

Abstract
Myelodysplastic syndromes (MDS) with ring sideroblasts are hematopoietic stem cell disorders with erythroid dysplasia and mutations in the SF3B1 splicing factor gene. MDS patients with SF3B1 mutations often accumulate excessive tissue iron, even in the absence of transfusions, but the mechanisms that are responsible for their parenchymal iron overload are unknown. Body iron content, tissue distribution, and the supply of iron for erythropoiesis are controlled by the hormone hepcidin, which is regulated by erythroblasts through secretion of the erythroid hormone erythroferrone (ERFE). Here, we identified an alternative ERFE transcript in MDS patients with the SF3B1 mutation. Induction of this ERFE transcript in primary SF3B1-mutated bone marrow erythroblasts generated a variant protein that maintained the capacity to suppress hepcidin transcription. Plasma concentrations of ERFE were higher in MDS patients with a SF3B1 gene mutation than in patients with SF3B1 wild-type MDS. Thus, hepcidin suppression by a variant erythroferrone is likely responsible for the increased iron loading in patients with SF3B1-mutated MDS, suggesting that ERFE could be targeted to prevent iron-mediated toxicity. The expression of the variant ERFE transcript that was restricted to SF3B1-mutated erythroblasts decreased in lenalidomide-responsive anemic patients, identifying variant ERFE as a specific biomarker of clonal erythropoiesis.
One Sentence Summary:
A variant erythroferrone contributes to hepcidin modulation and systemic iron accumulation in patients with SF3B1-mutated myelodysplastic syndrome.
Introduction
Myelodysplastic syndromes with ring sideroblasts (MDS-RS) are clonal hematopoietic stem cell (HSC) disorders with a prominent erythroid dysplasia of the bone marrow (BM) responsible for a macrocytic anemia. Mitochondrial iron accumulation and apoptosis of mature erythroblasts cause ineffective erythropoiesis (1, 2). In contrast to other MDS subtypes, patients with MDS-RS exhibit signs of systemic iron accumulation that is reflected by increased ferritin and non-transferrin bound iron concentrations even before they become transfusion-dependent and subsequently develop parenchymal iron overload (3, 4).
Splicing factor gene SF3B1 is mutated in ~90% of MDS-RS, and the diagnosis is considered whenever the gene is mutated, even if the percentage of RS is relatively low, between 5 and 15% (5–7). These mutations arise in the HSCs (8–10). Aberrant splicing events are reported in MDS and other SF3B1-driven cancers, including uveal melanoma and chronic lymphocytic leukemia (CLL) (11–14). The selection of an alternative branch site resulting in the use of a cryptic 3’ splice site (ss) is the most frequently detected abnormality. Computational analysis revealed that the majority of cryptic 3’ss are located upstream of canonical 3’ss at nucleotide distances that are not multiples of 3, suggesting that the aberrant transcripts would likely contain a premature termination codon (PTC) and be degraded by non-sense mediated decay (NMD). It has been predicted that half of the aberrantly spliced transcripts in SF3B1-mutated cells are NMD-sensitive and that the canonical isoforms are down-regulated (11). For instance, ABCB7 transcript encoding a mitochondrial transporter involved in the export of Fe-S clusters is aberrantly spliced and undergoes NMD. It is also possible that NMD-insensitive aberrant transcripts are translated into proteins with altered function (11). Globally, mis-splicing may contribute to defective mRNA production and deregulation of cellular pathways (17–18). How these aberrant splicing events contribute to the disease phenotype and in particular to systemic iron overload is unclear.
In contrast with other MDS subtypes, MDS-RS are associated with lower expression of the iron homeostasis regulator, hepcidin, and as a consequence, the absorption of iron by duodenal enterocytes and its release from erythrophagocytic macrophages may be increased (19–24). Inappropriately low hepcidin concentrations in MDS-RS could depend on the degree of ineffective erythropoiesis linked to impaired iron incorporation into heme because of mitochondrial iron trapping and/or to increased expression of hepcidin repressors (25). Growth differentiation factor 15 (GDF-15) and twisted gastrulation (TWSG1), two members of the transforming growth factor-β superfamily, have been proposed as pathological suppressors of hepcidin in ineffective erythropoiesis (26, 27). More recently, erythroferrone (ERFE), a C1q–tumor necrosis factor–related family of proteins (CTRP) member, has been described as a major erythroid regulator of hepcidin, involved in the pathological suppression of hepcidin in patients with β-thalassemia (28, 29).
In the present study, we identify a variant transcript of ERFE specific to SF3B1-mutated MDS. The expression of the variant ERFE is restricted to the erythroid lineage, and the variant contributes to increased concentration of ERFE protein, resulting in hepcidin suppression and systemic iron accumulation in patients. This variant appears to be a pertinent biomarker of clonal erythropoiesis with potential use for monitoring treatment response of anemic patients with SF3B1-mutated MDS.
Results
Upregulation of ERFE using an alternative 3’ splice site in SF3B1-mutated MDS
To investigate the mechanism of systemic iron overload, we established a cohort of 156 patients with lower risk MDS, including 60 MDS-RS with single lineage dysplasia (SLD), 17 MDS-RS with multilineage dysplasia (MLD), 2 5q-syndrome, 17 MDS-SLD, 42 MDS-MLD, and 18 MDS with type 1 excess of blasts (EB1) (table S1). The Revised-International Prognosis Scoring System score was very low in 15, low in 95, intermediate in 36, and high in 4 patients. Twenty-six genes commonly mutated in MDS were sequenced in the BM mononuclear cell (MNC) fraction. SF3B1 gene was mutated in 94 patients with MDS, including 63 (67%) affected by a SF3B1K700E mutation. Among the 62 patients with no SF3B1 mutation, other splicing genes, SRSF2, U2AF1, or ZRSR2 were mutated in 27 cases, and no splicing gene mutation was observed in 35 cases. Some patients presented with two splicing mutations (fig. S1). We evaluated the consequences of SF3B1 gene mutations on gene expression and splicing by sequencing the transcriptome of the BM MNCs isolated from 27 patients in this cohort, including 21 with SF3B1MUT MDS, 6 with SF3B1WT MDS, and 5 healthy controls. Differential analyses of gene expression and splice junctions were conducted using DESeq2 (30). We detected 6,343 genes as differentially expressed between SF3B1MUT MDS and SF3B1WT MDS with a P-value<0.05 (data file S1). Principal component analysis (PCA) of gene expression profiles separated SF3B1MUT and SF3B1WT MDS (Fig. 1A). The differentially expressed genes were enriched in 73 specific GO terms with an absolute log2 fold-change (FC)>1 and a Benjamini-Hochberg (BH)-adjusted P-value<0.05 (Fig. 1B). Genes with a log2(FC)< −1 were involved in serine/threonine kinase signaling, apoptosis, myeloid differentiation, inflammation, and cell-cell adhesion, whereas those with a log2(FC)>1 were involved in heme metabolism, erythrocyte differentiation, and iron homeostasis (Fig. 1C). We plotted the log2(FC) of 16 differentially expressed genes belonging to the IRON_ION_HOMEOSTASIS gene set (GO:0055072; http://amigo.geneontology.org) and showed that the FAM132B/ERFE transcript encoding erythroferrone was increased (Fig. 1D). The FAM132B/ERFE transcript was similarly expressed in samples with an epigenetic TET2MUT/DNMT3AWT/SF3B1WT genotype compared to healthy controls or with a DNMT3AMUT/TET2WT/SF3B1MUT compared to DNMT3AWT/TET2WT/ SF3B1MUT genotype, suggesting that the deregulation of ERFE transcript expression was linked to SF3B1 mutation (table S2).
Fig. 1: Differential gene expression in SF3B1MUT MDSs.
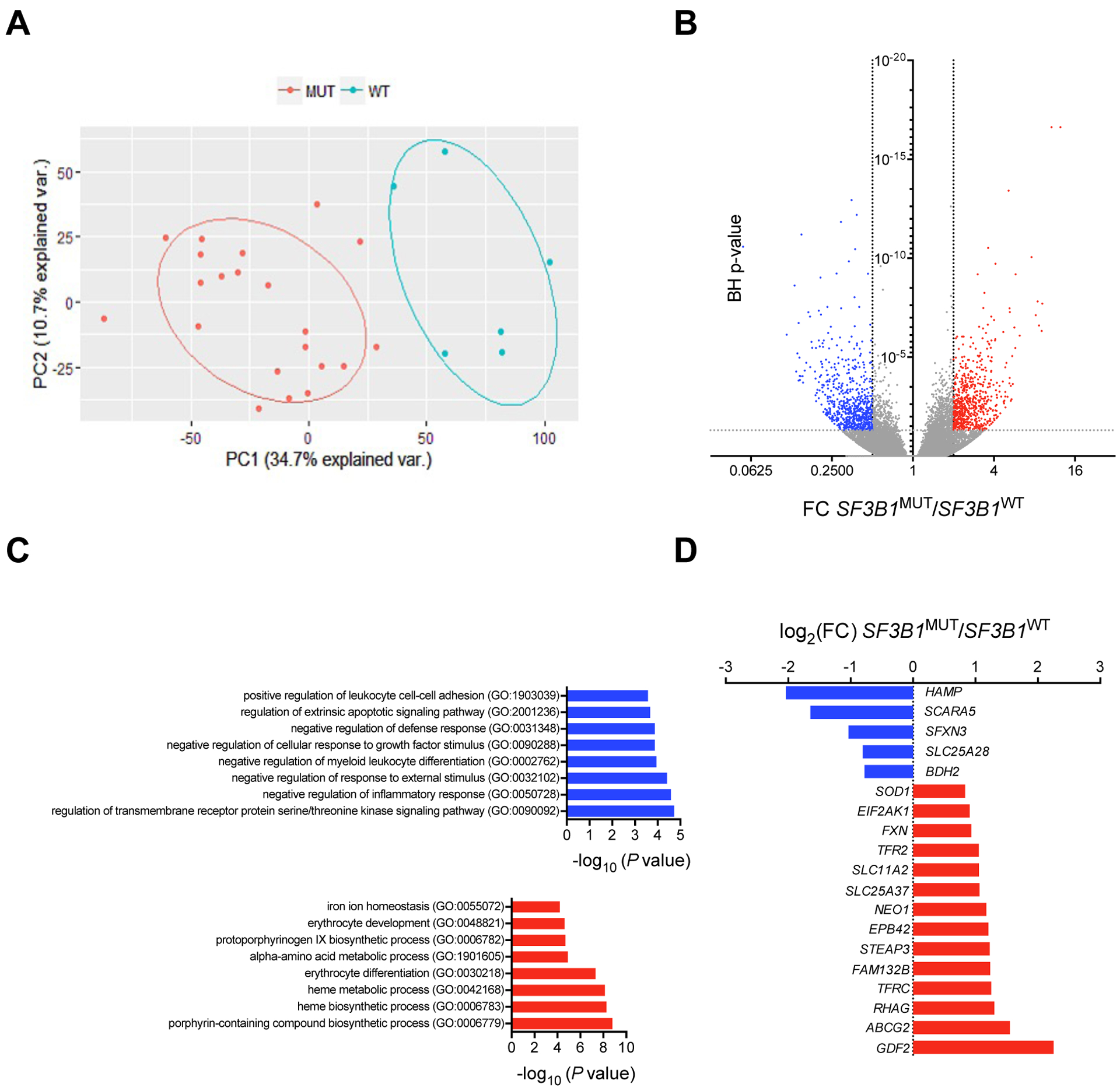
(A) Bi-plot of the first 2 principal components (PCs) showing 45.4% of the variability within the data (PC1, x-axis; PC2, y-axis). (B) Volcano plot showing differentially expressed transcripts in SF3B1MUT BM MNCs compared to SF3B1WT BM MNCs. Fold change (FC) on x-axis and negative log10 of Benjamini-Hochberg (BH) corrected P-values on y-axis. Dashed vertical and horizontal lines reflect the filtering criteria (FC <0.5 or >2.0 and BH-corrected P-value <0.05). The red dots represent differentially up-regulated transcripts, the blue dots represent differentially down-regulated transcripts, and the gray dots represent transcripts without differential expression. (C) GO enrichment analysis of differentially expressed genes with an absolute log2 (FC) >1 and a P-value <0.05 in SF3B1MUT MDS. The most down-regulated and up-regulated gene sets are represented. (D) Log2(FC) of the expression of 16 among the 96 genes of the IRON_ION_HOMEOSTASIS gene set (GO:005072) deregulated in SF3B1MUT MDS.
We then identified 1,528 differentially expressed 5’ and 3’ junctions, including annotated 5’ donor and 3’ acceptor ss, ambiguous junctions, and canonical junctions with BH-adjusted P-values ≤10−5 and absolute log2(FC) ≥1 (data file S2). These junctions allowed the hierarchical clustering of the 21 SF3B1MUT and 6 SF3B1WT MDS samples (Fig. 2A). After excluding differentially expressed canonical junctions, we then considered the 1,147 alternative junctions, among which we identified 786 3’ acceptor junctions (68.5%), 176 5’ donor junctions (15.3%), and 185 ambiguous junctions (16.1%) attributed to either the alternative 5’ or 3’ss or both alternative 5’ and 3’ss (Fig. 2B). The analysis of distances between the alternative and canonical 3’ss showed that the majority of alternative 3’ss (AG’) was located between −24 and −9 nucleotides preceding the canonical 3’ss (AG) (Fig. 2C). The proportion of alternative AG’ junctions in these novel splice variants was generally increased in SF3B1 mutants and less frequently in SF3B1WT samples (Fig. 2D). To detect alternative transcripts that were likely to generate substantial amounts of variant proteins with a modified length, we applied a filter selecting transcripts with an additional stretch of nucleotides numbering in multiples of 3, a ratio of alternative junction coverage (AG’) to alternative and canonical junctions coverage (AG’+AG) over 0.1, and an expression ratio of the alternative junction in SF3B1MUT versus SF3B1WT samples over 10 (fig. S2A). We obtained 66 alternative junctions in 63 genes (data file S3 & fig. S2B). These genes were involved in the epigenetic regulation and transcription, mRNA processing and translation, intracellular transport, cell cycle and migration, signaling and apoptosis, mitochondrial metabolism, and iron homeostasis (Fig. 2E). Among the 66 alternative junctions, 29 were related to an in-frame insertion of 9 to 27 nucleotides, and 26 of them were differentially expressed between SF3B1MUT and SF3B1WT MDS with an absolute log2(FC) >0.3 (Fig. 2F). FAM132B/ERFE gene was identified among the upregulated genes with an alternative junction due to the use of a cryptic 3’ss located between exons 2 and 3 (chr2: 239,070,357 – 239,071,364) and no PTC. The aberrant transcript contained 12 additional nucleotides in the open reading frame and from here on is referred to as ERFE+12 (Fig. 2G). It was systematically detected in all SF3B1MUT MDS samples and represented a mean percentage of 24.8% of FAM132B/ERFE transcripts in SF3B1MUT MDS versus 0.2% in SF3B1WT MDS. The mutation pattern of samples expressing ERFE+12 is shown in fig. S2C. This indicates that the expression of ERFE+12 was strongly linked to the presence of a mutation in SF3B1 gene.
Fig. 2: Differential splice junctions in SF3B1MUT MDSs.
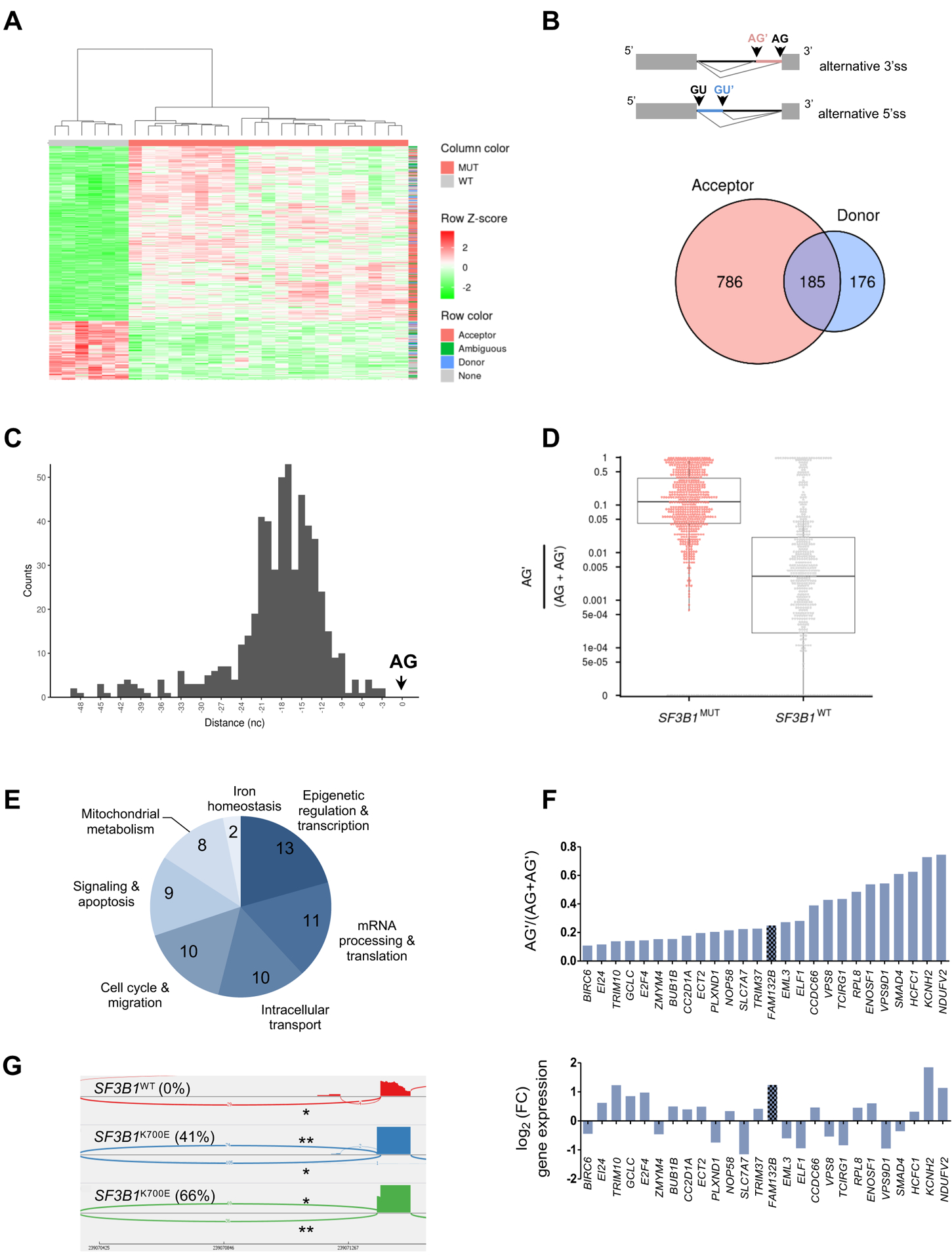
(A) Hierarchical clustering and heat-map of differential splice junctions between SF3B1MUT and SF3B1WT MDS samples. Values indicating percent usage of the differential splice junction versus all other junctions sharing the same splice site are normalized as Z scores across patients and limited to a maximum of |Z| = 2. Rows are splice junctions with indicated types: acceptor (red), donor (blue), ambiguous (green), and differentially expressed canonical junction (gray). Columns are patients. (B) Venn diagram of the number of differential alternative 5’ donor and 3’ acceptor junctions in SF3B1MUT compared to SF3B1WT MDS. The overlapping area represents ambiguous junctions (n = 185). (C) Distances between the alternative (AG’) and canonical (AG) 3’splice sites within the 50 nucleotides upstream of the AG plotted as a histogram. (D) Comparison of the expression of alternative junctions. The ratio AG’/AG’+AG for each junction in SF3B1MUT (red) versus SF3B1WT (gray) MDS is shown. (E) Distribution of the biological functions of 63 genes affected by one or two aberrant 3’ss junctions located at <50 bases of the canonical 3’ss, an additional sequence multiple of 3 nucleotides, a ratio AG’/(AG’+AG) >0.1 in SF3B1MUT samples, and a FC >10. (F) Ratio of AG’/(AG’+AG) in 26 genes whose expression was up- or down-regulated in SF3B1MUT patients. (G) Sashimi plot of 3’ss canonical (*) and aberrant (**) junctions in FAM132B/ERFE gene in three BM MNC samples, 1 SF3B1WT and 2 SF3B1K700E MDS. The ratio AG’/(AG’+AG) is indicated in parentheses.
SF3B1MUT-restricted expression of alternative ERFE+12 transcript
To ascertain that expression of the ERFE+12 transcript is dependent on the presence of a SF3B1 mutation, we transfected the erythro-megakaryocytic cell line UT7/EPO with a pLVX plasmid encoding a synthetic full-length SF3B1WT or mutant SF3B1K700E cDNA. We then designed a sensitive fluorescent PCR allowing the detection of ERFE+12 and ERFEWT transcripts as 162 nucleotides (nt) and 150 nt fragments by capillary electrophoresis (table S3). Twenty-four hours after transfection, ERFE+12 transcript was detected in SF3B1K700E transfected cells, but not in SF3B1WT cells (Fig. 3A). To validate the splice pattern induced in the context of SF3B1 mutation in ERFE pre-mRNA, we performed a minigene splicing assay. An ERFE sequence of about 200 nucleotides located on both sides of the cryptic 3’ss (AG’) was cloned in an ExonTrap vector. The alternative junction in ENOSF1 gene (chr18: 683,395–685,920) cloned in the same vector was used as a control (13). These minigenes were transfected into the murine G1E-ER4 cell line in which the SF3B1K700E mutation was edited by CRISPR-Cas9 technology. As expected due to the lack of intron homology between species, sequencing the transcriptome of G1E-ER4 SF3B1K700E cell line or isogenic SF3B1WT cell line demonstrated that endogenous murine ERFE and ENOSF1 did not undergo alternative splicing (12). After transfection, the alternative 3’ss AG’ ERFE+12 was detected by capillary electrophoresis in G1E-ER4 SF3B1K700E cells, but not in the isogenic G1E-ER4 SF3B1WT or parental cells (Fig. 3B). The usage of alternative 3’ss AG’ was detectable for ENOSF1 gene in SF3B1WT cell line and became predominant in SF3B1K700E cell line, suggesting that the mutation favored the usage of alternative AG’. Then, we performed a rescue experiment by adding a destabilization domain (DD)-tag to the mutant SF3B1R625G allele in Mel202 cell line, as described (31). The DD-tagged protein is stabilized by interaction with the DD ligand Shield-1 and is degraded upon Shield-1 withdrawal. In Mel202 clone 26 containing one DD-tagged SF3B1R625G allele, Shield-1 removal abrogated ERFE+12 expression. This supports a causal relationship between SF3B1 mutation and the aberrant splicing isoform of ERFE (fig. S3).
Fig. 3: SF3B1-dependent expression of 3’ss aberrant ERFE+12.
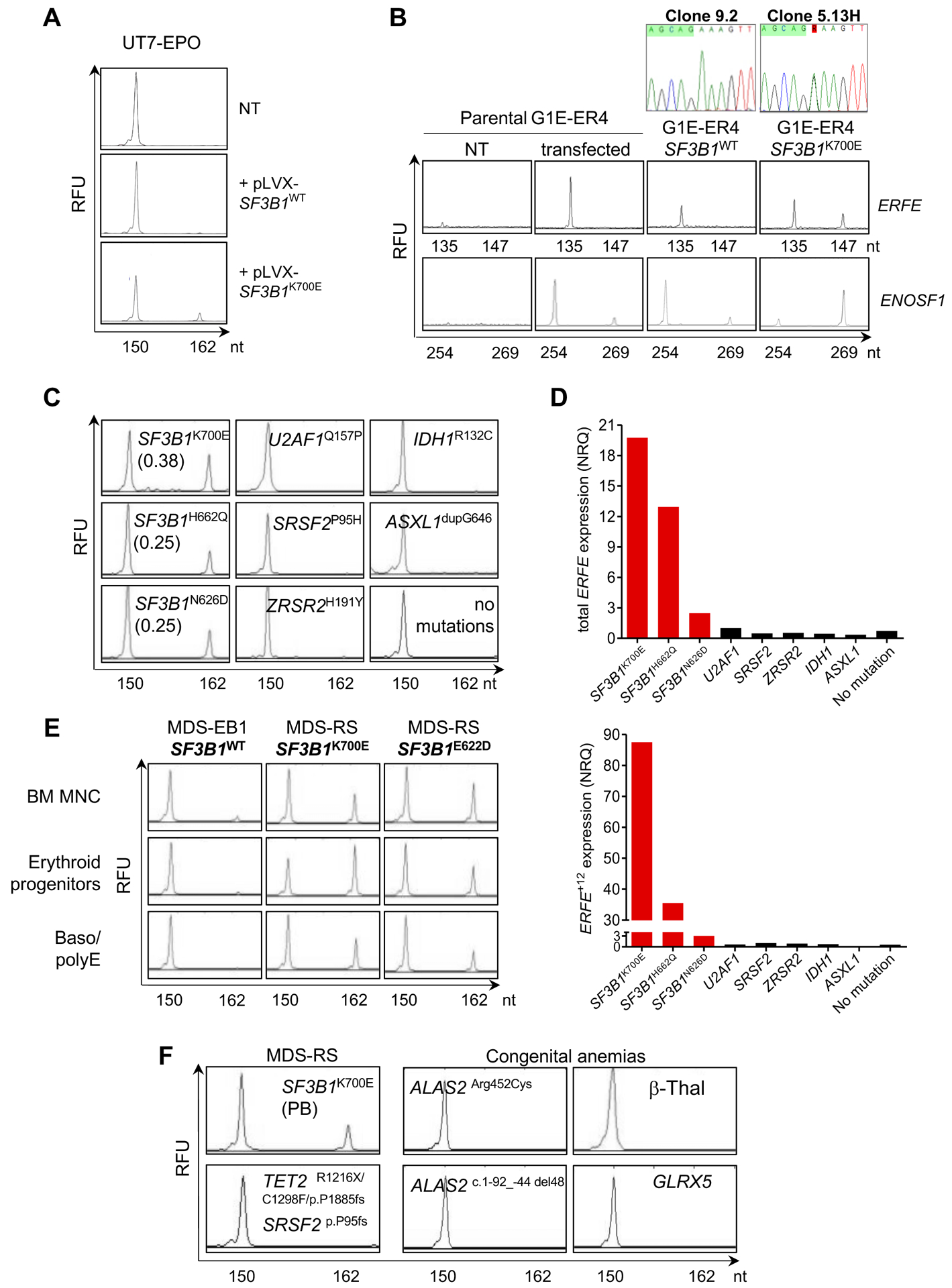
(A) Induction of ERFE+12 by expressing SF3B1K700E in human SF3B1WT UT-7/EPO cell line. Cells were transfected with a pLVX plasmid encoding a synthetic SF3B1WT or SF3B1K700E cDNA. Non-transfected (NT) UT-7/EPO cells are shown as a control. The canonical ERFE and aberrant ERFE+12 transcripts were detected by capillary electrophoresis of fluorescent PCR products. The x-axis represents molecular size (nt for nucleotides) of PCR amplicons, and the y-axis represents relative fluorescent units (RFU). The peak at 150 nt corresponds to the canonical transcript, whereas the peak at 162 nt refers to the alternative transcript due to cryptic AG’ usage. (B) Analysis of alternative AG’ and canonical AG usage of ERFE and ENOSF1 minigenes transfected into murine CRISPR-Cas9 SF3B1WT (clone 9.2) and SF3B1K700E (clone 5.13H) G1E-ER4 cells by fluorescent PCR. Transfected and NT parental G1E-ER4 cells are shown as controls. The peak at 135 nt corresponds to the transcript generated by a canonical AG usage, whereas the peak at 147 nt refers to the alternative transcript due to cryptic AG’ usage. (C) Detection of ERFE+12 depends on the presence of a SF3B1 mutant in MDS. BM MNC RNAs from 3 patients with SF3B1 mutations (SF3B1K700E, SF3B1H622Q, SF3B1N626D), 3 with mutations in other splice genes (U2AF1Q157P, SRSF2P95H, ZRSR2H191Y), and 3 with IDH1R132C, ASXL1dupG646 or no mutations were analyzed. ERFE+12/ERFE+12+ERFEWT ratios are indicated. (D) Quantification of ERFEWT and ERFE+12 transcripts by RT-qPCR in BM samples depicted in (C). Results are expressed as normalized ratio quantities (NRQ) to ACTB and B2M housekeeping genes. (E) Detection of ERFEWT and ERFE+12 transcripts in SF3B1MUT or SF3B1WT erythroid progenitors or basophilic/polychromatic erythroblasts (Baso/polyE) in culture in comparison with BM MNC. (F) Analysis of ERFE transcripts in SF3B1WT diseases with ineffective erythropoiesis. The image shows peripheral blood (PB) MNC from 1 patient with MDS-RS with TET2, SRSF2 and no SF3B1 mutations, samples from 3 patients with congenital sideroblastic anemias (2 BM samples with ALAS2 mutation and 1 PB sample with GLRX5 mutation) and one PB sample from a patient with severe β-thalassemia (β-Thal). Samples were analyzed by capillary electrophoresis of ERFE PCR products. One PB sample from a SF3B1K700E MDS patient was used as a positive control.
We then investigated the expression of ERFEWT and ERFE+12 in primary BM samples of MDS patients using fluorescent PCR and RT-qPCR (table S3). Among 46 lower risk MDS patients, SF3B1MUT was present in 25, including 20 with MDS-SLD-RS/MDS-MLD-RS, 3 with MDS-MLD, and 2 with MDS-EB1. By fluorescent PCR, ERFE+12 was detected in the BM MNC fractions of all SF3B1MUT MDS. ERFE+12 was not detected in any other cases of MDS with mutations in SRSF2 (n=10), U2AF1 (n=1), or ZRSR2 (n=1), or in 9 MDS patients with mutations in other genes, as shown in Fig. 3C and table S4. ERFEWT and ERFE+12 mRNA amounts measured by RT-qPCR were up-regulated in SF3B1MUT samples compared to any SF3B1WT samples with other splicing or epigenetic mutations or without recurrent mutation (Fig. 3D, table S4). In one patient with an SF3B1 monoallelic deletion and a G742D substitution on the remaining allele, the expression of alternative ERFE+12 exceeded that of the canonical ERFEWT (fig. S4). We then amplified primary erythroblasts from the BM CD34+ cells of two patients with SF3B1MUT and one patient with SF3B1WT MDS and showed that ERFE+12 was expressed in SF3B1MUT but not in SF3B1WT erythroid cells (Fig. 3E). This further supports the idea that ERFE+12 is related to SF3B1 mutation. SF3B1MUT MDS are characterized by the enrichment of the BM with erythroid cells. To investigate whether ERFE+12 could be detected in cells derived from erythroblastic BM with another genetic background, we collected samples from one patient with MDS-RS with 38% of BM erythroblasts, more than 15% of RS, a normal karyotype, and no SF3B1 mutation but one SRSF2 and one TET2 mutation, three patients with a congenital sideroblastic anemia due to an ALAS2 mutation/deletion or a GLRX5 mutation, one patient with a severe β-thalassemia, and one patient with SF3B1K700E MDS. By fluorescent PCR, ERFEWT was present, whereas ERFE+12 was not detectable in any sample except the SF3B1K700E MDS (Fig. 3F). This confirms that the onset of an aberrant ERFE+12 is dependent not on the amplification of the erythroid compartment or the presence of RS, but on the presence of a mutant SF3B1.
Translation of ERFE+12 into a variant protein that represses hepcidin
Human ERFE encodes a 354-aminoacid polypeptide. The addition of 12 nucleotides in ERFE mRNA generates an ERFE variant (further referred to as ERFEVPFQ) containing a valine-proline-phenylalanine-glutamine (VPFQ) insertion immediately upstream of the collagen domain (Fig. 4A & fig. S5A). To investigate whether the mutant ERFEVPFQ protein was produced in vivo, we prepared cell lysates of erythroblasts derived in culture from the BM CD34+ cells of patients with SF3B1MUT MDS. Through LC MS/MS protein identification, we obtained several peptide-matching propositions including the ALHELGVYYLPDAEGAFR peptide (fig. S5B) already reported in public databases (www.proteomicsdb.org/), and we identified a cryptic peptide VPFQFGLPGPPGPPGPQGPP GPIIPPEALLK corresponding to the VPFQ insertion at position 108 of ERFE (Fig. 4B). This confirms that the alternative ERFE+12 transcript is translated into ERFEVPFQ in SF3B1MUT erythroblasts.
Fig. 4: Identification of ERFEVPFQ peptide by mass spectrometry and hepcidin repression by recombinant ERFEVPFQ protein.
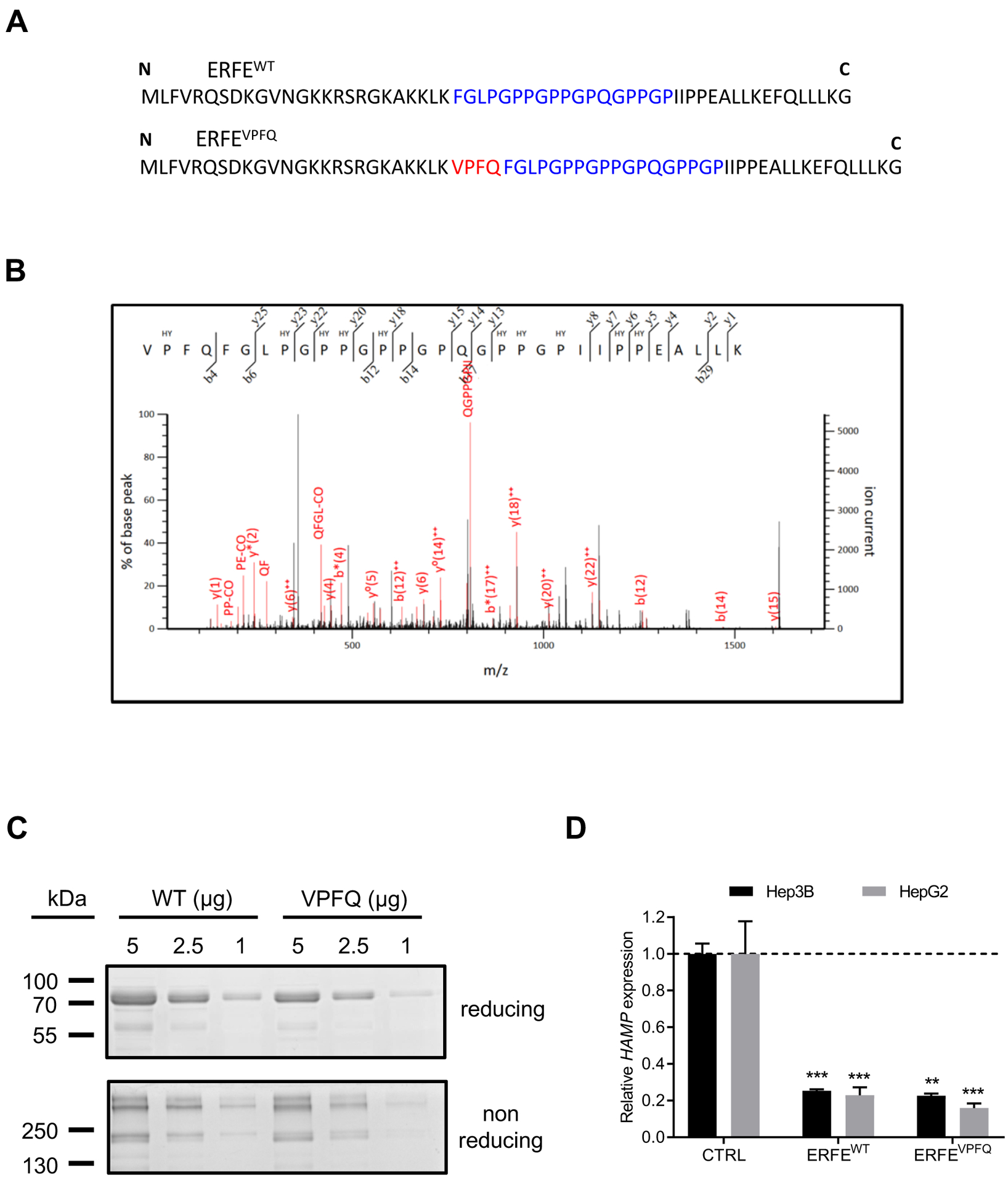
(A) Amino-acid sequence of ERFEWT and ERFEVPFQ peptides. VPFQ (red), collagen domain (blue). (B) Identification of a specific ERFEVPFQ peptide in erythroblast cell lysates by mass spectrometry using nano liquid chromatography coupled with a Q-Exactive Plus mass spectrometer. The calculated peptide mass was 3227.650782 from 3+ ion observed at m/z 1076.890870 with measured Δ = 3.6 ppm, Mascot Score = 17, expectation value = 0.069. HY: hydroxylated proline residues. (C) SDS-PAGE and Coomassie Blue staining of purified ERFEWT and ERFEVPFQ in reducing and non-reducing conditions. (D) Hep3B and HepG2 hepatocellular carcinoma cells were treated with 2 μg/ml of purified recombinant human ERFEWT or ERFEVPFQ for 16 hours. HAMP was quantified by RT-qPCR, and normalized to HPRT. Data shown are means ± SEM of three independent experiments and represent a fold change of hepcidin mRNA expression in ERFE-treated compared to untreated (CTRL) cells. Two-tailed Student t-test for P-values; *** P <0.0001; ** P <0.001.
ERFE represses hepcidin in mice and contributes to pathological hepcidin suppression in patients with transfused and non-transfused β-thalassemia (28, 29). Whether ERFEVPFQ also repressed hepcidin was tested. For this purpose, recombinant proteins ERFEWT and ERFEVPFQ were produced in HEK293F cells. SDS-PAGE analysis of ERFEWT and ERFEVPFQ in reducing and non-reducing conditions demonstrated that the insertion of 4 amino acids does not change the molecular weight of the protein or its multimerization pattern, such that both proteins were predominantly found in high molecular weight forms (>250 kDa) and in a trimeric form of ~200 kDa (Fig. 4C). Then, Hep3B and HepG2 hepatocellular carcinoma cells were treated for 16h with purified ERFEVPFQ or ERFEWT. Both proteins similarly caused a 5-fold reduction in the expression of HAMP mRNA encoding hepcidin compared to controls (Fig. 4D). These data indicate that insertion of 4 amino acids upstream of the collagen domain did not affect the bioactivity of the protein.
Prediction of hyperferritinemia by overproduction of erythroferrone in SF3B1MUT MDS
We then measured the plasma concentration of ERFE in the training cohort of 156 patients with MDS and 20 healthy non-blood donor controls using a validated immunoassay (29). We first verified that the ERFE immunoassay was able to detect both ERFEWT and ERFEVPFQ. Indeed, human ERFE ELISA detected similar amounts (1 μg/ml) of each recombinant protein in the supernatants of HEK293F cells transiently transfected with ERFEWT and ERFEVPFQ expression vectors. This established that both isoforms were detectable by ELISA. The mean concentration of ERFE in SF3B1MUT or SF3B1WT MDS was higher than in 20 non-blood donor healthy controls (P<0.0001; Fig. 5A). Among the MDS samples, the ERFE concentration was higher in SF3B1MUT (135.0±72.5 ng/ml) compared to SF3B1WT (62.1±36.7 ng/ml) MDS (P<0.0001; Fig. 5A). High concentrations of circulating ERFE correlated with high expression of ERFE+12 transcript (Pearson test; P<0.0001; fig. S6A). Consistently, the ERFE concentration was also higher in MDS-RS compared to all other WHO MDS subtypes (fig. S6B). Ferritin concentrations were significantly higher in SF3B1MUT patients compared to SF3B1WT patients (P<0.0001; Fig. 5B). We also measured plasma hepcidin and confirmed that the concentration of hepcidin in SF3B1MUT MDS was significantly lower compared to SF3B1WT MDS (P=0.031; Fig. 5C). We found no difference between SF3B1MUT MDS and healthy controls. The hepcidin/ferritin ratio was lower in SF3B1MUT than inSF3B1WT MDS (P<0.0001; Fig. 5D) and also in MDS-RS compared to other WHO subtypes (fig. S6C), resulting from both a lower concentration of hepcidin and a higher concentration of ferritin in SF3B1MUT patients. The plasma concentration of ERFE was inversely correlated to the hepcidin/ferritin ratio (Pearson test; P<0.0001; r=0.600; fig. S6D). Our analysis also highlights that an ERFE concentration above a threshold of 100 ng/ml repressed hepcidin more efficiently (fig. S6D). Serum erythropoietin (EPO) concentration was equally increased in SF3B1MUT and SF3B1WT patients compared to normal values (5 – 36 U/l; table S1), and although ERFE is regulated by erythropoietin in mice, we did not find any correlation between serum EPO and ERFE concentration (fig. S6E) (28). The increased concentration of plasma ERFE was associated with a more pronounced degree of ineffective erythropoiesis, as assessed by a significant increase in plasma concentration of soluble transferrin receptor (sTfR) in SF3B1MUT compared to SF3B1WT MDS patients (P<0.0001; fig. S6F).
Fig. 5: Increased plasma concentration of ERFE in SF3B1MUT MDS patients.
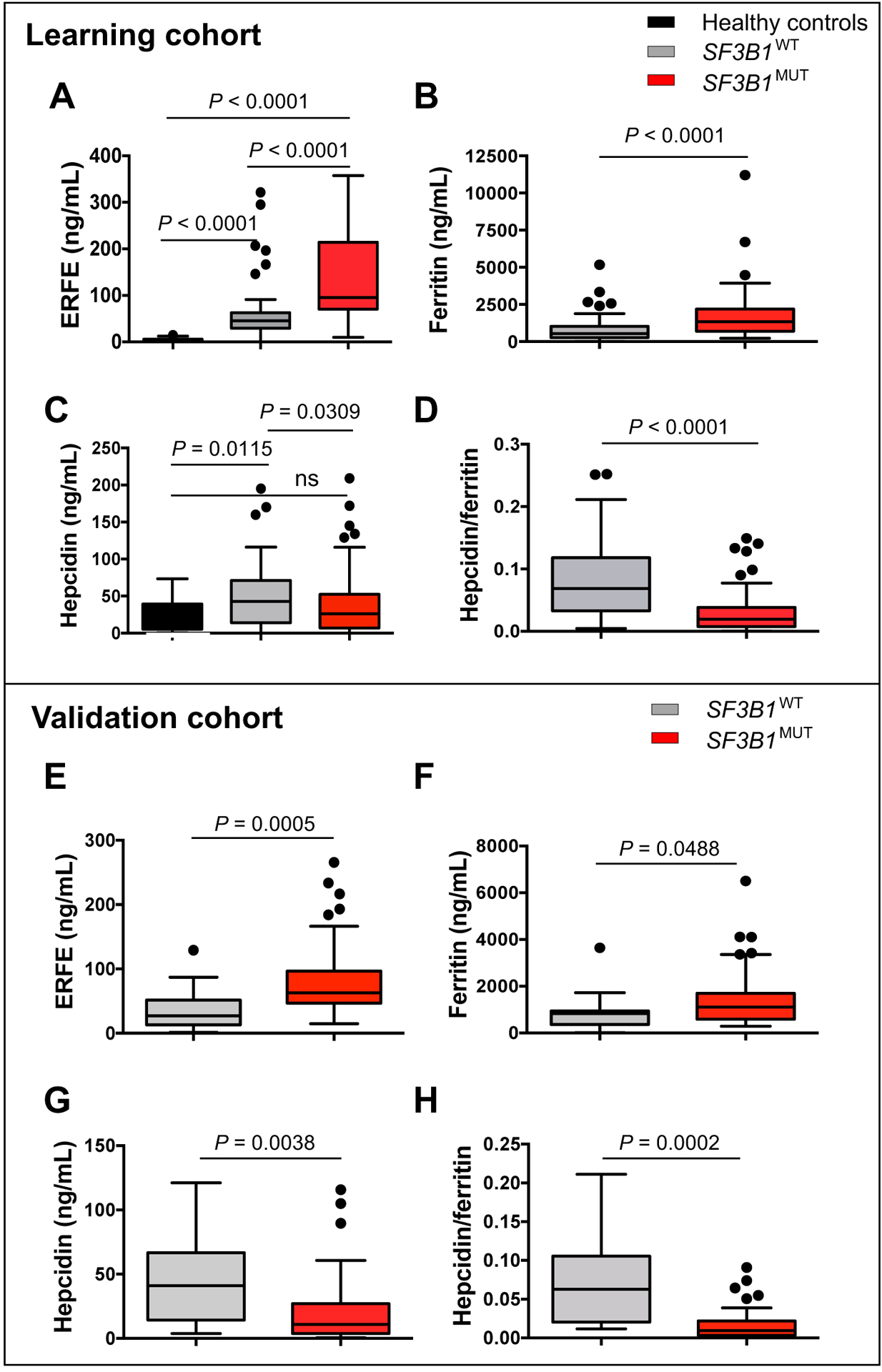
Quantitative analysis was performed in plasmas collected from 20 non-blood donor healthy volunteers (black), 156 patients with MDS including 94 SF3B1MUT (red) and 62 SF3B1WT (gray) representing the training cohort (A-D), and 55 patients with MDS including 42 SF3B1MUT (red) and 13 SF3B1WT (gray) representing the validation cohort (E-H). The graphs show quantification of erythroferrone (A, E), ferritin (B, F), hepcidin (C, G), and hepcidin/ferritin ratio (D, H). Results are expressed as medians and interquartile ranges (IQRs). The boxplots represent the median and the first and third quartiles, and the whiskers represent the lowest and the highest values still within the 1.5 IQR of the lower and upper quartiles. Mann-Whitney for P-values.
To validate these findings, we prospectively enrolled an external cohort of lower risk MDS patients in our study until the proportion of patients with SF3B1MUT MDS was comparable to that in the training cohort (table S5). This validation cohort consisted of 55 patients with MDS, 42 (76.3%) of whom had MDS with SF3B1 mutation. Notably, SF3B1MUT and SF3B1WT patients of this cohort received a similar transfusion burden with a mean number of 4 RBC units/8 weeks (table S5). The mean concentrations of ERFE and ferritin were significantly increased (P=0.0005 and P=0.0488, respectively; Fig. 5E, 5F), whereas hepcidin and hepcidin/ferritin ratio were significantly decreased in SF3B1MUT patients (P=0.0038 and P=0.0002, respectively; Fig. 5G, 5H). This confirms the results of the training cohort and suggests that the increase of ERFE concentration in SF3B1MUT MDS patients is independent of RBC transfusions.
Iron homeostasis changes after RBC transfusions, which ameliorate the anemia and increase the concentrations of circulating iron, with both effects expected to increase hepcidin. To assess the influence of RBC transfusion in our analysis, we delineated a subset of 61 patients with MDS in the training cohort, including 25 SF3B1MUT and 36 SF3B1WT patients with a low transfusion burden before inclusion (<4 RBC units per 8 weeks). In this subset, patients with SF3B1MUT or SF3B1WT MDS were equally transfused (mean 0.5 RBC unit/8 weeks), but ferritin and plasma iron concentrations remained higher in SF3B1MUT patients (fig. S7A, S7B). The hepcidin/ferritin and hepcidin/plasma iron ratios were significantly lower in SF3B1MUT patients (P<0.0001 and P=0.019, respectively; fig. S7C, S7D), and the circulating ERFE concentration remained significantly increased in low transfusion burden SF3B1MUT patients compared to SF3B1WT patients (P<0.0001; fig. S7E). Erythropoiesis-stimulating agents (ESA) are used as first line treatment of anemia, with low serum EPO and low transfusion burden as predictors of response (25). We investigated the impact of iron parameters on the response to epoetin ζ for 12 weeks in 59 patients with low-risk MDS included in a clinical trial of the Groupe Francophone des Myélodysplasies (GFM), GFM-Retacrit-2013 (NCT 03598582; table S6) (32). Plasma ERFE, ferritin, and hepcidin concentrations at enrollment were similar in responding and non-responding patients, suggesting that these parameters were not predictive of the erythropoietic response in this cohort (fig. S8). We then explored the determinants of hyperferritinemia >300 μg/ml in low transfusion burden patients. By univariate analysis, ERFE (P=0.005), hepcidin (P=0.013), and SF3B1 mutation (P=0.006), but not sTfR concentration or the number of transfused RBC units, were significantly linked to the concentration of serum ferritin (Table 1). By multivariate analysis, ERFE, hepcidin, and SF3B1 mutation remained independent predictors of high ferritin concentration (Table 1). Altogether, these results indicate that before patients reach a critical threshold of transfusion dependence, hyperferritinemia can be caused by SF3B1MUT-induced expression of ERFE, which in turn lowers hepcidin.
Table 1:
Erythroferrone, hepcidin and SF3B1 mutation as independent predictors of hyperferritinemia in low transfusion burden MDS patients. Erythroferrone, hepcidin, sTfR, number of RBC units per 8 weeks, and SF3B1 mutation were evaluated in the group of 60 patients receiving less than 4 RBC units per 8 weeks on the basis of ferritin with a cut-off value of 300 ng/ml. Parameters are indicated as means and 95% confidence intervals (95% CI) or ranges. For univariate analysis, Mann-Whitney and X2 tests were used to compare the variables between SF3B1MUT and SF3B1WT MDS. Multivariate logistic regression analysis was performed for variables with P-value <0.100 in univariate analysis.
| Parameters | Ferritin < 300 ng/ml | Ferritin ≥ 300 ng/ml | Univariate | Multivariate |
|---|---|---|---|---|
| n = 19 | n = 41 | P-value | P-value | |
| Erythroferrone (ng/ml), mean (95% CI) | 36.7 (26.8 – 46.6) | 72.7 (55.4 – 90.0) | 0.005 | 0.002 |
| Hepcidin (ng/ml), mean (95% CI) | 17.2 (11.2 – 23.2) | 35.9 (25.9 – 45.9) | 0.013 | <0.0001 |
| sTfR (ng/ml), mean (95% CI) | 1.21 (0.99 – 1.43) | 1.42 (1.18 – 1.65) | 0.484 | |
| Number of RBC units / 8 weeks, mean (range) | 0.2 (0 – 2) | 0.6 (0 – 3) | 0.104 | |
| SF3B1 mutation yes, n (%) | 3 (15.8) | 22 (53.6) | 0.006 | 0.023 |
Erythroid lineage-restricted expression of ERFE+12
In mice, ERFE mRNA expression in the BM is regulated by EPO and is predominant in basophilic and polychromatic erythroblasts (28). To investigate whether ERFE and ERFE+12 expression is restricted to the erythroid lineage, we amplified in parallel the erythroid and granulocytic precursors derived from the BM CD34+ cells of 1 SF3B1MUT and 1 SF3B1WT sample. The purity of each lineage was assessed by the cytological examination of May-Grünwald-Giemsa-stained cytospins and the quantification of lineage-restricted markers by RT-qPCR (Fig. 6A & fig. S9A). To compare the amount of each transcript isoform at the different stages of differentiation, we quantified the canonical ERFEWT and the aberrant ERFE+12 by RT-qPCR. The expression of the canonical transcript ERFEWT expressed as Normalized Ratio Quantities (NRQ) increased in both SF3B1MUT and SF3B1WT MDS erythroblasts. In granulocytes, the expression of ERFEWT was close to the limit of detection (Fig. 6B). The expression of ERFE+12 was restricted to the erythroid lineage, increased with the differentiation of SF3B1MUT erythroblasts, and was higher in SF3B1MUT compared to SF3B1WT erythroblasts (Fig. 6B). These results indicate that ERFE+12 expression is specific to the erythroid lineage.
Fig. 6: Erythroid cell-restricted expression of ERFE+12.
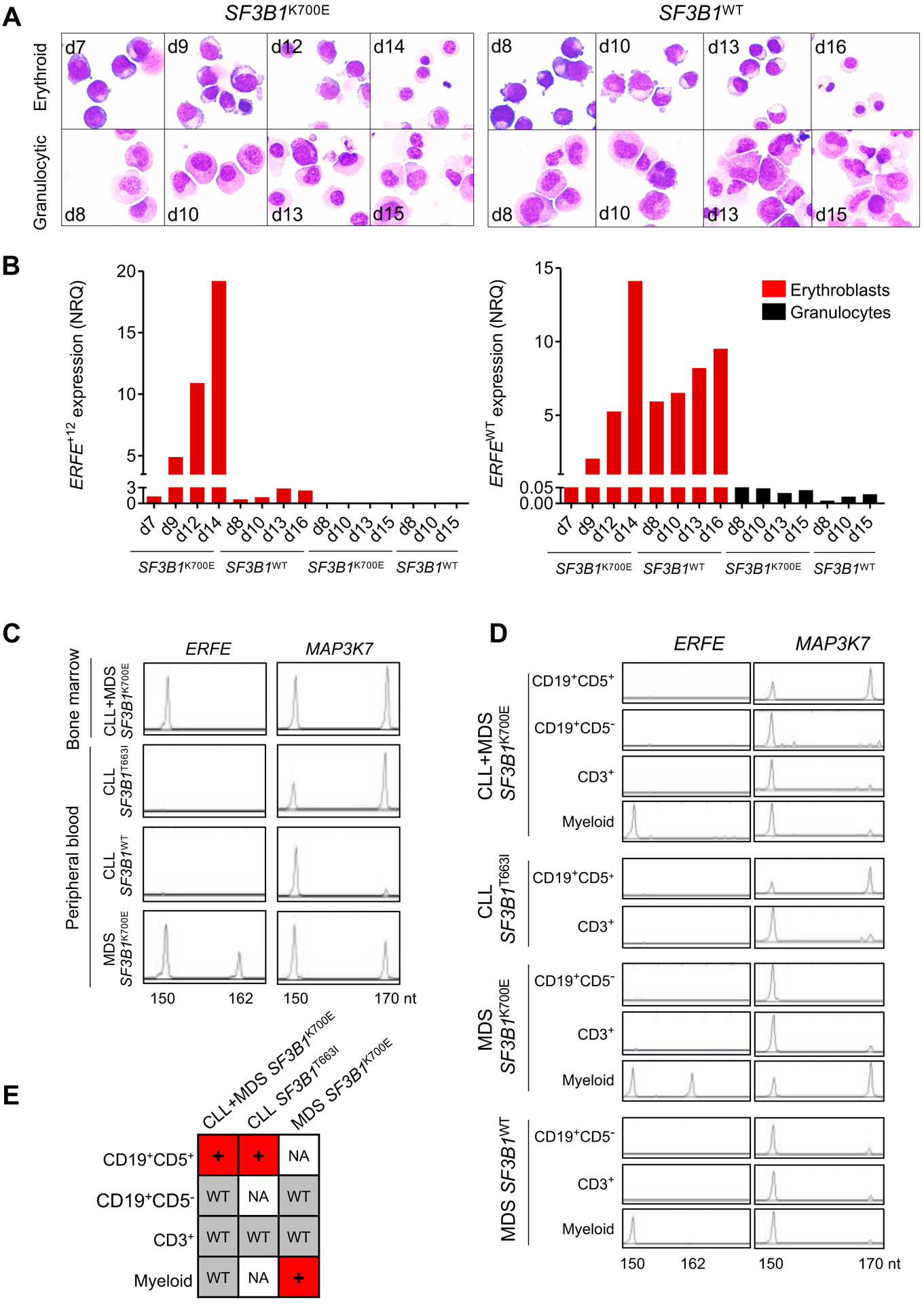
(A) May-Grünwald-Giemsa-stained cytospins of erythroid and granulocytic precursors obtained from liquid culture of BM SF3B1WT or SF3B1K700E CD34+ progenitors. (B) Quantification of ERFEWT and ERFE+12 in erythroid and granulocytic precursors by RT-qPCR. Results are expressed as NRQ ± SEM to ACTB and B2M housekeeping genes. (C) ERFE+12 is absent in SF3B1MUT CLL. BM MNCs from a patient with SF3B1K700E CLL+MDS or PB MNCs from patients with SF3B1T663I CLL, SF3B1WT CLL, and SF3B1K700E MDS were collected for analysis by capillary electrophoresis of ERFE and MAP3K7 fluorescent PCR products. ERFE+12 was detected as a 162-nt fragment and MAP3K7+20 as a 170-nt fragment. PB samples from one patient with an SF3B1WT CLL and one patient with an SF3B1MUT MDS are shown as controls. (D) ERFE+12 expression is restricted to SF3B1MUT myeloid lineage. CD19+CD5− B cells, CD19+CD5+ B CLL cells, CD3+ T cells, and myeloid cells were sorted from the BM MNC fraction of a SF3B1K700E CLL+MDS, a SF3B1K700E MDS, and a SF3B1WT MDS and from the PB MNCs of a SF3B1T663I CLL ERFE and MAP3K7 transcripts were analyzed by fluorescent PCR. (E) The sequencing of SF3B1 was performed on cDNA of each cell fraction. +: mutated; WT: wild type; NA: not available.
SF3B1 gene is mutated in 15% of patients suffering from CLL and the SF3B1MUT allele is present in CD19+ lymphocytes (14, 33). To investigate whether ERFE+12 was detectable in SF3B1MUT CLL, we collected one sample from a patient with CLL followed by MDS, whose BM MNC expressed a clonal SF3B1K700E mutation, and 3 peripheral blood (PB) MNC samples from 2 patients with CLL, one of whom harbored a clonal SF3B1T663I mutation with a variant allele frequency over 40% and the second had no mutation in SF3B1 gene, and from a patient with SF3B1K700E MDS. We analyzed the MAP3K7 transcript, which is alternatively spliced in SF3B1MUT CLL or MDS using a cryptic 3’ss (14, 16). A 170 nt fragment corresponding to the alternative MAP3K7 transcript was enriched in all SF3B1MUT samples compared to the SF3B1WT CLL (Fig. 6C). ERFE+12 was detected in the PB MNC of the SF3B1K700E MDS, but not in the SF3B1T663I or SF3B1WT CLL. ERFE+12 was not detected in BM MNC of the SF3B1K700E CLL+MDS sample. To get further insights on the cell types expressing ERFE+12, we sorted CD19+CD5− B cells, CD19+CD5+ pathological B cells, and CD3+ T cells and myeloid cells containing erythroblasts from the BM MNC fraction of the patients with SF3B1K700E CLL+MDS, SF3B1K700E MDS, and SF3B1WT MDS and from the PB MNC of the patient with SF3B1T663I CLL (fig. S9B). The number of cells in the myeloid fraction of SF3B1T663I CLL was too small for further studies. The 170 nt fragment of MAP3K7 was detected in CD19+CD5+ pathological B cells of SF3B1K700E CLL+MDS and SF3B1T663I CLL and also in the myeloid fraction, but not in the CD19+CD5− B cells of SF3B1K700E MDS (Fig. 6D). By sequencing SF3B1 RNA, we demonstrated that the mutation was present in the cell populations in which the alternative MAP3K7 transcript was detected (Fig. 6E). The alternative ERFE+12 transcript was not detected in the CD19+CD5+ pathological B cells of SF3B1K700E MDS+CLL or SF3B1T663I CLL, and its expression was restricted to SF3B1K700E myeloid MDS cells. Altogether, these results indicate that ERFE is expressed in erythroid cells and ERFE+12 is restricted to SF3B1MUT MDS myeloid cells.
Correlation between changes in ERFE+12 expression and the response to lenalidomide
Fifty percent of lower risk MDS patients, including patients with MDS-RS, experience primary resistance or secondary failure to treatments they receive to cure their anemia. Whether the mechanism of resistance involves the persistence of clonal erythropoiesis is always unknown. We have previously shown that lenalidomide administered to ESA-resistant non-del(5q) MDS patients targets the malignant clone and in some cases, eliminates the dominant SF3B1MUT clone for the duration of response (34). However, the frequency of the SF3B1 variant allele, which is expressed in erythroid and myeloid cells, does not only reflect the abundance of clonal erythroblasts. Here, we retrospectively monitored the expression of erythroid-specific ERFE+12 transcript for the follow-up of patients included in clinical trials of the GFM: GFM-Retacrit-2013 and GFM-LenEpo-2008 ((32,35). For this purpose, we performed a fluorescent PCR and integrated ERFE+12 and ERFEWT peak heights as a ratio ERFE+12/ERFE+12+ERFEWT in SF3B1MUT MDS patients. Then, the ratio was measured in paired RNA samples at enrollment and first evaluation. In GFM-Retacrit-2013, 4 non-responders and 6 responders were compared, and no significant variation was observed (Fig. 7A, fig. S10A). By contrast, in GFM-LenEpo-2008, the ratio ERFE+12/ERFE+12+ERFEWT decreased in 6 responding patients but remained stable in 8 non-responding patients (Fig. 7B, fig. S10B). The percent variation of ratios between samples obtained during screening and after 4 cycles of treatment was significantly different between responding and non-responding patients (Mann-Whitney test P=0.0013; Fig. 7B, right). The percent variation of SF3B1 variant allele frequency in BM mononuclear cells between screening and evaluation after 4 cycles was not significantly different between non-responding and responding patients (fig. S10C). This confirms that lenalidomide may target clonal erythropoiesis. By contrast, ERFE protein quantities did not vary with the response in both cohorts (fig. S11). Finally, we addressed the prognostic value of the ERFE+12/ERFE+12+ERFEWT ratio for overall survival (OS) in a cohort of 90 patients with low-risk MDS, including 24 patients with SF3B1MUT and 66 with SF3B1WT enrolled at diagnosis with a median follow-up of 36.9 months (table S7). In this cohort, a Receiver Operating Characteristic (ROC) analysis established that a value of 0.008 was the threshold of positivity of the ERFE+12/ERFE+12+ERFEWT ratio with a specificity and sensitivity of 100%. As shown in Fig. 7C, an ERFE+12/ERFE+12+ERFEWT ratio >0.008, was predictive of better OS (log-rank test; P=0.019) and was correlated with the presence of an SF3B1 mutation Among patients expressing the variant ERFE+12 transcript, there was no significant correlation between ERFE+12/ERFE+12+ERFEWT ratio and OS (log-rank test; P=0.064; fig. S12). Our results suggest that ERFE+12 expression may correlate with OS for SF3B1MUT MDS patients.
Fig. 7: ERFE+12 expression as a marker of clonal erythropoiesis and survival.
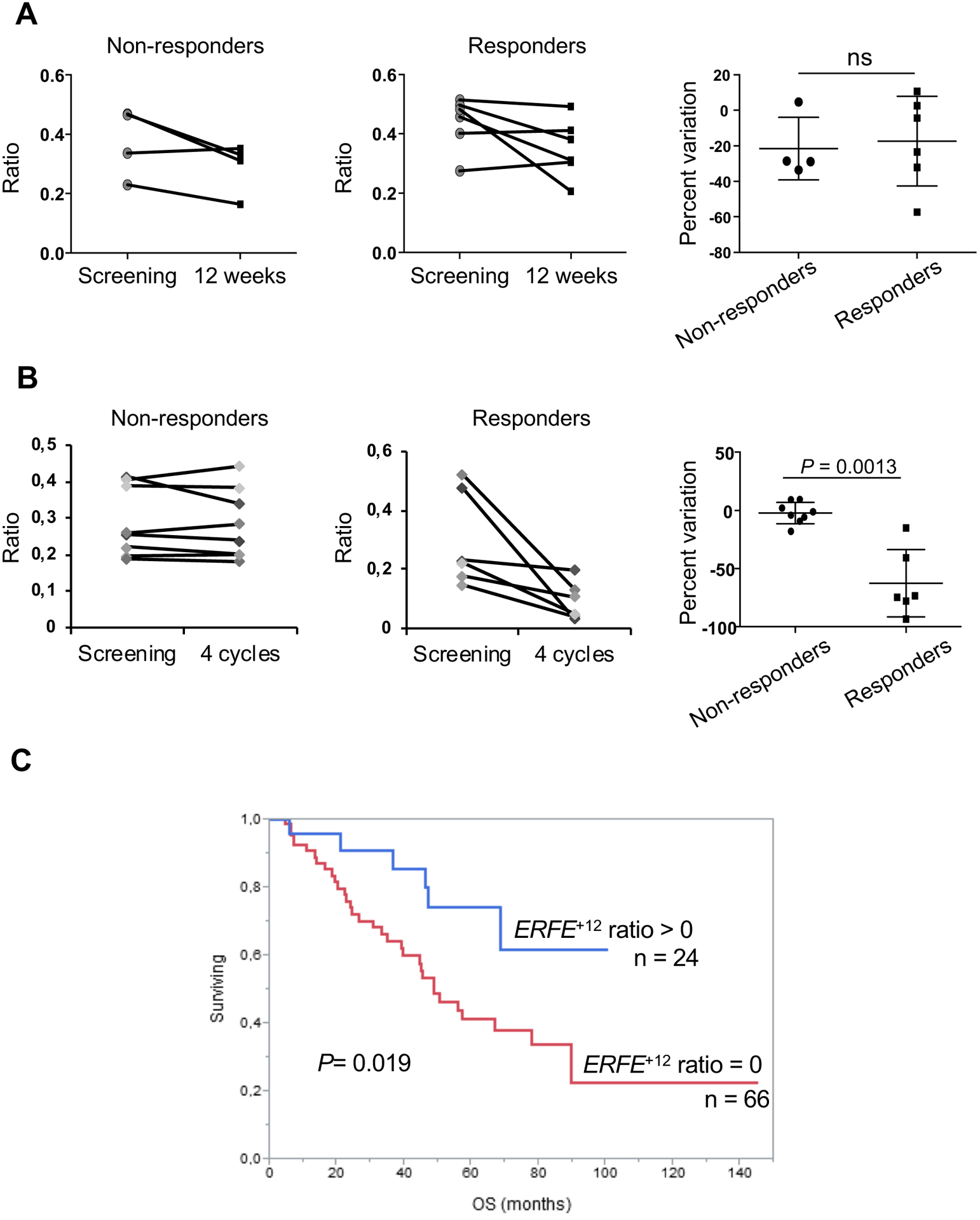
Fluorescent PCR was performed at screening and evaluation in (A) 10 paired samples from SF3B1MUT MDS patients enrolled in the GFM-Retacrit-2013 clinical trial (4 non-responding and 6 responding patients) and (B) 14 paired samples from patients with SF3B1MUT MDS enrolled in the GFM-LenEpo-2008 clinical trial (8 non-responding and 6 responding patients). Peak heights of ERFE+12 and ERFEWT signals were integrated as ERFE+12/ERFE+12+ERFEWT ratios. Percent variations of ratios are indicated (right) as medians and IQR (25 – 75%). Mann-Whitney test for P-values. (C) Overall survival according to ERFE+12/ERFE+12+ERFEWT ratio shown as a Kaplan-Meier curve. A threshold of positivity of 0.008 was determined by ROC analysis. Log-Rank test for P-value. ns: not significant.
Discussion
In this study, we show that in patients with SF3B1MUT MDS, an alternative FAM132B/ERFE+12 transcript is translated into an ERFEVPFQ protein and, together with the canonical transcript, contributes to the overexpression of ERFE. Similarly to ERFE, the ERFEVPFQ protein efficiently represses hepcidin. ERFE+12 is specifically induced by the clonal erythropoiesis.
In SF3B1MUT cancers, alternative 3’ss usages are the most frequent splicing aberrancies (11, 13, 14, 36). The recently resolved crystal structure of SF3b complex helps understanding this feature (37). In the spliceosome, SF3b complex interacts with the pre-mRNA and is involved in the branch site selection during splicing. Mutations in SF3B1 affect the structure of SF3b RNA-binding platform, resulting in the selection of alternative branch site and alternative transcripts, of which more than 50% are subjected to NMD (11, 13, 37). Based on these findings, we focused our attention on the transcripts generated by the use of an alternative AG’. The analysis of the distance separating AG from AG’ revealed strong peaks at 15, 18, and 21 nucleotides, showing that inserts could be multiples of 3 nucleotides. Among alternative in-frame junctions, we identified two regulators of iron homeostasis, ABCB7 and ERFE. As already reported, the alternative ABCB7 mRNA results from the addition of 21 nucleotides between exon 8 and 9 and, in our sample set, was slightly down-regulated in SF3B1MUT compared to SF3B1WT MDS (11, 15, 16). By contrast, the aberrant ERFE transcript modified by the addition of 12 nucleotides was up-regulated 2.3 fold in SF3B1MUT MDS. ERFE+12 has not been reported in other SF3B1MUT cancers despite some overlap between the sets of alternative 3’ss transcripts in SF3B1MUT uveal melanoma, CLL, and MDS (11, 13, 14, 38, 39). We showed the existence of a variant protein containing VPFQ sequence immediately upstream of the collagen domain putatively involved in protein-protein interactions in other CTRP family members (40). VPFQ may not influence protein conformation, because the apparent molecular weight was similar for recombinant ERFEWT and ERFEVPFQ proteins, which both repressed HAMP gene expression. However, we could not exclude an effect of the VPFQ insertion on protein stability and/or still unknown functions.
In MDS, serum EPO is increased but ineffective in stimulating erythropoiesis because maturing erythroid cells undergo apoptosis in terminal phases of differentiation (1, 2). Increased EPO and sTfR concentrations have been inversely correlated with hepcidin concentrations in MDS (19, 20, 41, 42). Our study indicates that plasma hepcidin concentration in SF3B1MUT MDS is similar to healthy non-blood donor controls and inappropriately low compared to SF3B1WT MDS, suggesting that its production could be dysregulated by severe ineffective erythropoiesis and/or a more specific mechanism (19). We showed the cell-autonomous SF3B1MUT-dependent expression of ERFE+12 and a role for increased concentrations of circulating ERFE in hepcidin reduction. This confirms the involvement of ERFE in human pathologies (29).
ERFE is the major erythroid regulator of hepcidin (28). Other candidate regulators of hepcidin linked to erythropoiesis have been proposed, including EPO and GDF-15. Although EPO administration decreases hepcidin concentration (43), this effect is predominantly indirect. The other candidate, GDF-15, is poorly induced by EPO in human volunteers. Although serum GDF-15 concentration was increased in β-thalassemia or congenital dyserythropoietic anemia, it was not inversely correlated with hepcidin in MDS (19, 26, 44). Low hepcidin preserves ferroportin on enterocytes and macrophages, causing increased iron intestinal absorption and macrophage release (45, 46). Compared to SF3B1WT MDS, the lower concentrations of hepcidin in SF3B1MUT MDS may explain early iron overload, before patients receive erythrocyte transfusions (19, 20). In patients with low transfusion burden, who had received a mean of 0.5 RBC unit, hepcidin, ERFE and SF3B1 mutation were independent predictors of hyperferritinemia. This demonstrates that iron overload is strongly related to the control of hepcidin through the SF3B1-regulated production of ERFE. Hepcidin increases with transfusion intensity in patients with MDS or β-thalassemia because of exogenous iron loading and transient suppression of ineffective erythropoiesis (29, 47, 48). Here, in regularly transfused SF3B1MUT MDS patients, hepcidin remained lower and ERFE higher than in SF3B1WT MDS patients. This establishes a driver role for SF3B1 mutation in the stimulation of ERFE expression and systemic iron overload that characterized patients.
The SF3B1 mutation confers a good prognosis in MDS (6, 49). However, when patients are transfusion-dependent, iron overload in cardiac and liver tissues becomes clinically evident and may impair life expectancy (50). Chelation efficiently reduces iron burden in regularly transfused MDS patients and serves as an adjuvant therapy for anemia, because deferasirox may improve hematopoiesis by protecting against oxidative stress (51–53). Promising substitutes for ESAs in resistant patients include lenalidomide, which may transiently reduce SF3B1 mutant allele burden, and activin receptor ligand traps such as luspatercept, which promotes late-stage erythropoiesis in mouse models of β-thalassemia and MDS and diminishes iron overload in β-thalassemia mice (34, 54–56). In lenalidomide-treated patients, but not in epoetin ζ-treated patients, the kinetics of ERFE+12 expression correlated with the response to treatment, indicating that in responding patients lenalidomide targeted clonal erythroid precursors either directly or indirectly. Of note, changes in plasma ERFE protein after lenalidomide were not correlated with the response to these treatments. The impact of transfusions on the regulation of ERFE expression at the post-transcriptional level should be examined in the future. ERFE+12 expression could also be useful for disease surveillance of patients with MDS-RS treated with other drugs, such as luspatercept. Further clinical studies will be required to validate ERFE+12 expression as a therapy-responsive biomarker of ineffective erythropoiesis in patients with SF3B1MUT MDS.
Finally, our findings open therapeutic avenues for preventing iron accumulation in MDS-RS patients. Increasing hepcidin could produce therapeutic benefits in SF3B1MUT MDS, because a moderate increase in hepcidin decreased iron and improved anemia in β-thalassemia mice (57). Therefore, either the administration of a hepcidin agonist or the targeting of erythroferrone overexpression may provide a potential strategy for preventing iron overload and improving erythropoiesis in SF3B1MUT MDS patients.
Materials and Methods
Study design
The study involved identifying a splicing variant of ERFE by RNA-sequencing human primary BM samples and the variant peptide by mass spectrometry. Each in vitro experiments transfection, minigene assay, degron-KI, transcriptional repression of hepcidin was repeated three times. Each experiment using primary cells was performed with at least three samples in each category. A ratio of ERFE variant transcript to total transcript was validated as a marker of clonal erythropoiesis based on the response to lenalidomide or ESA in two cohorts of MDS patients enrolled in clinical trials and as a prognostic marker of overall survival in a prospective multicenter cohort of MDS patients. Predictive value of plasma erythroferrone and hepcidin concentrations for hyperferritinemia was measured in training and validation cohorts of MDS patients. For these studies, randomization or blinding were not applicable
Patients
For the training cohort, MDS patients (n=156) were enrolled between 2008 and 2017 (ClinicalTrials.gov: GFM-LenEpo-2008, NCT01718379; GFM-Retacrit-2013, NCT 03598582). For the GFM-Retacrit-2013 cohort, the status of response to epoetin ζ was recorded. The prospective cohort of 90 MDS patients with survival data was enrolled between 2010 and 2018. BM aspirates and PB plasma samples were collected after each patient gave informed consent for biological investigations according to the recommendations of the institutional review boards (IRB): IdF X GFM-LenEpo-08 EudraCT 2008-008262-12; IdFII 2010-A00033-36; IdFIII: 2010-2753; IdFV 212-A01395-38 EudraCT 2012-002990-7338; OncoCCH 2015-08-11-DC). For the validation cohort, MDS patients (n=55) were enrolled prospectively for plasma collection in France (5 centers; IRB Onco-CCH 2015-08-11DC) and Germany (one center; IRB Ethikkommission an der TU Dresden; EK 115032015) between 2016 and 2018. BM samples from 5 age-matched controls and PB plasma samples from 20 healthy controls were collected. Patient characteristics are indicated in tables S1, S5, and S7 and according to the response to epoetin ζ in table S6. Low transfusion burden was defined as <4 RBC units per 8 weeks.
DNA and RNA-sequencing
Mutations in a panel of 26 genes were screened by next-generation sequencing (NGS) (fig. S1). RNA was sequenced on an Illumina HiSeq 2500 platform using a 100-bp paired-end sequencing strategy. TopHat (v2.0.6) was used to align the reads against the human reference genome Hg19 RefSeq (RNA sequences, GRCh37) downloaded from the UCSC Genome Browser (http://genome.ucsc.edu). Analyses of differential gene expression analysis and differential junction read counts were performed using DESeq2 (13, 30).
CRISPR/Cas9 generation of isogenic Sf3b1K700E and Sf3b1WT cell lines
The murine erythroid cell line G1E-ER4 (58) was used to generate isogenic Sf3b1K700E and Sf3b1WT cell lines using CRISPR/Cas9-stimulated homology-mediated repair.
ERFE and ENOSF1 minigenes
The ERFE minigene was synthesized by insertion of the ERFE alternative junction in pET01 Exontrap vector (Mobitec). G1E-ER4 9.2 (SF3B1WT) and G1E-ER4 5.13H (SF3B1K700E) cells were transfected and processed for fluorescent PCR.
Mass spectrometry analysis
Erythroblasts were lysed and peptides were obtained by trypsin digestion (Promega) and analyzed by nano liquid chromatography coupled with a Q-Exactive Plus mass spectrometer (Thermo). Data were analyzed using Mascot 2.5.1 (www.matrixscience.com).
Human erythroferrone quantification
Plasma erythroferrone concentration was determined as previously described (29).
Statistical analysis
For quantitative variables, values were compared using the Mann-Whitney test. Categorical variables were compared using X2 or Fisher’s exact tests. P-values<0.05 were considered significant. Receiver operating characteristic (ROC) analysis was used to calculate the thresholds of positivity. Multivariate logistic regression analysis was adjusted for selected variables chosen with a P-value<0.1 in univariate analysis (JMP version 10.0.2, SAS Institute Inc.).
Supplementary Material
Materials and methods
Fig. S1: Genomic characteristics of the training cohort of 156 MDS patients.
Fig. S2: Identification of in-frame alternative transcripts in SF3B1MUT MDS samples by RNA-sequencing.
Fig. S3: Allele-specific investigation of mutant SF3B1 causal relationship to aberrantly spliced ERFE+12 transcript using Degron-KI strategy.
Fig. S4: Massive use of FAM132B/ERFE cryptic junction in one MDS-RS with a bi-allelic alteration of SF3B1 gene.
Fig. S5: Identification of ERFE peptide by LC tandem mass spectrometry.
Fig. S6: Correlation of plasma ERFE concentrations with ERFE+12 transcript, WHO classification and iron homeostasis parameters
Fig. S7: Biological parameters of low transfusion burden MDS patients of the training cohort.
Fig. S8: Plasma ERFE, ferritin and hepcidin concentrations at enrollment in 59 patients with MDS according to the response to EPO.
Fig. S9: Erythroid-specific expression of ERFE variant transcript.
Fig. S10: Variations of ERFE+12 transcript expression SF3B1MUT low risk MDS after therapy.
Fig. S11: Variations of plasma concentration of ERFE protein in SF3B1MUT low risk MDS after therapy.
Fig. S12: Kaplan-Meier curve for the analysis of overall survival according to ERFE+12/ERFEWT+ERFE+12 ratio.
Table S1: Clinical and biological characteristics of the training cohort of 156 MDS patients according to SF3B1 status.
Table S2: Differential expression of ERFE transcripts according to the mutational status of SF3B1, TET2 and DNMT3A genes by RNA-seq.
Table S3: Primer sequences used for fluorescent PCR and RT-qPCR.
Table S4: Expression of total ERFE, ERFEWT and ERFE+12 transcripts by fluorescent PCR and RT-qPCR in 9 MDS bone marrow samples.
Table S5: Clinical and biological characteristics of the validation cohort of 55 MDS patients according to SF3B1 status.
Table S6: Clinical and biological characteristics of the cohort of 59 MDS patients of the GFM-Retacrit-2013 cohort according to response status.
Table S7: Clinical and biological characteristics of the cohort of 90 MDS patients with survival data according to SF3B1 and ERFE+12 status.
Data file S3: In-frame and differentially expressed 3’ cryptic splice site junctions in SF3B1MUT MDS
Data file S2: Differentially expressed 5’ and 3’ junctions between SF3B1MUT and SF3B1wT MDS patients by RNA-seq.
Data file S1: Differentially expressed transcripts between SF3B1MUT and SF3B1WT MDS patients by RNA-seq.
Acknowledgements
The authors want to thank Dr F. Pflumio (INSERM UMR967, CEA/DSV/iRCM, Fontenay-aux-Roses), Dr N. Taylor (Institut de Génétique Moléculaire, Montpellier) and Dr S. Vaulont (Institut Cochin), Dr E-F Gautier (3P5 proteomic platform), Dr J. Vinh (Ecole Supérieure de Physique et de Chimie Industrielles, Paris), B. Saint-Pierre (Institut Cochin) and D. Drubay (Institut Gustave Roussy) for fruitful discussions; Dr J-B Arlet, Service de Médecine Interne, Hôpital Européen Pompidou, Dr C. Kannengiesser, Service de Génétique, Hôpital Bichat, for providing samples; Dr F. Letourneur (Genom’IC, Institut Cochin), Dr S. Baulande (sequencing platform, Institut Curie), L. Zaroili, M. Dejean, A. Marcon, V. Verjus-Lisfranc (Cochin laboratory of hematology), E. Benana (3P5 platform) and B. Billoré (INSERM U1220) for technical assistance, Pr M. Weiss, St Jude Children’s research hospital (Memphis, TN) and Dr V. Paralkar, University of Pennsylvania (Philadelphia, PA) for providing the G1E-ER4 cell line and expert comments.
Funding: This study was funded by INSERM, by the Institut National du Cancer INCa PLBio 2015 (INCa_9290), by INCa and the Direction Générale de l’Offre de Soins (DGOS) of the French Ministry of Social Affairs and Health through the Programme Hospitalier de Recherche Clinique (PHRC MDS-04; INCa-DGOS_5480) and by the Site de Recherche Intégrée sur le Cancer (SIRIC) CAncer Research for PErsonalized Medicine CARPEM. CL and DR are the recipients of a salary funded by the Laboratory of Excellence on red cells GR-Ex. The Orbitrap Fusion MS was acquired with funds from the FEDER and “Canceropole Ile de France”. LK received a support from ANR-16-ACHN-0002-01 and from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant agreement N0 715491). TG received funding from NIH by R01 DK 065029 (and the UCLA Center for Accelerated Innovation, under NIH grant U54HL119893 (Palazzolo).
Footnotes
Competing interests: T.G., L.K., and E.N. are inventors on a patent application on ERFE. T.G. and E.N. are scientific founders of Intrinsic LifeSciences and Silarus Pharma, companies that have interests related to ERFE. M.F., O.K., L.K., F.G., M-H.S., S.A., A.H. are inventors on a patent application on variant ERFE. The other authors have no competing interests to disclose.
Data and materials availability: RNA-seq data are available in the Gene Expression Omnibus (GEO) repository (accession number GSE113433). Material transfer agreement with H3 Biomedicine Inc. (Dr S Buonamici) is required to obtain synthetic full length SF3B1WT or mutant SF3B1K700E cDNAs. All other data associated with this study are present in the paper or the Supplementary Materials.
References and notes
- 1.Tehranchi R, Invernizzi R, Grandien A, Zhivotovsky B, Fadeel B, Forsblom A-M, Travaglino E, Samuelsson J, Hast R, Nilsson L, Cazzola M, Wibom R, Hellström-Lindberg E, Aberrant mitochondrial iron distribution and maturation arrest characterize early erythroid precursors in low-risk myelodysplastic syndromes, Blood 106, 247–253 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Gyan E, Frisan E, Beyne-Rauzy O, Deschemin J-C, Pierre-Eugene C, Randriamampita C, Dubart-Kupperschmitt A, Garrido C, Dreyfus F, Mayeux P, Lacombe C, Solary E, Fontenay M, Spontaneous and Fas-induced apoptosis of low-grade MDS erythroid precursors involves the endoplasmic reticulum, Leukemia 22, 1864–1873 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Zhu Y, Li X, Chang C, Xu F, He Q, Guo J, Tao Y, Liu Y, Liu L, Shi W, SF3B1-mutated myelodysplastic syndrome with ring sideroblasts harbors more severe iron overload and corresponding over-erythropoiesis, Leukemia Research 44, 8–16 (2016). [DOI] [PubMed] [Google Scholar]
- 4.de Swart L, Reiniers C, Bagguley T, van Marrewijk C, Bowen D, Hellström-Lindberg E, Tatic A, Symeonidis A, Huls G, Cermak J, van de Loosdrecht AA, Garelius H, Culligan D, Macheta M, Spanoudakis M, Panagiotidis P, Krejci M, Blijlevens N, Langemeijer S, Droste J, Swinkels DW, Smith A, de Witte T, Labile plasma iron levels predict survival in patients with lower-risk myelodysplastic syndromes, Haematologica 103, 69–79 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawahata R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Ishiyama K, Mori H, Nolte F, Hofmann W-K, Miyawaki S, Sugano S, Haferlach C, Koeffler HP, Shih L-Y, Haferlach T, Chiba S, Nakauchi H, Miyano S, Ogawa S, Frequent pathway mutations of splicing machinery in myelodysplasia, Nature 478, 64–69 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, Godfrey AL, Rapado I, Cvejic A, Rance R, McGee C, Ellis P, Mudie LJ, Stephens PJ, McLaren S, Massie CE, Tarpey PS, Varela I, Nik-Zainal S, Davies HR, Shlien A, Jones D, Raine K, Hinton J, Butler AP, Teague JW, Baxter EJ, Score J, Galli A, Della Porta MG, Travaglino E, Groves M, Tauro S, Munshi NC, Anderson KC, El-Naggar A, Fischer A, Mustonen V, Warren AJ, Cross NCP, Green AR, Futreal PA, Stratton MR, Campbell PJ, Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium, Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts, N. Engl. J. Med 365, 1384–1395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Damm F, Kosmider O, Gelsi-Boyer V, Renneville A, Carbuccia N, Hidalgo-Curtis C, Della Valle V, Couronné L, Scourzic L, Chesnais V, Guerci-Bresler A, Slama B, Beyne-Rauzy O, Schmidt-Tanguy A, Stamatoullas-Bastard A, Dreyfus F, Prébet T, de Botton S, Vey N, Morgan MA, Cross NCP, Preudhomme C, Birnbaum D, Bernard OA, Fontenay M, Groupe Francophone des Myélodysplasies, Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes, Blood 119, 3211–3218 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Mian SA, Rouault-Pierre K, Smith AE, Seidl T, Pizzitola I, Kizilors A, Kulasekararaj AG, Bonnet D, Mufti GJ, SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment, Nature Communications 6, 10004 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chesnais V, Arcangeli M-L, Delette C, Rousseau A, Guermouche H, Lefevre C, Bondu S, Diop M, Cheok M, Chapuis N, Legros L, Raynaud S, Willems L, Bouscary D, Lauret E, Bernard OA, Kosmider O, Pflumio F, Fontenay M, Architectural and functional heterogeneity of hematopoietic stem/progenitor cells in non-del(5q) myelodysplastic syndromes, Blood 129, 484–496 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Mortera-Blanco T, Dimitriou M, Woll PS, Karimi M, Elvarsdottir E, Conte S, Tobiasson M, Jansson M, Douagi I, Moarii M, Saft L, Papaemmanuil E, Jacobsen SEW, Hellström-Lindberg E, SF3B1-initiating mutations in MDS-RSs target lymphomyeloid hematopoietic stem cells, Blood 130, 881–890 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, Corson L, Feala J, Fekkes P, Ichikawa K, Keaney GF, Lee L, Kumar P, Kunii K, MacKenzie C, Matijevic M, Mizui Y, Myint K, Park ES, Puyang X, Selvaraj A, Thomas MP, Tsai J, Wang JY, Warmuth M, Yang H, Zhu P, Garcia-Manero G, Furman RR, Yu L, Smith PG, Buonamici S, Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3’ Splice Site Selection through Use of a Different Branch Point, Cell Rep 13, 1033–1045 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, McConkey ME, Ali AM, Raza A, Yu L, Buonamici S, Smith PG, Mullally A, Wu CJ, Fleming MD, Ebert BL, Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation, Cancer Cell 30, 404–417 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, Dutertre M, Stern M-H, Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage, Nat Commun 7, 10615 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Brooks AN, Fan J, Wan Y, Gambe R, Li S, Hergert S, Yin S, Freeman SS, Levin JZ, Fan L, Seiler M, Buonamici S, Smith PG, Chau KF, Cibulskis CL, Zhang W, Rassenti LZ, Ghia EM, Kipps TJ, Fernandes S, Bloch DB, Kotliar D, Landau DA, Shukla SA, Aster JC, Reed R, DeLuca DS, Brown JR, Neuberg D, Getz G, Livak KJ, Meyerson MM, Kharchenko PV, Wu CJ, Transcriptomic Characterization of SF3B1 Mutation Reveals Its Pleiotropic Effects in Chronic Lymphocytic Leukemia, Cancer Cell 30, 750–763 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nikpour M, Scharenberg C, Liu A, Conte S, Karimi M, Mortera-Blanco T, Giai V, Fernandez-Mercado M, Papaemmanuil E, Högstrand K, Jansson M, Vedin I, Stephen Wainscoat J, Campbell P, Cazzola M, Boultwood J, Grandien A, Hellström-Lindberg E, The transporter ABCB7 is a mediator of the phenotype of acquired refractory anemia with ring sideroblasts, Leukemia 27, 889–896 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dolatshad H, Pellagatti A, Liberante FG, Llorian M, Repapi E, Steeples V, Roy S, Scifo L, Armstrong RN, Shaw J, Yip BH, Killick S, Kušec R, Taylor S, Mills KI, Savage KI, Smith CWJ, Boultwood J, Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes, Leukemia 30, 2322–2331 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pellagatti A, Armstrong RN, Steeples V, Sharma E, Repapi E, Singh S, Sanchi A, Radujkovic A, Horn P, Dolatshad H, Roy S, Broxholme J, Lockstone H, Taylor S, Giagounidis A, Vyas P, Schuh A, Hamblin A, Papaemmanuil E, Killick S, Malcovati L, Hennrich ML, Gavin AC, Ho AD, Luft T, Hellström-Lindberg E, Cazzola M, Smith CWJ, Smith S, Boultwood J. Blood 132, 1225–1240 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shiozawa Y, Malcovati L, Gallì A, Sato-Otsubo A, Kataoka K, Sato Y, Watatani Y, Suzuki H, Yoshizato T, Yoshida K, Sanada M, Makishima H, Shiraishi Y, Chiba K, Hellström-Lindberg E, Miyano S, Ogawa S, Cazzola M. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat Commun 9, 3649 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santini V, Girelli D, Sanna A, Martinelli N, Duca L, Campostrini N, Cortelezzi A, Corbella M, Bosi A, Reda G, Olivieri O, Cappellini MD, Hepcidin Levels and Their Determinants in Different Types of Myelodysplastic Syndromes, PLOS ONE 6, e23109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ambaglio I, Malcovati L, Papaemmanuil E, Laarakkers CM, Porta MGD, Gallì A, Vià MCD, Bono E, Ubezio M, Travaglino E, Albertini R, Campbell PJ, Swinkels DW, Cazzola M, Inappropriately low hepcidin levels in patients with myelodysplastic syndrome carrying a somatic mutation of SF3B1, Haematologica 98, 420–423 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weinstein DA, Roy CN, Fleming MD, Loda MF, Wolfsdorf JI, Andrews NC, Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease, Blood 100, 3776–3781 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S, The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation, J Clin Invest 110, 1037–1044 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T, Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein, Blood 101, 2461–2463 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Pak M, Lopez MA, Gabayan V, Ganz T, Rivera S, Suppression of hepcidin during anemia requires erythropoietic activity, Blood 108, 3730–3735 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Porta MGD, Pascutto C, Travaglino E, Groves MJ, Godfrey AL, Ambaglio I, Gallì A, Vià MCD, Conte S, Tauro S, Keenan N, Hyslop A, Hinton J, Mudie LJ, Wainscoat JS, Futreal PA, Stratton MR, Campbell PJ, Hellström-Lindberg E, Cazzola M, on behalf of the C. M. D. W. G. of the I. C. G. C. and of the A. I. per la R. sul C. G. I. M. Mieloproliferative, Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms, Blood 118, 6239–6246 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanno T, Bhanu NV, Oneal PA, Goh S-H, Staker P, Lee YT, Moroney JW, Reed CH, Luban NL, Wang R-H, Eling TE, Childs R, Ganz T, Leitman SF, Fucharoen S, Miller JL, High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin, Nature Medicine 13, 1096–1101 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Tanno T, Porayette P, Sripichai O, Noh S-J, Byrnes C, Bhupatiraju A, Lee YT, Goodnough JB, Harandi O, Ganz T, Paulson RF, Miller JL, Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells, Blood 114, 181–186 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T, Identification of erythroferrone as an erythroid regulator of iron metabolism, Nature Genetics 46, 678–684 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ganz T, Jung G, Naeim A, Ginzburg Y, Pakbaz Z, Walter PB, Kautz L, Nemeth E, Immunoassay for human serum erythroferrone, Blood 130, 1243–1246 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biology 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Q, Derti A, Ruddy D, Rakiec D, Kao I, Lira M, Gibaja V, Chan H, Yang Y, Min J, Schlabach MR, Stegmeier F, A chemical genetics approach for the functional assessment of novel cancer genes. Cancer Res 75, 1949–1958 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Park S, Kosmider O, Maloisel F, Drenou B, Chapuis N, Lefebvre T, Karim Z, Puy H, Alary AS, Ducamp S, Verdier F, Bouilloux C, Rousseau A, Jacob MC, Debliquis A, Charpentier A, Gyan E, Anglaret B, Leyronnas C, Corm S, Slama B, Cheze S, Laribi K, Amé S, Rose C, Lachenal F, Toma A, Pica GM, Carre M, Garban F, Mariette C, Cahn JY, Meunier M, Herault O, Fenaux P, Wagner-Ballon O, Bardet V, Dreyfus F, Fontenay M, Dyserythropoiesis evaluated by the RED score and hepcidin:ferritin ratio predicts response to erythropoietin in lower-risk myelodysplastic syndromes. Haematologica 104, 497–504 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Damm F, Mylonas E, Cosson A, Yoshida K, Valle VD, Mouly E, Diop M, Scourzic L, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Kikushige Y, Davi F, Lambert J, Gautheret D, Merle-Béral H, Sutton L, Dessen P, Solary E, Akashi K, Vainchenker W, Mercher T, Droin N, Ogawa S, Nguyen-Khac F, Bernard OA, Acquired Initiating Mutations in Early Hematopoietic Cells of CLL Patients, Cancer Discov 4, 1088–1101 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Chesnais V, Renneville A, Toma A, Lambert J, Passet M, Dumont F, Chevret S, Lejeune J, Raimbault A, Stamatoullas A, Rose C, Beyne-Rauzy O, Delaunay J, Solary E, Fenaux P, Dreyfus F, Preudhomme C, Kosmider O, Fontenay M, Effect of lenalidomide treatment on clonal architecture of myelodysplastic syndromes without 5q deletion, Blood 127, 749–760 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toma A, Kosmider O, Chevret S, Delaunay J, Stamatoullas A, Rose C, Beyne-Rauzy O, Banos A, Guerci-Bresler A, Wickenhauser S, Caillot D, Laribi K, De Renzis B, Bordessoule D, Gardin C, Slama B, Sanhes L, Gruson B, Cony-Makhoul P, Chouffi B, Salanoubat C, Benramdane R, Legros L, Wattel E, Tertian G, Bouabdallah K, Guilhot F, Taksin AL, Cheze S, Maloum K, Nimuboma S, Soussain C, Isnard F, Gyan E, Petit R, Lejeune J, Sardnal V, Renneville A, Preudhomme C, Fontenay M, Fenaux P, Dreyfus F. Lenalidomide with or without erythropoietin in transfusion-dependent erythropoiesis-stimulating agent-refractory lower-risk MDS without 5q deletion. Leukemia 30, 897–905 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Shiozawa Y, Sato-Otsubo S, Gallì A, Yoshida K, Yoshizato T, Sato Y, Kataoka K, Sanada M, Shiraishi Y, Chiba K, Miyano S, Malcovati L, Cazzola M, Ogawa S, Comprehensive Analysis of Aberrant RNA Splicing in Myelodysplastic Syndromes, Blood 124, 826–826 (2014). [Google Scholar]
- 37.Cretu C, Schmitzová J, Ponce-Salvatierra A, Dybkov O, De Laurentiis EI, Sharma K, Will CL, Urlaub H, Lührmann R, Pena V, Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations, Molecular Cell 64, 307–319 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, Ramsay AJ, Beà S, Pinyol M, Martínez-Trillos A, López-Guerra M, Colomer D, Navarro A, Baumann T, Aymerich M, Rozman M, Delgado J, Giné E, Hernández JM, González-Díaz M, Puente DA, Velasco G, Freije JMP, Tubío JMC, Royo R, Gelpí JL, Orozco M, Pisano DG, Zamora J, Vázquez M, Valencia A, Himmelbauer H, Bayés M, Heath S, Gut M, Gut I, Estivill X, López-Guillermo A, Puente XS, Campo E, López-Otín C, Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia, Nature Genetics 44, 47–52 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Gentien D, Kosmider O, Nguyen-Khac F, Albaud B, Rapinat A, Dumont AG, Damm F, Popova T, Marais R, Fontenay M, Roman-Roman S, Bernard OA, Stern M-H, A common alternative splicing signature is associated with SF3B1 mutations in malignancies from different cell lineages, Leukemia 28, 1355–1357 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Seldin MM, Tan SY, Wong GW, Metabolic function of the CTRP family of hormones, Rev Endocr Metab Disord 15, 111–123 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brada SJL, de Wolf JT, Hendriks D, Louwes H, van den Berg E, Vellenga E, Characterization of the erythropoiesis in myelodysplasia by means of ferrokinetic studies, in vitro erythroid colony formation and soluble transferrin receptor, Leukemia 12, 340–345 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Metzgeroth G, Rosée PL, Kuhn C, Schultheis B, Dorn‐Beineke A, Hehlmann R, Hastka J, The soluble transferrin receptor in dysplastic erythropoiesis in myelodysplastic syndrome, European Journal of Haematology 79, 8–16 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Ashby DR, Gale DP, Busbridge M, Murphy KG, Duncan ND, Cairns TD, Taube DH, Bloom SR, Tam FWK, Chapman R, Maxwell PH, Choi P, Erythropoietin administration in humans causes a marked and prolonged reduction in circulating hepcidin, Haematologica 95, 505–508 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tamary H, Shalev H, Perez-Avraham G, Zoldan M, Levi I, Swinkels DW, Tanno T, Miller JL, Elevated growth differentiation factor 15 expression in patients with congenital dyserythropoietic anemia type I, Blood 112, 5241–5244 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicolas G, Viatte L, Lou D-Q, Bennoun M, Beaumont C, Kahn A, Andrews NC, Vaulont S, Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis, Nature Genetics 34, 97–101 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J, Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization, Science 306, 2090–2093 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Cui R, Gale RP, Zhu G, Xu Z, Qin T, Zhang Y, Huang G, Li B, Fang L, Zhang H, Pan L, Hu N, Qu S, Xiao Z, Serum iron metabolism and erythropoiesis in patients with myelodysplastic syndrome not receiving RBC transfusions, Leukemia Research 38, 545–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pasricha S-R, Frazer DM, Bowden DK, Anderson GJ, Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with β-thalassemia major: a longitudinal study, Blood 122, 124–133 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Malcovati L, Karimi M, Papaemmanuil E, Ambaglio I, Jädersten M, Jansson M, Elena C, Gallì A, Walldin G, Della Porta MG, Raaschou-Jensen K, Travaglino E, Kallenbach K, Pietra D, Ljungström V, Conte S, Boveri E, Invernizzi R, Rosenquist R, Campbell PJ, Cazzola M, Hellström Lindberg E, SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts, Blood 126, 233–241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cazzola M, Barosi G, Gobbi PG, Invernizzi R, Riccardi A, Ascari E, Natural history of idiopathic refractory sideroblastic anemia, Blood 71, 305–312 (1988). [PubMed] [Google Scholar]
- 51.Gattermann N, Finelli C, Porta MD, Fenaux P, Stadler M, Guerci-Bresler A, Schmid M, Taylor K, Vassilieff D, Habr D, Marcellari A, Roubert B, Rose C, Hematologic responses to deferasirox therapy in transfusion-dependent patients with myelodysplastic syndromes, Haematologica 97, 1364–1371 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.List AF, Baer MR, Steensma DP, Raza A, Esposito J, Martinez-Lopez N, Paley C, Feigert J, Besa E, Deferasirox Reduces Serum Ferritin and Labile Plasma Iron in RBC Transfusion–Dependent Patients With Myelodysplastic Syndrome, JCO 30, 2134–2139 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Meunier M, Ancelet S, Lefebvre C, Arnaud J, Garrel C, Pezet M, Wang Y, Faure P, Szymanski G, Duployez N, Preudhomme C, Biard D, Polack B, Cahn J-Y, Moulis JM, Park S, Meunier M, Ancelet S, Lefebvre C, Arnaud J, Garrel C, Pezet M, Wang Y, Faure P, Szymanski G, Duployez N, Preudhomme C, Biard D, Polack B, Cahn J-Y, Marc Moulis J, Park S, Reactive oxygen species levels control NF-kappaB activation by low dose deferasirox in erythroid progenitors of low risk myelodysplastic syndromes, Oncotarget 8, 105510–105524 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Platzbecker U, Germing U, Götze KS, Kiewe P, Mayer K, Chromik J, Radsak M, Wolff T, Zhang X, Laadem A, Sherman ML, Attie KM, Giagounidis A, Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study, The Lancet Oncology 18, 1338–1347 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Suragani RNVS, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, Davies MV, Alexander MJ, Devine M, Loveday KS, Underwood KW, Grinberg AV, Quisel JD, Chopra R, Pearsall RS, Seehra J, Kumar R, Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis, Nat. Med 20, 408–414 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Dussiot M, Maciel TT, Fricot A, Chartier C, Negre O, Veiga J, Grapton D, Paubelle E, Payen E, Beuzard Y, Leboulch P, Ribeil J-A, Arlet J-B, Coté F, Courtois G, Ginzburg YZ, Daniel TO, Chopra R, Sung V, Hermine O, Moura IC, An activin receptor IIA ligand trap corrects ineffective erythropoiesis in β-thalassemia, Nat. Med 20, 398–407 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gardenghi S, Ramos P, Marongiu MF, Melchiori L, Breda L, Guy E, Muirhead K, Rao N, Roy CN, Andrews NC, Nemeth E, Follenzi A, An X, Mohandas N, Ginzburg Y, Rachmilewitz EA, Giardina PJ, Grady RW, Rivella S, Hepcidin as a therapeutic tool to limit iron overload and improve anemia in β-thalassemic mice, J Clin Invest 120, 4466–4477 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Welch JJ, Watts JA, Vakoc CR, Yao Y, Wang H, Hardison RC, Blobel GA, Chodosh LA, Weiss MJ, Global regulation of erythroid gene expression by transcription factor GATA-1, Blood 104, 3136–3147 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Roepstorff P, Fohlman J, Proposal for a common nomenclature for sequence ions in mass spectra of peptides, Biomed. Mass Spectrom 11, 601 (1984). [DOI] [PubMed] [Google Scholar]
- 60.Lefebvre T, Dessendier N, Houamel D, Ialy-Radio N, Kannengiesser C, Manceau H, Beaumont C, Nicolas G, Gouya L, Puy H, Karim Z, LC-MS/MS method for hepcidin-25 measurement in human and mouse serum: clinical and research implications in iron disorders, Clinical Chemistry and Laboratory Medicine (CCLM) 53, 1557–1567 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and methods
Fig. S1: Genomic characteristics of the training cohort of 156 MDS patients.
Fig. S2: Identification of in-frame alternative transcripts in SF3B1MUT MDS samples by RNA-sequencing.
Fig. S3: Allele-specific investigation of mutant SF3B1 causal relationship to aberrantly spliced ERFE+12 transcript using Degron-KI strategy.
Fig. S4: Massive use of FAM132B/ERFE cryptic junction in one MDS-RS with a bi-allelic alteration of SF3B1 gene.
Fig. S5: Identification of ERFE peptide by LC tandem mass spectrometry.
Fig. S6: Correlation of plasma ERFE concentrations with ERFE+12 transcript, WHO classification and iron homeostasis parameters
Fig. S7: Biological parameters of low transfusion burden MDS patients of the training cohort.
Fig. S8: Plasma ERFE, ferritin and hepcidin concentrations at enrollment in 59 patients with MDS according to the response to EPO.
Fig. S9: Erythroid-specific expression of ERFE variant transcript.
Fig. S10: Variations of ERFE+12 transcript expression SF3B1MUT low risk MDS after therapy.
Fig. S11: Variations of plasma concentration of ERFE protein in SF3B1MUT low risk MDS after therapy.
Fig. S12: Kaplan-Meier curve for the analysis of overall survival according to ERFE+12/ERFEWT+ERFE+12 ratio.
Table S1: Clinical and biological characteristics of the training cohort of 156 MDS patients according to SF3B1 status.
Table S2: Differential expression of ERFE transcripts according to the mutational status of SF3B1, TET2 and DNMT3A genes by RNA-seq.
Table S3: Primer sequences used for fluorescent PCR and RT-qPCR.
Table S4: Expression of total ERFE, ERFEWT and ERFE+12 transcripts by fluorescent PCR and RT-qPCR in 9 MDS bone marrow samples.
Table S5: Clinical and biological characteristics of the validation cohort of 55 MDS patients according to SF3B1 status.
Table S6: Clinical and biological characteristics of the cohort of 59 MDS patients of the GFM-Retacrit-2013 cohort according to response status.
Table S7: Clinical and biological characteristics of the cohort of 90 MDS patients with survival data according to SF3B1 and ERFE+12 status.
Data file S3: In-frame and differentially expressed 3’ cryptic splice site junctions in SF3B1MUT MDS
Data file S2: Differentially expressed 5’ and 3’ junctions between SF3B1MUT and SF3B1wT MDS patients by RNA-seq.
Data file S1: Differentially expressed transcripts between SF3B1MUT and SF3B1WT MDS patients by RNA-seq.