

Abstract
Acute myeloid leukemia (AML) is a cancer derived from the myeloid lineage of blood cells, characterized by overproduction of leukemic blasts. Although therapeutic improvements have made a significant impact on AML patient outcomes, survival rates remain low due to high incidence of relapse. Similar to how wildfires can reignite from hidden embers not extinguished from an initial round of firefighting, leukemic stem cells (LSCs) are the remaining embers after completion of traditional chemotherapeutic treatments. LSCs exhibit a unique metabolic profile and contain metabolically distinct subpopulations. In this review, we detail the metabolic features of LSCs and how these characteristics promote resistance to traditional chemotherapy. We also discuss new therapeutic approaches that target metabolic vulnerabilities of LSC to selectively eradicate them.
Keywords: AML, Acute myeloid leukemia, LSC, leukemic stem cells, oxidative phosphorylation, mitochondrial metabolism
Improved AML therapies require novel targets
Acute myeloid leukemia (AML) is a cancer derived from the myeloid lineage of blood cells, characterized by overproduction of leukemic blasts (see Glossary). Blasts replace normal hematopoietic cells, which begins in the bone marrow (BM) but can spill out into peripheral blood and spread to other organs.[1] AML is the most common acute leukemia in adults, with nearly 120,000 global cases per year. Though therapeutic improvements have positively impacted outcomes for younger patients, most cases present in patients over 65 and overall survival rates of individuals diagnosed before the age of 40 are five-fold higher than patients diagnosed at 65 years or older.[2] AML is a genetically complex and heterogeneous disease with many known drivers, and heterogenous mixtures of subclones with distinct evolutionary patterns can exist in the same individual [3]. Risk groups are often defined by the presence or absence of specific cytogenetic abnormalities and acquired genetic mutations in patient samples. Complex karyotypes are most frequently observed in elderly patients and are associated with poor prognosis.[4] Point mutations in driver genes are the most common lesion, and are enriched in intermediate risk AML patients[3]. Several point mutations have also been identified in genes that act as drivers of therapeutic resistance.[5] Autonomous proliferation of AML blast cells is also a predictor of survival, with increased proliferation resulting in decreased probability of survival due to enhanced growth rate and rapidity of relapse, as well as greater potential for acquiring additional mutations.[6]
Since 2012 there have been more drugs in development for leukemia than almost all solid tumors, but between 1979 and 2017 few new drugs were approved for use in AML.[7] Therapeutic developments have historically focused on optimization of existing traditional chemotherapeutics, so novel therapeutic targets are crucial for improved patient outcome. Since 2017, numerous novel therapeutics for AML have been approved, but most are not associated with widespread, deep, or durable responses. Leukemic stem cell (LSC) eradication is required to prevent relapse in AML, so this review discusses the unique metabolic characteristics and potential to therapeutically target LSCs: quiescent, chemotherapy-resistant cells with self-renewal capacity that regenerate AML. In addition, we discuss implications of metabolic reprogramming and comment on potential for therapeutic targeting.
LSCs are a therapy-resistant, long-term reservoir for AML
Hematopoiesis is a complex, highly regulated multi-step process through which hematopoietic cells are created from a small pool of self-renewing hematopoietic stem cells (HSC) in the BM. This hierarchical process ultimately gives rise to all blood lineages, including mature myeloid cells.[8] (Fig. 1). In AML, transforming mutations result in evolution of HSCs and/or their progeny into LSCs which lie at the top of their own unique hierarchy and compete against normal hematopoiesis. (Fig. 1). LSCs exhibit aberrant self-renewal capacity and indefinite proliferation. They also overproduce leukemic blasts, which are extremely proliferative[9], fail to fully mature, and exhibit limited function[10]. These blasts block the function and development of mature blood cells in all lineages.[11] Hence, one of the initial goals of treatment is the rapid removal, or ‘de-bulking’ of blast cells to promote hematopoietic recovery.[12]
Figure 1: Comparison of normal and leukemic myeloid hematopoietic hierarchies.
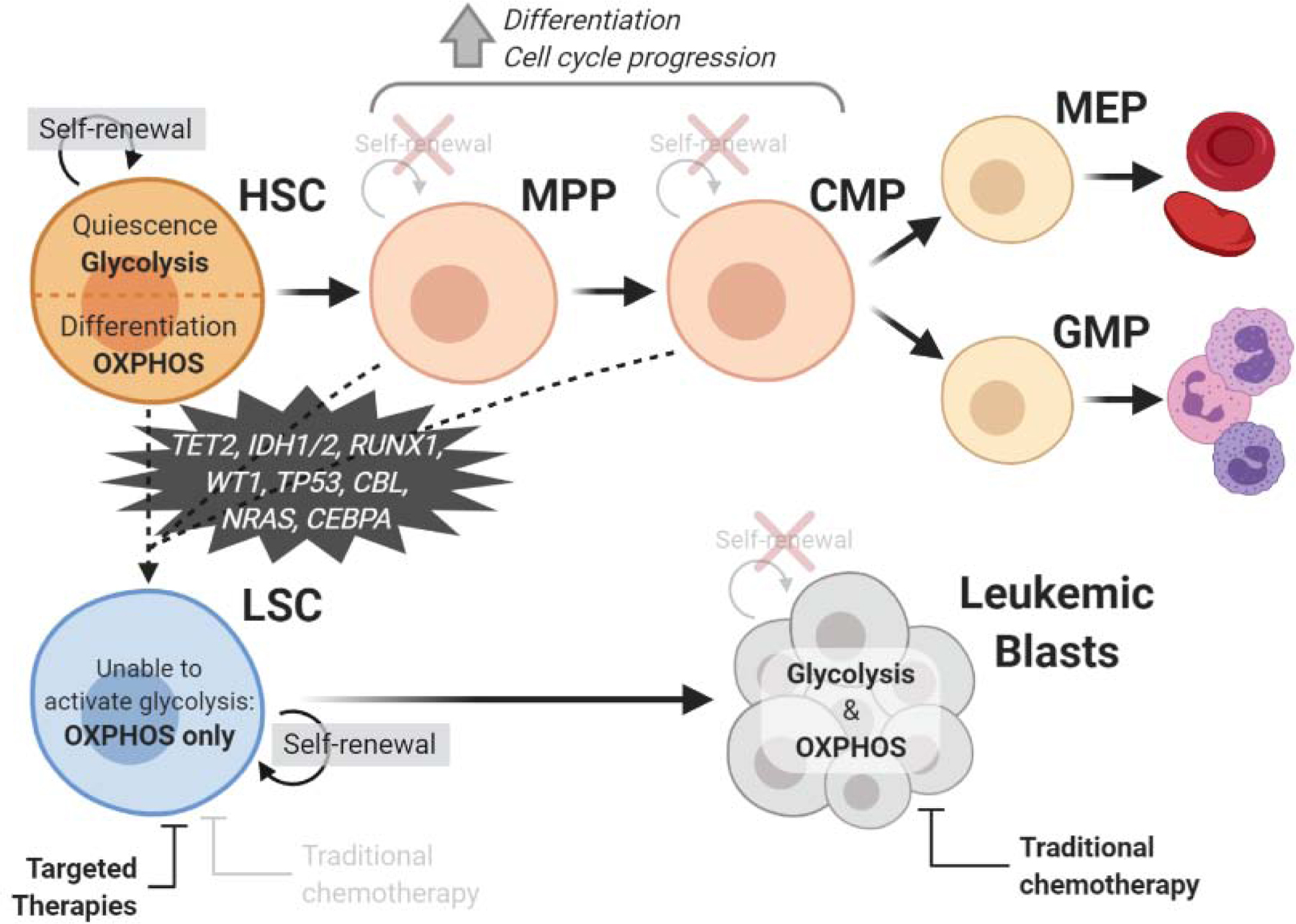
Hematopoietic stem cells (HSCs) self-renew and primarily exist in a glycolytic state during quiescence but switch to oxidative phosphorylation (OXPHOS) upon differentiation. HSCs differentiate into multipotent progenitors (MPPs), which lose the ability to self-renew but have increased frequency of cell cycle progression and differentiation activity. The MPP then differentiates to a common myeloid progenitor (CMP), which can become a megakaryocyte–erythroid progenitor (MEP), generating erythrocytes and platelets, or a granulocyte-macrophage progenitor (GMP), generating granulocytes. Both HSCs, MPPs, and CMPs can potentially become a leukemic stem cell (LSC) through the acquisition of transforming mutations. LSCs also have the capacity for self-renewal and are uniquely reliant on OXPHOS. They differentiate to produce leukemic blasts, which lose the ability to self-renew but can use both glycolysis and OXPHOS. While leukemic blasts are sensitive to traditional chemotherapy, LSCs require targeted therapies for their eradication. Created with BioRender.com.
The current standard of care for AML patients fit enough to receive it involves intensive chemotherapy, which is commonly comprised of a combination of cytarabine and an anthracycline, named “7+3” for 7 days of cytarabine treatment with 3 days of an anthracycline. Cytarabine functions as a DNA synthesis inhibitor, incorporating into DNA in place of deoxycytosine. Anthracyclines function through DNA intercalation.[13], [14] These traditional chemotherapeutics (Table 1) are widely cytotoxic to most hematopoietic cells, but as they especially target proliferating cells through inhibition of DNA synthesis, leukemic blasts are uniquely susceptible.
Table 1:
Traditional Chemotherapeutics.
| Name | Mechanism of action | Approved usage | Median OS |
|---|---|---|---|
| Cytarabine (AraC) [96] | Nucleoside analog; inhibitor of DNA polymerase | Remission induction in acute non-lymphocytic leukemia. | With standard-dose dau = 16.6 mo With high-dose dau = 25.4 mo |
| Daunorubicin [96] | Inhibitor of DNA topoisomerase II (also used to manufacture doxorubicin, idarubicin) | Remission induction in acute non-lymphocytic leukemia, with other approved anti-cancer drugs, in adults | With AraC, standard-dose = 16.6 mo With AraC, high-dose = 25.4 mo |
| 7+3 (cytarabine + anthracycline) [97] | Combination of cytarabine (7 days) + daunorubicin, doxorubicin, or idarubicin (3 days) | First line therapy for de novo AML | 5.95 mo |
| CPX-351 (Vyxeos) [97] | Liposomal formulation of cytarabine + daunorubicin at fixed 5:1 molar ratio | De novo AML-MRC and t-AML in patients unable to tolerate 7+3 | 9.56 mo |
| Fludarabine (Fludara) [98] | Inhibitor of DNA synthesis through DNA polymerase, ribonucleotide reductase, DNA primase and DNA ligase I | Off-label use: (1) de novo AML with cytarabine ± G-CSF ± idarubicin (FA, FLAG, or FLAG-IDA regimens) (2) R/R or high-risk patients with cytarabine and filgrastim ± idarubicin [FLAG, FLAG-IDA regimen] | 11 mo |
| Etoposide (VP-16) [99], [100] | Inhibitor of DNA replication and transcription through topoisomerase II | Off-label use: with mitoxantrone ± cytarabine in R/R AML | With LDAC = 8.7 mo With 7+3 = 8.4 mo |
| 6-thioguanine (6TG) [101] | Purine analog; inhibitor of DNA synthesis | Single agent for remission induction, remission consolidation, maintenance therapy in AML, also with 7+3 in DAT regimen | With AraC + Dau = 3.7 mo With etoposide + idarubicin = 9.9 mo |
| Hydroxyurea (Droxia) [102] | Purine analog; diphosphate reductase inhibitor | Off-label use: single agent for cytoreduction in adults with AML | 7.0 mo |
| Azacitidine (Vidaza) [103] | Pyrimidine analog; DNA methyltransferase inhibitor | Myelodysplastic syndromes | 10.3 mo |
| Decitabine (Dacogen) [104] | Pyrimidine analog; DNA methyltransferase inhibitor | Myelodysplastic syndromes | 8.09 mo |
However, while traditional chemotherapeutics may eliminate most bulk tumor cells, their use is limited to patients fit for chemotherapy, which excludes the majority of the AML patient population[15]. Further, they are rarely effective at eliminating LSCs, leading to disease relapse [16]. Relapse represents a significant clinical problem: disease recurs in ~60% AML patients in the favorable risk category and >85% patients in the adverse risk category, defined by the 2017 European LeukemiaNet (ELN) classification [17]. Within the last two decades a wide range of therapies have been proposed, including targeting immunophenotypic markers, specific mutations, epigenetic deregulation, microenvironmental alterations, proteasome activity, and others. Examples of targeted therapies commonly used in AML treatment are listed in Table 2. However, in the clinic, most approaches show limited potential to eradicate LSCs. A body of recent work has focused on identifying unique metabolic characteristics found in therapy-resistant relapse LSCs as well as in LSCs from refractory de novo AML.[18] Here, we describe key metabolic characteristics and potential targets of LSCs contributing to therapeutic resistance and relapse.
Table 2:
Targeted AML Therapies.
| Name | Mechanism of action | Target cell type | Approved for… | Combined with… | Median OS |
|---|---|---|---|---|---|
| Midostaurin (Rydapt) [105] | Inhibitor of FLT3, PKC, serine/ threonine + tyrosine kinases | Blasts + LSCs | De novo FLT3-mutated AML | 7+3 induction, HiDAC consolidation, azacitidine | With 7+3 = 74.7 mo. |
| Gilteritinib (Xospata) [106] | Inhibitor of FLT3 | Blasts + LSCs | R/R FLT3-mutated AML | Single agent | 9.3 mo. |
| Ivosidenib (Tibsovo) [107], [108] | Inhibitor of IDH1 | Blasts + LSCs | De novo AML in patients >75 years or R/R IDH1 mutated AML | Single agent |
De novo = 8.8 mo. R/R - 12.6 mo. |
| Enasidenib (Idhifa) [109] | Inhibitor of IDH2, induces terminal differentiation | Blasts | R/R IDH2-mutated AML | Single agent | 8.8 mo. |
| Gemtuzumab ozogamicin (Mylotarg) [110] | Anti-CD33 monoclonal humanized antibody | Blasts + CMPs | De novo or R/R CD33+ AML | Daunorubicin or cytarabine, or single agent | With dau/Ara-C = 17.3 mo. Single agent = 4.9 mo. |
| Venetoclax (Venclexta) [111] | Inhibitor of BCL-2 | Blasts + LSCs | De novo AML in patients >75 years or with comorbidities | Azacitidine | 17.5 mo. |
| Glasdegib (Daurismo) [112] | Inhibitor of the Hedgehog pathway | Blasts + LSCs | De novo AML in patients >75 years or with comorbidities | Low-dose cytarabine (LDAC) | 8.8 mo. |
LSCs tightly control ROS levels
One major similarity between HSC and LSC populations is their tight regulation of reactive oxygen species (ROS), highly reactive byproducts of aerobic metabolism that cause peroxidation of nucleic acids, amino acids, lipids, and carbohydrates[19]. (Fig. 2) ROS contribute to stem cell aging, force cells out of quiescence, and compromise their ability to maintain the stem cell population. ROS levels are regulated in part by localization to hypoxic microenvironments. There exists a decreasing oxygen gradient with increased depth in the BM[20]; the lowest oxygen levels correspond with deeper perisinusoidal regions.[21] HSC localization within this hypoxic microenvironment implies hypoxia is critical to stem cell function.
Figure 2: Mechanisms regulating ROS in LSCs.
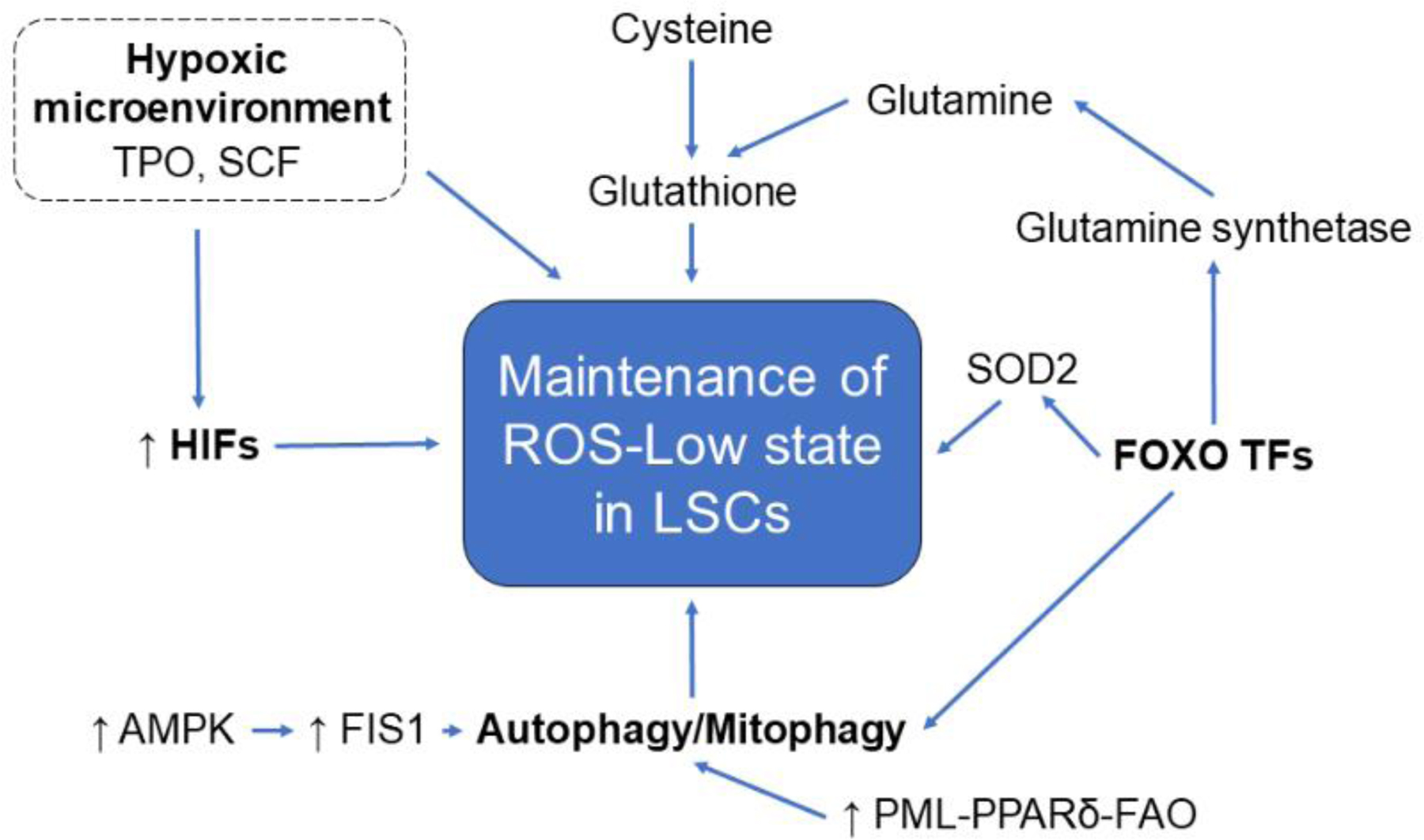
Leukemic stem cells (LSCs) display several mechanisms that function to maintain low levels of reactive oxygen species (ROS). LSCs reside in the hypoxic bone marrow niche, limiting oxidative stress. Hypoxia-inducible factors (HIFs), essential to the cellular response to hypoxia, are activated even in normoxia in LSCs and niche factors such as thrombopoietin (TPO) and stem cell factor (SCF) promote HIF stabilization. FOXO transcription factors also maintain low ROS levels by regulating mitochondrial expression of superoxide dismutase (SOD2) and glutamine synthetase. Glutamine, along with cysteine, is critical to the production of glutathione, which scavenges free radicals and other ROS. FOXO TFs are also required for autophagy and mitophagy, which mitigates oxidative stress through removal of toxic proteins and damaged mitochondria. AMP kinase (AMPK), which upregulates mitochondrial fission regulator FIS1 and therefore mitophagy, is intrinsically activated in LSCs. The PML-PPARδ-FAO pathway, upregulated in LSCs, also promotes mitophagy.
HSCs and LSCs also tightly regulate expression of hypoxia-inducible factors (HIFs), essential for the cellular hypoxic response. In the BM, factors such as thrombopoietin (TPO) and stem cell factor (SCF) promote HIF1α stabilization[22], [23]. HIFs bind hypoxia response elements (HREs) in regulatory regions of genes responsible for self-renewal, apoptosis, redox homeostasis, and metabolic reprogramming[24] (Text Box 1). In HSCs, HIF1α promotes glycolysis by binding HREs in the promoter region of genes coding for glycolytic enzymes such as pyruvate dehydrogenase kinases (PDKs), preventing entry of pyruvate into the mitochondrial TCA cycle[20] and acting as an enforcer of HSC quiescence through indirectly inhibiting oxidative phosphorylation (OXPHOS). Interestingly, LSCs are “pseudohypoxic” – HIF1α is activated even in normoxia, allowing LSC generation and maintenance.[25] However, HIF function in LSCs has not been fully elucidated. A recent study suggests HIFs exert transcriptional control of glycolysis but not OXPHOS in leukemic cells, and HIFs may not control a specific glycolytic step but instead ensure proper expression of necessary components during hypoxia[20]. Moreover, loss of HIF activity in leukemic cells does not affect glucose consumption or lactate production, suggesting glycolytic regulation through oncogenes can occur independently of HIF.[20] On the other hand, HIF1α knockout in MLL-AF9-driven AML enhanced disease progression after chemotherapy.[26] Thus HIF1a can serve as an oncogene or tumor suppressor,[27] and the outcome may be governed by autophagy. Autophagy, driven by HIF1a, in cancer cells represses tumor growth[28], but as will be discussed, autophagy is critical to LSC survival in AML. Therefore, additional work is needed to address the role of HIFs in LSC metabolism and understand the interplay between hypoxic signaling and LSC maintenance.
Text Box 1: HIF Target genes.
The target genes for HIF1α and HIF1β cover a wide array of cellular processes, many of which have been linked to poor outcomes in AML. One category of HIF target genes are those involved in stemness and self-renewal, critical for the maintenance of LSCs. Adrenomedullin (ADM) is a regulatory peptide involved in cellular growth that increases leukemic migration with excess levels in the bone marrow[113]. Aberrant signaling of vascular endothelial growth factor signaling (VEGF), which promotes angiogenesis, is also associated with poor outcome in AML[114], and insulin-like growth factor 2 (IGF2) is a critical component of the IGF2/IGF1R/Nanog signaling pathway, which regulates LSC proliferation.[115] HIF targets also include several genes regulating apoptosis. BNIP3L well-known as a regulator of apoptosis, but also mediates mitophagy, which is critical to LSC survival[52]. Further, Phorbol-12-myristate-13-acetate-induced protein 1 (also known as NOXA) is a member of the BCL-2 protein family and has been implicated in the mechanism by which the combination of azacitidine and venetoclax so effectively targets de novo LSCs.[116] The anti-apoptotic protein MCL1 has also been implicated for its involvement in the formation and persistence of AML.[117] Controlling ROS is also critical to LSC survival in AML, and HIF targets many genes involved in redox homeostasis. Glutathione peroxidase 3 (GPX3) detoxifies hydrogen peroxide, and increased expression is associated with poorer outcomes in AML. [118] Heme oxygenase-1 (HMOX1) has been shown to suppress apoptosis in AML cells[119], and superoxide dismutase (SOD2) is critical to cellular redox balance.[30]
FOXO transcription factors (FOXO1, 3, 4, 6) are also common ROS regulators in HSCs and LSCs. These factors govern HSC regenerative potential through regulating transcriptional responses to oxidative stress and controlling quiescence and survival.[29] FOXO3a regulates mitochondrial expression of superoxide dismutase (SOD2) [30], which converts the superoxide radical to oxygen and hydrogen peroxide. Further, FOXO3a is necessary for autophagy, which mitigates oxidative stress through removal of toxic proteins and damaged mitochondria.[31] The role of FOXOs in limiting oxidative stress is well defined in HSCs.[29] However, FOXOs are active in ~40% AML patient samples regardless of genetic subtype[32], suggesting their importance for maintaining low ROS in LSCs. In fact, FOXO3 knockdown increases expression of myeloid differentiation and apoptosis markers.[33] Further, glutamine synthetase is transcriptionally regulated by PI(3)K–PKB–FOXO signaling[34], and glutamine metabolism is critical to redox balance through glutathione (GSH) production.[35] GSH scavenges free radicals and other ROS. Additionally, ATP generation in OXPHOS is dependent on glutathionylation of succinate dehydrogenase (SDH), the second enzyme complex of the electron transport chain (ETC), and glutamine metabolism is vital to de novo LSC survival.[36] Despite FOXO protein involvement in leukemia-relevant pathways, like HIF proteins they can act as tumor suppressors due to their ability to arrest the cell cycle through p27 and p21 induction, and induce apoptosis through upregulation of pro-apoptotic genes including Bim and FasL.[37], [38] Further, elevated FOXO3A phosphorylation results in nuclear exclusion and degradation[39], and may be a targetable, independent adverse prognostic marker in AML.[40] Studies also show FOXO inactivation is crucial to the apoptosis arrest in AML[39]. The role of FOXOs in reducing oxidative stress in LSCs may be critical to disease progression. However, evidence for FOXO inhibition in reducing apoptosis in AML suggests FOXO transcription factors require additional investigation to disentangle their role in LSC metabolism and determine whether they offer a suitable therapeutic target.
Although not a transcription factor, the polycomb complex protein B cell-specific Moloney murine leukemia virus integration site 1 (BMI-1) is also critical to self-renewal and quiescence in HSCs and LSCs[41], as well as leukemic programming in myeloid progenitor cells[42]. Further, BMI-1 regulates both mitochondrial function and ROS homeostasis[41] and is overexpressed in AML and other hematologic malignancies.[43] As maintenance of low ROS levels as well as mitochondrial function are required for LSC stemness, BMI-1 overexpression may allow increased LSC survival through these mechanisms.
LSCs actively engage mitophagy
As mitochondrial metabolism differs between HSCs and LSCs, mitochondrial clearance does as well. Autophagy-mediated mitochondrial clearance, or mitophagy, is critical to cellular function and its dysregulation is implicated in a variety of hematopoietic disorders including AML.[44] In self-renewing HSCs, mitophagy is critical for HSC expansion [45] and function[46]. Active mitochondria, the main source of cellular ROS, can be detrimental to HSC maintenance so mitochondrial clearance is critical to allow activated HSCs to return to a low-ROS, quiescent state.[46] Indeed, impaired autophagy in aged HSCs is associated with high ROS and decreased stem cell capacity[47]. Strikingly however, HSCs not only have a larger mitochondrial mass than lineage-committed progenitors and mature cells, but also a relatively low mitochondrial turnover capacity.[48] As mitochondria drive OXPHOS and calcium-induced apoptosis[49], are the location of fatty acid oxidation[50], and are sources for ROS signaling[51], increased mitochondrial mass in HSCs may point to a much larger role of mitochondria in HSC biology than originally considered. This surprising discovery was made very recently, so implications in AML have yet to be elucidated. However, LSCs have increased dependence on mitophagy compared with HSCs[52]. As we have mentioned, LSCs are uniquely reliant on mitochondrial OXPHOS despite the necessity to remain ROS-low, so proper mitochondrial regulation and clearance are crucial to their survival. This is underscored by a study showing knockdown of mitochondrial fission regulator FIS1, a mitochondrial fission and apoptosis regulator, reduces mitophagy and increases myeloid differentiation, cell cycle arrest, and glycogen synthase kinase-3 (GSK3) inhibition, a serine/threonine protein kinase whose signaling is critical to self-renewal potential. AMP kinase (AMPK) upregulates FIS1 and is intrinsically activated in LSC populations. AMPK inhibition produces similar results to FIS1 knockdown. Through Ulk1 phosphorylation, AMPK contributes to mitochondrial targeting to the lysosome.[53] Together, these data suggest FIS1 critically mediates mitophagy and LSCs use AMPK/FIS1-mediated mitophagy as a survival mechanism by maintaining self-renewal potential.[54] Mitophagy is also implicated in the survival of other cancer stem cells (CSCs): hepatic CSC mitophagy mediates p53 degradation and favors proliferation, and BNIP3L-mediated mitophagy contributes to therapeutic resistance in colorectal and breast cancer CSCs.[55], [56] Further, the PML-PPARδ-FAO pathway, upregulated in LSCs[57], also promotes mitophagy[58] which suggests the they are interconnected and work together to promote self-renewal in HSCs and LSCs alike.
LSCs are uniquely reliant on OXPHOS
In low oxygen environments, such as the hypoxic BM niche, glycolysis oxidizes glucose to produce ATP and regenerate NADH[50]. Glucose can also enter the TCA cycle through acetyl-CoA oxidation[50]. While glucose is the most common fuel for the TCA cycle, amino acids can also be catabolized through the TCA cycle and fatty acids undergo fatty acid β-oxidation (FAO) to produce Acetyl-CoA[50]. (Fig. 3, Key Figure) The TCA cycle generates reducing equivalents that sustain formation of proton gradients and ATP synthesis via the ETC and OXPHOS. ETC Complex II (SDH) plays a dual role by transferring electrons to Complex III in the ETC as well as converting the TCA cycle intermediate succinate to fumarate[50]. (Fig. 3) These pathways are critical to cellular function, but they play especially important roles in hematopoietic regulation.
Figure 3: Metabolic features of LSCs.
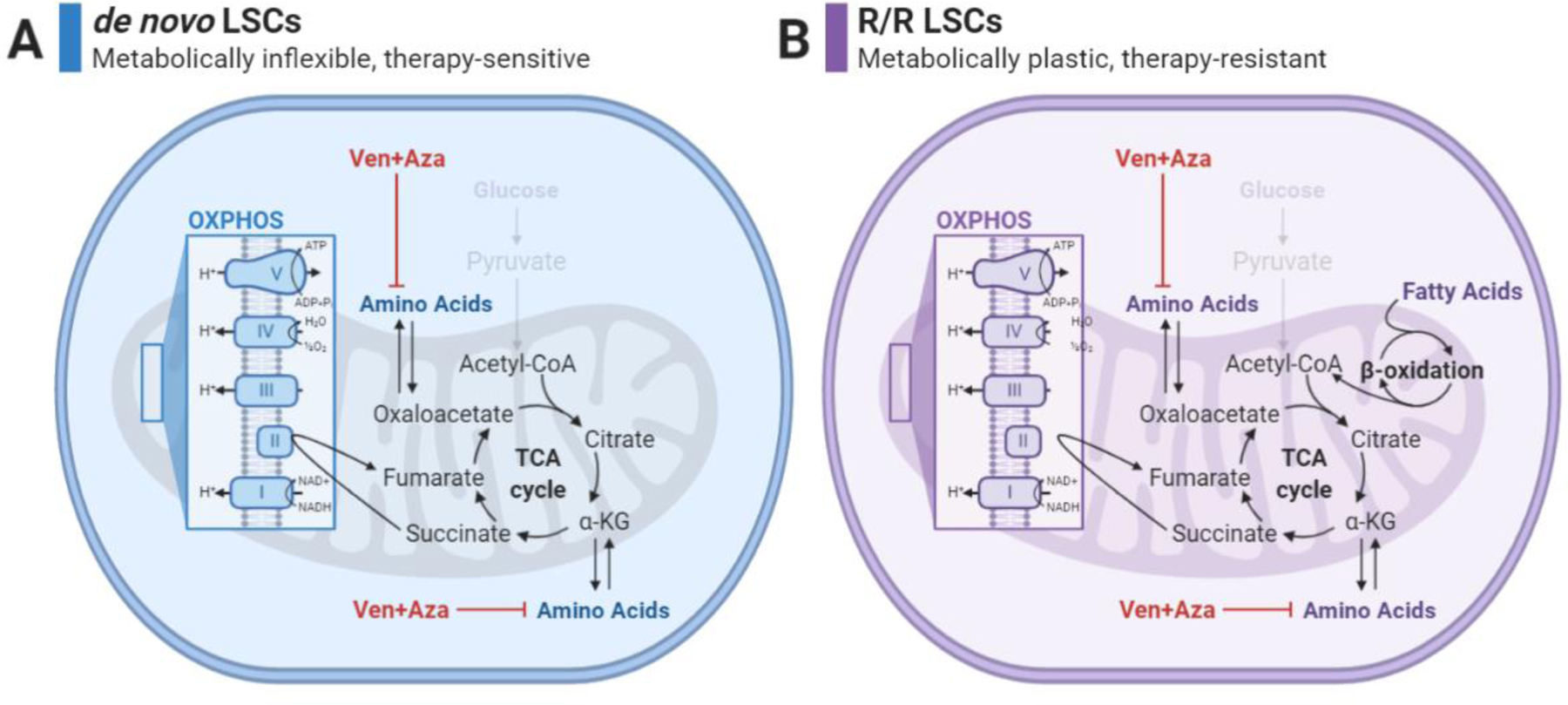
A) de novo LSCs are described as metabolically inflexible as they are unable to activate glycolysis and are uniquely reliant on OXPHOS for energy production. As a result, glucose can no longer be used as a fuel source for the TCA cycle and de novo LSCs are instead reliant on amino acids as a fuel source. Amino acids enter the TCA cycle through conversion to TCA cycle intermediates such as oxaloacetate and alpha-ketoglutarate. The combination of venetoclax and azacitidine (Ven+Aza) successfully inhibits amino acid import and metabolism as well as OXPHOS, allowing de novo LSCs to be eradicated.
B) Relapsed/refractory (R/R) LSCs are also unable to activate glycolysis and are also uniquely reliant on OXPHOS for energy production. While glucose is not available as a fuel source for the TCA cycle, R/R LSCs can use both amino acids and fatty acids to fuel the TCA cycle and OXPHOS. Fatty acids are converted to Acetyl-CoA through beta oxidation in the mitochondria. While Ven+Aza remains biologically active in these cells, the ability of R/R LSCs to use fatty acids as a fuel source allows for therapeutic resistance by compensating for the loss of amino acids. Created with BioRender.com.
While HSCs and LSC populations both use OXPHOS, they do so in distinct ways. HSCs generally avoid mitochondrial OXPHOS and instead rely on anaerobic glycolysis for ATP production. But during differentiation, HSCs activate OXPHOS to meet increased energetic requirements, then revert to glycolysis to prevent HSC pool exhaustion[59]. LSCs, however, are “metabolically inflexible”: they are uniquely reliant on OXPHOS for energy production and do not activate glycolysis [60]. This characteristic is seemingly inconsistent with limiting oxidative stress, as mitochondrial respiration is the primary source of ROS.[51] The purpose of this phenomenon has yet to be fully elucidated, although it is a unique metabolic property in CSCs[61]. One theory suggests OXPHOS acts as an adaptation to maintain survival through decreased metabolic rate[60], but a recent study suggests activation of the sublethal integrated stress response (ISR), highly activated in LSCs[62], suppresses glycolysis.[63] Due to decreased glycolytic activity, one of three metabolic fuels for the TCA cycle – glucose, amino acids, and fatty acids – is now unavailable, and LSCs must rely on amino acids and/or fatty acids to fuel OXPHOS. Despite OXPHOS being a slower energy production mechanism than glycolysis, it is significantly more efficient and may allow LSCs to survive by more effectively utilizing nutrients with limited availability[60].
The unique reliance on OXPHOS has driven many studies focusing on its pharmacological inhibition[16], [36], [60], [64]. Targeting mitochondrial function using tigecycline, a mitochondrial protein synthesis inhibitor, results in Ara-C sensitivity through induction of apoptosis, although human AML cells may metabolically adapt through increased glycolysis.[65] Another example is the selective sensitivity of LSCs from de novo AML patients to amino acid depletion, leading to OXPHOS inhibition.[16] This sensitivity is elicited through a combination of venetoclax, a BCL-2 inhibitor, and azacitidine, a DNA methyltransferase inhibitor (ven/aza)[36]. In LSCs from de novo AML patients, ven/aza significantly decreased OXPHOS through amino acid depletion and ETC complex II inhibition[36]. It is currently not understood why BCL-2 inhibition results in impairment of amino acid uptake and metabolism. One explanation lies in BCL-2 inhibition allowing increased formation of mitochondrial permeability transition pores[66], resulting in increased mitochondrial membrane leakage and decreased amino acid catabolism. Regardless, this combination is extremely effective in older AML patients, a population with historically poor outcomes due to the high toxicity of traditional chemotherapeutic agents. In fact, this regimen has been shown in a Phase 3 study to allow for deep and durable remissions and represents the new standard of care for older newly diagnosed AML patients [67]. Notably, the therapeutic effect of venetoclax is largely independent of LSC genetic background in de novo AML, indicating venetoclax targets a distinct metabolic state rather than a specific lesion.[67] This is unique relative to other targeted therapies that inhibit specific proteins with oncogenic roles such as mutant FLT3 and IDH1/2. Interestingly, excess 2-hydroxyglutarate (2-HG) produced by IDH mutant AML cells confer susceptibility to venetoclax due to ETC dysregulation; patients with IDH mutant AML in turn have more robust responses to ven/aza.[67] On the other hand, IDH inhibitors appear to reduce this sensitivity.[68] Conversely, FLT3 mutations have no known impact on outcomes of patients treated with ven/aza. Interestingly, BCL-2 upregulation is a mechanism of FLT3 inhibitor resistance, though the relevance to LSC metabolism has not been characterized[69]. Further studies will be needed to fully elucidate the relationship between targeted therapies such as FLT3 and IDH inhibitors and LSC metabolic function.
While ven/aza is an exciting option for de novo AML patients, its efficacy in the relapsed/refractory (R/R) AML setting is significantly lower.[70] This phenomenon is also observed ex vivo in LSCs from R/R AML patients. While ven/aza still inhibits BCL-2 and amino acid import and metabolism, R/R LSCs are able to metabolically compensate through upregulation of at least one alternative pathway to sustain mitochondrial demands: fatty acid metabolism (Fig. 3B). R/R LSCs show significantly increased overall fatty acid levels and OXPHOS function compared to de novo LSCs upon ven/aza treatment, suggesting fatty acids contribute to OXPHOS and their survival.[16] In a subsequent study, genetic and pharmacological targeting of fatty acid transport resensitized R/R LSCs to ven/aza both ex vivo and in a primary patient xenograft model.[71] LSC resistance to ven/aza may also result from loss of BCL2 expression occurring in phenotypically monocytic AML, which subsequently rely on MCL1, another BCL2 family member, to mediate OXPHOS.[72] While de novo LSCs are metabolically inflexible, R/R LSCs exhibit metabolic plasticity and alternative mechanisms of mediating OXPHOS, allowing for therapeutic resistance.
LSCs use amino acid metabolism to drive OXPHOS
Recent clinical and basic science studies have resulted in a major leap in understanding the metabolic underpinnings of LSC therapeutic resistance. As discussed, LSCs depend on OXPHOS for ATP production. However, until recently it had not been determined how OXPHOS is fueled in these cells. Recent work shows de novo AML LSCs display increased amino acid levels and uptake[16], and likewise rely on amino acid metabolism for their survival (Fig. 3A). When de novo LSCs from AML patients are treated with ven/aza, which inhibits amino acid uptake and metabolism, then supplemented with all common amino acids, LSC viability and relative oxygen consumption rate were rescued. This underscores the requirement of amino acids for OXPHOS in de novo AML LSCs.[36], [71] Amino acids fuel the TCA cycle, and therefore OXPHOS, through conversion to TCA cycle intermediates[16]. For example, arginine, glutamine, histidine, and proline are precursors to glutamate, which is converted to α-ketoglutarate by glutamate dehydrogenase.[50] The production of these intermediates drives TCA cycle function, which in turn drives OXPHOS through production of NADH and succinate, the substrate for ETC complex II[50]. While studies have hinted at the role of amino acids in leukemic pathogenesis, this is the first reported example of LSCs using amino acids to directly fuel OXPHOS.[16]
Cysteine, essential for intracellular redox balance, also plays a unique role in LSC function. Glutathione (GSH), crucial for regulating intracellular ROS[73], is a tripeptide consisting of glutamate, cysteine, and glycine[50], and multiple GSH regulatory pathways are overexpressed in AML[73]. As previously mentioned, ETC complex II glutathionylation is required for OXPHOS function in LSCs[36]. Cysteine is a non-essential amino acid[50], but synthesis of endogenous cysteine is often insufficient to maintain GSH levels. As a result, uptake of cysteine and cystine, the oxidized dimer form of cysteine[50], becomes essential to prevent oxidative stress[64]. Indeed, enzyme-mediated depletion of serum cysteine and cystine in mice and non-human primates suppresses AML proliferation through inactivation of the antioxidant cellular response.[64] Enzyme-mediated cysteine depletion has also succeeded in prostate cancer, breast cancer, and chronic lymphocytic leukemia models.[74] Interestingly, a recent study determined that decreased polyamine pathway activity, or the trans-sulfuration pathway, sensitized cancer cells to cysteine depletion[75] and may offer an additional therapeutic mechanism of cysteine depletion. Beyond what is described here, studies investigating cysteine depletion are limited, but with additional examination it may offer a novel therapeutic mechanism.
Branched-chain amino acids (BCAAs), leucine, isoleucine, and valine, also impact HSC and LSC survival. Valine is required for HSC maintenance[76], [77], but the metabolic role of valine in HSCs has yet to be determined, and limited research exists regarding the role of amino acid metabolism in HSCs. In de novo AML, however, BCAAs produced by BCAT1 (BCAA transaminase 1) contribute to malignant growth.[78] BCAT1 is a cytoplasmic aminotransferase that transfers α-amino groups from BCAAs to regulate cellular α-ketoglutarate, and is significantly overexpressed in LSCs[79]. The transfer of an amino group from a BCAA to α-ketoglutarate produces glutamate[80], which can be imported into mitochondria to fuel OXPHOS. It also results in hypermethylation of α-ketoglutarate-dependent dioxygenases such as Egl-9 family hypoxia inducible factor 1 (EGLN1) and the ten-eleven translocation (TET) family of DNA demethylases, similarly to IDH-mutant LSCs. Further, BCAT1 expression is significantly higher in relapse LSCs than de novo LSCs.[79] Mutations producing enhancer of zeste homolog 2 (EZH2) deficiency, the enzymatic component of polycomb repressive complex 2 which represses BCAT1, also result in progression of myeloproliferative neoplasms to highly aggressive AMLs in mice through enhanced mammalian target of rapamycin (mTOR) signaling.[81] This occurs by conversion of leucine to Acetyl-CoA, which positively regulates mTOR complex 1 (mTORC1) activity[82]. BCAA metabolism has been explored as a pharmacological target and shows promising primary AML cell inhibition[83], underlining the critical role of amino acids in AML.
Relapse LSC can switch to FAO to drive OXPHOS
FAO, the breakdown of fatty acids into Acetyl-CoA producing NADH and FADH2, fuels OXPHOS but regulates HSC fate. In HSCs, increased FAO promotes asymmetric division and HSC self-renewal.[84] This process is regulated via a PML-PPARδ-FAO axis, which acts as a metabolic switch where PML promotes PPARδ activation, which activates the FAO transcriptional program. [57]
However, FAO plays contrasting roles in R/R LSCs. R/R LSCs are resistant to venetoclax, and display increased fatty acid levels[16] and fatty acid transporter CD36 expression[89]. Based on studies inhibiting FAO through mitochondrial translocase carnitine palmitoyltransferase 1 (CPT1), FAO negatively regulates the BAK-dependent mitochondrial permeability transition, critical for cytochrome-c dependent promotion of apoptosis[85]. Venetoclax (Table 2) relies on mitochondrial apoptotic pathways, so aberrations within these pathways suggest a mechanism of therapeutic resistance[36], [86], [87]: venetoclax-resistant R/R LSCs compensate for amino acid loss by using fatty acids to fuel OXPHOS. Fatty acids enter mitochondria, then undergo beta oxidation to produce Acetyl-CoA. Beta oxidation also produces NADH and FADH2, used by the ETC to produce ATP.[50] While LSCs rely on autophagy for cell function, mitochondria-controlled autophagy also regulates fatty acid availability in OXPHOS through mitochondria-endoplasmic reticulum contact sites[88]. Recent studies have investigated LSC eradication through FAO inhibition: pharmacologic inhibition of CPT1 with etomoxir decreases refractory LSC function[71], and etomoxir restores eradication of ven/aza resistant LSCs.[89] Further, mitochondrial FAO inhibition and specific cytotoxic activity toward primary AML cells has been achieved using avocatin-B, although AML cells may adapt through fatty acid uptake from BM adipocytes[90], underlining the importance of performing studies within a relevant microenvironmental context. The inhibition of FAO and its role in fueling OXPHOS will likely become an exciting new direction in fighting therapeutic resistance in AML.
While fatty acids are critical to mitochondrial metabolism in R/R LSCs, they also contribute to proliferation, signaling, and other survival mechanisms. Aberrant lipid metabolism in cancer cells supports proliferation through increased availability of energy, structural cellular membrane components, and lipid-based signaling molecules. However, some cancer cells may rely on an alternative fatty acid desaturation pathway, increasing their metabolic plasticity and contributing to therapeutic resistance.[91] Alterations in lipid metabolism may also affect oncogenic signaling through the Wnt/β-catenin and Hippo/YAP pathways,[92] and proteins including fatty acid desaturases and mevalonate pathway enzymes have recently garnered attention for their roles in CSCs[93], [94]. Novel lipid detection methods are allowing for increased understanding of the role of fatty acids and lipids in cancer biology[93], and the discovery of targets involved in aberrant lipid metabolism in AML may offer a much needed mechanism of targeting R/R LSCs.
Concluding Remarks
Prior to 1995, AML patients faced survival rates ranging from 5–15% [95]. However, research within the last two decades has made impressive contributions to our understanding of leukemia biology and novel therapeutic targets, several of which have resulted in FDA approvals for AML treatment. (Clinician’s Corner, Text Box 2) Consequently, the overall survival rate has more than doubled, but gaps in our knowledge must be filled to continue this progress. (Outstanding Questions) Current treatments are successful in putting out the wildfire of leukemic blasts in AML. However, much like remaining embers can reignite the blaze, quiescent LSCs resistant to these therapies result in disease relapse and require a targeted approach to extinguish. Metabolic pathways fueling OXPHOS act as kindling for these outbreaks and will likely become key therapeutic targets in the fight to improve outcomes for patients with AML and other hematologic malignancies.
Text Box 2: Clinician’s Corner.
AML is the most common acute leukemia in adults. Since 1995, the overall survival rate has more than doubled. However, the median age at diagnosis is 68, and the five-year survival rate for this patient population remains very poor.
Traditional chemotherapeutics may eliminate the majority of bulk tumor cells, but they do not typically eliminate disease initiating LSCs, leading to relapse in over 65% of patients.
Recent developments in therapeutic targeting have allowed for the inhibition of unique metabolic characteristics found in LSCs, which contribute to therapeutic resistance in relapse as well as the initial refractory nature of some AML patients.
De novo LSCs are known to be “metabolically inflexible”: they are uniquely reliant on OXPHOS and cannot activate glycolysis. They also rely on amino acid metabolism to fuel OXPHOS, and the broad depletion of amino acid import by the combination of venetoclax and azacitidine has been extremely effective in clinical trials for elderly patients.
R/R LSCs are also uniquely reliant on OXPHOS but are resistant to many therapies, including venetoclax. They may be able to activate fatty acid metabolism to fuel OXPHOS in addition to amino acids, leading to therapeutic resistance.
Outstanding Questions.
LSCs activate HIF1α even in normoxia, but the role of HIFs in LSC survival is unclear. While HIFs are known to control glycolytic gene programs in HSCs, this may occur independently of HIFs in LSCs. Further, HIF1α can serve as an oncogene or tumor suppressor gene in different contexts depending on the importance of autophagy, but this connection is also not well understood.
FOXO transcription factors also play seemingly contrasting roles in both driving leukemic pathogenesis and acting as tumor suppressors. FOXO3a has been implicated in the prevention of myeloid differentiation and apoptosis, as well as the prevention of oxidative stress. However, FOXOs have also been shown to arrest the cell cycle and induce apoptosis, so further investigation is required to disentangle its role in LSC pathogenesis.
While the combination of venetoclax and azacitidine has proven to be effective in older de novo AML patients, their independent and combined mechanism of targeting LSCs remains unclear. Further, ~30% of AML patients have LSCs that do not respond to this regimen, so future studies will be critical to address this shortcoming.
The metabolism of R/R LSCs is unique, but their intricacies are only just beginning to be examined. The contributions of increased metabolic flexibility via fatty acid metabolism to therapeutic resistance as well as the role of aberrant lipid metabolism in LSCs requires additional investigation.
Aging, inflammation, and their role in the development and pathogenesis of AML have also been at the forefront of leukemia biology. However, the role of inflammatory cytokines in AML is not well understood. The immune response is closely linked to metabolism, so pro-inflammatory cytokines may have a significant impact on metabolic aberrations in AML.
Highlights.
Despite advances in our understanding of leukemia biology, rates of relapse and poor outcome for AML patients remain high as conventional treatments do not eliminate disease-initiating LSCs.
LSCs display unique metabolic features: they are resistant to traditional chemotherapeutics, employ a variety of molecular and metabolic mechanisms to maintain a ROS-Low state, and rely on OXPHOS for the production of high-energy compounds.
Successful treatment of AML requires the elimination of LSCs, and the recent successful therapies directly target de novo LSC metabolism.
R/R LSCs remain therapeutically resistant by utilizing alternative metabolic pathways to fuel OXPHOS, but recent advancements suggest targeting these pathways may provide novel therapies and improve patient outcome.
Acknowledgements
R.C.H. is supported by National Cancer Institute Fellowship (CA250361–01).
A.D. is supported by funds from the National Institute of General and Medical Sciences (RM1GM131968) and from the National Heart, Lung, and Blood Institute (R01HL146442, R01HL149714, R01HL148151, R21HL150032).
E.M.P. is supported by R01 DK119394 and the Cleo Meador and George Ryland Scott Endowed Chair in Hematology. E.M.P. and A.D. were also supported by Golfers Against Cancer.
Glossary
- B-cell lymphoma 2 (BCL-2)
An anti-apoptotic protein commonly upregulated in AML. BCL-2 localizes to the outer membrane of the mitochondria and acts to inhibit mitochondrial membrane permeabilization and release of cytochrome C and reactive oxygen species (ROS)
- de novo
Describes a patient or cells from a patient who has been diagnosed with AML for the first time and has not yet undergone chemotherapy
- Glycolysis
The process through which ATP is formed by the conversion of glucose into pyruvate, occurring in the cytosol
- Hematopoiesis
The multistep, highly regulated process by which the cellular components of blood are formed
- Leukemic blasts
Highly proliferative, immature myeloid blood cells that are unable to differentiate and function normally. Produced by leukemic stem cells (LSCs)
- Leukemic stem cells (LSCs)
A cell originating from a normal stem or progenitor cell after undergoing leukemic transformation. Like a normal stem cell, LSCs are quiescent and have the capacity for self-renewal but produce leukemic blasts and are resistant to traditional chemotherapies
- Mitophagy
The selective degradation of mitochondria by autophagy, the highly regulated cellular mechanism allowing removal of dysfunctional or unnecessary cellular components. It promotes mitochondrial turnover and prevents accumulation of dysfunctional or overactive mitochondria, limiting oxidative stress
- Oxidative Phosphorylation (OXPHOS)
The process through which ATP is formed by the transfer of electrons through the electron transport chain (ETC), located in the inner membrane of the mitochondria. It is fueled by the TCA cycle
- Quiescence
A reversible cellular state in which the cell does not proliferate but can be “activated” to become proliferative. This state allows resistance to traditional chemotherapies targeting actively proliferating cells
- Reactive Oxygen Species (ROS)
Chemically reactive molecules including peroxides, superoxide, hydroxyl radicals, etc. that are the normal metabolic byproducts. High levels can result in cellular damage
- Relapsed/Refractory (R/R)
Describes a patient or cells from a patient who has either been previously diagnosed with AML and has experienced disease recurrence or is resistant to treatment upon first diagnosis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Prada-Arismendy J, Arroyave JC, and Röthlisberger S, “Molecular biomarkers in acute myeloid leukemia,” Blood Rev, vol. 31, no. 1, pp. 63–76, January. 2017, doi: 10.1016/j.blre.2016.08.005. [DOI] [PubMed] [Google Scholar]
- [2].Yi M, Li A, Zhou L, Chu Q, Song Y, and Wu K, “The global burden and attributable risk factor analysis of acute myeloid leukemia in 195 countries and territories from 1990 to 2017: estimates based on the global burden of disease study 2017,” J. Hematol. Oncol.J Hematol Oncol, vol. 13, no. 1, p. 72, June. 2020, doi: 10.1186/s13045-020-00908-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Papaemmanuil E et al. , “Genomic Classification and Prognosis in Acute Myeloid Leukemia,” N. Engl. J. Med, vol. 374, no. 23, pp. 2209–2221, June. 2016, doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gupta M, Mahapatra M, and Saxena R, “Cytogenetics’ impact on the prognosis of acute myeloid leukemia,” J. Lab. Physicians, vol. 11, no. 2, pp. 133–137, 2019, doi: 10.4103/JLP.JLP_164_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Saultz JN and Garzon R, “Acute Myeloid Leukemia: A Concise Review,” J. Clin. Med, vol. 5, no. 3, March. 2016, doi: 10.3390/jcm5030033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lowenberg B, van Putten W, Touw IP, Delwel R, and Santini V, “Autonomous Proliferation of Leukemic Cells in Vitro as a Determinant of Prognosis in Adult Acute Myeloid Leukemia,” N. Engl. J. Med, vol. 328, no. 9, pp. 614–619, March. 1993, doi: 10.1056/NEJM199303043280904. [DOI] [PubMed] [Google Scholar]
- [7].Pollyea DA and Jordan CT, “Why are hypomethylating agents or low-dose cytarabine and venetoclax so effective?,” Curr. Opin. Hematol, vol. 26, no. 2, pp. 71–76, March. 2019, doi: 10.1097/MOH.0000000000000485. [DOI] [PubMed] [Google Scholar]
- [8].Zhang Y, Gao S, Xia J, and Liu F, “Hematopoietic Hierarchy – An Updated Roadmap,” Trends Cell Biol, vol. 28, no. 12, pp. 976–986, December. 2018, doi: 10.1016/j.tcb.2018.06.001. [DOI] [PubMed] [Google Scholar]
- [9].Thomas D and Majeti R, “Biology and relevance of human acute myeloid leukemia stem cells,” Blood, vol. 129, no. 12, pp. 1577–1585, March. 2017, doi: 10.1182/blood-2016-10-696054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].PDQ Adult Treatment Editorial Board, “Adult Acute Myeloid Leukemia Treatment (PDQ®): Patient Version,” in PDQ Cancer Information Summaries, Bethesda (MD): National Cancer Institute (US), 2002. [PubMed] [Google Scholar]
- [11].Zhang TY, Dutta R, Benard B, Zhao F, Yin R, and Majeti R, “IL-6 blockade reverses bone marrow failure induced by human acute myeloid leukemia,” Sci. Transl. Med, vol. 12, no. 538, April. 2020, doi: 10.1126/scitranslmed.aax5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Khwaja A et al. , “Acute myeloid leukaemia,” Nat. Rev. Dis. Primer, vol. 2, no. 1, Art. no. 1, March. 2016, doi: 10.1038/nrdp.2016.10. [DOI] [PubMed] [Google Scholar]
- [13].Dombret H and Gardin C, “An update of current treatments for adult acute myeloid leukemia,” Blood, vol. 127, no. 1, pp. 53–61, January. 2016, doi: 10.1182/blood-2015-08-604520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Heuser M et al. , “Acute myeloid leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up††Approved by the ESMO Guidelines Committee: August 2002, last update January 2020. This publication supersedes the previously published version—Ann Oncol. 2013;24(suppl 6):vi138–vi143.,” Ann. Oncol, vol. 31, no. 6, pp. 697–712, June. 2020, doi: 10.1016/j.annonc.2020.02.018. [DOI] [PubMed] [Google Scholar]
- [15].De Kouchkovsky I and Abdul-Hay M, “‘Acute myeloid leukemia: a comprehensive review and 2016 update,’” Blood Cancer J, vol. 6, no. 7, p. e441, 01 2016, doi: 10.1038/bcj.2016.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jones CL et al. , “Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells,” Cancer Cell, vol. 34, no. 5, pp. 724–740.e4, November. 2018, doi: 10.1016/j.ccell.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ferrara F, Lessi F, Vitagliano O, Birkenghi E, and Rossi G, “Current Therapeutic Results and Treatment Options for Older Patients with Relapsed Acute Myeloid Leukemia,” Cancers, vol. 11, no. 2, February. 2019, doi: 10.3390/cancers11020224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jordan CT, “Can we selectively target AML stem cells?,” Best Pract. Res. Clin. Haematol, vol. 32, no. 4, p. 101100, December. 2019, doi: 10.1016/j.beha.2019.101100. [DOI] [PubMed] [Google Scholar]
- [19].Kobayashi CI and Suda T, “Regulation of reactive oxygen species in stem cells and cancer stem cells,” J. Cell. Physiol, vol. 227, no. 2, pp. 421–430, February. 2012, doi: 10.1002/jcp.22764. [DOI] [PubMed] [Google Scholar]
- [20].Wierenga ATJ et al. , “HIF1/2-exerted control over glycolytic gene expression is not functionally relevant for glycolysis in human leukemic stem/progenitor cells,” Cancer Metab, vol. 7, p. 11, 2019, doi: 10.1186/s40170-019-0206-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Spencer JA et al. , “Direct measurement of local oxygen concentration in the bone marrow of live animals,” Nature, vol. 508, no. 7495, pp. 269–273, April. 2014, doi: 10.1038/nature13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kirito K, Fox N, Komatsu N, and Kaushansky K, “Thrombopoietin enhances expression of vascular endothelial growth factor (VEGF) in primitive hematopoietic cells through induction of HIF-1α,” Blood, vol. 105, no. 11, pp. 4258–4263, June. 2005, doi: 10.1182/blood-2004-07-2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pedersen M, Löfstedt T, Sun J, Holmquist-Mengelbier L, Påhlman S, and Rönnstrand L, “Stem cell factor induces HIF-1α at normoxia in hematopoietic cells,” Biochem. Biophys. Res. Commun, vol. 377, no. 1, pp. 98–103, December. 2008, doi: 10.1016/j.bbrc.2008.09.102. [DOI] [PubMed] [Google Scholar]
- [24].Dengler VL, Galbraith M, and Espinosa JM, “Transcriptional Regulation by Hypoxia Inducible Factors,” Crit. Rev. Biochem. Mol. Biol, vol. 49, no. 1, pp. 1–15, 2014, doi: 10.3109/10409238.2013.838205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang Y, Liu Y, Malek SN, Zheng P, and Liu Y, “Targeting HIF1α eliminates cancer stem cells in hematological malignancies,” Cell Stem Cell, vol. 8, no. 4, pp. 399–411, April. 2011, doi: 10.1016/j.stem.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Velasco-Hernandez T, Soneji S, Hidalgo I, Erlandsson E, Cammenga J, and Bryder D, “Hif-1α Deletion May Lead to Adverse Treatment Effect in a Mouse Model of MLL-AF9-Driven AML,” Stem Cell Rep, vol. 12, no. 1, pp. 112–121, December. 2018, doi: 10.1016/j.stemcr.2018.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Deynoux M, Sunter N, Hérault O, and Mazurier F, “Hypoxia and Hypoxia-Inducible Factors in Leukemias,” Front. Oncol, vol. 6, 2016, doi: 10.3389/fonc.2016.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chiavarina B et al. , “HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis,” Cell Cycle Georget. Tex, vol. 9, no. 17, pp. 3534–3551, September. 2010, doi: 10.4161/cc.9.17.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tothova Z and Gilliland DG, “FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system,” Cell Stem Cell, vol. 1, no. 2, pp. 140–152, August. 2007, doi: 10.1016/j.stem.2007.07.017. [DOI] [PubMed] [Google Scholar]
- [30].Ferber EC, Peck B, Delpuech O, Bell GP, East P, and Schulze A, “FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression,” Cell Death Differ, vol. 19, no. 6, pp. 968–979, June. 2012, doi: 10.1038/cdd.2011.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Warr MR et al. , “FoxO3a Directs a Protective Autophagy Program in Hematopoietic Stem Cells,” Nature, vol. 494, no. 7437, pp. 323–327, February. 2013, doi: 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sykes SM et al. , “AKT/FOXO Signaling Enforces Reversible Differentiation Blockade in Myeloid Leukemias,” Cell, vol. 146, no. 5, September. 2011, doi: 10.1016/j.cell.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Niemitz E, “A surprising role for FOXO,” Nat. Genet, vol. 43, no. 10, Art. no. 10, October. 2011, doi: 10.1038/ng.968. [DOI] [Google Scholar]
- [34].Wang Y, Zhou Y, and Graves DT, “FOXO transcription factors: their clinical significance and regulation,” BioMed Res. Int, vol. 2014, p. 925350, 2014, doi: 10.1155/2014/925350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Matés JM, Pérez-Gómez C, Núñez de Castro I, Asenjo M, and Márquez J, “Glutamine and its relationship with intracellular redox status, oxidative stress and cell proliferation/death,” Int. J. Biochem. Cell Biol, vol. 34, no. 5, pp. 439–458, May 2002, doi: 10.1016/s1357-2725(01)00143-1. [DOI] [PubMed] [Google Scholar]
- [36].Pollyea DA et al. , “Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia,” Nat. Med, vol. 24, no. 12, pp. 1859–1866, 2018, doi: 10.1038/s41591-018-0233-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lin S, Zhang J, and Mulloy JC, “Tumor Suppressor FOXO1 Serves As a Critical Oncogenic Mediator in AML1-ETO Leukemia,” Blood, vol. 124, no. 21, pp. 264–264, December. 2014, doi: 10.1182/blood.V124.21.264.264. [DOI] [Google Scholar]
- [38].Yuan C, Wang L, Zhou L, and Fu Z, “The function of FOXO1 in the late phases of the cell cycle is suppressed by PLK1-mediated phosphorylation,” Cell Cycle, vol. 13, no. 5, pp. 807–819, March. 2014, doi: 10.4161/cc.27727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cilloni D et al. , “FoxO Transcription Factor Is Delocalized and Inactivated in Acute Myeloid Leukaemia Patients.,” Blood, vol. 110, no. 11, pp. 1251–1251, November. 2007, doi: 10.1182/blood.V110.11.1251.1251.17452517 [DOI] [Google Scholar]
- [40].Kornblau SM, Singh N, Qiu Y, Chen W, Zhang N, and Coombes KR, “Highly Phosphorylated FOXO3A Is an Adverse Prognostic Factor in Acute Myeloid Leukemia,” Clin. Cancer Res, vol. 16, no. 6, pp. 1865–1874, March. 2010, doi: 10.1158/1078-0432.CCR-09-2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu J et al. , “Bmi1 regulates mitochondrial function and the DNA damage response pathway,” Nature, vol. 459, no. 7245, Art. no. 7245, May 2009, doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yuan J, Takeuchi M, Negishi M, Oguro H, Ichikawa H, and Iwama A, “Bmi1 is essential for leukemic reprogramming of myeloid progenitor cells,” Leukemia, vol. 25, no. 8, Art. no. 8, August. 2011, doi: 10.1038/leu.2011.85. [DOI] [PubMed] [Google Scholar]
- [43].Sahasrabuddhe AA, “BMI1: A Biomarker of Hematologic Malignancies,” Biomark. Cancer, vol. 8, pp. 65–75, May 2016, doi: 10.4137/BIC.S33376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Joshi A and Kundu M, “Mitophagy in hematopoietic stem cells,” Autophagy, vol. 9, no. 11, pp. 1737–1749, November. 2013, doi: 10.4161/auto.26681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ito K, Bonora M, and Ito K, “Metabolism as master of hematopoietic stem cell fate,” Int. J. Hematol, vol. 109, no. 1, pp. 18–27, January. 2019, doi: 10.1007/s12185-018-2534-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ianniciello A, Rattigan KM, and Helgason GV, “The Ins and Outs of Autophagy and Metabolism in Hematopoietic and Leukemic Stem Cells: Food for Thought,” Front. Cell Dev. Biol, vol. 6, September. 2018, doi: 10.3389/fcell.2018.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mattes K, Vellenga E, and Schepers H, “Differential redox-regulation and mitochondrial dynamics in normal and leukemic hematopoietic stem cells: A potential window for leukemia therapy,” Crit. Rev. Oncol. Hematol, vol. 144, p. 102814, December. 2019, doi: 10.1016/j.critrevonc.2019.102814. [DOI] [PubMed] [Google Scholar]
- [48].de Almeida MJ, Luchsinger LL, Corrigan DJ, Williams LJ, and Snoeck H-W, “Dye-Independent Methods Reveal Elevated Mitochondrial Mass in Hematopoietic Stem Cells,” Cell Stem Cell, vol. 21, no. 6, pp. 725–729.e4, December. 2017, doi: 10.1016/j.stem.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hajnóczky G et al. , “Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis,” Cell Calcium, vol. 40, no. 5–6, pp. 553–560, 2006, doi: 10.1016/j.ceca.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nelson D and Cox M, Eds., Lehninger Principes of Biochemistry, 6th ed. .
- [51].Li X, Fang P, Mai J, Choi ET, Wang H, and Yang X, “Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers,” J. Hematol. Oncol.J Hematol Oncol, vol. 6, p. 19, February. 2013, doi: 10.1186/1756-8722-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fay HRS et al. , “Mitophagy Plays a Key Role in the Anti-Leukemic Activity of Autophagy Inhibitors Under Hypoxia in Acute Myeloid Leukemia,” Blood, vol. 134, no. Supplement_1, pp. 1278–1278, November. 2019, doi: 10.1182/blood-2019-127024. [DOI] [Google Scholar]
- [53].“Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy | Nature Communications” https://www.nature.com/articles/s41467-017-00520-9 (accessed Aug. 06, 2020). [DOI] [PMC free article] [PubMed]
- [54].Pei S et al. , “AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells,” Cell Stem Cell, vol. 23, no. 1, pp. 86–100.e6, July. 2018, doi: 10.1016/j.stem.2018.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Nazio F, Bordi M, Cianfanelli V, Locatelli F, and Cecconi F, “Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications,” Cell Death Differ, vol. 26, no. 4, Art. no. 4, April. 2019, doi: 10.1038/s41418-019-0292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Vara-Perez M, Felipe-Abrio B, and Agostinis P, “Mitophagy in Cancer: A Tale of Adaptation,” Cells, vol. 8, no. 5, May 2019, doi: 10.3390/cells8050493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ito K et al. , “A PML-PPARδ pathway for fatty acid oxidation regulates haematopoietic stem cell maintenance,” Nat. Med, vol. 18, no. 9, pp. 1350–1358, September. 2012, doi: 10.1038/nm.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ito K et al. , “Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance,” Science, vol. 354, no. 6316, pp. 1156–1160, December. 2016, doi: 10.1126/science.aaf5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Snoeck H-W, “Mitochondrial regulation of hematopoietic stem cells,” Curr. Opin. Cell Biol, vol. 49, pp. 91–98, December. 2017, doi: 10.1016/j.ceb.2017.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lagadinou ED et al. , “BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells,” Cell Stem Cell, vol. 12, no. 3, pp. 329–341, March. 2013, doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Feng W et al. , “Targeting unique metabolic properties of breast tumor initiating cells,” Stem Cells Dayt. Ohio, vol. 32, no. 7, pp. 1734–1745, July. 2014, doi: 10.1002/stem.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].van Galen P et al. , “Integrated Stress Response Activity Marks Stem Cells in Normal Hematopoiesis and Leukemia,” Cell Rep, vol. 25, no. 5, pp. 1109–1117.e5, October. 2018, doi: 10.1016/j.celrep.2018.10.021. [DOI] [PubMed] [Google Scholar]
- [63].Sharon D et al. , “Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response,” Sci. Transl. Med, vol. 11, no. 516, 30 2019, doi: 10.1126/scitranslmed.aax2863. [DOI] [PubMed] [Google Scholar]
- [64].Jones CL et al. , “Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II,” Blood, vol. 134, no. 4, pp. 389–394, 25 2019, doi: 10.1182/blood.2019898114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jhas B et al. , “Metabolic Adaptation to Chronic Inhibition of Mitochondrial Protein Synthesis in Acute Myeloid Leukemia Cells,” PLoS ONE, vol. 8, no. 3, March. 2013, doi: 10.1371/journal.pone.0058367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chen Q et al. , “Inhibition of Bcl-2 Sensitizes Mitochondrial Permeability Transition Pore (MPTP) Opening in Ischemia-Damaged Mitochondria,” PLoS ONE, vol. 10, no. 3, March. 2015, doi: 10.1371/journal.pone.0118834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].DiNardo CD et al. , “Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia,” N. Engl. J. Med, vol. 383, no. 7, pp. 617–629, August. 2020, doi: 10.1056/NEJMoa2012971. [DOI] [PubMed] [Google Scholar]
- [68].Chan SM et al. , “Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia,” Nat. Med, vol. 21, no. 2, Art. no. 2, February. 2015, doi: 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kohl TM et al. , “BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts,” Leukemia, vol. 21, no. 8, Art. no. 8, August. 2007, doi: 10.1038/sj.leu.2404776. [DOI] [PubMed] [Google Scholar]
- [70].DiNardo CD et al. , “Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies,” Am. J. Hematol, vol. 93, no. 3, pp. 401–407, 2018, doi: 10.1002/ajh.25000. [DOI] [PubMed] [Google Scholar]
- [71].Jones CL et al. , “Inhibition of Fatty Acid Metabolism Re-Sensitizes Resistant Leukemia Stem Cells to Venetoclax with Azacitidine,” Blood, vol. 134, no. Supplement_1, pp. 1272–1272, November. 2019, doi: 10.1182/blood-2019-125773. [DOI] [Google Scholar]
- [72].Pei S et al. , “Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia,” Cancer Discov, vol. 10, no. 4, pp. 536–551, April. 2020, doi: 10.1158/2159-8290.CD-19-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Pei S et al. , “Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells,” J. Biol. Chem, vol. 288, no. 47, pp. 33542–33558, November. 2013, doi: 10.1074/jbc.M113.511170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Cramer SL et al. , “Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth,” Nat. Med, vol. 23, no. 1, pp. 120–127, 2017, doi: 10.1038/nm.4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhang T et al. , “Polyamine pathway activity promotes cysteine essentiality in cancer cells,” Nat. Metab, pp. 1–15, August. 2020, doi: 10.1038/s42255-020-0253-2. [DOI] [PMC free article] [PubMed]
- [76].Taya Y et al. , “Depleting dietary valine permits nonmyeloablative mouse hematopoietic stem cell transplantation,” Science, vol. 354, no. 6316, pp. 1152–1155, December. 2016, doi: 10.1126/science.aag3145. [DOI] [PubMed] [Google Scholar]
- [77].Wilkinson AC, Morita M, Nakauchia H, and Yamazaki S, “Branched-chain amino acid depletion conditions bone marrow for hematopoietic stem cell transplantation avoiding amino acid imbalance-associated toxicity,” Exp. Hematol, vol. 63, pp. 12–16.e1, July. 2018, doi: 10.1016/j.exphem.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kreitz J et al. , “Metabolic Plasticity of Acute Myeloid Leukemia,” Cells, vol. 8, no. 8, July. 2019, doi: 10.3390/cells8080805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Raffel S et al. , “BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation,” Nature, vol. 551, no. 7680, pp. 384–388, 16 2017, doi: 10.1038/nature24294. [DOI] [PubMed] [Google Scholar]
- [80].Tönjes M et al. , “BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1,” Nat. Med, vol. 19, no. 7, pp. 901–908, July. 2013, doi: 10.1038/nm.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gu Z et al. , “Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation,” Cancer Discov, vol. 9, no. 9, pp. 1228–1247, 2019, doi: 10.1158/2159-8290.CD-19-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Son SM et al. , “Leucine Signals to mTORC1 via Its Metabolite Acetyl-Coenzyme A,” Cell Metab, vol. 29, no. 1, pp. 192–201.e7, January. 2019, doi: 10.1016/j.cmet.2018.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hattori A et al. , “Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia,” Nature, vol. 545, no. 7655, pp. 500–504, 25 2017, doi: 10.1038/nature22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Oburoglu L, Romano M, Taylor N, and Kinet S, “Metabolic regulation of hematopoietic stem cell commitment and erythroid differentiation,” Curr. Opin. Hematol, vol. 23, no. 3, pp. 198–205, May 2016, doi: 10.1097/MOH.0000000000000234. [DOI] [PubMed] [Google Scholar]
- [85].Samudio I et al. , “Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction,” J. Clin. Invest, vol. 120, no. 1, pp. 142–156, January. 2010, doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Castelli G, Pelosi E, and Testa U, “Emerging Therapies for Acute Myelogenus Leukemia Patients Targeting Apoptosis and Mitochondrial Metabolism,” Cancers, vol. 11, no. 2, February. 2019, doi: 10.3390/cancers11020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Farge T et al. , “Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism,” Cancer Discov, vol. 7, no. 7, pp. 716–735, July. 2017, doi: 10.1158/2159-8290.CD-16-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Bosc C et al. , “Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites,” Nat. Commun, vol. 11, no. 1, Art. no. 1, August. 2020, doi: 10.1038/s41467-020-17882-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Stevens BM et al. , “Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells,” Nat. Cancer [DOI] [PMC free article] [PubMed]
- [90].Tabe Y et al. , “Inhibition of FAO in AML co-cultured with BM adipocytes: mechanisms of survival and chemosensitization to cytarabine,” Sci. Rep, vol. 8, no. 1, p. 16837, November. 2018, doi: 10.1038/s41598-018-35198-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Vriens K et al. , “Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity,” Nature, vol. 566, no. 7744, pp. 403–406, 2019, doi: 10.1038/s41586-019-0904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Yi M et al. , “Emerging role of lipid metabolism alterations in Cancer stem cells,” J. Exp. Clin. Cancer Res. CR, vol. 37, no. 1, p. 118, June. 2018, doi: 10.1186/s13046-018-0784-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Moon S-H et al. , “p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression,” Cell, vol. 176, no. 3, pp. 564–580.e19, January. 2019, doi: 10.1016/j.cell.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Mukherjee A, Kenny HA, and Lengyel E, “Unsaturated fatty acids maintain cancer cell stemness,” Cell Stem Cell, vol. 20, no. 3, pp. 291–292, March. 2017, doi: 10.1016/j.stem.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Howlader N et al. , “SEER Cancer Statistics Review, 1975–2017,” Natl. Cancer Inst, November. 2019.
- [96].Luskin MR et al. , “Benefit of high-dose daunorubicin in AML induction extends across cytogenetic and molecular groups,” Blood, vol. 127, no. 12, pp. 1551–1558, March. 2016, doi: 10.1182/blood-2015-07-657403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Lancet JE et al. , “CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia,” J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol, vol. 36, no. 26, pp. 2684–2692, 10 2018, doi: 10.1200/JCO.2017.77.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Carney DA et al. , “Therapy-related myelodysplastic syndrome and acute myeloid leukemia following fludarabine combination chemotherapy,” Leukemia, vol. 24, no. 12, Art. no. 12, December. 2010, doi: 10.1038/leu.2010.218. [DOI] [PubMed] [Google Scholar]
- [99].Baer M et al. , “Escalation of daunorubicin and addition of etoposide in the ADE regimen in acute myeloid leukemia patients aged 60 years and older: Cancer and Leukemia Group B Study 9720,” Leukemia, vol. 25, no. 5, May 2011, doi: 10.1038/leu.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Shin S-H et al. , “Comparison of the modified low-dose cytarabine and etoposide with decitabine therapy for elderly acute myeloid leukemia patients unfit for intensive chemotherapy,” Oncotarget, vol. 9, no. 5, pp. 5823–5833, December. 2017, doi: 10.18632/oncotarget.23629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].T R et al. , “Oral induction and consolidation of acute myeloid leukemia with etoposide, 6-thioguanine, and idarubicin (ETI) in elderly patients: a randomized comparison with 5-day TAD. Finnish Leukemia Group.,” Leukemia, vol. 8, no. 1, pp. 11–15, January. 1994. [PubMed] [Google Scholar]
- [102].Ma E et al. , “An Evaluation of Treatment Patterns and Outcomes in Elderly Patients Newly Diagnosed With Acute Myeloid Leukemia: A Retrospective Analysis of Electronic Medical Records From US Community Oncology Practices,” Clin. Lymphoma Myeloma Leuk, vol. 16, no. 11, pp. 625–636.e3, November. 2016, doi: 10.1016/j.clml.2016.08.006. [DOI] [PubMed] [Google Scholar]
- [103].Pleyer L et al. , “Azacitidine for Front-Line Therapy of Patients with AML: Reproducible Efficacy Established by Direct Comparison of International Phase 3 Trial Data with Registry Data from the Austrian Azacitidine Registry of the AGMT Study Group,” Int. J. Mol. Sci, vol. 18, no. 2, February. 2017, doi: 10.3390/ijms18020415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].He P-F et al. , “Efficacy and safety of decitabine in treatment of elderly patients with acute myeloid leukemia: A systematic review and meta-analysis,” Oncotarget, vol. 8, no. 25, pp. 41498–41507, April. 2017, doi: 10.18632/oncotarget.17241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Starr P, “Midostaurin the First Targeted Therapy to Improve Survival in AML: Potentially Practice-Changing,” Am. Health Drug Benefits, vol. 9, no. Spec Issue, pp. 1–21, February. 2016. [PMC free article] [PubMed] [Google Scholar]
- [106].Perl AE et al. , “Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML,” N. Engl. J. Med, vol. 381, no. 18, pp. 1728–1740, 31 2019, doi: 10.1056/NEJMoa1902688. [DOI] [PubMed] [Google Scholar]
- [107].Roboz GJ et al. , “Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia,” Blood, vol. 135, no. 7, pp. 463–471, 13 2020, doi: 10.1182/blood.2019002140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].McCafferty EH and Lyseng-Williamson KA, “Ivosidenib in relapsed or refractory acute myeloid leukemia: a profile of its use in the USA,” Drugs Ther. Perspect, vol. 35, no. 4, pp. 160–166, April. 2019, doi: 10.1007/s40267-019-00610-2. [DOI] [Google Scholar]
- [109].Stein EM et al. , “Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib,” Blood, vol. 133, no. 7, pp. 676–687, 14 2019, doi: 10.1182/blood-2018-08-869008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Jen EY et al. , “FDA Approval: Gemtuzumab Ozogamicin for the Treatment of Adults with Newly Diagnosed CD33-Positive Acute Myeloid Leukemia,” Clin. Cancer Res, vol. 24, no. 14, pp. 3242–3246, July. 2018, doi: 10.1158/1078-0432.CCR-17-3179. [DOI] [PubMed] [Google Scholar]
- [111].DiNardo CD et al. , “Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia,” Blood, vol. 133, no. 1, pp. 7–17, January. 2019, doi: 10.1182/blood-2018-08-868752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Cortes JE et al. , “Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome,” Leukemia, vol. 33, no. 2, pp. 379–389, 2019, doi: 10.1038/s41375-018-0312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Di Liddo R et al. , “Adrenomedullin in the growth modulation and differentiation of acute myeloid leukemia cells,” Int. J. Oncol, vol. 48, no. 4, pp. 1659–1669, April. 2016, doi: 10.3892/ijo.2016.3370. [DOI] [PubMed] [Google Scholar]
- [114].Kampen KR, Ter Elst A, and de Bont ESJM, “Vascular endothelial growth factor signaling in acute myeloid leukemia,” Cell. Mol. Life Sci. CMLS, vol. 70, no. 8, pp. 1307–1317, April. 2013, doi: 10.1007/s00018-012-1085-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Xu D et al. , “The IGF2/IGF1R/Nanog Signaling Pathway Regulates the Proliferation of Acute Myeloid Leukemia Stem Cells,” Front. Pharmacol, vol. 9, June. 2018, doi: 10.3389/fphar.2018.00687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Jin S et al. , “5-Azacitidine Induces NOXA to Prime AML Cells for Venetoclax-mediated Apoptosis.,” Clin. Cancer Res, January. 2020, doi: 10.1158/1078-0432.CCR-19-1900. [DOI] [PubMed]
- [117].Kadia TM, Kantarjian HM, and Konopleva M, “Myeloid cell leukemia-1 dependence in acute myeloid leukemia: a novel approach to patient therapy,” Oncotarget, vol. 10, no. 12, pp. 1250–1265, February. 2019, doi: 10.18632/oncotarget.26579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Zhou J-D et al. , “Down-regulation of GPX3 is associated with favorable/intermediate karyotypes in de novo acute myeloid leukemia,” Int. J. Clin. Exp. Pathol, vol. 8, no. 3, pp. 2384–2391, March. 2015. [PMC free article] [PubMed] [Google Scholar]
- [119].Lin X et al. , “Heme oxygenase-1 suppresses the apoptosis of acute myeloid leukemia cells via the JNK/c-JUN signaling pathway,” Leuk. Res, vol. 39, no. 5, pp. 544–552, May 2015, doi: 10.1016/j.leukres.2015.02.009. [DOI] [PubMed] [Google Scholar]