascular dysfunction, then halted treatment to remove the external driving forces and allow endogenous recovery. After one month of recovery (endpoint of the study), we examined the changes in the structure and function of the cerebrovascular network, integrity of cerebral white matter, and progression of AD pathology in the context of normal aging (in NTg rats) and an AD susceptible environment (in TgAD rats).
2. Results
2.1. Vascular reactivity
We have previously established that F344TgAD (TgAD) rats exhibit a significant reduction in vascular reactivity at nine months of age when both parenchymal plaques and cerebrovascular amyloid are established (Joo et al., 2017). Here, we initiated transient hypertension at the early stages of Aβ production and deposition (Cohen et al., 2013), seven months of age, and our study endpoint was nine months of age to correspond to the established vascular compromise we had previously reported (Joo et al., 2017). We induced hypertension by L-NAME treatment between seven and eight months of age, then removed L-NAME to restore normotension between eight and nine months of age (Fig. 1A). Blood pressure readings at seven, eight, and nine months of age demonstrated that one month of L-NAME treatment elevated systolic pressure from 133 ± 9 mmHg to 175 ± 4 mmHg in NTg rats (p = 0.001), and from 124 ± 4 mmHg to 185 ± 9 mmHg in TgAD rats (p = 0.001), while cessation of treatment for an additional month normalized systolic pressure to 131 ± 6 mmHg (p = 0.001) and 125 ± 5 mmHg (p = 0.001) in NTg and TgAD rats, respectively (Fig. 1B). Having established the transient nature of the hypertensive paradigm, we set out to investigate the relative contributions of transient hypertension and AD genotype on cerebrovascular function. We quantified vascular reactivity in the somatosensory cortex by comparing transit times of an injected dye bolus during hypercapnic versus normocapnic breathing conditions as described previously (Joo et al., 2017). Transit times are expressed as TTP i.e. time interval between bolus Texas Red-dextran injection and peak fluorescent signal in a measured vessel. As hypercapnic breathing leads to vasodilation and increased cerebral blood flow, TTP shortening reflects a more responsive vessel with higher vascular reactivity. We observed that as individual contributing factors, both AD genotype and transient hypertension reduced vascular reactivity by a similar magnitude (~ 50%) across the vascular bed (Fig. 1C). Specifically, transient hypertension significantly reduced vascular reactivity in all three vessel types: arterioles (p = 0.04), capillaries (p = 0.005) and venules (p = 0.002), while AD genotype decreased reactivity in capillaries (p = 0.03) and venules (p = 0.02) with a trend to significance in arterioles (p = 0.07) (Fig. 1C). Although we observed the stronger impact on capillaries and venules, loss of vascular reactivity measured from the draining vessels may reflect dysfunction of the cerebrovascular network as a whole: as the bolus dye traverses the vascular network from arterioles to draining vessels, effects of vessels’ impairments accumulate, making capillary and venular readouts sensitive to the sum total of the deficiencies encountered along the path. Our results overall demonstrate that both hypertension and AD pathogenesis facilitate the loss of vascular function.
Fig. 1.
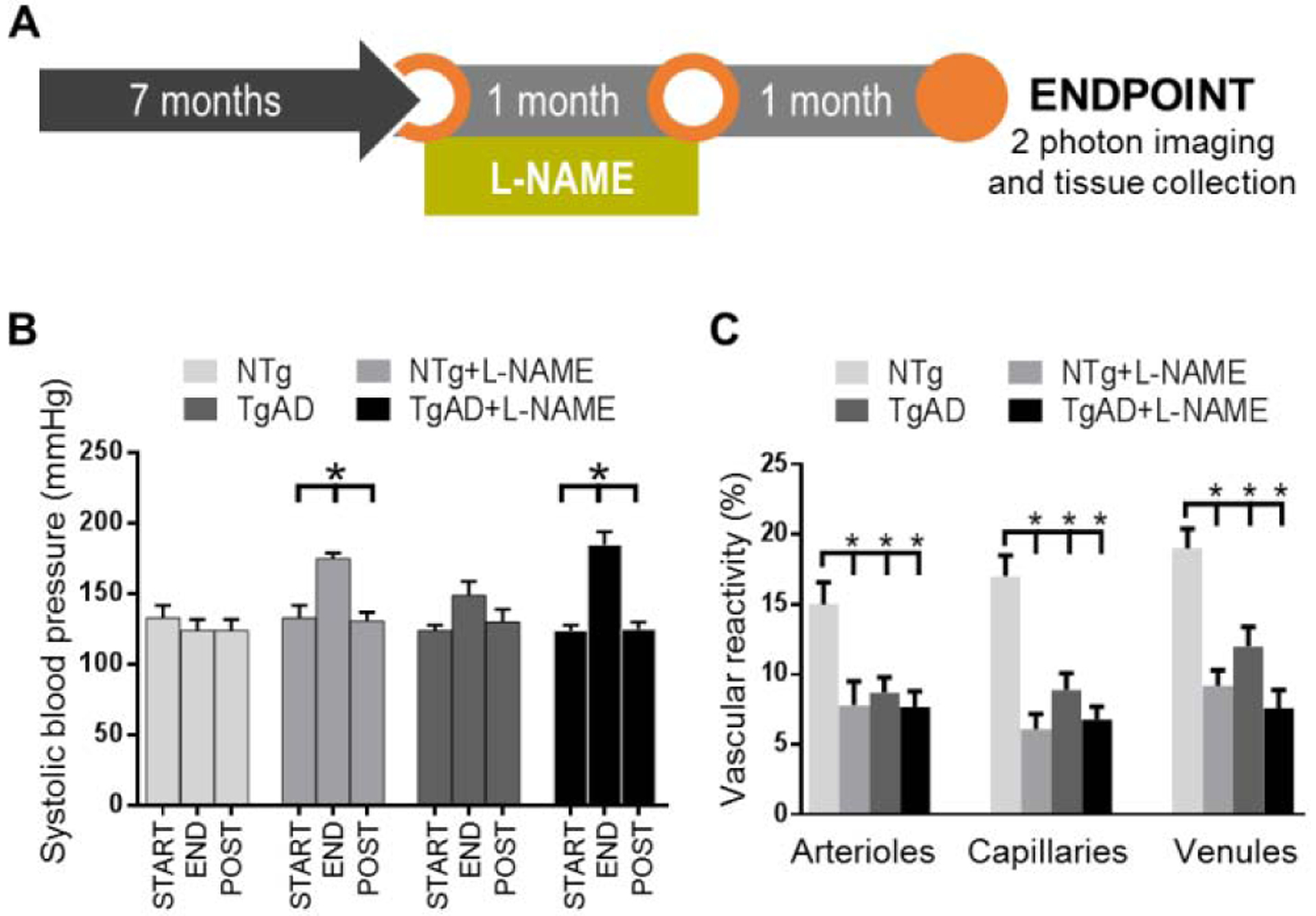
Vascular reactivity is compromised by both transient hypertension and AD. (A) Graphical depiction of the L-NAME treatment timeline. (B) Systolic blood pressure was measured at the start of L-NAME treatment (START), at four weeks after start of L-NAME treatment (END), and at four weeks after end of L-NAME treatment (POST). In both NTg and TgAD rats, increases in systolic blood pressure induced by L-NAME treatment returned to baseline levels (p = 0.001 and p = 0.001 respectively), demonstrating that the hypertensive event induced by the current treatment paradigm is transient. Significance was calculated by Holm-Sidak post hoc following one-way ANOVA. * denotes p < 0.05 in post hoc; n = 6 for NTg; n = 10 for NTg + L-NAME; n = 8 for TgAD; n = 8 for TgAD + L-NAME rats. (C) Measurements of vascular reactivity were taken from vessels in the somatosensory cortex including penetrating arterioles, capillaries, and penetrating venules. Significance was calculated by linear mixed effects regression followed by Holm-Sidak post hoc. P-values for L-NAME contributing effects = 0.04; 0.005; 0.002, AD contributing effects = 0.07; 0.03; 0.02, and interaction effects = 0.32; 0.08; 0.15 (arterioles; venules; capillaries). * denotes p < 0.05 in post hoc; n = 6 rats per group.
2.2. White matter injury
After age, unresolved hypertension is the most prevalent contributing factor to white matter lesions from middle age onwards (Basile et al., 2006; Liao et al., 1996). Injury to white matter compromises neuronal connectivity and thus may significantly impair vascular function and reactivity. To investigate whether transient hypertension is sufficient to induce white matter injury, we measured the density of myelin in the cortex in NTg and TgAD rats either treated or untreated with L-NAME. Both transient hypertension (p = 0.01) and AD genotype (p = 0.002) were significant contributors to decreased myelin density in the somatosensory cortex (Fig. 2A, B). Similar to deficits in vascular reactivity, myelin loss was the most prominent in L-NAME treated TgAD when both factors are present (Fig. 2A, B). Comparing untreated NTg to L-NAME treated TgAD rats, the loss of myelin in the cortex exhibited a gradient with the cingulate cortex exhibiting a higher percentage loss (59%; 0.27 ± 0.06 down to 0.11 ± 0.04; p = 0.02) than either the motor (33%; 0.36 ± 0.03 down to 0.24 ± 0.06; p = 0.04) or somatosensory (28%; 0.54 ± 0.02 down to 0.39 ± 0.07; p = 0.002) cortices (Fig. 2A, B). To further probe myelin loss, we measured immunoreactivity of myelin basic protein (MBP) in the same cortical regions. Across the cortical regions, transient hypertension and AD genotype were significant contributors to changes in MBP immunoreactivity (Fig. 2A, C). Surprisingly, rather than a loss, MBP immunoreactivity increased as an effect of hypertension and AD; L-NAME treated TgAD rats showed significantly higher MBP immunoreactivity compared to untreated NTg rats in somatosensory (p = 0.01), motor (p = 0.03), and cingulate (p = 0.046) cortices (Fig. 2A, C). Increased MBP immunoreactivity may reflect structural alterations in the myelin sheath concurrent with loss of myelin that resulted in increased antigen exposure. Together, these results demonstrate that the combined effects of transient hypertension and AD genotype accelerated myelin deterioration.
Fig. 2.
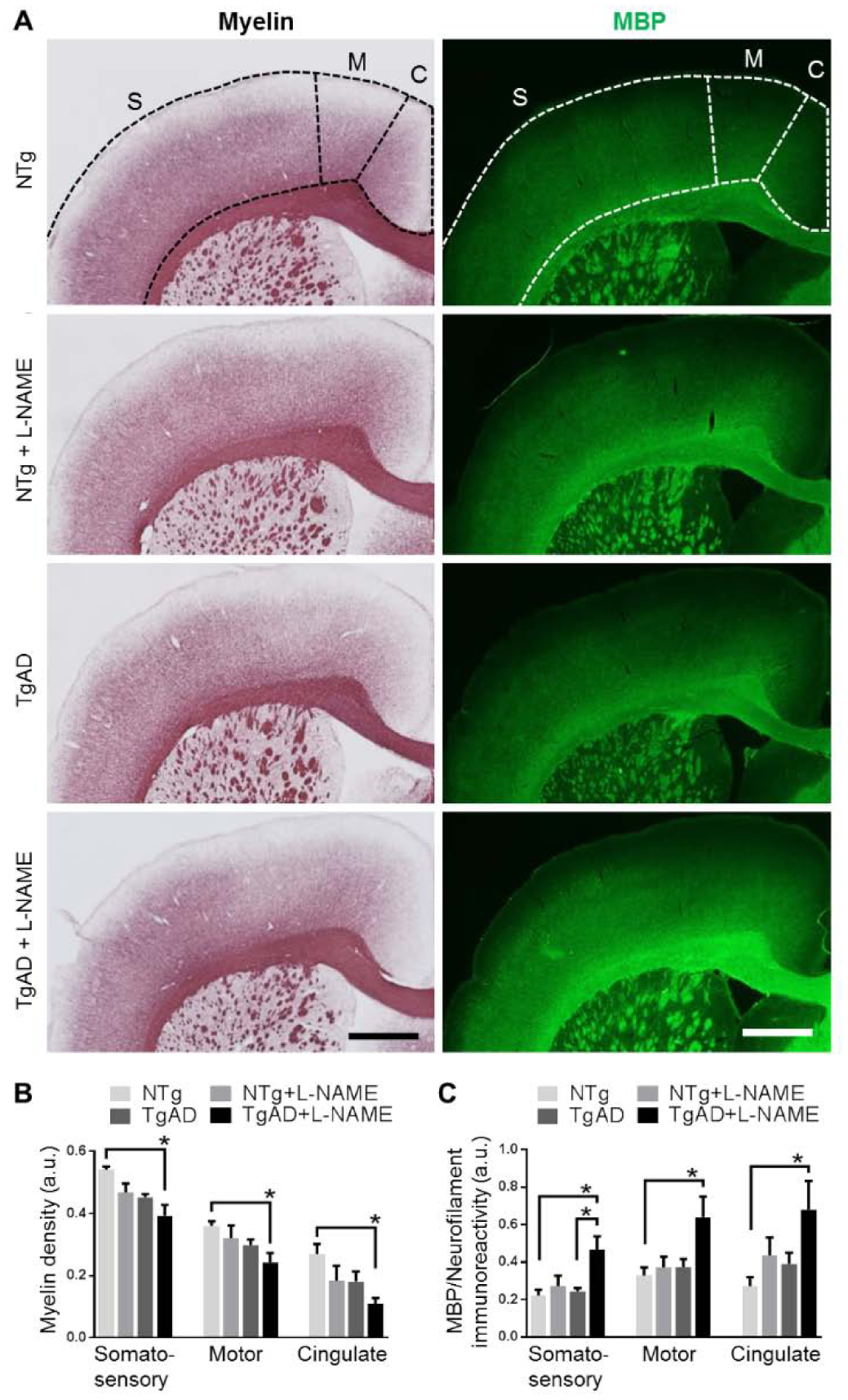
Transient hypertension and AD additively contribute to cortical myelin loss. Representative images (A) show a loss of myelin (left column) and an increase in MBP immunoreactivity (right column) in the somatosensory, motor, and cingulate cortices. Scale bar = 1 mm. Quantifications of myelin density (B) and MBP immunoreactivity (C) in the somatosensory, motor, and cingulate cortices show that myelin loss was the greatest in L-NAME treated TgAD rats (p = 0.002 compared to untreated NTg rats). Similarly, increased MBP immunoreactivity in all three cortical regions was also greatest in L-NAME treated TgAD rats (p = 0.01 compared to untreated NTg rats). MBP immunoreactivity was normalized to neurofilament immunoreactivity. Significance was calculated by Holm-Sidak post hoc following two-way ANOVA. P-values for L-NAME contributing effects (somatosensory, motor, cingulate), myelin = 0.01, 0.02, 0.03, MBP = 0.01, 0.04, 0.03; for AD contributing effects, myelin = 0.002, 0.11, 0.03, MBP = 0.04, 0.04, 0.08; for interaction effects, myelin = 0.78, 0.77, 0.82, MBP = 0.09, 0.13, 0.53. * denotes p < 0.05 in post hoc; n = 6 rats per group.
2.3. Vascular Amyloid
In patients, one of the most prominent pathological features comorbid with injury to cerebral white matter is cerebral amyloid angiopathy (CAA) (Charidimou et al., 2017; Smith, 2018). Previously, we demonstrated that in nine month-old TgAD rats, penetrating arterioles in the somatosensory cortex are laden with vascular Aβ deposits characteristic of CAA, correlating to deficits in vascular function (Joo et al., 2017). Here, in TgAD rats, transient hypertension significantly increased vascular Aβ/CAA coverage of penetrating arterioles in the somatosensory cortex (from 30.2 ± 4.3 % to 47.6 ± 7.6 %; p = 0.01) (Fig. 3A, B). A similar degree of increase in CAA coverage was observed in the cingulate cortex (from 33.2 ± 4.4 % to 45.7 ± 5.0 % 33.2 ± 4.4 %; p = 0.004) (Fig. 3B). CAA was not detected in NTg rats after transient hypertension (data not shown). Interestingly, transient hypertension did not affect coverage of parenchymal Aβ plaques in TgAD rats in either cortical region (Fig. 3C). These results are in partial agreement with large human cohort aging studies in which midlife hypertension was shown to result in post-mortem increase in both CAA and parenchymal amyloid plaques (Brickman et al., 2018; Shah et al., 2012). To determine whether increased CAA was due to increased production of Aβ, we measured the protein expression of amyloid precursor protein (APP), β-secretase (BACE), and CTFβ/CTFα, markers for APP production and processing, in the parenchyma-enriched fraction. We also measured the protein expression of neprilysin (NEP) and mature cathepsin B (CatB), two key enzymes involved in separate degradation pathways of Aβ, to see whether L-NAME affected Aβ degradation in either the vessel- or parenchyma-enriched fraction. We found that transient hypertension did not have an effect on either Aβ processing (Fig. 3D) or degradation (Fig. 3E), and thus the observed increase in CAA is likely a result of impaired vascular drainage.
Fig. 3.
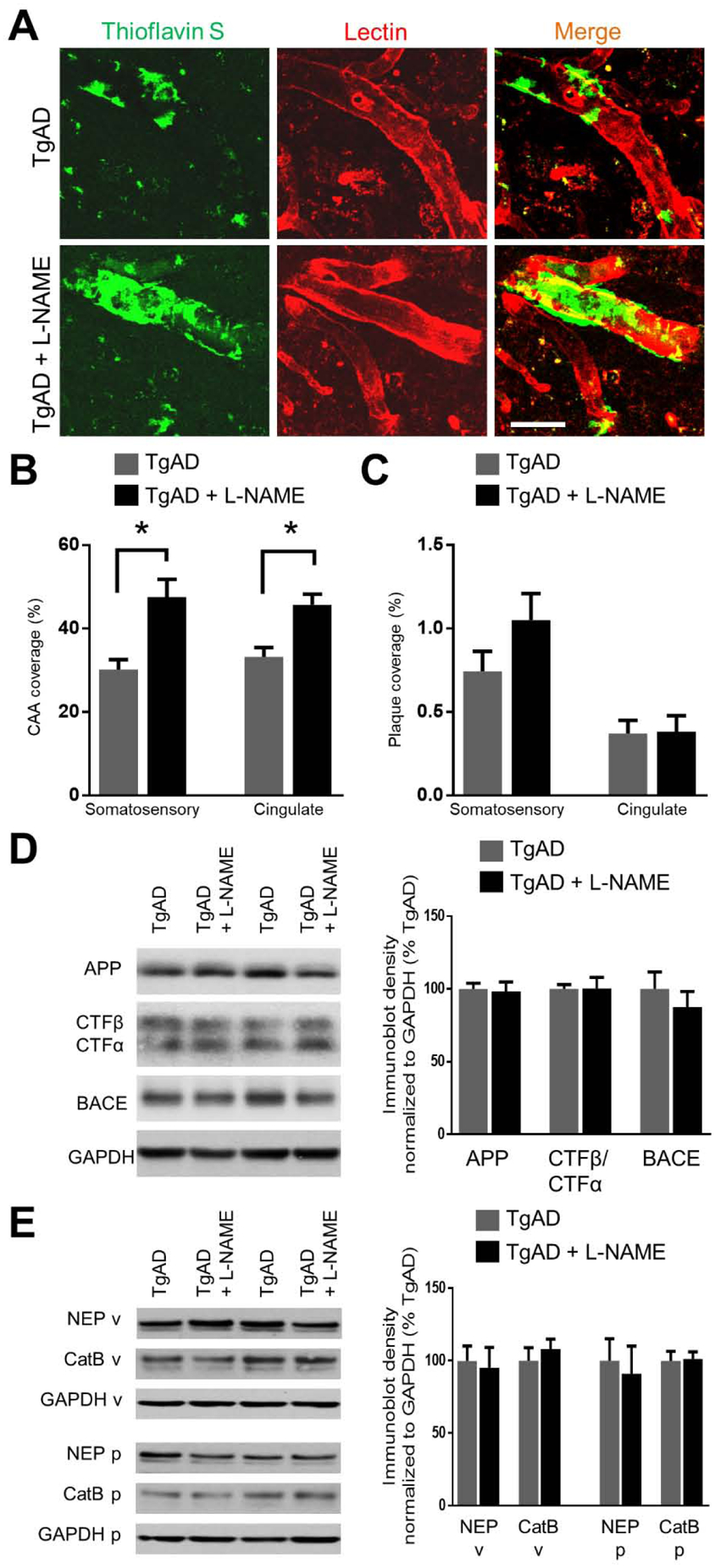
Transient hypertension promotes deposition of vascular Aβ in TgAD rats. Representative images (A) and quantification (B) of thioflavin S-positive vascular Aβ show that hypertension exacerbated the deposition of vascular Aβ in the penetrating arterioles of both somatosensory and cingulate cortices: Vascular Aβ increased from 30.2 ± 4.3 % to 47.6 ± 7.6 % area coverage (p = 0.01) in the somatosensory cortex and from and 33.2 ± 4.4 % to 45.7 ± 5.0 % area coverage (p = 0.004). Scale bar = 50 μm. Quantification (C) of 6F3D-positive parenchymal Aβ plaques in the somatosensory cortex. Area coverage of Aβ plaques was not significantly altered by transient hypertension in the somatosensory (p = 0.15) and cingulate (p = 0.93) cortices. Scale bar = 50 μm. Significance was calculated by t-test. * denotes p < 0.05; n = 6 rats per group. (D) Representative immunoblots and quantification of markers for Aβ processing and production in the parenchymal fraction: amyloid precursor protein (APP), CTFβ/CTFα, and β-secretase (BACE); p = 0.82, 0.97, 0.44 respectively. (E) Representative immunoblots and quantification of markers for Aβ degradation: neprilysin (NEP) and cathepsin B (CatB) in the vessel fraction (v) and the parenchymal fraction (p); p = 0.78, 0.72 for NEP and CatB in vessel fraction; p = 0.49, 0.88 in the parenchymal fraction; n = 11 rats per group.
2.4. Vascular structure
We probed for structural changes in the cerebrovasculature that may have contributed to the deficits in vascular reactivity and myelin loss after transient hypertension. Our previous work has shown that cortical vessels undergo extensive structural changes in response to AD pathogenesis (Joo et al., 2017). Similarly, hypertension is known to induce vascular remodeling including reduced wall elasticity and lowered capillary density (Pires et al., 2013). Brain capillaries comprise the majority of the area covered by the brain vascular bed; thus, we analyzed collagen IV (Col IV)-positive capillaries in the somatosensory and cingulate cortices and found that capillary density is unchanged in all of the treatment groups (data not shown). In contrast, hypertension and AD are significant contributing factors to decreased capillary diameter in the somatosensory (p = 0.03, 0.01 respectively) and cingulate (p = 0.04, 0.001 respectively) cortices (Fig. 4A). In addition, previous studies have shown that AD is associated with an increase in the number of string vessels in the capillary bed (Brown, 2010). String vessels are thin strands resembling remnants of capillaries that contain a shell of basement membrane but no endothelial cells (Brown, 2010). They carry no blood flow and are a sign of capillary regression and remodeling (Brown, 2010). We identified string vessels as Col IV-positive lectin-negative ‘threads’ in the capillary nexus (Fig. 4B). In both somatosensory and cingulate cortices, transient hypertension significantly increased the density of string vessels (p = 0.01 for both regions) while AD was not a significant contributing factor (Fig. 4B, C).
Fig. 4.
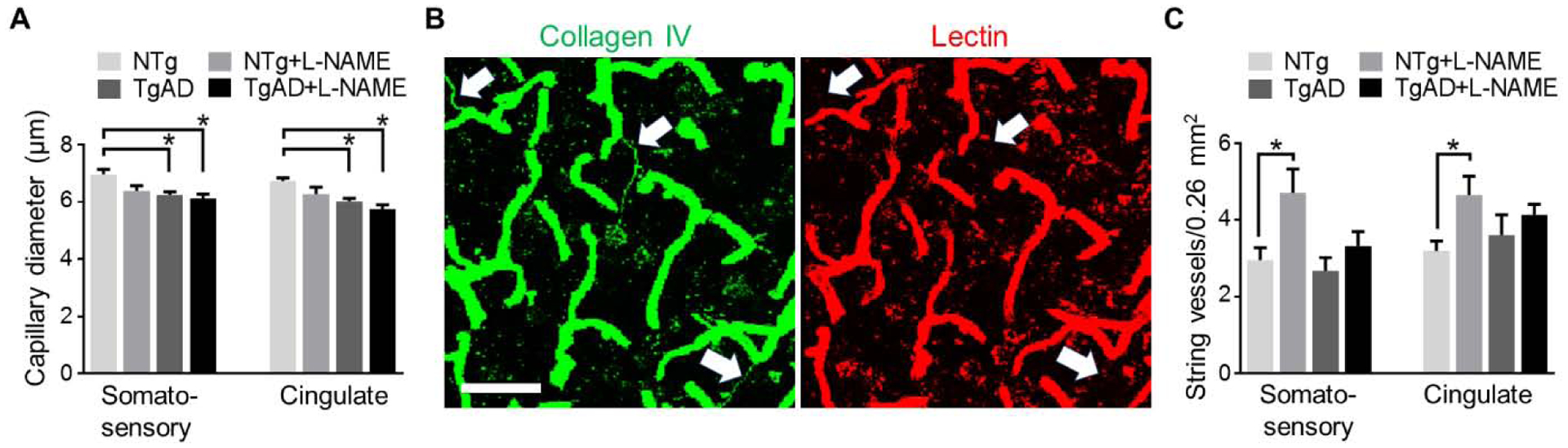
Structural remodeling of capillaries is modulated by both transient hypertension and AD. (A) L-NAME treated TgAD rats have the smallest capillary diameter of all four experimental groups inboth somatosensory (p = 0.01 vs. NTg) and cingulate (p = 0.002 vs. NTg) cortices. Representative images (B) and quantification (C) of string vessels (arrows) in the capillary bed show that in NTg rats, number of string vessels in both somatosensory (p = 0.049) and cingulate (p = 0.03) cortices increased in response to transient hypertension. Scale bar = 50 μm. Significance was calculated by Holm-Sidak post hoc following two-way ANOVA. P-values for L-NAME contributing effects (somatosensory, cingulate), capillary diameter = 0.03, 0.04, string vessels = 0.01, 0.01; for AD contributing effects, capillary diameter = 0.01, 0.001, string vessels = 0.07, 0.89; for interaction effects, capillary diameter = 0.17, 0.53, string vessels = 0.22, 0.18. * denotes p < 0.05 in post hoc; n = 6 rats per group.
We next examined structural changes in cortical penetrating arterioles and venules. Cortical penetrating vessels are a primary target of Aβ deposition and are subject to AD-related structural changes in caliber, tortuosity, and vessel wall integrity (Dorr et al., 2012a; Joo et al., 2017). Penetrating arterioles in particular undergo drastic structural changes in response to hypertensive insults: as an adaptive measure to withstand stress during hypertension, arteries and arterioles increase vessel wall thickness which in turn promotes vessel hardening and decreases overall vessel elasticity and function (Pires et al., 2013). To estimate the extent to which transient hypertension and AD pathology remodeled arteriolar walls, we measured the ratio of Col IV to elastin expression. Col IV is present in all layers of the basement membrane, whereas elastin is expressed only in the tunica media layer of arterial walls to regulate vessel elasticity and contractility (Wagenseil and Mecham, 2009) thus positively correlated with arterial wall hardening and stiffness (Wagenseil and Mecham, 2012). We found that in the somatosensory cortex, transient hypertension (p = 0.001) and AD (p = 0.009) significantly increased Col IV:elastin ratios (Fig. 5A, B). Similar to myelin loss, presence of both hypertension and AD resulted in the highest degree of arteriolar stiffening (p = 0.0004, NTg vs. TgAD+L-NAME). Echoing the region-specific trend observed with myelin loss, arteriolar stiffening in the cingulate cortex was larger in magnitude (0.83 ± 0.18 to 1.61 ± 0.33, NTg vs. TgAD+L-NAME) compared to that in the somatosensory cortex (0.88 ± 0.06 to 1.25 ± 0.14, NTg vs. TgAD+L-NAME) (Fig. 5B).
Fig. 5.
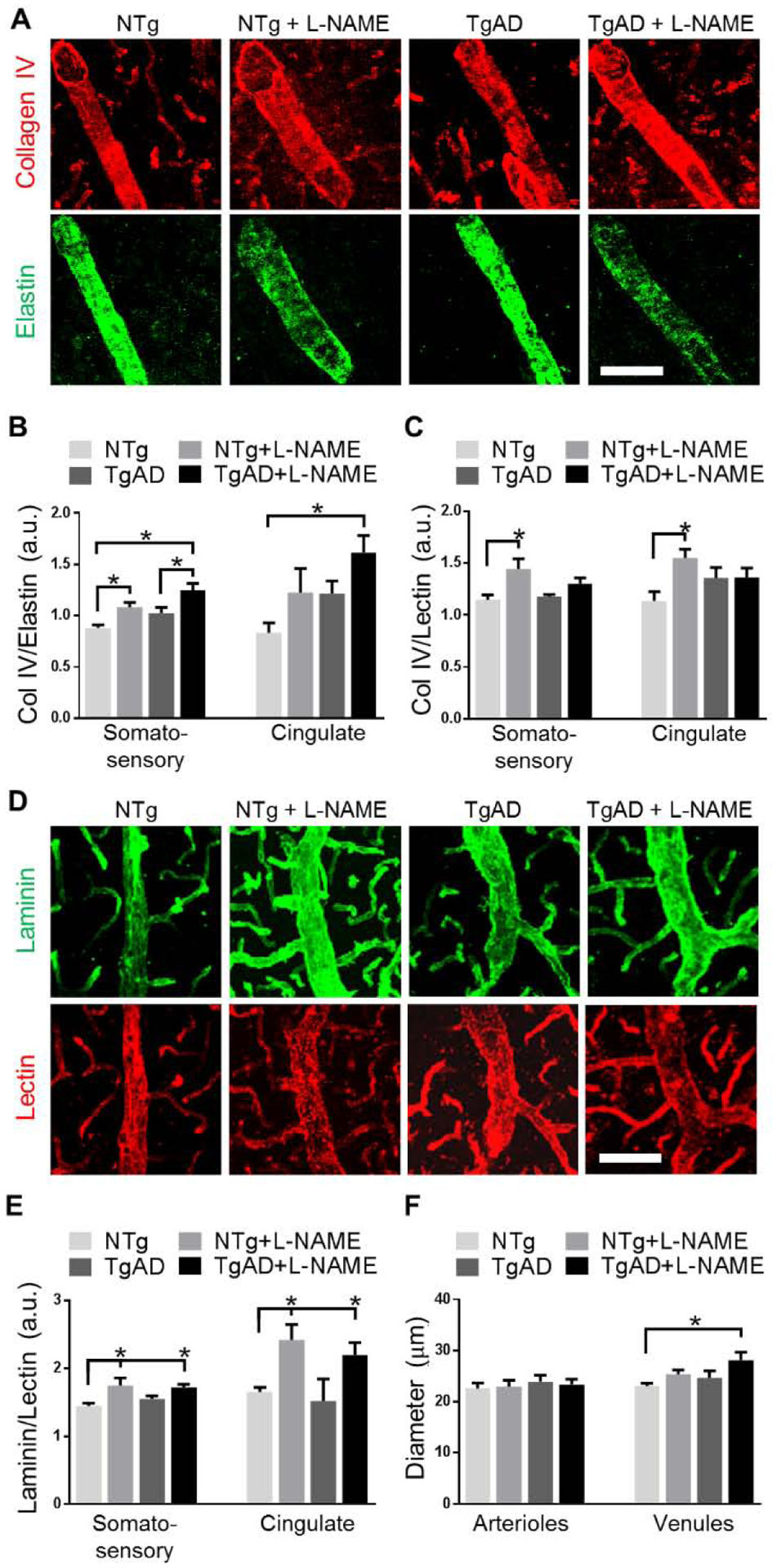
Differential contribution of transient hypertension and AD towards structural remodeling of penetrating arterioles and venules. Representative images (A) and quantification (B) of Col IV:elastin ratios in penetrating arterioles of the somatosensory and cingulate cortices: Transient hypertension and AD were both significant contributing factors to increased Col IV:elastin such that in L-NAME treated TgAD rats, Col IV:elastin ratio was the highest (p = 0.0004 vs. untreated NTg in somatosensory; p = 0.02 in cingulate). Quantification (C) of Col IV:lectin, representative images (D) and quantification (E) of laminin:lectin ratios in penetrating venules of the somatosensory and cingulate cortices: Transient hypertension but not AD contributed significantly to increased venular Col IV:lectin and laminin:lectin ratios. Quantification (F) of vessel diameter of penetrating arterioles and venules in the somatosensory cortex: Diameter of arterioles remained constant while diameter of venules increased (p = 0.03, TgAD vs. TgAD+L-NAME) when hypertension and AD are both present. Scale bar = 50 μm. Significance was calculated by Holm-Sidak post hoc following two-way ANOVA. P-values for L-NAME contributing effects (somatosensory, cingulate), Col IV:elastin = 0.001, 0.003, Col IV:lectin = 0.004, 0.03, laminin:lectin = 0.002, 0.0003; for AD contributing effects, Col IV:elastin = 0.01, 0.03, Col IV:lectin = 0.40, 0.85, laminin:lectin = 0.60, 0.40; for interaction effects, Col IV:elastin = 0.84, 0.98, Col IV:lectin = 0.19, 0.04, laminin:lectin = 0.35, 0.73. For diameter of penetrating vessels (arterioles, venules), L-NAME contributing effects = 0.93, 0.02, AD contributing effects = 0.47, 0.07, interaction effects = 0.70, 0.63. * denotes p < 0.05 in post hoc; n = 6 rats per group.
Venules, unlike arterioles, do not express elastin. To assess whether venules had undergone remodeling of the vascular wall, we analyzed the change in venular Col IV expression relative to the endothelium (labeled by lectin). In the somatosensory cortex, transient hypertension (p = 0.004) but not AD genotype (p = 0.40) contributed significantly to increased venular expression of Col IV suggesting venular wall remodeling (Fig. 5C). To buttress these analyses, we measured laminin expression, which like Col IV, is a structural protein expressed in all layers of the basement membrane (Chelladurai et al., 2012). Similarly, transient hypertension (p = 0.002) but not genotype (p = 0.60) contributed significantly to increased laminin expression in the somatosensory cortex thus confirming venular wall remodeling (Fig. 5D, E). Remodeling of venules was more pronounced in the cingulate cortex (1.65 ± 0.14 to 2.20 ± 0.35, NTg vs. TgAD+L-NAME) compared to that in the somatosensory cortex (1.45 ± 0.07 to 1.72 ± 0.09, NTg vs. TgAD+L-NAME) (Fig. 5E).
We then examined whether the diameter of venules increased after remodeling: Transient hypertension (p = 0.02) was a significant contributor while AD (p = 0.07) showed a trend towards increases in venular diameter (Fig. 5F). The combined exposure to hypertension and AD resulted in the largest magnitude of change in venular diameter (p = 0.03, NTg vs. TgAD+L-NAME; Fig. 5F). In contrast, the increase arteriolar wall thickening did not result in a corresponding increase in the diameter of the arterioles (Fig 5F). Since the diameter of arterioles was unchanged with decreased capillary and increased venular diameters suggest that the impact of remodeling of vessel caliber involves for the most part the draining vasculature.
2.5. Molecular substrates of vascular remodeling
To gain a fuller understanding of the mechanisms underlying hypertensive remodeling in both NTg and TgAD rats, we isolated cortical blood vessels from all treatment groups and examined the expression of signaling proteins implicated in clinical and experimental hypertension. Osteopontin (OPN) is a secreted protein synthesized by smooth muscle and endothelial cells; modulation of OPN expression has been associated with hypertension as well as with post-insult vascular remodeling (Caesar et al., 2017; deBlois et al., 1996; Lorenzen et al., 2011). Here, we found that transient hypertension (p = 0.01) but not AD (p = 0.18) contributed to decreased OPN levels (Fig. 6A, B). OPN expression was the lowest in hypertensive TgAD rats (p = 0.045, NTg vs. TgAD+L-NAME; Fig. 6A, B), a profile similar to that observed with vascular function, vessel diameter, and myelin loss. Important signaling targets of OPN are the Rho-associated protein kinases (ROCKs), ROCK1 and ROCK2. Previous studies have suggested that RhoA kinase pathway may be involved in cerebrovascular compromise (Chrissobolis and Sobey, 2001; Loirand, 2015). ROCKs have been implicated as both upstream and downstream modulators of hypertension (Dorr et al., 2012a). We found that ROCK1 and ROCK2 showed different expression profiles as a function of transient hypertension and AD: A significant interactive effect between hypertension and AD (p = 0.02) regulated ROCK1 expression such that transient hypertension increased ROCK1 expression in TgAD rats (p = 0.02) but not in NTg rats (p = 0.87; Fig. 6A, B). In contrast, only transient hypertension had a significant effect on ROCK2 expression (p = 0002) with the magnitude of change being of similar effect size in both NTg and TgAD rats (Fig. 6A, B). These results demonstrate that ROCK1 and ROCK2 modulate different aspects of vascular remodeling following transient hypertension. The data collectively suggest that transient hypertension activates distinct and lasting changes to molecular pathways in the context of both normal aging and AD.
Fig. 6.
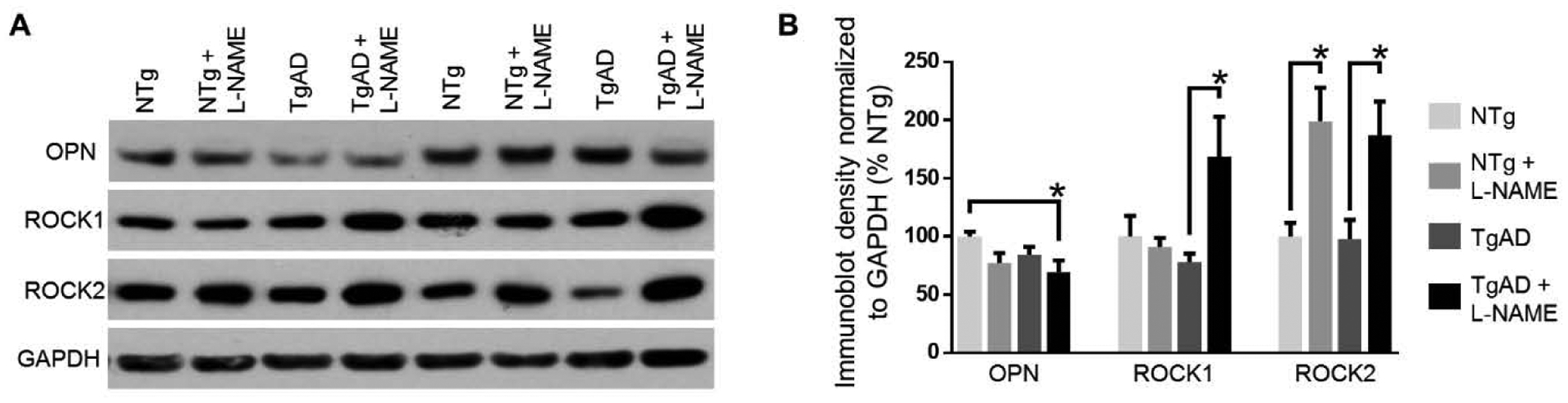
Signaling substrates involved in vascular remodeling. Representative immunoblots (A) and quantification (B) osteopontin (OPN), ROCK1, and ROCK2 protein expression in the vessel-enriched fraction. Transient hypertension alone increased expression of all three of OPN (p = 0.01), ROCK1 (p = 0.047), and ROCK2 (p = 0.0002) whereas AD as an individual factor did not have an effect on any of OPN (p = 0.18), ROCK1 (p = 0.17) or ROCK2 (p = 0.76). Hypertension and AD had an interaction effect with respect to ROCK1 expression (p = 0.02) but not OPN (p = 0.52) or ROCK2 (p = 0.83). The interaction effect resulted in differential increase in ROCK1 expression in TgAD rats (p = 0.02) but not in NTg rats (p = 0.87). Significance was calculated by Holm-Sidak post hoc following two-way ANOVA. * denotes p < 0.05 in post hoc; n =11 rats per group.
3. Discussion
Multiple longitudinal cohort studies provide evidence that the pathological process of AD may be a cumulative result of neurodegenerative and vascular pathologies (Akoudad et al., 2016; Kapasi et al., 2017; Rabinovici et al., 2017; White et al., 2016). Several studies have investigated the coexistence of vascular disease with AD pathology; some show an additive effect while others show a synergistic contribution (Esiri et al., 1999; Kapasi et al., 2017; Snowdon et al., 1997). The proportion of persons with a pathologic diagnosis of AD who exhibited both vascular and other degenerative pathologies increased from 24% in mild cognitive impairment to 47% in probable AD (Kapasi et al., 2017). In persons with probable AD and mixed pathologies, vascular disease is present in approximately 90% of all cases (Kapasi et al., 2017). Thus, the mechanism by which vascular pathologies contribute to AD remains an area of importance.
Vascular risk factors are one of the most tractable targets for treatment of late life dementia and AD, and effective prevention and intervention of vascular risk may potentially diminish the magnitude of the dementia epidemic. Although midlife hypertension is an important risk factor for AD, the mechanistic links that associate transient hypertensive insults to AD pathogenesis are not well defined. Our research demonstrated lasting loss of function to the brain vascular network even in the absence of ongoing hypertensive events. Moreover, we have identified pathological features in the cerebrovasculature that could underlie the observed loss of vascular function: white matter injury, accumulation of CAA, structural remodeling of brain blood vessels, and activation of vascular signaling substrates are key pathological changes we found as effects of transient hypertension and an AD-susceptible genotype.
Specifically, injury to the white matter has several key implications: As a consequence of hypertension-induced vascular remodeling, vascular cells including endothelial cells and pericytes may become dysfunctional; unhealthy endothelial cells and pericytes have been shown to directly impair oligodendrocyte function (Montagne et al., 2018; Rajani et al., 2018) resulting in myelin loss. Since white matter integrity positively correlates with integrity of neurovascular coupling (Sorond et al., 2013), degeneration of the white matter may feed a vicious cycle where an inefficient neurovascular coupling further compromises cerebral blood flow leading to exacerbation of AD pathologies, such as CAA. Therefore, the observed white matter injury may be both a cause and a consequence of hypertension-induced vascular remodeling. Interestingly, we show increased MBP immunoreactivity despite apparent loss of myelin density in hypertensive TgAD rats. Increased MBP immunoreactivity is a hallmark in the acute phases of brain injuries (Ajao et al., 2012; Michalski et al., 2018). In a chronic vascular disease such as diabetes, MBP immunoreactivity increases slowly but decreases over time (Nam et al., 2018). In some instances, chronic demyelination causes aggregation but not loss of MBP, significantly altering its immunoreactivity (Frid et al., 2015). The concurrent demyelination and increased MBP immunoreactivity observed in our study likely represent the end of the acute phase of myelin loss. Notably, the largest magnitude of myelin loss was observed in the cingulate cortex. The same trend was echoed by parameters of vascular remodeling such that the largest changes in arteriolar and venular basement membrane structure was also in the cingulate cortex. Vulnerability of the cingulate cortex to vascular insults has important clinical implications: its hypoperfusion was previously shown to be a strong predictor of conversion to AD from mild cognitive impairment (Huang et al., 2002).
It is of note that not all readout measures demonstrated a synergistic effect between transient hypertension and AD phenotype. This is not unexpected as we demonstrate that after 30 days of recovery from transient hypertension, our data provides evidence for vascular remodeling that could contribute to the diminished effect. Notably, in our colony, F344TgAD rats typically require sacrifice between 18 to 22 months of age due to spontaneous development of mammary tumors (unpublished observations); therefore at 9 months of age the F344TgAD are considered midlife. Nonetheless, with respect to loss of vascular reactivity/function, damage due to AD genotype at this age has already progressed to a level that subtle injuries due to transient hypertension are no longer detectable i.e. swamped out by the AD phenotype alone.
Clinical studies of association between hypertension and dementia define hypertension as systolic blood pressure of either > 160 mmHg or > 140 mmHg whereas strains of spontaneously hypertensive rats range from 145 mmHg to > 190 mmHg (Kihara et al., 1993; Lennon et al., 2019). Our transient hypertension model averaged 175 mmHg in NTg and 185 mmHg in TgAD rats, which should be considered moderate hypertension. Although L-NAME is frequently used to model hypertension, like any mono-factorial preclinical model of disease, L-NAME model of hypertension reduces multifactorial causes of a complex pathology to a single factor. In particular, L-NAME is a selective NO synthase inhibitor, and the decreased level of NO during L-NAME administration could be seen as confounding. However, NO synthase modulation has been reported in hypertensive patients and previous reports show that normal NO synthase activity resumes within 24–48 hours of L-NAME administration cessation (Ayers et al., 1997). The most common alternatives to L-NAME for induced hypertension include activation of the renin-angiotensin-aldosterone system (like the Ang II infusion model), and surgical models such as the renovascular and renoprival models. While these can induce increase in blood pressure, they also introduce other confounding factors, namely cardiac and renal injuries, which are typically absent in transient middle-age hypertension (Lerman et al., 2019).
Several findings in this model differ from those observed in models of ongoing hypertension. In rodent AD models exposed to persistent hypertension, overall brain capillary bed density was significantly decreased compared to normotensive AD animals (Cifuentes et al., 2017, 2015); in contrast, transient hypertension in the current study did not affect vascular density in the capillary beds. F344TgAD rats exposed to transient hypertension also had significantly higher vascular Aβ in the penetrating arterioles characteristic of CAA without significant increase in parenchymal Aβ. These results are in contrast to those of ongoing hypertension that have shown elevated parenchymal coverage of Aβ plaques in various animal models of AD (Cifuentes et al., 2017, 2015; Díaz-Ruiz et al., 2009; Shih et al., 2018). Notably, epidemiological data from human subjects report that although midlife hypertension predisposes cerebrovascular and parenchymal damage later in life, similar to our study, association to Aβ load was not present (Lane et al., 2019). Overall, although persistent hypertension may negatively affect more facets of cerebrovascular integrity, our results demonstrate that in an AD susceptible environment, transient hypertension induces sufficient vascular remodeling to result in deterioration of neuronal health.
We report genotype-specific upregulation of protein expression of ROCKs following transient hypertension. ROCKs signaling in the cerebrovasculature functions as both an upstream regulator and downstream effector of hypertension: ROCKs modulate dephosphorylation of myosin light chain subunits involved in vessel contractility and subsequently act as regulators of blood pressure; as an effector of hypertension, ROCKs mediate post-hypertension vascular remodeling (Bond et al., 2015; Huveneers et al., 2015; Lai and McLaurin, 2018; Pires et al., 2013). There is a wide range of cell-dependent functions attributed to ROCK1 and ROCK2 signaling including contraction, motility, polarity, proliferation, apoptosis and gene expression (Amano et al., 2010, 2003; Lai and McLaurin, 2018; Shimokawa et al., 2016): the downstream pathways that lead to vascular compromise will be investigated in future experiments.
In conclusion, we set out to characterize the effects of transient hypertension in the context of both normal aging and an AD-susceptible environment, characterizing the combined effects of hypertension and AD genotype on key aspects of vascular remodeling and cerebral damage. We demonstrate that even after normalization of systolic blood pressure from the initial hypertensive insult, deficits in the cerebrovasculature and brain parenchyma persist. Importantly, there is mounting evidence from human studies that modulating blood pressure earlier in life results in significantly improved later outcomes with regard to brain damage and dementia probability (SPRINT MIND Investigators for the SPRINT Research Group et al., 2019a, 2019b). Our results here outline several cellular processes directly targeted by hypertension that connect the hypertensive events to increased probability of dementia and henceforth may facilitate development of more precise therapeutic targets.
4. Methods and materials
4.1. Animals
F344TgAD rats overexpress human amyloid precursor protein containing the familial Swedish mutation (APPsw) and presenilin delta E9 (PS1ΔE9) under the mouse prion protein promoter on the Fischer-344 background (Cohen et al., 2013). We kept age-matched F344TgAD and their NTg littermates on a 12 hr:12 hr light/dark cycle with water and food ad libitum. Each cage contains two cage-mates and is monitored daily for health and normal grooming behaviors. Ethical approval of all experimental procedures was granted by The Animal Care Committee of the Sunnybrook Health Sciences Center, which adheres to the Policies and Guidelines of the Canadian Council on Animal Care, the Animals for Research Act of the Provincial Statute of Ontario, and the Federal Health of Animals Act.
4.2. Induction of transient hypertension
To induce hypertension, rats aged seven months were dosed at 10 mg/kg/day (males weighing 0.38 to 0.43 kg) and 7.5 mg/kg/day (females weighing 0.23 to 0.28 kg) L-NAME in drinking water for one month, respectively. Doses chosen were optimized such that hypertension is induced without visible effects on normal cage and grooming behavior. At their respective doses, we did not see measurable differences between sexes in any of the examined readouts. Water consumption was monitored to estimate drug exposure. We confirmed hypertension transience by measuring systolic blood pressure at seven, eight, and nine months of age using the CODA-HT2 tail-cuff system under isoflurane anesthesia (Kent Scientific). Two-way ANOVA followed by Holm-Sidak post hoc was used to measure statistical significance of the blood pressure data. Of the 82 rats used in the study (37 males and 45 females), random allocation was used to assign treatment across time and litters: A total of 24 sex-balanced rats were processed for immunohistochemical analyses, n = 6 per group of NTg±L-NAME and TgAD±L-NAME. Based on the relative sensitivities of the pathological assays in NTg and F344TgAD rats in our prior studies in this model (Joo et al., 2017; Morrone et al., 2020), the sample size n = 6 per group ensures the power of at least 80% across all contrasts at the significance level of 5%. A total of 47 rats were used for two-photon fluorescence microscopy, of which 24 yielded analyzable images. Of the 47 rats, 33 survived imaging and these brains were collected for immunoblotting. To correct for attrition, we included an additional 11 rats for immunoblotting analyses to balance sex and treatment group size.
4.3. Two-photon fluorescence microscopy
4.3.1. Surgery and data acquisition
Surgical procedures were adapted from previous studies (Dorr et al., 2012b; Joo et al., 2017; Lai et al., 2015). In brief, anaesthetized rats (2–3% isoflurane with 30–35% oxygen) rats were tracheostomized for delivery of hypercapnia. Cranial windows were centered at AP −3.0 mm and ML ± 2.5 mm relative to bregma and were covered by a coverslip and double-distilled water well to enable imaging by a water immersion objective (25X, NA 1.05; Olympus). Cerebral blood flow was measured by injecting 70 kDA Texas Red fluorescent dextran boluses into the tail vein (in PBS; 7 mg/kg per bolus; Thermo-Fisher).
We used an FV1000MPE multiphoton laser scanning microscope (Olympus) equipped with a Ti:Sapphire tunable laser (Mai Tai HP; 690–1040 nm; Newport). Texas Red was excited at 910 nm and the emission collected at 575–630 nm. Each acquisition consists of a 512 μm × 512 μm area at a cortical depth of 150–200 μm with nominal in-plane resolution of 0.5 μm × 0.5 μm. We collected free-hand line scans (10 μm/pixel, 20 ms/line, ~600 lines) over multiple cortical penetrating vessels and any capillaries found along the scan trajectory. Time of the bolus dye injection was triggered by the start of the line scans allowing for calculation of vascular transit times. The line scans lasted 11–13 seconds in order to track bolus passage through the selected vessels during normocapnia (0% FiCO2) and brief periods (~ 60 seconds) of hypercapnia (10% FiCO2). For each rat, up to six bolus injections were carried out and monitored at three different locations in the cortex. For structural analysis of the cortical microvasculature, we acquired a series of 133–300 slices parallel to the cortical surface every 1.5 μm to a depth of 200–450 μm, at 4 μs/pixel, 512 μm × 512 μm with nominal in-plane resolution of 1 μm × 1 μm. Rats that did not survive either the surgery or imaging were excluded from data analysis (mortality rate 5%).
4.3.2. Data analysis of bolus tracking and vascular structure
As described previously (Dorr et al., 2012b; Joo et al., 2017; Lai et al., 2015), vascular reactivity was calculated using time-to-peak (TTP), which represents the time interval between the line scan-triggered bolus injection and the time of peak fluorescence in the vessel. Vascular reactivity was expressed as the change in TTP during CO2 challenge normalized to TTP during normal breathing. Differentiation of penetrating arterioles, venules and capillaries was carried out by identifying the nearby pial vessels and tracing the connections between cortical penetrating vessels and their parent pial vessels. Vessel types were designated based on several morphological criteria: pial arteries have smaller diameter, and fewer branches while pial veins have larger diameter and more branches. Penetrating arterioles have fewer branches and maintain a constant diameter through the depth of the cortex whereas penetrating venules branch out more and exhibit a variable diameter. Vessels less than 10 μm in apparent diameter were deemed capillaries. Vessel designations based on morphology were additionally confirmed by examining the arrival times of the injected bolus.
Statistical significance was measured using linear mixed effect model (lmer function in lme4 package, R) which determines whether L-NAME treatment, genotype, and interaction between the two factors is a significant contributing factor. Significance between two specific treatment groups was determined using Holm-Sidak post hoc test.
4.4. Immunofluorescence and immunohistochemistry
Rat brains were sequentially perfused with PBS and 4% paraformaldehyde (in PBS). Extracted brains were post-fixed overnight in 4% paraformaldehyde (in PBS) then washed and embedded in 30% sucrose (in PBS). Sucrose-embedded brains were sectioned coronally at 40 μm of thickness between AP +2.5 to −7.5 mm.
For fluorescent labeling that includes collagen IV (Col IV) or laminin, the PBS-washed sections were mounted on HistoBond slides (16004–406, VWR) prior to labeling. To unmask antigens of Col IV and laminin, the mounted slides were immersed in 0.2% pepsin (P7000, Sigma, in 25 mM Tris pH 2.0) at 37°C for one hour. After three PBS washes, the slides were blocked for one hour (in PBS with 0.5% Triton and 5% donkey serum) and probed with primary antibodies overnight (in PBS with 0.5% Triton and 5% donkey serum). Slides were then washed and probed with both fluorescence-conjugated secondary antibodies and fluorescence-tagged tomato lectin (in PBS with 0.5% Triton and 0.5% bovine serum albumin) for two hours before coverslipping.
For labeling of vascular Aβ, floating sections were stained with 1% thioflavin S (in water) for 7 minutes followed by two 5 minute washes in 70% ethanol (in water), then probed with fluorescence-tagged tomato lectin (in PBS with 0.5% Triton and 0.5% bovine serum albumin; 1:200, DL-1178, Vector) for two hours before mounting.
Fluorescence labeled image were acquired using either the Leica TCS SP5 or the Zeiss Observer.Z1. For quantification of vascular Aβ, vessel morphology, and immunoreactivity of Col IV, elastin, laminin, and lectin, 20 images were acquired from three coronal sections between AP +1.50 mm and −0.50 mm. Each image contains two to five penetrating vessels. Penetrating arterioles and venules were defined as vessels > 10 μm in diameter. Capillaries were defined as vessels with a branching order > 2 and a diameter < 10 μm. Arterioles were distinguished from venules morphologically as they have a striated staining pattern with lectin. Reliability of using lectin morphology to identify arterioles was confirmed by co-labeling with the arteriole-specific marker α-smooth muscle actin (data not shown). Vessel diameter was measured by dividing the Col IV coverage area of a vessel by its total length. Vessel density was expressed as Col IV coverage area divided by total brain area. String vessels were defined as capillaries with a degenerating thread-like morphology that were positive for Col IV (basement membrane) but negative for lectin (endothelium). Immunoreactivity of Col IV, elastin, laminin, and lectin in penetrating vessels were measured using ImageJ. For quantification of myelin basic protein (MBP) immunoreactivity, whole brain images of three coronal sections between AP +1.50 mm and −0.50 mm were used. Immunoreactivity of MBP is normalized to that of neurofilament (measured using ImageJ) as a readout for myelin alterations. For all fluorescent labeling, image acquisition and quantification of each cortical region (somatosensory, motor, cingulate) were performed separately. Statistical significance was measured using two-way ANOVA followed by Holm-Sidak post hoc.
For labeling of parenchymal Aβ, we used the 6F3D anti-Aβ antibody as described previously (Joo et al., 2017). For visualization of myelin, we used Black Gold II Myelin Staining kit (AG105, Millipore) as per manufacturer protocol while omitting the cresyl violet step. For quantification of parenchymal Aβ, whole brain images of five coronal sections between AP +0 mm and −3.5 mm were used. For quantification of myelin density, whole brain images of three coronal sections between AP +1.50 mm and −0.50 mm were used. Images were acquired with the Zeiss Observer.Z1 and quantified using ImageJ. For both parenchymal Aβ and myelin staining, image acquisition and quantification of each cortical region (somatosensory, motor, cingulate) were performed separately. Statistical significance was measured using t-test. Refer to Supplementary Table S1 for list of antibodies used.
4.5. Isolation of vessel-enriched fractions and immunoblots
We adapted previously described methods for isolating vessel-enriched fractions from whole brains (Hawkes and McLaurin, 2009; Joo et al., 2017). Briefly, dry ice-frozen brains extracted from phosphate-buffered saline (PBS)-perfused rats were first dounce-homogenized in 0.1 M ammonium carbonate, 5 mM ethylenediaminetetraacetic acid, 0.01% sodium azide, and 1% protease inhibitor cocktail (539134, Millipore), then centrifuged for one hour (20,000 g, 4°C). Pellets were re-suspended in 0.1 M ammonium carbonate, 3% sodium dodecyl sulphate (SDS), and 2% protease inhibitor cocktail, stirred on ice for two hours, and filtered through 40 μm mesh filters. The parenchymal fraction filtered through the mesh while the vessel-enriched fraction stayed on the mesh. The vessel fraction was resuspended in 10 mM Tris pH 8.0, 1 mM ethylenediaminetetraacetic acid, 1 mM ethylene glycol tetraacetic acid, 0.8% Triton, 0.2% SDS, 140 mM NaCl, and 1% protease inhibitor cocktail. Both fractions were then sonicated. The resulting protein supernatants were subjected to bicinchoninic acid protein assays (23225, Thermo-Fisher), SDS-PAGE electrophoresis, and immunoblotting which have been previously described in detail (Joo et al., 2017). Densitometry analysis of immunoblots was carried out using ImageJ. Statistical significance was measured using two-way ANOVA followed by Holm-Sidak post hoc for experiments involving four treatment groups and t-test for those involving two treatment groups. Refer to Additional file 1: Table S1 for list of antibodies used.
Supplementary Material
Highlights.
Transient hypertension rat model is established to resemble clinical progression.
Its effects are compared between normal aging and an AD-susceptible brain.
Hypertension and AD each compromised cerebrovasculature and white matter.
Combined effects of the two factors yielded the largest degree of compromise.
Hypertension enhanced vascular Aβ deposition but not parenchymal plaque pathology.
Vascular remodeling proteins osteopontin, ROCK1, and ROCK2 are up-/down-regulated.
Acknowledgements
We thank Dr. Terrence Town and Dr. Tara M. Weitz for providing F344TgAD rat breeding pairs and Tina L. Beckett and Shirley Yang for technical assistance.
Funding
Funding: This work was supported in part by the Canadian Consortium on Neurodegeneration in Aging, which was supported by a grant from the Canadian Institute of Health Research with funding from several partners [grant number CAN-137794], Canadian Institutes of Health Research Project Grant [grant number PJY153101], Canada Research Chairs Program and National Institutes of Health R01 [grant number AG057665-02].
Abbreviations
- AD
Alzheimer’s disease
- Aβ
amyloid-beta peptide
- APP
amyloid precursor protein
- BACE
β-secretase
- CAA
cerebral amyloid angiopathy
- Col IV
collagen IV
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- L-NAME
L-NG-Nitroarginine methyl ester
- MBP
myelin basic protein
- NEP
neprilysin
- NO
nitric oxide
- NTg
non-transgenic littermate rats
- OPN
osteopontin
- PBS
phosphate-buffered saline
- ROCKs
rho-associated protein kinases
- SDS
sodium dodecyl sulfate
- TBS-T
tris-buffered saline with 0.1% Tween
- TgAD
F344TgAD rat
- TTP
time to peak
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare that they have no competing interests.
References
- Ajao DO, Pop V, Kamper JE, Adami A, Rudobeck E, Huang L, Vlkolinsky R, Hartman RE, Ashwal S, Obenaus A, Badaut J, 2012. Traumatic Brain Injury in Young Rats Leads to Progressive Behavioral Deficits Coincident with Altered Tissue Properties in Adulthood. J Neurotrauma 29, 2060–2074. 10.1089/neu.2011.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akoudad S, Wolters FJ, Viswanathan A, de Bruijn RF, van der Lugt A, Hofman A, Koudstaal PJ, Ikram MA, Vernooij MW, 2016. Association of Cerebral Microbleeds With Cognitive Decline and Dementia. JAMA Neurol 73, 934–943. 10.1001/jamaneurol.2016.1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M, Kaneko T, Maeda A, Nakayama M, Ito M, Yamauchi T, Goto H, Fukata Y, Oshiro N, Shinohara A, Iwamatsu A, Kaibuchi K, 2003. Identification of Tau and MAP2 as novel substrates of Rho-kinase and myosin phosphatase. J. Neurochem 87, 780–790. [DOI] [PubMed] [Google Scholar]
- Amano M, Nakayama M, Kaibuchi K, 2010. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 67, 545–554. 10.1002/cm.20472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers NA, Kapás L, Krueger JM, 1997. The inhibitory effects of N omega-nitro-L-arginine methyl ester on nitric oxide synthase activity vary among brain regions in vivo but not in vitro. Neurochem. Res 22, 81–86. 10.1023/a:1027385522859 [DOI] [PubMed] [Google Scholar]
- Basile AM, Pantoni L, Pracucci G, Asplund K, Chabriat H, Erkinjuntti T, Fazekas F, Ferro JM, Hennerici M, O’Brien J, Scheltens P, Visser MC, Wahlund L-O, Waldemar G, Wallin A, Inzitari D, 2006. Age, Hypertension, and Lacunar Stroke Are the Major Determinants of the Severity of Age-Related White Matter Changes. CED 21, 315–322. 10.1159/000091536 [DOI] [PubMed] [Google Scholar]
- Bond LM, Sellers JR, McKerracher L, 2015. Rho kinase as a target for cerebral vascular disorders. Future Med Chem 7, 1039–1053. 10.4155/fmc.15.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickman AM, Tosto G, Gutierrez J, Andrews H, Gu Y, Narkhede A, Rizvi B, Guzman V, Manly JJ, Vonsattel JP, Schupf N, Mayeux R, 2018. An MRI measure of degenerative and cerebrovascular pathology in Alzheimer disease. Neurology 91, e1402–e1412. 10.1212/WNL.0000000000006310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickman AM, Zahodne LB, Guzman VA, Narkhede A, Meier IB, Griffith EY, Provenzano FA, Schupf N, Manly JJ, Stern Y, Luchsinger JA, Mayeux R, 2015. Reconsidering harbingers of dementia: progression of parietal lobe white matter hyperintensities predicts Alzheimer’s disease incidence. Neurobiol. Aging 36, 27–32. 10.1016/j.neurobiolaging.2014.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WR, 2010. A review of string vessels or collapsed, empty basement membrane tubes. J. Alzheimers Dis 21, 725–739. 10.3233/JAD-2010-100219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caesar C, Lyle AN, Joseph G, Weiss D, Alameddine FMF, Lassègue B, Griendling KK, Taylor WR, 2017. Cyclic Strain and Hypertension Increase Osteopontin Expression in the Aorta. Cell Mol Bioeng 10, 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charidimou A, Boulouis G, Gurol ME, Ayata C, Bacskai BJ, Frosch MP, Viswanathan A, Greenberg SM, 2017. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 140, 1829–1850. 10.1093/brain/awx047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelladurai P, Seeger W, Pullamsetti SS, 2012. Matrix metalloproteinases and their inhibitors in pulmonary hypertension. Eur. Respir. J 40, 766–782. 10.1183/09031936.00209911 [DOI] [PubMed] [Google Scholar]
- Chrissobolis S, Sobey CG, 2001. Evidence that Rho-kinase activity contributes to cerebral vascular tone in vivo and is enhanced during chronic hypertension: comparison with protein kinase C. Circ. Res 88, 774–779. [DOI] [PubMed] [Google Scholar]
- Cifuentes D, Poittevin M, Bonnin P, Ngkelo A, Kubis N, Merkulova-Rainon T, Lévy BI, 2017. Inactivation of Nitric Oxide Synthesis Exacerbates the Development of Alzheimer Disease Pathology in APPPS1 Mice (Amyloid Precursor Protein/Presenilin-1). Hypertension. 10.1161/HYPERTENSIONAHA.117.09742 [DOI] [PubMed] [Google Scholar]
- Cifuentes D, Poittevin M, Dere E, Broquères-You D, Bonnin P, Benessiano J, Pocard M, Mariani J, Kubis N, Merkulova-Rainon T, Lévy BI, 2015. Hypertension accelerates the progression of Alzheimer-like pathology in a mouse model of the disease. Hypertension 65, 218–224. 10.1161/HYPERTENSIONAHA.114.04139 [DOI] [PubMed] [Google Scholar]
- Cohen RM, Rezai-Zadeh K, Weitz TM, Rentsendorj A, Gate D, Spivak I, Bholat Y, Vasilevko V, Glabe CG, Breunig JJ, Rakic P, Davtyan H, Agadjanyan MG, Kepe V, Barrio JR, Bannykh S, Szekely CA, Pechnick RN, Town T, 2013. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J. Neurosci 33, 6245–6256. 10.1523/JNEUROSCI.3672-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- deBlois D, Lombardi DM, Su EJ, Clowes AW, Schwartz SM, Giachelli CM, 1996. Angiotensin II induction of osteopontin expression and DNA replication in rat arteries. Hypertension 28, 1055–1063. [DOI] [PubMed] [Google Scholar]
- Delbosc S, Haloui M, Louedec L, Dupuis M, Cubizolles M, Podust VN, Fung ET, Michel J-B, Meilhac O, 2008. Proteomic Analysis Permits the Identification of New Biomarkers of Arterial Wall Remodeling in Hypertension. Mol Med 14, 383–394. 10.2119/2008-00030.Delbosc [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Ruiz C, Wang J, Ksiezak-Reding H, Ho L, Qian X, Humala N, Thomas S, Martínez-Martín P, Pasinetti GM, 2009. Role of Hypertension in Aggravating Aβ Neuropathology of AD Type and Tau-Mediated Motor Impairment. Cardiovasc Psychiatry Neurol 2009. 10.1155/2009/107286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr A, Sahota B, Chinta LV, Brown ME, Lai AY, Ma K, Hawkes CA, McLaurin J, Stefanovic B, 2012b. Amyloid-β-dependent compromise of microvascular structure and function in a model of Alzheimer’s disease. Brain 135, 3039–3050. 10.1093/brain/aws243 [DOI] [PubMed] [Google Scholar]
- Dupuis M, Soubrier F, Brocheriou I, Raoux S, Haloui M, Louedec L, Michel J-B, Nadaud S, 2004. Profiling of aortic smooth muscle cell gene expression in response to chronic inhibition of nitric oxide synthase in rats. Circulation 110, 867–873. 10.1161/01.CIR.0000138850.72900.FE [DOI] [PubMed] [Google Scholar]
- Esiri MM, Nagy Z, Smith MZ, Barnetson L, Smith AD, 1999. Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer’s disease. Lancet 354, 919–920. 10.1016/S0140-6736(99)02355-7 [DOI] [PubMed] [Google Scholar]
- Feldstein CA, 2012. Association between chronic blood pressure changes and development of Alzheimer’s disease. J. Alzheimers Dis 32, 753–763. 10.3233/JAD-2012-120613 [DOI] [PubMed] [Google Scholar]
- Frid K, Einstein O, Friedman-Levi Y, Binyamin O, Ben-Hur T, Gabizon R, 2015. Aggregation of MBP in chronic demyelination. Ann Clin Transl Neurol 2, 711–721. 10.1002/acn3.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CA, McLaurin J, 2009. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. U.S.A 106, 1261–1266. 10.1073/pnas.0805453106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Wahlund L-O, Svensson L, Winblad B, Julin P, 2002. Cingulate cortex hypoperfusion predicts Alzheimer’s disease in mild cognitive impairment. BMC Neurol 2, 9. 10.1186/1471-2377-2-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain K, 2002. Exercise conditioning attenuates the hypertensive effects of nitric oxide synthase inhibitor in rat. Mol. Cell. Biochem 231, 129–137. [DOI] [PubMed] [Google Scholar]
- Husain K, Hazelrigg SR, 2002. Oxidative injury due to chronic nitric oxide synthase inhibition in rat: effect of regular exercise on the heart. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1587, 75–82. 10.1016/S0925-4439(02)00070-4 [DOI] [PubMed] [Google Scholar]
- Huveneers S, Daemen MJAP, Hordijk PL, 2015. Between Rho(k) and a hard place: the relation between vessel wall stiffness, endothelial contractility, and cardiovascular disease. Circ. Res 116, 895–908. 10.1161/CIRCRESAHA.116.305720 [DOI] [PubMed] [Google Scholar]
- in’t Veld BA, Ruitenberg A, Hofman A, Stricker BH, Breteler MM, 2001. Antihypertensive drugs and incidence of dementia: the Rotterdam Study. Neurobiol. Aging 22, 407–412. [DOI] [PubMed] [Google Scholar]
- Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC, Alzheimer’s Disease Neuroimaging Initiative, 2016. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 7, 11934. 10.1038/ncomms11934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joffres M, Falaschetti E, Gillespie C, Robitaille C, Loustalot F, Poulter N, McAlister FA, Johansen H, Baclic O, Campbell N, 2013. Hypertension prevalence, awareness, treatment and control in national surveys from England, the USA and Canada, and correlation with stroke and ischaemic heart disease mortality: a cross-sectional study. BMJ Open 3, e003423. 10.1136/bmjopen-2013-003423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo IL, Lai AY, Bazzigaluppi P, Koletar MM, Dorr A, Brown ME, Thomason LAM, Sled JG, McLaurin J, Stefanovic B, 2017. Early neurovascular dysfunction in a transgenic rat model of Alzheimer’s disease. Sci Rep 7, 46427. 10.1038/srep46427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapasi A, DeCarli C, Schneider JA, 2017. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 134, 171–186. 10.1007/s00401-017-1717-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachaturian AS, Zandi PP, Lyketsos CG, Hayden KM, Skoog I, Norton MC, Tschanz JT, Mayer LS, Welsh-Bohmer KA, Breitner JCS, 2006. Antihypertensive medication use and incident Alzheimer disease: the Cache County Study. Arch. Neurol 63, 686–692. 10.1001/archneur.63.5.noc60013 [DOI] [PubMed] [Google Scholar]
- Kihara M, Horie R, Lovenberg W, Yamori Y, 1993. Comparative study of various genetic hypertensive rat strains: Blood pressure, body weight, growth and organ weights. Heart Vessels 8, 7–15. 10.1007/BF02630560 [DOI] [PubMed] [Google Scholar]
- Kruyer A, Soplop N, Strickland S, Norris EH, 2015. Chronic Hypertension Leads to Neurodegeneration in the TgSwDI Mouse Model of Alzheimer’s Disease. Hypertension 66, 175–182. 10.1161/HYPERTENSIONAHA.115.05524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AY, Dorr A, Thomason LAM, Koletar MM, Sled JG, Stefanovic B, McLaurin J, 2015. Venular degeneration leads to vascular dysfunction in a transgenic model of Alzheimer’s disease. Brain 138, 1046–1058. 10.1093/brain/awv023 [DOI] [PubMed] [Google Scholar]
- Lai AY, McLaurin J, 2018. Rho-associated protein kinases as therapeutic targets for both vascular and parenchymal pathologies in Alzheimer’s disease. J. Neurochem 144, 659–668. 10.1111/jnc.14130 [DOI] [PubMed] [Google Scholar]
- Lane CA, Barnes J, Nicholas JM, Sudre CH, Cash DM, Parker TD, Malone IB, Lu K, James S-N, Keshavan A, Murray-Smith H, Wong A, Buchanan SM, Keuss SE, Gordon E, Coath W, Barnes A, Dickson J, Modat M, Thomas D, Crutch SJ, Hardy R, Richards M, Fox NC, Schott JM, 2019. Associations between blood pressure across adulthood and late-life brain structure and pathology in the neuroscience substudy of the 1946 British birth cohort (Insight 46): an epidemiological study. Lancet Neurol 18, 942–952. 10.1016/S1474-4422(19)30228-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launer LJ, Ross GW, Petrovitch H, Masaki K, Foley D, White LR, Havlik RJ, 2000. Midlife blood pressure and dementia: the Honolulu-Asia aging study. Neurobiol. Aging 21, 49–55. [DOI] [PubMed] [Google Scholar]
- Lennon MJ, Makkar SR, Crawford JD, Sachdev PS, 2019. Midlife Hypertension and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimers Dis 71, 307–316. 10.3233/JAD-190474 [DOI] [PubMed] [Google Scholar]
- Lerman LO, Kurtz TW, Touyz RM, Ellison DH, Chade AR, Crowley SD, Mattson DL, Mullins JJ, Osborn J, Eirin A, Reckelhoff JF, Iadecola C, Coffman TM, 2019. Animal Models of Hypertension: A Scientific Statement From the American Heart Association. Hypertension 73, e87–e120. 10.1161/HYP.0000000000000090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Cooper L, Cai J, Toole JF, Bryan NR, Hutchinson RG, Tyroler HA, 1996. Presence and severity of cerebral white matter lesions and hypertension, its treatment, and its control. The ARIC Study. Atherosclerosis Risk in Communities Study. Stroke 27, 2262–2270. 10.1161/01.str.27.12.2262 [DOI] [PubMed] [Google Scholar]
- Loirand G, 2015. Rho Kinases in Health and Disease: From Basic Science to Translational Research. Pharmacol. Rev 67, 1074–1095. 10.1124/pr.115.010595 [DOI] [PubMed] [Google Scholar]
- Lorenzen JM, Nickel N, Krämer R, Golpon H, Westerkamp V, Olsson KM, Haller H, Hoeper MM, 2011. Osteopontin in patients with idiopathic pulmonary hypertension. Chest 139, 1010–1017. 10.1378/chest.10-1146 [DOI] [PubMed] [Google Scholar]
- Michalski D, Keck AL, Grosche J, Martens H, Härtig W, 2018. Immunosignals of Oligodendrocyte Markers and Myelin-Associated Proteins Are Critically Affected after Experimental Stroke in Wild-Type and Alzheimer Modeling Mice of Different Ages. Front Cell Neurosci 12. 10.3389/fncel.2018.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Nikolakopoulou AM, Zhao Z, Sagare AP, Si G, Lazic D, Barnes SR, Daianu M, Ramanathan A, Go A, Lawson EJ, Wang Y, Mack WJ, Thompson PM, Schneider JA, Varkey J, Langen R, Mullins E, Jacobs RE, Zlokovic BV, 2018. Pericyte degeneration causes white matter dysfunction in the mouse CNS. Nat Med 24, 326–337. 10.1038/nm.4482 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Morrone CD, Bazzigaluppi P, Beckett TL, Hill ME, Koletar MM, Stefanovic B, McLaurin J, 2020. Regional differences in Alzheimer’s disease pathology confound behavioural rescue after amyloid-β attenuation. Brain 143, 359–373. 10.1093/brain/awz371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam SM, Kwon HJ, Kim W, Kim JW, Hahn KR, Jung HY, Kim DW, Yoo DY, Seong JK, Hwang IK, Yoon YS, 2018. Changes of myelin basic protein in the hippocampus of an animal model of type 2 diabetes. Lab Anim Res 34, 176–184. 10.5625/lar.2018.34.4.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires PW, Dams Ramos CM, Matin N, Dorrance AM, 2013. The effects of hypertension on the cerebral circulation. Am J Physiol Heart Circ Physiol 304, H1598–H1614. 10.1152/ajpheart.00490.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM, 2010. Alzheimer’s disease. N. Engl. J. Med 362, 329–344. 10.1056/NEJMra0909142 [DOI] [PubMed] [Google Scholar]
- Rabinovici GD, Carrillo MC, Forman M, DeSanti S, Miller DS, Kozauer N, Petersen RC, Randolph C, Knopman DS, Smith EE, Isaac M, Mattsson N, Bain LJ, Hendrix JA, Sims JR, 2017. Multiple comorbid neuropathologies in the setting of Alzheimer’s disease neuropathology and implications for drug development. Alzheimers Dement (N Y) 3, 83–91. 10.1016/j.trci.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajani RM, Quick S, Ruigrok SR, Graham D, Harris SE, Verhaaren BFJ, Fornage M, Seshadri S, Atanur SS, Dominiczak AF, Smith C, Wardlaw JM, Williams A, 2018. Reversal of endothelial dysfunction reduces white matter vulnerability in cerebral small vessel disease in rats. Science Translational Medicine 10, eaam9507. 10.1126/scitranslmed.aam9507 [DOI] [PubMed] [Google Scholar]
- Santhanam AVR, d’Uscio LV, He T, Das P, Younkin SG, Katusic ZS, 2015. Uncoupling of endothelial nitric oxide synthase in cerebral vasculature of Tg2576 mice. J. Neurochem 134, 1129–1138. 10.1111/jnc.13205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NS, Vidal J-S, Masaki K, Petrovitch H, Ross GW, Tilley C, DeMattos RB, Tracy RP, White LR, Launer LJ, 2012. Midlife blood pressure, plasma β-amyloid, and the risk for Alzheimer disease: the Honolulu Asia Aging Study. Hypertension 59, 780–786. 10.1161/HYPERTENSIONAHA.111.178962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih Y-H, Wu S-Y, Yu M, Huang S-H, Lee C-W, Jiang M-J, Lin P-Y, Yang T-T, Kuo Y-M, 2018. Hypertension Accelerates Alzheimer’s Disease-Related Pathologies in Pigs and 3xTg Mice. Front Aging Neurosci 10, 73. 10.3389/fnagi.2018.00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa H, Sunamura S, Satoh K, 2016. RhoA/Rho-Kinase in the Cardiovascular System. Circ. Res 118, 352–366. 10.1161/CIRCRESAHA.115.306532 [DOI] [PubMed] [Google Scholar]
- Smith EE, 2018. Cerebral amyloid angiopathy as a cause of neurodegeneration. Journal of Neurochemistry 144, 651–658. 10.1111/jnc.14157 [DOI] [PubMed] [Google Scholar]
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR, 1997. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 277, 813–817. [PubMed] [Google Scholar]
- Sorond FA, Hurwitz S, Salat DH, Greve DN, Fisher NDL, 2013. Neurovascular coupling, cerebral white matter integrity, and response to cocoa in older people. Neurology 81, 904–909. 10.1212/WNL.0b013e3182a351aa [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPRINT MIND Investigators for the SPRINT Research Group, Nasrallah IM, Pajewski NM, Auchus AP, Chelune G, Cheung AK, Cleveland ML, Coker LH, Crowe MG, Cushman WC, Cutler JA, Davatzikos C, Desiderio L, Doshi J, Erus G, Fine LJ, Gaussoin SA, Harris D, Johnson KC, Kimmel PL, Kurella Tamura M, Launer LJ, Lerner AJ, Lewis CE, Martindale-Adams J, Moy CS, Nichols LO, Oparil S, Ogrocki PK, Rahman M, Rapp SR, Reboussin DM, Rocco MV, Sachs BC, Sink KM, Still CH, Supiano MA, Snyder JK, Wadley VG, Walker J, Weiner DE, Whelton PK, Wilson VM, Woolard N, Wright JT, Wright CB, Williamson JD, Bryan RN, 2019a. Association of Intensive vs Standard Blood Pressure Control With Cerebral White Matter Lesions. JAMA 322, 524–534. 10.1001/jama.2019.10551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPRINT MIND Investigators for the SPRINT Research Group, Williamson JD, Pajewski NM, Auchus AP, Bryan RN, Chelune G, Cheung AK, Cleveland ML, Coker LH, Crowe MG, Cushman WC, Cutler JA, Davatzikos C, Desiderio L, Erus G, Fine LJ, Gaussoin SA, Harris D, Hsieh M-K, Johnson KC, Kimmel PL, Tamura MK, Launer LJ, Lerner AJ, Lewis CE, Martindale-Adams J, Moy CS, Nasrallah IM, Nichols LO, Oparil S, Ogrocki PK, Rahman M, Rapp SR, Reboussin DM, Rocco MV, Sachs BC, Sink KM, Still CH, Supiano MA, Snyder JK, Wadley VG, Walker J, Weiner DE, Whelton PK, Wilson VM, Woolard N, Wright JT, Wright CB, 2019b. Effect of Intensive vs Standard Blood Pressure Control on Probable Dementia: A Randomized Clinical Trial. JAMA 321, 553–561. 10.1001/jama.2018.21442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabei K, Kida H, Hosoya T, Satoh M, Tomimoto H, 2017. Prediction of Cognitive Decline from White Matter Hyperintensity and Single-Photon Emission Computed Tomography in Alzheimer’s Disease. Front Neurol 8. 10.3389/fneur.2017.00408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, Monsell SE, Kukull WA, Trojanowski JQ, 2013. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 136, 2697–2706. 10.1093/brain/awt188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenseil JE, Mecham RP, 2012. Elastin in large artery stiffness and hypertension. J Cardiovasc Transl Res 5, 264–273. 10.1007/s12265-012-9349-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAGENSEIL JE, MECHAM RP, 2009. Vascular Extracellular Matrix and Arterial Mechanics. Physiol Rev 89, 957–989. 10.1152/physrev.00041.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker KA, Power MC, Gottesman RF, 2017. Defining the Relationship Between Hypertension, Cognitive Decline, and Dementia: a Review. Curr. Hypertens. Rep 19, 24. 10.1007/s11906-017-0724-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LR, Edland SD, Hemmy LS, Montine KS, Zarow C, Sonnen JA, Uyehara-Lock JH, Gelber RP, Ross GW, Petrovitch H, Masaki KH, Lim KO, Launer LJ, Montine TJ, 2016. Neuropathologic comorbidity and cognitive impairment in the Nun and Honolulu-Asia Aging Studies. Neurology 86, 1000–1008. 10.1212/WNL.0000000000002480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Bai F, Lin X, Wang Q, Wu Q, Sun S, Jiang C, Liang Q, Gao B, 2017. Association between Antihypertensive Drug Use and the Incidence of Cognitive Decline and Dementia: A Meta-Analysis of Prospective Cohort Studies. Biomed Res Int 2017, 4368474. 10.1155/2017/4368474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasar S, Xia J, Yao W, Furberg CD, Xue Q-L, Mercado CI, Fitzpatrick AL, Fried LP, Kawas CH, Sink KM, Williamson JD, DeKosky ST, Carlson MC, Ginkgo Evaluation of Memory (GEM) Study Investigators, 2013. Antihypertensive drugs decrease risk of Alzheimer disease: Ginkgo Evaluation of Memory Study. Neurology 81, 896–903. 10.1212/WNL.0b013e3182a35228 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.